

Murine cytomegalovirus (MCMV) is widely used as a model of human CMV (HCMV) infection. However, this model relies on strains of MCMV that have been serially passaged in the laboratory for over four decades. These laboratory strains have been cloned as bacterial artificial chromosomes (BACs), which permits rapid and precise manipulation. Low-passage strains of MCMV add to the utility of the mouse model of HCMV infection but do not exist as cloned BACs. This study describes the first such low-passage MCMV BAC. This BAC-derived G4 was initially attenuated in vivo, with subsequent full genomic sequencing revealing a novel spliced transcript required for salivary gland tropism. These data suggest that MCMV, like HCMV, undergoes tissue culture adaptation that can limit in vivo growth and supports the use of BAC clones as a way of standardizing viral strains and minimizing interlaboratory strain variation.
KEYWORDS: BAC, G4, MCMV, sgg1, sgg1.1, tissue tropism
ABSTRACT
The cloning of herpesviruses as bacterial artificial chromosomes (BACs) has revolutionized the study of herpesvirus biology, allowing rapid and precise manipulation of viral genomes. Several clinical strains of human cytomegalovirus (HCMV) have been cloned as BACs; however, no low-passage strains of murine CMV (MCMV), which provide a model mimicking these isolates, have been cloned. Here, the low-passage G4 strain of was BAC cloned. G4 carries an m157 gene that does not ligate the natural killer (NK) cell-activating receptor, Ly49H, meaning that unlike laboratory strains of MCMV, this virus replicates well in C57BL/6 mice. This BAC clone exhibited normal replication during acute infection in the spleen and liver but was attenuated for salivary gland tropism. Next-generation sequencing revealed a C-to-A mutation at nucleotide position 188422, located in the 3′ untranslated region of sgg1, a spliced gene critical for salivary gland tropism. Repair of this mutation restored tropism for the salivary glands. Transcriptional analysis revealed a novel spliced gene within the sgg1 locus. This small open reading frame (ORF), sgg1.1, starts at the 3′ end of the first exon of sgg1 and extends exon 2 of sgg1. This shorter spliced gene is prematurely terminated by the nonsense mutation at nt 188422. Sequence analysis of tissue culture-passaged virus demonstrated that sgg1.1 was stable, although other mutational hot spots were identified. The G4 BAC will allow in vivo studies in a broader range of mice, avoiding the strong NK cell responses seen in B6 mice with other MCMV BAC-derived MCMVs.
IMPORTANCE Murine cytomegalovirus (MCMV) is widely used as a model of human CMV (HCMV) infection. However, this model relies on strains of MCMV that have been serially passaged in the laboratory for over four decades. These laboratory strains have been cloned as bacterial artificial chromosomes (BACs), which permits rapid and precise manipulation. Low-passage strains of MCMV add to the utility of the mouse model of HCMV infection but do not exist as cloned BACs. This study describes the first such low-passage MCMV BAC. This BAC-derived G4 was initially attenuated in vivo, with subsequent full genomic sequencing revealing a novel spliced transcript required for salivary gland tropism. These data suggest that MCMV, like HCMV, undergoes tissue culture adaptation that can limit in vivo growth and supports the use of BAC clones as a way of standardizing viral strains and minimizing interlaboratory strain variation.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous betaherpesvirus with seroprevalence ranging from 50% to 60% in industrialized countries to almost 100% in developing countries (1). Typically, HCMV causes benign disease in immunocompetent individuals. However, if infection occurs in utero, or in immunosuppressed or compromised individuals, HCMV is a significant pathogen. In industrialized nations, HCMV is the leading infectious cause of congenital abnormalities and mental retardation (2). In adults, the group most at risk of HCMV infection are transplant recipients. In these patients, HCMV infection remains one of the most common complications following organ transplant (3) and is a major cause of morbidity and contributor to mortality (4).
HCMV, like other CMVs, is strictly species-specific, meaning the pathology of the virus cannot be readily studied in animal models. Murine cytomegalovirus (MCMV) has proven invaluable as a model of HCMV infection and has illuminated many aspects of CMV biology, including viral tropism, immune evasion, and persistence (5, 6). Most MCMV studies employ one of two strains of MCMV, Smith or K181, which have been serially passaged in the laboratory for at least 40 years (7). For HCMV, long-term passage, particularly on fibroblasts, leads to a series of gene deletions, point mutations, insertions, and rearrangements (8–12), which can be accompanied by the loss of tissue tropism (13–19). Most notably, the deletion of the UL/b′ region results in loss of the genes UL131A, UL130, and UL128, which confer tropism for epithelial cells (20), endothelial cells (13), and dendritic cells (21). The entry of HCMV into fibroblasts requires the trimolecular complex of gH/gL/gO (22–25), while entry into other cell types requires the pentameric gH/gL/UL131A–UL128 complex (13, 26–28).
In a bid to develop models of CMV infection better correlated with the circulating clinical strains of HCMV, we have isolated, sequenced, and characterized a series of low-passage MCMV strains from free-living mice. These strains have proven useful in expanding our knowledge of MCMV genetics, including that of the MCMV pangenome, the extent of recombination and negative selection operating on MCMV, and the allelic nature of MCMV immune evasion genes (29–31). MCMV does not appear to undergo the widespread genomic changes seen in serially passaged HCMV strains. Low-passage strains of MCMV display little evidence of the gene deletions or rearrangements relative to laboratory strains (29, 31). Furthermore, serial tissue culture passage of laboratory strains of MCMV does not lead to the wide-scale genomic rearrangements (32) seen in HCMV. Nevertheless, MCMV does undergo subtle tissue culture adaptation, as shown for Mck-2 (33). Mck-2, a positional homolog of UL131A-128, forms a trimeric gH/gL/Mck-2 complex that controls viral entry into macrophages (34) and is required for salivary gland tropism (35, 36). Serial tissue culture passage of the K181 and Smith strains of MCMV leads to the accumulation of mutations in Mck-2 (33). MCMV also rapidly mutates in vivo under strong selective pressure (37, 38). Taken together, these observations indicate that it is possible that laboratory strains of MCMV have undergone both in vivo and in vitro adaptation over the past decades.
The cloning of CMVs as bacterial artificial chromosomes (BACs) and the adoption of subsequent Escherichia coli-based mutagenesis techniques have significantly accelerated the speed and precision of herpesvirus mutagenesis. The application of rapid mutagenesis techniques to the field since the descriptions of these BACs, first for MCMV (39) and then for HCMV (40), has been so successful that a series of additional betaherpesviruses (41–44), as well as alphaherpesviruses (45) and gammaherpesviruses (46), have been cloned as BACs. Likewise, additional strains of CMV have been cloned as BACs, including the K181 strain of MCMV (47) and several more HCMV strains, including Towne (48) and a number of low-passage clinical strains (49, 50). The BAC cloning of clinical strains of HCMV is a significant advance, as it allows the rapid manipulation of viral strains that have not undergone continued rounds of tissue culture adaptation and allows the repair and continued control of mutations that affect cellular tropism (51). To date, no BAC clones of a low-passage MCMV strain are available. Cloning of low-passage MCMV isolates as BACs will broaden the utility of our low-passage MCMV strains substantially. We describe here the cloning of the low-passage MCMV strain G4 as a BAC. The G4 strain was selected, as it has been well characterized (31, 52) and replicates well in BALB/c and CBA mice and also, because it expresses an m157 protein that fails to ligate Ly49H (53), in C57BL/6 (B6) mice (26). Normal replication in B6 mice is important, as most recombinant mouse strains are on this background. This means that investigators, if they wish to avoid the confounding effects of robust NK cell activation, must delete m157 prior to working with MCMV strain K181 or Smith in B6 mice. Deletion of m157 may also lead to other unintended consequences if, like other MCMV genes, m157 has more than one function.
The initial BAC clone of G4, G4209, replicated like wild-type virus in vitro but was heavily attenuated for salivary gland replication. Next-generation sequencing was applied to determine the cause of this attenuation. Two mutations were confirmed within G4209. One mutation led to a nonsynonymous change in the tegument protein, M32, producing an alanine-to-valine substitution at amino acid (aa) 266, which, when repaired, failed to restore salivary gland tropism. The other mutation was located within the 3′ untranslated region (UTR) of sgg1. Repair of this erstwhile 3′ UTR mutation restored salivary gland tropism. Transcriptional analyses revealed a splice variant of sgg1, termed sgg1.1, that was disrupted by the mutation present in G4209. The repaired G4 BAC, ARG4, replicated like wild-type parental virus, while nonrepaired virus retained in vivo replication kinetics similar to those described for deletions of the canonical sgg1 spliced gene (54, 55). Serial passage of the repaired G4 BAC leads to the accumulation of mutations in several genes, including Mck-2, but not sgg1 or sgg1.1.
RESULTS
Strategy for cloning and production of the G4 BAC.
The G4 BAC cloning strategy used was similar to that followed for the K181 BAC, pARK25 (47) (Fig. 1A). The recombination vector pAJR15 contains regions homologous with nucleotides (nt) 5420 to 6642 and nt 12574 to 13660 of the G4 strain of MCMV (31). Homologous recombination results in the deletion of the majority of m07 up to and including the majority of m12, with the concomitant insertion of the BAC cassette. The truncated clone was repaired by two-step allele replacement using the plasmid pAJR30 to produce the full-length BAC clone pG4209. In both BACs, pG420 and pG4209, the expected DNA fragment patterns were obtained by restriction fragment length polymorphism (RFLP) analysis with reference to the published sequence of the G4 strain of MCMV (Fig. 1B).
FIG 1.

G4 BAC construction and in vitro characterization. (A) Schematic of BAC construction showing EcoRI restriction enzyme sites (E) and expected fragment sizes (double arrows). (B) RFLP of BAC constructs. The intermediate BAC (pG420) and repaired BAC (pG4209J) were digested with EcoRI and compared to DNA from the parental G4 virus. Solid arrows show DNA fragments unique to the parental virus; dashed arrows show DNA fragments unique to the BAC constructs. (C) Rescued virus (G4209) replicates like the wild type in vivo. Shown are the results of a multistep growth assay performed on MEFs; data are means ± standard deviations (SD) of duplicate data.
To assess in vitro and in vivo replication, G4209 was rescued from pG4209 by transfection of mouse embryonic fibroblasts (MEFs) and monitored for loss of green fluorescent protein (GFP) expression, an indicator of BAC cassette excision. BAC cassette excision is accomplished by homologous recombination between 230 bp of repeat sequence flanking the BAC cassette. Virus was limit cloned when GFP expression was 50%, usually within 2 or 3 passages. Non-GFP-expressing clones were tested for the complete excision of the BAC cassettes as previously described for the K181 BAC (47). The rescued virus G4209 was identical to parental G4 by RFLP analysis (data not shown). In vitro replication of G4209 was also indistinguishable from that of G4 (Fig. 1C).
In vivo replication and sequencing of G4209.
Low-passage strains of MCMV replicate to lower titers during acute infection (31); accordingly, virulent salivary gland virus (SGV) stocks were produced to assess the acute in vivo replication of G4209. Despite successive attempts, all G4209 SGV stocks were low titer (less than 1 × 102 PFU/ml) and were insufficient for in vivo experiments. To determine the cause of this attenuation, we undertook full genome sequencing of G4209. Using this approach, a total of 18 sequence conflicts between the G4209 and parental G4 were identified (Table 1). Conventional dye terminator (Sanger) sequencing revealed that 13 of these conflicts were due to errors in the original G4 sequence, probably reflecting improvements in the sequencing platforms used between the two studies. The sequence of G4 has been updated accordingly (EU579859.2). Three of the remaining five sequence differences were located within the GC-rich region between m58 and M69 and resisted PCR amplification, so they could not be confirmed. The two remaining sequence differences were novel mutations within G4209 and were located within M32 and the 3′ untranslated region (UTR) of sgg1 (Table 1).
TABLE 1.
Sequence differences identified between parental G4 and pG4209 BAC by next-generation sequencinga
| Position | Nucleotide(s) in: |
Gene or location | Comments | Sanger sequencing | |
|---|---|---|---|---|---|
| G4 | G4209 | ||||
| 40711 | G | A | M32 | A266V | Mutation in G4209 |
| 42459 | G | T | M33 | A248S | Error in EU579859 |
| 42745 | A | C | M33 | Extends reading frame | Error in EU579859 |
| 93356/7 | CA | AC | Between m58 and M69 | No known effect | NDb |
| 93366 | C | A | Between m58 and M69 | No known effect | NDb |
| 93369 | A | C | Between m58 and M69 | No known effect | NDb |
| 110801 | T | A | M77 | F615Y | Error in EU579859 |
| 161922 | T | G | 5′ of stable intron | No known effect | Error in EU579859 |
| 161923/4 | Ex G | 5′ of stable intron | No known effect | Error in EU579859 | |
| 182390/1 | Ex TA | m124.1 | New start site (nt 182380)c | Error in EU579859 | |
| 187887 | Ex C | Mck-2 | Intron, extends m130 | Error in EU579859 | |
| 188422 | C | A | Putative 3′ UTR of sgg1 | Premature stop, sgg1.1 | Mutation in G4209 |
| 203305 | C | T | m144 | G156R | Error in EU579859 |
| 203746 | A | C | m144 | L9V | Error in EU579859 |
| 215377 | C | G | m155 | W32C | Error in EU579859 |
| 215436 | C | T | m155 | A13T | Error in EU579859 |
| 215518 | G | T | 5′ of m155 | No known effect | Error in EU579859 |
| 227940/1 | AC | CA | m168 | No known effectd | Error in EU579859 |
All nucleotide positions relate to the newly deposited sequence of G4 (EU579859.2). All putative sequence errors were confirmed or refuted by standard dye terminal sequencing; those shown to be in error in G4 have been corrected in the deposited sequence. Ex, extra; ND, no data.
Confirmation of changes in final Illumina sequencing of ARG4.
The new start site for m124.1 uses new G4 annotation.
Lack of m168 ORF identified in G4 following updated annotation using new sequence data.
Repair of M32.
The mutation at nt 40711 in G4209 is nonsynonymous, causing an alanine-to-valine substitution at aa 266 in M32, which encodes the tegument protein pp150. Deletion of UL32, the HCMV homolog of M32, leads to defective cell-to-cell spread (56). Two-step allele replacement using the plasmid pG4M32repair was used to repair the single base pair (G-to-A) change in G4209 (see Fig. S1 in the supplemental material). SGV stocks of three independent, sequence-verified clones (9, 10, and 12) of pG4209.M32 were produced to allow in vivo characterization. However, as for parental G4209, all three clones were heavily attenuated in vivo. These data indicate that the replication defect in G4209 was not due to the A266V substitution in M32.
Repair of sgg1.
Failure to rescue the replication of G4209 with the repair of M32 indicated that attenuation of G4209 and G4209.M32 was due either to the mutations in the noncoding region between m58 to M69 or to the single mutation in the 3′ UTR of sgg1. The sgg1 gene product is required for salivary gland replication (54). To investigate if the mutation in the 3′ UTR of sgg1 in G4209.M32 is responsible for the salivary gland attenuation, the mutation was repaired by two-step allele replacement using the plasmid pG4sgg1repair to produce the BAC pARG4 (accession number MT886700). The repair of sgg1 was confirmed by sequence analysis (data not shown).
Two strains of mice, BALB/c and C57BL/6 (B6), are widely used for in vivo MCMV replication studies. BALB/c mice are susceptible to infection with laboratory strains of MCMV, whereas B6 mice are resistant to laboratory strains of MCMV (57–59) due to NK cell responses, driven upon engagement of Ly49H by the m157 alleles in these viral strains (60–65). The m157 allele present in the G4 strain of MCMV does not bind Ly49H (53), allowing this virus to replicate with similar kinetics in BALB/c and B6 mice (31). It was expected that virus rescued from pARG4 (ARG4) with a restored sgg1 3′ UTR should replicate well in both commonly used mouse strains.
To test this, female BALB/c or B6 mice were infected with either ARG4 or parental G4. Replication was assessed in the spleen and liver at day 3 and in the salivary glands at day 18 postinfection. In both mouse strains and in all organs tested, the replication of ARG4 was indistinguishable from that of G4 (Fig. 2). These data indicate that the attenuation of G4209 was due to the disruption in the 3′ UTR of sgg1. Repair of this mutation permitted the BAC cloning of a low-passage strain of MCMV that shows normal in vivo replication in the two most commonly used strains of mice.
FIG 2.
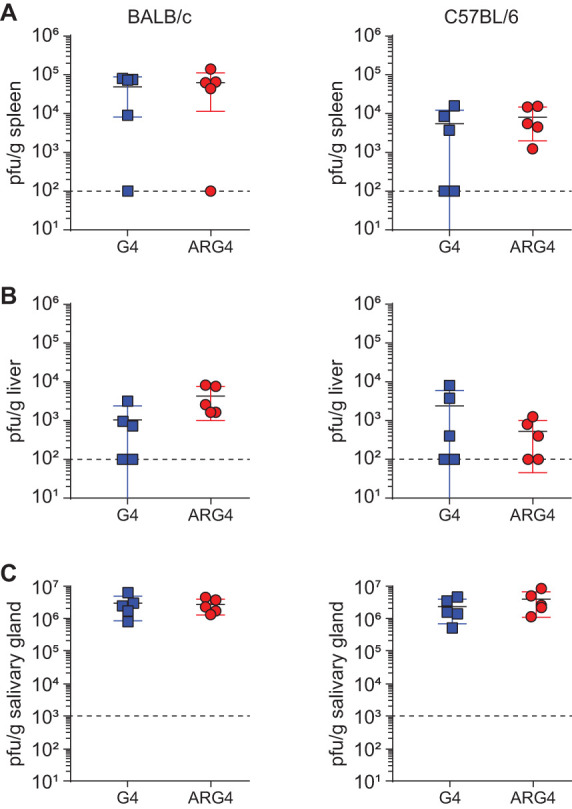
In vivo replication of rescued G4 BAC-derived virus, ARG4. Female BALB/c or C57BL/6 mice were infected with 5 × 103 PFU of SGV stocks of either G4 or ARG4. Titers of virus were assessed by plaque assay on day 3 postinfection in the spleen (A) and liver (B) and on day 17 postinfection in the salivary glands (C). No significant differences between titers of G4 versus ARG4 were noted in any tissue and at any time point. There were 5 mice per group. The dashed line indicates the limit of detection.
Transcriptional analysis of the 3′ UTR of sgg1.
The mutation within the putative 3′ UTR of sgg1 at nt 188422 in G4209.M32 led to an in vivo replication defect in the G4 strain of MCMV similar to that observed in sgg1 mutants. These data are consistent with the hypothesis that this mutation disrupts an essential regulatory element of sgg1 or alters an alternative transcript of sgg1 or a related gene. Four coding regions (Fig. 3A) are described within this region, m131, m132.1, m132 (exon 2), and m133 (66). The annotated sgg1 gene is composed of a large part of m133 (exon 1) and a small component of m132 (exon 2). The canonical two-exon sgg1 gene was predicted to encode a 312-aa protein, with the second exon encoding the last 7 aa (Fig. 3A) (55). The mutation in G4209 is located 3′ of the second exon of sgg1 (shown in red in Fig. 3A). The initial annotation of the Smith strain MCMV included a sequence error that prematurely truncated the m133 open reading frame (ORF). The full-length m133 extends from nt 188696 to 189799 in the Smith strain of MCMV. This extended ORF is highly conserved and present in all the sequenced strains of MCMV and extends the putative protein from 305 aa (66) to 367 aa (Fig. S2). However, this extended ORF does not extend to the mutation in G4209. Another ORF within this area, m132.1, shows some length polymorphisms, with the shortest predicted protein being 197 aa (188664 to 189257) in MCMV strain C4C, whereas most predicted proteins are 275 aa (Fig. S3). In G4, m132.1 encodes a predicted protein of 275 aa (188499 to 189326); however, this also does not include the mutation in G4209. Therefore, the mutation within the ssg1 region of MCMV does not map to a known ORF.
FIG 3.

The sgg1 locus encodes a novel spliced transcript. (A) Annotation of the sgg1 locus showing the locations of the ORFs from the previous MCMV genome annotation, i.e., m132.1 and sgg1, as well as the extension of the m133 ORF due to a correction to the original sequence of MCMV strain Smith. The red vertical bar denotes the mutation present in G4209 that introduces a premature stop codon into novel sgg1.1, defined by the results of Northern blotting, RNA-Seq, and RACE analysis, as described below in this legend. (B) Northern blot analysis of the sgg1 locus. Total RNA isolated at 6 hpi or 48 hpi from MEFs infected with the indicated viruses was probed with a strand-specific Northern blot probe (designated by a black bar in panel A and described in Materials and Methods). (C) RNA-Seq profile showing read coverage and splice junctions within the sgg1 region at a late stage of MCMV infection. The raw number of spliced reads spanning each splice junction is shown in the arc connecting the corresponding donor and acceptor splice sites. Total read coverage (Cov.) is shown on the y axis. (D) Sequences of the 5′ RACE and 3′ RACE PCR products were aligned to the G4 genome; the 5′ RACE sequences are shown in light blue, 3′ RACE sequences are in tan, and disagreements over the G4 genomic sequence are designated with a thin black rectangle within each RACE amplicon. The putative coding sequence in the newly identified sgg1.1 transcript is shown in light red, and the presumed poly(A) sequence is shown in yellow. An overview of the genomic region containing the sgg1.1 transcript, together with the genomic coordinates of the relevant features in the G4 genome, is shown at the bottom.
To determine whether a novel transcript or ORF could be present within the region affected by the mutation at 188422 in G4209.M32, we first performed Northern blot analysis of total RNA isolated from MEFs 6 and 48 h after infection with the G4, K181, or Smith MCMV strain. Membrane-immobilized total RNA was probed with a digoxigenin (DIG)-labeled, single-stranded RNA molecule that can hybridize to any RNA species which is synthesized in the direction toward the m131 ORF using nucleotides 188258 to 188457 of the G4 MCMV genome as a template. As expected, and consistent with previously published results (54, 55), we were able to detect the 1.5-kb and 1.8-kb viral sgg1 transcripts in infected MEFs 6 h postinfection (hpi), while the 1.5-kb sgg1 transcript continued to be expressed to a greater extent at the late stage of the infection (Fig. 3B). However, in addition to the 1.5-kb transcript, a distinct and robust signal in G4-infected MEFs at 48 hpi revealed the presence of a novel, previously unidentified viral RNA species containing approximately 600 nt which was also present in MEFs infected with MCMV strains Smith and K181 (Fig. 3B). Such a strong signal in a Northern blot is consistent with the results of our previous transcriptome sequencing (RNA-Seq) analysis, which demonstrated that the exon 2 of m132, including the putative 3′ UTR, is highly expressed in MCMV-infected fibroblasts (67). However, previous RNA-Seq analysis was performed using pooled total RNA from immediate early (IE), early (E), and late (L) phases of the infection, while the sequencing reads were mapped using splice-unaware aligners. Therefore, to further verify the high expression of exon 2 of m132 at the late stages of infection, we performed a remapping of raw RNA-Seq reads obtained from an unrelated experiment (our unpublished data) using a splice-aware read-mapper STAR v2.7.3a. As shown in the RNA-Seq coverage plot of the sgg1 region (Fig. 3C), this analysis not only confirmed the strong expression of the exon 2 of m132 and splice sites of the canonical sgg1 transcript but also revealed a novel spliced transcript whose intron spans nucleotides 188496 to 188589 of the G4 MCMV genome.
To investigate whether the newly identified splice sites are associated with the novel ∼600-nt transcript, we performed 5′ and 3′ rapid amplification of cDNA ends (RACE) using the gene-specific primers 5′-GSP2 and 3′-GSP2. Sequences of the primers were chosen in such a manner as to prevent them from annealing to cDNA of the canonical sgg1, while at the same time allowing them to pair to cDNA of any transcript that is spliced at novel donor and acceptor sites discovered by RNA-Seq (Fig. 3A). As shown in Fig. 3D, the longest 5′ RACE PCR products contained sequences extending from the 5′-GSP2 binding site up to an A-T base pair at coordinate 188789 of the G4 MCMV genome. Moreover, the longest 3′ RACE PCR products contained sequences extending from the 3′-GSP2 binding site, traversing the putative poly(A) site at coordinates 188177 to 188183 and reaching the T-A base pair at coordinate 188159 of the G4 MCMV genome. Importantly, all 3′ RACE PCR products lacked sequences corresponding to the putative intron discovered by RNA-Seq analysis. Taken together, the results strongly support the existence of a novel, spliced, two-exon transcript encoded by nucleotides 188159 to 188789 of the G4 MCMV genome, named here sgg1.1. Moreover, subsequent bioinformatic analysis demonstrated that spliced sgg1.1 transcript contains a coding sequence, encompassing nucleotides 188273 to 188495 of exon 2 and nucleotides 188590 to 188783 of G4, which has the potential to encode a small, 14.9-kDa, 138-amino-acid protein, which would be disrupted by the nonsense mutation at 188422 in G4209.M32. This novel the sgg1.1 ORF is highly conserved across MCMV strains (Fig. S4). The canonical sgg1 is also highly conserved across MCMV strains (Fig. S5).
In vivo replication of ARG4.
The canonical sgg1 gene affects MCMV salivary gland replication but has little effect on acute replication in the spleen or liver (54). To determine if premature truncation of sgg1.1 produced a phenotype similar to that initially described for sgg1, the replication of ARG4 was compared to that of G4209.M32, which is identical to ARG4 except for the single mutation in sgg1.1. Female BALB/c mice were infected with 2 × 105 PFU of tissue culture-derived G4209.M32 or ARG4 via the intraperitoneal route. Acute replication was assessed in the spleen and liver at day 3 of infection; chronic infection was assessed at day 17 in the salivary glands. Both the sgg1.1 mutant G4209.M32 and sgg1.1 intact ARG4 replicated to similar levels in the spleen and liver at day 3 postinfection. As seen previously, disruption of sgg1.1 in G4209.M32 inhibited viral replication and/or dissemination to the salivary glands of BALB/c mice (Fig. 4). The sgg1.1 mutant G4209.M32 retains in vivo replication kinetics similar to that described for the original sgg1 deletion mutants (36, 54). Taken together, these data indicate that sgg1.1 is a novel spliced gene that, like the canonical sgg1, modulates salivary gland tropism while having little effect on acute replication in the spleen and liver.
FIG 4.
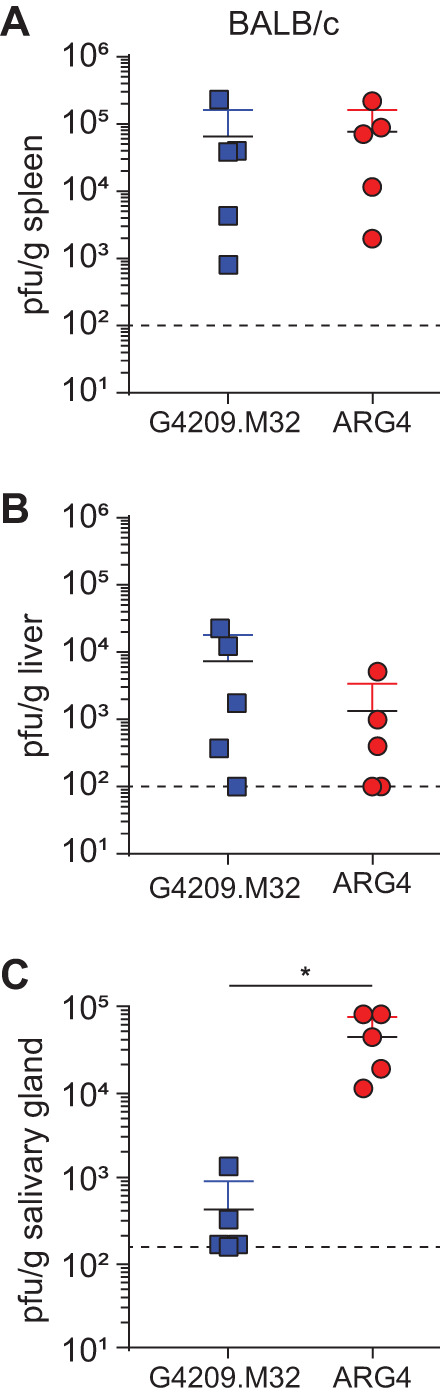
The sgg1.1 truncated virus replicates in vivo with similar kinetics to that seen for other sgg1 mutants. To determine if the in vivo phenotype of G4209.M32 (sgg1.1 mutant virus) was similar to that seen for other sgg1 mutant viruses, female BALB/c mice were infected with 5 × 103 PFU of SGV stocks of G4209.M32 or fully repaired ARG4. Titers of virus were assessed by plaque assay on day 3 postinfection in the spleen (A) and liver (B) and on day 17 postinfection in the salivary glands (C). There were 5 mice per group. The asterisk indicates a significant difference between ARG4 and G4209.M32 in the salivary glands (P = 0.0119 by Mann-Whitney U test). The dashed line indicates the limit of detection.
The sgg1 locus is stable in vitro.
The sgg1 locus is in close proximity to Mck-2, a gene important for salivary gland tropism, which is also unstable in tissue culture (33). To determine if sgg1 and sgg1.1 may also be under in vitro selective pressure, we serially passaged ARG4 on MEF and compared the sequences of passage 1 and passage 10 to the parental virus (EU579859.2). Sequences were assessed for the presence of single nucleotide polymorphisms (SNPs) (wild-type sequence equal to or less than 95% of total reads), insertions, or deletion. Multiple SNPs were detected across the MCMV genome. Shown in Table 2 are summary data from in vitro passaging (full data are shown in Tables S1 to S6). SNPs fell into two broad categories, early SNPs and amplified SNPs. Early SNPs were present by passage 1 and either declined or remained stable over 10 passages. Amplified SNPs were detected only at passage 10, suggesting that they confer a replication advantage to MCMV in tissue culture.
TABLE 2.
SNPs, indels, and deletions in in vitro-passaged ARG4a
| Gene | No. of: |
|||
|---|---|---|---|---|
| SNPs/kb | Amplified SNPs | Indels | Deletions | |
| m09 | 1 | |||
| m11 | 1 | |||
| m14 | 2.2 | 1 | ||
| m15 | 1 | |||
| m22 | 9.5 | |||
| m23.1 | 3.0 | |||
| m27 | 3.9 | 2 | ||
| M28/m29 | 1 | |||
| M34 | 1 | |||
| M36 | 1 | |||
| M44 | 1 | |||
| M45 | 4.2 | 4 | ||
| M47 | 1 | |||
| M48 | 1 | |||
| M49 | 5.6 | 3 | ||
| M50 | 8.4 | 1 | ||
| M55 | 1 | |||
| M56 | 14.9 | 6 | ||
| m60/M69 | 1 | |||
| m60/M70 | 2.1 | |||
| M77 | 2.6 | 2 | ||
| M80 | 2.4 | 2 | ||
| M86 | 1 | |||
| M87 | 3 | |||
| M89/m92 | 1 | |||
| M89/m94 | 1 | |||
| M97 | 1 | |||
| M102 | 1 | |||
| M103 | 1 | |||
| m117/m171.1 | 8.8 | |||
| m119 | 1 | |||
| m122 | 1 | |||
| m123 (ie3) | 1 | |||
| m125 | 24.2 | 3 | ||
| Mck-2 | 2 | 1 | 2 | |
| m136 | 1 | |||
| m154 | 8 | 2 | ||
| m165 | 3 | |||
| m166.5/m167 | 1 | |||
Shown are known ORFs that contain more than 2 SNPs/kb, one or more amplified SNPs, or one or more deletions or insertions.
Since multiple ORFs contained more than one SNP, we expressed SNP frequency (early plus amplified) per kilobase to account for gene length. Table 2 shows all ORFs that contain more than 2 SNPs/kb and those that contain at least one amplified SNP or one insertion or deletion. Most early SNPs were either purified during passage or remained stable over 10 passages and represented approximately 5% of the viral population. The exception was M56, which had multiple early SNPs, accounting for more than 20% of the population (Table S2) and a G-to-C SNP at nt 86884 in up to 27% of all reads; a novel G-to-T substitution was also seen in 5% of reads at this site in the P10 virus population (Tables S5 and S6). Other ORFs with a significant number of SNPs included M45, M49, M56, M87, m125, and m154. No SNPs were identified within m119; however, a 4-bp deletion following serial passage (171631 to 171634) was found within this ORF (Table S4). This 4-bp deletion potentially extends the reading frame from 171568 to 171927 (protein extended from 112 to 120 aa). Interestingly, a 3-bp deletion was also identified in m166.5/m167 that, while present at passages 1 and 10, declined as a percentage of the population during passage (Table S4).
Surprisingly, we noted no accumulation of mutations within the sgg1 locus, indicating that this region of the genome was stable. Likewise, we noted no mutations within M32. It is likely, therefore, that the mutations found within these ORFs in G4209 are due to random SNPs, as seen in other regions of the genome, that became fixed within the BAC clone. We did, however, note multiple mutations within the neighboring Mck-2 gene. Mck-2 contained two amplified-SNPs (at nt 188145 and nt 188162), a 1-bp indel and a 9-bp deletion. Both SNPs (at positions 188145 and 188162) introduce a premature stop codon. The 1-bp indel leads to a change in the consensus homopolymeric 7-bp G repeat (187402 to 187408) to a 6- or an 8-bp repeat, both of which introduce a frameshift mutation. The 9-bp deletion in Mck-2 maps to the 3′ end of the intron, likely affecting the splice site between the two introns of this ORF. A similar 9-bp deletion was found within serially passaged Smith strain of MCMV and the original Smith BAC (33).
DISCUSSION
In this report, we describe the production of a fully sequenced, replication-competent BAC clone of the low-passage MCMV strain G4. Cloning of this BAC revealed a novel splice variant, sgg1.1, that is essential for normal salivary gland tropism. MCMV infection of mice is a widely used animal model of HCMV infection, typically employing one of two serially passaged laboratory strains of MCMV: strain Smith, isolated in 1954 (68), and strain K181, isolated in the 1970s (69). These models have been invaluable in illuminating many aspects of MCMV biology, including viral tropism, immune evasion, and persistence (6). However, MCMV strains K181 and Smith are related viruses, and early stocks of Smith strain were contaminated with K181, suggesting that they were isolated from the same host (7). K181 remains a contaminant of some Smith strain stocks, evidenced by data showing that the Smith variant WT1 is a recombinant between these two strains (29). BAC cloning coupled with full genome sequencing provides the opportunity to standardize the MCMV strains Smith (33, 39), K181 (47), and in this study G4, thereby eliminating interlaboratory strain variability.
The rescued G4 BAC (ARG4) replicated like wild-type virus in MCMV-susceptible BALB/c and MCMV-resistant B6 mice. The resistance of B6 mice to MCMV is due to the expression of m157, a viral major histocompatibility complex (MHC) class I homologue that directly ligates the host NK cell-activating receptor Ly49H. Indeed, m157 is the cognate, and sole, ligand for this activating receptor. Viral engagement of m157 leads to heightened NK cell responses and attenuated viral replication (60–65). This elevated NK cell response has complex consequences for the control of immune responses to MCMV, leading to reduced T-cell responses and prolonged MCMV replication (70). However, resistance to MCMV is due to the interplay between the genetics of the host and virus. For example, BALB/c mice are resistant to infection with the WP15B strain of MCMV, while B6 mice are susceptible (31). Indeed, B6 mice are resistant only to MCMV strains that carry alleles of m157 capable of ligating Ly49H, and most MCMV strains do not contain such an allele (53). Because the G4 m157 allele does not bind Ly49H (53), the G4 BAC will permit studies in a broader range of mouse strains, in particular in situations where robust NK cell activation is to be avoided or where other more subtle NK cell-mediated effects or host immunity may be masked by the potent effects of the m157/Ly49H axis. Presently, in such situations, the m157 gene present in Smith or K181 is inactivated prior to the commencement of further study (71, 72). Because many MCMV genes have multiple functions (73), any additional undescribed roles for m157 will also form an unintentional component of such studies.
The G4 BAC, like the first Smith strain MCMV BAC, was initially attenuated for replication in the salivary glands of mice. This attenuation was due to a mutation in a novel two-exon transcript from the sgg1 region, termed sgg1.1. The intermediate virus, G209.M32, with a single point mutation in the second exon of sgg1.1, demonstrated in vivo replication similar to that seen for the canonical sgg1 mutant (54), with normal acute replication and attenuated replication and/or dissemination in the salivary glands. In the initial sgg1 studies, RQ401 contained a 323-bp deletion that encompassed the last exon of sgg1, an exon also shared by sgg1.1; consequently, both sgg1 and sgg1.1 are affected by this deletion. The deletion in RQ401 was proximal to the start of m131, which, when spliced to m129, encodes Mck-2 (74), a protein that also influences salivary gland tropism (35). Mck-2 has 4 potential start sites, the first of which is located at nt 188380 (in G4) and within the region deleted in RQ401. However, the transcription (nt 188295) and translation (nt 188180) start sites mapped by MacDonald and colleagues are not affected by this deletion (74), and the reading frame of Mck-2 was unaffected by the deletion in RQ401. In a follow-up study (75), an additional sgg1 mutant was employed (RM868 [55]) that carried a lacZ insertion in the first exon of sgg1. Like RQ401, the insertion in RM488 would functionally inactivate both sgg1 and sgg1.1. Therefore, this is the first study to demonstrate that disruption of a single transcript, sgg1.1, within the sgg1 locus alters salivary gland tropism. It is not clear if in isolation sgg1 also affects salivary gland tropism, although this appears likely given that sgg1 and sgg1.1 share the first exon. If so, three spliced transcripts, sgg1, sgg1.1, and Mck-2, modulate tropism for the salivary glands of mice.
MCMV, unlike HCMV, does not undergo widespread insertional and deletional mutation following serial in vitro culture. Despite this, the MCMV genome is not entirely stable following repeated in vitro or in vivo replication. In particular, Mck-2 was found to be mutated in the Smith strain MCMV BAC, pSM3fr, and is subject to mutational pressure in vitro (33). The immunomodulatory gene m157 mutates in vivo under pressure from Ly49H+ NK cells (37, 38). The stability of low-passage strains of MCMV remains unknown and may more closely resemble that seen for HCMV. Because we noted multiple potential mutations between m58 and M69 and confirmed mutations within mM32 and sggl.1, we sought to determine if tissue culture of the G4 strain MCMV led to the accumulation of mutations randomly across the genome or if mutational hot spots could be identified. Overall, our data indicate that MCMV stocks contain multiple low-level SNPs (5 to 10% of the population) within a single in vitro passage following rescue from the BAC clone. These “early SNPs” are either retained or purified following culture over 10 passages. It is possible that many of these SNPs represent variation that occurs within the carrier strain of E. coli. However, the location of multiple SNPs within a single ORF suggests that at least for these genes, this is not the case.
While random SNPs were present across the genome following in vitro replication, several mutational hot spots were noted. These hot spots were located in several ORFs, including M45, M49, M56, M87, m125, and m154, and were characterized by increased SNP frequency during in vitro passage. Insertions and deletions were also identified, most notably in Mck-2. These data suggest that G4, and likely other MCMV strains, are subjected to both random point mutations and also contain several mutational hot spots. Of note, we detected no mutations within either M32 or sgg1.1 following in vitro passage of the G4 BAC, suggesting that these two mutations fit within the category of random point mutations, outside the mutational hot spots, that were then fixed into the genome upon BAC cloning.
Three MCMV strains have been cloned as BACs; of these, two, namely, Smith and G4, were attenuated in vivo, specifically for salivary gland replication. Attenuation was due to mutations in the prototypical salivary gland tropism genes, Mck-2 and sgg1 (sgg1.1). Both of these ORFs affect salivary gland replication but have only moderate effects on acute replication and when cultured in fibroblasts have no obvious role. The situation is analogous to that seen in HMCV where tissue culture rapidly selects for viral strains with mutations in genes such as pUL128, pUL130, and pUL131A that are responsible for tropism on cells other than fibroblasts. Our early studies on low-passage strains of MCMV were undertaken at least in part to determine if the rapid mutation rate seen in HCMV when cultured in vitro would also be seen for MCMV. However, we noted no evidence for wide-scale mutations in MCMV. Nevertheless, it remains possible that the accumulation of errors at specific hot spots represents analogous, but subtle, tissue culture adaptation. In support of this, Smith strain stocks, including those from ATCC (ATCC-VR-1399), are an admixture of virions that contain different mutations in Mck-2 (33). K181 stocks are also described to be an admixture of virus with mutations that lead to truncations of this ORF (33). Taken together, these data suggest that multiple MCMV genes negatively impact the in vitro fitness of MCMV. However, unlike HCMV, the capacity to passage MCMV strains through their natural host means that such mutations may not necessarily come to dominate tissue culture stocks. Indeed, the Mck-2 mutations in Smith strain stocks are lost upon in vivo passage (33). These data may also in part explain the known enhanced virulence of salivary gland stocks of MCMV compared to tissue culture derived stocks.
In conclusion, we have, for the first time, produced a fully wild-type BAC of a low-passage strain of MCMV. This G4 BAC strain avoids strong NK cell responses mediated by the m157/Ly49H axis, thus providing an alternative strain of virus for in vivo studies in a broad range of mouse strains. During the in vivo characterization of this BAC, we identified a novel splice transcript, sgg1.1, that is required for normal salivary gland tropism. Finally, we suggest that BAC clones, following complete sequence analysis and in vivo characterization, may serve as a means of standardizing viral strains and minimizing intra- and interlaboratory variation.
MATERIALS AND METHODS
Virus and cells.
The previously described (52) G4 strain of MCMV was first isolated from the Geraldton area of Western Australia. Viral titers were determined in duplicate by plaque assay on mouse embryonic fibroblasts (MEFs). Plasmid BACs are represented by the prefix “p”; the prefix is removed to designate rescued virus. All genomic locations relate to the reannotated G4 sequence, accession number EU579859.2. MCMV strain K181 was derived from the MCMV BAC clone pARK25 (47). MCMV strain Smith was derived from the repaired BAC subclone (33) of the Smith strain BAC, pSM3fr (39). Tissue culture virus (TCV) stocks were prepared in BALB/c MEFs, while salivary gland virus (SGV) stocks were prepared in weanling BALB/c mice as previously described (6).
Isolation of viral DNA and BAC plasmids.
G4 MCMV DNA was extracted according to published methods (47). Briefly, MEFs were infected with the virus, and at 100% cytopathic effect (CPE) the cells were harvested by scraping; total DNA was extracted with phenol-chloroform. DNA was precipitated in 1 volume of isopropanol and spooled out of suspension. For the isolation of circular DNA from virus-infected cells, the Hirt method (76) was followed. Briefly, infected cells from a 10-cm dish were resuspended in 500 μl of 20 mM EDTA (pH 8.0) and then lysed with 500 μl of 1.2% SDS. Protein and high-molecular-weight DNA were precipitated by the addition of 660 μl of 5 M NaCl. The sample was left overnight at 4°C, and the proteins and high-molecular-weight DNA were removed by centrifugation at 15,000 × g at 4°C for 30 min. The supernatant was removed, and DNA was extracted with phenol-chloroform. DNA was precipitated with 1 volume of isopropanol, washed twice with 70% ethanol, and resuspended in 50 μl of Tris-EDTA (TE). Circular/low-molecular-weight DNA (10 μl) was electroporated into the E. coli strain DH10B. BAC DNA was isolated from E. coli using a NucleoBond Xtra midikit (Macherey-Nagel, Düren, Germany).
Plasmids.
For the construction of the recombination plasmid, pAJR15, two PCR fragments were cloned into the modified pPK18 plasmid (46). PCR primers (m12-For, 5′-GCAGAATTCTCGACCGCTTCAAATGATTGGGTTCG-3′, and m14-Rev, 5′-CGCGAATTCCTTCGAGTTTAGACAGCCGGTCAGTTG-3′) were designed to amplify the right homology arm (nt 12574 to 13660), which was then nondirectionally cloned (engineered EcoRI restriction enzyme sites are underlined) into pK18 to produce the plasmid pAJR13. PCR primers were designed to amplify the left homology arm with a NotI site on the forward primer and an AvrII site on the reverse primer (m06 For, 5′-TCTGCGGCCGCTTGGAGCGATACGTTGACAATG-3′, and m07 Rev, primer 5′-CCCCCTAGGGTACGCGAGCCATACGCAGAACACCA-3′). The PCR product containing nt 5420 to 6642 was directionally cloned into pAJR13 to produce pAJR14. Finally, the homologous recombination plasmid pAJR15 was produced by cloning the PacI fragment of pAJR02 (47) into pAJR14.
pAJR30 was used to repair the deletion in pG420 and restore the G4 BAC to full length. The plasmid pCMH512 (kindly provided by Chris Hardy) contains the entirety of m06 to m17 (nt 5335 to 18447) from G4 cloned into pBluescript. pCMH512 was digested with EcoRI and cloned into MfeI-digested pAJR04pl (47) to produce pAJR24. pAJR24 was digested with BglII and ligated into BamHI-digested pST76K-SR to produce the plasmid pAJR26. pAJR26 was SacII digested to remove 1,146 bp of excess G4 sequence and religated to produce pAJR30. Use of pAJR30 to repair G420 allowed the production of a BAC clone with 230 bp of repeat sequence flanking the BAC sequence, permitting spontaneous BAC cassette excision via homologous recombination.
pG4M32repair was made to repair the G-to-A mutation at nt 40 711 in the M32 open reading frame (ORF) of pG4209 (Table 1). First, the gateway destination plasmid pST76K-SR.DEST was created to allow the rapid construction of shuttle plasmids for later two-step allele replacement. AttP sites were amplified from pDONR201 and inserted into pGEM-T to produce pGem-T.AttP. These AttP sites were then removed from pGem-T.AttP using BglII and were inserted into the BamHI site in pST76K-SR to produce pST76K-SR.DEST. A 3,222-bp region encompassing the site of the G-to-A mutation site in M32 of pG420 was amplified from the parental G4 virus with the primers G4209.M32.F (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCTTTCTTTCTTTCTGCTGCT-3′) and G4209.M32.R (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCCATGTCATCATCACCACT-3′). The AttB sites are underlined. This PCR product was cloned into pST76K-SR.DEST using BP clonase (Life Technologies, Carlsbad, CA, USA), and repair of the mutation was confirmed by sequence analysis.
pG4sgg1repair was made to repair the C-to-A mutation at nt 188422 in pG4209.M32 (Table 1). A 1,567-bp region encompassing the mutation was amplified from parental G4 using the primers F187669 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCTCACAGACACACTCTATCCAG-3′) and R189215 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTCACCAGCCTTATTTCACGTG-3′). The AttB sites are underlined. This PCR product was cloned into pST76K-SR.DEST using BP clonase, and repair of the mutation was confirmed by sequence analysis.
Generation of the G4 BAC.
The G4 strain of MCMV used in the construction of the G4 BAC was plaque picked twice following isolation from a free-living mouse. This virus was passaged an additional three times before being used to infect MEFs at a multiplicity of infection (MOI) of 0.02. For homologous recombination, 18 μg of pAJR15 was digested with MluI, and the linear fragment (left homology arm, loxP, GFP gene, BAC vector, guanosine phosphoribosyltransferase (gpt), loxP, and right homology arm) was transfected by calcium phosphate precipitation 2 h after cells had been infected with G4. Clones containing the recombinant GFP+ virus were plaque picked three times under selection with xanthine and mycophenolic acid in the M210B4 cell line. Following plaque-picking, the recombinant virus was passaged twice more before circular viral DNA was extracted as described above. Viral DNA was transformed into DH10B cells, and resultant chloramphenicol resistant colonies were screened by RFLP analysis.
Sequencing and analysis.
The complete sequence of G4 was described previously (31). The rescued G4 BAC, G4209, was used to infect MEFs at an MOI of 5, and at 48 hpi, cells were frozen and thawed to release the cell-associated virus. Pelleted virions were DNase I treated before lysis to remove any contaminating murine genomic DNA. DNA was extracted from the virions by the addition of proteinase K/SDS prior to phenol-chloroform extraction. Samples were RNase treated prior to assessment of DNA quality by agarose gel electrophoresis. Genomic library preparation and sequencing were performed on the Roche FLX Next Generation sequencer, at the Lotterywest State Biomedical Facility at Royal Perth Hospital, Perth, Western Australia, Australia. The G4209 sequence was de novo assembled, and contigs were then assembled against the G4 reference genome, using the CLC Genomic Workbench. The sequence of G4209 was aligned to G4, allowing the detection of sequence discordance.
For in vitro passaging, repaired ARG4 was rescued by transfection of pARG4 and minimally passaged to allow cloning and BAC cassette deletion. This P1 virus was passaged a further 10 times on BALB/c MEF. Viral DNA was purified from passage 1 and passage 10 stocks of ARG4 as described above. Passaged genomes were sequenced on a MiSeq Illumina platform using 2 × 300-bp paired read chemistry targeting 100-fold sequencing depth of the target area. Demultiplexed data produced an average coverage of >837× across the reference CMV genome. Sequencing was performed at IIID, Murdoch University. SNP analysis was performed using VGAS software (http://www.iiid.com.au/software/vgas). A SNP was defined as variation from the reference in 5% or more of the reads at a particular nucleotide. All SNPs had a median forward/reverse balance of 0.493 (interquartile range, 0.012) and coverage greater than 100 (Table S1). Consensus changes not identified by previous 454-based sequencing (this publication and reference 31) were used to update the deposited G4 sequence (EU579859.2).
RNA-Seq analysis.
MEFs were infected at an MOI of 0.3 with MCMV strain Smith, followed by centrifugal enhancement at 800 × g for 30 min. Total RNA from infected cells was then extracted at 40, 60, and 80 hpi (late phase of the infection). Prior to library preparation, equal amounts of RNA from each time point were pooled, and sequencing libraries were prepared using a TruSeq Stranded Total RNA with Ribo-Zero Gold kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Single-end, 72-nt strand-specific reads were obtained by sequencing of libraries on an Illumina IIx genome analyzer. Following initial read QC, fastq files containing raw reads from independent replicates were concatenated into a single fastq file and mapped to the wild-type MCMV genome sequence (NCBI reference sequence NC_004065.1) using STAR v2.7.3a (77). Obtained alignment files were sorted and indexed using samtools (78), and splicing in the sgg1 region was then visualized using the Sashimi plot option in IGV 2.7.2 (79).
Northern blotting and RACE.
Approximately 2.5 × 106 MEFs were infected at an MOI of 0.5 with either the Smith, G4, or K181 strain of MCMV, followed by centrifugal enhancement at 800 × g for 30 min. Total RNA from infected cells was then extracted at 6 and 48 hpi using TRI reagent (Sigma-Aldrich) according to the manufacturer’s instructions and dissolved in 100 μl of RNase-free water plus 1 μl of Protector RNase inhibitor (Sigma-Aldrich). Following removal of contaminating DNA with RNase-free DNase I (New England Biolabs), RNA samples were stored at −80°C, and the stability of purified RNA was verified by incubating small aliquots of each sample for 2 h at 37°C and 80°C and running them alongside aliquots of original samples (stored at −80°C) on a 2% bleach gel (80). Preparation of samples for electrophoresis, electrophoresis, transfer of RNA to positively charged nylon membranes, cross-linking of RNA, prehybridization, hybridization, washing, and detection were performed using the NorthernMax-Gly kit according to the manufacturer’s instructions. Probe bound to target RNA molecules on the membrane was visualized and documented with an ImageQuant LAS 4000 series imager (GE Healthcare). A strand-specific Northern blot RNA probe was generated by PCR-amplifying the genomic region spanning nucleotides 188258 to 188457 of the MCMV G4 genome, using the primers SggP2-F (5′-TGGTCGGCTCATCTGACGCA-3′) and SggP2-R (TAATACGACTCACTATAGGGACGACACCTTCATCGTCACA-3′). The 220-bp PCR product was then excised from the agarose gel, column purified (NucleoSpin Gel and PCR Clean-up; Macherey-Nagel), and used as a template in the second PCR with the same primers as above. The 220-bp product of the second PCR was also excised from the agarose gel, column purified, and used as a template for in vitro RNA probe synthesis with a DIG RNA labeling kit (SP6/T7; Sigma-Aldrich), as instructed by the manufacturer. Gene-specific primers used for 5′ RACE and 3′ RACE were 5′-GSP2 (5′-GATTACGCCAAGCTTCCAGCACCCCGTCTCGGTCCGTCAC-3′) and 3′-GSP2 (GATTACGCCAAGCTTGTGACGGACCGAGACGGGGTGCTGG-3′). Primer design, generation of RACE-ready cDNA, RNA purity/stability/integrity assessment, rapid amplification of cDNA ends, and characterization, cloning, and sequencing of RACE PCR products were all performed according to the SMARTer RACE 5′/3′ kit manual (TaKaRa Bio Inc.). Sequences of obtained 5′ RACE and 3′ RACE PCR products were aligned to the G4 MCMV sequence (EU579859.2) using SnapGene v5.1.
Statistics.
Statistical analyses were performed using a Mann-Whitney U test in GraphPad Prism 5.0.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Health and Medical Research Council, the WA Department of Health FutureHealth WA Merit Award program (2013-14), and the grant “Strengthening the capacity of the Scientific Centre of Excellence CerVirVac for research in viral immunology and vaccinology,” KK.01.1.1.01.0006, financed by the European Regional Development Fund (S. Jonjić).
We thank Jerome Coudert and Nico Suárez for helpful discussions and Fran Borovečki and Kristina Gotovac Jerčić, School of Medicine, University of Zagreb, for technical assistance in RNA-Seq experiments.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Zuhair M, Smit GSA, Wallis G, Jabbar F, Smith C, Devleesschauwer B, Griffiths P. 2019. Estimation of the worldwide seroprevalence of cytomegalovirus: a systematic review and meta-analysis. Rev Med Virol 29:e2034. doi: 10.1002/rmv.2034. [DOI] [PubMed] [Google Scholar]
- 2.Ahlfors K, Ivarsson SA, Harris S. 1999. Report on a long-term study of maternal and congenital cytomegalovirus infection in Sweden. Review of prospective studies available in the literature. Scand J Infect Dis 31:443–457. doi: 10.1080/00365549950163969. [DOI] [PubMed] [Google Scholar]
- 3.Patel R, Paya CV. 1997. Infections in solid-organ transplant recipients. Clin Microbiol Rev 10:86–124. doi: 10.1128/CMR.10.1.86-124.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Keyzer K, Van Laecke S, Peeters P, Vanholder R. 2011. Human cytomegalovirus and kidney transplantation: a clinician's update. Am J Kidney Dis 58:118–126. doi: 10.1053/j.ajkd.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Reddehase MJ, Lemmermann NAW. 2018. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: predictions for medical translation that survived the “test of time.” Viruses 10:693. doi: 10.3390/v10120693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shellam GR, Redwood AJ, Smith LM, Gorman S. 2007. Murine cytomegaloviruses and other herpesviruses, p 1–48. In Fox JG, Barthold SW, Davisson MT, Newcomer CE, Quimby FW, Smith AL (ed), The mouse in biomedical research, 2nd ed, vol 2 Diseases. Academic Press, Amsterdam, The Netherlands. [Google Scholar]
- 7.Redwood A, Shellam G, Smith L. 2013. Molecular evolution of murine cytomegalovirus genomes, Chapter 2, p 23–37. In Reddehase M. (ed), Cytomegalovirus: from molecular pathogenesis to intervention, 2nd ed, vol 1 Caister Academic Press, Norwich, United Kingdom. [Google Scholar]
- 8.Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83. doi: 10.1128/JVI.70.1.78-83.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dargan DJ, Douglas E, Cunningham C, Jamieson F, Stanton RJ, Baluchova K, McSharry BP, Tomasec P, Emery VC, Percivalle E, Sarasini A, Gerna G, Wilkinson GWG, Davison AJ. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–1546. doi: 10.1099/vir.0.018994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dargan DJ, Jamieson FE, MacLean J, Dolan A, Addison C, McGeoch DJ. 1997. The published DNA sequence of human cytomegalovirus strain AD169 lacks 929 base pairs affecting genes UL42 and UL43. J Virol 71:9833–9836. doi: 10.1128/JVI.71.12.9833-9836.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley AJ, Lurain NS, Ghazal P, Trivedi U, Cunningham C, Baluchova K, Gatherer D, Wilkinson GWG, Dargan DJ, Davison AJ. 2009. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J Gen Virol 90:2375–2380. doi: 10.1099/vir.0.013250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown JM, Kaneshima H, Mocarski ES. 1995. Dramatic interstrain differences in the replication of human cytomegalovirus in SCID-hu mice. J Infect Dis 171:1599–1603. doi: 10.1093/infdis/171.6.1599. [DOI] [PubMed] [Google Scholar]
- 13.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahl M, Siegel-Axel D, Stenglein S, Jahn G, Sinzger C. 2000. Efficient lytic infection of human arterial endothelial cells by human cytomegalovirus strains. J Virol 74:7628–7635. doi: 10.1128/jvi.74.16.7628-7635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacCormac LP, Grundy JE. 1999. Two clinical isolates and the Toledo strain of cytomegalovirus contain endothelial cell tropic variants that are not present in the AD169, Towne, or Davis strains. J Med Virol 57:298–307. doi:. [DOI] [PubMed] [Google Scholar]
- 16.Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev Med Virol 20:136–155. doi: 10.1002/rmv.645. [DOI] [PubMed] [Google Scholar]
- 17.Sinzger C, Bissinger AL, Viebahn R, Oettle H, Radke C, Schmidt CA, Jahn G. 1999. Hepatocytes are permissive for human cytomegalovirus infection in human liver cell culture and In vivo. J Infect Dis 180:976–986. doi: 10.1086/315032. [DOI] [PubMed] [Google Scholar]
- 18.Waldman WJ, Sneddon JM, Stephens RE, Roberts WH. 1989. Enhanced endothelial cytopathogenicity induced by a cytomegalovirus strain propagated in endothelial cells. J Med Virol 28:223–230. doi: 10.1002/jmv.1890280405. [DOI] [PubMed] [Google Scholar]
- 19.Waldman WJ, Roberts WH, Davis DH, Williams MV, Sedmak DD, Stephens RE. 1991. Preservation of natural endothelial cytopathogenicity of cytomegalovirus by propagation in endothelial cells. Arch Virol 117:143–164. doi: 10.1007/BF01310761. [DOI] [PubMed] [Google Scholar]
- 20.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerna G, Percivalle E, Lilleri D, Lozza L, Fornara C, Hahn G, Baldanti F, Revello MG. 2005. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J Gen Virol 86:275–284. doi: 10.1099/vir.0.80474-0. [DOI] [PubMed] [Google Scholar]
- 22.Compton T, Nepomuceno RR, Nowlin DM. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395. doi: 10.1016/0042-6822(92)90200-9. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Nelson JA, Britt WJ. 1997. Glycoprotein H-related complexes of human cytomegalovirus: identification of a third protein in the gCIII complex. J Virol 71:3090–3097. doi: 10.1128/JVI.71.4.3090-3097.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber MT, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J Virol 72:8191–8197. doi: 10.1128/JVI.72.10.8191-8197.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanarsdall AL, Chase MC, Johnson DC. 2011. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J Virol 85:11638–11645. doi: 10.1128/JVI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. doi: 10.1099/vir.0.81921-0. [DOI] [PubMed] [Google Scholar]
- 28.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith LM, McWhorter AR, Shellam GR, Redwood AJ. 2013. The genome of murine cytomegalovirus is shaped by purifying selection and extensive recombination. Virology 435:258–268. doi: 10.1016/j.virol.2012.08.041. [DOI] [PubMed] [Google Scholar]
- 30.Smith LM, Shellam GR, Redwood AJ. 2006. Genes of murine cytomegalovirus exist as a number of distinct genotypes. Virology 352:450–465. doi: 10.1016/j.virol.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 31.Smith LM, McWhorter AR, Masters L, Shellam GR, Redwood AJ. 2008. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J Virol 82:6689–6696. doi: 10.1128/JVI.00160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng TP, Valentine MC, Gao J, Pingel JT, Yokoyama WM. 2010. Stability of murine cytomegalovirus genome after in vitro and in vivo passage. J Virol 84:2623–2628. doi: 10.1128/JVI.02142-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan S, Krause J, Prager A, Mitrovic M, Jonjic S, Koszinowski UH, Adler B. 2011. Virus progeny of MCMV bacterial artificial chromosome pSM3fr shows reduced growth in salivary glands due to a fixed mutation of MCK-2. J Virol 85:10346–10353. doi: 10.1128/JVI.00545-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagner FM, Brizic I, Prager A, Trsan T, Arapovic M, Lemmermann NAW, Podlech J, Reddehase MJ, Lemnitzer F, Bosse JB, Gimpfl M, Marcinowski L, MacDonald M, Adler H, Koszinowski UH, Adler B. 2013. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog 9:e1003493. doi: 10.1371/journal.ppat.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fleming P, Davis-Poynter N, Degli-Esposti M, Densley E, Papadimitriou J, Shellam G, Farrell H. 1999. The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J Virol 73:6800–6809. doi: 10.1128/JVI.73.8.6800-6809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saederup N, Aguirre SA, Sparer TE, Bouley DM, Mocarski ES. 2001. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol 75:9966–9976. doi: 10.1128/JVI.75.20.9966-9976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HRC, Yokoyama WM, Scalzo aa. 2003. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc Natl Acad Sci U S A 100:13483–13488. doi: 10.1073/pnas.2233572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.French AR, Pingel JT, Wagner M, Bubic I, Yang L, Kim S, Koszinowski U, Jonjic S, Yokoyama WM. 2004. Escape of mutant double-stranded DNA virus from innate immune control. Immunity 20:747–756. doi: 10.1016/j.immuni.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 39.Messerle M, Crnkovic I, Hammerschmidt W, Ziegler H, Koszinowski UH. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A 94:14759–14763. doi: 10.1073/pnas.94.26.14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borst EM, Hahn G, Koszinowski UH, Messerle M. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J Virol 73:8320–8329. doi: 10.1128/JVI.73.10.8320-8329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGregor A, Schleiss MR. 2001. Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli. Mol Genet Metab 72:15–26. doi: 10.1006/mgme.2000.3102. [DOI] [PubMed] [Google Scholar]
- 42.Chang WL, Barry PA. 2003. Cloning of the full-length rhesus cytomegalovirus genome as an infectious and self-excisable bacterial artificial chromosome for analysis of viral pathogenesis. J Virol 77:5073–5083. doi: 10.1128/jvi.77.9.5073-5083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borenstein R, Frenkel N. 2009. Cloning human herpes virus 6A genome into bacterial artificial chromosomes and study of DNA replication intermediates. Proc Natl Acad Sci U S A 106:19138–19143. doi: 10.1073/pnas.0908504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang H, Kawabata A, Yoshida M, Oyaizu H, Maeki T, Yamanishi K, Mori Y. 2010. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 407:360–367. doi: 10.1016/j.virol.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 45.Smith GA, Enquist LW. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc Natl Acad Sci U S A 97:4873–4878. doi: 10.1073/pnas.080502497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol 74:6964–6974. doi: 10.1128/jvi.74.15.6964-6974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redwood AJ, Messerle M, Harvey NL, Hardy CM, Koszinowski UH, Lawson MA, Shellam GR. 2005. Use of a murine cytomegalovirus, K181-derived, bacterial artificial chromosome as a vaccine vector for immunocontraception. J Virol 79:2998–3008. doi: 10.1128/JVI.79.5.2998-3008.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marchini A, Liu H, Zhu H. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 75:1870–1878. doi: 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hahn G, Khan H, Baldanti F, Koszinowski UH, Revello MG, Gerna G. 2002. The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J Virol 76:9551–9555. doi: 10.1128/jvi.76.18.9551-9555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hahn G, Rose D, Wagner M, Rhiel S, McVoy MA. 2003. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Towne long as BACs and site-directed mutagenesis using a PCR-based technique. Virology 307:164–177. doi: 10.1016/S0042-6822(02)00061-2. [DOI] [PubMed] [Google Scholar]
- 51.Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, Davison AJ, Wilkinson GWG. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Booth TW, Scalzo aa, Carrello C, Lyons PA, Farrell HE, Singleton GR, Shellam GR. 1993. Molecular and biological characterization of new strains of murine cytomegalovirus isolated from wild mice. Arch Virol 132:209–220. doi: 10.1007/BF01309855. [DOI] [PubMed] [Google Scholar]
- 53.Corbett AJ, Coudert JD, Forbes CA, Scalzo aa. 2011. Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J Immunol 186:1713–1722. doi: 10.4049/jimmunol.1003308. [DOI] [PubMed] [Google Scholar]
- 54.Manning WC, Stoddart CA, Lagenaur LA, Abenes GB, Mocarski ES. 1992. Cytomegalovirus determinant of replication in salivary glands. J Virol 66:3794–3802. doi: 10.1128/JVI.66.6.3794-3802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lagenaur LA, Manning WC, Vieira J, Martens CL, Mocarski ES. 1994. Structure and function of the murine cytomegalovirus sgg1 gene: a determinant of viral growth in salivary gland acinar cells. J Virol 68:7717–7727. doi: 10.1128/JVI.68.12.7717-7727.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.AuCoin DP, Smith GB, Meiering CD, Mocarski ES. 2006. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J Virol 80:8199–8210. doi: 10.1128/JVI.00457-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scalzo aa, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. 1990. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med 171:1469–1483. doi: 10.1084/jem.171.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chalmer JE, Mackenzie JS, Stanley NF. 1977. Resistance to murine cytomegalovirus linked to the major histocompatibility complex of the mouse. J Gen Virol 37:107–114. doi: 10.1099/0022-1317-37-1-107. [DOI] [PubMed] [Google Scholar]
- 59.Grundy JE, Mackenzie JS, Stanley NF. 1981. Influence of H-2 and non-H-2 genes on resistance to murine cytomegalovirus infection. Infect Immun 32:277–286. doi: 10.1128/IAI.32.1.277-286.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 61.Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo aa, Yokoyama WM. 2001. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 62.Daniels KA, Devora G, Lai WC, O'Donnell CL, Bennett M, Welsh RM. 2001. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med 194:29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee SH, Girard S, Macina D, Busa M, Zafer A, Belouchi A, Gros P, Vidal SM. 2001. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat Genet 28:42–45. doi: 10.1038/ng0501-42. [DOI] [PubMed] [Google Scholar]
- 64.Lee SH, Zafer A, de Repentigny Y, Kothary R, Tremblay ML, Gros P, Duplay P, Webb JR, Vidal SM. 2003. Transgenic expression of the activating natural killer receptor Ly49H confers resistance to cytomegalovirus in genetically susceptible mice. J Exp Med 197:515–526. doi: 10.1084/jem.20021713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo aa, Fremont DH, Yokoyama WM. 2002. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A 99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 70:8833–8849. doi: 10.1128/JVI.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Juranic Lisnic V, Babic Cac M, Lisnic B, Trsan T, Mefferd A, Das Mukhopadhyay C, Cook CH, Jonjic S, Trgovcich J. 2013. Dual analysis of the murine cytomegalovirus and host cell transcriptomes reveal new aspects of the virus-host cell interface. PLoS Pathog 9:e1003611. doi: 10.1371/journal.ppat.1003611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith MG. 1954. Propagation of salivary gland virus of the mouse in tissue cultures. Proc Soc Exp Biol Med 86:435–440. doi: 10.3181/00379727-86-21123. [DOI] [PubMed] [Google Scholar]
- 69.Hudson JB, Misra V, Mosmann TR. 1976. Cytomegalovirus infectivity: analysis of the phenomenon of centrifugal enhancement of infectivity. Virology 72:235–243. doi: 10.1016/0042-6822(76)90326-3. [DOI] [PubMed] [Google Scholar]
- 70.Andrews DM, Estcourt MJ, Andoniou CE, Wikstrom ME, Khong A, Voigt V, Fleming P, Tabarias H, Hill GR, van der Most RG, Scalzo aa, Smyth MJ, Degli-Esposti MA. 2010. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J Exp Med 207:1333–1343. doi: 10.1084/jem.20091193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsuda Y, Caposio P, Parkins CJ, Botto S, Messaoudi I, Cicin-Sain L, Feldmann H, Jarvis MA. 2011. A replicating cytomegalovirus-based vaccine encoding a single Ebola virus nucleoprotein CTL epitope confers protection against Ebola virus. PLoS Negl Trop Dis 5:e1275. doi: 10.1371/journal.pntd.0001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haynes LD, Verma S, McDonald B, Wu R, Tacke R, Nowyhed HN, Ekstein J, Feuvrier A, Benedict CA, Hedrick CC. 2015. Cardif (MAVS) regulates the maturation of NK cells. J Immunol 195:2157–2167. doi: 10.4049/jimmunol.1402060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lisnić B, Lisnić VJ, Jonjić S. 2015. NK cell interplay with cytomegaloviruses. Curr Opin Virol 15:9–18. doi: 10.1016/j.coviro.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 74.MacDonald MR, Burney MW, Resnick SB, Virgin HI. 1999. Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a beta chemokine of novel structure. J Virol 73:3682–3691. doi: 10.1128/JVI.73.5.3682-3691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vieira J, Farrell HE, Rawlinson WD, Mocarski ES. 1994. Genes in the HindIII J fragment of the murine cytomegalovirus genome are dispensable for growth in cultured cells: insertion mutagenesis with a lacZ/gpt cassette. J Virol 68:4837–4846. doi: 10.1128/JVI.68.8.4837-4846.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 77.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aranda PS, LaJoie DM, Jorcyk CL. 2012. Bleach gel: a simple agarose gel for analyzing RNA quality. Electrophoresis 33:366–369. doi: 10.1002/elps.201100335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.