

Abstract
We describe outcomes from the first‐in‐human study of garetosmab (a fully human monoclonal antibody that inhibits activin A) under development for the treatment of fibrodysplasia ossificans progressiva (FOP). In a double‐blind, placebo‐controlled phase 1 study, 40 healthy women of nonchildbearing potential were randomized to receive a single dose of intravenous garetosmab 0.3, 1, 3, or 10 mg/kg; subcutaneous garetosmab 300 mg; or placebo. Serum concentrations of functional garetosmab (with ≥1 arm free to bind to target), total activin A, and antidrug antibodies were measured predose and up to 113 days post–first dose. Garetosmab demonstrated an acceptable safety profile with no dose‐limiting toxicities. Garetosmab displayed nonlinear pharmacokinetics with target‐mediated elimination. With increasing doses of intravenous garetosmab, mean peak concentration increased in a dose‐proportional manner; mean steady‐state estimates ranged from 41.4 to 47.8 mL/kg. A greater than dose‐proportional increase in mean area under the concentration‐time curve from time zero extrapolated to infinity (range, 72.2‐7520 mg*day/L) was observed, consistent with decreasing mean clearance (range, 4.35‐1.34 mL/day/kg). Following administration of intravenous garetosmab, mean concentrations of total activin A increased in a dose‐dependent manner. At 10 mg/kg, total activin A levels reached a state of little or no change between weeks 4 and 12, suggesting saturation of the target‐mediated pathway. No safety signals were seen in this study to preclude investigation in patients. Following intravenous administration, garetosmab concentrations decreased quickly, then decreased over time (reflecting linear elimination), and finally decreased in a nonlinear phase, reflecting target‐mediated elimination. Results here support further investigation. Garetosmab 10 mg/kg every 4 weeks intravenously is being evaluated in patients with FOP (NCT03188666).
Keywords: clinical trial, monoclonal antibody, activin A, fibrodysplasia ossificans progressiva, garetosmab
Fibrodysplasia ossificans progressiva (FOP), a rare and severely disabling life‐threatening disease that affects approximately 800 people worldwide, is associated with progressive multifocal heterotopic bone formation of skeletal muscle, ligaments, tendons, and fascia. 1 , 2 FOP is characterized by cumulative heterotopic ossification, which leads to significant physical disability (commonly by the second decade of life), respiratory insufficiency, and death. 1 The median age of survival for patients with FOP is 40 years. 2 No effective treatment currently exists. 1 , 2
FOP is caused by mutations in the intracellular domain of the activin A type I receptor, ACVR1 (also known as activin receptor‐like kinase 2). 3 The mutations confer the receptor with the abnormal ability to recognize activin A as an agonistic ligand, inducing signaling through the SMAD 1/5/8 pathway and resulting in heterotopic bone formation. 4 Garetosmab (REGN2477) is a fully human monoclonal antibody (containing a human immunoglobulin G4 constant region) that specifically binds to and blocks signaling of activin proteins containing the inhibin βα subunit (activins A, AB, and AC). In vitro, garetosmab has high affinity and specificity for activins A, AB, and AC, 5 but does not bind or functionally inhibit other transforming growth factor‐β family ligands that share the same activin receptor pathways, including activin B, inhibin A, bone morphogenetic protein (BMP)2, BMP6, BMP9, BMP10, growth differentiation factor (GDF) 8, and GDF11.
In a humanized animal model of FOP conditionally expressing the most common human ACVR1 mutation (R206H) that recapitulates heterotopic ossification, garetosmab‐mediated inhibition of activin A prevented the onset of heterotopic ossification and halted the progression of preexisting heterotopic ossification. 4
Here, we report data from a randomized, double‐blind, placebo‐controlled, first‐in‐human phase 1 study that was undertaken to evaluate the safety, tolerability, pharmacokinetics, and immunogenicity of intravenous and subcutaneous formulations of garetosmab administered to healthy women of nonchildbearing potential.
Methods
Study Design
This study was undertaken at SGS Clinical Pharmacology Unit (Antwerp, Belgium) between June 2016 and January 2017 in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. Approval was obtained from the site's Independent Ethics Committee. Written informed consent was obtained from all volunteers prior to participation in the trial.
In this double‐blind, placebo‐controlled phase 1 study (NCT02870400), healthy women of nonchildbearing potential were randomized to receive a single intravenous or subcutaneous dose of garetosmab or placebo. Eligible participants were aged 18‐65 years with a body mass index (BMI) ≥18 to ≤30 kg/m2. The age range of 18‐65 years is standard for first‐in‐human studies in healthy subjects of this type, as people aged older than 65 years are more likely to have background medical conditions that may confound assessment of drug safety. Exclusion criteria included clinically significant abnormal complete blood count, clinical chemistry, or urinalysis; positive urine drug test results during screening; history of drug or alcohol abuse in the year prior to screening; history of diabetes; abnormal blood pressure (diastolic blood pressure > 90 mm Hg and/or systolic blood pressure > 140 mm Hg or orthostatic hypotension); reduced renal function (estimated glomerular filtration rate < 70 mL/min/1.73 m2); history of osteoporosis requiring treatment; and use of hormone or thyroid replacement therapy (unless treatment doses had been stable for ≥6 months prior to screening and were expected to remain unchanged for the study duration).
Women were randomized (6:2) through an interactive voice response system to garetosmab or placebo. Five dose cohorts were planned (n = 8 for each): cohort 1, intravenous garetosmab 0.3 mg/kg; cohort 2, intravenous garetosmab 1 mg/kg; cohort 3, intravenous garetosmab 3 mg/kg; cohort 4, intravenous garetosmab 10 mg/kg; and cohort 5, subcutaneous garetosmab 300 mg. Dosing began with cohort 1, which was split into 3 blocks (block 1, n = 2; block 2, n = 2; block 3, n = 4). The 2 subjects assigned to block 1 were dosed first. Dosing of blocks 2 and 3, which was performed on different days, occurred only after the 2 subjects in block 1 were assessed for safety on day 2 and the blinded data reviewed. Cohorts 2‐5 were divided into 2 blocks of 4 women each (garetosmab, n = 3; placebo, n = 1). Dosing of each block was performed on different days. Escalation to cohorts 2‐5 proceeded only once all subjects in the preceding dose cohort had been assessed for safety on day 8, the blinded safety data had been reviewed, and the decision to dose‐escalate had been approved. The starting dose level of 0.3 mg/kg and subsequent escalations up to 10 mg/kg were supported by the results of studies in female rats and monkeys, which showed that garetosmab was well tolerated at intravenous doses up to 50 mg/kg/week when administered for 5 weeks, with an 8‐week recovery period (unpublished data, Regeneron Pharmaceuticals Inc.). Based on equivalent dose conversion for humans, the threshold for no adverse effects in humans was determined to be 50 mg/kg, with the conservative starting dose of 0.3 mg/kg determined to give a large margin of safety. The 300 mg subcutaneous dose was predicted to provide a level of exposure similar to the 3 mg/kg intravenous dose. The subcutaneous dose was included to determine the bioavailability of garetosmab via the subcutaneous route of administration and to assess local tolerability of garetosmab subcutaneous injection.
Doses could be adjusted downwards based on safety, tolerability, and pharmacokinetic data collected during the study. If 1 or more dose‐limiting toxicities in a cohort were observed, dosing would be suspended until a safety review had been conducted. Dose‐limiting toxicity was defined as a treatment‐related severe or serious adverse event (AE) or a severe infusion reaction. If a potentially life‐threatening AE were observed in any cohort, dosing would be suspended, and a comprehensive safety review would be conducted prior to any consideration of further dosing.
Study Objectives
The primary objective of this phase 1 study was to assess the safety and tolerability of single ascending doses of garetosmab. The secondary objectives were to characterize the pharmacokinetic profile (assessed through measurement of functional garetosmab serum concentrations over time) and immunogenicity of garetosmab (determined by the presence of antidrug antibodies [ADAs] to garetosmab over 113 days). Functional garetosmab refers to garetosmab with at least 1 arm free to bind to the target. To explore the relationship between concentrations of functional garetosmab and total activin A, total activin A serum concentrations were also measured.
Study Procedures
Safety assessments included AEs, laboratory data, vital signs, and physical examinations. AEs were coded to the Medical Dictionary for Regulatory Activities version 19.0.
Blood for the assessment of concentrations of functional garetosmab and total activin A in serum was drawn on day 1 at predose (baseline), immediately after the dose, 1, 2, 4, and 8 hours postdose, and on days 2, 4, 8, 15, 22, 29, 43, 57, 85, 99, and 113 (end of study) or at early termination.
The functional garetosmab assay was used because activin A is a soluble target and the pharmacologic effect of garetosmab is determined by the concentration of functional garetosmab. Serum samples were analyzed to quantify “functional garetosmab” using a validated enzyme‐linked immunosorbent assay (ELISA) with an upper limit of quantitation of 5 mg/L and a lower limit of quantitation (LLOQ) of 0.078 mg/L in undiluted human serum. This method was thoroughly tested, with acceptable inter‐ and intra‐assay accuracy, precision, and linearity that were within the preestablished acceptance criteria. Specificity was demonstrated as the assay quantified functional garetosmab (garetosmab with at least 1 arm free to bind target) and does not detect garetosmab fully bound to target activin A. Under the conditions tested, the assay was demonstrated to be selective for the quantitation of functional garetosmab, with no interference from human serum matrix.
Serum concentrations of total activin A were measured using an ELISA, with an LLOQ in neat human serum of 313 ng/L. The assay uses a mouse anti–activin A monoclonal antibody as the capture reagent and recombinant human activin A as the standard. Captured activin A is detected using a biotinylated mouse anti–activin A monoclonal antibody.
Blood for the assessment of the presence of ADAs in serum was drawn on day 1 at baseline and on day 113 (end of study) or on the day of early termination.
The electrochemiluminescence bridging immunoassay used to determine ADAs involves a 3‐tier approach: an initial screen to identify samples that are potentially positive, a confirmation step to determine whether positive responses in the screen assay are specific for garetosmab, and a titer procedure to assess the level of ADA in the confirmed ADA‐positive samples. The sensitivity of the assay in neat serum is 4.0 ng/mL of the monoclonal antibody‐positive control. The drug tolerance limit in neat serum is 969 μg/mL of garetosmab with 250 ng/mL of monoclonal antibody‐positive control. ADA responses were categorized into low (<1000), moderate (≥1000 to ≤10 000), and high (>10 000) titers. The ADA end points in this study are standard for initial evaluation of biological compounds in clinical development.
Statistical and Pharmacokinetic Analysis
No formal hypothesis testing was planned for this first‐in‐human safety study. The number of subjects enrolled in each dose group was deemed sufficient for pharmacokinetic analysis and assessment of any common adverse effects, and was in line with previous studies of this type. No sample size calculation was performed. Approximately 40 women were planned for enrollment.
Dose proportionality was determined for area under the concentration‐time curve (AUC) or peak concentration (Cmax) by AUC/dose or Cmax/dose. There was no statistical test conducted to evaluate the differences among doses.
Descriptive statistics were generated using SAS version 9.2 (Cary, North Carolina). Noncompartmental pharmacokinetic parameters (eg, area under the concentration‐time curve from time zero extrapolated to infinity [AUCinf], clearance [CL], Cmax, time to Cmax, and volume of distribution at steady state [Vss]) were calculated using Phoenix WinNonlin version 6.4 (Certara, LP, Princeton, New Jersey).
Results
Study Participants
A total of 118 women of nonchildbearing potential were screened for study eligibility, and 40 were randomized and treated. Of the 40 treated study participants, 24 received intravenous garetosmab (0.3 mg/kg, n = 6; 1 mg/kg, n = 6; 3 mg/kg, n = 6; 10 mg/kg, n = 6), 6 received subcutaneous garetosmab, and 10 received placebo (intravenous, n = 8; subcutaneous, n = 2). All study participants were white. The mean ± standard deviation (SD) age was 57.1 ± 4.0 years, and the mean ± SD BMI was 25.4 ± 3.1 kg/m2.
Safety and Tolerability
No dose‐limiting toxicities were observed. A total of 80.0% (24 of 30) and 70.0% (7 of 10) of women randomized to garetosmab and placebo, respectively, reported an AE (Supplementary Table 1). Most AEs reported by both garetosmab and placebo recipients were of mild intensity. Six of 30 subjects treated with garetosmab (20%) experienced at least 1 treatment‐emergent AE (TEAE) of moderate severity compared with 2 of the 10 subjects (20%) in the placebo group. Most of these TEAEs were transient and resolved by the end of the study. No TEAEs of severe intensity were observed. TEAEs in the system organ classes “nervous system disorders” (43.3% [13 of 30] versus 30.0% [3 of 10]) and “gastrointestinal disorders” (30.0% [9 of 30] versus 10.0% [1 of 10]) occurred more frequently in those administered garetosmab versus placebo. The most common TEAE was headache (garetosmab, 36.7% [11 of 30]; placebo, 30.0% [3 of 10]). AEs deemed by the investigator to be related to the study drug occurred in 46.7% of subjects administered garetosmab (14 of 30) and 20% of subjects administered placebo (2 of 10). Headache was also the most frequently reported treatment‐related AE (garetosmab, 23.3% [7 of 30]; placebo, 20.0% [2 of 10]). A review of AE preferred terms by treatment groups did not show a clear relationship of a specific TEAE with garetosmab or an AE dose‐response.
One serious AE was reported; this subject was hospitalized for hyperthyroidism of moderate severity 110 days after receipt of garetosmab 0.3 mg/kg. The subject did not have any medical history involving thyroid function, and the AE was considered by the investigator as not related to garetosmab. There were no clinically significant or apparent dose‐related changes in laboratory values (including reproductive hormones), vital signs, or electrocardiograms.
Pharmacokinetics of Functional Garetosmab
Following intravenous administration, garetosmab concentrations quickly decreased first (when drug initially bound to target and distributed into the peripheral compartment), then decreased over time in a manner reflecting linear elimination (when drug concentrations were sufficient to saturate the binding target), and elimination was mainly by non‐target‐related pathways, and finally decreased in a nonlinear phase reflecting target‐mediated elimination (driven by antibody binding to the target molecule; Figure 1). Although both the parallel linear and nonlinear elimination processes are active at all concentrations studied, linear elimination processes generally appear to dominate the observed concentration‐time profiles at values > 30 mg/L. At lower systemic concentrations of functional garetosmab, the observed concentration‐time profiles appear to be dominated by capacity‐limited (nonlinear), target‐mediated elimination processes.
Figure 1.
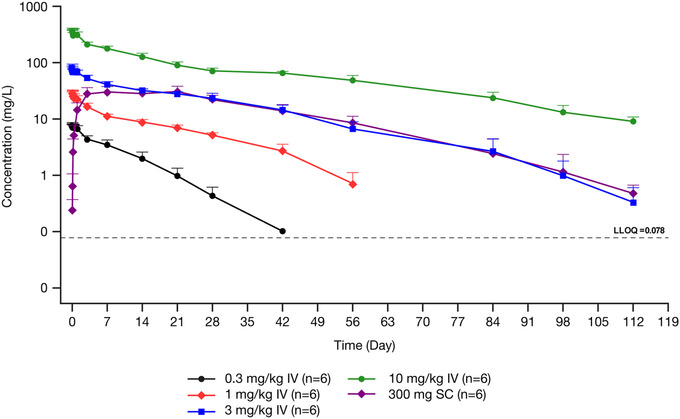
Mean ± SD log‐scaled concentrations of functional garetosmab in serum versus nominal time in healthy women administered a single dose. Dashed line indicates the LLOQ. Concentrations below the LLOQ were excluded. IV, intravenous; LLOQ, lower limit of quantification; SC, subcutaneous; SD, standard deviation.
Consistent with the target‐mediated kinetics described by the concentration‐time profiles, the pharmacokinetics of functional garetosmab were described as nonlinear. Nonlinearity was illustrated by the greater than dose‐proportional increase in mean AUCinf (ranging from 72.2 to 7520 mg*day/L) and the dose‐dependent decrease in CL (ranging from 4.35 to 1.34 mL/day/kg), with increasing doses of intravenous garetosmab studied (Table 1).
Table 1.
Pharmacokinetic Parameters for Functional Garetosmab in Healthy Women Administered a Single Intravenous Dose
| Intravenous Garetosmab 0.3 mg/kg (n = 6) | Intravenous Garetosmab 1 mg/kg (n = 6) | Intravenous Garetosmab 3 mg/kg (n = 6) | Intravenous Garetosmab 10 mg/kg (n = 6) | |
|---|---|---|---|---|
| Cmax, mg/L | ||||
| Mean (SD) | 8.10 (0.929) | 29.6 (5.01) | 86.5 (9.74) | 378 (39.7) |
| Median (Min‐Max) | 8.08 (6.96‐9.44) | 32.2 (22.4‐33.6) | 84.9 (73.0‐98.0) | 382 (327‐421) |
| CV% | 11.5 | 16.9 | 11.3 | 10.5 |
| Cmax/dose, kg/L | ||||
| Mean (SD) | 27.0 (3.10) | 29.6 (5.01) | 28.8 (3.25) | 37.8 (3.97) |
| Median (Min‐Max) | 26.9 (23.2‐31.5) | 32.2 (22.4‐33.6) | 28.3 (24.3‐32.7) | 38.2 (32.7‐42.1) |
| CV% | 11.5 | 16.9 | 11.3 | 10.5 |
| tmax, days | ||||
| Median (Min‐Max) | 0.10 (0.04‐1.04) | 0.08 (0.04‐0.21) | 0.08 (0.04‐0.21) | 0.06 (0.04‐0.38) |
| AUCinf, mg · day/L | ||||
| Mean (SD) | 72.2 (15.4) | 379 (50.3) | 1597 (176) | 7520 (809) |
| Median | 75.8 (46.8‐91.8) | 387 (301‐439) | 1581 (1408‐1905) | 7464 (6319‐8417) |
| CV% | 21.3 | 13.3 | 11.0 | 10.8 |
| AUCinf/dose, mg · day/L | ||||
| Mean (SD) | 241 (51.2) | 379 (50.3) | 532 (58.6) | 752 (80.9) |
| Median (Min‐Max) | 253 (156‐306) | 387 (301‐439) | 527 (469‐635) | 746 (632‐842) |
| CV% | 21.3 | 13.3 | 11.0 | 10.8 |
| CL, mL/day/kg | ||||
| Mean (SD) | 4.35 (1.11) | 2.68 (0.382) | 1.90 (0.196) | 1.34 (0.149) |
| Median (Min‐Max) | 3.96 (3.27‐6.41) | 2.59 (2.28‐3.32) | 1.90 (1.57‐2.13) | 1.34 (1.19‐1.58) |
| CV% | 25.5 | 14.3 | 10.3 | 11.1 |
| Vss, mL/kg | ||||
| Mean (SD) | 41.4 (4.66) | 47.8 (4.70) | 45.3 (4.86) | 46.6 (5.40) |
| Median (Min‐Max) | 42.1 (32.9‐46.0) | 47.9 (41.3‐53.7) | 44.3 (39.4‐51.8) | 45.5 (40.8‐54.6) |
| CV% | 11.2 | 9.83 | 10.7 | 11.6 |
AUCinf, area under the concentration‐time curve from time zero extrapolated to infinity; CL, clearance; CL/F, apparent clearance; Cmax, peak concentration; CV, coefficient of variation; SD, standard deviation; tmax, time to peak concentration; Vss, volume of distribution at steady state.
Following administration of intravenous garetosmab, mean Cmax values increased in a dose‐proportional manner with increasing dose (Table 1), consistent with an initial volume of distribution of approximately 33 mL/kg (similar to volume of serum). Mean Vss was similar for the different intravenous doses studied, ranging from 41.4 to 47.8 mL/kg (Table 1).
Pharmacokinetics following subcutaneous administration of garetosmab 300 mg are shown in Table 2. Because the concentration‐time profiles of functional garetosmab following the 300‐mg subcutaneous dose showed nonlinear concentration‐dependent CL, a dose correction could not be appropriately conducted, and bioavailability was not determined here. Following subcutaneous administration, peak concentrations were achieved between 7 and 21 days.
Table 2.
Pharmacokinetic Parameters for Functional Garetosmab in Healthy Women Administered a Single Subcutaneous Dose
| Subcutaneous Garetosmab 300 mg (n = 6) | |
|---|---|
| Cmax, mg/L | |
| Mean (SD) | 31.6 (7.94) |
| Median (Min‐Max) | 33.2 (21.8‐41.4) |
| CV% | 25.1 |
| Cmax/dose, 1/L | |
| Mean (SD) | 0.105 (0.0265) |
| Median (Min‐Max) | 0.111 (0.0727‐0.138) |
| CV% | 25.1 |
| tmax, days | |
| Median (Min‐Max) | 20.9 (6.88‐21.0) |
| AUCinf, mg · day/L | |
| Mean (SD) | 1334 (376) |
| Median (Min‐Max) | 1432 (824‐1694) |
| CV% | 28.2 |
| AUCinf/dose, mg · day/L | |
| Mean (SD) | 4.45 (1.25) |
| Median (Min‐Max) | 4.77 (2.75‐5.65) |
| CV% | 28.2 |
| CL/F, mL/day | |
| Mean (SD) | 243 (79.0) |
| Median (Min‐Max) | 211 (177‐364) |
| CV% | 32.5 |
AUCinf, area under the concentration‐time curve from time zero extrapolated to infinity; CL/F, apparent clearance; Cmax, peak concentration; CV, coefficient of variation; SD, standard deviation; tmax, time to peak concentration.
Concentrations of Total Activin A
At baseline, total activin A concentrations were below the LLOQ for all treatment groups. Following intravenous administration, total activin A concentrations increased, reflecting binding of garetosmab to activin A (Figure 2). Among the intravenous doses studied, mean Cmax increased in a dose‐dependent manner from 0.0117 to 0.0538 mg/L.
Figure 2.
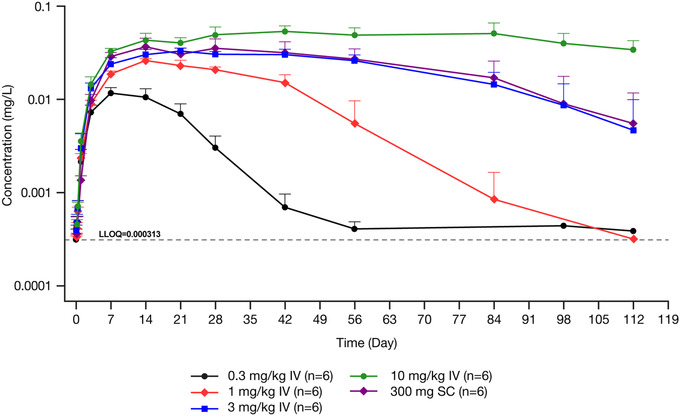
Mean ± SD log‐scaled concentrations of total activin A in serum versus nominal time in healthy women administered a single dose of garetosmab. Dashed line indicates the LLOQ. Concentrations below the LLOQ were excluded. IV, intravenous; LLOQ, lower limit of quantification; SC, subcutaneous; SD, standard deviation.
The highest concentration of total activin A was observed with the highest dose of intravenous garetosmab studied (10 mg/kg). The higher concentrations of total activin A appeared to reach a state of little or no change between weeks 4 and 12 (Figure 3). Decline in total activin A concentrations from this state of little or no change coincided with the onset of the concentration‐dependent decline of functional garetosmab.
Figure 3.
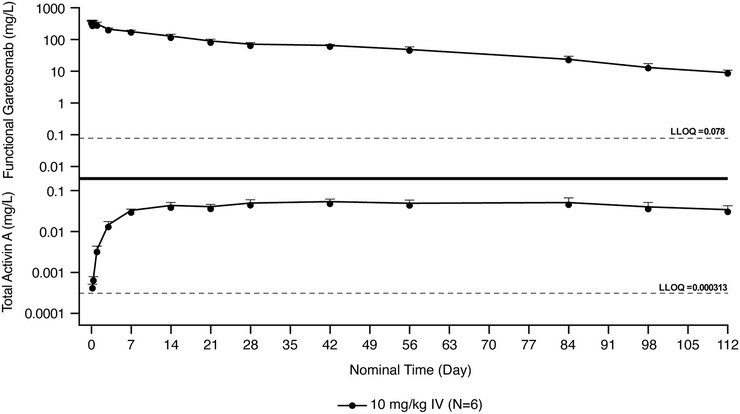
Mean ± SD log‐scaled concentrations of functional garetosmab in serum and total activin A versus nominal time in healthy women administered a single intravenous dose of 10 mg/kg. Dashed line indicates the LLOQ. Concentrations below the LLOQ were excluded. IV, intravenous; LLOQ, lower limit of quantification; SD, standard deviation.
Antidrug Antibodies
One subject who was administered intravenous garetosmab (1 mg/kg) showed a positive response in the ADA assay on day 113. The titer was low (< 1000) and had no effect on the pharmacokinetics of functional garetosmab. No safety events were associated with this ADA response.
Discussion
Here, we report the safety and pharmacokinetics of garetosmab, a fully human monoclonal antibody that has high affinity and specificity to activin A, in healthy women of nonchildbearing potential (18‐65 years). In this double‐blind, placebo‐controlled, first‐in‐human phase 1 study of a single dose of garetosmab, no safety issues were identified that precluded dose escalation, and there were no clinically significant or apparent dose‐related changes in laboratory values, vital signs, or electrocardiograms. These safety findings support further investigation of garetosmab in patients with FOP.
Analyses conducted in this study indicated that garetosmab was well tolerated in healthy nonpregnant women, eliciting no serious safety concerns, with a similar AE profile as placebo. No safety signals were seen in the healthy subjects in this study that would preclude testing the drug in patients. This study did not include healthy male subjects or patients with FOP and only involved a limited number of participants who received a single dose; hence, safety assessments are limited.
Following intravenous administration, garetosmab concentrations quickly decreased first (when drug initially bound to target and distributed into the peripheral compartment), then decreased over time in a manner reflecting linear elimination (when drug concentrations were sufficient to saturate the binding target), and elimination was mainly by non‐target‐related pathways, and finally decreased in a nonlinear phase reflecting target‐mediated elimination (driven by antibody binding to the target molecule). The results are consistent with pharmacokinetic behavior of target‐mediated drug disposition. 6 , 7 The absorption of garetosmab administered via the subcutaneous route is consistent with results seen previously with other antibodies. 8 , 9 , 10
At baseline, the total activin A concentrations were comparable across treatment groups. Total activin A concentrations increased from baseline following administration of garetosmab, indicating binding of garetosmab with the target. Mean maximum concentrations increased from 0.0117 to 0.0538 mg/L in a dose‐dependent manner following intravenous administration of 0.3 to 10 mg/kg garetosmab. The highest concentrations of total activin A were seen with garetosmab 10 mg/kg, the highest dose studied. These higher concentrations of total activin A remained relatively stable over weeks 4‐12, and their decline coincided with the onset of a faster, concentration‐dependent decline of garetosmab, indicative of target‐mediated clearance. This likely suggests that a single dose of garetosmab 10 mg/kg saturates the target‐mediated pathway for approximately 12 weeks. The overall pharmacokinetic data from this study (for garetosmab and the activin A target) support a dosing regimen of every 4 weeks, provided that garetosmab does not display time‐dependent pharmacokinetics. The current analysis is limited by the single‐dose nature of the study design.
Only 1 study participant developed ADAs; however, the titer was low and did not impact the safety or pharmacokinetics of functional garetosmab. The effects of multiple doses of garetosmab over time on the development of ADAs remain to be explored in future studies.
There is a critical unmet need for a definitive therapy option for FOP, as there is no cure for the condition; current treatment options, including corticosteroids, are only used to relieve the pain and swelling associated with acute flare‐ups. 11 In our study, we show that inhibition of activin A with garetosmab was generally well tolerated in healthy nonpregnant women, with predictable pharmacokinetics following a single dose. There were no safety signals seen in the healthy subjects in this study that would preclude testing the drug in patients. The current study supported the testing of garetosmab in patients with FOP. The highest dose studied here (10 mg/kg) was well tolerated, and the target‐mediated pathway was saturated by this dose in week 12; therefore, the 10 mg/kg dose was chosen to move forward into the LUMINA‐1 study (NCT03188666).
Conclusion
The initial safety, tolerability, and pharmacokinetics of garetosmab support further investigation. Garetosmab 10 mg/kg administered intravenously every 4 weeks is currently being evaluated in patients with FOP in the phase 2 LUMINA‐1 study (NCT03188666).
Conflicts of Interest
Dr. F. Vanhoutte has no disclosures to report. All other authors are salaried employees of Regeneron Pharmaceuticals, Inc.
Funding
The study was funded by Regeneron Pharmaceuticals, Inc., Tarrytown, New York. The sponsor participated in the design and conduct of the trial, analysis of the data, and preparation of the article.
Data Accessibility Statement
Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, and statistical analysis plan) that support the methods and findings reported in this article. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant reidentification. Submit requests to https://vivli.org/.
Medical Writing Support
Medical writing support under the direction of the authors was provided by Melissa Purves, PhD, and Rob Campbell, PhD, of Prime (Knutsford, UK), according to Good Publication Practice guidelines and was funded by Regeneron Pharmaceuticals, Inc.
Supporting information
Table S1. Treatment‐Emergent Adverse Events Reported in >1 Subject in Either Treatment Group by System Organ Class and Preferred Term.
Acknowledgments
The authors acknowledge the volunteers who participated in this study, the study's medical officer, Xiaobing Qian (now at Boston Pharmaceuticals), and the staff at SGS Clinical Pharmacology Unit (Antwerp, Belgium).
References
- 1. Huning I, Gillessen‐Kaesbach G. Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype‐phenotype correlation. Mol Syndromol. 2014;5(5):201‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: diagnosis, management, and therapeutic horizons. Pediatr Endocrinol Rev. 2013;10(Suppl 2):437‐448. [PMC free article] [PubMed] [Google Scholar]
- 3. Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38(5):525‐527. [DOI] [PubMed] [Google Scholar]
- 4. Hatsell SJ, Idone V, Wolken DM, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015;7(303):303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Latres E, Mastaitis J, Fury W, et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun. 2017;8:15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dua P, Hawkins E, van der Graaf PH. A tutorial on target‐mediated drug disposition (TMDD) models. CPT Pharmacometrics Syst Pharmacol. 2015;4(6):324‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507‐532. [DOI] [PubMed] [Google Scholar]
- 8. Kovalenko P, DiCioccio AT, Davis JD, et al. Exploratory population PK analysis of dupilumab, a fully human monoclonal antibody against IL‐4Ralpha, in atopic dermatitis patients and normal volunteers. CPT Pharmacometrics Syst Pharmacol. 2016;5(11):617‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lunven C, Paehler T, Poitiers F, et al. A randomized study of the relative pharmacokinetics, pharmacodynamics, and safety of alirocumab, a fully human monoclonal antibody to PCSK9, after single subcutaneous administration at three different injection sites in healthy subjects. Cardiovasc Ther. 2014;32(6):297‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol. 2013;53(3):314‐325. [DOI] [PubMed] [Google Scholar]
- 11. Kaplan FS, Al Mukaddam M, Baujat G, et al. The medical management of fibrodysplasia ossificans progressiva: current treatment considerations. Proc Intl Clin Council FOP. 2019;1:1‐111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Treatment‐Emergent Adverse Events Reported in >1 Subject in Either Treatment Group by System Organ Class and Preferred Term.
Data Availability Statement
Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, and statistical analysis plan) that support the methods and findings reported in this article. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant reidentification. Submit requests to https://vivli.org/.