

Abstract
Following the recent discovery of stable octa‐coordinated alkaline earth metals with N2 and CO, the role of group II metals in the catalytic reduction of these ligands by means of density functional theory (DFT) calculations and conceptual DFT‐based reactivity indices is investigated. Cubic group IV and octahedral group VI transition metal complexes as well as the free ligands are computed for reference. The outer and most accessible atoms of N2 and CO become much more nucleophilic and electrophilic in all complexes, relevant for N2 fixation, as probed by the Fukui function and local softness. Within one row of the periodic table, the alkaline earth complexes often show the strongest activation. On the contrary, the electrostatic character is found to be virtually unaffected by complexation. Trends in the soft frontier orbital and hard electrostatic character are in agreement with calculated proton affinities and energy decomposition analyses of the protonated structures, demonstrating the dominance of the soft (HOMO–LUMO) orbital interactions.
Keywords: alkaline earth metals, conceptual DFT, density functional calculations, energy decomposition analysis, nitrogen fixation
Alkaline earth complexes: The viability of alkaline earth metal‐based N2 fixation is investigated. It is shown that the activation of N2 through complexation originates from the d–π back‐donation. Within one row of the periodic table, the alkaline earth metal shows the strongest nucleophilic activation of the N2 ligand, confirming clear trends in proton affinities.
Introduction
Until recently, the heavy alkaline earth metals calcium, strontium, and barium were considered to be involved in bond formation through their ns and np valence orbitals, (n=4, 5, 6) typical for main‐group elements, in contrast to transition metals for which the (n−1)d orbitals enrich the bonding pattern, leading to an increased variety in their bonding/coordination behavior.1, 2, 3, 4 Carbonyl and dinitrogen complexes with varying coordination behaviors are well‐known examples of this situation.5
However, in 2018, the isolation and spectroscopic characterization of octa‐coordinated carbonyl complexes of Ca, Sr, and Ba in a low‐temperature neon matrix, revealing a cubic (O h) symmetry, were reported in a Chinese/German experimental/theoretical collaborative work.6 Analysis of the electronic structure through DFT calculations7 by two of the present authors revealed that the electronic reference state of the heavy alkaline earth atom (MII) in the complexes is a triplet state with ns0 (n−1)d2 electronic configuration, similar in its d‐orbital occupation to the corresponding group IV transition metals (Ti, Zr, Hf) or the anionic complexes of the group III transition metals (Sc, Y, La). The synthesis and characterization as cubic octa‐carbonyl complexes of both the anionic group III complexes and the neutral group IV complexes was also recently reported by the same authors.8, 9 In the abovementioned heavy alkaline earth main‐group metal complexes, the metal–CO bonding was scrutinized, and was shown to be dominated by the M(dπ)→(CO)8 π back‐donation of the degenerate (eg) set of singly occupied (n−1)d atomic orbitals of the metal into the antibonding π* molecular orbitals (MOs) of CO.8 Further analysis shows that the complexes obey the 18‐electron rule if only those electrons are counted that are engaged in the metal–ligand interactions.1, 2, 3 This bonding pattern accounts for the experimentally observed large redshift of the CO stretching mode of the octa‐carbonyls, as confirmed by the DFT calculations.
These results smoothed the border between (the behavior of) main‐group and transition elements, a paradigm connected with the structure of Mendeleev's Table.1, 2, 3 This issue was already put forward before by Gagliardi and Pyykkö on promoting, for example, Ba to an honorary d‐element in their studies on the stability of CsN7Ba and the analogy of its dissociation mechanism with ScN7.10, 11 Their findings were a hint that the heavier alkaline earth elements might play a role as substitutes for the transition metals, and that they might display a much richer chemistry and reactivity than hitherto thought.
This paradigm shift was confirmed very recently by similar studies by the same collaboration on the octa‐coordinated heavy alkaline earth metal dinitrogen complexes MII(N2)8 (MII=Ca, Sr, Ba). Isolation and spectroscopic identification revealed a similar cubic (O h) symmetry, again with a large redshift in the N−N stretching frequency hypothesized on the basis of d–π back‐donation as confirmed by DFT calculations. These complexes were also seen to obey the 18‐electron rule.12 Besides their intrinsic importance as a further indicator of the blurring of the main‐group/transition metal border, these results might be of interest in the quest for (di)nitrogen fixation. For decades, the activation of the strong, nonpolar bond in N2 has been challenging chemists. The search for milder conditions for nitrogen fixation than the extreme ones in the Haber–Bosch process,13 inspired by Nature's nitrogenase catalysts, invariably built around transition metal centers, essentially Mo and Fe,14 has led to countless studies on the use of N2 transition metal complexes for activating the N2 ligand (for reviews see Ref. 15). Just two examples are the highly detailed studies on the catalytic reduction of N2 at a single Mo center by stepwise additions of protons and electrons by Yandulov and Schrock,16, 17 and the recent studies of the Ta2 +‐mediated cleavage of N2 and reaction with H2, yielding NH3.18 In the context of the recent advances in transition‐metal‐based N2 fixation and taking into account the abovementioned results on the similarity of behavior between the heavy alkaline earth metals and transition metals, the question arises whether N2 can be activated by these main‐group alkaline earth metals to the same extent, or even more so, compared with their transition metal congeners. In other words, these considerations incited us to probe the reactivity of alkaline earth metal complexes in a comparative context with their transition metal congeners. Needless to say, in view of the issues of abundancy and low cost of the main‐group elements considered as compared with the transition elements, such experimental and theoretical studies are also of societal importance.19
Several authors of this paper have been active in the field of chemical reactivity for quite some time, adopting the Conceptual Density Functional Theory (CDFT) ansatz, introduced by Parr and co‐workers in the late 1970s/early 1980s.20, 21 In this sub‐branch of DFT (for reviews see Ref. 22), neither an explicit ab initio nor DFT study is performed on the reaction path of a number of possible reactions; instead, a number of well‐chosen indicators of reactivity are scrutinized, which, on the basis of their physical and chemical significance, can give an overall, first‐order insight into what can be expected from the activation of the N2 (or CO) ligand in the main‐group metal complexes as compared with the transition metal ones. Conceptual DFT, as this branch of DFT was named (sometimes also called Chemical Reactivity theory), undertakes this endeavor by evaluating a number of response functions.23 These quantities reflect the sensitivity of the energy E of a given system to perturbations in the number of electrons N and/or the external potential ν(r) (i.e., the potential felt by the electrons attributed to the nuclei), the changes at stake in a chemical reaction.
The response functions, which are nowadays sometimes used up to third order,24 then boil down to a number of (sometimes mixed) partial and functional derivatives of E with respect to N and ν(r). Ever since that period, not only has a remarkable deepening and extension of the theory been witnessed in the literature, but countless studies have also been published on its application in nearly all fields of chemistry, varying from organic to inorganic and materials chemistry, from catalysis to medicinal and biochemistry, and so on (examples can be found in the reviews in Ref. 22; for a very recent report on the status, prospects, and issues of Conceptual DFT we refer to Ref. 25).
In the present study, we will concentrate on two particular descriptors, which have been used extensively and deliberately in the literature: the Fukui function26 and the local softness,27 both in their nucleophilic and electrophilic form. At first sight, electrophilic attacks at the terminal atoms of the N2 and CO ligands (the terminal N or the O atom, respectively) are expected in view of their σ lone pair pointing away from the center of the complex. These atoms and the N2 or CO ligands as a whole are expected to become more nucleophilic in view of the metal‐to‐ligand π back‐donation, which was shown by some of the present authors to be more important than the ligand σ‐electron donation to the metal.6 The so‐called nucleophilic Fukui function f −(r) and the corresponding local softness s −(r) (as different systems are compared with each other rather than different sites on one complex22b, 28) were selected for this purpose. In particular, the evolution of these quantities upon passing from transition metal complexes (the cases of group IV transition metals (Ti, Zr, Hf) also yielding cubic octa‐coordination,9 and group VI transition metals (Cr, Mo, W) yielding octahedral coordination) to main‐group metal complexes will be investigated in detail. The fundamental question pertains to the similarity in activation of the ligand, increasing its reactivity, between main‐group and transition metals. In view of their particular relevance (vide supra) the N2 results will be highlighted in more detail; the overall tendencies will, however, be checked for parallelism between the N2 and CO complexes. A word of caution should be given here. It is well known that in the context of chemical softness,29, 30 here applied at local level,27 the local softness essentially probes molecular regions prone to soft interactions;22b therefore, a study of the charges, molecular electrostatic potential31 (MEP; much more indicative for hardness‐dominated interactions),22b, 32 and proton affinity will also be pursued to probe the hard character of the ligands. Thus, we avoid the caveat regarding the applicability of a local hard and soft acids and bases (HSAB) principle.28, 33, 34, 35
A similar approach involving the Fukui functions and the local softness will be followed for nucleophilic attack on the complexes. Although at first sight perhaps less relevant, the reductive mechanism of N2 fixation involves a reduction in which electrons play a clear role, and this led us to investigate the electrophilic Fukui function f +(r) as a distributor of electrophilic behavior over a molecule, as it is with the total softness in the local softness s +(r) (vide infra).36
Focusing again mainly on the N2 complexes but reassuring ourselves on similar general trends for N2 and CO complexes, the combined results of two parts of this study should yield answers to the following two questions: 1) Do heavy main‐group alkaline earth metals activate, in a similar way to transition metals, ligands of the CO and N2 types for similar reactions (electrophilic, nucleophilic, with varying hardness/softness character)?; and 2) Can we reconcile the overall results with what is known about the role of transition metals in N2 fixation and a possible replacement of transition metals by heavy main‐group alkaline earth metals?
This paper is structured as follows. In the section “Theoretical and Computational Details”, some basic elements are presented on CDFT and the energy decomposition analysis (EDA), and the working equations applied in the study of the N2 and CO complexes. Particular attention is devoted to intricacies for treating spin and degeneracy. In the section “Results and Discussion”, a systematic overview of the results is given on possible activation of the main‐group metal complexes for both nucleophilic (hard and soft) and electrophilic attacks, with particular emphasis on the comparison between transition metal and main‐group metal complexes. The final part is more speculative in nature, and attempts to see how the overall results might shed further light on the N2 fixation process.
Theoretical and Computational Details
Conceptual DFT
Conceptual DFT is a perturbational approach to rationalize and quantify chemical reactivity through reactivity indices originating from the functional derivatives in a Taylor expansion of the E[N,ν(r)] functional,22 in which N and ν(r) are the number of electrons and the external potential between the electrons and nuclei, respectively. The Fukui function in Equation (1a) and (1b) is the second‐order mixed derivate of E with respect to N and ν(r) and probes the change in the electron density when the number electrons in a system is increased or decreased.26 Therefore, regions in which f +(r) is large tend to be electrophilic, whereas regions with large f −(r) are nucleophilic.
| (1a) |
| (1b) |
As nitrogen fixation concerns the processes converting N2 to ammonia or other reduced forms of nitrogen, and thus, the reduction of the N atoms as well as binding electrophilic substrates, the electrophilic and nucleophilic Fukui functions can be relevant descriptors for quantifying the tendency of a N atom to be reduced. The Fukui function is used to study different sites within one system, whereas the local softness is more appropriate for comparing sites in different molecules.22b, 28 The local softness s(r) is defined in Equation (2) and integrates to the global softness (S), which is the inverse of the difference between I and A, which are the vertical ionization potential and electron affinity, respectively [Eq. (3)].
| (2) |
| (3) |
Hard sites, that is, the molecular regions prone to strong electrostatic interactions within the HSAB context, were probed by means of the molecular electrostatic potential (MEP). In a next step, proton affinities (PA) were calculated to quantify the interaction between H+, as a hard electrophile, and N2 upon isolation or binding in complexes by group II, IV, and VI metals.
Energy decomposition analysis
The interaction energy between two chemical species (or fragments) can be decomposed into physically meaningful terms within Kohn–Sham MO theory37 [Eq. (4)].
| (4) |
In Equation (4), is the quasi‐classical electrostatic interaction between the unperturbed charge distributions, and , of two fragments A and B, respectively. The Pauli interaction is the result of anti‐symmetrization and renormalization of the wavefunction corresponding to the superposition of the fragment densities. This term accounts for the repulsion between electrons with identical spin. The term originates from orbital interactions, charge transfer, and polarization. In the case of a metahybrid density functional, an additional correction term ( ) can be added.
Electronic structure of the M(L)8 and M(L)6 complexes
The MII(L)8 (with L=N2, CO) complexes have a triplet ground state electronic structure, whereas the MIV(L)8 and MVI(L)6 (with L=N2, CO) were more stable with singlet multiplicity at the M06‐2X‐D3/Def2‐TZVPP level of theory.38, 39, 40 For Ti(N2)8, the cubic singlet structure is only a local minimum, which is thermodynamically unstable for loss of one N2 ligand leading to Ti(N2)7 as the coordinatively saturated stable complex, similar to the octa‐coordinated CO complexes.9 The MVI(N2)6 complexes are also singlet species. All neutral complexes have O h symmetry in their ground state, and all geometry optimizations were performed using the M06‐2X‐D3 functional and the def2‐TZVPP basis set for all atoms using the Gaussian41 software conforming with recent work on octa‐coordinated alkaline earth complexes.6, 12 All charged complexes were computed at the same geometry as the neutral compounds (to obtain vertical ionization potentials and electron affinities) with doublet spin state, except one: Cr(N2)6 + is more stable in the sextet spin state with half‐occupied spatial t2g and eg orbitals (see Figure 1). CDFT descriptors, MEPs, and proton affinities were calculated at the ZORA‐M06‐2X/TZ2P level of theory using the ADF software.42
Figure 1.
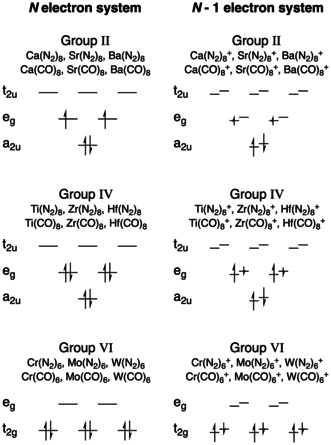
Fractional electron approach for the N−1 electron systems with doublet spin state to maintain the symmetry of the electron density (the same approach was used for the N+1 electron systems). Smaller arrows indicate electron for the charged group II and IV complexes or electron for the charged group VI complexes.
Importantly, CDFT descriptors require the calculation of ionic species. The electron system of each metal complex was calculated using fractional electron occupations, as implemented in the ADF software package (see Figure 1), maintaining the degeneracy of α‐orbitals and β‐orbitals. In doing so, the O h point‐group symmetry not only matches the nuclear constellation but also the molecular orbital occupations and electron density.
The Fukui functions were atom‐condensed using the Hirshfeld population analysis.43 The Hirshfeld method is commonly used for computing condensed Fukui functions and provides reliable charges that very often lead to identical conclusions to other population analyses such as the natural population analysis (NPA) method.44 In its foundation, the Hirshfeld atoms‐in‐molecules partitioning resembles neutral ground state atoms as much as possible in the information theoretic sense.25, 45 Moreover, in a very recent, extensive contribution by Liu, Chattaraj, and co‐workers, it was shown that Hirshfeld charges are able to describe regioselectivity and simultaneously quantify both electrophilicity and nucleophilicity accurately (vide infra).46
Results and Discussion
Fukui function and local softness analysis
In view of the recent advances in transition metal‐based N2 fixation described above and the discovery of stable octa‐coordinated alkaline earth N2 complexes, the role of the metal center in these intriguing group II complexes was studied. In this section, we present an approach within the framework of conceptual density functional theory (CDFT) to scrutinize the potency of group II metals, MII (Ca, Sr and Ba), to activate the N2 ligand in cubic MII(N2)8 complexes, and directly compare their reactivity to more traditional MIV(N2)8 (MIV=Ti, Zr, Hf) and MVI(N2)6 (MVI=Cr, Mo, W) transition metal complexes.
The strong redshift in the N−N and C−O stretching frequencies of MII(N2)8 and MII(CO)8 complexes, as well as bond‐order analyses, indicate a significant π back‐donation from the d orbitals of the alkaline earth metal to the π* orbitals of the ligands. This phenomenon, which is typically associated with transition metal bonds, is the foundation for our decision to focus on the electron‐donating properties of the complexes, and thus, the nucleophilic Fukui function f −(r). That is, the metal relieving its excess negative charge to the ligand will make the ligand more prone to donate electrons and less likely to interact with electron‐rich substrates.
The back‐donation from the alkaline earth metal to the ligand was recently questioned by Koch et al.;47 however, EDA‐NOCV calculations conclusively demonstrate the importance of alkaline earth metal d orbitals in the metal–ligand bonds.6, 12 Moreover, the cubic geometry of MII(N2)8 complexes is even dictated by the mixing of d orbitals into the bonding mechanism.48
To understand the reactivity of the complexes, we considered a cage‐assembly approach, as shown schematically for a cubic complex in Figure 2.49 In this approach, the ligands are first considered at infinite distance from one another. Then, the ligands are frozen into the relative orientation they adopt in the complex and this system is referred to as the ligand cage. In a final step, the metal is inserted into the cage, turning on the metal–ligand bonding, and in particular, the d–π back‐donation.
Figure 2.
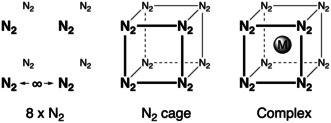
Cage‐assembly approach for constructing a cubic metal complex. The same approach with six ligands was used for the octahedral group VI complexes.
Table 1 summarizes the condensed Fukui function values for each type of atom in all the calculated complexes, in which Nin is the N atom bound to the metal, and Nout is the outer N atom that is most accessible to any substrate. Electron densities were atom‐condensed by means of the Hirshfeld population method.43 In Table S1 (Supporting Information), the same data for the CO complexes can be found.
Table 1.
Condensed nucleophilic Fukui functions f −(r), according to the Hirshfeld population analysis, for all N2 complexes along the stepwise cage‐assembly approach.
|
|
|
8×N2 |
N2 cage |
Complex |
|
|
8×N2 |
N2 cage |
Complex |
|
|
6×N2 |
N2 cage |
Complex |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Ca(N2)8 |
Nout |
0.063 |
0.086 |
0.083 |
Ti(N2)8 |
Nout |
0.063 |
0.088 |
0.088 |
Cr(N2)6 |
Nout |
0.083 |
0.115 |
0.116 |
|
|
Nin |
0.063 |
0.039 |
0.024 |
|
Nin |
0.063 |
0.037 |
0.019 |
|
Nin |
0.083 |
0.052 |
0.033 |
|
|
Ca |
– |
– |
0.145 |
|
Ti |
– |
– |
0.146 |
|
Cr |
– |
– |
0.107 |
|
Sr(N2)8 |
Nout |
0.063 |
0.085 |
0.080 |
Zr(N2)8 |
Nout |
0.063 |
0.088 |
0.085 |
Mo(N2)6 |
Nout |
0.083 |
0.114 |
0.108 |
|
|
Nin |
0.063 |
0.040 |
0.024 |
|
Nin |
0.063 |
0.037 |
0.019 |
|
Nin |
0.083 |
0.053 |
0.024 |
|
|
Sr |
– |
– |
0.173 |
|
Zr |
– |
– |
0.169 |
|
Mo |
– |
– |
0.210 |
|
Ba(N2)8 |
Nout |
0.063 |
0.083 |
0.071 |
Hf(N2)8 |
Nout |
0.063 |
0.088 |
0.087 |
W(N2)6 |
Nout |
0.083 |
0.114 |
0.109 |
|
|
Nin |
0.063 |
0.042 |
0.019 |
|
Nin |
0.063 |
0.037 |
0.020 |
|
Nin |
0.083 |
0.053 |
0.023 |
|
|
Ba |
– |
– |
0.282 |
|
Hf |
– |
– |
0.146 |
|
W |
– |
– |
0.209 |
In the case of eight infinitely separated N2 ligands, and maintaining the O h symmetry of the electronic configuration, each ligand will lose one eighth of an electron if a total of one electron is removed from the system. As there is no interaction between the ligands, each N atom will lose 1/16 (≈0.063) electrons. The same reasoning applies for the hexa‐coordinated group VI complexes (1/12≈0.083). It should be remarked that the Fukui function is normalized, and thus, the sum of all atom‐condensed values should always be equal to one, which is indeed the case for all complexes in Table 1 considering there are eight (or six) ligands in the group II and IV (or group VI) complexes.
Bringing the ligands from infinity to the empty ligand cage configuration (i.e., their respective orientation in the metal complexes) polarizes the N−N bonds, which is clearly illustrated by the Fukui function in Table 1. Interestingly, the outer N atoms become more nucleophilic than the inner N atoms owing to the interaction between the N2 ligands in the cage configuration. The condensed f −(r) values for Nout and Nin are typically 0.085 and 0.040, respectively, and are barely affected by the type of complex because all cages are identical except for different ligand–ligand distances and marginally different N−N bond lengths. For the hexavalent complexes, the condensed f −(r) values for Nout and Nin are typically 0.115 and 0.050, respectively. The same polarization of the CO cages, leading to more nucleophilic O atoms, was found for the CO complexes (Table S1).
In the metal complexes, the same trend is found: Nout is more nucleophilic than Nin, regardless of the metal center. For a comparison of the nucleophilic centers in all metal complexes, and in particular, the Nout atoms, the condensed local softness s −(r) values in Table 2 are discussed (see Table S4, Supporting Information, for the s −(r) data of the CO complexes).
Table 2.
Condensed nucleophilic local softness s −(r), according to the Hirshfeld population analysis, for all N2 complexes along the stepwise cage‐assembly approach.
|
|
|
8 × N2 |
N2 cage |
Complex |
|
|
8 × N2 |
N2 cage |
Complex |
|
|
6 × N2 |
N2 cage |
Complex |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Ca(N2)8 |
Nout |
0.120 |
0.168 |
0.438 |
Ti(N2)8 |
Nout |
0.120 |
0.180 |
0.386 |
Cr(N2)6 |
Nout |
0.159 |
0.213 |
0.408 |
|
|
Nin |
0.120 |
0.076 |
0.125 |
|
Nin |
0.120 |
0.075 |
0.085 |
|
Nin |
0.159 |
0.096 |
0.117 |
|
|
Ca |
– |
– |
0.765 |
|
Ti |
– |
– |
0.641 |
|
Cr |
– |
– |
0.376 |
|
Sr(N2)8 |
Nout |
0.120 |
0.163 |
0.436 |
Zr(N2)8 |
Nout |
0.120 |
0.175 |
0.360 |
Mo(N2)6 |
Nout |
0.159 |
0.209 |
0.351 |
|
|
Nin |
0.120 |
0.078 |
0.130 |
|
Nin |
0.120 |
0.075 |
0.079 |
|
Nin |
0.159 |
0.098 |
0.077 |
|
|
Sr |
– |
– |
0.950 |
|
Zr |
– |
– |
0.714 |
|
Mo |
– |
– |
0.681 |
|
Ba(N2)8 |
Nout |
0.120 |
0.157 |
0.387 |
Hf(N2)8 |
Nout |
0.120 |
0.177 |
0.369 |
W(N2)6 |
Nout |
0.159 |
0.210 |
0.355 |
|
|
Nin |
0.120 |
0.080 |
0.106 |
|
Nin |
0.120 |
0.075 |
0.085 |
|
Nin |
0.159 |
0.097 |
0.073 |
|
|
Ba |
– |
– |
1.548 |
|
Hf |
– |
– |
0.622 |
|
W |
– |
– |
0.680 |
From Table 2, the same polarization of the N−N bond is observed upon constraining the N2 molecules in the ligand cages because the Fukui function is multiplied by the global softness, that is, a system‐dependent constant. For the metal complexes, the attention will be devoted to the Nout atoms as these are the most accessible positions for interactions with electrophilic substrates, even though the shielded metal is clearly the most nucleophilic center in each of the complexes.
In our approach, it was hypothesized that the π back‐donation from the (alkaline earth) metal to the ligand can have important consequences for the activation of N2. Detailed analysis of the Fukui function and (local) softness showed that this activation is essentially the Nout atom becoming more nucleophilic, and, from Table 2, the nucleophilicity of Nout is similar for all metal complexes: the condensed s −(r) values for this atom are all between 0.350 and 0.450. Remarkably, within one row of the periodic table, the ligand is most activated by the alkaline earth metal, indicating that the back‐donation from the metal to the ligand is not exclusive for the transition metals, and that alkaline earth chemistry could be a viable alternative to the use of transition metals in the search for efficient N2 fixation.
From the condensed Fukui functions in Table 1, however, the smallest values were obtained for the alkaline earth metals. Therefore, the group II complexes must be softer than the group IV and VI complexes. Indeed, according to Equation (3), the softer character is apparent from their lower ionization potential, or, from a molecular orbital perspective, a higher HOMO energy (Table 3). None of the negatively charged species in this table were stable, so the electron affinity was set to zero.50 Similar trends were obtained for the CO complexes (see Table S7, Supporting Information).
Table 3.
Frontier orbital energies (in eV) and I values (in kcal mol−1) used for the calculation of the softness of N2 and the N2 complexes.[a] Values in brackets were obtained from the geometry optimization at the M06‐2X‐D3/def2‐TZVPP level.
|
|
E HOMO |
E LUMO |
I |
|---|---|---|---|
|
N2 |
−13.89 (−13.93) |
1.04 (1.03) |
370.4 |
|
Ca(N2)8 |
−3.68 (−3.71) |
−0.76 (−0.85) |
119.1 |
|
Sr(N2)8 |
−3.52 (−3.56) |
−0.71 (−0.84) |
114.5 |
|
Ba(N2)8 |
−3.57 (−3.50) |
−1.21 (−1.19) |
114.4 |
|
Ti(N2)8 |
−5.35 (−5.28) |
−0.35 (−0.29) |
142.4 |
|
Zr(N2)8 |
−5.20 (−5.15) |
−0.45 (−0.41) |
148.5 |
|
Hf(N2)8 |
−5.16 (−5.12) |
−0.56 (−0.53) |
147.7 |
|
Cr(N2)6 |
−6.60 (−6.54) |
−0.13 (−0.06) |
177.8 |
|
Mo(N2)6 |
−6.69 (−6.65) |
−0.37 (−0.32) |
193.3 |
|
W(N2)6 |
−6.71 (−6.66) |
−0.56 (−0.53) |
192.9 |
[a] The vertical electron affinities (A) were all negative and were set to zero.
Despite the negative vertical electron affinities, the electrophilic Fukui function f +(r) and local softness s +(r) were calculated using the same cage‐assembly approach for the N2 complexes (Tables S2 and S5, Supporting Information) and CO complexes (Tables S3 and S6, Supporting Information) because the reduction of the ligands involves the addition of electrons after all. Conveniently, all trends for the nucleophilic descriptors are maintained for the electrophilic counterparts. That is, the Nout atoms (or O atoms) become more electrophilic in the cage configuration as probed by the Fukui function. In the metal complexes, the atom‐condensed local softness of the outer atoms is again very similar, regardless of the central metal atom. The group II metals prevail for the activation of the O atoms in the CO complexes and the Nout atoms in the N2 complexes.
Alternatively, one could rationalize the reactivity of these complexes within the framework of CDFT by means of philicity indices.51 These are based on the electrophilicity index,36, 52 a derived descriptor25 representing the energy gain upon an optimal charge transfer from the environment to the system. As this is a global descriptor probing the reactivity of an entire molecule similar to the global softness, a localized formulation was proposed involving the electrophilic or nucleophilic Fukui function, yielding a measure for local electrophilicity or nucleophilicity, respectively.51 As the electrophilicity index and the electron affinity are both measures of the potency of a molecular system to accept electrons, the instability of most of the anionic complexes with respect to the neutral complexes (reflected by negative electron affinities) indicates that an analysis based on the electrophilicity index would be less relevant.52 The Fukui function itself is, in this case, more adequate for assessing the local electrophilic and nucleophilic behavior, as it is a difference in electron density upon an increase or decrease in the number of electrons in the system, respectively, allowing a comparison of different regions in a molecule. Additionally, Hirshfeld charges were recently shown to correlate well with experimental electrophilicity and nucleophilicity scales.46, 53 Table S8 (Supporting Information) summarizes the Hirshfeld charges of each atom in each of the studied complexes and reveals marginal differences in the charge on the outer atom (of the order of 0.01) if the ligands are either isolated or bound to the metal, thus indicating that variations in philicity upon complexation would be extremely low, leading to non‐conclusive results.
(The lack of) electrostatic activation
From the CDFT analysis, all complexes are more prone to soft interactions compared with the isolated N2 molecule, with the alkaline earth complexes slightly outperforming the transition metal complexes owing to their higher global softness. For the evaluation of hard interactions, we first analyze the MEPs. These isosurfaces were calculated at the electron density isovalue that corresponds to the local minimum of the electrostatic potential values, V min. Figure 3 shows the MEPs of N2, Ca(N2)8, Ti(N2)8, and Cr(N2)6, as well as the potential range, V min value, and the corresponding density isovalue.
Figure 3.
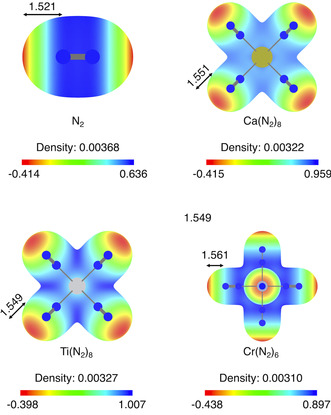
Molecular electrostatic potential (MEP) of N2, Ca(N2)8, Ti(N2)8, and Cr(N2)6 mapped on the electron density isosurface corresponding to V min. V min is always the minimum of the potential range and the distance to this point is given in Å. The MEP scales are in eV.
Interestingly, the MEPs in Figure 3 reveal that the (hard) electrostatic character of N2 is barely affected by complexation, in contrast to the soft frontier orbital character. For all metal complexes as well as isolated N2, a V min of about −0.400 eV is found along the N−N axis. Furthermore, the density isovalue as well as the Euclidian distance to this point are all very similar. This trend is also found for all the heavier N2 complexes (Figure S1, Supporting Information) as well as the CO complexes (Figure S2, Supporting Information). Therefore, the N2 (and CO) ligands are not activated electrostatically by any of the central metal atoms, despite the strong d–π* back‐donation. Also in contrast to the MEPs, the Fukui function does not have a strong component along the N−N axes but a toroidal shape around the N2 ligands, owing to the strong frontier orbital (HOMO) density character if one electron is removed or if the complex acts as the electron donor in reactions with electrophiles (vide infra). For an illustration of the degenerate HOMO in the MII(N2)8 complexes, see Figure 3 in Ref. 12, and for isovalue plots of the Fukui function see Figure S3 (Supporting Information).
Proton affinities: H+ seeks the HOMO and soft interactions
Finally, we explore the catalytic power of alkaline earth and transition metals for dinitrogen activation by means of proton affinities, because in the reduction of N2 to NH3, the addition of protons to the N2 ligand is an inevitable step. Table 4 summarizes the proton affinities of the group II, IV, and VI complexes, and interestingly, two types of protonated complexes were found (see Figure 4). In the type A complexes, the N2H+ ligand is attached end‐on, and all [MII(N2)H]+ complexes as well as [Ti(N2)8H]+ now have antiprism octavalent coordination, whereas ligands in [Zr(N2)8H]+ and [Hf(N2)8H]+ maintain the cubic coordination of the metal. In the type B complexes, the N2H+ ligand is attached side‐on, and they are more stable than type A for the [MII(N2)H]+ and [MIV(N2)H]+ complexes. Also note that the energetically lowest‐lying protonated isomers A and B have an electronic singlet state; the triplet states of the protonated group II complexes are higher in energy (see Table S12, Supporting Information).
Table 4.
Proton affinities [kcal mol−1] of N2 and all studied N2 complexes.
|
|
Type A |
Type B |
|---|---|---|
|
[N2H]+ |
120.8 |
– |
|
[Ca(N2)8H]+ |
237.7 |
250.8 |
|
[Sr(N2)8H]+ |
237.5 |
250.9 |
|
[Ba(N2)8H]+ |
238.4 |
250.5 |
|
[Ti(N2)8H]+ |
209.4 |
230.3[a] |
|
[Zr(N2)8H]+ |
209.9 |
227.2[a] |
|
[Hf(N2)8H]+ |
212.5 |
231.2[a] |
|
[Cr(N2)6H]+ |
195.1 |
192.6 |
|
[Mo(N2)6H]+ |
193.9 |
184.0 |
|
[W(N2)6H]+ |
197.0 |
186.6 |
[a] Cations [MIV(N2)8H]+ (MIV=Ti, Zr, Hf) with side‐on bonded N2H ligand (type B) are better described as [MIV(N2)6(N2H)]+—N2 because of one weakly bound N2 ligand.
Figure 4.
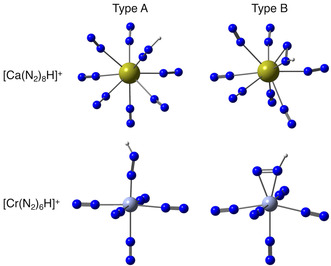
Protonated complexes [Ca(N2)8H]+ (top) and [Cr(N2)6H]+ (bottom) with an end‐on N2H+ ligand (type A) or a side‐on N2H+ (type B) ligand.
One can conceive proton affinities as an improvement of the MEP, in the sense that optimized protonated structures account for orbital interactions and geometric relaxation, whereas the MEP is the electrostatic interaction between a point charge of +1e mapped on a density isosurface. However, the MEPs and V min data in Figure 3 turn out to be a very bad approximation for the proton affinities in Table 4 for a few reasons. First, the proton affinity of isolated N2 is much lower (≈100 kcal mol−1) than the proton affinity of the complexes, whereas the V min values are marginally different, indicating that besides the electrostatic interaction, the proton is also involved in significant other (attractive) interactions, such as orbital (HOMO–LUMO) interactions. Furthermore, the proton is not aligned with the N2 axis in either type A or type B protonated complexes (see Figure 4), unlike the point at which V min is found, which is again an indication that the electrostatic attraction is dominated by orbital interactions. Indeed, the HOMO for all of the metal complexes has N2–π character, and the proton affinity also increases from the group VI (≈195 kcal mol−1) to the group IV (≈210 kcal mol−1) to the group II (≈238 kcal mol−1) type A complexes. The same increasing trend was found for the HOMO energies and global softness (which is the inverse of the vertical ionization potential if A is set to zero) in Table 3. In other words, the energy and geometry of a hard electrophilic proton and the metal complexes is determined by the soft frontier orbital interactions. In the case of isolated N2, both the electrostatic and orbital interactions lead to a linear geometry as the HOMO of N2 is the bonding σ orbital.
In order to gain quantitative insight into the importance of orbital and electrostatic interactions, we performed energy decomposition (EDA) calculations on the type A protonated complexes, as they are geometrically most similar to the neutral complexes. In each case, the two fragments were the proton and the neutral complex (or N2 in the case of N2H+), and the interaction energy of the fourth‐row complexes between these fragments is decomposed in Table 5. All trends are conserved for the heavier metal complexes (see Tables S9 and S10, Supporting Information, for the EDA of the row 5 and 6 complexes, respectively).
Table 5.
Energy decomposition analysis for [N2H]+ and the end‐on (type A) protonated fourth‐row complexes.[a] All energies are in kcal mol−1 and computed at the ZORA‐M06‐2X/TZ2P//M06‐2X‐D3//def2‐TZVPP level of theory.
|
|
[N2H]+ |
[Ca(N2)8H]+ |
[Ti(N2)8H]+ |
[Cr(N2)6H]+ |
|---|---|---|---|---|
|
ΔE int |
−120.6 |
−272.3 |
−249.3 |
−217.3 |
|
ΔE Metahybrid |
−0.3 |
−43.6 |
−40.8 |
−36.9 |
|
ΔE elstat |
19.4 |
−4.8 |
−0.9 |
18.0 |
|
ΔE Pauli |
0.0 |
0.0 |
0.0 |
0.0 |
|
ΔE orb |
−139.7 |
−223.9 |
−207.7 |
−198.4 |
|
ΔE prep |
0.0 |
33.1 |
41.5 |
22.7 |
[a] Trends are always very similar for all complexes with a central metal atom within one group of the periodic table.
The quantitative EDA analysis confirms the dominance of orbital interactions over the electrostatic term. For all complexes, the electrostatic interaction energy between the proton and the metal complexes, ΔE elstat, is positive or minutely negative, showing that the strongly negative orbital interaction energy, ΔE orb, outbalances the electrostatic term. Also, the N−H bond lengths are typically 1.0 Å, which is significantly shorter than the 1.5 Å found for V min. Elegantly, ΔE orb becomes more stabilizing upon moving from right to left in the periodic table: ΔE orb is −198.4, −207.7, and −223.9 kcal mol−1 in [Cr(N2)6H]+, [Ti(N2)8H]+, and [Ca(N2)8H]+, respectively. This trend is in agreement with the trends in softness, ionization potential, and HOMO energies, favoring the interaction with an alkaline earth metal complex as compared to a transition metal complex.
Interestingly, the results of [N2H]+ are very similar to those of the protonated complexes in the sense that the strongly negative ΔE orb compensates the positive electrostatic term. The ΔE orb term, however, is much less stabilizing (about 100 kcal mol−1) for [N2H]+ compared with the complexes, conforming with the softness, ionization potential, and HOMO energy. The proton was also systematically moved away from the N2 moiety (Table S11, Supporting Information), showing that the gain in ΔE orb by moving the proton closer to the N2 fragment outweighs the increase in ΔE elstat at shorter N−H lengths. The equilibrium N−H length is 1.039 Å, which is again significantly shorter than the distance to V min (1.521 Å) in Figure 3, similar to the protonated complexes. The minimum in ΔE elstat in Table S11 was also found to be around 1.5 Å. Finally, ΔE prep is the energy required to deform the fragments to the geometry from their equilibrium distance (also called the strain energy in the activation strain/distortion interaction model54), and ΔE Pauli originates from the repulsive interaction between electrons with identical spin. As one of the fragments (H+) does not have any electrons, this term is zero for each protonated species.
Conclusions
The recent discovery of stable cubic alkaline earth complexes of N2 and CO incited us to investigate the viability of alkaline earth metal‐based N2 fixation. DFT calculations and DFT‐based reactivity indices were used to determine the intrinsic reactivities of MII(N2)8 (MII=Ca, Sr, Ba), MIV(N2)8 (MIV=Ti, Zr, Hf), and MVI(N2)6 (MVI=Cr, Mo, W) complexes, and to connect them to trends in proton affinities. It was shown that the activation of N2 through complexation originates from the d–π back‐donation and the consequent enhanced (soft) frontier orbital character, rather than the (hard) electrostatic properties of the outer N atom. The HOMO energies, ionization potential, and chemical softness correctly predict the increased proton affinity of N2 complexes compared with isolated N2, whereas the molecular electrostatic potential falls short. Quantitative energy decomposition analyses confirm the dominance of orbital interactions over the electrostatic term. Significant activation of the N2 ligand is found for all central metal atoms, and within one row of the periodic table, the alkaline earth metal shows the strongest nucleophilic activation of the N2 ligand, confirming clear trends in proton affinities. The electrophilic character of the outer atoms of all complexes is also enhanced with very similar activation for all metals.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
SP and GF thank the Nanjing Tech University and the Deutsche Forschungsgemeinschaft for financial support. FDP and PG acknowledge Vrije Universiteit Brussel (VUB) and the Research Foundation Flanders (FWO) for continuous support to the ALGC research group. In particular, the Strategic Research Program funding of the VUB is thanked for financial support. FDP also wishes to acknowledge the Francqui Foundation for a position of Francqui Research Professor for the period 2016–2019. GF but also PG and FDP are very grateful to the International Solvay Institutes for Physics and Chemistry for honoring GF with the Solvay Chemistry Chair for 2019 and his concomitant stay at VUB, during which this work was initiated.
T. Bettens, S. Pan, F. De Proft, G. Frenking, P. Geerlings, Chem. Eur. J. 2020, 26, 12785.
Contributor Information
Gernot Frenking, Email: frenking@chemie.uni-marburg.de.
Paul Geerlings, Email: pgeerlin@vub.be.
References
- 1. Huheey J. E., Keiter E. A., Keiter R. L., Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed., Harper Collins, New York, 1993. [Google Scholar]
- 2. Cotton F. A., Wilkinson G., Murillo C. A., Bochmann M., Advanced Inorganic Chemistry, 6th ed., Wiley, New York, 1999. [Google Scholar]
- 3. Housecroft C. E., Sharpe A. G., Inorganic Chemistry , 3rd ed., Pearson, Harlow, Essex, UK, 2008. [Google Scholar]
- 4.
- 4a. Frenking G., Fröhlich N., Chem. Rev. 2000, 100, 717–774; [DOI] [PubMed] [Google Scholar]
- 4b. Zhao L., Pan S., Holzmann N., Schwerdtfeger P., Frenking G., Chem. Rev. 2019, 119, 8781–8845. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Elschenbroich C., Organometallics , 3rd ed., Wiley-VCH, Weinheim, 2006; [Google Scholar]
- 5b. Fryzuk M. D., Acc. Chem. Res. 2009, 42, 127–133. [DOI] [PubMed] [Google Scholar]
- 6. Wu X., Zhao L., Jin J., Pan S., Li W., Jin X., Wang G., Zhou M., Frenking G., Science 2018, 361, 912–916. [DOI] [PubMed] [Google Scholar]
- 7. Kohn W., Sham L. J., Phys. Rev. A 1965, 140, A1133–A1138. [Google Scholar]
- 8. Hin J. J., Yang T., Xin K., Wang G., Jing X., Zhou M., Frenking G., Angew. Chem. Int. Ed. 2018, 57, 6236–6241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6344–6349. [Google Scholar]
- 9. Wang Q., Pan S., Wu Y., Deng G., Wang G., Zhao L., Zhou M., Frenking G., Chem. Eur. J. 2019, 10.1002/chem.201905552. [DOI] [Google Scholar]
- 10. Gagliardi L., Pyykkö P., Theor. Chem. Acc. 2003, 110, 205–210. [Google Scholar]
- 11. Gagliardi L., Pyykkö P., J. Am. Chem. Soc. 2001, 123, 9700–9701. [DOI] [PubMed] [Google Scholar]
- 12. Wang Q., Pan S., Lei S., Jin J., Deng G., Wang G., Zhao L., Zhou M., Frenking G., Nat. Commun. 2019, 10, 3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smil V., Enriching the Earth: Fritz Haber, Carl Bosch and the Transformation of World Food Production, MIT Press, Cambridge, 2004. [Google Scholar]
- 14. Burgess B. K., Lowe D. J., Chem. Rev. 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Hidai M., Mizobe Y., Chem. Rev. 1995, 95, 1115–1133; [Google Scholar]
- 15b. Hidai M., Coord. Chem. Rev. 1999, 185–186, 99–108; [Google Scholar]
- 15c. Chirik P. J., Dalton Trans. 2007, 16–25; [DOI] [PubMed] [Google Scholar]
- 15d. Schrock R. R., Angew. Chem. Int. Ed. 2008, 47, 5512–5522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5594–5605; [Google Scholar]
- 15e. McWilliams S. F., Holland P. L., Acc. Chem. Res. 2015, 48, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yandulov D. V., Schrock R. R., Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]
- 17. Schrock R. R., Acc. Chem. Res. 2005, 38, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Geng C., Li J., Weiske T., Schwarz H., Proc. Natl. Acad. Sci. USA 2018, 115, 11680–11687; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Geng C., Li J., Weiske T., Schwarz H., Proc. Natl. Acad. Sci. USA 2019, 116, 21416–21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.See for example the Element Scarcity EuChemS Periodic Table, available at the EuChemS website; https://www.euchems.eu/euchems-periodic-table/.
- 20. Parr R. G., Donnelly R. A., Levy M., Palke W. E., J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar]
- 21. Parr R. G., Yang W., Density Functional Theory of Atoms and Molecules, Oxford University Press and Clarendon Press, New York and Oxford, 1989. [Google Scholar]
- 22.
- 22a. Chermette H., J. Comput. Chem. 1999, 20, 129–154; [Google Scholar]
- 22b. Geerlings P., De Proft F., Langenaeker W., Chem. Rev. 2003, 103, 1793–1873; [DOI] [PubMed] [Google Scholar]
- 22c. Chattaraj P. K., Nath S., Maiti B., Computational Medicinal Chemistry for Drug Discovery (Eds.: J. Tollenaere, P. Bultinck, H. D. Winter, W. Langenaeker), Marcel Dekker, New York, 2003, pp. 295–322; [Google Scholar]
- 22d. Ayers P. W., Anderson J., Bartolotti L. J., Int. J. Quant. Chem. 2005, 101, 520–534; [Google Scholar]
- 22e. Gázquez J. L., J. Mex. Chem. Soc. 2008, 52, 3–10; [Google Scholar]
- 22f. Liu S. B., Acta Phys.-Chim. Sin. 2009, 25, 590–600. [Google Scholar]
- 23. Parr R. G., Yang W., Annu. Rev. Phys. Chem. 1995, 46, 701–728. [DOI] [PubMed] [Google Scholar]
- 24. Geerlings P., De Proft F., Phys. Chem. Chem. Phys. 2008, 10, 3028–3042. [DOI] [PubMed] [Google Scholar]
- 25. Geerlings P., Chamorro E., Chattaraj P. K., De Proft F., Gàzquez J. L., Liu S., Morell C., Toro-Labbé A., Vela A., Ayers P., Theor. Chem. Acc. 2020, 139, 36. [Google Scholar]
- 26. Parr R. G., Yang W., J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar]
- 27. Yang W., Parr R. G., Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geerlings P., De Proft F., Int. J. Quant. Chem. 2000, 80, 227–235. [Google Scholar]
- 29. Pearson R. G., Acc. Chem. Res. 1993, 26, 250–255. [Google Scholar]
- 30. Pearson R. G., Chemical Hardness: Applications from Molecules to Solids, Wiley-VCH, Weinheim, Chapter 1, 1997. [Google Scholar]
- 31. Bonaccorsi R., Scrocco E., Tomasi J., J. Chem. Phys. 1970, 52, 5270–5284. [Google Scholar]
- 32. Melin J., Aparicio F., Subramanian V., Galván M., Chattaraj P. K., J. Phys. Chem. A 2004, 108, 2487–2491. [Google Scholar]
- 33. Gazquez J. L., Mendez F., J. Phys. Chem. 1994, 98, 4591–4593. [Google Scholar]
- 34. Mendez F., Gazquez J. L., J. Am. Chem. Soc. 1991, 113, 1855–1856. [Google Scholar]
- 35. Chattaraj P. K., J. Phys. Chem. A 2001, 105, 511–513. [Google Scholar]
- 36. Chattaraj P. K., Sarkar U., Roy D. R., Chem. Rev. 2006, 106, 2065–2091. [DOI] [PubMed] [Google Scholar]
- 37. Ziegler T., Rauk A., Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar]
- 38. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 39. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 40. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 41.Gaussian 16, Revision A.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 42.ADF, SCM Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands, 2019; http://www.scm.com.
- 43. Hirshfeld F. L., Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar]
- 44.
- 44a. Wiberg K. B., Rablen P. R., J. Comput. Chem. 1993, 14, 1504–1518; [Google Scholar]
- 44b. De Proft F., Van Alsenoy C., Peeters A., Langenaeker W., Geerlings P., J. Comput. Chem. 2002, 23, 1198–1209; [DOI] [PubMed] [Google Scholar]
- 44c. Oláh J., Van Alsenoy C., Sannigrahi A. B., J. Phys. Chem. A 2002, 106, 3885–3890; [Google Scholar]
- 44d. Saha S., Roy R. K., Int. J. Quantum Chem. 2009, 109, 1790–1806; [Google Scholar]
- 44e. Wiberg K. B., Rablen P. R., J. Org. Chem. 2018, 83, 15463–15469; [DOI] [PubMed] [Google Scholar]
- 44f. Bettens T., Alonso M., Geerlings P., De Proft F., Chem. Sci. 2020, 11, 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.
- 45a. Nalewajski R. F., Parr R. G., Proc. Natl. Acad. Sci. USA 2000, 97, 8879–8882; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45b. Liu S., J. Chem. Phys. 2007, 126, 244103. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Liu S., Rong C., Lu T., J. Phys. Chem. A 2014, 118, 3698–3704; [DOI] [PubMed] [Google Scholar]
- 46b. Liu S., Acta Phys. Chim. Sin. 2016, 32, 98–118; [Google Scholar]
- 46c. Rong C., Wang B., Zhao D., Liu S., WIREs Comput. Mol. Sci. 2019, e1461; [Google Scholar]
- 46d. Wang B., Rong C., Chattaraj P. K., Liu S., Theor. Chem. Acc. 2019, 138, 124. [Google Scholar]
- 47. Koch D., Chen Y., Golub P., Manzhos S., Phys. Chem. Chem. Phys. 2019, 21, 20814–20821. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Koch D., Chen Y., Golub P., Manzhos S., Phys. Chem. Chem. Phys. 2020, 22, 5380–5382; [DOI] [PubMed] [Google Scholar]
- 48b. Pan S., Frenking G., Phys. Chem. Chem. Phys. 2020, 22, 5377–5379. [DOI] [PubMed] [Google Scholar]
- 49. Davidson E. R., Kunze K. L., Machado F. B. C., Chakravorty S. J., Acc. Chem. Res. 1993, 26, 628–635. [Google Scholar]
- 50. Cárdenas C., Ayers P. W., De Proft F., Tozer D., Geerlings P., Phys. Chem. Chem. Phys. 2011, 13, 2285–2293. [DOI] [PubMed] [Google Scholar]
- 51. Chattaraj P., Maiti B., Sarkar U., J. Phys. Chem. A 2003, 107, 4973–4975. [Google Scholar]
- 52. Parr R. G., Szentpály L. V., Liu S., J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar]
- 53.
- 53a. Mayr H., Patz M., Angew. Chem. Int. Ed. Engl. 1994, 33, 938–957; [Google Scholar]; Angew. Chem. 1994, 106, 990–1010; [Google Scholar]
- 53b. Mayr H., Bug T., Gotta M. F., Hering N., Irrgang B., Janker B., Kempf B., Loos R., Ofial A. R., Remennikov G., Schimmel H., J. Am. Chem. Soc. 2001, 123, 9500–9512; [DOI] [PubMed] [Google Scholar]
- 53c. Lucius R., Loos R., Mayr H., Angew. Chem. Int. Ed. 2002, 41, 91–95; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 97–102. [Google Scholar]
- 54. Bickelhaupt F. M., Houk K. N., Angew. Chem. Int. Ed. 2017, 56, 10070–10086; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10204–10221. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary