

Abstract
A series of novel 2-pyridyl, 4-morpholinyl substituted thiazolo[5,4-b]pyridine analogues have been designed and synthesized in this paper. These thiazolo[5,4-b]pyridines were efficiently prepared in seven steps from commercially available substances in moderate to good yields. All of these N-heterocyclic compounds were characterized by nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HRMS) analysis and tested for phosphoinositide 3-kinase (PI3K) enzymatic assay. The results indicated that these N-heterocyclic compounds showed potent PI3K inhibitory activity, and the IC50 of a representative compound (19a) could reach to 3.6 nm. The structure−activity relationships (SAR) study showed that sulfonamide functionality was important for PI3Kα inhibitory activity, and 2-chloro-4-florophenyl sulfonamide (19b), or 5-chlorothiophene-2-sulfonamide (19c) showed potent inhibitory activity with a nanomolar IC50 value. The pyridyl attached to thiazolo[5,4-b]pyridine was another key structural unit for PI3Kα inhibitory potency, and replacement by phenyl lead to a significant decrease in activity. Enzymatic Inhibition results showed that compound 19a inhibited PI3Kα, PI3Kγ, or PI3Kδ with a nanomolar IC50 value, but its inhibitory activity on PI3Kβ was approximately 10-fold reduced. Further docking analysis revealed that the N-heterocyclic core of compound 19a was directly involved in the binding to the kinase through the key hydrogen bonds interaction.
Keywords: heterocycle; thiazolo[5,4-b]pyridine; phosphoinositide 3-kinase; inhibitory potency; docking analysis
1. Introduction
Heterocycles are important organic chemical structures commonly found in a large number of agrochemicals, materials, and pharmaceutical molecules [1,2,3]. Heterocyclic structural units are especially prevalent in synthetic drug molecules because they can improve the interaction between drug molecules and proteins and regulate the physicochemical properties of drugs [4,5,6]. It is estimated that about 70% of pharmaceutical products contain heterocyclic substructures, which clearly shows that heterocycles play an extremely important role in drug development and discovery [7,8].
Over the past few decades, many biological compounds containing bicyclic heterocycles have been found with potent pharmacological activities; moreover, some of them have also been successfully developed into drugs approved for marketing [9,10]. As a typical [5,6]-fused bicyclic scaffold, thiazolo[5,4-b]pyridine has been regarded as an important privileged structure in medicinal chemistry because of its structural similarity to thiazolo[4,5-d]pyrimidine, which is a classic biologically useful skeleton [11]. Furthermore, thiazolo[5,4-b]pyridine analogues have been found to exhibit a range of biological activities, such as S1p1 and S1p5 agonist (1) [12], H3 receptor antagonist (2) [13], DNA gyrase B inhibitor (3) [14], anticancer agent (4) [15], and glucokinase activator (5) [16], among others (Figure 1). A few of them show promising drug-like properties, which has led to increased interest in the design, synthesis, and bioactivity evaluation of thiazolo[5,4-b]pyridine analogues. In this paper, the novel 2-pyridyl, 4-morpholinyl substituted thiazolo[5,4-b]pyridine has been designed and utilized as a template to afford multi-heterocyclic phosphoinositide 3-kinase (PI3K) inhibitors with high potency.
Figure 1.

Selected biological compounds with thiazolo[5,4-b]pyridine skeleton.
The PI3Ks signaling pathway plays a crucial biological function in the process of cell growth, survival, proliferation, and differentiation, which has been proven to be an important target for tumor-targeted therapy [17]. In many tumor cells, the amplification, overexpression, or activating mutation of PI3Kα can lead to abnormal activation of the PI3K signaling pathway, which is a key driving force for tumor occurrence. Given that the PI3K signaling pathway is closely related to tumorigenesis, chemotherapy by inhibiting the PI3K signaling pathway has become an important means of tumor treatment [18], and some potent PI3K inhibitors have been discovered in recent years. It is worth noting that heteroaryl morpholines are common structural cores of PI3K inhibitor molecules—such as PI-103 (6) [19], BKM120 (7) [20], and GDC-0980 (8) [21]—and are representative PI3K inhibitors for the treatment for cancer. Another important core is the 2-methoxyl pyridine unit, which has been seen in PF-04691502 (9) [22] and GSK2126458 (10) [23] as PI3K/mTOR dual inhibitors. In this paper, we designed a new structural template consisting of morpholinyl and 2-methoxyl pyridine to obtain a series of thiazolo[5,4-b]pyridines with high PI3K inhibitory activity. Further analysis of the isoform inhibitory selectivity showed that compound 19a showed highly potent inhibition of PI3kα, PI3kγ, and PI3kδ with an IC50 of 3.4 nM, 1.8 nM, and 2.5 nM, respectively. Furthermore, the IC50 value of PI3kβ was approximately 10-fold higher than the other three (Figure 2).
Figure 2.

Design of 2-pyridyl, 4-morpholinyl substituted thiazolo[5,4-b]pyridine PI3K inhibitor.
