

INTRODUCTION
Lipodystrophies are a group of rare disorders of diverse cause characterized by variable loss of body fat. The loss of body fat may affect nearly the entire body (generalized), only certain body regions (partial), or small areas under the skin (localized). Depending on the severity and extent of body fat loss, patients may be predisposed to metabolic complications associated with insulin resistance.1,2 These metabolic complications include early onset of diabetes mellitus, hypertriglyceridemia, and hepatic steatosis.1–3 In some patients, these metabolic complications are challenging to manage and can lead to complications including diabetic nephropathy and retinopathy, acute pancreatitis (from extreme hypertriglyceridemia and chylomicronemia), hepatic cirrhosis, and premature cardiovascular disease. Other common clinical manifestations include polycystic ovarian syndrome (PCOS), acanthosis nigricans as a result of severe insulin resistance, and eruptive xanthomas caused by extreme hypertriglyceridemia.1–3
The loss of body fat can result from underlying genetic defects (genetic lipodystrophies including autosomal-recessive or autosomal-dominant subtypes) or from autoimmune mechanisms (acquired lipodystrophies including generalized or partial subtypes) or drugs (eg, highly active antiretroviral therapy [HAART]-induced partial lipodystrophy in human immunodeficiency virus [HIV]-infected patients or localized lipodystrophies from insulin and other injected drugs).1–3 The localized lipodystrophies and lipodystrophy in HIV-infected patients are the most prevalent subtype of lipodystrophies, whereas the other genetic and acquired lipodystrophies are rare.2 Localized lipodystrophies do not predispose to metabolic complications because the loss of fat is trivial; however, other partial or generalized lipodystrophies cause variable predisposition to metabolic complications.
The major subtypes of lipodystrophy are described in Table 1 and shown in Fig. 1. However, given the heterogeneity of manifestations, variable patterns of fat loss, and genetic basis that have yet to be identified, all lipodystrophy syndromes cannot be classified into these categories.4 Regardless of the cause, patients with generalized lipodystrophy have extremely low serum levels of adipocytokines, such as leptin and adiponectin,5,6 whereas serum leptin and adiponectin levels in those with partial lipodystrophies can range from low to high. Marked hypoleptinemia may induce excessive appetite and can exacerbate metabolic complications of insulin resistance.3 This article covers the major types of lipodystrophy syndromes.
Table 1.
General classification of major lipodystrophy subtypes
| Lipodystrophy Subtype | Main Characteristics |
|---|---|
| Congenital generalized lipodystrophy | Presents with near total loss of body fat at birth or during infancy. Autosomal-recessive inheritance. |
| Familial partial lipodystrophy | Presents with variable loss of subcutaneous fat from the upper and lower extremities and the truncal region at puberty or later. Autosomal-dominant inheritance. |
| Acquired generalized lipodystrophy | Characterized by gradual loss of subcutaneous fat from nearly all over the body. Associated with autoimmune diseases. |
| Acquired partial lipodystrophy | Characterized by gradual loss of fat from the upper body, including head, neck, upper extremities, and truncal region during childhood. Associated with autoantibodies called complement 3 nephritic factor and in ~20% of patients with membranoproliferative glomerulonephritis. |
| HAART-induced lipodystrophy in HIV patients | Associated with therapy including HIV protease inhibitors or nucleoside analogues. |
| Localized lipodystrophy | Usually caused by insulin injections or other injectables, such as steroids. |
Fig. 1.
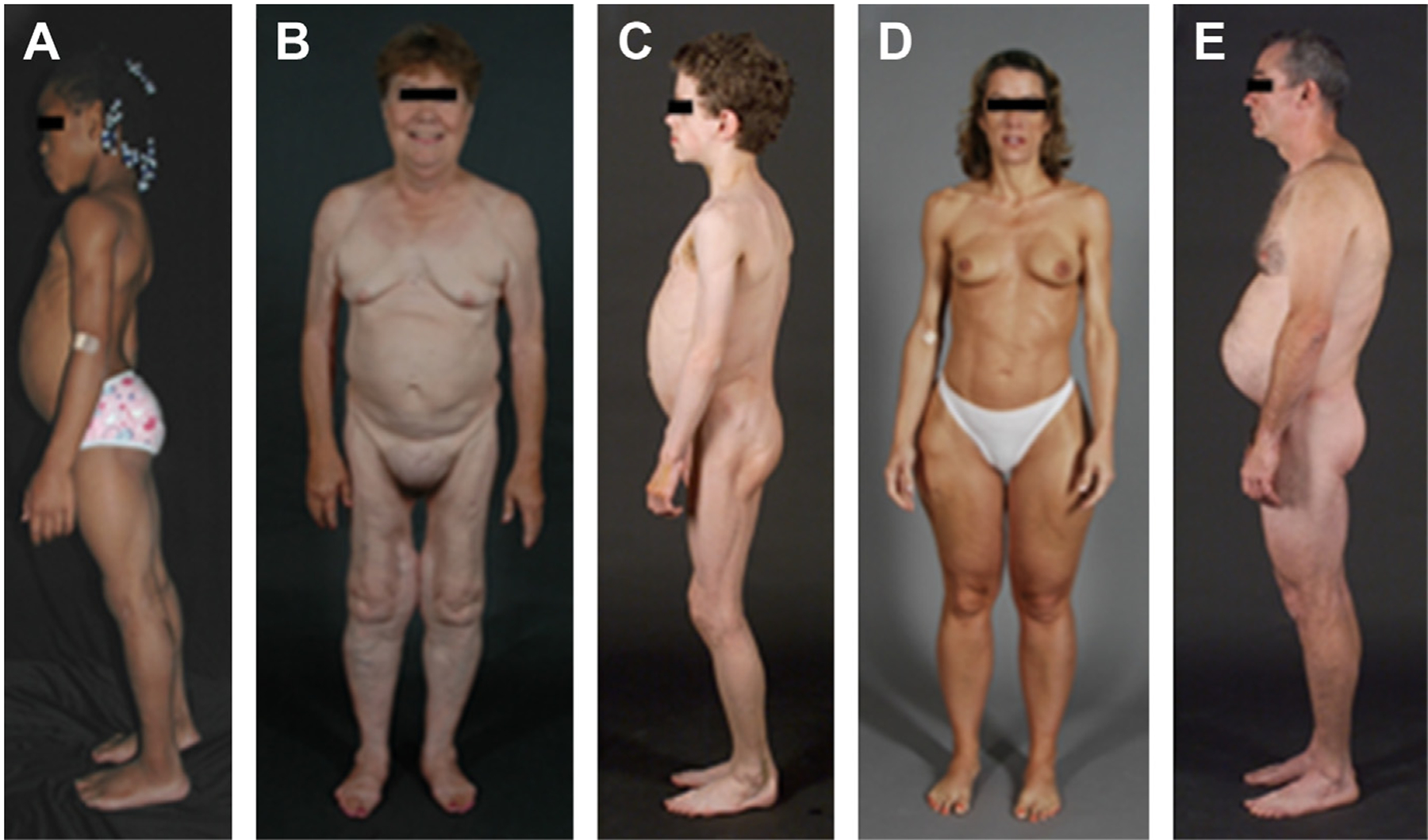
Clinical features of patients with various types of lipodystrophies. (A) Lateral view of an 8-year-old African American girl with congenital generalized lipodystrophy (also known as Berardinelli-Seip congenital lipodystrophy) type 1 caused by homozygous c.377insT (p.Leu126fs*146) mutation in AGPAT2. The patient had generalized loss of subcutaneous fat at birth and developed mild acanthosis nigricans in the axillae and neck later during childhood. She had umbilical prominence and acromegaloid features (enlarged mandible, hands, and feet). (B) Anterior view of a 65-year-old white woman with familial partial lipodystrophy of the Dunnigan variety caused by heterozygous p.Arg482Gln mutation in LMNA. She noticed loss of subcutaneous fat from the limbs at the time of puberty and later lost subcutaneous fat from the anterior truncal region. The breasts were atrophic. She had increased subcutaneous fat deposits in the face, anterior neck, suprapubic and vulvar region, and medial parts of the knees. (C) Lateral view of an 8-year-old German boy with acquired generalized lipodystrophy. He started experiencing generalized loss of subcutaneous fat at age 3 with marked acanthosis nigricans in the neck, axillae, and groin. He developed Crohn’s disease at age 11 requiring hemicolectomy at age 13. (D) Anterior view of a 39-year-old white woman with acquired partial lipodystrophy (Barraquer-Simons syndrome). She noticed marked loss of subcutaneous fat from the face, neck, upper extremities, chest, and abdomen at the age of 12 years but later developed increased subcutaneous fat deposition in the lower extremities. (E) Lateral view of a 39-year-old white man infected with HIV with protease inhibitor–containing highly active antiretroviral therapy–induced lipodystrophy. He had marked loss of subcutaneous fat from the face and limbs but had increased subcutaneous fat deposition in the neck region anteriorly and posteriorly showing buffalo hump. Abdomen was protuberant because of excess intra-abdominal fat. He had been on protease inhibitor–containing antiretroviral therapy for more than 7 years. ([A] From Simha V, Garg A. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol 2006;17:162–9; with permission; [B–E] Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3317; with permission.)
GENETIC LIPODYSTROPHIES
The two main types of genetic lipodystrophies are congenital generalized lipodystrophy (CGL), an autosomal-recessive syndrome (Tables 2 and 3), and familial partial lipodystrophy (FPLD), mostly an autosomal-dominant syndrome (Table 4). There are other extremely rare types that have been reported in approximately 30 patients or less (Table 5). These extremely rare types of genetic lipodystrophies are not discussed further in this article.
Table 2.
Subtypes of CGL
| Subtype | Gene | Molecular Basis | Prevalence |
|---|---|---|---|
| CGL1 | AGPAT2 | AGPAT enzymes play a key role in biosynthesis of triglycerides and phospholipids in various organs. AGPAT isoform 2 is highly expressed in the adipose tissue. | Most common subtype7,8,10 |
| CGL2 | BSCL2 | Seipin, encoded by BSCL2, plays a key role in fusion of small lipid droplets in the adipocytes and in adipocyte differentiation. | Second most common subtype7–9 |
| CGL3 | CAV1 | Caveolin 1 is an integral component of caveolae, which are present on adipocyte membranes. Caveolae translocate fatty acids and other lipids to lipid droplets. | Only one patient reported11 |
| CGL4 | PTRF | PTRF (also known as cavin-1) is involved in biogenesis of caveolae and regulates expression of caveolins 1 and 3. | About 20 patients reported12,43,44 |
Abbreviations: AGPAT2, 1-acylglycerol-3-phosphate O-acyltransferase 2; BSCL2, Berardinelli-Seip congenital lipodystrophy 2; CAV1, caveolin 1; PTRF, polymerase I and transcript release factor.
Table 3.
Unique clinical features in CGL subtypes
| Affected Feature | CGL Type 1 (AGPAT2) | CGL Type 2 (BSCL2) | CGL Type 3 (CAV1) | CGL Type 4 (PTRF) |
|---|---|---|---|---|
| Body fat loss | Only metabolically active adipose tissue is lost Mechanical adipose tissue preserved |
Both metabolically active and mechanical adipose tissues are lost | Absent metabolically active adipose tissue Preserved mechanical and bone marrow adipose tissue |
Absent metabolically active adipose tissue Preserved mechanical and bone marrow adipose tissue |
| Cardiovascular complications | N/A | Cardiomyopathy | N/A | Cardiomyopathy, catecholaminergic polymorphic ventricular tachycardia, prolonged QT, and sudden death |
| Lytic bone lesions in long bones | Most frequent | Occasional | Not reported | Not reported |
| Gastrointestinal complications | N/A | N/A | Functional megaesophagus | Congenital pyloric stenosis achalasia |
| Skeletal muscle | N/A | N/A | N/A | Congenital myopathy Developmental delay Muscle weakness, Percussion-induced myotonia |
| Other features | N/A | Teratozoospermia in one patient | Short stature, hypocalcemia, vitamin D resistance | Low bone density for age, distal metaphyseal deformation with joint stiffness, atlantoaxial instability Late onset of lipodystrophy in infancy |
Abbreviation: N/A, not applicable.
Table 4.
Subtypes of FPLD
| Subtype | Genetic Mutation | Prevalence |
|---|---|---|
| FPLD1 (Kobberling-type) | Molecular basis unknown | Rare16 |
| FPLD2 (Dunnigan-type) | Missense mutations in LMNA | Most common subtype; more than 500 patients reported17–19 |
| FPLD3 | Heterozygous mutations in PPARG | Second most common subtype; about 30–50 patients reported20,21 |
| FPLD4 | Heterozygous mutations in PLIN1 | Reported in three families22 |
| FPLD5 | Homozygous nonsense mutation in CIDEC (autosomal recessive) | One patient reported23 |
| FPLD6 | Homozygous mutation in LIPE (autosomal recessive) | Six patients reported24,25 |
| FPLD7 | Heterozygous mutation in ADRA2A | Reported in one family27 |
| AKT2-linked lipodystrophy | Heterozygous mutation in AKT2 | Reported in one family26 |
Abbreviations: AKT2, v-akt murine thymoma viral oncogene homolog 2; CIDEC, cell death-inducing DFFA-like effector c; LIPE, hormone sensitive lipase; LMNA, lamin A/C; PLIN1, perilipin 1; PPARG, peroxisome proliferator-activated receptor gamma.
Table 5.
Extremely rare genetic lipodystrophy syndromes
| Lipodystrophy Type | Gene | Molecular Basis | Clinical Features |
|---|---|---|---|
| MAD type A | LMNA | Mutations may disrupt nuclear function resulting in premature cell death in many tissues. | Mandibular and clavicular hypoplasia, acro-osteolysis. Partial lipodystrophy affecting the extremities and trunk.45,46 |
| MAD type B | ZMPSTE24 | Mutations result in accumulation of farnesylated prelamin A that can disrupt nuclear function in several tissues. | Mandibular and clavicular hypoplasia, acro-osteolysis. More generalized loss of fat, premature renal failure, progeroid features.47 |
| JMP/CANDLE | PSMB8 | PSMB8 encodes subunit of immunoproteasomes that degrade abnormal/excess proteins in cells. | Joint contractures, muscle atrophy, microcytic anemia and panniculitis-induced lipodystrophy. Recurrent fevers, annular erythematous skin lesions, violaceous eyelid swelling, partial lipodystrophy.48,49 |
| SHORT syndrome | PIK3R1 | PIK3R1 plays a role in metabolic actions of insulin, mutations associated with insulin resistance. | Variable loss of subcutaneous fat, short stature, hyperextensibility, ocular depression, teething delay.50 |
| MDP syndrome | POLD1 | Critical for DNA replication and repair. | Mandibular hypoplasia, deafness, and progeroid features.51,52 |
| Neonatal progeroid syndrome, type A | FBN1 | Fibrillin 1 | Generalized loss of body fat and muscle mass, and progeroid appearance at birth. Marfanoid habitus.53,54 |
| Neonatal progeroid syndrome, type B | CAV1 | Caveolin 1, present on adipocyte membranes, binds fatty acids and translocates them to lipid droplets. | Generalized loss of body fat and muscle mass, and progeroid appearance at birth.55 |
| Atypical progeroid syndrome | LMNA | Different heterozygous, mostly de novo mutations cause nuclear dysfunction. | Partial or generalized loss of subcutaneous fat, progeroid features.56 |
| Hutchinson-Gilford progeria | LMNA | Specific de novo mutations induce abnormal splicing and accumulation of truncated farnesylated prelamin A. | Generalized loss of subcutaneous fat, progeroid features.57 |
Abbreviations: CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; CAV1, caveolin 1; JMP, joint contractures, muscle atrophy, microcytic anemia and panniculitis-induced lipodystrophy; LMNA, lamin A/C; MAD, mandibuloacral dysplasia; MDP, mandibular hypoplasia, deafness, progeroid features; PIK3R1, phosphoinositide-3-kinase regulatory subunit 1; POLD1, polymerase (DNA) delta 1, catalytic subunit; PSMB8, proteasome subunit beta 8; SHORT, short stature, hyperextensibility or inguinal hernia, ocular depression, Rieger anomaly, and teething delay; ZMPSTE24, zinc metalloprotease STE24.
Congenital Generalized Lipodystrophy
CGL, or Berardinelli-Seip syndrome, is an autosomal-recessive disorder characterized by generalized lack of adipose tissue either at birth or within the first year of life. Patients have prominent musculature and subcutaneous veins.1,7,8 Most cases are diagnosed at birth or early in childhood because of the striking fat loss, but a few patients without access to regular medical care may be identified later in life.
Patients with CGL can develop hyperphagia as a result of profound leptin deficiency in early childhood, and may have accelerated linear growth; advanced bone age; and features suggestive of acromegaly, such as enlarged hands, feet, and jaw.5,6 Severe metabolic complications, along with hepatomegaly and splenomegaly, develop at an early age. Hyperinsulinemia leads to development of widespread acanthosis nigricans, followed by onset of diabetes mellitus during adolescence.7,8 Diabetes is generally ketosis-resistant. Some patients develop extreme hypertriglyceridemia especially after the onset of insulin-resistant diabetes mellitus and are prone to recurrent attacks of acute pancreatitis.7,8
Hepatic steatosis is common and severe, and can progress to steatohepatitis, cirrhosis, and liver failure.4 Female patients with CGL have additional clinical features including hirsutism, clitoromegaly, irregular menstrual periods, polycystic ovaries, and/or infertility.1 There are four genetically distinct subtypes of CGL7–12 and besides common clinical features listed previously, each one has some peculiar clinical features (see Tables 2 and 3).
Familial Partial Lipodystrophy
FPLD is mostly inherited as an autosomal-dominant disorder and is characterized by subcutaneous fat loss from the upper and lower extremities and variable fat loss from the trunk.13,14 These patients have normal fat distribution during childhood, followed by onset around late childhood or puberty of progressive and variable subcutaneous fat loss typically from the extremities (causing the musculature to appear prominent), but variably from the anterior abdomen and chest.13,14 Some patients may have small size of the breasts because of reduced or lack of overlying subcutaneous fat. At the same time, there is often fat accumulation in the face, neck, perineal, and intra-abdominal areas, particularly in women. Excess fat accumulation in the dorsocervical (causing a buffalo-hump), supraclavicular, and submental regions gives these patients a cushingoid appearance and many of these patients may be confused with having Cushing’s syndrome. These patients may be clinically hard to detect if the fat loss is subtle, especially in males because many normal men are also quite muscular.13
FPLD in women may present with masculinization and menstrual irregularity and metabolic complications. Women with FPLD have a high prevalence of PCOS compared with the 6% to 8% prevalence observed in the general population; however, infertility is not common.13 This increased prevalence of PCOS and metabolic complications occurs more frequently in those women who have excess fat accumulation in nonlipodystrophic regions.
As compared to patients with generalized lipodystrophies, hepatic steatosis and acanthosis nigricans is less pronounced; however, hypertriglyceridemia is common and severe, with high risk of acute pancreatitis. In addition, these patients may also develop myopathy, cardiomyopathy, and/or conduction system abnormalities.15 There are several genetically distinct varieties of FPLD16–26; however, the clinical differences among these various subtypes have not been clear so far (see Table 4).
ACQUIRED LIPODYSTROPHIES
Acquired Generalized Lipodystrophy
Acquired generalized lipodystrophy (AGL), or Lawrence syndrome, is characterized by generalized loss of subcutaneous fat that occurs gradually in individuals who are born with a normal fat distribution. The fat loss typically begins in childhood or adolescence, but can rarely begin after 30 years of age.28 It can occur over a variable time period, ranging from a few weeks to months or years, and affects all subcutaneous areas of the body especially the face and extremities and may include the palms and soles. Orbital and bone marrow fat depots seem to be preserved, whereas intra-abdominal fat loss is variable. AGL is more frequent in the females than males (3:1).28 Patients with AGL are predisposed to the same metabolic complications as other patients with lipodystrophies, such as insulin resistance associated with diabetes mellitus and hypertriglyceridemia. Hypoleptinemia is thought to contribute to the metabolic complications. Usually these complications are quite severe. Most of the patients have associated autoimmune diseases, especially juvenile dermatomyositis, or panniculitis (pathologically infiltration of adipose tissue with inflammatory cells of various types resulting in loss of subcutaneous fat) (Table 6). In some patients, the underlying mechanism of fat loss is not clear (idiopathic variety). Usually the metabolic complications are less severe in patients with panniculitis-associated AGL compared with the other two subtypes.
Table 6.
Classification of AGL
| Subtype | Prevalence | Clinical Features |
|---|---|---|
| Panniculitis-associated AGL | ~ 25% | Initial development of panniculitis (subcutaneous inflammatory nodules) followed by localized fat loss when these lesions heal. Ongoing panniculitis later results in generalized loss of subcutaneous fat.28 |
| Autoimmune AGL | ~ 25% | Gradual generalized fat loss associated with autoimmune diseases, especially juvenile dermatomyositis. Some patients have low levels of serum complement 4.28,58 |
| Idiopathic AGL | ~ 50% | Gradual generalized subcutaneous fat loss of unclear etiology.28 |
Acquired Partial Lipodystrophy (Barraquer-Simons Syndrome)
Acquired partial lipodystrophy is characterized by gradual loss of subcutaneous fat from the upper body (ie, the face, neck, upper extremities, and upper trunk).29 Usually the lower abdomen, hips, and lower extremities are spared. In fact, after puberty, patients, especially females, may accumulate excess fat there. Acquired partial lipodystrophy is more frequent in females than males (4:1). It is often associated with autoimmune diseases. Most patients have a circulating autoantibody called complement 3 nephritic factor, and have low circulating levels of serum complement 3.29 Approximately 20% of these patients develop membranoproliferative glomerulonephritis and some develop end-stage renal disease requiring renal transplantation. Rare patients have drusen on fundus examination. Metabolic complications are not seen as frequently as in other types of lipodystrophy.29
Highly Active Antiretroviral Therapy–Induced Lipodystrophy in Patients Infected with Human Immunodeficiency Virus
Lipodystrophy in HIV-infected patients usually occurs after approximately 2 to 4 years of HAART consisting of HIV-1 protease inhibitors or nucleoside reverse transcriptase inhibitors (Table 7).30,31 It is characterized by the loss of subcutaneous fat from the upper and lower extremities and from the face, with increased fat accumulation in the neck, anteriorly and posteriorly, and in the upper trunk and intra-abdominal region.30,31 Many protease inhibitors have been shown to inhibit zinc metalloprotease, the key enzyme involved in posttranslation processing of prelamin A to mature lamin A.32 Thus, protease inhibitor–based HAART may result in accumulation of toxic prelamin A. Nucleoside reverse transcriptase inhibitors may induce lipodystrophy by causing mitochondrial dysfunction.33
Table 7.
Etiology of drug-induced lipodystrophy in HIV-infected patients
| Type/Etiology | Pathogenesis and Molecular Basis |
|---|---|
| PI-induced | PIs inhibit ZMPSTE24, which is important for the correct maturation and processing of prelamin A. Thus, PIs result in accumulation of toxic farnesylated prelamin A.32 May also cause dysregulation of transcription factors involved in adipogenesis. They may also inhibit glucose transporter 4 expression leading to insulin resistance. |
| NRTI-induced | NRTIs (especially stavudine and zidovudine) inhibit mitochondrial polymerase-γ and subsequently cause mitochondrial toxicity.33 |
Abbreviations: NRTI, nucleoside reverse transcriptase inhibitor; PI, protease inhibitor; polymerase-γ, polymerase gamma; ZMPSTE24, zinc mellatoproteinase STE24.
Localized Lipodystrophies
Localized lipodystrophies are characterized by loss of fat from small areas, either single or multiple. Sometimes it can affect portions of the limbs or large contiguous areas on the trunk. Patients with localized lipodystrophies do not develop any metabolic abnormalities. There are several etiologies of localized lipodystrophies (Table 8).34
Table 8.
Characteristics of different types of localized lipodystrophies
| Type | Etiology | Clinical Features |
|---|---|---|
| Drug-induced localized lipodystrophy | Insulin therapy (more common before purified/human insulin was available), steroids, and antibiotics. High local production of TNF-α may cause dedifferentiation of adipocytes. Other mechanisms include presence of lipases, repeated trauma and/or autoimmune processes. | More common in patients with high titers of anti-insulin antibodies. May have deposition of IgA and C3 locally. Sometimes responds to local corticosteroids. |
| Pressure-induced localized lipodystrophy | Trauma and decreased perfusion caused by repeated pressure to the same area over a long period of time. | Fat atrophy localized to the area exposed to repeated pressure. This tends to improve when the pressure is avoided. |
| Panniculitis-associated localized lipodystrophy | Associated with serum ANA or anti dsDNA antibodies; may also have autoimmune diseases, such as SLE. | Initial development of panniculitis (subcutaneous inflammatory nodules in several areas) followed by localized fat loss when these lesions heal. |
| Centrifugal lipodystrophy (lipodystrophia centrifugalis abdominalis infantalis) | Cause is unknown and most patients recover spontaneously with no intervention. | More common in Asians. Fat loss spreads in a centrifugal pattern from abdomen and groin area and is associated with peripheral panniculitis. It begins in infancy, stops spreading between the ages of 3 and 8 and then in most cases, resolves by itself. |
| Idiopathic localized lipodystrophy | Undetermined etiology. |
Abbreviations: ANA, antinuclear antibodies; anti dsDNA Ab, anti-double-stranded deoxyribonucleic acid antibodies; C3, complement 3; SLE, systemic lupus erythematosus; TNF-α, tumor necrosis factor alpha.
Data from Garg A. Lipodystrophies. Am J Med 2000;108(2):143–52.
MANAGEMENT
The treatment of lipodystrophy is focused on managing the metabolic abnormalities to prevent complications, and cosmetic appearance. Although there is no cure for lipodystrophy, morbidity and mortality are improved through early intervention. Diet and exercise form an integral part of the treatment plan, although clinical trial data are not available.
A diet with a well-balanced macronutrient composition of about 50% to 60% carbohydrates, 20% to 30% fat, and about 10% to 20% protein is appropriate for most patients. Overfeeding should be avoided, especially in infants and children (despite their lack of weight gain), because this can accelerate hepatic steatosis and worsen diabetes and hyperlipidemia. Energy-restricted diets are more appropriate in adults, because children with growth and developmental needs may otherwise develop deficiencies.
Exercise, in the absence of contraindications, can help improve metabolic parameters, so patients should be encouraged to be physically active. Those who are predisposed to cardiomyopathy, such as patients with CGL4, FPLD2, and progeroid syndromes, should undergo a cardiac evaluation before engaging in an exercise program, and should avoid strenuous exercise. To avoid traumatic injuries, patients with severe hepatosplenomegaly and patients with CGL with lytic lesions in the bones should avoid contact sports.
Strategies to reduce hypertriglyceridemia include medium-chain triglyceride-based formulas in infants,35 and very-low-fat diets in older individuals. Any fat intake should be in the form of cis-monounsaturated fats and long-chain omega-3 fatty acids. In patients who have developed acute pancreatitis secondary to hypertriglyceridemia, parental nutrition should be administered until they recover and they should subsequently be on an extremely low-fat (total dietary fat <20 g/day) diet. In patients who have not reached lipid-lowering goals after diet and lifestyle intervention, lipid-lowering drugs may be used.
Patients with insulin resistance and diabetes mellitus should be treated with conventional therapies, including oral agents (metformin is the first-line drug) and insulin. Insulin therapy often provides the mainstay of treatment, and many patients require concentrated forms (eg, 500 U regular insulin) because of severe insulin resistance. Whether thiazolidinediones are particularly efficacious in patients with FPLD with PPARG mutations remains unclear. Simple sugars should be avoided in favor of high-fiber complex carbohydrates consumed throughout the day in combination with protein and/or fat, to avoid blood glucose spikes. The treatment goals are similar to patients with diabetes without lipodystrophy.
Hypertension, if uncontrolled, may be treated with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, because these medications also have favorable effects on proteinuria. No specific treatments have been shown to be particularly effective for hepatic steatosis or steatohepatitis associated with lipodystrophy.
Generalized lipodystrophies are characterized by extremely low serum leptin levels,5 which led to research into recombinant human leptin (metreleptin) as a treatment option,36 and since then several long-term studies have shown beneficial effects.37–40
Metreleptin therapy has been shown to improve metabolic abnormalities in patients with generalized lipodystrophy, including decreased serum triglyceride levels, increased insulin sensitivity, and reduced hepatic steatosis.3 It is currently the only drug specifically approved for treatment of generalized lipodystrophy.3 It is administered as a daily subcutaneous injection,41 and dose adjustments are made every 3 to 6 months based on metabolic parameters and weight change. The most common side effects include hypoglycemia and injection site reactions, such as erythema and/or urticaria. The other side effects include development of neutralizing antibodies to metreleptin, and development of cutaneous T-cell lymphomas especially in patients with AGL.42 The precise significance of neutralizing antibodies to leptin remains unclear at this time and some patients with AGL who have never received metreleptin therapy have also been reported to develop lymphomas. Because of paucity of data, approval of metreleptin for different types of lipodystrophy varies by country, depending on their regulatory boards (Table 9).
Table 9.
Approval and indications of metreleptin therapy
| Type of Lipodystrophy | Approvals | Indications | Clinical Considerations |
|---|---|---|---|
| Generalized lipodystrophy (both CGL and AGL) | United States: approved as adjunct to diet for treatment of metabolic complications. Japan: approved Europe: available through compassionate care programs. |
First-line drug treatment (after diet/exercise intervention) for metabolic and endocrine abnormalities. May prevent comorbidities and metabolic complications in young children. |
Decreases hyperphagia, leading to weight loss. May need to be discontinued if excessive weight loss occurs. |
| Partial lipodystrophy (both FPLD and APL) | United States: not approved. Japan: approved as an adjunct to diet Europe: through compassionate care programs. |
May be considered for patients with hypoleptinemia (leptin <4 ng/mL) who have severe metabolic abnormalities, such as HbA1c >8% and/or triglycerides >500 mg/dL. | Clinical response not as good as in generalized lipodystrophy. Patients with lower leptin levels show the most benefit. |
Abbreviations: APL, acquired partial lipodystrophy; HbA1c, glycated hemoglobin.
Change in body shape caused by lipodystrophy can often lead to psychological distress, and sometimes even physical discomfort, such as from absent fat pads on the feet and buttocks. Patients should be referred to appropriate mental health providers for emotional distress. Plastic surgery may improve appearance in some people, although data are limited. Possible interventions include autologous fat transfer, dermal fillers, or muscle grafts to treat facial lipoatrophy; surgical reduction or liposuction of areas with excessive fat; and breast implants for improved cosmetics in women.
KEY POINTS.
Lipodystrophies are a group of heterogeneous disorders characterized by varying degrees of body fat loss and predisposition to insulin resistance–related metabolic complications.
They are classified as generalized, partial or localized by extent of fat loss; and genetic and acquired by etiology.
Highly active antiretroviral therapy–induced lipodystrophy in HIV-infected patients and drug-induced localized lipodystrophy are more prevalent subtypes, followed by genetic and acquired autoimmune lipodystrophies.
Common metabolic abnormalities and complications include insulin resistance and diabetes mellitus, hypertriglyceridemia, and hepatic steatosis.
Management options include diet and exercise; conventional antihyperglycemic agents and lipid-lowering therapy; and metreleptin therapy, which is the only drug approved specifically for generalized lipodystrophy.
ACKNOWLEDGMENTS
The authors thank Pei-Yun Tseng, BS, for help with illustrations.
Grant Support: This work was supported by the National Institutes of Health grant RO1 DK105448, CTSA grants UL1RR024982 and UL1TR001105, and the Southwest Medical Foundation.
Disclosure Statement: Dr I. Hussain has no disclosures. Dr A. Garg coholds a patent regarding use of leptin for treating human lipoatrophy and the method of determining predisposition to this treatment but receives no financial compensation. He receives research grant support from Aegerion, Pfizer, and Ionis Pharmaceuticals and is a consultant for Aegerion.
REFERENCES
- 1.Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004;350(12): 1220–34. [DOI] [PubMed] [Google Scholar]
- 2.Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96(11):3313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown RJ, Gorden P. Leptin therapy in patients with lipodystrophy and syndromic insulin resistance In: Dagogo-Jack S, editor. Leptin: regulation and clinical applications. New York: Springer International Publishing; 2015. p. 225–36. [Google Scholar]
- 4.Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy: an AACE consensus statement. Endocr Pract 2013; 19(1):107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haque WA, Shimomura I, Matsuzawa Y, et al. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 2002;87(5):2395. [DOI] [PubMed] [Google Scholar]
- 6.Antuna-Puente B, Boutet E, Vigouroux C, et al. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-o-acyltransferase-2 deficiency. J Clin Endocrinol Metab 2010;95(3):1463–8. [DOI] [PubMed] [Google Scholar]
- 7.Van Maldergem L, Magre J, Khallouf TE, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet 2002;39(10):722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal AK, Simha V, Oral EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab 2003;88(10): 4840–7. [DOI] [PubMed] [Google Scholar]
- 9.Magre J, Delepine M, Khallouf E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet 2001;28(4):365–70. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet 2002;31(1): 21–3. [DOI] [PubMed] [Google Scholar]
- 11.Kim CA, Delepine M, Boutet E, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab 2008;93(4):1129–34. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest 2009;119(9):2623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab 2000;85(5): 1776–82. [DOI] [PubMed] [Google Scholar]
- 14.Garg A, Peshock RM, Fleckenstein JL. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab 1999;84(1):170–4. [DOI] [PubMed] [Google Scholar]
- 15.Subramanyam L, Simha V, Garg A. Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin Genet 2010;78(1):66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobberling J, Dunnigan MG. Familial partial lipodystrophy: two types of an X linked dominant syndrome, lethal in the hemizygous state. J Med Genet 1986; 23(2):120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 2000;9(1):109–12. [DOI] [PubMed] [Google Scholar]
- 18.Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 2000;24(2):153–6. [DOI] [PubMed] [Google Scholar]
- 19.Speckman RA, Garg A, Du F, et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet 2000;66(4):1192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal AK, Garg A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab 2002;87(1):408–11. [DOI] [PubMed] [Google Scholar]
- 21.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest 2006;116(3):581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandotra S, Le Dour C, Bottomley W, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med 2011;364(8):740–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubio-Cabezas O, Puri V, Murano I, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med 2009;1(5):280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albert JS, Yerges-Armstrong LM, Horenstein RB, et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N Engl J Med 2014;370(24): 2307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farhan SM, Robinson JF, McIntyre AD, et al. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol 2014;30(12):1649–54. [DOI] [PubMed] [Google Scholar]
- 26.George S, Rochford JJ, Wolfrum C, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004;304(5675):1325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg A, Sankella S, Xing C, et al. Whole-exome sequencing identifies ADRA2A mutation in atypical familial partial lipodystrophy. JCI Insight 2016;1(9):e86870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore) 2003;82(2):129–46. [DOI] [PubMed] [Google Scholar]
- 29.Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore) 2004;83(1):18–34. [DOI] [PubMed] [Google Scholar]
- 30.Chen D, Misra A, Garg A. Clinical review 153: Lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab 2002;87(11):4845–56. [DOI] [PubMed] [Google Scholar]
- 31.Grinspoon S, Carr A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N Engl J Med 2005;352(1):48–62. [DOI] [PubMed] [Google Scholar]
- 32.Hudon SE, Coffinier C, Michaelis S, et al. HIV-protease inhibitors block the enzymatic activity of purified Ste24p. Biochem Biophys Res Commun 2008;374(2): 365–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee H, Hanes J, Johnson KA. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 2003; 42(50):14711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg A. Lipodystrophies. Am J Med 2000;108(2):143–52. [DOI] [PubMed] [Google Scholar]
- 35.Wilson DE, Chan IF, Stevenson KB, et al. Eucaloric substitution of medium chain triglycerides for dietary long chain fatty acids in acquired total lipodystrophy: effects on hyperlipoproteinemia and endogenous insulin resistance. J Clin Endocrinol Metab 1983;57(3):517–23. [DOI] [PubMed] [Google Scholar]
- 36.Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002;346(8):570–8. [DOI] [PubMed] [Google Scholar]
- 37.Chong AY, Lupsa BC, Cochran EK, et al. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010;53(1):27–35. [DOI] [PubMed] [Google Scholar]
- 38.Javor ED, Cochran EK, Musso C, et al. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes 2005;54(7):1994–2002. [DOI] [PubMed] [Google Scholar]
- 39.Park JY, Javor ED, Cochran EK, et al. Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism 2007; 56(4):508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simha V, Subramanyam L, Szczepaniak L, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab 2012;97(3):785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez AJ, Mastronardi CA, Paz-Filho GJ. New advances in the treatment of generalized lipodystrophy: role of metreleptin. Ther Clin Risk Manag 2015;11: 1391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown RJ, Chan JL, Jaffe ES, et al. Lymphoma in acquired generalized lipodystrophy. Leuk Lymphoma 2016;57(1):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shastry S, Delgado MR, Dirik E, et al. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A 2010;152A(9):2245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajab A, Straub V, McCann LJ, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet 2010;6(3):e1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simha V, Agarwal AK, Oral EA, et al. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab 2003;88(6):2821–4. [DOI] [PubMed] [Google Scholar]
- 46.Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet 2002;71(2):426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agarwal AK, Fryns JP, Auchus RJ, et al. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet 2003;12(16):1995–2001. [DOI] [PubMed] [Google Scholar]
- 48.Garg A, Hernandez MD, Sousa AB, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab 2010;95(9):E58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet 2010;87(6): 866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thauvin-Robinet C, Auclair M, Duplomb L, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet 2013;93(1):141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shastry S, Simha V, Godbole K, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab 2010; 95(10):E192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet 2013;45(8):947–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graul-Neumann LM, Kienitz T, Robinson PN, et al. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3’ terminus of the FBN1-gene. Am J Med Genet A 2010;152A(11): 2749–55. [DOI] [PubMed] [Google Scholar]
- 54.Garg A, Xing C. De novo heterozygous FBN1 mutations in the extreme C-terminal region cause progeroid fibrillinopathy. Am J Med Genet A 2014;164A(5):1341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garg A, Kircher M, Del Campo M, et al. , University of Washington Center for Mendelian Genomics. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A 2015;167A(8):1796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garg A, Subramanyam L, Agarwal AK, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab 2009;94(12): 4971–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 2008;358(6):592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Savage DB, Semple RK, Clatworthy MR, et al. Complement abnormalities in acquired lipodystrophy revisited. J Clin Endocrinol Metab 2009;94(1):10–6. [DOI] [PubMed] [Google Scholar]