

Abstract
A homoleptic organometallic FeIV complex that is stable in both solution and in the solid state at ambient conditions has been synthesized and isolated as [Fe(phtmeimb)2](PF6)2 (phtmeimb=[phenyl(tris(3‐methylimidazolin‐2‐ylidene))borate]−). This FeIV N‐heterocyclic carbene (NHC) complex was characterized by 1H NMR, HR‐MS, elemental analysis, scXRD analysis, electrochemistry, Mößbauer spectroscopy, and magnetic susceptibility. The two latter techniques unequivocally demonstrate that [Fe(phtmeimb)2](PF6)2 is a triplet FeIV low‐spin S=1 complex in the ground state, in agreement with quantum chemical calculations. The electronic absorption spectrum of [Fe(phtmeimb)2](PF6)2 in acetonitrile shows an intense absorption band in the red and near IR, due to LMCT (ligand‐to‐metal charge transfer) excitation. For the first time the excited state dynamics of a FeIV complex was studied and revealed a ≈0.8 ps lifetime of the 3LMCT excited state of [Fe(phtmeimb)2](PF6)2 in acetonitrile.
Keywords: high-valent iron, N-heterocyclic carbenes, organometallic complexes, photophysics, transient absorption spectroscopy
A stable homoleptic organometallic FeIV complex, [Fe(phtmeimb)2](PF6)2 was synthesized and isolated. It is stable in the solid state and in acetonitrile at ambient temperature. The excited state of the 3GS [Fe(phtmeimb)2](PF6)2 was shown to be of 3LMCT nature, and shows a 0.8 ps lifetime as determined by transient absorption spectroscopy.
High‐valent iron complexes, both heme‐ and non‐heme, function as active key intermediates in various biological catalytic cycles and important organic transformations.1 This has inspired the development of synthetic chemistry of FeIV complexes.1, 2 Several heteroleptic high‐valent FeIV complexes have been reported, where the high oxidation state is stabilized by terminal π‐donating auxiliary ligands (PDALs) such as oxide, nitride, imide, isocyanide, and ketimide, with various stabilities.3, 4, 5, 6, 7 In particular, FeIV‐oxo complexes have been found capable of various oxidative transformations.8 Examples of FeIV coordination compounds without stabilizing PDALs are scarce however. Besides FeF4, that could be isolated in a matrix,9 there are only few examples of more traditional Werner complexes using for example, electron‐donating dithiocarbamate ligands to stabilize the iron center in oxidation state FeIV,10, 11, 12 or FeIV complexes containing multidentate macrocyclic tetraamide ligands,13 a FeIV cyclam–azide complex14 and a FeIV cyano‐complex based on a tridentate imino‐ligand,15 the two latter electrochemically generated and studied in situ. To date, the exceptional example of an entirely stable FeIV complex belongs to the class of coordination cage compounds; FeIV hexahydrazide clathrochelate.16 This compound shows infinite stability in both aqueous and non‐aqueous solutions as well as in the solid state, unique to synthetic FeIV complexes.