2. Results
2.1. Synthesis
An effective and economical method for preparing the thiazolo[5,4-b]pyridine analogues is shown in Figure 3. A commercially available and inexpensive 2,4-dichloro-3-nitropyridine (11) was used as a starting material, and the designed seven-step synthetic route was simple and easy to handle. 2,4-dichloro-3-nitropyridine (11) could be smoothly transformed into 4-morpholinyl pyridine derivative 12 via selective substation with morpholine in the presence of triethylamine. Molecule 12 underwent another substitution with a thiocyanate by treatment with KSCN in acetic acid at 80 °C to successfully afford 4-(3-nitro-2-thiocyanatopyridin-4-yl)morpholine (13). The nitro group was reduced by treatment with Fe powder in acetic acid at 60 °C; the subsequent intramolecular cyclization occurred in one-pot to construct the thiazolo[5,4-b]pyridine skeleton, and the corresponding amino thiazolo[5,4-b]pyridine derivative (14) was obtained in moderate yield. A copper bromide mediated bromination effectively gave the bromothiazolo[5,4-b]pyridine derivative (15) at room temperature, which underwent a Suzuki reaction with aryl borates at 100 °C to afford the desired heterocycle substituted thiazolo[5,4-b]pyridine analogues (19a–19f) in good yields. Sulfonamide substituted aryl borates were prepared from the corresponding amino-substituted aryl bromide through sulfonamidation and boronization in two steps.
Figure 3.

Synthesis of thiazolo[5,4-b]pyridine analogues. (a) Morpholine, TEA, THF, 0 °C. (b) KSCN, HOAc, 80 °C. (c) Fe powder, HOAc, 60 °C. (d) CuBr2, tert-butyl nitrite, CH3CN, rt. (e) DMAP, pyridine, DCM, rt. (f) PdCl2(dppf), KOAc, bis(pinacolato)diboron, 1,4-dioxane, 100 °C. (g) PdCl2(dppf), K2CO3, 1,4-dioxane/H2O, aryl borate, 100 °C. dppf = 1,1′-Bis(diphenylphosphino)ferrocene.
2.2. Enzymatic Assay
All of the N-heterocycle substituted thiazolo[5,4-b]pyridine analogues were evaluated for their PI3Kα inhibitory activity through enzymatic assay. The structure−activity relationships (SAR) are summarized in Table 1. We firstly designed and synthesized an N-heterocyclic compound (19a) composed of methoxypyridine and morpholinyl thiazolo[5,4-b]pyridine. As expected, this thiazolo[5,4-b]pyridine compound exhibited extremely strong PI3kα inhibitory activity with an IC50 of 3.6 nM. In further SAR studies, the sulfonamide functionality was proved to be a key structural unit that affected activity. For example, despite a 2–3 fold drop in potency compared with that of 2,4-difluorophenyl sulfonamide, 2-chloro-4-florophenyl sulfonamide (19b), or 5-chlorothiophene-2-sulfonamide (19c) showed high inhibitory activity with an IC50 of 4.6 nM and 8.0 nM, respectively. This activity was attributed to the fact that the electron-deficient aryl group resulted in a more acidic sulfonamide NH proton being able to make a stronger charged interaction with Lys802 in PI3Kα [24]. Consistent with this finding, when 2,4-difluorophenyl was replaced with methyl (19d), the PI3Kα inhibitory potency dropped over 10-fold (IC50 = 53 nM). Removal of the methoxyl group attached to the pyridine ring lead did not significantly affect the potency (IC50 = 4.0 nM). Replacement of pyridyl with phenyl resulted in an apparent decrease in PI3Kα inhibitory activity (IC50 = 501 nM), demonstrating that the pyridyl attached to thiazolo[5,4-b]pyridine was a necessary moiety for enzymatic potency. We also evaluated the inhibitory selectivity of compound 19a against four isoforms of class I PI3K (Table 2). It potently inhibited PI3Kα, PI3Kγ, and PI3Kδ with nanomolar IC50 values, which is approximately 10-fold higher than that of PI3Kβ, clearly proving that it is a novel and potent PI3K inhibitor.
Table 1.
SAR studies of 2-substitutions on the thiazolo[5,4-b]pyridine.
| Compd | X | R1 | R2 | cLogP a | PSA a | PI3Kα (nm) b |
|---|---|---|---|---|---|---|
| 19a | N | OMe |
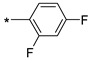
|
4.1 | 105.0 | 3.6 |
| 19b | N | OMe |
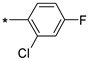
|
4.4 | 105.0 | 4.6 |
| 19c | N | OMe |
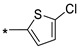
|
4.3 | 105.0 | 8.0 |
| 19d | N | OMe | Me | 2.1 | 105.0 | 53 |
| 19e | CH | OMe |
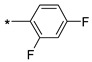
|
4.4 | 92.6 | 501 |
| 19f | N | H |
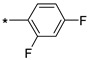
|
3.3 | 95.7 | 4.0 |
a Calculated from ChemBioDraw Ultra 14.0. b Mean of at least three separate experiments.
Table 2.
Enzymatic inhibition by compound 19a a.
| Enzymatic Assay | IC50 (nM) |
|---|---|
| PI3Kɑ | 3.6 |
| PI3Kβ | 34 |
| PI3Kγ | 1.6 |
| PI3Kδ | 2.9 |
a Mean of at least three separate experiments.
2.3. Molecular Docking Study
Because of its superior PI3K potency, thiazolo[5,4-b]pyridine 19a was selected for a molecular docking study. As shown in Figure 4, the 2-pyridyl thiazolo[5,4-b]pyridine scaffold fits well into the ATP binding pocket of the PI3Kα kinase, forming a hydrogen bond interaction with the Val851 residue of the hinge region and a water bridge with Typ836 and Asp810. It is worth noting that another hydrogen bond interaction was observed between the sulfonamide group and Lys802. These interaction networks are the key factors for the PI3K bonding of these pyridyl thiazolo[5,4-b]pyridine analogues.
Figure 4.

Predicted binding mode of 19a with PI3Kα.
3. Materials and Methods
3.1. General
Acetic acid, 1,4-dioxane, acetonitrile and dried THF were purchased from domestic corporations and used without further purification. Aryl borates were synthesized according to our previous work [24]. Nuclear magnetic resonance spectroscopy (1H NMR and 13C NMR) was performed on Bruker Advance 400M NMR spectrometers, and high-resolution LC-MS was carried out by Agilent LC/MSD TOF. Purity of all compounds tested for PI3K activity were determined to be >95% by LCMS analysis. The kinase inhibitory activity assay was performed by Shanghai ChemPartner Co., Ltd. (Shanghai, China). For details, see our previously published protocols [24]. Molecular docking study was conducted in the Schrodinger software.
3.2. Synthesis
3.2.1. Synthesis of 4-(2-chloro-3-nitropyridin-4-yl)morpholine (12)
Morpholine (50 mmol, 4.36 mL) was added dropwise to the mixture of 2,4-dichloro-3-nitropyridine (11, 50 mmol, 9.6 g) and trimethylamine (50 mmol, 7.0 mL) in THF (100 mL) at 0 °C, then the reaction mixture was stirred at 0 °C for 1 h, and the reaction was monitored by LC-MS. When the starting material was consumed completely, solvent was removed under reduced pressure, and the residue was purified with flash column chromatography on silica gel to afford compound 7 (9.1 g, 75% yield). Yellow solid, Rf: 0.5 (EtOAc/Petroleum ether = 1:1), m.p.: 138–140 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (d, J = 6.0 Hz, 1H), 7.25 (d, J = 6.1 Hz, 1H), 3.69–3.64 (m, 4H), 3.25–3.20 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 149.9, 149.7, 142.3, 128.0, 113.9, 65.5, 48.6. HRMS (ESI): m/z (M + H+) calcd for C9H11ClN3O3, 244.0483, found: 244.0492. IR (ATR): 3155, 2965, 2865, 1524, 1356, 1215, 1117, 964, 848, 719 cm−1. The data is consistent with the data reported in the literature. [25]
3.2.2. Synthesis of 4-(3-nitro-2-thiocyanatopyridin-4-yl)morpholine (13)
A mixture of KSCN (48 mmol, 4.7 g) and compound 12 (37 mmol, 9 g) in acetic acid was stirred at 80 °C for 2h. Solvent was removed under reduced pressure, and the residue was purified with flash column chromatography on silica gel to afford compound 8 (5.1 g, 52% yield). Yellow solid, Rf: 0.4 (EtOAc/Petroleum ether = 1:1), m.p.: 177–179 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, J = 6.1 Hz, 1H), 7.32 (d, J = 6.2 Hz, 1H), 3.72–3.64 (m, 4H), 3.35–3.29 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 151.5, 150.2, 149.8, 130.5, 113.4, 109.4, 65.5, 50.2. HRMS (ESI): m/z (M + H+) calcd for C10H11N4O3S, 267.0546, found: 267.0541. IR (ATR): 3109, 2985, 2861, 1585, 1504, 1312, 1209, 1108, 961, 822 cm−1.
3.2.3. Synthesis of 7-morpholinothiazolo[5,4-b]pyridin-2-amine (14)
A mixture of Fe powder (75.2 mmol, 4.2 g) and compound 13 (18.8 mmol, 5 g) in acetic acid was stirred at 60 °C for 2h and filtered. The filtrate was concentrated under reduced pressure, and the residue was purified with flash column chromatography on silica gel to afford compound 9 (2.44 g, 55% yield). Colorless solid, Rf: 0.4 (DCM/MeOH = 20:1). m.p.: 230–232 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, J = 5.5 Hz, 1H), 7.55 (s, 2H), 6.65 (d, J = 5.6 Hz, 1H), 3.78–3.70 (m, 4H), 3.56–3.50 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.1, 155.6, 146.0, 143.0, 135.3, 107.0, 66.0, 48.3. HRMS (ESI): m/z (M + H+) calcd for C10H13N4OS, 237.0805, found: 237.0807. IR (ATR): 3346, 3121, 2961, 1659, 1558, 1297, 1113, 973, 799, 700 cm−1.
3.2.4. Synthesis of 4-(2-bromothiazolo[5,4-b]pyridin-7-yl)morpholine (15)
A mixture of CuBr2 (15 mmol, 3.4 g), tert-butyl nitrite (20 mmol, 2.0 g) and compound 14 (10 mmol, 2.4 g) in acetonitrile (20 mL) was stirred at room temperature for 2h. Water (20 mL) was added, and aqueous layer was extracted by DCM (20 mL × 2). The organic layer was combined and washed with brine and dried over anhydrous MgSO4. DCM was then removed under reduced pressure and the residue was purified with flash column chromatography on silica gel to afford the compound 10 (1.2 g, 40% yield. Colorless solid, Rf: 0.4 (EtOAc/Petroleum ether = 1:1). m.p.: 168–170 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, J = 5.8 Hz, 1H), 6.88 (d, J = 5.9 Hz, 1H), 3.82–3.74 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 161.1, 148.8, 148.2, 134.2, 132.7, 106.9, 65.8, 48.3. HRMS (ESI): m/z (M + H+) calcd for C10H11BrN3OS, 299.9801, found: 299.9802. IR (ATR): 3014, 2948, 2865, 1568, 1463, 1259, 1120, 1008, 956, 803, 669 cm−1.
3.2.5. General Procedure for the Synthesis of Aryl Borates 18a–18f
Sulfonyl chloride (1.2 equiv.) was added to a mixture of compound 16 (20 mmol), DMAP (2 mmol) and pyridine (30 mmol) in the DCM (50 mL), and the reaction; mixture was stirred at room temperature overnight. Solvent was then removed under reduced pressure and the residue was purified with flash column chromatography on silica gel to give the compound 17a–17f. KOAc (25 mmol) was added to a mixture of PdCl2(dppf) (0.63 mmol), compound 17a–17f (12.5 mmol) and bis(pinacolato)diboron (15 mmol) in 1,4-dioxane (25 mL), and the reaction mixture was stirred at 100 °C for 5 h. Solvent was removed under reduced pressure, and DCM (20 mL) was added. Then the mixture was filtered, and the filtrate was concentrated under reduced pressure and the residue was purified with flash column chromatography on silica gel to give the compound 18a–18f.
2,4-Difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (18a). Colorless solid (3.5 g, 66% yield), m.p.: 163–165 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 8.21 (s, 1H), 7.72 (s, 1H), 7.71–7.65 (m, 1H), 7.60–7.52 (m, 1H), 7.22–7.16 (m, 1H), 3.62 (s, 3H), 1.29 (s, 12H). 13C NMR (101 MHz, DMSO-d6). δ 165.0 (dd, J = 255.2, 12.1 Hz), 160.2, 159.3 (dd, J = 258.6, 14.1 Hz), 150.9, 141.0, 131.8 (d, J = 11.1 Hz), 125.0 (dd, J = 15.2, 4.0 Hz), 119.2, 116.7, 111.8 (dd, J = 22.2, 4.0 Hz), 105.7 (t, J = 26.3 Hz), 84.0, 53.4, 24.6. HRMS (ESI): m/z (M + H+) calcd for C18H22BF2N2O5S, 427.1305, found: 427.1314. IR (ATR): 3264, 2989, 2883, 1600, 1406, 1340, 1139, 1019, 969, 850, 670 cm−1.
2-Chloro-4-fluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (18b). Colorless solid (3.4 g, 61% yield), m.p.: 138–140 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.18 (d, J = 1.6 Hz, 1H), 7.85 (dd, J = 8.8, 6.0 Hz, 1H), 7.73 (dd, J = 8.7, 2.5 Hz, 1H), 7.68 (d, J = 1.5 Hz, 1H), 7.34 (td, J = 8.5, 2.5 Hz, 1H), 3.63 (s, 3H), 1.28 (s, 12H). 13C NMR (101 MHz, DMSO-d6) δ 163.8 (d, J = 255.9 Hz), 159.9, 150.6, 140.1, 134.5 (d, J = 3.4 Hz), 133.2 (d, J = 11.6 Hz), 132.9 (d, J = 10.3 Hz), 119.5, 119.2 (d, J = 24.7 Hz), 117.1, 114.5 (d, J = 21.9 Hz), 84.0, 53.4, 24.6. HRMS (ESI): m/z (M + H+) calcd for C18H22BClFN2O5S, 443.1010, found: 443.1001. IR (ATR): 3288, 3103, 2981, 1586, 1341, 1154, 908, 850, 679 cm−1.
5-Chloro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)thiophene-2-sulfonamide (18c). Colorless solid (3.6 g, 67% yield), m.p.: 156–158 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.24 (s, 1H), 7.73 (s, 1H), 7.29 (d, J = 4.1 Hz, 1H), 7.20 (d, J = 4.1 Hz, 1H), 3.71 (s, 3H), 1.29 (s, 12H). 13C NMR (101 MHz, DMSO-d6) δ 160.4, 151.2, 140.4, 139.4, 135.5, 132.5, 128.2, 119.9, 117.2, 84.5, 54.1, 25.1. HRMS (ESI): m/z (M + H+) calcd for C16H21BClN2O5S2, 431.0668, found: 431.0680. IR (ATR): 3251, 3088, 2924, 1599, 1397, 1344, 1253, 1153, 992, 852, 679 cm−1.
N-(2-Methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)methanesulfonamide (18d). Colorless solid (2.4 g, 58% yield), 1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 8.20 (s, 1H), 7.77 (s, 1H), 3.93 (d, J = 3.6 Hz, 3H), 3.01 (s, 3H), 1.29 (s, 12H). 13C NMR (101 MHz, DMSO-d6) δ 158.8, 149.24, 137.0, 121.0, 116.6, 83.9, 73.5, 53.8, 40.4, 24.9, 24.6. HRMS (ESI): m/z (M + H+) calcd for C13H22BN2O5S, 329.1337, found: 329.1318. IR (ATR): 3263, 2979, 1603, 1497, 1392, 1256, 1138, 970, 850, 771, 670 cm−1.
2,4-Difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)benzenesulfonamide (18e). Colorless solid (3.3 g, 62% yield), m.p.: 150–152 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H), 7.64 (dd, J = 14.9, 8.4 Hz, 1H), 7.57–7.47 (m, 3H), 7.16 (td, J = 8.6, 2.1 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 3.49 (s, 3H), 1.29 (s, 12H). 13C NMR (101 MHz, DMSO-d6) δ 164.8 (dd, J = 254.5, 12.1 Hz), 159.4 (dd, J = 258.6, 13.1 Hz), 156.5, 134.8, 134.5, 131.8 (d, J = 11.1 Hz), 125.3 (dd, J = 14.1, 4.0 Hz), 123.7, 120.1, 111.4 (dd, J = 22.2, 3.0 Hz), 111.3, 105.5 (t, J = 30.3 Hz), 83.6, 55.2, 24.7. HRMS (ESI): m/z (M + H+) calcd for C19H23BF2NO5S, 426.1353, found: 426.1369. IR (ATR): 3300, 2925, 2850, 1601, 1340, 1258 1167, 1129, 850, 669 cm−1.
3.2.6. General Procedure for the Synthesis of Target Compounds 19a–19f
K2CO3 aqueous solution (2N, 0.39 mL) was added to a mixture of PdCl2(dppf) (0.03 mmol), compound 15 (0.26 mmol) and Aryl borate 18 (0.31 mmol) in 1,4-dioxane (2.5 mL), and the reaction mixture was stirred at 100 °C for 5h. Solvent was removed under reduced pressure, and DCM (20 mL) was added. Then the mixture was filtered, and the filtrate was concentrated under reduced pressure and the residue was purified with flash column chromatography on silica gel to give compound 19. 1H NMR and 13CNMR spectra of the compounds 19a–19f, and the HPLC spectra of compound 19a can be found in the Supplementary Materials.
2,4-Difluoro-N-(2-methoxy-5-(7-morpholinothiazolo[5,4-b]pyridin-2-yl)pyridin-3-yl)benzenesulfonamide (19a). Colorless solid (103 mg, 76% yield), Rf: 0.5 (DCM/MeOH = 30:1), m.p.: 175–177 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 8.62 (d, J = 2.1 Hz, 1H), 8.21 (d, J = 5.7 Hz, 1H), 8.11 (d, J = 2.1 Hz, 1H), 7.80 (dd, J = 14.9, 8.5 Hz, 1H), 7.64–7.56 (m, 1H), 7.25 (td, J = 8.6, 2.1 Hz, 1H), 6.86 (d, J = 5.7 Hz, 1H), 3.88–3.78 (m, 8H), 3.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.1 (dd, J = 255.5 Hz, 12.1 Hz), 159.4, 159.3 (dd, J = 258.6 Hz, 14.1 Hz), 158.9, 157.2, 149.3, 147.9, 142.7, 135.2, 131.8 (d, J = 11.1 Hz), 131.2, 124.8 (dd, J = 14.1 Hz, 3.0 Hz), 123.4, 120.5, 112.1 (dd, J = 22.2 Hz, 3.0 Hz), 106.4, 105.9 (t, J = 26.3 Hz), 65.9, 54.0, 48.5. HRMS (ESI): m/z (M + H+) calcd for C22H20F2N5O4S2, 520.0919, found: 520.0909. IR (ATR): 3183, 2922, 2854, 1602, 1423, 1261, 1149, 971, 864, 715, 668 cm−1.
2-Chloro-4-fluoro-N-(2-methoxy-5-(7-morpholinothiazolo[5,4-b]pyridin-2-yl)pyridin-3-yl)benzenesulfonamide (19b). Colorless solid (102 mg, 73% yield), Rf: 0.5 (DCM/MeOH = 30:1), m.p.: 188–190 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 8.60 (d, J = 2.1 Hz, 1H), 8.20 (d, J = 5.7 Hz, 1H), 8.07 (d, J = 2.1 Hz, 1H), 7.97 (dd, J = 8.9, 5.9 Hz, 1H), 7.77 (dd, J = 8.7, 2.4 Hz, 1H), 7.40 (td, J = 8.5, 2.5 Hz, 1H), 6.86 (d, J = 5.7 Hz, 1H), 3.88–3.78 (m, 8H), 3.77 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.9 (d, J = 255.5 Hz), 159.3, 158.6, 157.2, 149.3, 148.0, 142.4, 135.2, 134.3 (d, J = 3.0 Hz), 133.1 (d, J = 6.1 Hz), 133.0 (d, J = 4.0 Hz), 130.3, 123.3, 120.9, 119.4 (d, J = 26.3 Hz), 114.7 (d, J = 22.2 Hz), 106.4, 65.9, 54.0, 48.5. HRMS (ESI): m/z (M + H+) calcd for C22H20ClFN5O4S2, 536.0624, found: 536.0612. IR (ATR): 3376, 3008, 2921, 1568, 1471, 1341, 1253, 1157, 977, 803, 661 cm−1.
5-Chloro-N-(2-methoxy-5-(7-morpholinothiazolo[5,4-b]pyridin-2-yl)pyridin-3-yl)thiophene-2-sulfonamide (19c). Colorless solid (95 mg, 70% yield), Rf: 0.5 (DCM/MeOH = 30:1), m.p.: 116–118 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.63 (d, J = 1.9 Hz, 1H), 8.21 (d, J = 5.6 Hz, 1H), 8.14 (d, J = 2.0 Hz, 1H), 7.44 (d, J = 4.1 Hz, 1H), 7.25 (d, J = 4.1 Hz, 1H), 6.87 (d, J = 5.7 Hz, 1H), 3.91–3.80 (m, 11H). 13C NMR (101 MHz, DMSO-d6) δ 159.3, 158.5, 157.4, 149.3, 148.0, 142.3, 138.8, 135.4, 135.3, 132.4, 129.4, 128.1, 123.4, 121.0, 106.5, 66.0, 54.2, 48.5. HRMS (ESI): m/z (M + H+) calcd for C20H20ClN5O4S3, 524.0282, found: 524.0285. IR (ATR): 3350 3015 2921, 1566, 1406, 1341, 1256, 1149, 976, 802, 676 cm−1.
N-(2-Methoxy-5-(7-morpholinothiazolo[5,4-b]pyridin-2-yl)pyridin-3-yl)methanesulfonamide (19d). Colorless solid (73 mg, 67% yield), Rf: 0.5 (DCM/MeOH = 30:1),m.p.: 230–232 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 8.63 (d, J = 2.2 Hz, 1H), 8.24–8.20 (m, 2H), 6.89 (d, J = 5.7 Hz, 1H), 4.02 (s, 3H), 3.92–3.80 (m, 8H), 3.13 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 159.4, 157.7, 157.6, 149.3, 148.0, 140.9, 135.3, 128.1, 123.5, 122.4, 106.5, 65.9, 54.3, 48.5, 40.6. HRMS (ESI): m/z (M + H+) calcd for C17H20N5O4S2, 422.0951, found: 422.0944. IR (ATR): 3215, 2998, 2882, 1585, 1464, 1393, 1287, 1137, 1003, 841, 771, 684 cm−1.
2,4-Difluoro-N-(2-methoxy-5-(7-morpholinothiazolo[5,4-b]pyridin-2-yl)phenyl)benzenesulfonamide (19e). Colorless solid (101 mg, 75% yield), Rf: 0.5 (DCM/MeOH = 30:1), m.p.: 195–197 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.19 (d, J = 5.6 Hz, 1H), 7.84–7.80 (m, 2H), 7.74 (dd, J = 14.9, 8.5 Hz, 1H), 7.61–7.54 (m, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H), 6.86 (d, J = 5.7 Hz, 1H), 3.89–3.79 (m, 8H), 3.62 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.0 (dd, J = 253.5 Hz, 14.1 Hz), 160.0, 159.3, 159.2 (dd, J = 258.6 Hz, 15.2 Hz), 155.6, 149.3, 147.7, 135.6, 131.8 (d, J = 1.1 Hz), 126.6, 125.5, 125.3, 125.1 (dd, J = 14.1 Hz, 4.0 Hz), 124.7, 112.5, 111.8 (dd, J = 23.2 Hz, 4.0 Hz), 106.5,105.8 (t, J = 26.3 Hz), 65.9, 55.8, 48.5. HRMS (ESI): m/z (M + H+) calcd for C23H21F2N4O4S2, 519.0967, found: 519.0970. IR (ATR): 3268, 2922, 2844, 1567, 1426, 1336, 1272, 1122, 969, 816, 670 cm−1.
2,4-Difluoro-N-(5-(7-morpholinothiazolo[5,4-b]pyridin-2-yl)pyridin-3-yl)benzenesulfonamide (19f). Colorless solid (57 mg, 45% yield), Rf: 0.3 (DCM/MeOH = 30:1). m.p.: 99–101 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H), 8.87 (s, 1H), 8.47 (d, J = 2.0 Hz, 1H), 8.23 (d, J = 5.7 Hz, 1H), 8.08 (s, 1H), 8.00 (dd, J = 14.8, 8.5 Hz, 1H), 7.61–7.54 (m, 1H), 7.32 (t, J = 8.4 Hz, 1H), 6.89 (d, J = 5.7 Hz, 1H), 3.92–3.80 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 165.5 (dd, J = 256.5, 12.1 Hz), 159.6, 159.0 (dd, J = 258.6, 14.1 Hz), 156.9, 149.6, 148.5, 142.9, 142.5, 135.3, 134.6, 132.4 (d, J = 10.1 Hz), 129.1, 123.4 (dd, J = 14.1, 3.0 Hz), 123.2, 112.7 (dd, J = 22.2, 4.0 Hz), 106. (t, J = 25.3 Hz), 65.9, 48.5. HRMS (ESI): m/z (M + H+) calcd for C21H18F2N5O3S2, 490.0814, found: 490.0805. IR (ATR): 3000, 2928, 1557, 1476, 1425, 1343, 1266, 1068, 961, 846, 669 cm−1.
4. Conclusions
In summary, we have designed and synthesized a series of 2-pyridyl, 4-morpholinyl substituted thiazolo[5,4-b]pyridines by pharmacophore splicing of methoxyl pyridine and morpholinyl heterocyclic inhibitors. These molecules were evaluated for the inhibitory activity of kinases. It was found that compound 19a exhibited nanomolar inhibitory activity against the three isoforms of PI3Kα, PI3Kγ, and PI3Kδ. Further docking study demonstrated that compound 19a fit well into the ATP binding pocket of the PI3Kα kinase. Overall, these novel thiazolo[5,4-b]pyridines may be potentially used as the potent PI3K inhibitors for the treatment of disease mediated by the PI3K signaling pathway after further activity evaluation in vivo and optimization of their pharmacological properties.
Acknowledgments
We thank Xudong Wu for his help on molecular modeling.
Supplementary Materials
1H NMR and 13CNMR spectra of the compounds 19a–19f, and the HPLC spectra of compound 19a.
Author Contributions
L.X., Y.Z., J.Z., S.L., K.Z., and H.T. performed the experiments and analyzed the data; H.X. and Y.D. conceived and designed the study; all authors contributed to writing and editing of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was Funded by the National Natural Science Foundation of China (81703329 and 21702234), the CAMS Innovation Fund for Medical Sciences (2017-I2M-3-011 and 2019-I2M-1-005), the Drug Innovation Major Project (2018ZX09711-001-005), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018PT35003 and 2019RC-HL-008), and the Fundamental Research Funds for the Central Universities (3332020041).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Balaban A.T. Aromaticity as a Cornerstone of Heterocyclic Chemistry. Chem. Rev. 2004;104:2777–2812. doi: 10.1021/cr0306790. [DOI] [PubMed] [Google Scholar]
- 2.Taylor R.D., MacCoss M., Lawson A.D.G. Rings in Drugs. J. Med. Chem. 2014;57:5845–5859. doi: 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- 3.Mullard A. 2012 FDA Drug Approvals. Nat. Rev. Drug Discov. 2013;12:87–90. doi: 10.1038/nrd3946. [DOI] [PubMed] [Google Scholar]
- 4.Das P., Delost M.D., Qureshi M.H., Smith D.T., Njardarson J.T. A Survey of the Structures of US FDA Approved Combination Drugs. J. Med. Chem. 2019;62:4265–4311. doi: 10.1021/acs.jmedchem.8b01610. [DOI] [PubMed] [Google Scholar]
- 5.Ertl P., Jelfs S., Mühlbacher J., Schuffenhauer A., Selzer P. Quest for the Rings. In Silico Exploration of Ring Universe To Identify Novel Bioactive Heteroaromatic Scaffolds. J. Med. Chem. 2006;49:4568–4573. doi: 10.1021/jm060217p. [DOI] [PubMed] [Google Scholar]
- 6.Pitt W.R., Parry D.M., Perry B.G., Groom C.R. Heteroaromatic Rings of the Future. J. Med. Chem. 2009;52:2952–2963. doi: 10.1021/jm801513z. [DOI] [PubMed] [Google Scholar]
- 7.Gibson S., McGuire R., Rees D.C. Principal Components Describing Biological Activities and Molecular Diversity of Heterocyclic Aromatic Ring Fragments. J. Med. Chem. 1996;39:4065–4072. doi: 10.1021/jm960058h. [DOI] [PubMed] [Google Scholar]
- 8.Kalaria P.N., Karad S.C., Raval D.K. A Review on Diverse Heterocyclic Compounds as the Privileged Scaffolds in Antimalarial Drug Discovery. Eur. J. Med. Chem. 2018;5:917–936. doi: 10.1016/j.ejmech.2018.08.040. [DOI] [PubMed] [Google Scholar]
- 9.Vitaku E., Smith D.T., Njardarson J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 10.Taylor A.P., Robinson R.P., Fobian Y.M., Blakemore D.C., Jones L.H., Fadeyi O. Modern Advances in Heterocyclic Chemistry in Drug Discovery. Org. Biomol. Chem. 2016;14:6611–6637. doi: 10.1039/C6OB00936K. [DOI] [PubMed] [Google Scholar]
- 11.Kuppast B., Fahmy H. Thiazolo[4,5-d]pyrimidines as a Privileged Scaffold in Drug Discovery. Eur. J. Med. Chem. 2016;113:198–213. doi: 10.1016/j.ejmech.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 12.Cee V.J., Frohn M., Lanman B.A., Golden J., Muller K., Neira S., Pickrell A., Arnett H., Buys J., Gore A., et al. Discovery of AMG 369, a Thiazolo[5,4-b]pyridine Agonist of S1P1 and S1P5. ACS Med. Chem. Lett. 2011;2:107–112. doi: 10.1021/ml100306h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao A.U., Palani A., Chen X., Huang Y., Aslanian R.G., West R.E., Jr., Williams S.M., Wu R., Hwa J., Sondey C., et al. Synthesis and Structure–activity Relationships of 2-(1,4′-bipiperidin-1′-yl)thiazolopyridine as H3 Receptor Antagonists. Bioorg. Med. Chem. Lett. 2009;19:6176–6180. doi: 10.1016/j.bmcl.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Kale M.G., Raichurkar A., Hameed P.S., Waterson D., McKinney D., Manjunatha M.R., Kranthi U., Koushik K., Jena L.K., Shinde V., et al. Thiazolopyridine Ureas as Novel Antitubercular Agents Acting through Inhibition of DNA Gyrase, B. J. Med. Chem. 2013;56:8834–8848. doi: 10.1021/jm401268f. [DOI] [PubMed] [Google Scholar]
- 15.Xie X., Li H., Wang J., Mao S., Xin M., Lu S., Mei Q., Zhang S. Synthesis and Anticancer Effects Evaluation of 1-alkyl-3-(6-(2-methoxy-3-sulfonylaminopyridin-5-yl)benzo[d]thiazol-2-yl)urea as Anticancer Agents with Low Toxicity. Bioorg. Med. Chem. 2015;23:6477–6485. doi: 10.1016/j.bmc.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 16.Bebernitz G.R., Beaulieu V., Dale B.A., Deacon R., Duttaroy A., Gao J., Grondine M.S., Gupta R.C., Kakmak M., Kavana M., et al. Investigation of Functionally Liver Selective Glucokinase Activators for the Treatment of Type 2 Diabetes. J. Med. Chem. 2009;52:6142–6152. doi: 10.1021/jm900839k. [DOI] [PubMed] [Google Scholar]
- 17.Liu P., Cheng H., Roberts T.M., Zhao J.J. Targeting the Phosphoinositide 3-kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M.D. Cellular Function of Phosphoinositide 3-kinases: Implications for Development, Homeostasis, and Cancer. Annu. Rev. Cell Dev. Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 19.Fan Q.W., Knight Z.A., Goldenberg D.D., Yu W., Mostov K.E., Stokoe D., Shokat K.M., Weiss W.A. A Dual PI3 Kinase/mTOR Inhibitor Reveals Emergent Efficacy in Glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burger M.T., Pecchi S., Wagman A., Ni Z.J., Knapp M., Hendrickson T., Atallah G., Pfister K., Zhang Y., Bartulis S., et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011;2:774–779. doi: 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutherlin D.P., Bao L., Berry M., Castanedo G., Chuckowree I., Dotson J., Folks A., Friedman L., Goldsmith R., Gunzner J., et al. Discovery of a Potent, Selective, and Orally Available Class I Phosphatidylinositol 3-kinase (PI3K)/mammalian Target of Rapamycin (mTOR) Kinase Inhibitor (GDC-0980) for the Treatment of Cancer. J. Med. Chem. 2011;54:7579–7587. doi: 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- 22.Cheng H.M., Bagrodia S., Bailey S., Edwards M., Hoffman J., Hu Q.Y., Kania R., Knighton D.R., Marx M.A., Ninkovic S., et al. Discovery of the Highly Potent PI3K/mTOR Dual Inhibitor PF-04691502 Through Structure Based Drug Design. Med. Chem. Comm. 2010;1:139–144. doi: 10.1039/c0md00072h. [DOI] [Google Scholar]
- 23.Knight S.D., Adams N.D., Burgess J.L., Chaudhari A.M., Darcy M.G., Donatelli C.A., Luengo J.I., Newlander K.A., Parrish C.A., Ridgers L.H., et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010;1:39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin S., Wang C., Ji M., Wu D., Lv Y., Zhang K., Dong Y., Jin J., Chen J., Zhang J., et al. Discovery and Optimization of 2-Amino-4-methylquinazoline Derivatives as Highly Potent Phosphatidylinositol 3-Kinase Inhibitors for Cancer Treatment. J. Med. Chem. 2018;61:6087–6109. doi: 10.1021/acs.jmedchem.8b00416. [DOI] [PubMed] [Google Scholar]
- 25.McDonald S.L., Hendrick C.E., Wang Q. Copper-Catalyzed Electrophilic Amination of Heteroarenes and Arenes by C−H Zincation. Angew. Chem. Int. Ed. 2014;53:4667–4670. doi: 10.1002/anie.201311029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.