

Abstract
Bone repair is a complex process involving the sophisticated interplay of osteogenic stem cells, extracellular matrix, and osteoinductive factors, and it is affected by bacterial toxins and oxidative stress. Inspired by the nature of plant-derived phytochemicals and inorganic-organic analogues of the bone extracellular matrix, we report herein the facile design of a nanoclay-organic hydrogel bone sealant (NoBS) that integrates multiple physico-chemical cues for bone regeneration into a single system. Assembly of phytochemical-modified organic chitosan and silica-rich inorganic nanoclay serves as highly biocompatible and osteoconductive extracellular matrix mimics. The decorated phytochemical exerts inherent bactericidal and antioxidant activities, and acts as an intermolecular networking precursor for gelation with injectable and self-healing capabilities. Moreover, the NoBS exerts osteoinductive effects mediated by the nanoclay, which regulates the Wnt/β-catenin pathway, along with the addition of osteoinductive signals, resulting in bone regeneration in a non-healing cranial defect. Engineering of this integrated bone graft substitute with multifunctional properties inspired by natural materials may suggest a promising and effective approach for creating a favorable microenvironment for optimal bone healing.
Keywords: Nature-inspired hydrogel, bone sealant, nanoclay-organic composite, phytochemical, bone regeneration
Graphical Abstract
A nature-inspired nanoclay-organic hydrogel bone sealant is developed by co-assembly of inorganic nanoclay and organic phytochemical-reinforced chitosan, thereby mimicking the plant and bone microenvironment. The system serves as a biocompatible and osteoconductive microenvironment with multiple desirable physico-chemical properties, including self-healing and antibacterial and antioxidant activities, for robust bone repair in a single system.
1. Introduction
Treatment of bone fractures and defects is a major challenge from the standpoints of clinical and socio-economic perspectives. Limitations of autogenous or allogenous bone have spurred the development of many different bone substitutes.[1] The ideal material should exhibit suitable properties for promoting the healing of bone defect, such as biocompatibility, osteoconductivity, osteoinductivity, mechanical integrity, ease of use, and affordability. Inspired by biomineralization in living organisms, biological and synthetic organic-inorganic composites have been explored in the hope of revolutionizing bone graft substitutes.[2] Recent studies report nanocomposite hydrogels in which silicate nanoclays (NC) such as LAPONITE® (a trademark of BYK Additives Ltd.; hectorite) are incorporated into various synthetic or naturally derived polymers including polyethylene glycol, gelatin, chitosan or hyaluronic acid to improve their physical and mechanical properties.[3] These clay nanocomposite systems have been investigated as suitable candidates for bioadhesives, tissue engineering scaffolds, and vehicles for drug delivery.[3a, 3f, 4] Although each system exhibits beneficial effects on its specific purpose, no single device meets all requirements for bone regeneration.
The healing of bone fractures is a complex physiological process relying on concerted events involving interactions of osteogenic stem cells, cytokines, and matrix, and the process is often disrupted by bacterial toxins and inflammatory responses.[5] The infection of a bone graft device is a devastating complication requiring surgical debridement and antibiotic treatment, leading to high patient morbidity and economic burden resulting from multiple surgeries and adverse antibiotic reactions.[6] Moreover, elevated levels of reactive oxygen species (ROS) during defect healing can lead to oxidative stress and cellular damage, inhibiting mesenchymal cell osteogenic differentiation while promoting adipogenesis.[7] Current approaches involve delivery of antioxidant agents to reduce the harmful effects of ROS, but are often unsuccessful because of cytotoxicity and adverse side effects.[8] Lastly, there exists a clinical need for an advanced bone graft formulation allowing better handling, one that can be injected through a syringe following gelation and then self-assembles into an integral object that seals the complex contour of defect sites and effects robust bone repair.
Catechol chemistry has been extensively explored for the development of nature-inspired polymers and adhesives since the discovery that catechol functional groups are present in mussel adhesive proteins.[9] Catecholamine, in particular, polydopamine is one of the most studied catechol-containing polymers, because of its robust and durable surface adhesion.[10] Phytochemicals are phenolic components produced by plants and have recently emerged as an alternative to catecholamine. Plant-derived catechol derivatives such as caffeic acid have many advantages relative to dopamine, including high availability, low cost and desirable esthetics, and they have been shown to exhibit excellent inherent biological properties such as antibacterial, anti-inflammatory, and antioxidant effects.[10c, 11] In addition, phenolic hydroxyl-rich moieties in caffeic acid are able to form networks, alone or with a wide range of organic and inorganic substrates and proteins, and these may involve diverse interactions such hydrogen bonding, aromatic or hydrophobic interactions, and metal coordination. Consequently, their favorable nature inspired us to devise a multifunctional bone graft nanocomposite to address critical gaps in the use of bone substitutes, thereby using an integrated strategy to create a favorable microenvironment for bone regeneration. In this study inspired by the nature of plant-derived phytochemicals and inorganic-organic analogues of the bone extracellular matrix, we describe the facile design of a phytochemical-reinforced NC-organic hydrogel bone sealant (NoBS) exhibiting self-healing, antibacterial and antioxidant activities, osteoinductivity, and controlled drug delivery for robust bone regeneration (Figure 1). The NoBS hydrogel is formed by the self-assembly of phytochemical-grafted chitosan (PGC) with nanosilicate NCs via multiple covalent and non-covalent interactions involving the catechol groups in phytochemicals, resulting in a malleable and self-healing hydrogel with enhanced mechanical properties that can be injected into defects with complex geometries. Furthermore, the NoBS exerts antimicrobial and antioxidant activities resulting from the phenolic hydroxyl-rich moieties of the phytochemicals. Lastly, NCs serve as attractive crosslinkers that enhance the osteoinductivity of NoBS by regulating the Wnt/β-catenin pathway, and as nanocarriers for controlled drug delivery; drug delivery was evaluated by loading smoothened agonist (SAG) as a model drug. SAG is a small molecule that is known to activate hedgehog signaling and osteogenesis to promote bone regeneration.[12] The engineered nanocomposite hydrogels may serve as an integrative bone graft device with multifunctional and modular components for clinical bone repair.
Figure 1. Schematic illustration of nanoclay-organic bone sealant (NoBS).
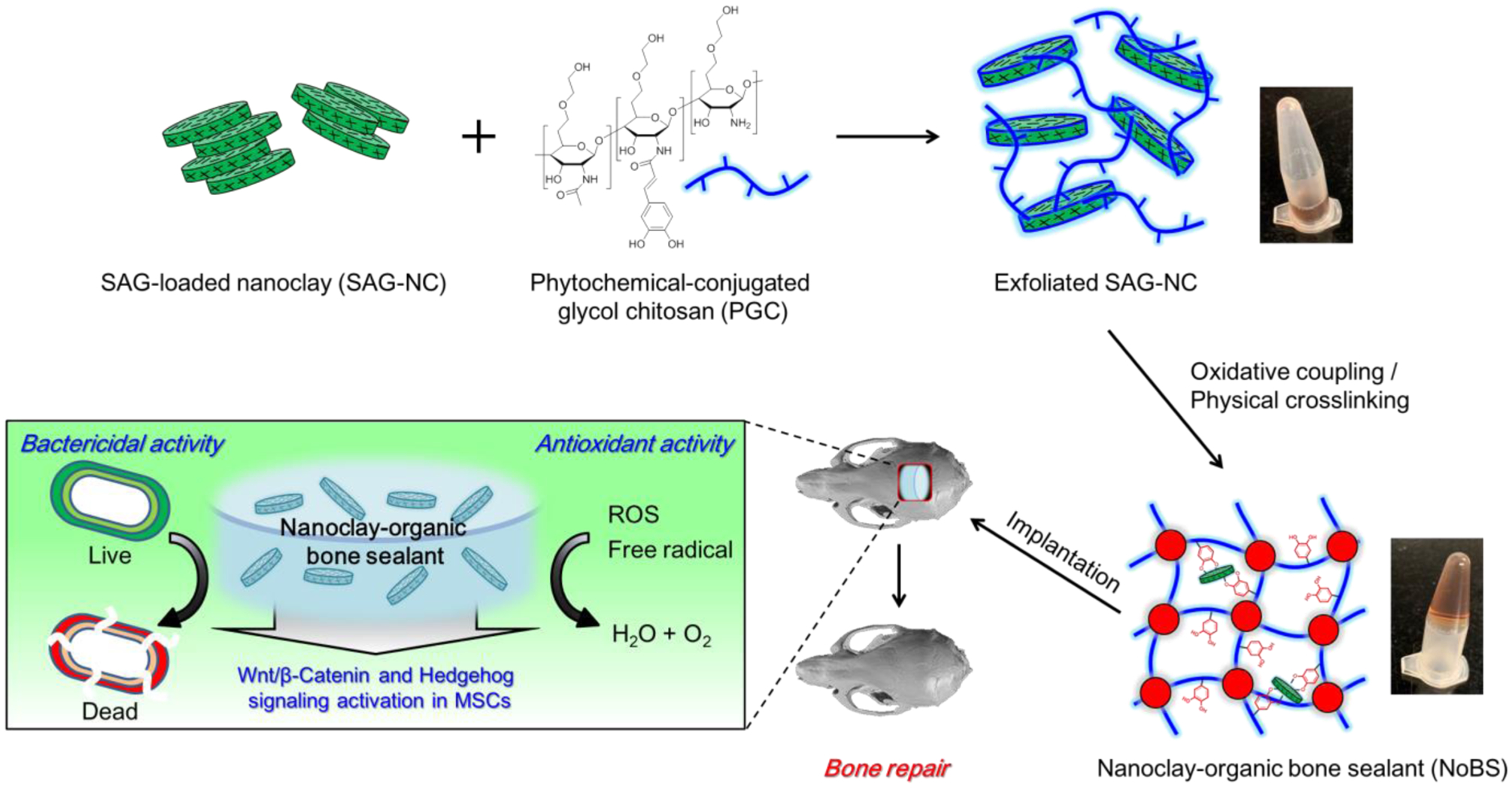
Preparation of SAG-loaded nanoclay (SAG-NC) via intercalation of SAG between nanoclay (NC) layers. Exfoliation of SAG-NC by electrostatic interactions between phytochemical-conjugated glycol chitosan (PGC) and SAG-NC. The mixture of PGC and SAG-NC was crosslinked to develop the NoBS via oxidative coupling and physical crosslinking upon the addition of oxidizer (NaIO4). After implantation of the NoBS at the site of bone defect, NoBS exhibited bone regeneration by upregulating Wnt/β-catenin and Hedgehog signaling pathways in mesenchymal stem cells (MSCs) with bactericidal and antioxidant activities and controlled SAG delivery.
2. Results and Discussion
2.1. Preparation and characterization of NoBS
Caffeic acid, a phenolic hydroxyl-rich phytochemical, was grafted to the amino groups of glycol chitosan (GC) to yield the PGC using carbodiimide chemistry.[13] The degree of catechol graft on GC was calculated as 9.7% of the 100 glucose rings of the GC by 1H-NMR spectroscopy (Figure S1). This result was reached by comparison of the peaks at 6.2–7.5 ppm with the peaks at 1.8–2.1 ppm, based on 12.1% of the acetylated units in the GC backbone. As shown in Figure S2, the Fourier-transform infrared (FT-IR) spectrum of GC showed peaks at 1652 cm−1 (amide I vibration band) and 1601 cm−1 (amide II vibration band).[14] These peaks in the spectrum of the PGC were shifted to 1635 cm−1 (aromatic C=O stretch), indicating successful coupling between the carboxyl group of caffeic acid and the amino group of GC. In addition, an aromatic C=C peak at 1522 cm−1 appeared in the spectrum of PGC and was assigned to the phenolic hydroxyl-rich catechol group of PGC.
NC has been used as an effective drug carrier because of its large surface area and specific nanosheet-layered structure, which lead to good stability, dispersibility, and drug carrying capacity.[15] The present study investigated the feasibility of NC for delivery of osteoinductive molecules, with SAG as a model drug. The SAG was loaded into the inner space of the NC, with intercalation occurring via cation exchange between the cations within NC (Na+) and the cationic form of SAG.[16] The x-ray diffraction (XRD) patterns of NC and SAG-loaded NC are shown in Figure S3. The peak of the (001) plane was shifted from 2θ=7.83° in free NC to 7.65° in SAG-loaded NC, suggesting that the distance between 001 planes of the NC layer (d-spacing) was increased when SAG was loaded via intercalation (Table S1). This result was supported by FT-IR analysis, which indicated that the SAG was successfully intercalated into the layers of the NC (Figure S4). The peaks for SAG, at around 1200–1500 cm−1, were clearly observed in the FT-IR spectrum of SAG-loaded NC. The negligible shift of the Si-O stretch (approximately 1000 cm−1) of NC was evidence of by the molecular interactions with SAG. To optimize the composition of NoBS for gel-formation, the gelation time was determined with varying oxidizer/catechol molar ratios and NC contents (Figure S5). No significant shrinkage was observed after gelation for all experimental groups. The gelation time was prolonged by a decrease of oxidizer/catechol molar ratios and an increase in NC content. It is possible that a catechol moiety of the phytochemical is oxidized to form the PGC polymer network via intermolecular crosslinking, but that this interaction was disturbed by coordination between the catechol group and NC.[17] Based on the gelation results for the NoBS with 1% NC, we used this level as the maximum NC content in further investigations. The compressive modulus of NoBSs was characterized in the presence or absence of NC or SAG (Figure 2A). The addition of NC significantly enhanced the mechanical property (2-fold) of NoBS, suggesting strong interfacial interactions between dispersed NC particles and the polymer chains of the crosslinked NoBSs. However, varying the NC content of 0.5 or 1.0% in the presence or absence of SAG resulted in no obvious changes in the mechanical strength of NoBS. A possible explanation for the observed lack of concentration effect on the compressive modulus is that the increase in nanoclay concentration may induce catechol-nanoclay coordination, while simultaneously reducing the availability of the catechol groups required for crosslinking between polymer chains. It is well known that NC addition can enhance the mechanical properties of hydrogels.[17b, 18] In addition, the high stiffness (~24 kPa) of the hydrogel, which is almost identical to that of NoBS hydrogel, promotes the osteogenic differentiation of MSCs.[19]
Figure 2. Characterization of nanoclay-organic bone sealant (NoBS).
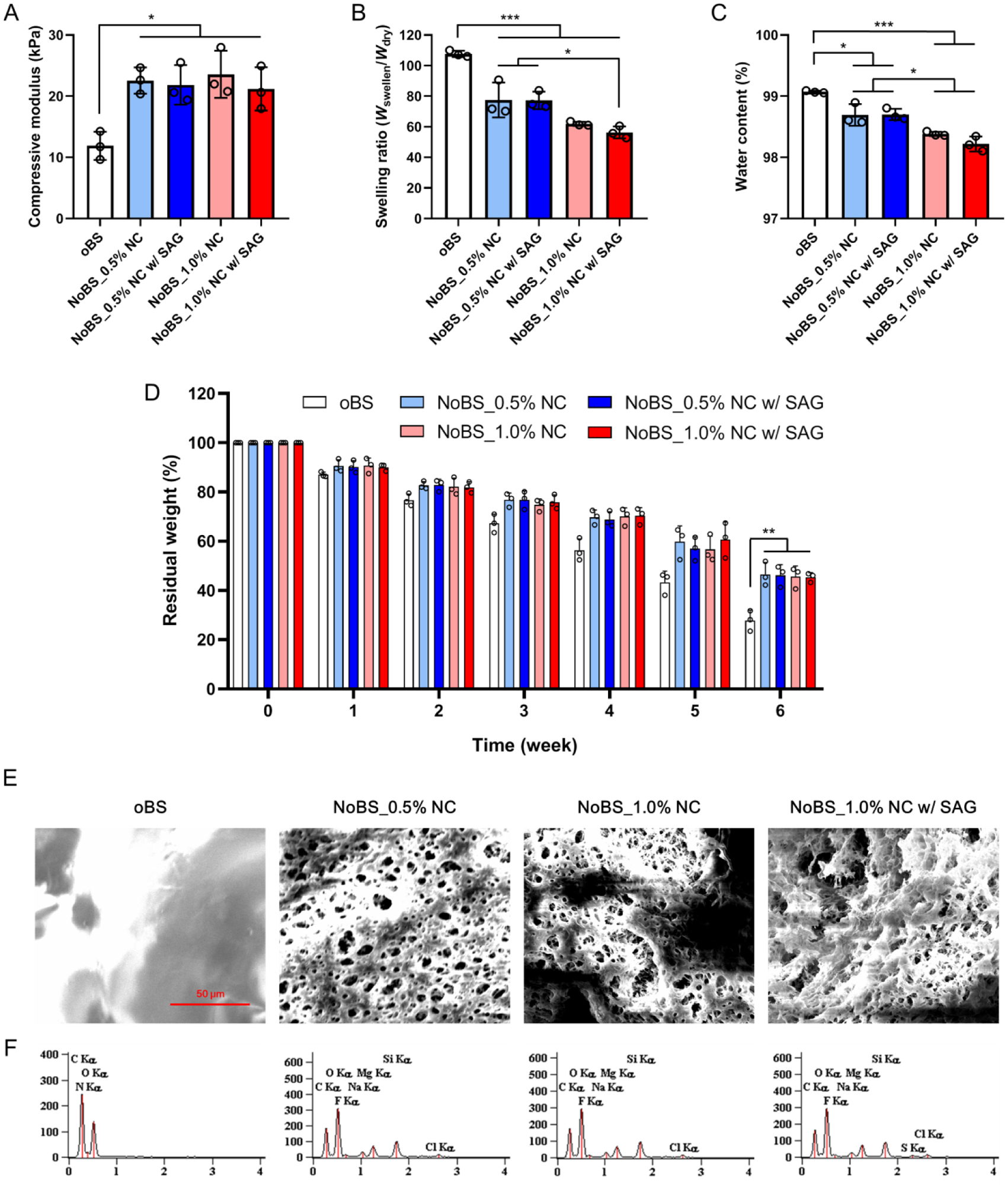
A) Compressive modulus of oBS and NoBSs with various amounts of NCs in the presence or absence of SAG (10 μM). B) Swelling ratios of oBS and NoBSs with various amounts of NCs at the presence or absence of SAG (10 μM). C) Equilibrium water contents of oBS and NoBSs with various amounts of NCs at the presence or absence of SAG (10 μM). D) Residual weight of oBS and NoBSs in the presence of 0.5 and 1.0% of NCs with or without 10 μM of SAG in PBS at 37 °C for 6 weeks. Error bars indicate standard deviation (n = 3), *p < 0.05, **p < 0.01, and ***p < 0.001 (ANOVA followed by Tukey’s post hoc test). E) SEM images and F) EDX spectra of oBS and NoBSs with various amounts of NCs at the presence or absence of SAG (10 μM). Scale bar indicates 50 μm in E).
The adhesive property of NoBS with different NC contents was evaluated by performing ex vivo adhesion tests with bone (Figure S6). The adhesive strength was decreased with the addition of NC. Adhesion of catechol-containing hydrogels involves the interaction between the catechol group and reactive groups (-NH2 and –SH groups) or minerals on the tissue surface.[4e] Increasing the NC content will increase the number of catechol-NC interactions formed, which disturbs the interactions between catechol groups and bone tissue. As with previously reported chitosan-based composite hydrogels, the swelling ratio and equilibrium water content were decreased with NC content as a result of physical crosslinking between PGC polymer and NC (Figure 2B and C).[4c, 20] Both measurements on NoBS suggest the enhanced mechanical strength of the composite hydrogels with NC.
The residual weight of the NoBS was measured for 6 weeks (Figure 2D). All of the NoBS hydrogels showed time-dependent degradation. The degradation of the NoBS groups with NC and SAG, regardless of their content levels, was significantly diminished at week 6, relative to that of the oBS group. We have evaluated the trends in the mechanical properties resulting from degradation (Figure S7). To facilitate degradation, the hydrogels were also tested in the presence of lysozyme, a chitosan lytic enzyme. The compressive modulus was decreased by the degradation of hydrogels. The modulus of NoBS groups was higher than that of oBS group after degradation.
Scanning electron microscopy (SEM) was employed to confirm the microstructure of NoBS in the wet state, which was tuned by the addition of NC and SAG (Figure 2E). NoBSs exhibit a highly porous microstructure, whereas a smooth non-porous surface was observed for the oBS group. The microporous structure of the NoBS may result because the PGC, a catechol-rich and positively-charged chitosan polymer, was coordinated to, and distributed inside of, the NC, as seen in our previous study.[4c] This feature can enhance the permeability of essential components and support cell functions such as adhesion, proliferation, and differentiation for tissue engineering.[21] Energy-dispersive X-ray spectroscopy (EDX) analysis was used to confirm that the chemical composition of the NoBS was altered by NC and SAG (Figure 2F and Table S2). The peaks for Si and Mg atoms, resulting from the NC, were obviously appeared in the spectra of all of the NoBS groups. Furthermore, a signal for sulfur was observed in the SAG-containing NoBS. The results of SEM and EDX indicate that NC and SAG were successfully incorporated into the PGC polymer networks with microporous structures, thereby providing great potential for delivery of bioactive molecules. The drug release profile of hydrogels was investigated in the presence or absence of NC (Figure S8). More than 80% of the initially loaded SAG was released from oBS hydrogels at day 1, with significantly higher initial bursts as compared with NoBS hydrogels, suggesting the potential of nanoclays as efficient drug carriers. During the initial 14 days, the amount of SAG released from the 10 or 50 μм SAG-containing NoBSs was 72.6±7.1% and 64.9±1.2%, respectively. These results indicate that NC as a drug carrier prevents drug release bursts and enables controlled drug release via ion exchange of biological cations such as Na+, K+, Ca2+, and Mg2+ with the intercalated SAG.[16] While there was approximately a 10% mass loss of the hydrogels seen after 1 week, 60% to 100% of the SAG was released from the NoBS and oBS, respectively, indicating that drug release is dependent on ion exchange and/or diffusion rather than on matrix degradation.
2.2. Moldable and self-healing properties of NoBS
The NoBS described was injectable, and gels were extruded through a 23 gauge needle without clogging and reassembled in water as hydrogels approached each other (Figure 3A and B). As shown in Figure 3C, the NoBS kept its round shape when it was held by tweezers.
Figure 3. Moldable and self-healing properties of nanoclay-organic bone sealant (NoBS).
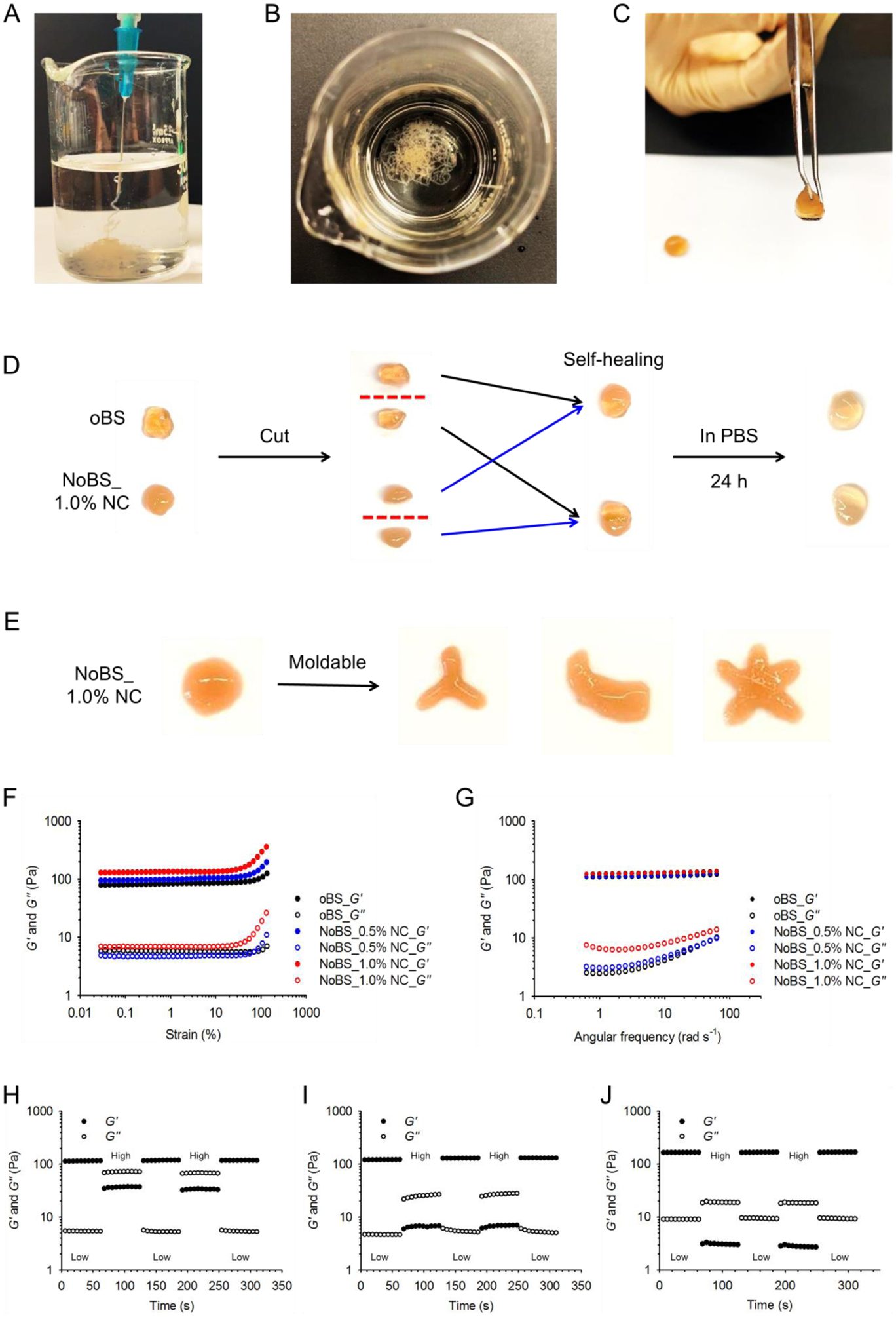
A) Image of NoBS (1.0% NC) injected from a 23 gauge needle into 10 mM (pH 7.4) PBS. B) Image after NoBS (1.0% NC) was injected from a 23 gauge needle into 10 mM (pH 7.4) PBS. C) Image of NoBS (1.0% NC) held by tweezers. D) Self-healing process of oBS and NoBS (1.0% NC). Hydrogels were cut into two pieces that were then reassembled to form completely integrated gels at room temperature. These two new hydrogels were immersed in 10 mM PBS (pH 7.4) for 24 h. E) Photographic images of NoBS (1.0% NC) demonstrating the moldability to different shapes after injection. F) Strain-dependent (ω = 10 rad s−1, 25 °C) and G)frequency-dependent (ε = 1%, 25 °C) oscillatory shear rheology of oBS and NoBS containing various concentrations of NCs. H-J) Step-strain measurements of H) oBS and NoBS containing I) 0.5 and J) 1.0% NC with high strains (destructive; 700%) and low strains (restorative; 1%) to characterize the extent and rate of stationary self-healing.
To evaluate the self-healing ability, the oBS and the NoBS with 1% NC were prepared and cut into two pieces. The pieces of the hydrogels were held together along the cut surface without any external forces (Figure 3D). After 24 h in phosphate buffered saline, the boundaries between the two pieces completely blended and the result was an integral form. These results suggested that both hydrogels (oBS and NoBS), in the presence or absence of NC, had excellent self-healing ability resulting from dynamic non-covalent and covalent interactions involving NC-polymer and polymer-polymer networks of the hydrogel. In addition, the NoBS can be remolded to different shapes after injection (Figure 3E). The self-healing and moldable nature of NoBS may prove useful for facile and minimally invasive treatments.[22]
Further, to investigate the self-healing property of the NoBS, a strain-dependent oscillatory rheological test was carried out to determine the critical point of the fluid state and the solid state of the matrix. The oBS and NoBS measured while controlling the concentrations of NC showed that the storage modulus (G′) was higher than the loss modulus (G″), consistent with solid-like behavior. The hydrogels with different NC concentrations also showed linear viscoelastic responses up to 100% strains (Figure 3F). These measurements suggest that the solid-like properties of oBS and NoBS are retained over a broad range of strains. The addition of nanoclays increased the storage modulus of the hydrogels from 85 Pa to 100 Pa or 130 Pa at 0.5% or 1% NC, respectively, indicating increased mechanical strength. A frequency sweep measurement was also performed to study the gel-like behavior of oBS and NoBS (Figure 3G). The G′ was larger than the G″ over the entire range of frequency-dependent oscillatory shear measurements studied, and both moduli showed a weak dependence on the angular frequency. Rheological characterization was also undertaken to demonstrate the self-healing properties of oBS and NoBS. Step-strain measurements were used to evaluate the repeated recovery of the dynamic matrix response of oBS and NoBS with 0.5 and 1.0% NC, following network rupture by a mechanical shearing force (Figure 3H–J). A small-amplitude oscillatory shear (strain=1%) was applied to destroy the hydrogel. Under this condition, the value of G′ is larger than that of G″, and both moduli remained constant as time passed. The hydrogel was then subjected to a large-amplitude oscillatory shear (strain=700%). The G′ and G″ for all of the hydrogels were inverted at high strain, indicating destruction of the hydrogels and fluid-like behavior, but immediately reverted to the initial solid-like state after the shear was decreased. This property also was maintained over several repeatable cycles, regardless of NC content, confirming the capacity for self-healing.
2.3. Antibacterial activity of NoBS
Bone substitute material should exhibit minimal risk of infection when implanted into an injured area. Therefore, antibacterial property is one of the important clinical necessities for stable healing and accelerated bone regeneration. The caffeic acid and chitosan comprising the PGC are known antibacterial materials because of the hydroxyl-rich structure of caffeic acid and the positively-charged protonated amino group of chitosan, and they operate via formation of a barrier on the bacterial surface that prevents entry of nutrients and leads to the destruction of the bacterial cell.[23] The antibacterial activity was evaluated with the use of S. aureus (gram positive) and E. coli (gram negative) as model microorganisms.[24] The various concentrations of caffeic acid, glycol chitosan, and PGC were exposed to S. aureus and E. coli for 24 h (Figure S9). The caffeic acid showed an antibacterial effect toward both microorganisms. In addition, the PGC was more effective toward both microorganisms than was glycol chitosan, indicating that the introduced caffeic acid reinforced the antibacterial activity of chitosan. An agar petri dish cultured with S. aureus and E. coli was placed with the hydrogels and the petri dish was incubated at 37 °C for one day so as to analyze the inhibition zone (Figure 4A and B). The growth of strains on the agar plate was observed to determine the antibacterial activity of hydrogels, including methacrylated chitosan (MeGC) hydrogel (phytochemical- and NC- free), oBS, and NoBS. It was observed that, after a 24 h incubation, the bacterial strain totally covered the plates into which no gels had been placed. The inhibition zones for oBS and NoBS were also present and showed no significant differences. The chitosan hydrogel (MeGC) showed little or no antibacterial effect (Figure S10), indicating that the antibacterial effects observed for phytochemical grafted chitosan hydrogels (oBS and NoBS) are largely associated with the phytochemical constituent introduced. When compared with the NoBS, with or without SAG, there was no difference in the antibacterial activity (Figure S11). These results suggest that bacterial growth was significantly inhibited by the effect of PGC polymer and not by NC and SAG. Moreover, the antibacterial activities of the MeGC hydrogel, oBS, and NoBS for 24 h were tested by microplate proliferation assay (Figure 4C and D). The outstanding antibacterial activity of oBS and NoBS, compared to that of phytochemical-free MeGC hydrogel, was due to the PGC polymers. Thus, these findings demonstrate that the NoBS has great potential for reducing the risks of bacterial infections in clinical biomedical applications.
Figure 4. Bactericidal activity of nanoclay-organic bone sealant (NoBS).
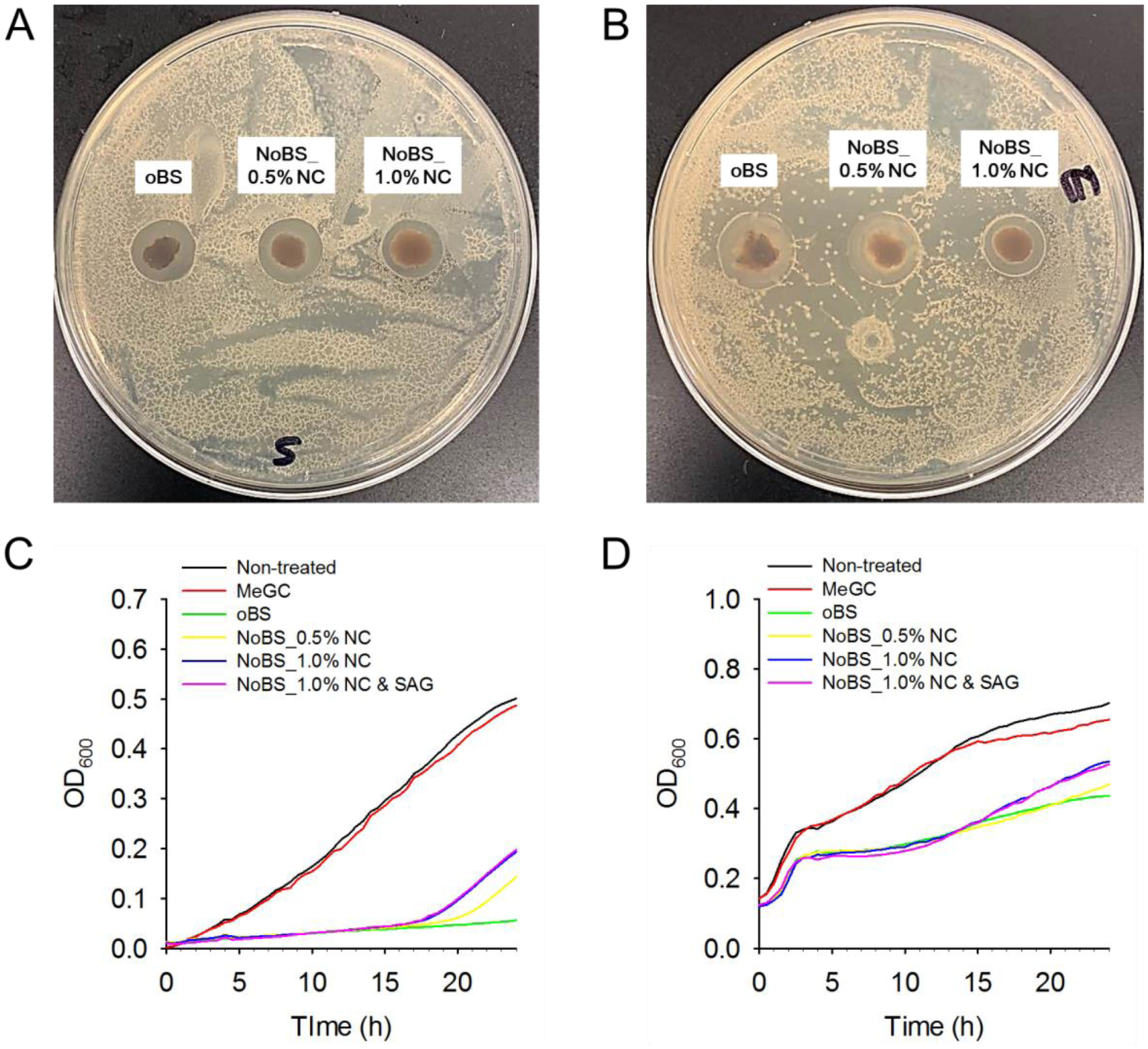
Representative image of bacterial colonies formed by A) S. aureus and B) E. coli with oBS (NC-free), and NoBS placed on the agar petri dish for a day to analyze the inhibition zone. Antibacterial activity of methacrylated chitosan (MeGC) hydrogel (phytochemical- and NC-free), oBS and various NoBSs to C) S. aureus and D) E. coli over 24 h tested by microplate proliferation assay.
2.4. Antioxidant activity of NoBS
Since phytochemicals are known to possess antioxidant activity and tend to prevent oxidative stress on biological components, it is possible that NoBS is able to scavenge or quench free radicals and that ROS are generated during cellular metabolic responses. Therefore, NoBS could be able to enhance the survival, proliferation, and differentiation of osteoblasts. To substantiate the antioxidant nature of NoBS, we studied the scavenging of H2O2 and 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radicals; this suggested the capacity to turn over the ROS and radicals (including electrons or hydrogens) (Figure 5A and B). The oBS and NoBS showed H2O2 and free radical scavenging activity after incubation for 30 min. The proportions of H2O2 and DPPH free radicals scavenged were 75% and 88%, respectively, compared to < 10% with phytochemical-free MeGC hydrogel. The intracellular ROS was visualized using dichlorofluorescein diacetate (DCFDA) as an ROS-sensitive cell-permeable fluorogenic marker; this initially shows no fluorescence but, once oxidized to dichlorofluorescein (DCF) by ROS, it emits green fluorescence (Figure 5C).[25] Strong green fluorescence due to DCF was observed when cells were treated with or without MeGC hydrogel in the presence of H2O2. However, negligible green fluorescence was observed for the cells incubated with oBS and NoBS hydrogels. This staining resulted in the efficient scavenging of ROS affected by phytochemicals on the PGC comprising the NoBS, regardless of incorporation of NC and SAG. Thus, our results indicate that the NoBS is able to scavenge ROS, and hence possesses excellent antioxidant property.
Figure 5. Antioxidant activity of nanoclay-organic bone sealant (NoBS).
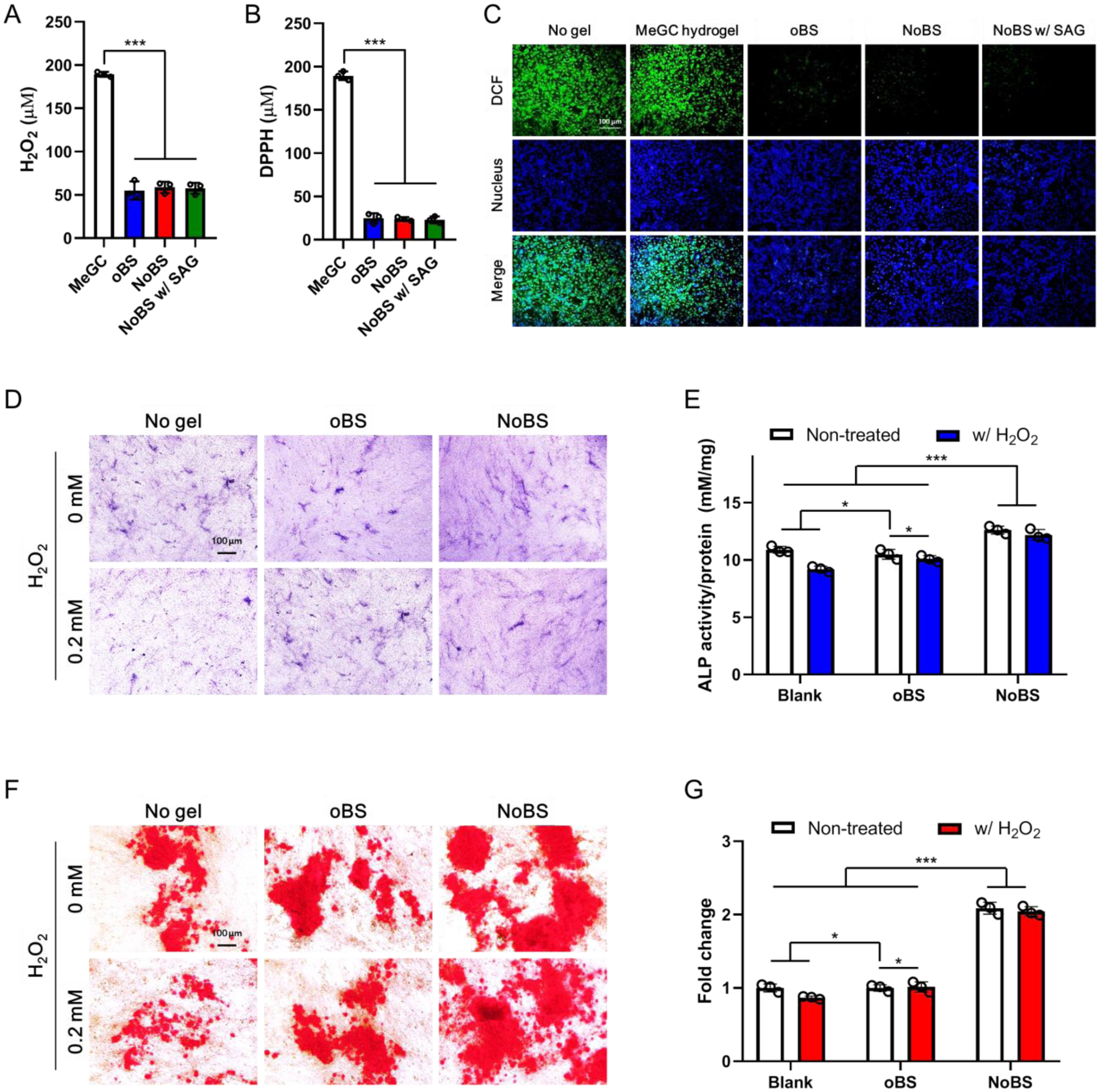
A) Hydrogen peroxide and B) DPPH radical scavenging activity of methacrylated chitosan (MeGC) hydrogel, oBS, and NoBS (1.0% NC) with or without SAG (10 μM). C) Fluorescence microscopy images of H2DCFDA-treated BMSCs grown with MeGC hydrogel, oBS, and NoBS (1.0% NC) with or without SAG (10 μM) in 0.2 mM H2O2-containing growth medium. Green fluorescence indicates DCF, which is the product resulting from the reaction between ROS and H2DCFDA ROS indicator. Nuclei were stained with Hoechst 33342 (blue). Scale bar indicates 100 μm. Error bars indicate standard deviation (n = 3), ***p < 0.001 (ANOVA followed by Tukey’s post hoc test). D-G) Antioxidant activity of oBS, and NoBS (1.0% NC) on osteogenic activity of BMSCs was evaluated in the presence or absence of 0.2 mM, hydrogen peroxide. D) ALP staining and E) ALP activity were performed at day 4. F) Alizarin red S staining was carried out at day 14, and G) their quantification was also evaluated. Scale bars indicate 100 μm in D) and F). Error bars indicate standard deviations (three independent cultures, n = 3), *p < 0.05 (ANOVA followed by Tukey’s post hoc test)
Antioxidant biomolecules are known to induce osteoblast differentiation and the differentiation of osteoblastic progenitor cells is inhibited by oxidative stress.[26] We determined further the antioxidant potential of NoBS so as to understand the possible means of cellular protection against oxidative stress. Towards this aim, we performed alkaline phosphatase (ALP) and alizarin red S staining in the presence of H2O2 (as a model ROS) on bone marrow stromal cells (BMSCs). The ALP staining was weakened upon exposure to H2O2, whereas there was no obvious difference between oBS and the untreated group (Figure 5D). Once the oBS was cultured with cells, and upon treatment with H2O2, the level of ALP was recovered to the level of untreated cells. When compared with the oBS, the levels of ALP for the NoBS, with or without H2O2, were increased and there was no obvious difference between the NoBS groups. We further confirmed the activity of cellular ALP in vitro (Figure 5E). The ALP activity was reduced 0.8-fold (p< 0.05) upon H2O2 exposure, as compared to untreated cells, but was maintained with the oBS and NoBS. The cells exposed to H2O2 showed an obviously lower degree of staining by Alizarin red S (Figure 5F). Conversely, mineralized nodules maintained in untreated cells and in the cells treated with hydrogels (oBS and NoBS), regardless of H2O2 exposure, indicating that the oBS and NoBS exhibited ROS scavenging activity. The quantification of the stained hydrogels was also undertaken and is presented in Figure 5G. The results revealed a 13% negative effect of H2O2 for osteogenic differentiation of the MSCs, and this effect was protected by incubating with the oBS and NoBS.
A quantitative real time polymerase chain reaction (qRT-PCR) analysis was performed to detect the level of gene expression related to osteogenesis, including ALP, Runx2, and OCN, in the presence or absence of H2O2 and hydrogel (Figure S12). When we analyzed at day 4 and day 14, the mRNA levels of a series of genes in the cells treated with H2O2 were significantly down-regulated by factors of 0.5-, 0.8-, and 0.5 for ALP, Runx2, and OCN, respectively. The negative effect of ROS on the osteogenic differentiation of MSCs was dramatically attenuated upon treatment with oBS and NoBS. Regulation of ROS production by NoBS is necessary to optimize the performance of endogenous osteoblasts for bone regeneration. A series of results indicates that our functional hydrogel sealant has great antioxidant potential and protects osteoblast differentiation because of the specific antioxidant activity originating in the phytochemicals.
2.5. Biocompatibility and osteoinductivity of NoBS
The biocompatibility of NoBS was determined in a 2D cell culture and analyzed using alamarBlue assay and live/dead fluorescence staining for 7 days. There was no significant difference in cytotoxicity and cell growth rate in oBS and NoBS with 0.5 and 1.0% NC in the presence or absence of SAG, supporting the notion that products derived from NoBS show no toxic effect and are biocompatible toward cells (Figure S13). Consistent with the metabolic assay, all of the hydrogels also showed remarkable cell density increases and no difference in the appearance of cells after incubation for 7 days, as evidenced by the fact that most of the cells were stained green (live cell) with well spread cells and rarely stained red (dead cell) in the images (Figure S14). These results indicate that the NoBS is highly biocompatible and able to support cell growth and proliferation.
To evaluate the osteoinductive activity of NoBS, the osteogenic potential was investigated by analyzing the activity of ALP, an early marker of osteogenesis, and mineralization in BMSCs at day 4 and 14, respectively. The oBS showed little increase in ALP expression (Figure 6A). The ALP expression of oBS was intensified as Hedgehog agonist SAG was incorporated. In contrast, the ALP staining of NoBS was obviously intensified with the increase of NC concentration, and particularly showed the best performance in the presence of SAG. We also determined ALP activity to provide for quantitative analysis (Figure 6B). Consistent with prior results of ALP staining, ALP expression was enhanced by up to 3-fold with NoBS. With NoBS containing SAG, it was significantly elevated, and was 2-fold higher in comparison with the NoBS without SAG, and with the oBS containing SAG. Alizarin red S was used to detect mineralization as a late osteogenic marker by staining calcium (Figure 6C). The staining of hydrogels was dramatically intensified as the amount of NC was increased and SAG was introduced. The highest level of staining was observed for the NoBS with SAG, indicating enhanced osteogenic capacity. When the stain on the samples was quantified and normalized relative to blank group, the NoBS with SAG showed the highest change (~ 6-fold) over other samples with statistically significant changes (Figure 6D). These colorimetric analyses indicate that the NoBS has the ability to promote osteogenic differentiation of MSCs. The gene expression related to osteogenesis, Wnt/β-catenin signaling, and Hedgehog signaling was evaluated with qRT-PCR on a molecular level. The NoBS with NC concentrations of 0.5 and 1% exhibited higher gene expression levels of ALP (an early osteogenic marker, Figure 6E), Runx2 (an osteoblast-specific marker, Figure 6F), and OCN (a late osteogenic marker, Figure 6G).[4c, 27] These gene levels of NoBS with 1% NC and SAG were higher than those of oBS containing SAG and NoBSs containing NC with significant differences. The NoBS with 1% NC and SAG had higher ALP and Runx2 levels at day 4, increased 3.6-fold and 5.2-fold, respectively, and the level of OCN was increased 5.0-fold at day 14, as compared to the non-treated group. Overall, the gene levels were up-regulated with an increase in NC concentration and significantly elevated by SAG loading, suggesting that the osteogenic capacity of NoBS was amplified with the osteoinductive activity of NC and the pro-osteogenic agonist SAG. Furthermore, the mRNA levels of several key proteins in Wnt/β-catenin signaling including Gsk3 (Figure 6H) and β-catenin (Figure 6I), were also determined using qRT-PCR. The levels of Gsk3, which negatively regulates β-catenin signal, were decreased to 0.9-fold, while the level of β-catenin in the NoBS with 1.0% NC and SAG was increased up to 1.9-fold (compared to the non-treated and oBS groups). The activation of transcription of Hedgehog target genes, including PTCH (Figure 6J) and Gli 1 (Figure 6K), was also analyzed. The qRT-PCR results showed that the expression levels of both PTCH and Gli 1 for SAG-containing oBS and NoBS were significantly higher than those of other groups. Collectively, the results indicate that NoBS combined with SAG delivery led to up-regulated Hedgehog signaling and Wnt/β-catenin signaling mediated by the small molecular activator SAG and NC-derived byproducts (lithium, magnesium, and orthosilicic acid), respectively, resulting in enrichment of mesenchymal cell osteogenic differentiation (Figure 6L).[28]
Figure 6. Osteoinductive activity of nanoclay-organic bone sealant (NoBS) in 2D cell culture.
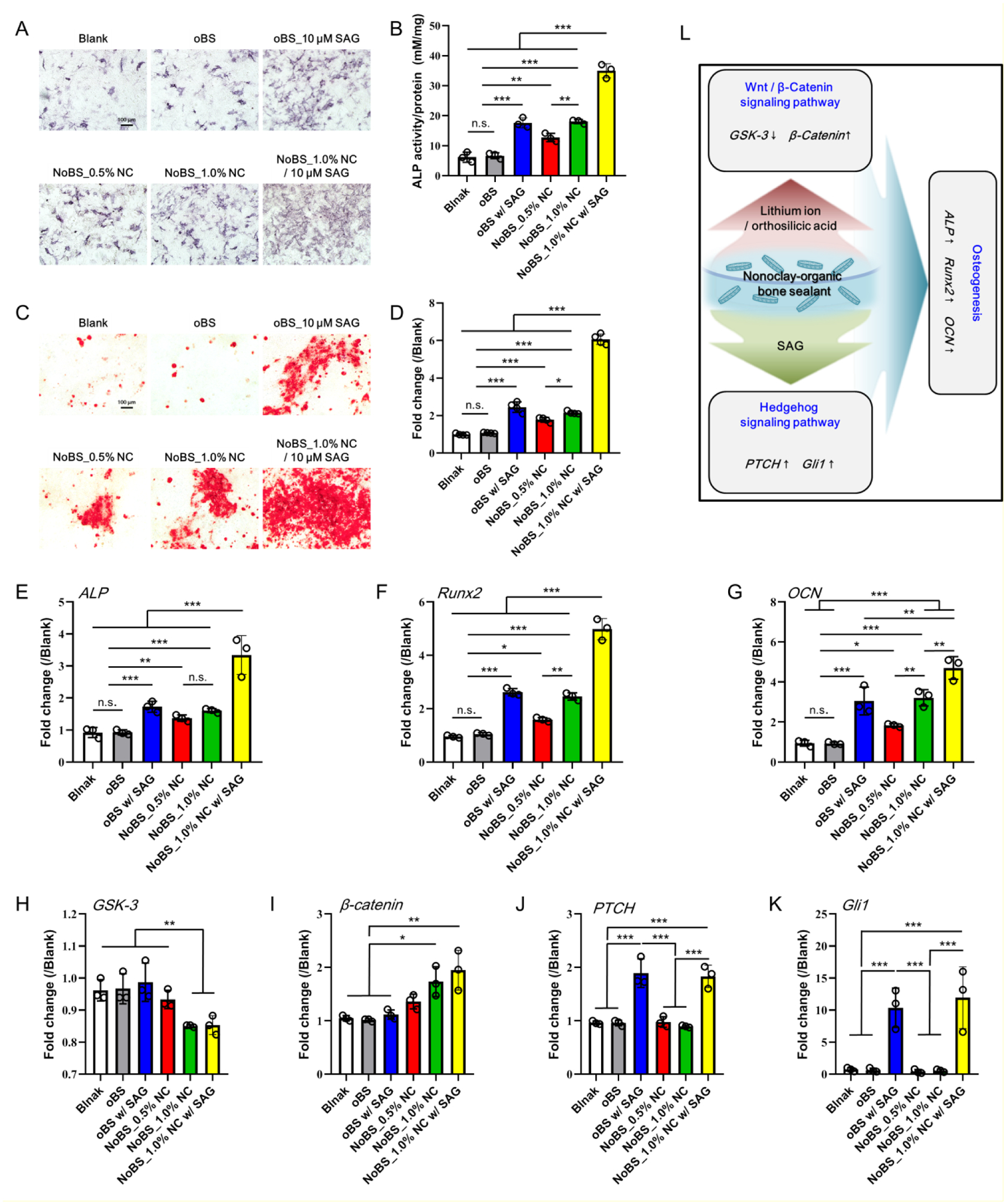
A) ALP staining and B) ALP activity were performed and determined at day 4. C) Alizarin red S staining was carried out at day 14, and D) quantification was also evaluated. Scale bars indicate 100 μm in A) and C). Error bars indicate standard deviations (three independent cultures, n = 3). The gene expression related to osteogenesis, Wnt/β-catenin signaling, and Hedgehog signaling was evaluated with qRT-PCR. E) ALP and F) Runx2 were examined at day 4, and G) OCN was measured at day 14 for osteogenesis. H) GSK-3 and I) β-catenin were examined at day 4 for Wnt/β-catenin signaling, and J) PTCH and K) Gli1 were examined at day 4 for Hedgehog signaling. Error bars indicate standard deviations (n = 3), *p < 0.05, **p < 0.01, and ***p < 0.001 (ANOVA followed by Tukey’s post hoc test). n.s.= not significant. L) Schematic illustration of osteogenesis through Wnt/β-catenin and Hedgehog signaling induced by NoBS. The concentration of SAG for SAG-containing groups was 10 μM
2.6. 3D cell encapsulation and osteoinductivity of NoBS
Cell encapsulation in a 3D construct plays a major role in cell-based transplantation therapies and can mimic the native cell microenvironment. Encapsulation of cells in suitable scaffolds has attracted considerable attention and exhibits advantages such as ease of handling, highly hydrated tissue-like microenvironment, uniform cell distribution, and spatial localization.[29] Based on the excellent biocompatibility of NoBS in the aforementioned 2D determinations, we performed an alamarBlue assay and live/dead fluorescence staining for 14 days to evaluate biocompatibility in a 3D hydrogel construct. BMSCs were encapsulated into oBS and NoBS and cultured in the growth medium. The number of cells in the hydrogels increased by factors of up to 3.8 over time, as compared to initial levels (day 1). There were no distinct differences in cell proliferation in the hydrogels with various levels of NC incorporation, indicating that the hydrogels in the presence or absence of NC are capable of supporting the cells and promoting cell proliferation (Figure 7A).
Figure 7. Cell encapsulation, proliferation, and osteoinductivity evaluation of nanoclay-organic bone sealant (NoBS) in 3D cell culture.
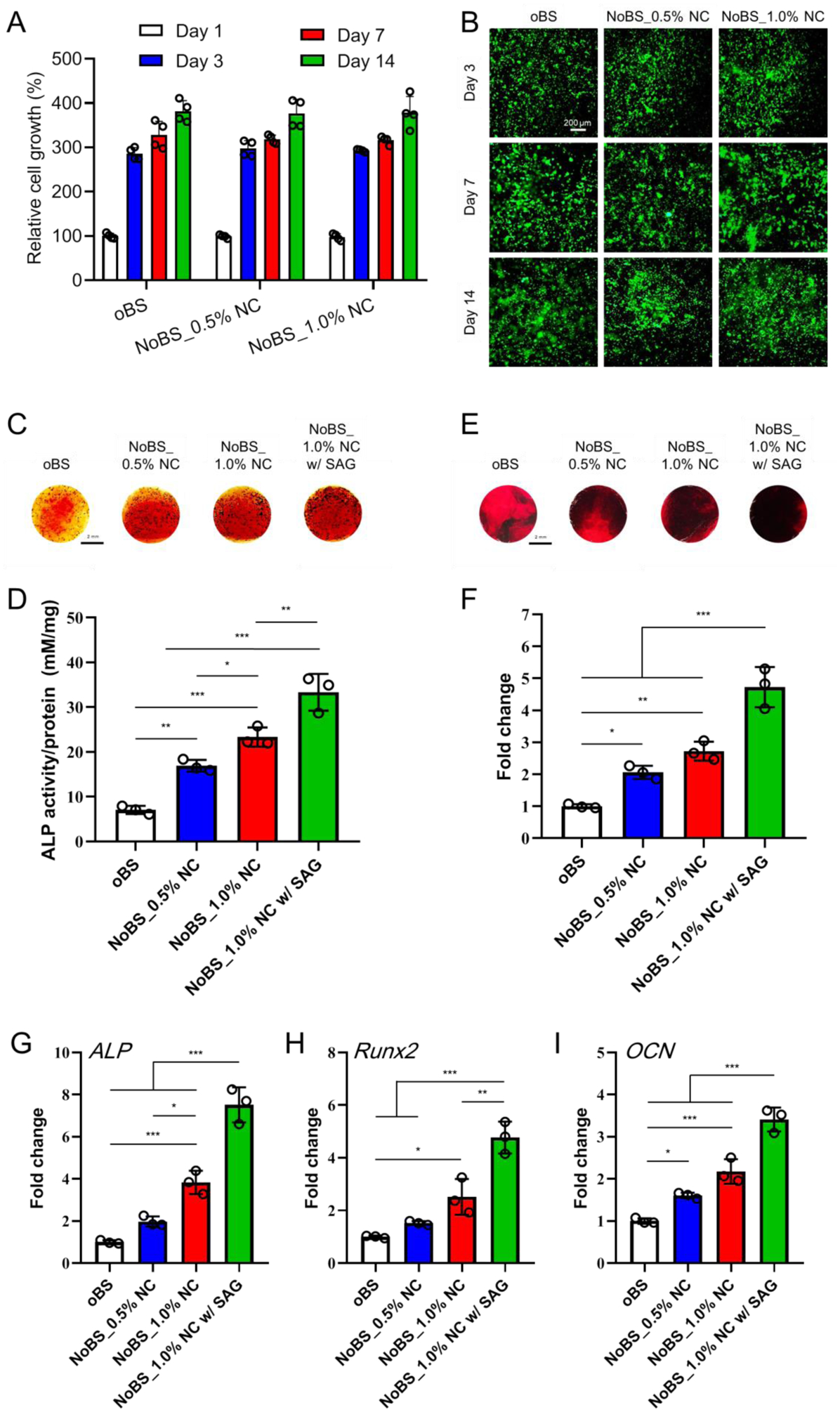
A) In vitro cell proliferation in oBS and NoBSs after 1, 4, 7 and 14 days. The values were normalized with the oBS group on day 1 (n=4). B) Representative fluorescence images of BMSCs stained with calcein AM (live cells, green fluorescence) and ethidium homodimer (dead cells, red fluorescence) for 14 days. Scale bar indicates 200 μm. C) ALP staining and D) colorimetric quantification (n=3) of cell-encapsulated oBS and NoBSs treated in osteogenic medium at day 4. E) Alizarin red S staining and F) quantified mineralized extracellular matrix (n=3) of cell-encapsulated oBS and NoBSs after incubation with osteogenic medium at day 14. Scale bar represents 2 mm in D) and F). Gene expression of cell-encapsulated oBS and NoBSs (n=3). G) ALP and H) Runx2 were evaluated to assess the osteogenic differentiation after 4 days of incubation. I) OCN was evaluated to assess the osteogenic differentiation after 14 days of incubation. Data are presented as mean ± SD. Statistical analyses were done with one-way ANOVA with Tukey’s post hoc test; *P < 0.05, **P < 0.01, and ***P < 0.001. The concentration of SAG for SAG-containing group was 10 μM.
The live and dead staining was conducted at the time points of interest (day 3, 7, and 14) to evaluate the visualization analysis. Consistent with the metabolic activity, the fluorescence images demonstrated that most of the cells encapsulated in the hydrogel matrix remained alive and well spread, with only a few dead cells observed after 14 days of cultivation (Figure 7B). As time passed, the cell density increased in all of the hydrogels, either in the presence or absence of NC. These results show that the NoBS can serve as an excellent microenvironment for promoting cell support, survival, and proliferation, and thus can be utilized as a matrix for safe cell delivery. Although cells were found to be homogeneously distributed in the hydrogels and the live/dead staining indicated high viability, we recognize that the cells may experience shear stress upon mixing in the hydrogels that can lead to cell damage. Further study will be needed to encapsulate cells in a safer manner, using more biocompatible catechol crosslinking methods such as enzymatic oxidation.
To quantify the osteoinductive ability of BMSC-laden NoBS, ALP expression and activity were determined at day 4. The ALP staining of NoBS groups was intensified by increased concentrations of NC and further enhanced with SAG loading, as compared with the oBS group (Figure 7C). In addition, the activity of ALP expressed in the NoBS was gradually promoted by increasing NC and exhibited the best performance in NoBS with 1% NC and SAG (Figure 7D), which was consistent with the ALP staining results. Furthermore, a similar tendency was determined with Alizarin red S staining, verifying the formation of a mineralized extracellular matrix (Figure 7E). Staining also was remarkably intensified as the content of NC was increased, even though the staining of cell-free hydrogels was increased by the addition of NC (because of the high affinity of NC to cations including Ca2+), as presented in Figure S15. The darkest alizarin red S staining was observed in NoBS with 1% NC and SAG (as compared with the other groups), indicating the deposition of extracellular minerals. When the level of alizarin red S staining was evaluated by comparison to the stained hydrogels, a significant increment in the amount of mineralized matrix was observed for the NoBS with 1% NC and SAG, as compared to the other groups (Figure 7F).
Subsequently, we used qRT-PCR to confirm the expression of osteogenic genes, including ALP (Figure 7G), Runx2 (Figure 7H), and OCN (Figure 7I), in BMSC-laden NoBS. After culturing in osteogenic medium for 4 days, the mRNA level of ALP in NoBS with 0.5 and 1.0% NC was up-regulated 2.0- and 3.8-fold, respectively, as compared with NC-free oBS, and SAG-containing NoBS showed the highest gene expression (approximately 7.5-fold). The levels of Runx2 for NoBS with NC were also increased 1.5-fold (0.5% NC) and 2.5-fold (1.0% NC), most notably in the SAG-containing NoBS. Similarly, the expression of OCN, a key osteogenic marker of non-collagenous bone matrix proteins, was significantly increased by NC and SAG incorporation on day 14. Collectively, the results indicate that the biocompatible NoBS holds great potential as a cell delivery carrier and also enhances the osteoinductive ability, an effect aided by incorporation of NC and SAG.
2.7. In vivo bone regenerative capacity of NoBS
We investigated the bone regenerative capacity of the NoBS in a critical-sized mouse calvarial defect model (Figure 8). We evaluated the oBS and two types of NoBS in parallel so as to compare results obtained in the presence or absence of NC and SAG. During 8 weeks of the experiment, none of the mice showed any defect complications such as inflammatory soft tissue swelling or ectopic bone formation. The micro-computed tomography (microCT) scanning images and quantitative analyses were gathered to evaluate the extent of the re-ossification. As shown in Figure 8A, newly generated bone in the original defect area was observed for the NoBS groups, and was significantly enhanced with SAG, whereas no obvious regenerated bone was found in the oBS group. When the remaining defect area was normalized with the original defect area in Figure 8B, the bone healing area of NoBS with SAG (62.0 ± 11.6%) was considerably increased in comparison with that of the oBS group (24.0 ± 4.5%) and the NoBS without SAG (40.1 ± 6.0%). We next performed a quantitative 3D analysis to determine the total amount of bone tissue in the defect sites. Figure 8C shows that the NoBS led to a significant increase (20.8 ± 4.1%, P < 0.01 for NoBS without SAG; 30.0 ± 5.5%, P < 0.001 for NoBS with SAG) in bone volume density, as compared to the control oBS group (8.0 ± 4.1%). Furthermore, SAG-containing NoBS showed significantly higher bone volume density than did the NoBS without SAG (P < 0.05). Similarly, bone mineral density (Figure 8D) was significantly greater in the SAG-containing NoBS group (0.56 ± 0.06 g cm−3) than it was in either the NoBS without SAG (0.40 ± 0.05 g cm−3) or the oBS group (0.33 ± 0.08 g cm−3). Additionally, the trabecular number of the SAG-containing NoBS group was significantly higher than was that of the oBS group (P < 0.01), even though the NoBS without SAG group showed incremental increases (Figure 8E). The results of microCT and quantitative analysis revealed significantly better re-ossification in the SAG-containing NoBS group relative to the other groups, suggesting that NoBS combined with SAG, an osteoinductive compound, could lead to faster and more efficient healing of bone defects.
Figure 8. In vivo bone regeneration after 8 weeks of implantation of nanoclay-organic bone sealant (NoBS) on calvarial defect.
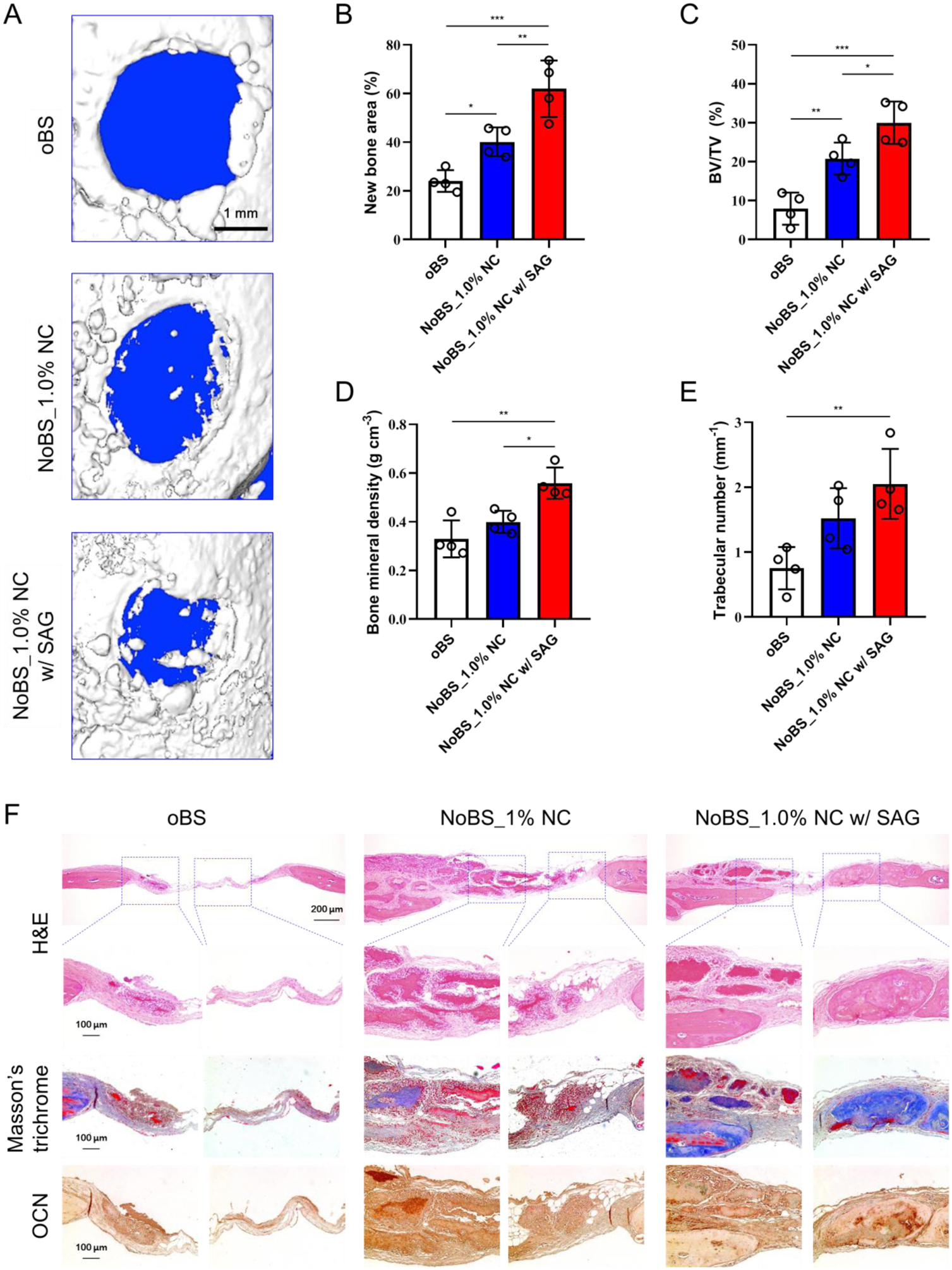
A) Representative 3D reconstructed images of calvarial defect at 8 weeks after implantation of oBS and NoBSs taken with superficial view. Quantified parameters of the regenerated bone including B) new bone area, C) bone volume density (BV/TV, bone volume; BV, tissue volume; TV), D) bone mineral density, and E) trabecular number taken at the three-millimeter diameter cylindrical defect. All data were presented as mean ± SD (n=4). Statistical analyses were done with one-way ANOVA with Tukey’s post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001. F) H&E- and Masson’s trichrome-stained histologic sections of calvarial decalcified sections, and immunohistochemical analysis of osteocalcin (OCN) after 8 weeks of implantation. The concentration of SAG for SAG-containing group was 10 μM.
Histological evidence further supported the radiographic findings and confirmed bone regeneration with the use of Haematoxylin & Eosin (H&E), Masson’s trichrome, and immunohistochemical staining (Figure. 8F). Histological analysis indicated small amounts of bone tissue and less bone formation in the oBS group. However, the formation of bony structures was clearly observed for the SAG-containing NoBS group, with nearly complete osseous closure along the junction of the defects as well as inside the defects; this exceeded the moderate bone formation seen for the NoBS without SAG group. Bone regeneration efficacy of the SAG-containing NoBS group was superior to those of NoBS without SAG and the oBS groups, with noticeable covered spaces, thick soft tissue, and formation of bone similar to native bone. To analyze further the extent of de novo ossification, immunohistochemistry was performed to determine the expression of osteogenic marker OCN (Figure 8G). A strong expression of OCN was observed along the areas of new bone formation in the NoBS groups, whereas no obvious positive staining was observed for the oBS group. In addition, the NoBS with SAG demonstrated the highest expression levels of OCN in the osteoblasts and osteocytes, higher than those of NoBS without SAG. These findings indicate that the NoBS combined with SAG reinforces the osteoinductive capability for enhanced bone regeneration.
3. Conclusion
We have demonstrated the development of a nature-inspired hydrogel bone sealant (NoBS) system that serves as a powerful biomaterial bone substitute. The NoBS exhibits biocompatibility and multifunctional properties including injectability, self-healing, moldability, antibacterial and antioxidant activities, and osteoinductivity for robust bone regeneration. Our system was fabricated with an inorganic NC-organic polymer composition for facile and minimally invasive application at the site of an arbitrary defect. Therefore, this hydrogel bone sealant avoids many of the barriers associated with conventional antibiotics and prevents the dysfunction of MSCs during the bone healing process. Additionally, based on the nature-inspired approach, the NoBS displayed excellent biocompatibility and even potential as a cell delivery carrier. Significantly, the NoBS exhibited great osteoinductive capability by regulating the Wnt/β-catenin pathway, and loading of the smoothened agonist SAG into NC led to enhanced performance resulting from activation of Hedgehog pathway. Finally, the NoBS facilitated adequate bone regeneration in critical-sized mouse calvarial defects. The design of this nature-inspired hydrogel bone sealant with multimodality allows for safer and faster bone regeneration and avoids the typical barriers impeding regeneration processes, and presents an optimal bone substitute for new therapeutic approaches in bone tissue engineering and regenerative medicine.
4. Experimental Section
Materials:
Glycol chitosan was purchased from MP Biomedicals (Irvine, CA). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, N-hydroxysuccinimide (NHS), 5-bromo-4-chloro-3-indoxylphosphate (BCIP), Nitro Blue tetrazolium (NBT), dimethyl sulfoxide (DMSO), Alizarin red S, L-ascorbic acid, p-nitrophenol phosphate, β-glycerophosphate, and dexamethasone were purchased from Sigma Aldrich (St. Louis, MO). Caffeic acid was obtained from TCI chemicals (Portland, OR). 2,2-Diphenyl-1-picrylhydrazyl (DPPH) was purchased from Alfa Aesar (Haverhill, MA). Coumarin boronic acid (CBA) was purchased from Cayman chemical (Ann Arbor, MI). Hectorite NC was obtained from Best of Chemicals (Shirley, NY). SAG was purchased from Tocris Bioscience (Bristol, UK). AlamarBlue, Trizol reagents, BCA protein assay kit, hydrogen peroxide, Calcein AM, and Ethidium homodimer-1 (EthD-1) were purchased from Thermo Fisher Scientific (Waltham, MA). The RNeasy Mini kit was obtained from Qiagen (Valencia, CA). All other chemicals were analytical grade and were used without further purification.
Synthesis of PGC:
PGC was synthesized using standard EDC chemistry. Briefly, glycol chitosan (1 g, 4.87 mmol unit) was dissolved in distilled water/DMSO (5:2, 70 mL, pH 5). Caffeic acid (2.44 mg, 1.37 mmol), EDC (514 mg, 2.68 mmol), and NHS (308 mg, 2.68 mmol) were dissolved in DMSO (10 mL), and this solution was slowly added to the chitosan solution. The pH of the solution was adjusted to 4.5 to prevent the oxidation of catechol groups. After reacting overnight, the product was purified by membrane dialysis (MWCO: 3,500, Spectra/Por®, Los Angeles, CA) against pH 4.0 deionized water for 2 days and deionized water for 4 h, and was then freeze dried. The degree of phytochemical conjugation was determined using 1H-NMR spectrometry (Bruker Avance, 400 MHz, D2O) and Fourier-transform infrared (FT-IR) spectroscopy (Jasco 420 FTIR). The degree of catechol substitution was approximately 9.7 per 100 glucose rings of chitosan.
Preparation of SAG-loaded NC:
One hundred mg of NC was suspended in 8.0 mL of milli-Q water. One mg of SAG was dissolved in 1 mL of ethanol. After the SAG was completely dissolved, 1 mL of Milli-Q water was added. The SAG solution was added to the NC suspension, and then the final solution was stirred vigorously overnight. The final solution was purified and concentrated with a 30 kDa centrifugal filter (EMD Millipore, Temecula, CA, USA) at 2,500 rpm for 15 min at room temperature with PBS (pH 7.4), and this was repeated three times with an equivalent volume of Milli-Q water. The final sample was lyophilized for 3 days and then stored at −20 °C. SAG-NC was characterized using FT-IR spectroscopy (Jasco 420 FTIR). The powder X-ray Diffraction (XRD, Bruker Corporation, Germany) pattern was collected from 2 to 10° or 70° (2θ range) with a diffractometer using Ni-filtered CuKα X-ray radiation (λ = 1.5418 Å).
Preparation and characterization of NoBS:
To prepare hydrogels, PGC was dissolved in 0.02 M pH 7.4 PBS solution so that a 4% (w/v) PGC solution was obtained. NC was dispersed in Milli-Q water and yielded a 5% (w/v) stock dispersion. Various amounts of NC (w/v), in the presence or absence of SAG-NC, were mixed with 4% (w/v) PGC solution and sonicated using a high-power 500 W sonic dismembrator (5 s on, 1 s off, 20% amplitude, 25 W cm−2 of power intensity) for 1 min. The NoBSs were fabricated with oxidizer (NaIO4, 1:1 final molar ratio versus catechol groups of PGC). The final concentration of PGC was 2% (w/v), with various amounts of NC in the presence or absence of SAG-NC.
The gelation times of NoBS solutions with different concentrations of NaIO4 were investigated via a vial-tilting method.[30] The NoBS solutions in the presence of varying NaIO4 concentrations or NC contents were put into 2 ml vials. The sol–gel transition time was determined by inverting the vial until the point at which no fluidity was observed. Each data point was derived from an average of three measurements.
A 400 μL sample of NoBS was placed in a 48-well plate and the compressive modulus of NoBS was measured via an indentation experiment done with an Instron Electro-Mechanical Testing Machine (Instron, Model 5564, Norwood, MA, USA), using a flat-ended indenter 1.6 mm in diameter. Three measurements were performed and three parallel samples were utilized. The compressive modulus was calculated as described previously, using a Poisson’s ratio of 0.25.[31]
Ex vivo bone adhesion of NoBS was evaluated using chicken bones. Two specimens were placed into an Instron Electro-Mechanical Testing Machine for tensile loading with a strain rate of 1 mm/min. The adhesive strength was determined by the point of detachment.
Hydrogels were equilibrated in PBS for 24 h and lyophilized for 16 h to obtain dry gels. Equilibrium water content was calculated using equation (1):
| (1) |
where Ww and Wd refer to the weight of wet and dry hydrogels, respectively.
The swelling ratio of NoBS was quantified with equation (2):
| (2) |
where Ws and Wd refer to the swollen and dry weights, respectively.
The degradation of NoBS was run for 6 weeks. The NoBS was incubated at 37 °C in 0.01 M (pH 7.4) PBS, which was replaced every 7 days. The NoBS was lyophilized and the weight was determined according to equation (3):
| (3) |
where W0 and Wt refer to the weight of hydrogels at times 0 and t, respectively.
NoBSs were imaged at low vacuum using scanning electron microscopy with X-ray microanalysis (SEM/EDS, FEI Nova NanoSEM 230, Hillsboro, OR, USA) to observe the microstructure and chemical compositions.
To determine the release profile of SAG from NoBS, the hydrogels were immersed in 1.5 ml of 10 mM PBS solution (pH 7.4) with 0.1% Tween 20 added. The scaffolds were then incubated at 70 rpm at 37 °C. At the predetermined time, the medium was exchanged with fresh buffer. SAG was determined from the absorbance at 299 nm, using ultraviolet spectrophotometry.
Characterization of self-healing:
NoBSs comprising 2% (w/v) PGC in the presence of 1.0% NC were fabricated as described above. Both oBS and NoBS samples were cut into 2 pieces. A piece of oBS and a piece of NoBS with 1.0% NC were reformed into an integrated gel. These two healed hydrogels were then incubated in 0.01 M (pH 7.4) PBS solution for 24 h to evaluate their stability. A photograph of each step was taken with a digital camera.
Rheological characterization was performed with a TA Instruments HR-3 hybrid rheometer fitted with a Peltier stage. All measurements were performed using a 50 mm cone plate geometry (cone angle: 2 degrees) and analyzed using TA Instruments TA Orchestrator software.
Characterization of bactericidal activity:
A fresh culture of gram-positive bacteria, vancomycin-resistant Staphylococcus aureus Mu50 (S. aureus), and gram-negative bacteria, Escherichia coli K-12 (E. coli) were prepared by suspending a single colony from an LB agar culture in 5 mL of sterile LB medium. The counts of bacteria were determined from reading the optical density of the medium at 600 nm (OD600), measured with a UV/Vis spectrophotometer (Beckman Coulter). The OD600 value was used to calculate the number of bacterial colony-forming units (CFU) per mL.
The measurements of NoBS inhibition zones were done with the disc diffusion method. A 100 μL sample with a bacterial suspension of 106 CFU mL−1 was spread on an LB agar plate, 100 μL of NoBS was added, and the plate was incubated at 37 °C for 24 h.
An in vitro antimicrobial activity test was performed by microplate proliferation assay.[32] NoBS and oBS were incubated with 500 μL of 107 CFU bacterial suspensions in LB broth medium at 37 °C for 1 h. Subsequently, 200 μL of the solution was transferred to a 96 well plate and incubated for 24 h at 37 °C to monitor the bacterial proliferation, as indicated by the absorbance at 600 nm, with a UV/Vis spectrophotometer (Beckman Coulter).
Characterization of antioxidant activity:
The DPPH radical scavenging activity of NoBS was evaluated. A 0.25 mM solution of DPPH was prepared in 95% ethanol prior to use. The DPPH solution (800 μL) was mixed with 200 μL of NoBSs, and then gently shaken in the dark for 30 min. The radical scavenging activity was measured by monitoring the absorbance decrease at 516 nm.
Hydrogen peroxide scavenging activity was confirmed with the use of CBA. CBA was dissolved in DMSO at a 0.1 M concentration, and the solution diluted in 10 mM PBS (pH7.4) containing 0.2 mM H2O2 was added directly to NoBS for 30 min. The desired concentration of CBA was 0.8 mM. The excitation and emission wavelengths were set at 360 and 460 nm, respectively, so as to monitor the fluorescence of 7-hydroxycoumarin that resulted from the oxidation of CBA.
For visualization of intracellular ROS, BMSCs were grown in a glass-bottom dish in hydrogen peroxide-containing growth medium (DMEM, 10% FBS, 1% P/S), with or without NoBS, in which BMSCs and NoBS were separated by insertion of a 70 μm pore mesh. After 24 h, the cells were incubated with H2DCFDA and Hoechst 33342 to visualize ROS and stain the nuclei for 30 min at 37 °C in the dark, then rinsed twice with PBS and imaged with the use of fluorescence microscopy (Olympus IX 71 microscope, Center Valley, PA).
Cell culture and cell viability:
The mouse bone marrow stromal cell line (BMSCs, D1 ORL UVA [D1], D1 cell, CRL-12424) was purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. Cell culture media DMEM, penicillin, streptomycin, and FBS were purchased from Gibco (Manassas, VA).
Cell viability was evaluated using alamarBlue assay (Life Technologies). Briefly, BMSCs were cultured with oBS or NoBS with various concentrations of NC (0.5 or 1.0%, w/v), and hydrogel and BMSCs were separated by insersion of a 70 μm pore mesh in the growth medium (DMEM, 10% FBS, 1% P/S). The cells were rinsed with PBS and incubated for 3 h with sterilized 10% alamarBlue solution. AlamarBlue fluorescence was measured at 530 nm (excitation) and 590 nm (emission) at day 1, 4, and 7. The cells were stained with a Live/Dead staining kit (Life Technologies) and observed with fluorescence microscopy (Olympus IX 71 microscope, Center Valley, PA) at day 1 and day 7.
Cell encapsulation in NoBS:
NoBS was incubated with PBS to eliminate excess NaIO4. NoBSs were mixed with 2×107 BMSCs in 50 μL of medium. The cell-encapsulated NoBS was shaken and pipetted immediately. A 100 μL sample of cell-encapsulated NoBS was placed in a 24 well plate and cultured with 2 mL of culture medium. The culture medium was changed every 2 days.
To estimate the biocompatibility of NoBS in a 3D cell culture, cell viability was detected using alamarBlue assay (Life Technologies). Briefly, cell-encapsulated NoBSs were rinsed with PBS and incubated for 3 h with sterilized 10% alamarBlue solution. AlamarBlue fluorescence was measured at 530 nm (excitation) and 590 nm (emission). To visualize cell proliferation, cell-encapsulated NoBSs were stained with a Live/Dead staining kit (Life Technologies) and observed under a fluorescence microscope (Olympus IX 71 microscope, Center Valley, PA) at day 3, 7, and 14.
Staining and quantification of alkaline phosphatase (ALP):
For 2D assays, BMSCs were cultured in 6-well plates with growth medium (DMEM, 10% FBS, 1% P/S). After 100% confluence, the medium was replaced with an osteogenic medium containing 10% FBS, 1%, P/S, 10 mM β-glycerophosphate, 50 g L−1 L-ascorbic acid, 100 nM dexamethasone, and hydrogel (oBS or NoBS), and hydrogel and BMSCs were separated by insertion of a 70 μm pore mesh. After 4 days of culture, cells were fixed in 10% formalin, rinsed with 10 mM PBS (pH 7.4), and incubated in a solution containing Nitro Blue Tetrazolium (Sigma-Aldrich, St. Louis, MO) and 5-bromo-4-chloro-3-indoxylphosphate (Sigma-Aldrich, St. Louis, MO) stock solutions in ALP buffer (100 mM Tris, pH 8.5, 50 mM MgCl2, 100 mM NaCl) for 1 h. The stained samples were viewed with an Olympus IX 71 microscope (Center Valley, PA).
For 3D assays, a 100 μL sample of cell-encapsulated NoBS was placed in a 24 well plate and cultured with 2 mL of culture medium for a day. The medium was then replaced with an osteogenic medium. After 4 days of culture, cells were fixed in 10% formalin, rinsed with PBS, and incubated for 1 h in a solution containing Nitro Blue Tetrazolium and 5-bromo-4-chloro-3-indoxylphosphate stock solutions in ALP buffer. The stained NoBSs were viewed with the Olympus SZX 16 stereomicroscope. For the colorimetric measurements of ALP activity, cells were lysed in a lysis buffer (0.1% Tween-20 in 10 mM PBS, pH 7.4) and the absorbance at 405 nm was measured by using ρ-nitrophenol phosphate substrate. Measurements were normalized to total protein content evaluated by the BCA Assay (Thermo scientific, Rockford. IL).
Staining and quantification of mineralization:
Mineralized extracellular matrix was evaluated with the use of Alizarin red S staining. At 14 days post-incubation, the cells or cell-encapsulated NoBSs were immersed in 10% formalin for 15 min and rinsed three times with Milli-Q water. The fixed cells were incubated in 1% Alizarin red S solution for 5 or 60 min for 2D or 3D culture, respectively. The stained cells or NoBSs were observed with an Olympus IX 71 microscope or an Olympus SZX 16 stereomicroscope. The quantification of mineralized extracellular matrix was performed with acetic acid extraction and neutralization with ammonium hydroxide. The colorimetric measurement was carried out at a wavelength of 405 nm and statistical analyses were performed on the resulting data. The data are presented as fold change normalized to non-treated (osteogenic medium, OM or oBS) samples.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR):
Total RNA was extracted from cells cultured on the cell culture plate or in NoBSs using Trizol reagent (Life Technologies) and the RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The concentration of purified total RNA was determined spectrophotometrically using aNanoDrop™ Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). The ratio of the absorbances at 260 nm and 280 nm, which represents the purity of RNA, was 1.5–1.8. Total RNA (0.5 μg) was reverse-transcribed utilizing the SuperScript® III First-Strand (Invitrogen, Carlsbad, CA), and then 1 μL of cDNA was used for each reaction in the presence of 20 μL of LightCycler 480 SYBR Green master mix (Roche, Indianapolis, IN). Real-time PCR (RT-PCR) was performed using LightCycler 480 PCR (Roche, Indianapolis, IN) under the following PCR conditions: 95°C for 10 min, followed by 45 cycles of 95 °C for 15 sec, and 58 °C for 45 sec. Gene expression was calculated using the CT value, and fold-changes in expression were determined with the 2−ΔΔCT method. The relative gene levels were calculated with reference to the housekeeping gene (GAPDH). The values were normalized to the average of control values (Blank or oBS); delta-CT. The specific forward and reverse primer sequences are shown in Table S3.
Calvarial Defect Model:
All surgical experiments strictly abided by the guidelines of the Chancellor’s Animal Research Committee (ARC) at the University of California, Los Angeles. All full-thickness craniotomy defects (3 mm) were drilled in the parietal bone of 8–10-week-old male CD-1 mice. Each defect was cleaned and then implanted with NoBSs. Following the surgery, all animals were monitored until they regained sternal recumbency, and they were transported to the vivarium for postoperative care. Buprenorphine was injected subcutaneously in all animals, using a concentration of 0.1 mg kg−1 for 3 days for pain management. All animals had free access to water containing trimethoprim/sulfamethoxazole for 7 days in order to avoid infection.
Micro computed tomography (microCT) Scanning and Analysis:
The calvarial tissues from all experimental animals were harvested at 8 weeks post-implantation and immersed in 4% paraformaldehyde solution at 25 °C for 48 h with gentle shaking. High-resolution microCT images were acquired for all of the samples on a SkyScan 1172 (Kontich, Belgium) with 0.5 mm Al filtration (184 μA, 57 kVp). The exposure time was 190 ms, and 475 projections were acquired at an angle of 190°. The resolution was 10 μm for both the pixel size of projections and the voxel size of the reconstructed data set. Visualization and reconstruction of the data were carried out with the OsiriX MD imaging software. The new bone area was determined using ImageJ (National Institutes of Health, Bethesda, MD) and normalized to the original defect surface area (3 mm in diameter). Bone volume density (BV/TV, %), trabecular number (mm−1), and bone mineral density (BMD, g cm−3) were measured with the SkyScan CT-Analyzer program (Bruker microCT).
Histological Evaluation:
After microCT scanning, all fixed tissues were incubated in 10% ethylenediaminetetraacetic acid (EDTA) solution for 7 days with gentle shaking, with one refresh of the EDTA solution on day 3. After decalcification, samples were embedded into paraffin and cut to a thickness of 5 μm. H&E and Masson’s trichrome staining were performed to determine the extent of new bone formation. The blue color, indicative of regenerated or mature bone, was observed with an Olympus IX71 microscope (Tokyo, Japan).
The size of the remaining defect was determined with ImageJ and compared to the blank control group. Additional sections underwent immunohistochemical analysis. The deparaffinized sections were processed with citric acid for antigen retrieval and thereafter incubated with the primary antibody of osteocalcin (OCN, FL-95, sc-30045, Santa Cruz Biotechnology), then studied with the HRP/DAB Detection IHC kit (ab64261, Abcam). The sections were further counterstained with Mayer’s hematoxylin.
Statistical Analysis:
Statistical analyses were performed using SigmaPlot 12.5 software. Data were presented as mean ± SD. for all results. Statistical significance was determined by two-tailed t-test or one-way ANOVA with Tukey. *P < 0.05, **P < 0.01, ***P < 0.001 were considered significant.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01 DE027332), the Department of Defense (W81XWH-18-1-0337), and the MTF Biologics.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Chung-Sung Lee, Division of Advanced Prosthodontics, University of California Los Angeles, CA 90095, USA.
Hee Sook Hwang, Division of Advanced Prosthodontics, University of California Los Angeles, CA 90095, USA.
Soyon Kim, Division of Advanced Prosthodontics, University of California Los Angeles, CA 90095, USA.
Jiabing Fan, Division of Advanced Prosthodontics, University of California Los Angeles, CA 90095, USA.
Tara Aghaloo, Division of Diagnostic and Surgical Sciences, University of California Los Angeles, CA 90095, USA.
Min Lee, Division of Advanced Prosthodontics, University of California Los Angeles, CA 90095, USA; Department of Bioengineering, University of California Los Angeles, CA 90095, USA.
References
- [1].a) Xu B, Zheng P, Gao F, Wang W, Zhang H, Zhang X, Feng X, Liu W, Advanced Functional Materials 2017, 27, 1604327; [Google Scholar]; b) Pina S, Oliveira JM, Reis RL, Advanced Materials 2015, 27, 1143. [DOI] [PubMed] [Google Scholar]
- [2].a) Wei Z-L, Yang J, Li F-M, Peng W-Q, Xie D, Qiu L-B, Zhao J-M, Journal of Biomaterials and Tissue Engineering 2017, 7, 696; [Google Scholar]; b) Park J-W, Hwang J-U, Back J-H, Jang S-W, Kim H-J, Kim P-S, Shin S, Kim T, Composites Part B: Engineering 2019, 178, 107449; [Google Scholar]; c) Guarino V, Causa F, Taddei P, di Foggia M, Ciapetti G, Martini D, Fagnano C, Baldini N, Ambrosio L, Biomaterials 2008, 29, 3662; [DOI] [PubMed] [Google Scholar]; d) Kalteis T, Lüring C, Gugler G, Zysk S, Caro W, Handel M, Grifka J, Zeitschrift fur Orthopadie und ihre Grenzgebiete 2004, 142, 666. [DOI] [PubMed] [Google Scholar]
- [3].a) Okesola BO, Ni S, Derkus B, Galeano CC, Hasan A, Wu Y, Ramis J, Buttery L, Dawson JI, D’Este M, Advanced Functional Materials 2020, 30, 1906205; [Google Scholar]; b) Kim Y-H, Yang X, Shi L, Lanham SA, Hilborn J, Oreffo RO, Ossipov D, Dawson JI, Nature communications 2020, 11, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gaharwar AK, Rivera CP, Wu C-J, Schmidt G, Acta biomaterialia 2011, 7, 4139; [DOI] [PubMed] [Google Scholar]; d) Li C, Mu C, Lin W, RSC advances 2016, 6, 43663; [Google Scholar]; e) Rajabi N, Kharaziha M, Emadi R, Zarrabi A, Mokhtari H, Salehi S, Journal of Colloid and Interface Science 2020, 564, 155; [DOI] [PubMed] [Google Scholar]; f) Cebe T, Ahuja N, Monte F, Awad K, Vyavhare K, Aswath P, Huang J, Brotto M, Varanasi V, Journal of Materials Research 2020, 35, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Ordikhani F, Dehghani M, Simchi A, Journal of Materials Science: Materials in Medicine 2015, 26, 269; [DOI] [PubMed] [Google Scholar]; b) Zhang X, Fan J, Lee C-S, Kim S, Chen C, Lee M, ACS applied materials & interfaces 2020, 12, 16088; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cui Z-K, Kim S, Baljon JJ, Wu BM, Aghaloo T, Lee M, Nature communications 2019, 10, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dhivya S, Saravanan S, Sastry T, Selvamurugan N, Journal of nanobiotechnology 2015, 13, 40; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Liu Y, Meng H, Qian Z, Fan N, Choi W, Zhao F, Lee BP, Angewandte Chemie International Edition 2017, 56, 4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Okike K, Bhattacharyya T, JBJS 2006, 88, 2739; [DOI] [PubMed] [Google Scholar]; b) Yang Y, Chu L, Yang S, Zhang H, Qin L, Guillaume O, Eglin D, Richards RG, Tang T, Acta biomaterialia 2018, 79, 265. [DOI] [PubMed] [Google Scholar]
- [6].a) Brown ED, Wright GD, Nature 2016, 529, 336; [DOI] [PubMed] [Google Scholar]; b) Brooks BD, Brooks AE, Advanced drug delivery reviews 2014, 78, 14. [DOI] [PubMed] [Google Scholar]
- [7].a) Kawazoe N, Chen G, Biomaterials 2015, 54, 226; [DOI] [PubMed] [Google Scholar]; b) Kim WK, Meliton V, Bourquard N, Hahn TJ, Parhami F, Journal of cellular biochemistry 2010, 111, 1199; [DOI] [PubMed] [Google Scholar]; c) Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC, Journal of Biological Chemistry 2007, 282, 27298; [DOI] [PubMed] [Google Scholar]; d) Li J, Zhang J, Chen Y, Kawazoe N, Chen G, ACS applied materials & interfaces 2017, 9, 35683; [DOI] [PubMed] [Google Scholar]; e) Atashi F, Modarressi A, Pepper MS, Stem cells and development 2015, 24, 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Meng H, Li Y, Faust M, Konst S, Lee BP, Acta biomaterialia 2015, 17, 160; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Z, Wang F, Roy S, Sen CK, Guan J, Biomacromolecules 2009, 10, 3306; [DOI] [PubMed] [Google Scholar]; c) Chen W, Shen X, Hu Y, Xu K, Ran Q, Yu Y, Dai L, Yuan Z, Huang L, Shen T, Biomaterials 2017, 114, 82; [DOI] [PubMed] [Google Scholar]; d) Wang L, Zhao X, Wei B.-y., Liu Y, Ma X.-y., Wang J, Cao P.-c., Zhang Y, Yan Y.-b., Lei W, Biochimie 2015, 108, 85. [DOI] [PubMed] [Google Scholar]
- [9].a) Krogsgaard M, Nue V, Birkedal H, Chemistry–A European Journal 2016, 22, 844; [DOI] [PubMed] [Google Scholar]; b) Faure E, Falentin-Daudré C, Jérôme C, Lyskawa J, Fournier D, Woisel P, Detrembleur C, Progress in polymer science 2013, 38, 236; [Google Scholar]; c) Ryu JH, Hong S, Lee H, Acta biomaterialia 2015, 27, 101. [DOI] [PubMed] [Google Scholar]
- [10].a) Kang SM, Hwang NS, Yeom J, Park SY, Messersmith PB, Choi IS, Langer R, Anderson DG, Lee H, Advanced Functional Materials 2012, 22, 2949; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lim C, Huang J, Kim S, Lee H, Zeng H, Hwang DS, Angewandte Chemie International Edition 2016, 55, 3342; [DOI] [PubMed] [Google Scholar]; c) Ryu JH, Messersmith PB, Lee H, ACS applied materials & interfaces 2018, 10, 7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Dillard CJ, German JB, Journal of the Science of Food and Agriculture 2000, 80, 1744; [Google Scholar]; b) King A, Young G, Journal of the American Dietetic Association 1999, 99, 213; [DOI] [PubMed] [Google Scholar]; c) Lee JY, Aguilar LE, Park CH, Kim CS, Polymers 2019, 11, 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) McKenzie JA, Maschhoff C, Liu X, Migotsky N, Silva MJ, Gardner MJ, J Orthop Res 2019, 37, 51; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dohle E, Fuchs S, Kolbe M, Hofmann A, Schmidt H, Kirkpatrick CJ, Tissue Eng Part A 2010, 16, 1235; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee C-S, Kim S, Fan J, Hwang HS, Aghaloo T, Lee M, Science Advances 2020, 6, eaaz7822; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lee S, Wang C, Pan HC, Shrestha S, Meyers C, Ding C, Shen J, Chen E, Lee M, Soo C, Plastic and reconstructive surgery 2017, 139, 1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Park E, Lee J, Huh KM, Lee SH, Lee H, Advanced healthcare materials 2019, 8, 1900275; [DOI] [PubMed] [Google Scholar]; b) Ryu JH, Lee Y, Kong WH, Kim TG, Park TG, Lee H, Biomacromolecules 2011, 12, 2653. [DOI] [PubMed] [Google Scholar]
- [14].Soliman GM, Zhang YL, Merle G, Cerruti M, Barralet J, European Journal of Pharmaceutics and Biopharmaceutics 2014, 88, 1026. [DOI] [PubMed] [Google Scholar]
- [15].Tomás H, Alves CS, Rodrigues J, Nanomedicine: Nanotechnology, Biology and Medicine 2018, 14, 2407. [DOI] [PubMed] [Google Scholar]
- [16].Ghadiri M, Hau H, Chrzanowski W, Agus H, Rohanizadeh R, RSC advances 2013, 3, 20193. [Google Scholar]
- [17].a) Lee BP, Dalsin JL, Messersmith PB, Biomacromolecules 2002, 3, 1038; [DOI] [PubMed] [Google Scholar]; b) Skelton S, Bostwick M, O’Connor K, Konst S, Casey S, Lee BP, Soft Matter 2013, 9, 3825. [Google Scholar]
- [18].Liu Y, Meng H, Konst S, Sarmiento R, Rajachar R, Lee BP, ACS applied materials & interfaces 2014, 6, 16982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kim TH, An DB, Oh SH, Kang MK, Song HH, Lee JH, Biomaterials 2015, 40, 51. [DOI] [PubMed] [Google Scholar]
- [20].a) Rohindra DR, Nand AV, Khurma JR, The South Pacific Journal of Natural and Applied Sciences 2004, 22, 32; [Google Scholar]; b) Im O, Li J, Wang M, Zhang LG, Keidar M, International journal of nanomedicine 2012, 7, 2087; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Anseth KS, Bowman CN, Brannon-Peppas L, Biomaterials 1996, 17, 1647. [DOI] [PubMed] [Google Scholar]
- [21].a) Salerno A, Di Maio E, Iannace S, Netti P, Journal of Porous Materials 2012, 19, 181; [Google Scholar]; b) Kim S, Cui Z-K, Koo B, Zheng J, Aghaloo T, Lee M, ACS applied materials & interfaces 2018, 10, 41138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Dong R, Zhao X, Guo B, Ma PX, ACS applied materials & interfaces 2016, 8, 17138; [DOI] [PubMed] [Google Scholar]; b) Tseng TC, Tao L, Hsieh FY, Wei Y, Chiu IM, Hsu S. h., Advanced Materials 2015, 27, 3518; [DOI] [PubMed] [Google Scholar]; c) Rodell CB, MacArthur JW Jr, Dorsey SM, Wade RJ, Wang LL, Woo YJ, Burdick JA, Advanced Functional Materials 2015, 25, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Liu X, Song L, Li L, Li S, Yao K, Journal of applied polymer science 2007, 103, 3521; [Google Scholar]; b) Kong M, Chen XG, Xing K, Park HJ, International journal of food microbiology 2010, 144, 51; [DOI] [PubMed] [Google Scholar]; c) Chung Y-C, Chen C-Y, Bioresource technology 2008, 99, 2806. [DOI] [PubMed] [Google Scholar]
- [24].a) Liang Y, Zhao X, Hu T, Chen B, Yin Z, Ma PX, Guo B, Small 2019, 15, 1900046; [DOI] [PubMed] [Google Scholar]; b) Tao Y, Ju E, Ren J, Qu X, Advanced Materials 2015, 27, 1097; [DOI] [PubMed] [Google Scholar]; c) Kim S, Fan J, Lee C-S, Lee M, ACS Applied Bio Materials 2020, 3, 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Zheng Y, Lu H, Jiang Z, Guan Y, Zou J, Wang X, Cheng R, Gao H, Journal of Materials Chemistry B 2017, 5, 6277; [DOI] [PubMed] [Google Scholar]; b) Seo EH, Lee CS, Na K, Advanced healthcare materials 2015, 4, 2822. [DOI] [PubMed] [Google Scholar]
- [26].a) Mody N, Parhami F, Sarafian TA, Demer LL, Free Radical Biology and Medicine 2001, 31, 509; [DOI] [PubMed] [Google Scholar]; b) Chaturvedi R, Singha PK, Dey S, PLoS One 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cui Z-K, Kim S, Baljon JJ, Doroudgar M, Lafleur M, Wu BM, Aghaloo T, Lee M, ACS nano 2017, 11, 8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang X, Fan J, Lee C-S, Kim S, Chen C, Lee M, ACS applied materials & interfaces 2020, 12, 16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Gerecht S, Townsend SA, Pressler H, Zhu H, Nijst CL, Bruggeman JP, Nichol JW, Langer R, Biomaterials 2007, 28, 4826; [DOI] [PubMed] [Google Scholar]; b) Wu D-Q, Sun Y-X, Xu X-D, Cheng S-X, Zhang X-Z, Zhuo R-X, Biomacromolecules 2008, 9, 1155. [DOI] [PubMed] [Google Scholar]
- [30].Arakawa C, Ng R, Tan S, Kim S, Wu B, Lee M, Journal of tissue engineering and regenerative medicine 2017, 11, 164. [DOI] [PubMed] [Google Scholar]
- [31].Amsden BG, Sukarto A, Knight DK, Shapka SN, Biomacromolecules 2007, 8, 3758. [DOI] [PubMed] [Google Scholar]
- [32].Zheng Z, Yin W, Zara JN, Li W, Kwak J, Mamidi R, Lee M, Siu RK, Ngo R, Wang J, Biomaterials 2010, 31, 9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.