For organometallic iron complexes the electron‐donating effect of carbanions can be exploited to stabilize higher metal oxidation states which has led to a current interest in homoleptic FeIV organometallic complexes (Figure 1).17 Three of the reported complexes constitute FeIV tetra alkyl species (Figure 1, 1–3, Table S4).18a, 18b, 18c The FeIV state of these compounds is however only accessible via disproportionation reactions and suffers from thermal instability and air sensitivity at ambient conditions (Figure 1, Table S4).18a, 18b, 18c Similarly, also the recently described organometallic FeIV complex [Fe(Cp*)2]2+ (Cp*=pentamethylcyclopentadienyl) (Figure 1, 4) is thermally stable at room temperature only in the solid state under inert atmosphere.18d We have recently reported that the strongly σ‐donating N‐heterocyclic carbene (NHC) ligands form homoleptic FeII and FeIII complexes with a hexa‐NHC coordination sphere leading to long photo‐induced charge‐transfer states.19, 20, 21 The complexes being [Fe(btz)3](PF6)2 19 and [Fe(btz)3](PF6)3 20 (btz22=3,3′‐dimethyl‐1,1′‐bis(p‐tolyl)‐4,4′‐bis(1,2,3‐triazol‐5‐ylidene), and [Fe(phtmeimb)2](PF6)21 (phtmeimb23 = [phenyl(tris(3‐methylimidazolin‐2‐ylidene))borate]−), the latter complex building on Fehlhammer's scorpionate ligand (htmeimb=[hydrido(tris(3‐methylimidazolin‐2‐ylidene))borate]−) and corresponding FeIII complex, [Fe(htmeimb)2](X) (X=BPh4 −, BF4 −),24 using the analogous phtmeimb ligand developed by Smith.23 Another effect of the strong σ‐donor NHC ligands above is a pronounced stabilization of higher metal oxidation states apparent from the remarkably low FeIII/II reduction potentials.20, 21, 24b In case of the scorpionate NHC ligand (phtmeimb), we obtained indications for the formation of a stable FeIV complex at modestly oxidizing potentials,21 something also observed by Fehlhammer for the FeIII complex of htmeimb.24b
Figure 1.
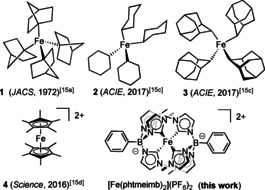
All reported organometallic homoleptic FeIV complexes.
These observations attracted our attention as examples of FeIV‐NHCs, outside the electrochemical observation of the FeIV scorpionates above, are exclusively found among heteroleptic complexes, having PDALs as additionally stabilizing groups (Table S3), resulting in complexes of various stabilities.4a, 4c, 5b, 25 Here, we present the synthesis and isolation, of the stable homoleptic hexa‐coordinated, [Fe(phtmeimb)2](PF6)2 (Figure 1). The combined spectroscopic, electrochemical, magnetic and theoretical investigations characterization univocally characterize this complex as a FeIV complex. Its S=1 ground state undergoes a ligand‐to‐metal charge transfer (LMCT) transition and we present the dynamics of the 3LMCT excited state that complements our previous observations of 3MLCT and 2LMCT states with FeII and FeIII NHC complexes, respectively.19, 20, 21
As previously reported, the FeIII precursor [Fe(phtmeimb)2]PF6 undergoes a reversible one‐electron electrochemical oxidation (E 1/2=0.25 V vs. ferrocene) that was assigned to the FeIV/III redox couple based on the spectroelectrochemical changes.21 Chemical oxidation of the red solution of [Fe(phtmeimb)2]PF6 in acetonitrile with thianthrenylhexafluorophosphate radical (E 1/2=0.85 V vs. ferrocene)26 produced a dark green colored solution of the FeIV compound [Fe(phtmeimb)2](PF6)2, which could be isolated pure in 95 % yield as green crystals (see Scheme 1), as proven by the combination of 1H NMR spectroscopy, HR‐MS, IR, elemental analysis, scXRD analysis, electrochemistry, 57Fe Mößbauer spectroscopy, magnetic susceptibility and quantum chemical calculations. [Fe(phtmeimb)2](PF6)2 is stable in the solid state exposed to air as well as in acetonitrile solution for days at ambient temperature. In addition, the crystals of [Fe(phtmeimb)2](PF6)2, are stable when washed with water, however slow decomposition was observed when water was added to a solution of the complex in acetonitrile.
Scheme 1.
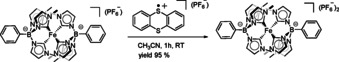
Synthesis of complex [Fe(phtmeimb)2](PF6)2 from [Fe(phtmeimb)2](PF6).
The 1H spectrum of [Fe(phtmeimb)2](PF6)2 shows highly deshielded resonances in comparison to its FeIII congener and the resonances are rather well‐resolved (See Supporting Information).21 Dark‐green single crystals of [Fe(phtmeimb)2](PF6)2 suitable for scXRD analysis were grown in a saturated anhydrous acetonitrile solution of [Fe(phtmeimb)2](PF6)2 by slow diffusion of anhydrous diethyl ether at room temperature exposed to air (see section S4, Supporting Information, for details). The molecular structure shows an octahedral iron center surrounded by two fac‐tridentate phtmeimb ligands (Figure 2). The Fe−C bond lengths are 1.99 to 2.01 Å, which are close to that of the FeIII congener (1.96 to 2.01 Å).21 Similarly, the C–Fe–C bite angles (87.3° to 87.7°) for [Fe(phtmeimb)2](PF6)2 are similar to the FeIII congener (86.2° to 87.6°).21 Thus, [Fe(phtmeimb)2](PF6)2 exhibits a close to perfect octahedral geometry that is virtually identical to its FeIII salt (details in section S4, Supporting Information). This indicates that very little structural re‐organization energy is needed when altering between the FeIV/FeIII oxidation states in [Fe(phtmeimb)2], thus facilitating rapid electron transfer processes involving this couple. The very similar Fe−C distances between the FeIV and FeIII oxidation states, as well as the Fe−C distances themselves, have also been observed for a heteroleptic macrocyclic tetra‐NHC complex in the two oxidation states, as FeIV=O and FeIII–O–FeIII complexes.25b We suggest that the very minor difference in Fe−C bond lengths is due to the rigidity of the tridentate phtmeimb ligand and/or the covalent nature of this bond, where the influence of the formal charge differences on iron is of little importance for this bond length in the latter case.
Figure 2.
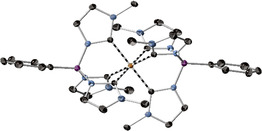
scXRD Molecular structure of the cation in [Fe(phtmeimb)2](PF6)2. Thermal ellipsoids are shown at 50 % probability. Hydrogen atoms, counter ions and solvent molecules are omitted for clarity. Orange=Fe; purple=B; blue=N, and grey=C.
The 57Fe Mößbauer spectrum of [Fe(phtmeimb)2](PF6)2 is shown in Figure 3 a). The isomer shift δ and electric quadrupole splitting Δ, of the doublet at 80 K are −0.23(1) and 3.04(1) mm s−1, respectively, the former in the same range as for complexes 1–3, and the latter in the same range as for complex 4,18 and differ from the doublet at 87 K of the FeIII congener, [Fe(phtmeimb)2](PF6), −0.09 and 1.54 mm s−1, respectively.21 The combination of an unusual large Δ‐value and a negative δ‐value for the doublet supports, that this pattern emanates from FeIV triplet low spin S=1 in a quasi‐octahedral coordination (Section S5, Supporting Information).27, 28 The magnetic susceptibility and magnetization data for [Fe(phtmeimb)2](PF6)2 are reported in Figure 3 b. The distinct nesting of the magnetization curves (Figure 3 b, insert) differs from the response of the FeIII precursor and clearly demonstrates the system to have an effective S> with a significant zero field splitting. The formulation of the complex as a low‐spin FeIV is corroborated by these magnetic data (Section S6, Supporting Information). From the magnetization data, a sizeable ZFS of D≈22 cm−1 was deduced. EPR of [Fe(phtmeimb)2](PF6)2, generated electrochemically from the FeIII congener, does not show any EPR signal at X‐band frequencies, in either perpendicular or parallel mode (Section S7, Supporting Information). This is explained by the magnitude and likely positive sign of the determined d‐value which indeed precludes detection of EPR signals at any accessible frequency.
Figure 3.
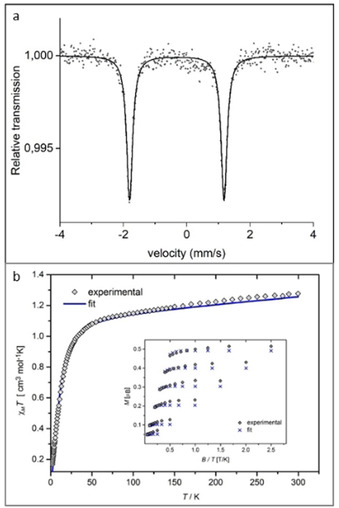
(a) 57Fe Mößbauer spectrum of [Fe(phtmeimb)2](PF6)2 at 80 K. (b) Experimental magnetic data and model fits for [Fe(phtmeimb)2](PF6)2. Main panel: magnetic susceptibility, represented by the χT product in the temperature range 2–300 K. Insert: magnetization data for the same sample recorded at T=2–10 K and B=0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 T.
The electronic absorption spectrum of [Fe(phtmeimb)2](PF6)2 in deaerated acetonitrile (Figure 4) is dominated by a broad, intense absorption band peaking at 715 nm (ϵ=6850 m −1 cm−1) with a shoulder around 810 nm, in excellent agreement with the reported spectrum obtained upon electrochemical one‐electron oxidation of the FeIII precursor [Fe(phtmeimb)2](PF6).21 Based on electrochemical potentials of the FeIV/III couple and ligand oxidation, the low energy absorption band of the oxidized complex was previously attributed to a LMCT transition.21 This assignment can now be supported form the experimental data of the isolated complex and computational data (vide infra) that the transition occurs from the triplet ground state (3GS) with a (t2g 4) electronic configuration to a 3LMCT state (t2g 5πL 1).
Figure 4.
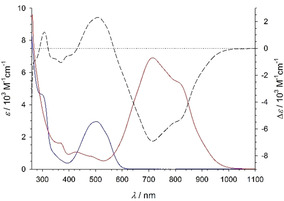
Electronic absorption spectra of [Fe(phtmeimb)2](PF6)2 (−) and [Fe(phtmeimb)2](PF6) (−) in deaerated acetonitrile, and the FeIII–FeIV differential spectrum (– – –).
In previous studies of complexes [Fe(btz)3]3+ and [Fe(btz)3]2+ we showed that the same NHC ligand set furnishes both the FeIII and FeII oxidation states with exceptional lifetimes (hundreds of picoseconds) of their 2LMCT and 3MLCT excited states, respectively.19, 21 With the relative photostability of [Fe(phtmeimb)2](PF6)2 in acetonitrile solution (Section S8, Supporting Information), we have the opportunity to compare the excited state dynamics following LMCT excitation of a FeIV complex to the recently reported record 2.0 ns lifetime of the 2LMCT state of a Fe complex, featured by the FeIII congener. Transient absorption spectra following 800 nm excitation of [Fe(phtmeimb)2](PF6)2 are shown in Figure 5. The pronounced ground state bleach (GSB), peaking at 715 nm, and the excited state absorption (ESA) at wavelengths below around 600 nm are readily rationalized in terms of the spectral differences arising from the FeIV to FeIII reduction. The additional excited state absorption at wavelengths above around 900 nm can be attributed to the oxidation of the phtmeimb ligand in analogy to the spectrum of the LMCT excited state of [Fe(phtmeimb)2](PF6) that involves the same ligand oxidation.21 The transient‐absorption spectrum hence corroborates the assignment of the 715 nm absorption band of [Fe(phtmeimb)2](PF6)2 to an LMCT transition from the t2g 4 low‐spin ground state (3GS) to a t2g 5 πL 1 (3LMCT) excited state. Most of the transient absorption decays with a ≈0.8 ps lifetime, indicating fast deactivation of the 3LMCT state accompanied by similarly fast ground state recovery. Only a minor component with a 16 ps lifetime that contributes to the ground state recovery and the transient absorption between 850 and 1000 nm points to the involvement of additional states in the deactivation of the 3LMCT excited state.
Figure 5.
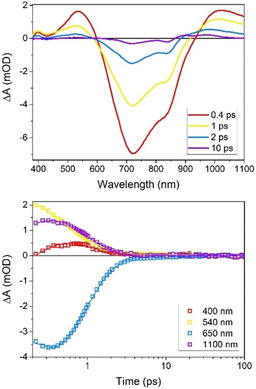
Transient absorption results for [Fe(phtmeimb)2](PF6)2 in deaerated acetonitrile—full spectral decays (top), and kinetics at 400 nm, 540 nm, 650 nm, and 1100 nm (bottom).
Quantum chemical calculations of different relaxed spin states corroborate the nature and structure of the ground state as a triplet state. Figure 6 shows the calculated ground state spin density of the 3[Fe(phtmeimb)2]2+ complex, which, together with the calculated Mulliken spin density on the iron for this state (Table 1) supports an assignment of the ground state as a triplet FeIV complex with (t2g)4 character, with some admixing of the frontier molecular orbitals with NHC‐π contributions.
Figure 6.
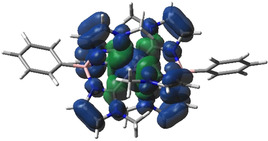
Calculated spin density for the optimized lowest energy state of 3[Fe(phtmeimb)2]2+. Spin‐up density is in blue and spin down in green.
Table 1.
Quantum chemically calculated properties of [Fe(phtmeimb)2]2+ for fully optimized states of different overall spin multiplicities. Calculated properties include relative total energies (E rel), average Fe−C bond lengths (R av(Fe‐C)), and Mulliken Spin densities on the central metal ion (Fe Spin)
|
State |
E rel [eV] |
R av(Fe−C) [Å] |
Fe Spin |
|---|---|---|---|
|
1[Fe(phtmeimb)2]2+ |
1.26 |
2.000 |
0.00 |
|
3[Fe(phtmeimb)2]2+ |
0.00 |
2.021 |
2.00 |
|
5[Fe(phtmeimb)2]2+ |
1.84 |
2.129 |
3.41 |
The synthesized and isolated [Fe(phtmeimb)2](PF6)2 complex, is stable in the solid state and acetonitrile solution at ambient conditions. The facile tunability of the NHC ligand systems provide potential for stabilization of high‐valent metal complexes in general. We have built on Fehlhammer's initial observation,24 and developed an organometallic high‐valent FeIV NHC complex without additional stabilizing π‐donating ligands,4a, 4c, 5b, 25a, 25b, 25d stable at ambient conditions, utilizing the strongly σ‐donating mono‐anionic facial tris‐NHC scorpionate ligand phtmeimb developed by Smith.23 The excited state dynamics of homoleptic [Fe(phtmeimb)2](PF6)2 was studied, constituting the first of its kind study of a FeIV complex. We observed the fundamental difference in excited state lifetimes for FeIII‐2LMCT (2 ns, 2.1 eV) vs. FeIV‐3LMCT (0.8 ps, <1.5 eV) states, which deserves further investigation beyond the scope of the present paper. Moreover, while advanced designs and studies are needed for a better understanding of FeIV NHC complexes, modifications that aim at further prolonging the 3LMCT state through subtle tuning of the energy levels are also in progress. Finally, having access to stable FeIV species, with long lived 3LMCT state that can take part in redox/photocatalytic processes/cycles, is an appealing area for future exploration.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Swedish Research Council (VR), the Swedish Foundation for Strategic Research (SSF), the Knut and Alice Wallenberg Foundation (KAW) and the Swedish Energy Agency (Energimyndigheten) are acknowledged for financial support. K.W. thanks the LMK‐foundation for a generous grant. P.P. acknowledges support from eSSENCE, and the computing centers LUNARC and NSC, through support via SNIC. O.P. thanks the Carl Tryggers Stiftelse for support. N.W.R. gratefully acknowledges funding from the Alexander von Humboldt Foundation within the Feodor‐Lynen Fellowship program.
O. Prakash, P. Chábera, N. W. Rosemann, P. Huang, L. Häggström, T. Ericsson, D. Strand, P. Persson, J. Bendix, R. Lomoth, K. Wärnmark, Chem. Eur. J. 2020, 26, 12728.
Contributor Information
Prof. Dr. Petter Persson, Email: petter.persson@teokem.lu.se.
Prof. Dr. Jesper Bendix, Email: bendix@chem.ku.dk.
Dr. Reiner Lomoth, Email: reiner.lomoth@kemi.uu.se.
Prof. Dr. Kenneth Wärnmark, Email: kenneth.warnmark@chem.lu.se.
References
- 1.
- 1a. Groves J. T., J. Inorg. Biochem. 2006, 100, 434–447; [DOI] [PubMed] [Google Scholar]
- 1b. Hohenberger J., Ray K., Meyer K., Nat. Commun. 2012, 3, 720; [DOI] [PubMed] [Google Scholar]
- 1c. Moody P. C. E., Raven E. L., Acc. Chem. Res. 2018, 51, 427–435; [DOI] [PubMed] [Google Scholar]
- 1d. L. Que, Jr. , Tolman W. B., Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. McDonald A. R., L. Que, Jr. , Coord. Chem. Rev. 2013, 257, 414–428; [Google Scholar]
- 2b. Nam W., Acc. Chem. Res. 2007, 40, 522–531; [DOI] [PubMed] [Google Scholar]
- 2c. Krebs C., Fujimori D. G., Walsh C. T., J. M. Bollinger, Jr. , Acc. Chem. Res. 2007, 40, 484–492; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Kryatov S. V., Rybak-Akimova E. V., Chem. Rev. 2005, 105, 2175–2226. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Rohde J.-U., In J.-H., Lim M. H., Brennessel W. W., Bukowski M. R., Stubna A., Münck E., Nam W., L. Que, Jr. , Science 2003, 299, 1037–1039; [DOI] [PubMed] [Google Scholar]
- 3b. Kupper C., Mondal B., Serrano-Plana J., Klawitter I., Neese F., Costas M., Ye S., Meyer F., J. Am. Chem. Soc. 2017, 139, 8939–8949; [DOI] [PubMed] [Google Scholar]
- 3c. Bigi J. P., Harman W. H., Lassalle-Kaiser B., Robles D. M., Stich T. A., Yano J., Britt R. D., Chang C. J., J. Am. Chem. Soc. 2012, 134, 1536–1542; [DOI] [PubMed] [Google Scholar]
- 3d. Monte Pérez I., Engelmann X., Lee Y.-M., Yoo Mi., Kumaran E., Farquhar E. R., Bill E., England J., Nam W., Swart M., Ray K., Angew. Chem. Int. Ed. 2017, 56, 14384–14388; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14576–14580. [Google Scholar]
- 4.
- 4a. Vogel C., Heinemann F. W., Sutter J., Anthon C., Meyer K., Angew. Chem. Int. Ed. 2008, 47, 2681–2684; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2721–2724; [Google Scholar]
- 4b. Maity A. K., Murillo J., Metta-Magaña A. J., Pinter B., Fortier S., J. Am. Chem. Soc. 2017, 139, 15691–15700; [DOI] [PubMed] [Google Scholar]
- 4c. Scepaniak J. J., Fulton M. D., Bontchev R. P., Duesler E. N., Kirk M. L., Smith J. M., J. Am. Chem. Soc. 2008, 130, 10515–10517. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Klinker E. J., Jackson T. A., Jensen M. P., Stubna A., Juh-sz G., Bominaar E. L., Münck E., L. Que, Jr. , Angew. Chem. Int. Ed. 2006, 45, 7394–7397; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7554–7557; [Google Scholar]
- 5b. Nieto I., Ding F., Bontchev R. P., Wang H., Smith J. M., J. Am. Chem. Soc. 2008, 130, 2716–2717; [DOI] [PubMed] [Google Scholar]
- 5c. Thomas C. M., Mankad N. P., Peters J. C., J. Am. Chem. Soc. 2006, 128, 4956–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collins T. J., Fox B. G., Hu Z. G., Kostka K. L., Münck E., Rickard C. E. F., Wright L. J., J. Am. Chem. Soc. 1992, 114, 8724–8725. [Google Scholar]
- 7. Lewis R. A., Wu G., Hayton T. W., J. Am. Chem. Soc. 2010, 132, 12814–12816. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Collins T. J., Ryabov A. D., Chem. Rev. 2017, 117, 9140–9162; [DOI] [PubMed] [Google Scholar]
- 8b. Chantarojsiri T., Sun Y., Long J. R., Chang C. J., Inorg. Chem. 2015, 54, 5879–5887; [DOI] [PubMed] [Google Scholar]
- 8c. Serrano-Plana J., Oloo W. N., Acosta-Rueda L., Meier K. K., Verdejo B., García-España E., Basallote M. G., Münck E., L. Que, Jr. , Company A., Costas M., J. Am. Chem. Soc. 2015, 137, 15833–15842; [DOI] [PubMed] [Google Scholar]
- 8d. Dantignana V., Serrano-Plana J., Draksharapu A., Magallón C., Banerjee S., Fan R., Gamba I., Guo Y., L. Que, Jr. , Costas M., Company A., J. Am. Chem. Soc. 2019, 141, 15078–15091; [DOI] [PubMed] [Google Scholar]
- 8e. Klein J. E. M. N., Mandal D., Ching W.-M., Mallick D., L. Que, Jr. , Shaik S., J. Am. Chem. Soc. 2017, 139, 18705–18713. [DOI] [PubMed] [Google Scholar]
- 9. Schlöder T., Vent-Schmidt T., Riedel S., Angew. Chem. Int. Ed. 2012, 51, 12063–12067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12229–12233. [Google Scholar]
- 10.
- 10a. Pasek E. A., Straub D. K., Inorg. Chem. 1972, 11, 259–263; [Google Scholar]
- 10b. Petridis D., Niarchos D., Kanellakopulos B., Inorg. Chem. 1979, 18, 505–509; [Google Scholar]
- 10c. Pasek E. A., Straub D. K., Inorg. Chim. Acta 1977, 21, 23–27; [Google Scholar]
- 10d. Raston C. L., White A. H., Petridis D., Taylor D., J. Chem. Soc. Dalton Trans. 1980, 1928–1931. [Google Scholar]
- 11.
- 11a. Martin R. L., Rohde N. M., Robertson G. B., Taylor D., J. Am. Chem. Soc. 1974, 96, 3647–3649; [Google Scholar]
- 11b. Chant R., Hendrickson A. R., Martin R. L., Rohde N. M., Inorg. Chem. 1975, 14, 1894–1902; [Google Scholar]
- 11c. Petrouleas V., Petridis D., Inorg. Chem. 1977, 16, 1306–1309. [Google Scholar]
- 12.
- 12a. Pignolet L. H., Lewis R. A., Holm R. H., Inorg. Chem. 1972, 11, 99–104; [Google Scholar]
- 12b. Lewis G. R., Dance I., J. Chem. Soc. Dalton Trans. 2000, 3176–3185; [Google Scholar]
- 12c. Milsmann C., Sproules S., Bill E., Weyhermüller T., George S. D., Wieghardt K., Chem. Eur. J. 2010, 16, 3628–3645. [DOI] [PubMed] [Google Scholar]
- 13. Collins T. J., Kostka K. L., Münck E., Uffelman E. S., J. Am. Chem. Soc. 1990, 112, 5637–5639. [Google Scholar]
- 14. Berry J. F., Bill E., Bothe E., Weyhermüller T., Weighardt K., J. Am. Chem. Soc. 2005, 127, 11550–11551. [DOI] [PubMed] [Google Scholar]
- 15. England J., Farquhar E. R., Guo Y., Cranswick M. A., Ray K., Münck E., L. Que, Jr. , Inorg. Chem. 2011, 50, 2885–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tomyn S., Shylin S. I., Bykov D., Ksenofontov V., Gumienna-Kontecka E., Bon V., Fritsky I. O., Nat. Commun. 2017, 8, 14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schilter D., Nat. Rev. Chem. 2017, 1, 0036. [Google Scholar]
- 18.
- 18a. Bower B. K., Tennent H. G., J. Am. Chem. Soc. 1972, 94, 2512–2514; [Google Scholar]
- 18b. Lewis R. A., Smiles D. E., Darmon J. M., Stieber S. C. E., Wu G., Hayton T. W., Inorg. Chem. 2013, 52, 8218–8227; [DOI] [PubMed] [Google Scholar]
- 18c. Casitas A., Rees J. A., Goddard R., Bill E., DeBeer S., Fürstner A., Angew. Chem. Int. Ed. 2017, 56, 10108–10113; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10242–10247; [Google Scholar]
- 18d. Malischewski M., Adelhardt M., Sutter J., Meyer K., Seppelt K., Science 2016, 353, 678–682. [DOI] [PubMed] [Google Scholar]
- 19. Chábera P., Kjær K. S., Prakash O., Honarfar A., Liu Y., Fredin L. A., Harlang T. B., Lidin S., Uhlig J., Sundström V., Lomoth R., Persson P., Wärnmark K., J. Phys. Chem. Lett. 2018, 9, 459–463. [DOI] [PubMed] [Google Scholar]
- 20. Chábera P., Liu Y., Prakash O., Thyrhaug E., El Nahhas A., Honarfar A., Essén S., Fredin L. A., Harlang T. C. B., Kjær K. S., Handrup K., Ericson F., Tatsuno H., Morgan K., Schnadt J., Häggström L., Ericsson T., Sobkowiak A., Lidin S., Huang P., Styring S., Uhlig J., Bendix J., Lomoth R., Sundström V., Persson P., Wärnmark K., Nature 2017, 543, 695–699. [DOI] [PubMed] [Google Scholar]
- 21. Kjær K. S., Kaul N., Prakash O., Chábera P., Rosemann N. W., Honarfar A., Gordivska O., Fredin L. A., Bergquist K.-E., Häggström L., Ericsson T., Lindh L., Yartsev A., Styring S., Huang P., Uhlig J., Bendix J., Strand D., Sundström V., Persson P., Lomoth R., Wärnmark K., Science 2019, 363, 249–253. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Liu Y., Kjær K. S., Fredin L. A., Chábera P., Harlang T., Canton S. E., Lidin S., Zhang J., Lomoth R., Bergquist K.-E., Persson P., Sundström V., Chem. Eur. J. 2015, 21, 3628–3629; [DOI] [PubMed] [Google Scholar]
- 22b. Guisado-Barrios G., Bouffard J., Donnadieu B., Bertrand G., Organometallics 2011, 30, 6017–6021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Fletcher J. T., Bumgarner B. J., Engels N. D., Skoglund D. A., Organometallics 2008, 27, 5430–5433. [Google Scholar]
- 23. Forshaw A. P., Bontchev R. P., Smith J. M., Inorg. Chem. 2007, 46, 3792–3794. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Kernbach U., Ramm M., Luger P., Fehlhammer W. P., Angew. Chem. Int. Ed. Engl. 1996, 35, 310–312; [Google Scholar]; Angew. Chem. 1996, 108, 333–335; [Google Scholar]
- 24b. Fränkel R., Kernbach U., Bakola-Christianopoulou M., Plaia U., Suter M., Ponikwar W., Nöth H., Moinet C., Fehlhammer W. P., J. Organomet. Chem. 2001, 617–618, 530–545. [Google Scholar]
- 25.
- 25a. Scepaniak J. J., Young J. A., Bontchev R. P., Smith J. M., Angew. Chem. Int. Ed. 2009, 48, 3158–3160; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3204–3206; [Google Scholar]
- 25b. Meyer S., Klawitter I., Demeshko S., Bill E., Meyer F., Angew. Chem. Int. Ed. 2013, 52, 901–905; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 935–939; [Google Scholar]
- 25c. Cramer S. A., Sncáhez R. H., Brakhagea D. F., Jenkins D. M., Chem. Commun. 2014, 50, 13967–13970; [DOI] [PubMed] [Google Scholar]
- 25d. Anneser M. R., Elpitiya G. R., Townsend J., Johnson E. J., Powers X. B., DeJesus J. F., Vogiatzis K. D., Jenkins D. M., Angew. Chem. Int. Ed. 2019, 58, 8115–8118; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8199–8202. [Google Scholar]
- 26. Becka J., Bredowb T., Tjahjantoa R. T., Z. Naturforsch. B 2009, 64, 145–152. [Google Scholar]
- 27. Gütlich P., Bill E., Trautwein A. X., Mößbauer Spectroscopy and Transition Metal Chemistry, Springer, Heidelberg: 2011. [Google Scholar]
- 28. Ingalls R., Phys. Rev. 1964, 133, A787–A795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary