

Abstract
Microbiota live in a closely regulated interaction with their environment, and vice versa. The presence and absence of microbial entities is greatly influenced by features of the niche in which they thrive. Characteristic of this phenomenon is that different human skin sites harbor niche‐specific communities of microbes. Microbial diversity is considerable, and the current challenge lies in determining which microbes and (corresponding) functionality are of importance to a given ecological niche. Furthermore, as there is increasing evidence of microbial involvement in health and disease, the need arises to fundamentally understand microbiome processes for application in health care, nutrition and personal care products (e.g. diet, cosmetics, probiotics). This review provides a current overview of state‐of‐the‐art sequencing‐based techniques and corresponding data analysis methodology for profiling of complex microbial communities. Furthermore, we also summarize the existing knowledge regarding cutaneous microbiota and their human host for a wide range of skin diseases.
Keywords: bioinformatics, cutaneous diseases, microbiomics, next‐generation sequencing, skin microbiota
Microorganisms are Omnipresent
Consortia of microbes are found in many niches of the earth, like on various sites of animals and plants, in soil, in water and in the atmosphere, but also in industrial fermentations and biofilms. In this review, we will focus on the human skin microbiota (mainly on bacteria), their currently known relevance in health and disease, and provide an overview of main sequencing‐based methods and bioinformatic tools to measure them. The relevance of bacteria for human life becomes clear by looking at the numbers, as bacteria colonize our body sites with a ratio of 1.3:1 bacterium to human cell. 1 Even if they did not evoke an effect on bodily processes – but which many of them do – their sheer number in members, genes and variation relative to their host alone, make them interesting subjects of research.
Next‐Generation Sequencing of Microbiota
Currently, next‐generation sequencing (NGS) advances allow for high‐throughput, massively parallel and deep sequencing of DNA samples, thereby dismissing the need for vector‐based cloning of sequences. NGS has given an enormous boost to the field of genomics, microbiomics and bioinformatics, amongst others, mainly due to its substantially reduced sequencing costs and ultra‐high‐throughput application. Bacterial consortia are currently mostly analyzed either by marker gene sequencing (MGS) metataxonomics or by whole‐genome sequencing (WGS, in this review also referred to as “shotgun”) metagenomics (Fig. 1). Metagenomics is defined as the study of the collection of genomes and genes from the members of a microbiome. 2 It is not to be confused with MGS initiatives such as bacterial 16S (or its fungal counterpart internal transcribed spacer) amplicon sequencing, which are better described as metataxonomics: the high‐throughput identification, classification and naming of microbiota.
Figure 1.
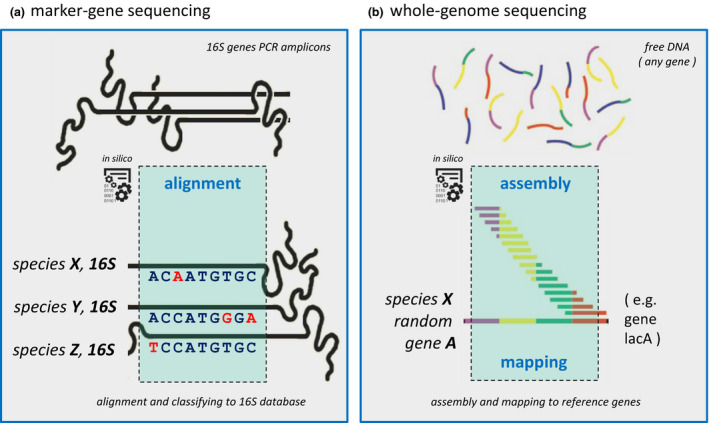
Principal microbiota sequencing approaches. We roughly distinguish two main applications for next‐generation sequencing (NGS) of microbial communities: marker‐gene sequencing (MGS) for metataxonomics and whole‐genome sequencing (WGS) for metagenomics. (a) In the MGS example, 16S is selected as marker gene, which is extracted from a mixed microbial population by polymerase chain reaction (PCR, not shown), and sequenced by NGS techniques. After MGS sequencing, reads (±500 bp) are aligned, and based on informative positional differences in the 16S gene, known reference microbiota can be assigned, or novel taxonomies can be inferred. With WGS, one can extract genomic potential and function information, in contrast with MGS, as with the latter, one can only extract taxonomic information. (b) In the WGS example, typically small sequences (100–150 bp), derived randomly from the full genomic content (i.e. all genes present, not focusing only on 16S) of a mixed microbial population, are assembled into genes of all microbiota present.
MGS
There are many types of marker genes that can be utilized for bacterial MGS, but the gene which is most widely chosen for this purpose is the universal 16S rRNA gene (16S, in short). In this section we will therefore focus on 16S, but the principles and application of alternative marker genes are similar; for example, see Scholz et al. 3 who use an alternative gene to specifically type Cutibacterium acnes species in high resolution. For more information on the use of alternative bacterial marker genes, we refer to our recently published generic workflow for discovery and analysis of single‐locus sequence typing (SLST) marker genes. 4 Please note that a marker gene in the context of bacterial MGS should not be confused with marker genes in the context of molecular cloning, where the term “marker gene” has been adopted for genes used to indicate successful genome editing. 16S MGS focuses on the 16S rRNA genes present in all prokaryotes and archaea. In theory, the 16S rRNA gene can be targeted by universal polymerase chain reaction (PCR) primers, and the technique does therefore not require bacterial reference genomes for analysis. However, for classifying 16S sequencing reads, prior knowledge in the form of 16S rRNA gene databases with corresponding taxonomy information is required. Most notable, the Ribosomal Database Project, 5 Greengenes 6 and SILVA 7 are well‐established examples of such databases. 16S allows for confident profiling of bacteria down to the genus level. The process of analyzing 16S sequencing reads usually involves clustering of sequencing reads in operational taxonomic units (OTU), where they are classified (Fig. 1a). Although OTU picking is very commonly applied when analyzing 16S data, there are some non‐OTU‐based alternatives for analyzing microbiomes, such as by oligotyping or by using exact sequence variants as suggested in a recent review by Knight et al. 8 Oligotyping allows for discriminating between closely related but distinct taxa by looking at position‐specific information in 16S rRNA sequences. In practice, for 16S, there is a delicate trade‐off between taxonomic sensitivity and resolution (specificity) in terms of microbiota classification (the “sensitivity‐to‐classification” problem). Hence, determining which 16S primers to use is crucial (Fig. 2a). The 16S rRNA gene has multiple alternating conserved and variable (V1–V9) regions, with a total length of roughly 1.5 kbp (Fig. 2b). For application in sequencing, the longer the 16S sequencing read the more confidently microbial classification can be performed. Therefore, depending on the sequencing platform, one best selects those V‐regions that maximize potential of their sequencing platform with regard to read length. In case of low input DNA for 16S amplification by PCR, which is a notorious problem for skin samples, one has the option to choose a nested PCR (or two‐step PCR). First performing a PCR to amplify a large 16S regions such as V3–V6, and a second round of PCR to amplify another smaller region within the large region, such as V3–V4, can be beneficial to boost MGS amplicon yield as input for sequencing. Furthermore, every 16S primer (combination) has its pros and cons when it comes to sensitivity and specificity to different bacterial families (genera). 9 Primer choice also depends on the target niche of interest, such as for gut, skin or oral samples, which therefore is additionally motivated by bacteria typically present on those sites. 10 To analyze MGS data, many different methods and bioinformatic tools are available. OTU methods like QIIME, 11 which by default uses the UCLUST OTU clustering algorithm, 12 and Mothur, 13 based on average linkage OTU clustering, are widely used and commonly accepted. 14 , 15
Figure 2.
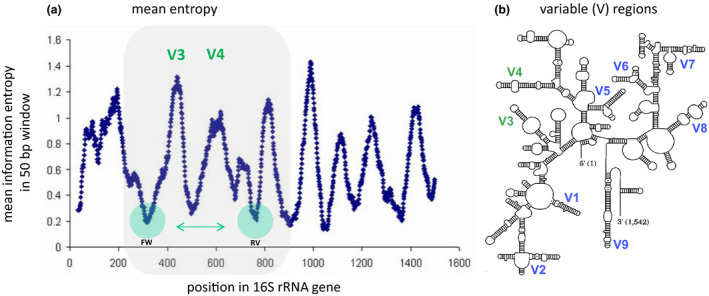
16S rRNA marker gene characteristics. The 16S rRNA gene in bacteria is widely used for metataxonomics. Between different clades of phylogenetically‐related bacteria, this gene varies strongly in terms of conservation and variation, as shown in the left panel by the consecutive peaks and valleys in that graph. (a) Visualization of the mean information entropy for each position of the 16S gene (±1.5 kbp in length), based on all known 16S genes present in the Ribosomal Database Project. One can see peaks (strong variation) and valleys (strong conservation) in different regions of the 16S gene sequence, regions which can be used for taxonomic discrimination and primer design, respectively. In this example, we observe nine peaks (variable regions), of which V3 and V6 show the largest peaks and deepest valleys. Therefore, the gene region from V3 up to V6 is very suitable for primer design and marker‐gene sequencing. Nonetheless, as most currently applied short‐read next‐generation sequencing applications are not able to sequence for more than 500 bp, like for Illumina MiSeq, one has to choose for a shorter region length, such as for V1–V2, or for V3–V4 as illustrated here. Figure adapted from Andersson et al. 81 . (b) The nine different variable regions, structurally visualized over the full length of the 16S gene. The locations of V3 and V4 are indicated in green.
Metagenome Sequencing
Shotgun metagenomics, in contrast to MGS, does not suffer from the aforementioned sensitivity‐to‐classification problem as it sequences all (free) DNA present in a sample (Fig. 1b). 16 The major advantage of shotgun over MGS is that it provides insight into gene and metabolic function potential of a sample based on its metagenome. This allows for pathway analysis and even mining for virulence factors, antibiotic resistance, (pathogen) lineage‐specific markers or novel enzymes for catalysis of reactions in food, pharmaceutical or industrial processes. Alternatively, complete bacterial genomes can be extracted from shotgun samples if the metagenomics data is assembled, provided that the genomes are present in high enough numbers to achieve a minimum feasible reads‐to‐genome coverage to allow for confident assembly. 17 Furthermore, apart from bacteria and fungi, shotgun enables retrieval of DNA sequences from viruses (i.e. non‐RNA), bacteriophages, eukaryotes and even host cells. The latter can be problematic in terms of contamination. In cutaneous microbiome studies, this can be a notorious problem, as skin has a low microbe‐to‐host ratio resulting in few bacteria and a large number of dead host cells that are easily taken along upon skin sampling. 16 Therefore, minimizing host DNA contamination during microbial DNA sampling is of importance to yield high‐quality sequencing data. For skin, depending on the specific research question at hand, one may want to minimize mechanical disruption of the skin by taking skin (wet) swabs, in contrast to the skin scraping alternative. Instead, if measuring microbiota from deeper skin layers is desirable (e.g. to inspect deeper layers or to minimize measuring transient microbiota), one may want to choose from methods that use skin scraping by a sterile scalpel or by tape stripping. Unfortunately, shotgun needs much more microbial DNA input than MGS, is very labor intensive for both personnel and computation, and should therefore ideally be applied only when there is a specific hypothesis to be tested: for example, is a particular (new) functionality such as antibiotic resistance or virulence factors present, and is it linked to certain bacteria? Typically, shotgun metagenomic dataset resolution allow for classification to the level of genus/species with tools such as MetaPhlAn, 18 MEGAN, 19 AMPHORA 20 and mOTUs, 21 or even down to subspecies level with tools such as ConStrains, 22 PanPhlAn 23 and StrainPhlAn. 24 However, these methods can be used confidently only for (sub)species with an overall relative abundance of 1% or above. With shotgun, it is in principle possible to determine the presence of entities close to the strain level, but this requires complex methods and typically large datasets are involved.
Recently, the concept of multi‐locus sequence typing has been adopted for high‐resolution bacterial classification in NGS efforts by initiatives such as PathoScope 25 and PhyloPhlAn 26 that provide sets of representative marker genes and algorithms for phylogenetic inference of sequencing reads. Likewise, metagenomic analysis methods such as AMPHORA, ConStrains, MetaPhlAn and StrainPhlAn effectively apply in high throughput the concept of single nucleotide variant patterns in marker genes. For pathway reconstruction and analysis of potential functions of microbiota, various bioinformatic tools exist and can be adopted, most notably MG‐RAST 27 and HUMAnN (includes ChocoPhlAn), 18 although some of the aforementioned tools for microbial classification also offer a function analysis to some extent. Noteworthy, methods such as PICRUSt, 28 PanFP 29 and Tax4Fun 30 are alternatives to shotgun in order to yield information about microbial function from 16S sequencing data. It deduces the presence of bacteria based on 16S reads, and infers from this information the total metabolic potential of a microbiome with a taxon‐to‐function reference database. Albeit conceivably inferior to shotgun metagenomics, it does provide a coarse‐grained overview of the metabolic capacity of a microbiome. In opposite fashion, one is able to “mine” 16S rRNA or other marker genes from shotgun data, which could be interesting for a quick screen, or when there is specific interest in one particular phylogenetic clade or (associated) function.
Microbial strains are the most specific source of information with regard to metabolic and regulatory potential of a microbiome, and follow‐up experiments are straight‐forward to perform with (combinations of) microbial strains. Hence, pinpointing the right candidate strains from studies with access to shotgun data is crucial; however, getting enough (DNA) material for WGS can be a challenge for some biological sources like low microbial density skin sites. Nevertheless, new WGS techniques with larger data output are emerging rapidly, and altogether the microbiome community seems to be on the verge of a shift from MGS to shotgun as a general application for profiling of microbiota.
Cutaneous Microbiome
Simply put, the human skin is a physical barrier of the body that has one main purpose: to keep the inside in, and the outside out. In addition, the skin functions as an immunological barrier with processes like microbial colonization resistance by the skin microbiome, and host immune sensing and surveillance. 31 The skin is anatomically comprised of several layers with different cells and properties. The epidermis largely (>90%) consists of keratinocytes, an epithelial cell type with barrier and host defense functions, and can be divided into different strata. The stratum basale is the germinative layer from which the cells migrate to the skin surface in approximately 20 days. Ultimately, in the last living cell layer of the epidermis (stratum granulosum), cells excrete large amounts of lipids, form a cross‐linked cell envelope and lose their nuclei. These enucleated cornified envelopes form the stratum corneum. The stratum corneum is where most of the skin microorganisms reside, and the deeper into the stratum corneum the less microbes are present. 16 The estimated microbial density on the total skin surface (from all body parts combined) is very low compared with the entire large intestine, but it is comparable with quantities found in the complete small intestine, with estimates of 1011 bacterial cells in total. 1 Note that this calculation does not take into account microbiota from the deeper layers, only from the skin surface. Normally, the epidermal layers beneath the stratum corneum and the dermis are considered sterile with the exception of sweat and sebaceous glands and hair follicles, which are also colonized by microbes. 32
Microbial make up of skin niches is highly dependent on characteristics based on skin type and location. We distinguish three main physiological skin sites: (i) oily/sebaceous skin sites such as the forehead, the upper back and the skin behind the ear; (ii) dry skin sites such as the forearm and lower back; and (iii) moist skin sites such as the armpits, backs of knees, nostrils and groin. But also acidity (pH), salt content and temperature of the microenvironments are important drivers for microbial inhabitants. Despite site‐to‐site compositional variation, common skin commensals typically found on humans are the genera Corynebacterium, Cutibacterium, Staphylococcus, Micrococcus, Actinomyces, Streptococcus and Prevotella. 16 , 33 Even though interindividual variation between healthy volunteers is high, microbial communities are largely stable over time, despite exposure of the skin to the external environment. 34 , 35 However, mechanical disruption of the skin barrier results in temporary change in microbial composition up until recovery of the skin. 16
Both the stratum corneum and the differentiated cell layers of the epidermis are essential to control microbial invasion by providing a physical barrier and an antimicrobial protein (AMP) shield. Keratinocytes express many AMP in order to control skin microbiota colonization and infection. Notable examples are psoriasin (S100A7) and human β‐defensins (hBD‐2) that target Gram‐negative bacteria such as Escherichia coli and Pseudomonas aeruginosa. Other important skin AMP are SKALP/elafin, hBD‐3, SLPI, calprotectin (complex of S100A8/S100A9), LL37 (CAMP), lysozyme, RNase‐7, and the recently reported group of late cornified envelope (LCE) proteins. 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43
Cutaneous Microbiome and Inflammatory Skin Conditions
Over the past years, many studies have been conducted on lesional skin of common skin diseases to identify which species are most abundant and whether this microbial composition differs from healthy skin. Although bacteria are normally presented as commensal microbes, in certain situations (e.g. when the skin barrier is compromised or the immune system is failing), some of these bacterial species can become opportunistic pathogens that give rise to infections. Perturbations of “normal” microbial communities where homeostasis between the host and its microbiota are disturbed is called “dysbiosis”. Many common skin conditions are associated with a dysbiotic state of the skin microbiota, with the most research conducted on the inflammatory skin disease atopic dermatitis (AD). 44
AD
Atopic dermatitis is a common chronic skin condition mostly affecting infants, and is characterized by pruritic inflammatory skin patches. The risk factors for development of AD are multifactorial, ranging from environmental to genetic susceptibility factors. The disease pathophysiology is complex, with impaired epidermal barrier function, T‐helper (Th)2 cell‐mediated inflammation and neuroimmune interactions which have been reviewed extensively by Weidinger et al. 45 In addition to the aforementioned contributing factors to AD development, dysbiosis of the skin microbiota has been described already in the 1970s, where it was shown that Staphylococcus aureus is overrepresented on the skin of AD patients. In addition, whereas the healthy cutaneous microbiota composition is diverse, the composition of lesional AD skin is far less diverse and consists mainly of S. aureus, a bacterium that in healthy situations can be a commensal on the skin and mucous membranes. 46 S. aureus can become an opportunistic pathogen with a plethora of weaponry to make sure it adheres to the skin, 47 weakens the skin barrier 48 and triggers the immune system, 49 which eventually exacerbates the cutaneous inflammation (as reviewed recently by Paller et al.). 50 Interestingly, a study by Chng et al. 51 recently showed that the bacterial composition of AD‐prone, non‐flare skin is more diverse than the composition of healthy skin. An enriched abundance of Streptococcus, Gemella and Haemophilus species and decreased abundance of Dermacoccus species was presented on AD‐prone, non‐flare skin versus normal healthy skin. Whereas it is known that Staphylococcus species can invoke a strong cytokine and chemokine response in order to induce inflammation, the bacteria present in AD flare‐prone skin seem to mute this response, suggesting a protective role for these bacteria. 51 Recently, a large cohort study was presented that combined skin microbiomes (AD and psoriasis) and associated host transcriptomes. 52 Unique gene profiles were identified that distinguish healthy from inflamed skin, and colonization of AD skin by S. aureus was associated with dysregulation of genes involved in epithelial barrier function, immune activation and tryptophan metabolism. 52 Furthermore, lesional AD skin could be more amenable for colonization by S. aureus because of an imbalanced antimicrobial response. Where healthy skin expresses many different antimicrobial molecules (e.g. β‐defensins, cathelicidins, free fatty acids and reactive oxygen species), the skin of AD patients has been shown to have a reduced expression of defensins and cathelicidins. 53 This could partly be explained by Th2 cytokines which have been shown to suppress the expression of such AMP. 54 , 55 Recently, it was reported that coal tar treatment of AD lesions induces AMP production via canonical aryl hydrocarbon receptor (AHR) signaling. 56 By application of a novel SLST MGS method, 4 a shift in microbiota composition toward that of healthy controls was observed, which suggests that restoring AMP levels in AD skin via AHR‐dependent transcription regulation can be beneficial by creating an (anti)microbial milieu that is less prone to infection and inflammation. However, more studies are required to address the causal relation between AMP profiles and their effect on the cutaneous microbiota composition of AD skin. Furthermore, colonization resistance has been described in AD, where certain coagulase‐negative Staphylococcus strains hamper the growth of S. aureus by expressing antimicrobial peptides. 57 Our group also showed that the skin microbiota of filaggrin (FLG)‐deficient patients (FLG–/–), compared with healthy controls (FLG+/+), contain a lower relative abundance of Gram‐positive anaerobic cocci (GPAC). This is thought to be the result of the absence of FLG and its degradation products, namely natural moisturizing factors which GPAC use as a carbon source. 58 In addition, we showed that GPAC microbes, such as Finegoldia spp., can quickly induce the expression of the antimicrobial proteins hBD‐2, hBD‐3 and LL37 by keratinocytes. 58 In the absence of GPAC, the antimicrobial response may be hampered or delayed in AD patients with FLG mutations, which can favor S. aureus colonization and infection.
Psoriasis
Research on the psoriasis microbiome has shown that microbiota composition differs widely between healthy and psoriatic skin; however, the results between the studies are contradictory. Firmicutes were reported to be overrepresented while Actinobacteria were underrepresented in one study, 59 and another study claimed that Staphylococcus species were overrepresented in an overall less diverse microbiome. 60 The next study claimed a decrease in the relative abundance of Cutibacterium and Staphylococcus species, and an increase in Corynebacterium. 61 Overall, due to these conflicting results, it is safe to conclude that a general psoriasis microbiome has not been deciphered yet, or perhaps a characteristic psoriasis‐like microbiome does not exist and the current observations are best explained by strong interindividual variation within the psoriasis phenotype. 62 , 63
Acne Vulgaris
Acne vulgaris is a chronic skin disease of the pilosebaceous unit, which are sebaceous glands that are connected to hair follicles, and important for the secretion of sebum. Although C. acnes is highly common in healthy adults, its presence and formation of biofilms are associated with acne vulgaris. 64 The anaerobic and lipid‐rich milieu of the sebaceous gland provides the optimal environment for C. acnes to thrive, especially when the follicle shaft is blocked. Recent metagenomic studies on several C. acnes strains have shown that genetic differences can possibly explain why some C. acnes strains act as commensals and other strains act as pathogens. 65 Furthermore, it was shown that skin microbiome differences relate to the grade of acne vulgaris. 66 These studies underline the importance and added value of performing metagenomic studies to go up to strain‐level depth.
Rosacea
Characteristics of rosacea, a common inflammatory condition of the facial skin, are facial flushing, redness, papules and pustules of which the pathogenesis is largely unknown. 67 Evidence of rosacea‐associated microorganisms has been shown for Staphylococcus epidermidis, Helicobacter pylori and Chlamydophila pneumonia. Colonization with Demodex folliculorum mites (and the microbiota they carry) also positively correlates with disease severity. 68 Furthermore, 16S rRNA gene sequencing showed that different subtypes of rosacea harbor Demodex mites with different microbiota. 69
Seborrheic Dermatitis
Seborrheic dermatitis (SD) and dandruff are chronic skin conditions that are often displayed on skin that is rich in sebaceous glands, like the upper back, nose and scalp. It is thought that both conditions are within the spectrum of the same disease with a different severity and location. 70 The pathophysiology of both conditions is not understood completely, but it has been shown that fungal colonization is a predisposing factor. Malassezia fungi Malassezia globosa and Malassezia restricta were identified as the predominant fungi on both normal skin and the scalp of SD and dandruff patients, and the amount of fungi was shown to correlate with disease severity. 71 , 72
Skin Wounds and Cutaneous Infections
Chronic wounds, such as diabetic foot ulcers, postsurgical wounds or decubitus ulcers, are an ideal place for bacterial overgrowth, and frequently these ulcers show impaired cutaneous wound healing. 73 Similar to acne vulgaris, biofilms are regularly formed by monocolonization of one or a few bacterial species, thereby further fostering pathogenic growth and making these wounds particularly difficult to heal. 74 Bacterial species from the Staphylococcus genus (e.g. S. aureus and S. epidermidis) and Pseudomonas (e.g. P. aeruginosa) genus are often identified in chronic wounds, 75 just like bacteria from other anaerobic genera (e.g. Finegoldia, Peptoniphilus, Peptostreptococcus). 76 Acute cutaneous infections (e.g. resulting from burn wounds) frequently display an altered bacterial composition that is characterized by elevated abundances of thermophilic bacteria like Aeribacillus, Caldalkalibacillus, Nesterenkonia and Halomonas, and by a decrease in the relative abundance of skin commensals such as Cutibacterium and Corynebacterium. 77 It is speculated that these thermophiles are introduced during debridement procedures, or that their increase is the result of the disruption of the skin barrier, thereby supplying nutrients to these bacteria. More clinical observations are needed to further validate these theories. 78
Concluding Remarks
In conclusion, commensal bacteria can play a pathogenic role under certain conditions, which further underlines the significance of studying skin diseases in the context of host genetics, immune responses, skin barrier function and the complete microbiome. Furthermore, microbe–microbe interactions can play an additional role in the pathogenesis of skin conditions. For identifying (skin) microbiota, MGS is currently still the most accessible approach for most applications, when it comes down to technical and methodological complexity. Nevertheless, we foresee that WGS will take the stage in the following years, which will enable functional analysis and a higher taxonomical resolution profiling of the abundant microbial fractions. Importantly, it should be pointed out that microbiome sequencing‐based studies can in principle only find correlations and associations, and cannot directly elucidate causal relations. In addition, we observe that the majority of microbiome studies still report association‐based results, whereas validation of associations and a mechanistic understanding thereof is what we should ideally strive for (Fig. 3). This will require the use of models such as in vitro (3‐D) skin systems 79 or (germ‐free) animal models, where the effect of specific candidate microbial isolates or a synthetic consortium of microbes (minimal microbiome) on the model can be tested (hence, microbial candidates identified in pilot/initial sequencing‐based studies). For in vivo modulation of cutaneous microbiota as a tool for keeping a healthy skin microbiome, skin microbiota transplantation is finding its way into research applications, as are pre‐ and probiotics for topical application. 80 In the end, our goal should be functional applications that could potentially be devised from our microbiome study findings (e.g. organisms, proteins, compounds, protease inhibitors), ultimately leading to novel therapeutic interventions for treatment of skin disease.
Figure 3.
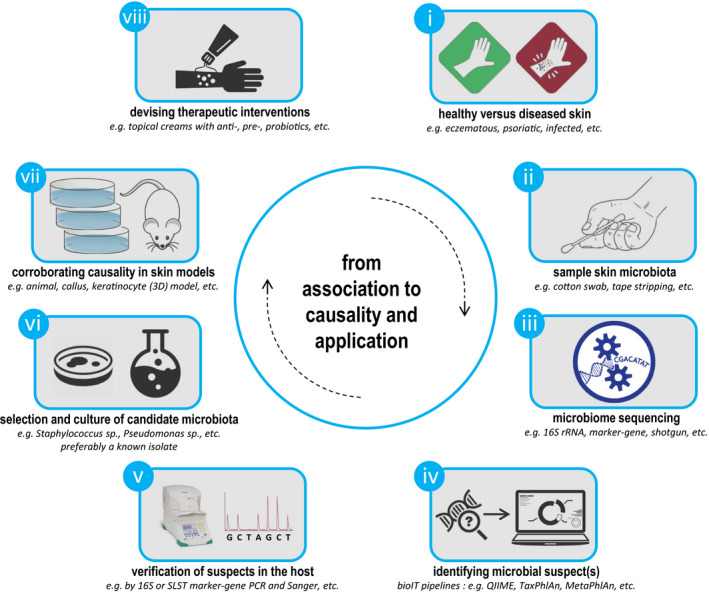
Studies do not stop at association. Schematic typical workflow/study approach in order to potentially go from association to causality, with specific focus on skin (micro)biology. (i) Healthy versus affected skin is evaluated, with suspected involvement of the microbiome as causative driver. (ii) Skin samples are collected through standard protocols, and (iii) are sequenced by a suitable platform depending on research question and study budget. (iv) Microbial suspects are identified by available data analysis pipelines, and (v) their specific presence and (differential) abundance are validated in the host by alternative (conventional) methods. Thereafter, (vi) candidates are selected and cultured for (vii) corroboration of microbiota‐associated effects of initial study findings by relevant in vitro or in vivo (disease) models. Finally, if applicable, (viii) functional applications could potentially be devised from study findings (e.g. organisms, proteins, compounds, protease inhibitors), ultimately leading to novel therapeutic interventions for treatment of skin disease. PCR, polymerase chain reaction; SLST, single‐locus sequence typing.
Conflict of Interest
None declared.
Acknowledgments
Author contributions are as follows: T. H. A. E, J. P. H. S., J. B., J. S., E. H. B. and P. L. J. M. Z. wrote the paper, and all authors read and approved the final manuscript.
References
- 1. Sender R, Fuchs S, Milo R. revised estimates for the number of human and bacteria cells in the body. PLOS Biology 2016; 14(8): e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marchesi JR, Ravel J. The vocabulary of microbiome research: a proposal. Microbiome 2015; 3(1): 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scholz CF, Jensen A, Lomholt HB, Brüggemann H, Kilian M. A novel high‐resolution single locus sequence typing scheme for mixed populations of Propionibacterium acnes in vivo. PLoS One 2014; 9(8): e104199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ederveen THA, Smits JPH, Hajo K et al A generic workflow for Single Locus Sequence Typing (SLST) design and subspecies characterization of microbiota. Sci Rep 2019; 9(1): 19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cole JR, Wang Q, Fish JA et al Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 2014; 42(D1): D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeSantis TZ, Hugenholtz P, Larsen N et al Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72(7): 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pruesse E, Quast C, Knittel K et al SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 2007; 35(21): 7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knight R, Vrbanac A, Taylor BC et al Best practices for analysing microbiomes. Nat Rev Microbiol 2018; 16(7): 410–422. [DOI] [PubMed] [Google Scholar]
- 9. Zeeuwen PLJM, Boekhorst J, Ederveen THA et al Reply to Meisel et al. Journal of Investigative Dermatology 2017; 137(4): 961–962. [DOI] [PubMed] [Google Scholar]
- 10. Meisel JS, Hannigan GD, Tyldsley AS et al Skin microbiome surveys are strongly influenced by experimental design. Journal of Investigative Dermatology 2016; 136(5): 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caporaso JG, Kuczynski J, Stombaugh J et al QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 2010; 7(5): 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26(19): 2460–2461. [DOI] [PubMed] [Google Scholar]
- 13. Schloss PD, Westcott SL, Ryabin T et al Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009; 75(23): 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes–application to the identification of mycorrhizae and rusts. Mol Ecol 1993; 2(2): 113–118. [DOI] [PubMed] [Google Scholar]
- 15. Abarenkov K, Henrik Nilsson R, Larsson K‐H et al The UNITE database for molecular identification of fungi – recent updates and future perspectives. New Phytologist 2010; 186(2): 281–285. [DOI] [PubMed] [Google Scholar]
- 16. Zeeuwen PL, Boekhorst J, van den Bogaard EH et al Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biology 2012; 13(11): R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luo C, Tsementzi D, Kyrpides NC, Konstantinidis KT. Individual genome assembly from complex community short‐read metagenomic datasets. ISME J 2012; 6(4): 898–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abubucker S, Segata N, Goll J et al Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 2012; 8(6): e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huson DH, Weber N. Microbial community analysis using MEGAN. Methods Enzymol 2013; 531: 465–85. [DOI] [PubMed] [Google Scholar]
- 20. Wu M, Eisen JA. A simple, fast, and accurate method of phylogenomic inference. Genome Biol 2008; 9(10): R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sunagawa S, Mende DR, Zeller G et al Metagenomic species profiling using universal phylogenetic marker genes. Nat Meth 2013; 10(12): 1196–1199. [DOI] [PubMed] [Google Scholar]
- 22. Luo C, Knight R, Siljander H, Knip M, Xavier RJ, Gevers D. ConStrains identifies microbial strains in metagenomic datasets. Nat Biotechnol 2015; 33(10): 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scholz M, Ward DV, Pasolli E et al Strain‐level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods 2016; 13(5): 435–438. [DOI] [PubMed] [Google Scholar]
- 24. Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. Microbial strain‐level population structure and genetic diversity from metagenomes. Genome Res 2017; 27(4): 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Francis OE, Bendall M, Manimaran S et al Pathoscope: species identification and strain attribution with unassembled sequencing data. Genome Res 2013; 23(10): 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segata N, Börnigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nature Communications 2013; 4: 2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meyer F, Paarmann D, D'Souza M et al The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008; 9: 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langille MG, Zaneveld J, Caporaso JG et al Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31(9): 814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jun S‐R, Robeson MS, Hauser LJ, Schadt CW, Gorin AA. PanFP: pangenome‐based functional profiles for microbial communities. BMC Research Notes 2015; 8: 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Asshauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015; 31(17): 2882–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science 2014; 346(6212): 954–959. [DOI] [PubMed] [Google Scholar]
- 32. Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol 2011; 9(4): 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grice EA, Kong HH, Conlan S et al Topographical and temporal diversity of the human skin microbiome. Science 2009; 324(5931): 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oh J, Byrd AL, Park M, Kong HH, Segre JA. Temporal stability of the human skin microbiome. Cell 2016; 165(4): 854–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oh J, Byrd AL, Deming C, Conlan S, Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature 2014; 514(7520): 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niehues H, Tsoi LC, van der Krieken DA et al Psoriasis‐associated late cornified envelope (LCE) proteins have antibacterial activity. J Invest Dermatol 2017; 137(11): 2380–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol 2012; 12(7): 503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Molhuizen HO, Alkemade HAC, Zeeuwen PLJM, de Jongh GJ , Wieringa B, Schalkwijk J. SKALP/elafin: an elastase inhibitor from cultured human keratinocytes. Purification, cDNA sequence, and evidence for transglutaminase cross‐linking. J Biol Chem 1993; 268(16): 12028–12032. [PubMed] [Google Scholar]
- 39. Alkemade JA, Molhuizen HO, Ponec M et al SKALP/elafin is an inducible proteinase inhibitor in human epidermal keratinocytes. J Cell Sci 1994; 107(Pt 8): 2335–2342. [DOI] [PubMed] [Google Scholar]
- 40. Simpson AJ, Maxwell AI, Govan JRW, Haslett C, Sallenave J‐M. Elafin (elastase‐specific inhibitor) has anti‐microbial activity against gram‐positive and gram‐negative respiratory pathogens. FEBS Lett 1999; 452(3): 309–313. [DOI] [PubMed] [Google Scholar]
- 41. Hiemstra PS, Maassen RJ, Stolk J, Heinzel‐Wieland R, Steffens GJ, Dijkman JH. Antibacterial activity of antileukoprotease. Infect Immun 1996; 64(11): 4520–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harder J, Schroder JM. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J Biol Chem 2002; 277(48): 46779–46784. [DOI] [PubMed] [Google Scholar]
- 43. Schroder JM. Antimicrobial peptides in healthy skin and atopic dermatitis. Allergol Int 2011; 60(1): 17–24. [DOI] [PubMed] [Google Scholar]
- 44. Zeeuwen PL, Kleerebezem M, Timmerman HM, Schalkwijk J. Microbiome and skin diseases. Curr Opin Allergy Clin Immunol 2013; 13(5): 514–520. [DOI] [PubMed] [Google Scholar]
- 45. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers 2018; 4(1): 1. [DOI] [PubMed] [Google Scholar]
- 46. Kong HH, Oh J, Deming C et al Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 2012; 22(5): 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feuillie C, Vitry P, McAleer MA et al Adhesion of Staphylococcus aureus to Corneocytes from atopic dermatitis patients is controlled by natural moisturizing factor levels. Mbio 2018; 9(4): e01184‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams MR, Nakatsuji T, Gallo RL. Staphylococcus aureus: master manipulator of the skin. Cell Host Microbe 2017; 22(5): 579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bekeredjian‐Ding I, Stein C, Uebele J. The innate immune response against Staphylococcus aureus . Curr Top Microbiol Immunol 2017; 409: 385–418. [DOI] [PubMed] [Google Scholar]
- 50. Paller AS, Kong HH, Seed P et al The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol 2019; 143(1): 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chng KR, Tay ASL, Li C et al Whole metagenome profiling reveals skin microbiome‐dependent susceptibility to atopic dermatitis flare. Nat Microbiol 2016; 1(9): 16106. [DOI] [PubMed] [Google Scholar]
- 52. Fyhrquist N, Muirhead G, Prast‐Nielsen S et al Microbe‐host interplay in atopic dermatitis and psoriasis. Nature Communications 2019; 10(1): 4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ong PY, Ohtake T, Brandt C et al Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 2002; 347(15): 1151–1160. [DOI] [PubMed] [Google Scholar]
- 54. Howell MD, Gallo RL, Boguniewicz M et al Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity 2006; 24(3): 341–348. [DOI] [PubMed] [Google Scholar]
- 55. Zeeuwen PL, de Jongh GJ, Rodijk‐Olthuis D et al Genetically programmed differences in epidermal host defense between psoriasis and atopic dermatitis patients. PLoS One 2008; 3(6): e2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smits JPH, Ederveen THA, Rikken G et al Targeting the cutaneous microbiota in atopic dermatitis by coal tar via AHR‐dependent induction of antimicrobial peptides. Journal of Investigative Dermatology 2020; 140: 415–424. [DOI] [PubMed] [Google Scholar]
- 57. Nakatsuji T, Chen TH, Narala S et al Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med 2017; 9(378): eaah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeeuwen PL, Ederveen THA, van der Krieken DA et al Gram‐positive anaerobe cocci are underrepresented in the microbiome of filaggrin‐deficient human skin. J Allergy Clin Immunol 2017; 139(4): 1368–1371. [DOI] [PubMed] [Google Scholar]
- 59. Gao Z, Tseng C‐H, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3(7): e2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tett A, Pasolli E, Farina S et al Unexplored diversity and strain‐level structure of the skin microbiome associated with psoriasis. NPJ Biofilms Microbiomes 2017; 3: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Loesche MA, Farahi K, Capone K et al Longitudinal study of the psoriasis‐associated skin microbiome during therapy with ustekinumab in a randomized phase 3b clinical trial. J Invest Dermatol 2018; 138(9): 1973–1981. [DOI] [PubMed] [Google Scholar]
- 62. Alekseyenko AV, Perez‐Perez GI, De Souza A et al Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 2013; 1(1): 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Statnikov A, Alekseyenko AV, Li Z et al Microbiomic signatures of psoriasis: feasibility and methodology comparison. Sci Rep 2013; 3: 2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tomida S, Nguyen L, Chiu B‐H et al Pan‐genome and comparative genome analyses of Propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. Mbio 2013; 4(3): e00003–00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fitz‐Gibbon S, Tomida S, Chiu B‐H et al Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol 2013; 133(9): 2152–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li CX, You Z‐X, Lin Y‐X, Liu H‐Y, Su J. Skin microbiome differences relate to the grade of acne vulgaris. J Dermatol 2019; 46(9): 787–790. [DOI] [PubMed] [Google Scholar]
- 67. Lazaridou E, Giannopoulou C, Fotiadou C, Vakirlis E, Trigoni A, Ioannides D. The potential role of microorganisms in the development of rosacea. J Dtsch Dermatol Ges 2011; 9(1): 21–25. [DOI] [PubMed] [Google Scholar]
- 68. Holmes AD. Potential role of microorganisms in the pathogenesis of rosacea. J Am Acad Dermatol 2013; 69(6): 1025–32. [DOI] [PubMed] [Google Scholar]
- 69. Murillo N, Aubert J, Raoult D. Microbiota of Demodex mites from rosacea patients and controls. Microb Pathog 2014; 71–72: 37–40. [DOI] [PubMed] [Google Scholar]
- 70. Schwartz JR, Messenger A, Tosti A et al A comprehensive pathophysiology of dandruff and seborrheic dermatitis ‐ towards a more precise definition of scalp health. Acta Derm Venereol 2013; 93(2): 131–137. [DOI] [PubMed] [Google Scholar]
- 71. Schwartz RA, Janusz CA, Janniger CK. Seborrheic dermatitis: an overview. Am Fam Physician 2006; 74(1): 125–130. [PubMed] [Google Scholar]
- 72. Findley K, Oh J, Yang J et al Topographic diversity of fungal and bacterial communities in human skin. Nature 2013; 498(7454): 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Loesche M, Gardner SE, Kalan L et al Temporal stability in chronic wound microbiota is associated with poor healing. J Invest Dermatol 2017; 137(1): 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhao G, Usui ML, Lippman SI et al Biofilms and inflammation in chronic wounds. Adv Wound Care (New Rochelle) 2013; 2(7): 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wolcott RD, Hanson JD, Rees EJ et al Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen 2016; 24(1): 163–174. [DOI] [PubMed] [Google Scholar]
- 76. Choi Y, Banerjee A, McNish S et al Co‐occurrence of Anaerobes in Human Chronic Wounds. Microb Ecol 2019; 77: 808–820. [DOI] [PubMed] [Google Scholar]
- 77. van Rensburg JJ, Lin H, Gao X et al The human skin microbiome associates with the outcome of and is influenced by bacterial infection. Mbio 2015; 6(5): e01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Plichta JK, Gao X, Lin H et al Cutaneous burn injury promotes shifts in the bacterial microbiome in autologous donor skin: implications for skin grafting outcomes. Shock 2017; 48(4): 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Niehues H, Bouwstra JA, Ghalbzouri AE et al 3D skin models for 3R research: The potential of 3D reconstructed skin models to study skin barrier function. Exp Dermatol 2018; 27(5): 501–511. [DOI] [PubMed] [Google Scholar]
- 80. Myles IA, Williams KW, Reckhow JD et al Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight 2016; 1(10): e86955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Andersson AF, Lindberg M, Jakobsson H et al Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 2008; 3(7): e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]