

Abstract
The modified point charge plus continuum (mPC) model [Constantin L. A.; Phys. Rev. B 2019, 99, 085117] solves the important failures of the original counterpart, namely, the divergences when the reduced gradient of the density is large, such as in the tail of the density and in quasi-dimensional density regimes. The mPC allows us to define a modified interaction-strength interpolation (mISI) method inheriting these good features, which are important steps toward the full self-consistent treatment. Here, we provide an assessment of mISI for molecular systems (i.e., considering thermochemistry properties, correlation energies, vertical ionization potentials, and several noncovalent interactions), harmonium atoms, and functional derivatives in the strong-interaction limit. For all our tests, mISI provides a systematic improvement over the original ISI method. Semilocal approximations of the second-order Görling–Levy (GL2) perturbation theory are also considered in the mISI method, showing considerable worsening of the results. Possible further development of mISI is briefly discussed.
1. Introduction
The exact exchange–correlation (XC) functional can be formally defined using the adiabatic connection formalism as1−7
| 1 |
where ρ is the electron density, λ is the electron–electron interaction strength, and
| 2 |
is the density-fixed linear adiabatic connection integrand, with Ψλ[ρ] being the antisymmetric wave function that minimizes T̂ + λV̂ee while yielding the density ρ (T̂ and V̂ee are the kinetic and electron–electron interaction operators, respectively), and U[ρ] being the Hartree energy.
Many accurate hybrid XC functionals are based on, and explained by, this method,4,6,8−10 which provides a rationale for mixing the Hartree–Fock (HF) exchange with semilocal XC functionals.11 Moreover, the basic Görling–Levy (GL) perturbation theory12−14 uses a generalized adiabatic connection formula13 to obtain the Taylor expansion of the correlation energy at a small coupling constant (λ → 0). The adiabatic connection formalism also stays behind the more recent and sophisticated double hybrids15 that are using either the second-order Møller–Plesset16 (MP2) correlation mixed with fractions of HF exchange and semilocal XC functionals17−23 into the generalized Kohn–Sham (KS) density functional theory (DFT) scheme24,25 or GL2 correlation13 combined with fractions of KS-DFT exact exchange and semilocal XC functionals26 into the optimized effective potential (OEP) scheme of the true KS-DFT.26−29 The adiabatic connection is also of utmost importance for the ground-state calculations of linear-response time-dependent DFT, being part of the so-called adiabatic connection fluctuation-dissipation theorem that provides a framework for high-level, orbital-dependent methods based on XC kernel approximations.30−45
Here, we focus on the interaction-strength interpolation (ISI) method46−48 that accurately interpolates the weak (λ → 0)- and strong (λ → ∞)-interaction limits that are well known. Thus, in the weak-interaction limit, the GL perturbation theory becomes exact46
| 3 |
where W0[ρ] = Ex[ρ] is the exact KS-DFT exchange functional and W0′[ρ] = 2Ec[ρ]. On the other hand, the strong-interaction limit is
| 4 |
where W∞[ρ] and W∞′[ρ] can be found in the strictly correlated electron (SCE) approach,49 but in practice, they have been approximated from the second-order gradient expansion (GE2) of the point charge plus continuum (PC) model.46−48 Starting from the ISI method, several improvements and variants have been proposed.49−65 In particular, we mention the methods of refs (49, 50), which remove the spurious λ–1 term in the large λ limit of Wλ, keeping the almost same accuracy of the XC energy as the original ISI method. Moreover, correlation energy densities have been constructed from local interpolations along the adiabatic connection curve.57,62,63,65−67
Recently, the modified ISI (mISI) method has been developed,65 which solves important failures of the original ISI approach in the tail of the density and in quasi-low dimensional density regimes. The mISI method has been tested on very few atoms showing improved correlation energies over its initial variant. In this article, we assess mISI for molecular systems, using the OEP exact exchange (OEPx) orbitals and densities. Noting that mISI and ISI are pure KS-DFT functionals and not wave function methods, the choice of OEPx orbitals is the most physical, presumably being very close to the true self-consistent ISI orbitals.
The GL2 correlation energy EcGL2[ρ] enters the expressions of ISI and mISI functionals, as shown, e.g., in eq 17 of ref (60). Because GL2 depends on all occupied and unoccupied orbitals and orbital energies, its evaluation even with OEPx orbitals is still very expensive. In this sense, semilocal approximations of Ec[ρ] are of interest not only for simplification of the ISI and mISI methods but also for the development of correlation functionals compatible with exact exchange.68,69 Hence, in our assessment, we also consider the mISI@TPSS-GL2 method that replaces the true GL2 with the TPSS-GL2 meta-generalized-gradient approximation (meta-GGA) correlation functional,68 which is one of the most accurate GL2 approximations available nowadays (see Table S12 of ref (70) for a comparison of few GL2 models).
This paper is organized as follows: In Section 2, we show a brief theoretical overview of the methods; in Section 3, we report the computational details; and in Section 4, we present and analyze the results. Finally, in Section 5, we summarize our conclusions.
2. Theoretical Overview of the Methods
Both ISI and mISI functionals have the same Wλ[ρ] integrand in eq 1
| 5 |
where X = xy2/z2, Y = x2y2/z4, Z = xy2/z3 – 1, and x = −4EcGL2, y = W∞, z = W0 – W∞ which, in turn, lead to following XC functional expression
| 6 |
The ISI functional approximates W∞ with the GE2 of the PC model, and W∞′ has been fitted to the PKZB meta-GGA71 counterpart such that
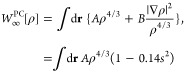 |
7 |
and
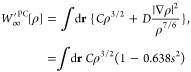 |
8 |
where s = |∇ρ|/[2(3π2)1/3ρ4/3] is the reduced gradient of the density, A = −1.451 and C = 1.535 are the LDA low-density-limit coefficients, B = 0.005317 is the second-order gradient expansion coefficient of the PC model, and D = −0.02558 was fixed from W∞′PKZB[ρ] of the He atom.
On the other hand, mISI uses the following expressions65
| 9 |
and
| 10 |
Here, a = 2 is the smallest integer that ensures W∞mISI[ρ] ≤ sLL[ρ], where sLL[ρ] = A ∫dr ρ4/3 – 0.245 ∫dr ρ4/3s1/4 is the simplified gradient-dependent bound of Lewin and Lieb.72,73 Note that W∞[ρ] still recovers the GE2 of the PC model. Finally, b = 1.3 has been fitted to the correlation energy of the He atom.
By construction, mISI permits local interpolations along the adiabatic connection57,62 and the adiabatic connection semilocal correlation (ACSC) functional formalism has been constructed.65 Thus, for completeness, we will also show results with ACSC GGA correlation functional combined with OEPx.
The ACSC energy per particle ϵcACSC(r) is defined by Ec[ρ] = ∫dr ρϵcACSC(r), where65
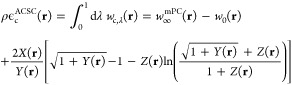 |
11 |
while
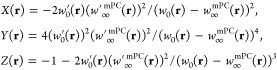 |
12 |
Here, all energy densities wλ(r), wλ′(r) are defined by Wλ = ∫drwλ(r) and Wλ = ∫drwλ′(r).
The ACSC energy density is a local interpolation along the adiabatic connection, and it is meaningful when all of the energy densities are in the same gauge. Moreover, eq 11 is well defined only when w0(r) > w∞mPC(r),65 which is violated by the exact exchange energy density in the conventional gauge of the electrostatic potential of the XC hole,55,74 but it is satisfied by the exact exchange energy density in the TPSS gauge.65,74 Thus, all energy densities should be in the TPSS gauge, which is more appropriate for semilocal expressions. Because of their semilocal nature, both w∞(r) and w∞′mPC(r) can be considered to be in the TPSS gauge.
For the ACSC GGA correlation functional, we use w0′(r) = 2ρϵc and w0(r) = ρϵxPBE(r) as in ref (65), where ϵc and ϵxPBE are the GL2 and exchange energy densities of the popular PBE GGA functional.65,75
The nonlinear form of the ISI correlation functional expression leads to the increase of size-consistency error,58,61i.e., the total energy of two nonidentical, distant systems is not equal to the sum of energies of subsystems A and B separately, Exc(m)ISI(AB)R→∞ ≠ Exc(A) + Exc(m)ISI(B). As is generally known, the lack of size consistency may lead to wrong predictions, e.g., for atomization or binding energies of molecular systems.76 This feature can be restored, without additional cost, utilizing the sum over fragments idea from ref (61), which allows us to compute the correlation energies of the infinitely far fragments (Exc(AB)R→∞) utilizing as ingredients in eq 6 the sum of energies of isolated parts. Then, the size-consistency correction can be written (for considered case) as
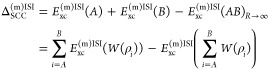 |
13 |
where W[ρ] = {Ex[ρ], EcGL2[ρ], W∞[ρ], W∞[ρ]} is a compact notation for all ISI input ingredients (for more technical details, we refer the reader to ref (61)).
3. Computational Details
All methods considered in the present study have been implemented in a locally modified version of the ACES II77 program. The ISI, mISI, as well as ACSC and mISI with TPSS-GL2 results have been obtained in a post-self-consistent-field (SCF) manner, using as a reference OEPx SCF converged quantities (i.e., orbitals, orbital energies, and densities). As in our previous studies,26,29,60,78−81 to solve the OEPx equation, we have employed the finite-basis set procedure of refs (82, 83). To enable the comparison with GL2 results and avoid problems related to full SCF treatment78−80,84 (see also the discussion in refs (29, 85, 86)), the reported GL2 results are obtained in the same manner. As reference data, we have utilized the coupled-cluster single double and perturbative triple [CCSD(T)]87 results obtained in the same basis set, to make a comparison on the same footing and to reduce basis set-related errors. Additionally, for comparison, we also report the results of the MP216 and ab initio DFT OEP2-sc methods.88
To assess all methods, we have considered several test cases:
Correlation energies: evaluated for the set of 9 atoms and 27 small closed- and open-shell molecules from refs (81, 89).
Thermochemistry data: This set contains atomization energies, i.e., AE6,90,91 G2 subset89 (see the Supporting Information of ref (81) for more details), small radicals,92 K9 barrier heights,91,93 21 isomerization/reaction energies (ISO21),76 closed- and open-shell reaction energies listed in refs (81, 94), and the hydrogen transfer reactions (HTR).81,95 All of the aforementioned calculations have been performed using uncontracted cc-pVTZ basis sets of Dunning96 without counterpoise corrections for basis set superposition error (BSSE). In the case of atomization energies, the size-consistency correction from ref (61) was employed.
Vertical ionization potentials: 32 vertical ionization potentials (VIP)79 computed as the energy difference between the neutral and the ionic species.97 The computational setup, namely, basis sets and geometries (in the case of molecules), is identical to that in ref (79).
Noncovalent interactions data: The interaction energies of several types of noncovalent interacting molecular systems also used in our previous studies,98−101 such as weak interaction (WI), dipole–dipole interaction (DI6), hydrogen-bond interaction (HB6), double-hydrogen-bond interaction (DHB), and charge transfer interaction (CT7). The energies have been obtained using an uncontracted aug-cc-pVTZ102 basis sets together with the geometries from refs (103−106). All quantities have been calculated without counterpoise corrections for BSSE. In all cases, the size-consistency correction from ref (61) was employed.
Harmonium atoms: We have performed calculation for various values of ω in the Hooke’s atom model107 ranging between 0.03 (strong interaction) to 1000 (weak interaction) using a even-tempered Gaussian basis set from ref (108) (N = 8, L = 0, 1). The accuracy of full configuration interaction (FCI) results employing this basis set for all values of ω was cross-checked with the exact ones reported in ref (108) yielding, in total, a mean absolute error (MAE) of 0.002 Ha and a mean absolute relative error (MARE) of 0.06%. Because the ISI formula interpolates between weak- and strong-correlation limits, this test seems to be essential for the assessment of XC formula accuracy.
Potentials for the strong-interaction limit: We have computed the functional derivatives of original and modified PC W∞ and W∞′ models in a post-SCF manner using OEPx densities. This approach was already successfully utilized in some studies60,109−111 to investigate the quality of the potentials.
4. Results
Here, we present and analyze results for thermochemistry, noncovalent interactions, and Hooke’s atom112−115 at various frequencies ω of the harmonic potential. We also investigate the potentials for the strong-interaction limit for a few systems.
4.1. Thermochemistry Results
In Table 1, we present the error statistics (MAE and MARE) of the thermochemistry results. We observe that the mISI method shows a systematic improvement over the ISI method, for all of the tests. On the other hand, GL2 fails badly, being the worst method here. We recall that the GL213 correlation energy expression is
| 14 |
where the indices i, j, a, and b are used for occupied and virtual KS orbitals, ε’s are the KS eigenvalues, and vxKS and v̂x are the local, multiplicative KS OEPx and the nonlocal HF exchange potentials, respectively. The first term on the right-hand side of eq 14 is the same as the MP2 correlation energy expression, the only difference being computed using KS OEPx orbitals and orbital energies instead of the HF ones. The last term of eq 14 is small80 in comparison to the first one such that EcGL2 is approximately Ec evaluated with KS orbitals. A comparison between the MP2 and GL2 results of Table 1 shows huge accuracy differences. In this respect, the ISI and mISI methods show significantly improved performance over GL2. This is a consequence of attenuating the GL2 energy term60,79,80 in the ISI functional expression. Nevertheless, in general, they are still worse than MP2.
Table 1. Error Statistics (MAE and MARE) of Several Thermochemistry Propertiesg.
| @OEPx |
|||||||
|---|---|---|---|---|---|---|---|
| MP2 | OEP2-sc | GL2 | ISIf | mISIf | mISIe | ACSC | |
| Correlation Energies (9 Atoms and 27 Molecules) | |||||||
| MAEa | 24.4 | 18.4 | 95.4 | 42.3 | 34.9 | 85.9 | 73.6 |
| MAREb | 10.5 | 8.7 | 26.6 | 10.4 | 8.3 | 21.4 | 16.9 |
| Atomization Energies (27 Molecules) | |||||||
| MAEcI | 5.5 | 8.0 | 37.8 | 16.8 | 15.1 | 14.8 | 17.8 |
| MAREb | 5.2 | 7.3 | 31.6 | 15.2 | 13.4 | 13.2 | 17.2 |
| Atomization Energies (AE6 Test) | |||||||
| MAEcI | 12.2 | 17.1 | 82.1 | 32.7 | 23.6 | 25.5 | 21.0 |
| MAREb | 3.7 | 5.6 | 23.6 | 13.6 | 11.7 | 7.5 | 6.9 |
| Atomization Energies (Small Radicals) | |||||||
| MAEcI | 6.1 | 15.7 | 53.0 | 21.7 | 16.6 | 30.3 | 34.2 |
| MAREb | 3.9 | 12.1 | 89.1 | 17.0 | 12.2 | 31.1 | 46.4 |
| Kinetics (K9 Test) | |||||||
| MAEcI | 2.8 | 14.9 | 18.9 | 16.0 | 15.0 | 15.9 | 14.8 |
| MAREb | 22.5 | 46.5 | 75.9 | 45.5 | 37.8 | 68.3 | 68.6 |
| HTR Test | |||||||
| MAEcI | 13.5 | 2.2 | 25.4 | 14.8 | 12.2 | 2.9 | 2.8 |
| MAREb | 143.5 | 32.6 | 259.5 | 150.2 | 119.0 | 29.5 | 34.0 |
| 21 Isomerization Energies (ISO21 Test) | |||||||
| MAEcI | 0.89 | 0.39 | 20.71 | 12.48 | 10.97 | 7.16 | 6.61 |
| MAREb | 7.97 | 16.59 | 96.60 | 42.74 | 23.87 | 82.39 | 84.89 |
| 31 Closed-Shell Reaction Energies | |||||||
| MAEcI | 2.0 | 3.0 | 10.6 | 4.7 | 4.6 | 11.4 | 10.3 |
| MAREb | 46.8 | 34.5 | 106.3 | 54.5 | 57.3 | 144.8 | 125.7 |
| 26 Open-Shell Reaction Energies | |||||||
| MAEcI | 4.3 | 3.4 | 18.3 | 11.5 | 10.4 | 12.2 | 11.6 |
| MAREb | 12.3 | 12.7 | 51.5 | 34.6 | 29.5 | 35.6 | 30.2 |
| 32 Vertical Ionization Potentials (VIP) | |||||||
| MAEd | 0.2 | 0.2 | 1.2 | 0.6 | 0.5 | 0.4 | 0.4 |
| MAREb | 2.4 | 1.2 | 9.0 | 4.9 | 4.3 | 3.8 | 3.6 |
In mHa.
In %.
n kcal/mol.
In eV.
With TPSS-GL2.
Corrected for size-consistency error (see ref (61)).
The best result between ISI and mISI of each line is highlighted in bold style. The full results of all tests are reported in ref (70). The OEP2-sc results are fully self-consistent, the MP2 and (reference) CCSD(T) results are calculated on top of HF orbitals, and the results of the other methods (GL2, ISI, mISI, mISI@TPSS-GL2, ACSC) are computed on top of OEPx orbitals.
On the other hand, mISI@TPSS-GL2, which replaces into the mISI expression the GL2 term with the semilocal TPSS-GL2 variant, is usually worse than mISI, especially for the correlation energies of atoms and molecules. Indeed, more reliable semilocal approximations of GL2 are needed. Finally, the ACSC GGA correlation combined with OEPx exact exchange has a similar accuracy to mISI@TPSS-GL2.
Next, in Figure 1, we show the atomization energy errors versus correlation energy errors for the set of 27 small molecules, used also in Table 1. For almost all of the molecules, the mISI method performs better than the ISI for both properties. We also mention that the worst results are found from F2 and P2, which are difficult cases in quantum chemistry, due to their multiconfiguration character.116
Figure 1.

Absolute relative errors (in %) of the atomization energies (AE) versus the ones of the correlation energies, for a set of 27 small molecules BeH, OH, NH2, NH, NO, PH2, O2, S2, SiH2, C2H5, CH2, CH3, CN, COH, CH4, LiF, Li2, F2, CO, CH2, H2, CS, LiH, N2, P2, NaCl, and H2O. For the worst cases F2 and P2, GL2 fails badly for atomization energies, with errors of 188 and 73%, respectively.
4.2. Noncovalent Interaction Results
In Figure 2, we report the binding energy curves for three representative cases, namely, Ne2, Ar–Ne, and H2S–HCl dimers. In the case of ISI and mISI curves, to enable the comparison with reference results, we have applied the size-consistency correction from ref (61).
Figure 2.

Binding energy curves of Ne2 (top), Ne–Ar (middle), and H2S–HCl (bottom) computed using different theoretical methods. In the case of ISI and mISI methods, the size-consistency correction from ref (61) was employed.
First of all, we note that the ACSC gives too shallow binding curves similar to most semilocal functionals. Almost same behavior (not reported) was observed for the mISI@TPSS-GL2 method. The ISI and mISI methods, in turn, reduce the huge overestimation of the GL2 method, with one exception of Ne dimer where ISI gives slightly worse predictions. Nevertheless, also in this case, the utilization of modified point charge plus continuum (mPC) variants of W∞ and W∞′ corrects the predictions. The mISI method, in general, performs better than the ISI method, the results being much closer to reference CCSD(T) and to the ab initio OEP2-sc data.
This finding is further confirmed by the data gathered in Table 2, where we report the error statistics obtained for several classes of noncovalent interacting molecular systems. The overall statistics confirm previous findings. The mISI outperforms the ISI method, giving in the same time results comparable to the OEP2-sc method. The only exception is for the HB6 test, where we observe an inverse trend. However, a closer look at the total energies (used to calculate HB6 binding energies) shows also here very good performance of the mISI method. The MAEs of total energies, calculated for GL2, ISI, and mISI with respect to the CCSD(T) reference, follow the general observed trend mISI (0.11 Ha) < ISI (0.14 Ha) < GL2 (0.28 Ha). This may indicate that the relatively good performance of ISI for the HB6 binding energies can be related to some error balancing between W∞PC and W∞ GE2 terms.
Table 2. MAE and MARE for Noncovalent Interaction Test Setse.
| @OEPx |
|||||||
|---|---|---|---|---|---|---|---|
| MP2 | OEP2-sc | GL2 | ISId | mISId | mISIc | ACSC | |
| WI | |||||||
| MAEa | 0.04 | 0.05 | 0.51 | 0.36 | 0.24 | 1.96 | 0.32 |
| MAREb | 18.53 | 17.08 | 598.63 | 455.11 | 426.50 | 899.71 | 150.26 |
| HB6 | |||||||
| MAEa | 0.17 | 0.27 | 0.75 | 0.39 | 0.70 | 0.64 | 0.66 |
| MAREb | 1.81 | 4.32 | 9.27 | 4.92 | 7.28 | 7.50 | 6.49 |
| DI6 | |||||||
| MAEa | 0.45 | 0.71 | 2.16 | 1.41 | 1.14 | 1.01 | 1.09 |
| MAREb | 12.85 | 20.24 | 64.05 | 42.53 | 34.80 | 38.97 | 34.77 |
| CT7 | |||||||
| MAEa | 0.55 | 1.35 | 2.54 | 1.71 | 1.39 | 1.94 | 1.64 |
| MAREb | 14.99 | 40.25 | 80.36 | 54.94 | 44.61 | 69.62 | 60.33 |
| DHB | |||||||
| MAEa | 0.54 | 0.85 | 1.62 | 0.98 | 0.79 | 0.91 | 1.01 |
| MAREb | 8.19 | 13.18 | 30.31 | 18.52 | 15.17 | 20.54 | 20.53 |
| Overall | |||||||
| MAEa | 0.38 | 0.74 | 1.63 | 1.05 | 0.91 | 1.37 | 1.03 |
| MAREb | 12.58 | 24.28 | 154.22 | 113.00 | 103.06 | 195.24 | 58.49 |
In kcal/mol.
In %.
With TPSS-GL2.
Corrected for size-consistency error (see ref (61)).
The best result between ISI and mISI of each line is highlighted in bold style. The OEP2-sc results are fully self-consistent, the MP2 and (reference) CCSD(T) results are calculated on top of HF orbitals, and the results of the other methods (GL2, ISI, mISI, mISI@TPSS-GL2, ACSC) are computed on top of OEPx orbitals.
We also note that in the case of WI systems, the ISI and mISI methods yield quite large MAE and MARE. This is caused by large errors given by two dimers, namely, He–Ne and He–Ar. We have found that the large overestimation of ISI binding energies is inherited from the GL2 method, which gives energies of 0.90 and 0.83 kcal/mol for He–Ne and He–Ar, respectively. At the same time, the reference CCSD(T) results yield −0.04 and −0.06 kcal/mol for these two cases. To explain this, we have had a closer look at the GL2 total energies involved in the calculation of the binding energy of the He–Ne dimer. It has turned out that while the Ne and He–Ne dimer total GL2 energies largely overestimate the reference CCSD(T) data (approximately by about 80 mHa), the He total GL2 energy gives quite accurate results, being only 6.4 mHa off CCSD(T) data, in consequence, causing the rise of large error in the binding energy. Nevertheless, we note that both the ISI and mISI methods slightly reduce the value of GL2 binding energies for both dimers.
In Table S13 of ref (70), we report additionally the comparison of binding energies obtained from the ISI and mISI methods using HF reference orbitals. Because the MP2 method already provides quite accurate results (MAE = 0.38 kcal/mol, MARE = 12.58%), the improvement seen in ISI and mISI results, which give here overall MAEs of 0.25 kcal/mol (MARE = 10.26%) and 0.29 kcal/mol (MARE = 17.11%), respectively, is not so significant as observed in Table 2.
To conclude, the mISI shows a systematic improvement over the ISI method, and most importantly, both these methods provide significant correction over GL2, which fails badly, being the worst method for all of the tests.
4.3. Hooke’s Atom
Figure 3 reports the errors in the total energies of harmonium atoms calculated for various values of ω ranging between 0.03 and 1000 for several methods. Because both ISI formulas interpolate between weak- and strong-correlation limits, this type of test can be of utmost importance for checking their accuracy and smooth transition between these two regimes. An inspection of the figure reveals that the best performance is given by the mISI method closely followed by ISI, OEP2-sc, and MP2 results. This indicates that the modified GGA PC formula, which fixes the behavior of ISI functional in the density tail, has a strong impact on the proper description of both regimes. In medium- and weakly correlated limits, we observe really good performance of the GL2 method. This is not surprising because GL2 becomes exact in this regime. On the other hand, in the strongly correlated limit, the GL2 method shows the worst performance of all methods. The ACSC and mISI@TPSS-GL2 give similar performance to the ab initio OEP2-sc method, namely, a small error for ω1/2 < 1 and a large overestimation for larger values of ω.
Figure 3.

Absolute error on total energies of harmonium atoms for various values of ω.
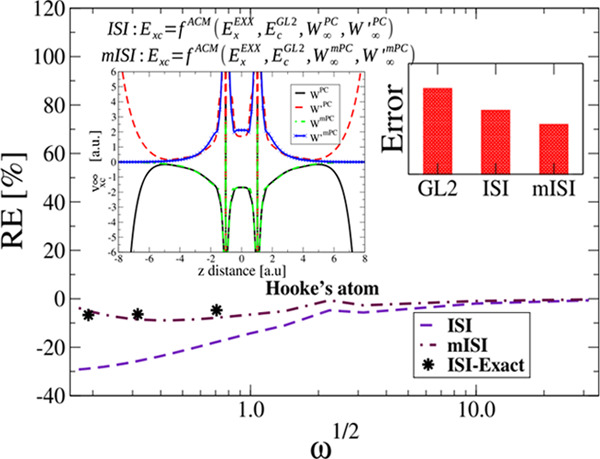
To better see the functional performance at the strong-correlation limit (ω → 0), we report in Figure 4 the relative errors (RE) on the correlation energy. When ω → 0, mISI is the most accurate method being in good agreement with the ISI method computed with exact W∞ and W∞′.62 On the other hand, the original ISI, which uses the PC model, gives quite large errors of about 30%, but still much better than GL2, which fails badly, with an error of about 120%. Note that when ω → 0, the density varies rapidly almost everywhere such that the reduced density gradient s is large, and the gradient expansion of the PC model cannot describe this regime.
Figure 4.

Relative error on correlation energies of harmonium atoms for various values of ω. The black dots (denoted with ISI-Exact) show the results of the ISI method computed with exact W∞ and W∞′, taken from ref (62).
4.4. Analysis of Functional Derivatives of W∞ and W∞′
Finally, in Figures 5 and 6, we report the comparison of the functional derivatives of W∞PC and W∞ and their modified counterparts for two representative systems, namely, H– ion and N2 molecule.
Figure 5.

(Top) Comparison between the exact (SCE) functional derivative of W∞ and the one computed from PC and modified PC model. (Bottom) Functional derivative of W∞′ computed from PC and modified PC model. Both figures are done for H–.
Figure 6.

Functional derivative of PC and modified PC for N2 molecule plotted along the bond axis.
Let us turn first our attention to the H– ion. We note that both mPC and PC W∞ and W∞′ potentials, like any other GGA, diverge on the nuclei, contrary to the exact SCE potential. Furthermore, in the tail of the density, both PC potentials diverge.60 This is not the case for mPC potentials where we observe smooth and fast decay to zero in this region. We recall here that the exact SCE potential decays as −1/r in the tail of the density. On the other hand, it was shown recently117 that the functional derivative of an exact W∞ diverges in the tail for one-dimensional systems. This might indicate that the divergence is an exact feature of W∞′ and W∞ is a more accurate model.
A similar behavior is observed for the N2 molecule. In the nuclei, both potentials diverge, whereas in the far asymptotic region, the mPC potentials decay quite fast to zero in contrast to the PC potential which diverges.
We note, however, that the utilization of ISI-like functionals in practical KS calculations requires that the functional derivative of the XC functional is finite in the tail. Thus, one needs to remove the divergences of the functional derivative118 despite whether this is an exact or model feature. This step is important toward the fully self-consistent implementation of ISI functionals.118
5. Conclusions
We have presented a small assessment and comparison of ISI and mISI methods showing that the utilization of the modified PC model in ISI formula leads always to the improvement in the results. Moreover, the inspection of functional derivatives of W∞ and W∞′ obtained from the mPC model shows a lack of divergence in the tail of the density, which is a crucial step toward the self-consistent implementation of ISI functionals.
We note that the overall accuracy of the results provided by all ISI-like functionals is not better than other semilocal, hybrid, and double-hybrid functionals.58 This is due to the dominant role of the GL2 or MP2 (depending on the reference orbitals used in the evaluation of energy) term in the ISI formula60 for most of the chemically important applications. Thus, one possible way to improve the results is to include more terms in eq 3, i.e., third-order GL/MP or higher. This, however, will significantly increase the cost of the method. Another possible path is to substitute the GL2/MP2 term by rescaled GL2/MP276 or one of spin-component-scaled variant80,119−122 of GL or MP perturbation theory.
Finally, we mention that the mPC, and implicitly the mISI method, can be further improved by considering the Pauli kinetic energy enhancement factor123−128 as an additional ingredient, which is relevant for one- and two-electron systems,129 atomic core,129 asymptotic behavior in the tail of the density,130 and superior overall performance.128 These will be a subject of further studies.
Acknowledgments
S.Ś. is thankful to the Polish National Science Center (Grant No. 2016/21/D/ST4/00903) and CANALETTO Project (Nos. PPN/BIL/2018/2/00004 and PO19MO06) for partial financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.0c00328.
The authors declare no competing financial interest.
Supplementary Material
References
- Langreth D. C.; Perdew J. P. The exchange-correlation energy of a metallic surface. Solid State Commun. 1975, 17, 1425–1429. 10.1016/0038-1098(75)90618-3. [DOI] [Google Scholar]
- Gunnarsson O.; Lundqvist B. I. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys. Rev. B 1976, 13, 4274 10.1103/PhysRevB.13.4274. [DOI] [Google Scholar]
- Savin A.; Colonna F.; Pollet R. Adiabatic connection approach to density functional theory of electronic systems. Int. J. Quantum Chem. 2003, 93, 166–190. 10.1002/qua.10551. [DOI] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P.; Yang W. Assessment and formal properties of exchange-correlation functionals constructed from the adiabatic connection. J. Chem. Phys. 2007, 127, 034101 10.1063/1.2749510. [DOI] [PubMed] [Google Scholar]
- Ernzerhof M. Construction of the adiabatic connection. Chem. Phys. Lett. 1996, 263, 499–506. 10.1016/S0009-2614(96)01225-0. [DOI] [Google Scholar]
- Burke K.; Ernzerhof M.; Perdew J. P. The adiabatic connection method: a non-empirical hybrid. Chem. Phys. Lett. 1997, 265, 115–120. 10.1016/S0009-2614(96)01373-5. [DOI] [Google Scholar]
- Colonna F.; Savin A. Correlation energies for some two-and four-electron systems along the adiabatic connection in density functional theory. J. Chem. Phys. 1999, 110, 2828–2835. 10.1063/1.478234. [DOI] [Google Scholar]
- Kohn W.; Becke A. D.; Parr R. G. Density functional theory of electronic structure. J. Phys. Chem. A 1996, 100, 12974–12980. 10.1021/jp960669l. [DOI] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Adamo C.; Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys. 1998, 108, 664–675. 10.1063/1.475428. [DOI] [Google Scholar]
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Görling A.; Levy M. Exact Kohn-Sham scheme based on perturbation theory. Phys. Rev. A 1994, 50, 196 10.1103/PhysRevA.50.196. [DOI] [PubMed] [Google Scholar]
- Görling A.; Levy M. Correlation-energy functional and its high-density limit obtained from a coupling-constant perturbation expansion. Phys. Rev. B 1993, 47, 13105 10.1103/PhysRevB.47.13105. [DOI] [PubMed] [Google Scholar]
- Görling A.; Levy M. Hardness of molecules and the band gap of solids within the Kohn-Sham formalism: A perturbation-scaling approach. Phys. Rev. A 1995, 52, 4493 10.1103/PhysRevA.52.4493. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Møller C.; Plesset M. S. Note on an approximate treatment for many-electron systems. Phys. Rev. 1934, 36, 618–622. 10.1103/PhysRev.46.618. [DOI] [Google Scholar]
- Grimme S.; Neese F. Double-hybrid density functional theory for excited electronic states of molecules. J. Chem. Phys. 2007, 127, 154116 10.1063/1.2772854. [DOI] [PubMed] [Google Scholar]
- Hui K.; Chai J.-D. SCAN-based hybrid and double-hybrid density functionals from models without fitted parameters. J. Chem. Phys. 2016, 144, 044114 10.1063/1.4940734. [DOI] [PubMed] [Google Scholar]
- Brémond É.; Sancho-Garcia J. C.; Perez-Jimenez A. J.; Adamo C. Communication: Double-hybrid functionals from adiabatic-connection: The QIDH model. J. Chem. Phys. 2014, 141, 031101 10.1063/1.4890314. [DOI] [PubMed] [Google Scholar]
- Sharkas K.; Toulouse J.; Savin A. Double-hybrid density-functional theory made rigorous. J. Chem. Phys. 2011, 134, 064113 10.1063/1.3544215. [DOI] [PubMed] [Google Scholar]
- Souvi S. M.; Sharkas K.; Toulouse J. Double-hybrid density-functional theory with meta-generalized-gradient approximations. J. Chem. Phys. 2014, 140, 084107 10.1063/1.4865963. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Hansen A.; Bauer C.; Ehrlich S.; Najibi A.; Grimme S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. 10.1039/C7CP04913G. [DOI] [PubMed] [Google Scholar]
- Cornaton Y.; Franck O.; Teale A. M.; Fromager E. Analysis of double-hybrid density functionals along the adiabatic connection. Mol. Phys. 2013, 111, 1275–1294. 10.1080/00268976.2013.783640. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Seidl A.; Görling A.; Vogl P.; Majewski J. A.; Levy M. Generalized Kohn-Sham schemes and the band-gap problem. Phys. Rev. B 1996, 53, 3764–3774. 10.1103/PhysRevB.53.3764. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Franck O.; Mussard B.; Buksztel A.; Grabowski I.; Luppi E.; Toulouse J. Self-consistent double-hybrid density-functional theory using the optimized-effective-potential method. J. Chem. Phys. 2016, 145, 144102 10.1063/1.4964319. [DOI] [PubMed] [Google Scholar]
- Sharp R. T.; Horton G. K. A Variational Approach to the Unipotential Many-Electron Problem. Phys. Rev. 1953, 90, 317. 10.1103/PhysRev.90.317. [DOI] [Google Scholar]
- Talman J. D.; Shadwick W. F. Optimized effective atomic central potential. Phys. Rev. A 1976, 14, 36–40. 10.1103/PhysRevA.14.36. [DOI] [Google Scholar]
- Śmiga S.; Grabowski I.; Witkowski M.; Mussard B.; Toulouse J. Self-Consistent Range-Separated Density-Functional Theory with Second-Order Perturbative Correction via the Optimized-Effective-Potential Method. J. Chem. Theory Comput. 2020, 16, 211–223. 10.1021/acs.jctc.9b00807. [DOI] [PubMed] [Google Scholar]; PMID: 31816237.
- Dobson J. F.; Wang J.; Gould T. Correlation energies of inhomogeneous many-electron systems. Phys. Rev. B 2002, 66, 081108 10.1103/PhysRevB.66.081108. [DOI] [Google Scholar]
- Constantin L. A.; Pitarke J. M. Adiabatic-connection-fluctuation-dissipation approach to long-range behavior of exchange-correlation energy at metal surfaces: A numerical study for jellium slabs. Phys. Rev. B 2011, 83, 075116 10.1103/PhysRevB.83.075116. [DOI] [Google Scholar]
- Terentjev A. V.; Constantin L. A.; Pitarke J. M. Gradient-dependent exchange-correlation kernel for materials optical properties. Phys. Rev. B 2018, 98, 085123 10.1103/PhysRevB.98.085123. [DOI] [Google Scholar]
- Constantin L. A. Simple effective interaction for dimensional crossover. Phys. Rev. B 2016, 93, 121104 10.1103/PhysRevB.93.121104. [DOI] [Google Scholar]
- Corradini M.; Del Sole R.; Onida G.; Palummo M. Analytical expressions for the local-field factor G (q) and the exchange-correlation kernel K xc (r) of the homogeneous electron gas. Phys. Rev. B 1998, 57, 14569 10.1103/PhysRevB.57.14569. [DOI] [Google Scholar]
- Toulouse J. Simple model of the static exchange-correlation kernel of a uniform electron gas with long-range electron-electron interaction. Phys. Rev. B 2005, 72, 035117 10.1103/PhysRevB.72.035117. [DOI] [Google Scholar]
- Richardson C. F.; Ashcroft N. W. Dynamical local-field factors and effective interactions in the three-dimensional electron liquid. Phys. Rev. B 1994, 50, 8170 10.1103/PhysRevB.50.8170. [DOI] [PubMed] [Google Scholar]
- Bates J. E.; Laricchia S.; Ruzsinszky A. Nonlocal energy-optimized kernel: Recovering second-order exchange in the homogeneous electron gas. Phys. Rev. B 2016, 93, 045119 10.1103/PhysRevB.93.045119. [DOI] [Google Scholar]
- Bates J. E.; Sensenig J.; Ruzsinszky A. Convergence behavior of the random phase approximation renormalized correlation energy. Phys. Rev. B 2017, 95, 195158 10.1103/PhysRevB.95.195158. [DOI] [Google Scholar]
- Ruzsinszky A.; Constantin L. A.; Pitarke J. M. Kernel-corrected random-phase approximation for the uniform electron gas and jellium surface energy. Phys. Rev. B 2016, 94, 165155 10.1103/PhysRevB.94.165155. [DOI] [Google Scholar]
- Dobson J. F.; Wang J. Energy-optimized local exchange-correlation kernel for the electron gas: Application to van der Waals forces. Phys. Rev. B 2000, 62, 10038 10.1103/PhysRevB.62.10038. [DOI] [Google Scholar]
- Görling A. Exact exchange kernel for time-dependent density-functional theory. Int. J. Quantum Chem. 1998, 69, 265–277. 10.1002/(SICI)1097-461X(1998)69:3<265::AID-QUA6>3.0.CO;2-T. [DOI] [Google Scholar]
- Kim Y.-H.; Görling A. Excitonic optical spectrum of semiconductors obtained by time-dependent density-functional theory with the exact-exchange kernel. Phys. Rev. Lett. 2002, 89, 096402 10.1103/PhysRevLett.89.096402. [DOI] [PubMed] [Google Scholar]
- Erhard J.; Bleiziffer P.; Görling A. Power Series Approximation for the Correlation Kernel Leading to Kohn-Sham Methods Combining Accuracy, Computational Efficiency, and General Applicability. Phys. Rev. Lett. 2016, 117, 143002 10.1103/PhysRevLett.117.143002. [DOI] [PubMed] [Google Scholar]
- Patrick C. E.; Thygesen K. S. Adiabatic-connection fluctuation-dissipation DFT for the structural properties of solidsThe renormalized ALDA and electron gas kernels. J. Chem. Phys. 2015, 143, 102802 10.1063/1.4919236. [DOI] [PubMed] [Google Scholar]
- Görling A. Hierarchies of methods towards the exact Kohn-Sham correlation energy based on the adiabatic-connection fluctuation-dissipation theorem. Phys. Rev. B 2019, 99, 235120 10.1103/PhysRevB.99.235120. [DOI] [Google Scholar]
- Seidl M.; Perdew J. P.; Kurth S. Simulation of all-order density-functional perturbation theory, using the second order and the strong-correlation limit. Phys. Rev. Lett. 2000, 84, 5070 10.1103/PhysRevLett.84.5070. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Perdew J. P.; Kurth S. Density functionals for the strong-interaction limit. Phys. Rev. A 2000, 62, 012502 10.1103/PhysRevA.62.012502. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Kurth S.; Seidl M. Exploring the adiabatic connection between weak-and strong-interaction limits in density functional theory. Int. J. Mod. Phys. B 2001, 15, 1672–1683. 10.1142/S0217979201006197. [DOI] [Google Scholar]
- Gori-Giorgi P.; Vignale G.; Seidl M. Electronic zero-point oscillations in the strong-interaction limit of density functional theory. J. Chem. Theory Comput. 2009, 5, 743–753. 10.1021/ct8005248. [DOI] [PubMed] [Google Scholar]
- Liu Z.-F.; Burke K. Adiabatic connection in the low-density limit. Phys. Rev. A 2009, 79, 064503 10.1103/PhysRevA.79.064503. [DOI] [Google Scholar]
- Liu Z.-F.; Burke K. Adiabatic connection for strictly correlated electrons. J. Chem. Phys. 2009, 131, 124124 10.1063/1.3239472. [DOI] [PubMed] [Google Scholar]
- Magyar R.; Terilla W.; Burke K. Accurate adiabatic connection curve beyond the physical interaction strength. J. Chem. Phys. 2003, 119, 696–700. 10.1063/1.1579465. [DOI] [Google Scholar]
- Sun J. Extension to Negative Values of the Coupling Constant of Adiabatic Connection for Interaction-Strength Interpolation. J. Chem. Theory Comput. 2009, 5, 708–711. 10.1021/ct800515w. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Gori-Giorgi P. Adiabatic connection at negative coupling strengths. Phys. Rev. A 2010, 81, 012508 10.1103/PhysRevA.81.012508. [DOI] [Google Scholar]
- Mirtschink A.; Seidl M.; Gori-Giorgi P. Energy densities in the strong-interaction limit of density functional theory. J. Chem. Theory Comput. 2012, 8, 3097–3107. 10.1021/ct3003892. [DOI] [PubMed] [Google Scholar]
- Gori-Giorgi P.; Seidl M. Density functional theory for strongly-interacting electrons: perspectives for physics and chemistry. Phys. Chem. Chem. Phys. 2010, 12, 14405–14419. 10.1039/c0cp01061h. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Irons T. J.; Savin A.; Teale A. M.; Gori-Giorgi P. Exchange-correlation functionals via local interpolation along the adiabatic connection. J. Chem. Theory Comput. 2016, 12, 2598–2610. 10.1021/acs.jctc.6b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiano E.; Gori-Giorgi P.; Seidl M.; Della Sala F. Interaction-strength interpolation method for main-group chemistry: benchmarking, limitations, and perspectives. J. Chem. Theory Comput. 2016, 12, 4885–4896. 10.1021/acs.jctc.6b00713. [DOI] [PubMed] [Google Scholar]
- Giarrusso S.; Gori-Giorgi P.; Della Sala F.; Fabiano E. Assessment of interaction-strength interpolation formulas for gold and silver clusters. J. Chem. Phys. 2018, 148, 134106 10.1063/1.5022669. [DOI] [PubMed] [Google Scholar]
- Fabiano E.; Śmiga S.; Giarrusso S.; Daas T. J.; Della Sala F.; Grabowski I.; Gori-Giorgi P. Investigation of the Exchange-Correlation Potentials of Functionals Based on the Adiabatic Connection Interpolation. J. Chem. Theory Comput. 2019, 15, 1006–1015. 10.1021/acs.jctc.8b01037. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Gori-Giorgi P.; Della Sala F.; Fabiano E. Restoring size consistency of approximate functionals constructed from the adiabatic connection. J. Phys. Chem. Lett. 2018, 9, 3137. 10.1021/acs.jpclett.8b01054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooi D. P.; Gori-Giorgi P. Local and global interpolations along the adiabatic connection of DFT: a study at different correlation regimes. Theor. Chem. Acc. 2018, 137, 166 10.1007/s00214-018-2354-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Bahmann H.; Ernzerhof M. Construction of exchange-correlation functionals through interpolation between the non-interacting and the strong-correlation limit. J. Chem. Phys. 2015, 143, 124103 10.1063/1.4931160. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Giarrusso S.; Vuckovic S.; Fabiano E.; Gori-Giorgi P. Communication: Strong-interaction limit of an adiabatic connection in Hartree-Fock theory. J. Chem. Phys. 2018, 149, 241101 10.1063/1.5078565. [DOI] [PubMed] [Google Scholar]
- Constantin L. A. Correlation energy functionals from adiabatic connection formalism. Phys. Rev. B 2019, 99, 085117 10.1103/PhysRevB.99.085117. [DOI] [Google Scholar]
- Vuckovic S.; Irons T. J.; Wagner L. O.; Teale A. M.; Gori-Giorgi P. Interpolated energy densities, correlation indicators and lower bounds from approximations to the strong coupling limit of DFT. Phys. Chem. Chem. Phys. 2017, 19, 6169–6183. 10.1039/C6CP08704C. [DOI] [PubMed] [Google Scholar]
- Vuckovic S.; Gori-Giorgi P. Simple Fully Nonlocal Density Functionals for Electronic Repulsion Energy. J. Phys. Chem. Lett. 2017, 8, 2799–2805. 10.1021/acs.jpclett.7b01113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J. P.; Staroverov V. N.; Tao J.; Scuseria G. E. Density functional with full exact exchange, balanced nonlocality of correlation, and constraint satisfaction. Phys. Rev. A 2008, 78, 052513 10.1103/PhysRevA.78.052513. [DOI] [Google Scholar]
- Śmiga S.; Constantin L. A. Unveiling the Physics behind Hybrid Functionals. J. Phys. Chem. A 2020, PMID: 32551627 10.1021/acs.jpca.0c04156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śmiga S.; Constantin L. A.. Supporting Information.
- Perdew J. P.; Kurth S.; Zupan A.; Blaha P. Accurate density functional with correct formal properties: A step beyond the generalized gradient approximation. Phys. Rev. Lett. 1999, 82, 2544 10.1103/PhysRevLett.82.2544. [DOI] [Google Scholar]
- Constantin L. A.; Terentjevs A.; Della Sala F.; Fabiano E. Gradient-dependent upper bound for the exchange-correlation energy and application to density functional theory. Phys. Rev. B 2015, 91, 041120 10.1103/PhysRevB.91.041120. [DOI] [Google Scholar]
- Lewin M.; Lieb E. H. Improved Lieb-Oxford exchange-correlation inequality with a gradient correction. Phys. Rev. A 2015, 91, 022507 10.1103/PhysRevA.91.022507. [DOI] [Google Scholar]
- Tao J.; Staroverov V. N.; Scuseria G. E.; Perdew J. P. Exact-exchange energy density in the gauge of a semilocal density-functional approximation. Phys. Rev. A 2008, 77, 012509 10.1103/PhysRevA.77.012509. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Fabiano E. Approximate solution of coupled cluster equations: application to the coupled cluster doubles method and non-covalent interacting systems. Phys. Chem. Chem. Phys. 2017, 19, 30249–30260. 10.1039/C7CP06417A. [DOI] [PubMed] [Google Scholar]
- Stanton J. F.et al. ACES II; Quantum Theory Project: Gainesville, Florida, 2007. [Google Scholar]
- Grabowski I.; Teale A. M.; Śmiga S.; Bartlett R. J. Comparing ab initio density-functional and wave function theories: The impact of correlation on the electronic density and the role of the correlation potential. J. Chem. Phys. 2011, 135, 114111 10.1063/1.3636114. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Della Sala F.; Buksztel A.; Grabowski I.; Fabiano E. Accurate Kohn-Sham ionization potentials from scaled-opposite-spin second-order optimized effective potential methods. J. Comput. Chem. 2016, 37, 2081–2090. 10.1002/jcc.24436. [DOI] [PubMed] [Google Scholar]
- Grabowski I.; Fabiano E.; Teale A. M.; Śmiga S.; Buksztel A.; Della Sala F. D. Orbital-dependent second-order scaled-opposite-spin correlation functionals in the optimized effective potential method. J. Chem. Phys. 2014, 141, 024113 10.1063/1.4887097. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Marusiak V.; Grabowski I.; Fabiano E. The ab initio density functional theory applied for spin-polarized calculations. J. Chem. Phys. 2020, 152, 054109 10.1063/1.5128933. [DOI] [PubMed] [Google Scholar]
- Görling A. New KS method for molecules based on an exchange charge density generating the exact local KS exchange potential. Phys. Rev. Lett. 1999, 83, 5459–5462. 10.1103/PhysRevLett.83.5459. [DOI] [Google Scholar]
- Ivanov S.; Hirata S.; Bartlett R. J. Exact exchange treatment for molecules in finite-basis-set Kohn-Sham theory. Phys. Rev. Lett. 1999, 83, 5455–5458. 10.1103/PhysRevLett.83.5455. [DOI] [Google Scholar]
- Grabowski I.; Teale A. M.; Fabiano E.; Smiga S.; Buksztel A.; Della Sala F. D. A density difference based analysis of orbital-dependent exchange-correlation functionals. Mol. Phys. 2014, 112, 700–710. 10.1080/00268976.2013.854424. [DOI] [Google Scholar]
- Mori-Sánchez P.; Wu Q.; Yang W. Orbital-dependent correlation energy in density-functional theory based on a second-order perturbation approach: Success and failure. J. Chem. Phys. 2005, 123, 062204 10.1063/1.1904584. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Engel E. Second-order Kohn-Sham perturbation theory: Correlation potential for atoms in a cavity. J. Chem. Phys. 2005, 123, 224102 10.1063/1.2128674. [DOI] [PubMed] [Google Scholar]
- Raghavachari K.; Trucks G. W.; Pople J. A.; Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. 10.1016/S0009-2614(89)87395-6. [DOI] [Google Scholar]
- Bartlett R. J.; Grabowski I.; Hirata S.; Ivanov S. The exchange-correlation potential in ab initio density functional theory. J. Chem. Phys. 2005, 122, 034104 10.1063/1.1809605. [DOI] [PubMed] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Redfern P. C.; Pople J. A. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys. 1997, 106, 1063–1079. 10.1063/1.473182. [DOI] [Google Scholar]
- Lynch B. J.; Truhlar D. G. Small representative benchmarks for thermochemical calculations. J. Phys. Chem. A 2003, 107, 8996–8999. 10.1021/jp035287b. [DOI] [Google Scholar]
- Haunschild R.; Klopper W. Theoretical reference values for the AE6 and BH6 test sets from explicitly correlated coupled-cluster theory. Theor. Chem. Acc. 2012, 131, 1112 10.1007/s00214-012-1112-3. [DOI] [Google Scholar]
- Tentscher P. R.; Arey J. S. Geometries and Vibrational Frequencies of Small Radicals: Performance of Coupled Cluster and More Approximate Methods. J. Chem. Theory Comput. 2012, 8, 2165–2179. 10.1021/ct300194x. [DOI] [PubMed] [Google Scholar]; PMID: 26593847.
- Lynch B. J.; Truhlar D. G. Robust and Affordable Multicoefficient Methods for Thermochemistry and Thermochemical Kinetics: The MCCM/3 Suite and SAC/3. J. Phys. Chem. A 2003, 107, 3898–3906. 10.1021/jp0221993. [DOI] [Google Scholar]
- Soyda E.; Bozkaya U. Assessment of Orbital-Optimized MP2.5 for Thermochemistry and Kinetics: Dramatic Failures of Standard Perturbation Theory Approaches for Aromatic Bond Dissociation Energies and Barrier Heights of Radical Reactions. J. Chem. Theory Comput. 2015, 11, 1564–1573. 10.1021/ct501184w. [DOI] [PubMed] [Google Scholar]; PMID: 26574366.
- Bozkaya U.; Sherrill C. D. Analytic energy gradients for the orbital-optimized second-order Møller-Plesset perturbation theory. J. Chem. Phys. 2013, 138, 184103 10.1063/1.4803662. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Śmiga S.; Grabowski I. Spin-Component-Scaled ΔMP2 Parametrization: Toward a Simple and Reliable Method for Ionization Energies. J. Chem. Theory Comput. 2018, 14, 4780–4790. 10.1021/acs.jctc.8b00638. [DOI] [PubMed] [Google Scholar]; PMID: 30040889.
- Constantin L. A.; Fabiano E.; Śmiga S.; Della Sala F. Jellium-with-gap model applied to semilocal kinetic functionals. Phys. Rev. B 2017, 95, 115153 10.1103/PhysRevB.95.115153. [DOI] [Google Scholar]
- Śmiga S.; Fabiano E.; Constantin L. A.; Della Sala F. Laplacian-dependent models of the kinetic energy density: Applications in subsystem density functional theory with meta-generalized gradient approximation functionals. J. Chem. Phys. 2017, 146, 064105 10.1063/1.4975092. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Fabiano E.; Laricchia S.; Constantin L. A.; Della Sala F. Subsystem density functional theory with meta-generalized gradient approximation exchange-correlation functionals. J. Chem. Phys. 2015, 142, 154121 10.1063/1.4917257. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Constantin L. A.; Della Sala F.; Fabiano E. The Role of the Reduced Laplacian Renormalization in the Kinetic Energy Functional Development. Computation 2019, 7, 65 10.3390/computation7040065. [DOI] [Google Scholar]
- Kendall R. A.; Dunning T. H. Jr.; Harrison R. J. Electron affinities of the first row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. 10.1063/1.462569. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. Design of Density Functionals That Are Broadly Accurate for Thermochemi stry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 109, 5656–5667. 10.1021/jp050536c. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Benchmark Databases for Nonbonded Interactions and Their Use To Test Density Functional Theory. J. Chem. Theory Comput. 2005, 1, 415–432. 10.1021/ct049851d. [DOI] [PubMed] [Google Scholar]
- Wesolowski T. A.; Chermette H.; Weber J. Accuracy of approximate kinetic energy functionals in the model of Kohn-Sham equations with constrained electron density: The FH [center-dot] [center-dot] [center-dot] NCH complex as a test case. J. Chem. Phys. 1996, 105, 9182–9190. 10.1063/1.472823. [DOI] [Google Scholar]
- Fabiano E.; Constantin L. A.; Della Sala F. Wave Function and Density Functional Theory Studies of Dihydrogen Complexes. J. Chem. Theory Comput. 2014, 10, 3151–3162. 10.1021/ct500350n. [DOI] [PubMed] [Google Scholar]
- Kestner N. R.; Sinanog̅lu O. Study of Electron Correlation in Helium-Like Systems Using an Exactly Soluble Model. Phys. Rev. 1962, 128, 2687–2692. 10.1103/PhysRev.128.2687. [DOI] [Google Scholar]
- Matito E.; Cioslowski J.; Vyboishchikov S. F. Properties of harmonium atoms from FCI calculations: Calibration and benchmarks for the ground state of the two-electron species. Phys. Chem. Chem. Phys. 2010, 12, 6712–6716. 10.1039/b926389f. [DOI] [PubMed] [Google Scholar]
- Grabowski I.; Lotrich V. Accurate orbital-dependent correlation and exchange-correlation potentials from non-iterative ab initio dft calculations. Mol. Phys. 2005, 103, 2085–2092. 10.1080/00268970500131462. [DOI] [Google Scholar]
- Fabiano E.; Della Sala F. Localized exchange-correlation potential from second-order self-energy for accurate Kohn-Sham energy gap. J. Chem. Phys. 2007, 126, 214102 10.1063/1.2735300. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Siecińska S.; Fabiano E. Methods to generate reference total and Pauli kinetic potentials. Phys. Rev. B 2020, 101, 165144 10.1103/PhysRevB.101.165144. [DOI] [Google Scholar]
- Taut M. Two electrons in a homogeneous magnetic field: particular analytical solutions. J. Phys. A: Math. Gen. 1994, 27, 1045. 10.1088/0305-4470/27/3/040. [DOI] [Google Scholar]
- Ludeña E. V.; Gómez D.; Karasiev V.; Nieto P. Exact analytic total energy functional for Hooke’s atom generated by local-scaling transformations. Int. J. Quantum Chem. 2004, 99, 297–307. 10.1002/qua.10858. [DOI] [Google Scholar]
- Amovilli C.; March N. Exact density matrix for a two-electron model atom and approximate proposals for realistic two-electron systems. Phys. Rev. A 2003, 67, 022509 10.1103/PhysRevA.67.022509. [DOI] [Google Scholar]
- O’Neill D. P.; Gill P. M. Wave functions and two-electron probability distributions of the Hooke’s-law atom and helium. Phys. Rev. A 2003, 68, 022505 10.1103/PhysRevA.68.022505. [DOI] [Google Scholar]
- Das G.; Wahl A. C. Theoretical study of the F2 molecule using the method of optimized valence configurations. J. Chem. Phys. 1972, 56, 3532–3540. 10.1063/1.1677728. [DOI] [Google Scholar]
- Grossi J.; Seidl M.; Gori-Giorgi P.; Giesbertz K. J. H. Functional derivative of the zero-point-energy functional from the strong-interaction limit of density-functional theory. Phys. Rev. A 2019, 99, 052504 10.1103/PhysRevA.99.052504. [DOI] [Google Scholar]
- Grossi J.; Musslimani Z. H.; Seidl M.; Gori-Giorgi P.. Kohn-Sham Equations with Functionals from the Strictly-Correlated Regime: Investigation with a Spectral Renormalization Method. arXiv:2004.10436. arXiv.org e-Print archive. https://arxiv.org/abs/2004.10436 (submitted April 22, 2020). [DOI] [PubMed]
- Grimme S. Improved second-order Møller-Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. 10.1063/1.1569242. [DOI] [Google Scholar]
- Jung Y.; Lochan R. C.; Dutoi A. D.; Head-Gordon M. Scaled opposite-spin second order Møller-Plesset correlation energy: An economical electronic structure method. J. Chem. Phys. 2004, 121, 9793–9802. 10.1063/1.1809602. [DOI] [PubMed] [Google Scholar]
- Witkowski M.; Śmiga S.; Grabowski I.. Density-Based Analysis of Spin-Resolved MP2 Method. In Adv. Quantum Chem., Hoggan P. E., Ed.; Novel Electronic Structure Theory: General Innovationsand Strongly Correlated Systems 1st Edition; Academic Press, 2018; Vol. 76. [Google Scholar]
- Buksztel A.; Śmiga S.; Grabowski I.. Chapter Fourteen - The Correlation Effects in Density Functional Theory Along the Dissociation Path. In Adv. Quantum Chem., Hoggan P. E.; Ozdogan T., Eds.; Electron Correlation in Molecules - Ab Initio Beyond Gaussian Quantum Chemistry; Academic Press, 2016; Vol. 73, pp 263–283. [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Sun J.; Ruzsinszky A.; Perdew J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402 10.1103/PhysRevLett.115.036402. [DOI] [PubMed] [Google Scholar]
- Tao J.; Mo Y. Accurate semilocal density functional for condensed-matter physics and quantum chemistry. Phys. Rev. Lett. 2016, 117, 073001 10.1103/PhysRevLett.117.073001. [DOI] [PubMed] [Google Scholar]
- Constantin L. A.; Fabiano E.; Della Sala F. Semilocal Pauli-Gaussian kinetic functionals for orbital-free density functional theory calculations of solids. J. Phys. Chem. Lett. 2018, 9, 4385–4390. 10.1021/acs.jpclett.8b01926. [DOI] [PubMed] [Google Scholar]
- Constantin L. A. Semilocal properties of the Pauli kinetic potential. Phys. Rev. B 2019, 99, 155137 10.1103/PhysRevB.99.155137. [DOI] [Google Scholar]
- Patra B.; Jana S.; Constantin L. A.; Samal P. Relevance of the Pauli kinetic energy density for semilocal functionals. Phys. Rev. B 2019, 100, 155140 10.1103/PhysRevB.100.155140. [DOI] [Google Scholar]
- Perdew J. P.; Tao J.; Staroverov V. N.; Scuseria G. E. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. J. Chem. Phys. 2004, 120, 6898–6911. 10.1063/1.1665298. [DOI] [PubMed] [Google Scholar]
- Constantin L. A.; Fabiano E.; Pitarke J. M.; Della Sala F. Semilocal density functional theory with correct surface asymptotics. Phys. Rev. B 2016, 93, 115127 10.1103/PhysRevB.93.115127. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.