

Abstract
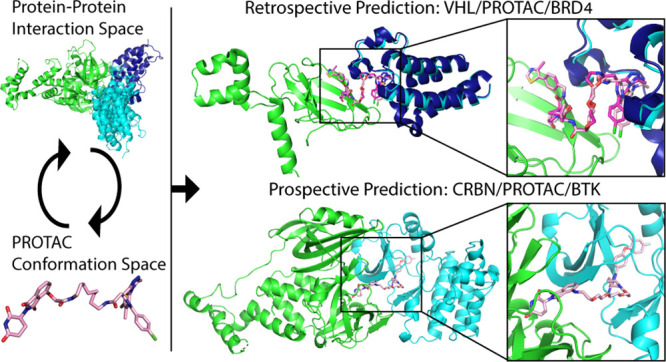
Proteolysis-targeting chimeras (PROTACs), which induce degradation by recruitment of an E3 ligase to a target protein, are gaining much interest as a new pharmacological modality. However, designing PROTACs is challenging. Formation of a ternary complex between the protein target, the PROTAC, and the recruited E3 ligase is considered paramount for successful degradation. A structural model of this ternary complex could in principle inform rational PROTAC design. Unfortunately, only a handful of structures are available for such complexes, necessitating tools for their modeling. We developed a combined protocol for the modeling of a ternary complex induced by a given PROTAC. Our protocol alternates between sampling of the protein–protein interaction space and the PROTAC molecule conformational space. Application of this protocol—PRosettaC—to a benchmark of known PROTAC ternary complexes results in near-native predictions, with often atomic accuracy prediction of the protein chains, as well as the PROTAC binding moieties. It allowed the modeling of a CRBN/BTK complex that recapitulated experimental results for a series of PROTACs. PRosettaC generated models may be used to design PROTACs for new targets, as well as improve PROTACs for existing targets, potentially cutting down time and synthesis efforts. To enable wide access to this protocol, we have made it available through a web server (https://prosettac.weizmann.ac.il/).
Introduction
Proteolysis-targeting chimeras (PROTACs) are modular small molecules that induce the degradation of a target protein.1−3 PROTACs can be conceptually divided into three parts: A target binding moiety, an E3 ubiquitin-ligase binding moiety, and a linker that connects the two. Through simultaneous binding of their target and E3 ligase, they induce the formation of a ternary complex that facilitates the ubiquitination of the target, which then leads to its proteasomal degradation.
PROTACs offer several advantages over traditional small molecule inhibitors. These include: a complete inhibition of function, which is not limited to enzymatic function or a specific site on the target; long duration-of-action that is proportional to the protein turnover rate; and enhanced selectivity.4−7 Finally, since a single PROTAC molecule can degrade multiple copies of the target, PROTACs can work at a substoichiometric concentration.8 These advantages propelled wide interest in their development as chemical tools and potential drugs.
Most reported PROTACs to date are based on a known and potent small-molecule target binding ligand, typically one for which a cocrystal structure was available to define a suitable exit vector for the installation of the linker. From the E3 binding side, the vast majority of PROTACs are limited to just two ligases: CRBN, which is targeted by various immunomodulatory drugs (IMiDs) such as thalidomide,9 pomalidomide10 or lenalidomide,11,12 and VHL, for which high-affinity small molecule ligands were developed.13,14
A key challenge in PROTAC design to date is the selection of the optimal linker to connect these two binding components. In most cases, linkers of various lengths are screened using synthetically accessible chemistry.15−19 In some cases, it was shown that arbitrarily long linkers can successfully degrade their targets.20 However, for other systems, a protein–protein interface between the target and the E3 ligase, including interactions with the linker, is important for cooperativity.4 Evidence is accumulating that a stable ternary complex involving protein–protein interactions is important for efficient degradation.4,21,22 Still, there is currently no way to predict or design linkers which would allow, or promote, ternary complex formation, a likely key parameter to a successful PROTAC. This gap necessitates significant synthetic effort.
Despite the increasing popularity of PROTACs and the many examples of experimentally validated, cell-active PROTACs, structural information on Target/E3/PROTAC ternary complexes is still very limited. The ability to model such ternary complexes can in theory inform the design of more potent PROTACs, significantly reduce the number of linker designs required to achieve efficient degradation, and rationalize the structure–activity relationship of already known series of PROTACs. However, such modeling efforts are still rare.
A pioneering work in modeling PROTAC mediated ternary complexes23 explored four methods to generate ensembles of ternary complexes and had some success in recapitulating the very few available crystal structures of such complexes. Recently, this work was extended by the addition of clustering to improve its performance.24 Very recently, a new Rosetta-based protocol was also reported that successfully rationalized PROTAC structure–activity relationships.25 Some system specific modeling efforts were also reported. For instance, docking followed by short molecular dynamics simulations was used to model p38 isoforms in complex with PROTACs and VHL and to predict residues that contribute to linker interactions and selectivity of the PROTACs.5,6 PROTAC ensembles in the absence of the protein complex were used to define distance distribution between the binding partners.26 HADDOCK27 was used to dock Sirt2 and CRBN;28 subsequent docking of a PROTAC into the complex recapitulated the known binding modes of the separate protein binding moieties. Notably, RosettaDock was used by Nowak et al.29 to inform the design of selective PROTACs. Molecular dynamics simulations were used to design a macrocyclic PROTAC,30 and two additional relevant computational approaches for linker design were recently introduced31,32 that can be used to design PROTAC linkers based on the protein–protein binary complex. While these may be suitable as a subprocedure within a general protocol, they cannot in and of themselves model ternary complexes.
Here, we present PRosettaC, a holistic protocol for modeling PROTAC mediated ternary complexes. It combines global docking with PatchDock33,34 under PROTAC derived distance constraints and local docking with RosettaDock,35 followed by modeling of the PROTAC into the ternary complex.36 The protocol was able to accurately recover published structures of ternary complexes to near atomic resolution and recapitulate experimental trends for two model systems. This general protocol should be useful in the design of new PROTACs for a wide variety of targets. To enable wide access to this protocol, we have made available both the code (https://github.com/LondonLab/PRosettaC) as well as a Web server for running PRosettaC (https://prosettac.weizmann.ac.il/).
Results
The PRosettaC Protocol
The protocol includes several consecutive steps, each intended to decrease the large conformational search space which includes the protein–protein interaction degrees of freedom, the PROTAC internal conformation and its pose relative to the protein–protein complex (Figure 1).
Figure 1.

An overview of the PRosettaC protocol. The protocol consists of the following consecutive steps: 1. Sampling of the distance between the two ligand anchor points. 2. Constrained global protein–protein docking with PatchDock. 3. Local docking with RosettaDock. 4. Generating constrained PROTAC conformations compatible with the local docking solutions. 5. Clustering of the top scoring results.
The input for the protocol is structures, or models, of the protein target and E3 ligase, each in complex with its own binding ligand, as well as a SMILES string, representing the entire PROTAC (Figure 2). Similar to Drummond and Williams,23 we use two anchor atoms in the two ligands which are defined as part of the input. In most cases, we arbitrarily chose the anchor atoms as the atoms through which the PROTAC’s linker is attached to the two ligands, with the exception of thalidomide, where the attachment atom is not uniquely defined within the SMILES string, and thus we chose a more central atom. Since the choice of anchor atoms is arbitrary, it should not affect the results greatly, as long as the atom is uniquely defined by SMILES.
Figure 2.
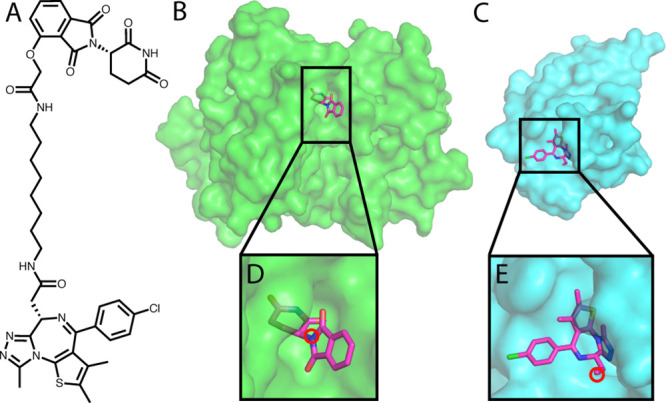
The input for the PRosettaC protocol. A. The chemical structure of a BRD4 PROTAC molecule including a CRBN ligand (thalidomide), a BRD4 ligand (JQ1), and an alkane linker. B. Structure of CRBN with thalidomide. C. Structure of BRD4 with JQ1. D. Thalidomide with the anchor atom marked. E. JQ1 with the anchor atom marked. All structures are based on PDB: 6BOY.
Step 1. Rough Sampling of the Distance between the Anchor Points
To estimate the distance between the anchor atoms, we do the following: for each value starting from 1 Å and in increments of 1 Å, we generate 200 random pairs of ligand positions with the anchor distance set to that value. Then, for each pair of ligand positions, we generate a random conformation of the entire PROTAC which incorporates the “fixed” ligand positions. Due to geometrical constraints of the molecule, some of the pairs fail to generate a valid PROTAC conformation while others succeed. We sum up the successfully generated PROTAC conformations for each distance bin (Figure 3A) and then pick distance constraints according to the distribution of successful conformations (see the Methods, Figure S1 and Table S1, for a detailed explanation of the choice of distance constraints).
Figure 3.
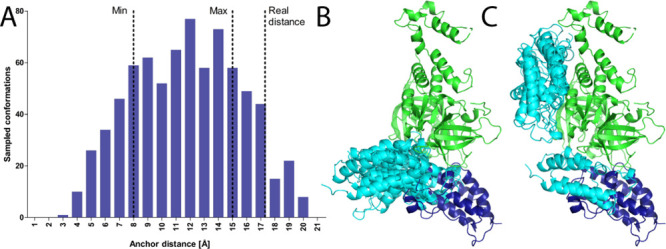
Sampling the PROTAC anchor points distance distribution successfully constrains global docking. A. For each distance (in 1 Å increments), we generate 200 random orientations of the target and E3 ligands and try to generate PROTAC conformations to match these random orientations. The histogram represents successfully generated conformations of an example PROTAC (PDB: 6BOY). On the basis of this histogram, the constraints chosen for the following step were 8—15 Å. Despite the crystallographic distance falling outside the constraints boundaries in this example, the protocol eventually is able to recapitulate the correct binding mode. See Figure S1 and Table S1 for more details on boundary selection and the crystallographic distances. B. Top 5 solutions of global protein–protein docking, constrained by the range calculated by the previous step of the protocol. C. Top 5 solutions of unconstrained protein–protein docking. Green, E3 ligase CRBN; dark blue, crystallographic BRD4 (6BOY); cyan, docked BRD4.
Step 2. Global Protein–Protein Docking
We use PatchDock, a very efficient global docking algorithm,33 to sample the protein–protein interaction space. The distance between the two anchors is forced to be within the constraints calculated in the previous step. This limits the docking search space considerably (Figure 3B,C).
Step 3: Local Docking Refinement
After using PatchDock to rapidly sample the protein–protein interaction space, we use RosettaDock37 local docking to produce 50 high-resolution models for each PatchDock global docking solution. The anchor distance constraints are not incorporated in this step. This allows the protocol to find a native solution, even if the crystallographic anchor distance is slightly outside the range of the constraints, like in the case of PDB: 6BOY (Figure 3A).
Step 4: Modeling the PROTACs into the Docking Solutions
We treat each of the local docking solutions as a hypothesis for the final ternary complex (Figure 4A). Thus, we constrain the ligands to be in the appropriate positions, derived from the protein–protein docking (Figure 4B), and construct 100 conformations of the full PROTAC that would fit these positions (Figure 4C). In other words, we create up to 100 random linker conformations which could connect the two ligands, given their position in the docking solution. For a significant portion of the local docking solutions, after 1,000 trials, no conformations could be found. Thus, no linker can attach the two ligands in these conformations (Table S1). For example, for 6BOY, out of ∼36,000 local docking solutions, only ∼28,000 got at least one generated conformation (Table S1).
Figure 4.
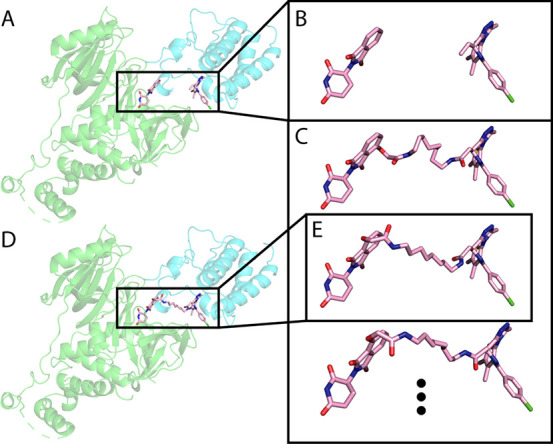
Generating PROTAC conformations in the context of the protein–protein interaction. Using RDKit, we generate up to 100 PROTAC conformations, to match the ligand orientation of each local docking solution. Then, we use Rosetta Packer to choose the best conformation to fit the protein–protein docking model. A. An example local docking solution for 6BOY. B. The ligand orientation, extracted from the local docking solution. C. Constrained conformations that bridge between the ligand orientation. D. Output model of Rosetta after choosing the best constrained conformation. E. The conformation chosen by Rosetta. Green, E3 ligase CRBN; cyan, predicted BRD4; light pink, predicted PROTAC conformation.
We use the Rosetta Packer38 to choose the optimal linker conformation of those generated (if one exists) that could connect the two ligands in the context of the docking solution (Figure 4E). This step outputs possible ternary complexes (Figure 4D) and prevents clashes between the PROTAC and the protein–protein complex. At this point, we filter complexes with high a Rosetta energy score and exclude them from further analysis.
Step 5: Clustering Top Scoring Complexes
After scoring the ternary complexes from the last step, we cluster them, under the assumption that near-native solutions will be sampled many times. From the 1,000 models with the lowest Rosetta score, we choose the 200 with the best interface score (energy of the complex less the energy of the separate components after side-chain minimization). We cluster these 200 complexes with a threshold of 4 Å RMSD for the moving chain using the DBSCAN39 clustering method and rank the clusters by the number of models in each cluster. Between clusters of the same size, the ranking is based on the average score of the final models. The 4 Å RMSD threshold was chosen based on a manual inspection of similarity with different thresholds (Figure S2) and is consistent with accepted criteria in protein–protein docking.40 The “near-native” cluster is defined as the top cluster which contains at least one near-native structure—a structure with a target Cα RMSD below 4 Å to the crystallographic conformation.
Optimization of Hyper Parameters
The protocol contains several parameters that require optimization. These include choosing whether to use the entire E3 ligase complex from the crystal structure or only the CRBN or VHL monomer; the RMSD threshold for clustering the global docking results; the number of top scoring models to consider for clustering; and the number of top interface scores within the top scoring models. Choosing the optimal parameters was not straightforward since there are only a handful of ternary structures available. We chose to optimize them on a set of five structures which were previously used for benchmarking23 (PDB IDs: 6BOY, 6BN7, 6BN8, 6BN9,295T354). We tested a matrix of different combinations for the five aforementioned parameters (Table S2). The choice of docking the E3 monomer or complex had little effect, likely since the distance constraints to the global docking already restrict the possible interaction interfaces, we hence chose to use the monomer to reduce runtime. Increasing the number of RosettaDock local docking models from 5 to 10 to 50 improved performance.
Accurate Recapitulation of Ternary Complexes Using Bound Structures
With the best combination of parameters, we were able to predict near-native models for four out of the five cases in the train set. In all four cases, the near-native models were found in the first or second ranked clusters (Table 1; see Figure 5 for the lowest RMSD solution and Figure S3 for representatives of the top three clusters for each of the four structures). Most of the native interactions were recapitulated in these models (see Table S3 for a comparison of the protein–protein interactions between the crystal and model complexes, calculated using the PIC server41).
Table 1. PRosettaC Performance against Known Ternary Complex Structures.
| train |
test |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PDB | 5T35 | 6BOY | 6BN7 | 6BN8 | 6BN9 | 6BNB | 6SIS | 6HAX | 6HAY | 6HR2 |
| ranka | 2/46 | 2/22 | 1/29 | NA/51c | 2/40 | 1/20 | 3/17 | 71/76d | 20/35d | 18/61 |
| RMSD < 10 Åb | 3.5% | 17.0% | 62.0% | 0.5% | 22.0% | 47.0% | 19.0% | 2.5% | 1.0% | 5% |
Rank of the near-native cluster/total number of clusters.
Percent of models that show less than 10 Å RMSD to the crystal structure (performance measure reported by Drummond and Williams23).
No near-native clusters were found for this structure.
These clusters were singletons, i.e., contained a single structure.
Figure 5.

PRosettaC can recapitulate known ternary complexes to atomic accuracy. Upper panels: near native predictions for four structures of the training set: 5T35, 6BOY, 6BN7 and 6BN9, with reported Cα RMSD of the moving chain (BRD4), protein interface RMSD which is calculated on all backbone atoms of residues which are within 8 Å of any residue on the other protein, the RMSD over the small molecule ligands (excluding the linker), and the RMSD over the entire PROTAC. Lower panels: zoom in on the ligand binding predictions. In all four, PRosettaC achieved predictions of atomic accuracy for the target ligands. Green, E3 ligase CRBN; dark blue, X-ray BRD4; cyan, docked BRD4; magenta, X-ray PROTAC conformation; light pink, predicted PROTAC conformation. *Note that the PROTAC molecule is not resolved in 6BN9 and was modeled by PRosettaC. The RMSD values for the ligands are based on alignment to 6BN7. Since the ligand–protein interactions are highly conserved, the ligand RMSD, based on the alignment, should still be representative.
Comparison of PRosettaC to Previously Reported Methods
Drummond and Williams23 reported the results of their methods against five structures. To be able to compare our results, we first had to realign the models, since they chose the degradation target as the static chain, while we chose the E3 ligase, due to the large size of CRBN compared to BRD4. The results (Table S4) are therefore based on the E3 Cα RMSD. As a metric for success, they report the percent of models with RMSD to the X-ray structure below 10 Å. In three cases (6BOY, 6BN7, and 6BN9), PRosettaC outperformed their method. In one case (5T35), their method did better. Both methods performed poorly on 6BN8. We should note that while Drummond and Williams were not able to generate successful models for 6BOY, our method generated 17% such models, ranking the native cluster in second place. Also, even though we could not match the overall fraction of near native solutions for 5T35, we did rank the cluster which includes the native structure in second place. To our understanding, Drummond and Williams do not report ranking.
Application to a New Set of Test Structures
After optimizing the hyper parameters on the training set (Table S2), we tested PRosettaC on five additional structures which were not modeled in previous work (PDBs: 6BNB, 6SIS, 6HAX, 6HAY, 6HR2) nor optimized against. The most successful out of these was 6BNB, a complex of BRD4 with CRBN (Figure 6A), in which CRBN shows an “open” conformation, substantially different from the closed conformation seen in most structures. The top ranking cluster, which included 67.7% of the final models, was the near-native cluster. The PROTAC molecule (dBET57) is not modeled in the original structure, likely due to the low resolution of the structure (6.3 Å). We placed the ligands by aligning structures with the individual ligands onto 6BNB (using domains from 6BOY as templates). On the basis of our prediction, we were able to easily model the PROTAC (Figure 6A). A second successful prediction was for a complex of VHL with BRD4 (PDB: 6SIS), bridged by a nonconventional macrocyclic PROTAC.30 The near native cluster was ranked third, and the ligand binding moieties were recapitulated with subangstrom accuracy (Figure 6B). For the rest of the test cases, all complexes of VHL with SMARCA2/4,42 while near-native clusters were found, they were not ranked highly (Table 1).
Figure 6.
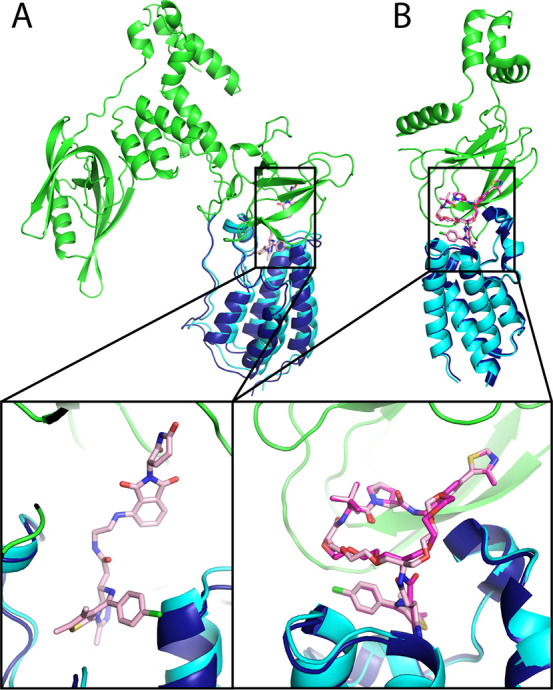
Near native predictions for two nontraditional PROTAC ternary complexes. A. Near native prediction of PDB: 6BNB of a BRD4 PROTAC with an open conformation of CRBN. The target Cα RMSD is 2.35 Å. The native cluster was ranked 1, containing 67.7% of the final models. The PROTAC molecule is not modeled in the original structure, but could be easily modeled in using ProsettaC. B. Near native prediction of 6SIS, a complex of BRD4 and CRBN with a macrocyclic PROTAC. The target Cα RMSD is 1.3 Å, with a target ligand RMSD of 0.5 Å and a full PROTAC RMSD of 0.72 Å. The native cluster was ranked 3. Green, E3 ligase; dark blue, crystallographic BRD4; cyan, predicted BRD4; magenta, X-ray PROTAC conformation; light pink, predicted PROTAC conformation.
PRosettaC Models Can Help to Explain Structure–Activity Relationships
The BRD4[BD2] triple mutant K378Q, E383V, and A384K mimics the first bromodomain of BRD2. MZ1, the PROTAC from 5T35, selectively degrades BRD4[BD2] over BRD2[BD1]. It was predicted and shown experimentally that this triple mutant reduces ternary complex formation with VHL4 and was previously used as a negative test case for modeling.23 We introduced these three mutations in silico (based on 5T35) and reran the protocol. While for the wild-type BRD4, the native cluster was ranked second out of 46, containing 9% of the final models, none of 34 clusters proposed for the triple mutant were near-native. This suggests that indeed these three positions were crucial for the formation of the productive ternary complex.
To test our method’s ability to predict unknown ternary complexes, we next turned to a systematic study of PROTACs against BTK.20 The authors tested 11 different PROTACs with increasing linker lengths, between 5 and 21 atoms, for their ability to degrade BTK, as well as for their ability to form a ternary complex (using a TR-FRET experiment). PROTACs 1–4 cannot degrade BTK efficiently nor lead to ternary complex formation at low concentrations. PROTACs 6–11 were all efficient in both degrading BTK and forming the ternary complex. The authors used computational modeling to recapitulate their experimental results. Using a proprietary pipeline to create models of the ternary complex they were able to produce nonclashing models for PROTACs with longer linkers (PROTACs 7–11) but not for PROTACs with shorter linkers (1–4).
We applied PRosettaC to this model system. We used a structure of CRBN bound to lenalidomide (PDB: 4TZ4) after switching the lenalidomide to thalidomide and a structure of mouse BTK (98% identical to human BTK) bound to the inhibitor on which the PROTACs were based (PDB: 6MNY). Our results (Table S5) correlated very well with the experimental data. The first distinction between the active and nonactive PROTACs was the number of generated models. For PROTACs 1–3, our protocol was not able to generate any ternary structure. For PROTACs 4 and 5, it generated less than 250 models and, for 6–11, more than 1,000 models. While the number of models itself was not indicative of a successful prediction in our training and test sets, we noticed that the top ranked clusters of PROTACs 5–11 were very similar, but different for that of PROTAC 4. This suggests that the complex represented by the top clusters of PROTACs 5–11 is a low energy solution that may lead to efficient degradation. In order to find a consensus structure within these top clusters, we pooled together and clustered the top cluster from the separate runs of PROTACs 5–11 with a lower RMSD threshold of 1 Å. The top cluster was a tight ensemble of 213 similar structures, still containing representatives of PROTACs 5–11. To further fine-tune our prediction, we clustered this top overall cluster with an even lower RMSD threshold of 0.5 Å. While the first subcluster did not contain representatives of 5–11, the second one did, and included 33 structures, forming a very tight ensemble (Figure 7A).
Figure 7.
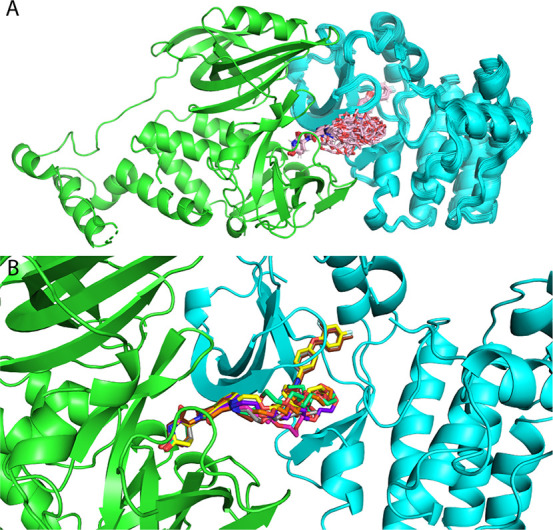
High confidence prediction of a BTK–CRBN ternary complex. Modeling of a BTK–CRBN ternary complex, with a series of 11 PROTACs, recapitulates which PROTACs are active and which are not and suggests a high-confidence model for the interaction. A. A cluster (with 0.5 Å threshold) of 33 models from various PRosettaC runs contains representatives from active PROTACs 5–11. B. Representative of PROTACs 5–11: 5, brown; 6, gray; 7, magenta; 8, yellow; 9, orange; 10, purple; 11, green.
Discussion
The field of PROTAC mediated targeted degradation is gaining tremendous momentum, as highlighted by the recent positive results from the first PROTAC (ARV-110) to enter clinical trials.43 Structure-based design is a key paradigm in drug discovery, and PROTAC design can clearly benefit from structural insights, as evidenced by the few cases in which ternary complex structures were actually determined. Gadd et al.4 used the structure of the BRD4/VHL/MZ1 ternary complex to design a new PROTAC (AT1) in which the linker uses a different exit vector from the VHL ligand.4 Testa et al.30 used the same structure in order to create a macrocyclic PROTAC with improved selectivity. Farnaby et al.42 used the complex of the SMARCA2/VHL/PROTAC-1 complex to rigidify the PROTAC linker and introduce a new π-stacking interaction, resulting in a significantly improved molecule. In the absence of ternary complex experimental structures, PROTAC designers can turn to modeling in order to generate design hypotheses.
Modeling of PROTAC mediated ternary complexes, however, is a challenging task for multiple reasons. First is the very small number of available structures of such interactions, which complicates the benchmarking of new methods. Second, is the fact that the E3 ligase and protein target are not cognate interaction partners, thus any binding interface that may be stabilized by the PROTAC is likely transient with weak affinity.44−46 Such low affinity PPIs are known to be challenging for docking methods.47 Another difficulty is the need to simultaneously model protein–protein interactions and protein–ligand interactions, the latter often with a significant number of rotatable bonds.
Here, we presented a general approach which overcomes some of the challenges by using the PROTAC’s distance distribution to restrict the protein–protein docking search space and then uses the docking solution to restrict the ligand conformational search space. PRosettaC was very successful in recapitulating the ternary complexes in 6 out of 10 cases. Moreover, the near-native models were ranked in the top three solutions. It was further able to recapitulate trends for a series of BTK targeting PROTACs and propose a high-confidence model for the BTK/CRBN ternary complex, by combining the experimental results from various PROTACs.20
On the basis of these successful results, we suspect that PRosettaC would be a useful tool to enable structure-based PROTAC design. The fact that in most cases the near-native complex was ranked in the top three clusters suggests that in the future a small number of PROTACs that are designed based on a limited number of the top clusters could be used to validate which model captures the productive complex that leads to degradation.
One design prediction already emerged based on the modeling of the BRD4/CRBN/dBET23 complex (PDB: 6BN7), for which PRosettaC identified the native cluster as the top ranking solution, including 65.5% of the final structures. In this case, the protein–protein interaction was accurately recapitulated, as were the thalidomide and BRD4 ligand moieties. However, a flip of the thalidomide phthalimide, which scored better, forced the linker to exit out of the opposite atom compared to the one observed in the crystal structure (Figure 5). Testa et al. previously used the structure of BRD4/VHL/MZ1 elegantly to create a macrocyclic PROTAC.30 The 6BN7 modeling result suggests a similar strategy would work for the BRD4/CRBN complex as well (Figure S4).
Despite its success, our protocol still suffers from some challenges. In the set of the original five test structures, PRosettaC was least successful for 6BN8, a complex of BRD4/CRBN and the PROTAC dBET55 for which the native solution did not belong to any of the clusters. dBET55 contains a very long PEG9 linker. The length and flexibility of the linker presents a problem for the anchor distance sampling step, and later to global docking, which runs with very loose constraints. Indeed, global docking produced an exceptionally large number of structures, and most near-native solutions were not ranked among the top 1,000 (Figure S5). Since such long and flexible linkers are rare for PROTACs, we do not consider this a significant failure. For another three cases involving complexes of VHL with SMARCA2/4, PRosettaC was able to sample near-native solutions but did not rank them as one of the top clusters. Analysis of the global docking results (Figure S5) suggests that not sampling a near-native conformation for these complexes in the global docking step may be the reason for these failures. Future developments may investigate optimization or alternatives to the global docking step. One possibility is that the crystal structure represents a single possible conformation for the interaction, stabilized by crystal contacts (Figure S6), while other conformations proposed by our protocol may be sampled in the solution state. Indeed, Nowak et al. used such a “non-native” docking prediction to guide the design of a new and more selective PROTAC,29 strongly suggesting that such additional conformations are sampled in solution and can be accessed by a suitable PROTAC. Hence, even if the top predicted clusters do not correspond to the crystal structure, they may still represent attractive opportunities for PROTAC design.
Another important caveat of this (and previous23) benchmark is the use of the bound structures. Ideally, the protocol should be able to reproduce the ternary complex starting from the structures of the unbound monomers. Currently, PRosettaC fails to rank near-native clusters when starting from unbound structures. Future improvements to the protocol may address some of the aforementioned challenges. Introduction of backbone flexibility may significantly improve complex prediction based on unbound structures as was reported for various docking approaches.48−56 Incorporation of additional biological constraints such as the location of the target lysine for ubiquitination, or known mutations that enhance/decrease complex formation, may further restrict the conformational space.
Despite these caveats, we present an end-to-end approach to model target/E3-ligase/PROTAC ternary complexes. Our method leverages the advantages of the highly constrained search space of this unique problem. We demonstrated how the use of various experimental results can lead to high confidence in our prediction. While still lacking prospective experimental proof, this work may contribute to future in-silico design of new PROTACs and may significantly reduce the time and resources that are currently required for the design of PROTACs against a new target.
Methods
Preparation of Input Files
The PDB files were downloaded from the PDB (https://www.rcsb.org/). The chains of the E3 ligase and the target protein were cleaned from any nonprotein atoms. The ligands were extracted manually from the PROTAC in the PDB. In cases where the PROTAC was not modeled into the structure (6BN8, 6BN9, 6BNB), we aligned another structure(s) with the same domains and ligands and copied the ligand coordinates. We added hydrogens to the ligands using OpenBabel (http://openbabel.org/wiki/Main_Page). We added the protonated ligands to the structures, which then underwent side-chain repacking and minimization.36 Next, in order to keep the original coordinates and atom numbers, we replaced the ligand with their preminimized version. The linker SMILES representation was taken from the PDB. In cases where it was not modeled in the PDB, it was taken from the paper where they were reported. Input files for the bound examples (Table 1) are available at https://zenodo.org/record/3967246#.XyLML_gzZQJ.
Estimating Linker Distance Constraints
We sample the length of the PROTAC in the following fashion. For each bin starting with 1 Å, with increments of 1 Å, we made 200 random trials to produce a PROTAC conformation. In each trial, a random orientation of the two ligands is produced, while keeping the anchors in the distance set by the bin “b.” This is done by generating a random conformation of the two ligands and transforming them such that the anchors are at (0,0,0), (b,0,0), followed by a random rotation of both of them around the anchors. Then, using RDKit, we attempt to create a valid PROTAC conformation based on the randomized ligand orientation. Once an orientation is generated, we remeasure the distance between the two anchors, to make sure it stays closer than 0.5 Å from the distance set by the bin. We stopped this procedure when at least 10 bins had been sampled, and we reached a bin for which no conformations were generated. We sum up the number of successful conformations generated in each bin, to generate a histogram. From the histogram, we take the highest value, multiply it by 0.75, and set it as a threshold. Then, we take the minimum and maximum bin values that achieve that threshold: minA and maxA. Due to some cases where only a few bins were populated, we also take the average bin value of all generated conformations plus and minus 2 Å as minB and maxB. The final distance constraints for the two anchors are min(minA, minB) and max(maxA, maxB).
Constrained Global Protein–Protein Docking
Global docking is performed with PatchDock,33 using the downloadable linux version of the program. The constraints from the previous step are incorporated in the parameters file. The output includes solutions of the protein–protein docking problem, clustered by a cutoff of 2 Å. Up to 1,000 global docking solutions are considered for the next step.
Local Docking of Selected Global Docking Solutions
RosettaDock35 local docking is used to create up to 50 local docking results for each global docking solution produced by PatchDock, treating the ligands as extra residues with a fixed conformation.
Constrained PROTAC Conformation Generation in the Context of the Docking Solution
We use RDKit to produce up to 100 conformations of the PROTAC, with the constraints of the two ligands to fit the local docking solution. Therefore, only the linker’s conformation is being sampled, in regard to a fixed conformation and position of the two ligands. Due to the constraints being atom-distance-based, the conformation that is generated is not aligned with the position of the ligands, and another alignment step is necessary. After the alignment, a threshold of 0.5 Å RMSD between each ligand independently and its X-ray conformation is used to ensure that the conformation of the ligands is really the same as their native conformation. This is necessary because RDKit generation of constrained conformations, which is based on atom distances, can result in conformations not fully within the desired constraints. If after 1,000 trials no valid conformation has been generated, the local docking pose is discarded. For the unbound and BTK-CRBN runs, this number was reduced to 100 for efficiency. For 6SIS, 6HAX, 6HAY, and 6HR2, an extra atom was added to the E3 binding ligand, in order to uniquely define the position of the exit vector, making sure the constrained conformation attaches the linker to the right atom.
Modeling the PROTAC within the Ternary Complex
We use Rosetta’s repack36 protocol to choose the best PROTAC conformation from the set generated in the previous step. In the repack protocol, the side-chains of the residues are allowed to switch rotamers. We supplied the constrained conformations which we generated using RDKit as rotamers for the PROTAC, using the first one as the initial rotamer. Since the Rosetta Packer aligns conformations based on a single atom (called the neighboring atom), we added three virtual atoms with fixed coordinates to the set of generated residue conformations. The center virtual atom has the coordinates of the center of mass of the two ligands, while the two other virtual atoms are 1 Å away from it, on the x and y axes. The two other atoms are connected to the center virtual atom, which is defined as the neighboring atom, thus defining a fixed 3D alignment, which does not translate the sampled conformations and avoids lever-arm effects. Repacking was applied on the PROTAC, as well as any residue that is within 10 Å distance from it. After repacking, a final step of side-chain minimization for the entire structure, excluding the PROTAC, was applied. Models with a final score ≥0 are excluded from further analysis.
Clustering Top Scoring Complexes
We used the DBSCAN39 clustering method and ranked the clusters by the number of models in each cluster, assuming the highly populated clusters would represent the best solutions. The DBSCAN method can work either with coordinates of points in n dimensions or with a distance matrix of precomputed distances between each pair of points. Since we used Cα RMSD values of the moving chain (defined always as the target protein), we precalculated all the pairwise RMSDs between the final solutions and fed it to DBSCAN as the distance matrix. We used 4 Å RMSD as the clustering threshold. We clustered the top 200 solutions based on the interface score reported by Rosetta (energy of the complex less the energy of the separate components after side-chain minimization), out of the top 1,000 scoring final solutions, created by Rosetta’s repacking protocol. We ranked the clusters according to the number of structures in each cluster. Between clusters of the same size, the ranking was based on the average score of the final models. We define a native cluster as having at least one member whose Cα RMSD from the native conformation is lower than 4 Å. The reported rank is the top ranked cluster among the native clusters.
Software and Files
https://zenodo.org/record/3967246#.XyLML_gzZQJ includes the input files for the bound results, the final clusters of the train and test sets (Table 1), the unsuccessful unbound results for the train set, the CRBN-BTK final models, and the best RMSD models after renumbering the residues, for which a detailed analysis of the protein–protein interaction was performed (Table S3). The Rosetta version used in this study is 2019.27.post.dev+132.master.966c9eb966c9eb6b3ab993de7aa3af5988125b7c2e464af git@github.com: RosettaCommons/main.git 2019–07–18T11:43:25. Rosetta was applied using RosettaScripts.57 The Python version used in the study is 2.7.14. However, for the Web server and git repository, it was implemented in Python version 3.7.6. The RDKit version used in the study is 2018–09–03. However, for the Web server, the version was updated to 2020–03–03. The PatchDock executable was downloaded from https://bioinfo3d.cs.tau.ac.il/PatchDock.
Acknowledgments
We would like to thank the Rosetta Commons, and in particular Drs. Rocco Moretti and Steven Lewis, for much help with algorithmic details. N.L. is the incumbent of the Alan and Laraine Fischer Career Development Chair. N.L. would like to acknowledge funding from the Israel Science Foundation (grant no. 2462/19), The Israel Cancer Research Fund, the Israeli Ministry of Science and Technology (grant no. 3-14763), the Moross Integrated Cancer Center, and the Dr. Barry Sherman institute for Medicinal Chemistry. N.L. is also supported by the Estate of Emile Mimran, Rising Tide Foundation, Honey and Dr. Barry Sherman Lab, and Nelson P. Sirotsky. D.Z. would like to acknowledge the Pearlman Grant for student-initiated research in chemistry from the Faculty of Chemistry, the Weizmann Institute.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00589.
Supplementary figures and tables (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Toure M.; Crews C. M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem., Int. Ed. 2016, 55, 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Neklesa T. K.; Winkler J. D.; Crews C. M. Targeted Protein Degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. Proteolysis TArgeting Chimeras (PROTACs)—past, Present and Future. Drug Discovery Today: Technol. 2019, 31, 15. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87. e5 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-T.; Dobrovolsky D.; Paulk J.; Yang G.; Weisberg E. L.; Doctor Z. M.; Buckley D. L.; Cho J.-H.; Ko E.; Jang J.; Shi K.; Choi H. G.; Griffin J. D.; Li Y.; Treon S. P.; Fischer E. S.; Bradner J. E.; Tan L.; Gray N. S. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell Chem. Biol. 2018, 25, 88–99. e6 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. S.; Böhm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thomä N. H. Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C.; Hu Y.; Zhou B.; Fernandez-Salas E.; Yang C.-Y.; Liu L.; McEachern D.; Przybranowski S.; Wang M.; Stuckey J.; Meagher J.; Bai L.; Chen Z.; Lin M.; Yang J.; Ziazadeh D. N.; Xu F.; Hu J.; Xiang W.; Huang L.; Li S.; Wen B.; Sun D.; Wang S. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61, 6685–6704. Others. 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain P. P.; Lopez-Girona A.; Miller K.; Carmel G.; Pagarigan B.; Chie-Leon B.; Rychak E.; Corral L. G.; Ren Y. J.; Wang M.; Riley M.; Delker S. L.; Ito T.; Ando H.; Mori T.; Hirano Y.; Handa H.; Hakoshima T.; Daniel T. O.; Cathers B. E. Structure of the Human Cereblon–DDB1–lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- Van Molle I.; Thomann A.; Buckley D. L.; So E. C.; Lang S.; Crews C. M.; Ciulli A. Dissecting Fragment-Based Lead Discovery at the von Hippel-Lindau Protein:Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem. Biol. 2012, 19, 1300–1312. 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohbeck J.; Miller A. K. Practical Synthesis of a Phthalimide-Based Cereblon Ligand to Enable PROTAC Development. Bioorg. Med. Chem. Lett. 2016, 26, 5260–5262. 10.1016/j.bmcl.2016.09.048. [DOI] [PubMed] [Google Scholar]
- Krajcovicova S.; Jorda R.; Hendrychova D.; Krystof V.; Soural M. Solid-Phase Synthesis for Thalidomide-Based Proteolysis-Targeting Chimeras (PROTAC). Chem. Commun. 2019, 55, 929–932. 10.1039/C8CC08716D. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus K.; Wehenkel M.; Choi E.-Y.; Lee H.; Swanson H.; Kim K.-B. Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. ChemMedChem 2010, 5, 979–985. 10.1002/cmdc.201000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M.; Xiong Y.; Safaee N.; Nowak R. P.; Donovan K. A.; Yuan C. J.; Nabet B.; Gero T. W.; Feru F.; Li L.; Gondi S.; Ombelets L. J.; Quan C.; Jänne P. A.; Kostic M.; Scott D. A.; Westover K. D.; Fischer E. S.; Gray N. S. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–31. e6 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7285–E7292. 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S. J.; Ciulli A. Molecular Recognition of Ternary Complexes: A New Dimension in the Structure-Guided Design of Chemical Degraders. Essays Biochem. 2017, 61, 505–516. 10.1042/EBC20170041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M. J.; Winkler S.; Hughes S. J.; Whitworth C.; Galant M.; Farnaby W.; Rumpel K.; Ciulli A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond M. L.; Williams C. I. In Silico Modeling of PROTAC-Mediated Ternary Complexes: Validation and Application. J. Chem. Inf. Model. 2019, 59, 1634–1644. 10.1021/acs.jcim.8b00872. [DOI] [PubMed] [Google Scholar]
- Drummond M. L.; Henry A.; Li H.; Williams C. I.. Improved Accuracy for Modeling PROTAC-Mediated Ternary Complex Formation and Targeted Protein Degradation via New In Silico Methodologies, J. Chem. Inf. Model. 2020, 10.1021/acs.jcim.0c00897. [DOI] [PubMed] [Google Scholar]
- Bai N.; Kirubakaran P.; Karanicolas J.. Rationalizing PROTAC-Mediated Ternary Complex Formation Using Rosetta, bioRxiv 2020, 10.1101/2020.05.27.119347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H.; Wright D. J.; Johnson C. N.; Heightman T. D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert G. C. P.; Rodrigues J. P. G. L. M.; Trellet M.; Schmitz C.; Kastritis P. L.; Karaca E.; Melquiond A. S. J.; van Dijk M.; de Vries S. J.; Bonvin A. M. J. J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Schiedel M.; Herp D.; Hammelmann S.; Swyter S.; Lehotzky A.; Robaa D.; Oláh J.; Ovádi J.; Sippl W.; Jung M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Nowak R. P.; DeAngelo S. L.; Buckley D.; He Z.; Donovan K. A.; An J.; Safaee N.; Jedrychowski M. P.; Ponthier C. M.; Ishoey M.; Zhang T.; Mancias J. D.; Gray N. S.; Bradner J. E.; Fischer E. S. Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat. Chem. Biol. 2018, 14, 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa A.; Hughes S. J.; Lucas X.; Wright J. E.; Ciulli A. Structure-Based Design of a Macrocyclic PROTAC. Angew. Chem., Int. Ed. 2020, 59, 1727–1734. 10.1002/anie.201914396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Benito L.; Henry A.; Matsoukas M.-T.; Lopez L.; Pulido D.; Royo M.; Cordomí A.; Tresadern G.; Pardo L. The Size Matters? A Computational Tool to Design Bivalent Ligands. Bioinformatics 2018, 34, 3857–3863. 10.1093/bioinformatics/bty422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrie F.; Bradley A. R.; van der Schaar M.; Deane C. M. Deep Generative Models for 3D Linker Design. J. Chem. Inf. Model. 2020, 60, 1983. 10.1021/acs.jcim.9b01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhovny D.; Nussinov R.; Wolfson H. J.. Efficient Unbound Docking of Rigid Molecules. In Algorithms in Bioinformatics; Springer: Berlin, 2002; pp 185–200. [Google Scholar]
- Schneidman-Duhovny D.; Inbar Y.; Nussinov R.; Wolfson H. J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. J.; Moughon S.; Wang C.; Schueler-Furman O.; Kuhlman B.; Rohl C. A.; Baker D. Protein–Protein Docking with Simultaneous Optimization of Rigid-Body Displacement and Side-Chain Conformations. J. Mol. Biol. 2003, 331, 281–299. 10.1016/S0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A.; Tyka M.; Lewis S. M.; Lange O. F.; Thompson J.; Jacak R.; Kaufman K.; Renfrew P. D.; Smith C. A.; Sheffler W.; Davis I. W.; Cooper S.; Treuille A.; Mandell D. J.; Richter F.; Ban Y.-E. A.; Fleishman S. J.; Corn J. E.; Kim D. E.; Lyskov S.; Berrondo M.; Mentzer S.; Popović Z.; Havranek J. J.; Karanicolas J.; Das R.; Meiler J.; Kortemme T.; Gray J. J.; Kuhlman B.; Baker D.; Bradley P. ROSETTA3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S.; Gray J. J. The RosettaDock Server for Local Protein–protein Docking. Nucleic Acids Res. 2008, 36, W233–W238. 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B.; Baker D. Native Protein Sequences Are close to Optimal for Their Structures. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10383–10388. 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M.; Kriegel H.-P.; Sander J.; Xu X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Kdd 1996, 96, 226–231. [Google Scholar]
- Lensink M. F.; Méndez R.; Wodak S. J. Docking and Scoring Protein Complexes: CAPRI 3rd ed. Proteins 2007, 69, 704–718. [DOI] [PubMed] [Google Scholar]
- Tina K. G.; Bhadra R.; Srinivasan N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007, 35, W473–W476. 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnaby W.; Koegl M.; Roy M. J.; Whitworth C.; Diers E.; Trainor N.; Zollman D.; Steurer S.; Karolyi-Oezguer J.; Riedmueller C.; Gmaschitz T.; Wachter J.; Dank C.; Galant M.; Sharps B.; Rumpel K.; Traxler E.; Gerstberger T.; Schnitzer R.; Petermann O.; Greb P.; Weinstabl H.; Bader G.; Zoephel A.; Weiss-Puxbaum A.; Ehrenhöfer-Wölfer K.; Wöhrle S.; Boehmelt G.; Rinnenthal J.; Arnhof H.; Wiechens N.; Wu M.-Y.; Owen-Hughes T.; Ettmayer P.; Pearson M.; McConnell D. B.; Ciulli A. BAF Complex Vulnerabilities in Cancer Demonstrated via Structure-Based PROTAC Design. Nat. Chem. Biol. 2019, 15, 672–680. 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T.; Snyder L. B.; Willard R. R.; Vitale N.; Pizzano J.; Gordon D. A.; Bookbinder M.; Macaluso J.; Dong H.; Ferraro C.; Wang G.; Wang J.; Crews C. M.; Houston J.; Crew A. P.; Taylor I. ARV-110: An Oral Androgen Receptor PROTAC Degrader for Prostate Cancer. J. Clin. Oncol. 2019, 37, 259–259. 10.1200/JCO.2019.37.7_suppl.259. [DOI] [Google Scholar]
- Daniels D. L.; Riching K. M.; Urh M. Monitoring and Deciphering Protein Degradation Pathways inside Cells. Drug Discovery Today: Technol. 2019, 31, 61–68. 10.1016/j.ddtec.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Crews C. M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. 10.1016/j.chembiol.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons C.; Grosdidier S.; Solernou A.; Pérez-Cano L.; Fernández-Recio J. Present and Future Challenges and Limitations in Protein-Protein Docking. Proteins: Struct., Funct., Genet. 2010, 78, 95–108. 10.1002/prot.22564. [DOI] [PubMed] [Google Scholar]
- Marze N. A.; Roy Burman S. S.; Sheffler W.; Gray J. J. Efficient Flexible Backbone Protein–protein Docking for Challenging Targets. Bioinformatics 2018, 34, 3461–3469. 10.1093/bioinformatics/bty355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda D.; Gray J. J. Pushing the Backbone in Protein-Protein Docking. Structure 2016, 24, 1821–1829. 10.1016/j.str.2016.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach E.; Nussinov R.; Wolfson H. J. FiberDock: Flexible Induced-Fit Backbone Refinement in Molecular Docking. Proteins: Struct., Funct., Genet. 2010, 78, 1503–1519. 10.1002/prot.22668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Bradley P.; Baker D. Protein–Protein Docking with Backbone Flexibility. J. Mol. Biol. 2007, 373, 503–519. 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- Zacharias M. Accounting for Conformational Changes during Protein–protein Docking. Curr. Opin. Struct. Biol. 2010, 20, 180–186. 10.1016/j.sbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Ehmann U.; Zacharias M. Monte Carlo Replica-Exchange Based Ensemble Docking of Protein Conformations. Proteins: Struct., Funct., Genet. 2017, 85, 924–937. 10.1002/prot.25262. [DOI] [PubMed] [Google Scholar]
- Kurkcuoglu Z.; Bonvin A. M. Pre-and Post-Docking Sampling of Conformational Changes Using ClustENM and HADDOCK for Protein-Protein and Protein-DNA Systems. Proteins: Struct., Funct., Genet. 2020, 88, 292–306. 10.1002/prot.25802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvin A. M. Flexible Protein–protein Docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. 10.1016/j.sbi.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Torchala M.; Moal I. H.; Chaleil R. A. G.; Fernandez-Recio J.; Bates P. A. SwarmDock: A Server for Flexible Protein–protein Docking. Bioinformatics 2013, 29, 807–809. 10.1093/bioinformatics/btt038. [DOI] [PubMed] [Google Scholar]
- Fleishman S. J.; Leaver-Fay A.; Corn J. E.; Strauch E.-M.; Khare S. D.; Koga N.; Ashworth J.; Murphy P.; Richter F.; Lemmon G.; Meiler J.; Baker D. RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLoS One 2011, 6, e20161 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.