

Abstract

In this study, the synthesis of N-alkyl-2-halophenazin-1-ones has been established. Six N-alkyl-2-halophenazin-1-ones, including WS-9659 B and marinocyanins A and B, were synthesized by the direct oxidative condensation of 4-halo-1,2,3-benzenetriol with the corresponding N-alkylbenzene-1,2-diamines. One of the most significant features of the present method is that it can be successfully applied to the synthesis of N-alkyl-2-chlorophenazin-1-ones. The traditional chlorination of N-alkyl-phenazin-1-ones with N-chlorosuccinimide selectively occurs at the 4-position to afford the undesired N-alkyl-4-chlorophenazin-1-ones. Our synthetic route successfully circumvents this problem, culminating in the first chemical synthesis of WS-9659 B. The cytotoxicity of six N-alkyl-2-halophenazin-1-ones and three N-alkylphenazin-1-ones against human promyelocytic leukemia HL-60, human lung cancer A549, and normal MRC-5 cells was evaluated. Among the compounds tested in this study, 2-chloropyocyanin possesses significant selectivity toward A549 cells. The cytotoxic evaluation provides structural insights into the potency and selectivity of these compounds for cancer cells.
Introduction
Phenazines are a large group of nitrogen-containing heterocycles. To date, more than 100 phenazine derivatives have been identified in nature and over 6000 phenazine compounds synthesized.1−4N-Alkylphenazin-1-ones are a minor group of the phenazines. To the best of our knowledge, only four N-alkylphenazin-1-ones [pyocyanin,5−10 lavanducyanin (WS-9659 A),11−15 phenazinomycin,16,17 and 1-hydroxy-7-oxolavanducyanin18 and seven N-alkyl-2-halophenazin-1-ones (WS-9659 B12−14 and marinocyanins A-F15,19) have been identified as natural products. In the present study, we focused on N-alkylphenazin-1-ones and N-alkyl-2-halophenazin-1-ones, which are depicted in Figure 1. Among them, pyocyanin (1a), lavanducyanin (WS-9659 A) (2a), WS-9659 B (2c), and marinocyanins A (2b) and B (3b) are natural products, which were isolated from Pseudomonas and Streptomyces species (Figure 1).3,4 One of the simplest and best-known N-alkylphenazin-1-ones is pyocyanin (1a) isolated from Pseudomonas aeruginosa.5,6 Pyocyanin shows several biological activities including antibacterial, antifungal, and cytotoxic activities.5 This compound plays an important role in biofilm formation and quorum sensing in P. aeruginosa.6−10 Lavanducyanin (2a), isolated from the Streptomyces sp., shows cytotoxic activities against mouse leukemia P388 and mouse lymphoid leukemia L1210 cells with half-maximal inhibitory concentration (IC50) values of 0.09 and 0.10 μg/mL, respectively.11 The same compound and its 2-chlorinated derivative, respectively, named as WS-9659 A (2a) and WS-9659 B (2c), were isolated from the Streptomyces sp. as testosterone 5α-reductase inhibitors with IC50 values of 0.5 and 10 μM, respectively.12−14 Marinocyanins A (2b) and B (3b), 2-brominated derivatives, were isolated from a marine-derived Streptomyces sp. and actinomycete strains CNS-284 and CNY-960.15,19 Compounds 2b and 3b inhibited TNF-α-induced NFκB activity (IC50 values of 4.1 and 24.2 μM, respectively) and LPS-induced nitric oxide production (IC50 values of >48.6 and 15.1 μM, respectively).15 They blocked PGE2 production with greater efficacy (IC50 values of 7.5 and 0.89 μM, respectively).15 Compounds 2b and 3b also showed strong cytotoxic activity against human colon carcinoma HCT116 cells with IC50 values of 0.049 and 0.029 μM, respectively.19 In addition, both these compounds display modest antimicrobial activity against Staphylococcus aureus and the pathogenic yeast Candida albicans.19
Figure 1.

Structures of our target N-alkylphenazin-1-ones.
Because of these diverse biological activities, N-alkylphenazin-1-ones are attractive synthetic targets.3,4,20−22 Our group recently reported the synthesis of 1a, 2a, 2b, 3a, and 3b.22 The oxidative condensation of pyrogallol with N-alkylbenzene-1,2-diamines (4, 5, and 6) under an oxygen atmosphere in 2-propanol afforded 1a, 2a, and 3a, respectively (Scheme 1). Bromination of the resultant phenazin-1-ones (2a and 3a) proceeded at the 2-position in a regioselective manner to afford 2b and 3b, respectively.
Scheme 1. Syntheses of Pyocyanin (1a), Lavanducyanin (2a), and Marinocyanins A (2b) and B (3b).

In the present study, we describe the synthesis of N-alkyl-2-halophenazin-1-ones by coupling 4-halo-1,2,3-benzenetriol with the corresponding N-alkylbenzene-1,2-diamines. The first synthesis of WS-9659 B (2c) was achieved by oxidative condensation of 4-chloro-1,2,3-benzenetriol with the corresponding N-alkylbenzene-1,2-diamine. Furthermore, the cytotoxic activities of synthetic N-alkylphenazin-1-ones and N-alkyl-2-halophenazin-1-ones were evaluated.
Results and Discussion
Halogenation of Pyocyanin and Lavanducyanin
To obtain 2-chlorinated N-alkylphenazin-1-ones, halogenation of pyocyanin (1a) was examined. A drastic change in the regioselectivity between bromination and chlorination was observed (Scheme 2). Bromination of 1a with N-bromosuccinimide (NBS) in CH2Cl2 afforded 2-bromopyocyanin (1b) in 51% yield. However, chlorination of 1a with N-chlorosuccinimide (NCS) gave 4-chloropyocyanin (7) in 65% yield. The position of the chlorine atom in 7 was confirmed by nuclear Overhauser effect spectroscopy (NOESY) correlations. NOESY correlations between H-2 (δH 6.47) and H-3 (δH 7.44) and between the H-6 (δH 7.40) and N-methyl group (δH 4.07) are observed (Figures 2, S32 and S33 in the Supporting Information). The difference in outcome can presumably be attributed to steric hindrance between the N-methyl group in 1a and the halogen atom. The van der Waals radius of a chlorine atom (1.75 Å) is smaller than that of a bromine atom (1.85 Å).23,24 Consequently, electrophilic substitution with the sterically large bromine atom at the 4-postion is disfavored. The chlorination of lavanducyanin (2a) with NCS also proceeded at the 4-position in a regioselective manner to give 8 in 48% yield (Scheme 3). The position of the chlorine atom in 8 was also confirmed by NOESY correlations. NOESY correlations between H-2 (δH 6.49) and H-3 (δH 7.46) and between the H-6 (δH 7.27–7.25) and N-methylene group (δH 5.29) are observed (Figures 2 and S39 in the Supporting Information). Thus, an alternative methodology for the synthesis of WS-9659 B (2c) is needed.
Scheme 2. Selective Halogenation of Pyocyanin with NBS and NCS.

Figure 2.

Selected NOESY correlations observed in 7 and 8.
Scheme 3. Chlorination of Lavanducyanin (2a).

Oxidative Condensation of 4-Halo-1,2,3-benzenetriol with N-Alkylbenzene-1,2-diamines
To obtain N-alkyl-2-chlorophenazin-1-ones, reaction conditions for the oxidative condensation between the N-methylbenzene-1,2-diamine (4)22 and 4-chloro-1,2,3-benzenetriol (9)25 were examined (Table 1). When 4 and 9 were reacted under an oxygen atmosphere, the desired 2-chloropyocyanin (1c) was obtained in 12% yield (entry 1). NOESY correlations between the H-4 (δH 6.27) and N-methyl group (δH 4.04) and between the H-6 (δH 8.10) and N-methyl group in 1c indicate the presence of the chlorine atom at the 2-position (Figures 3 and S11 in the Supporting Information). The use of 1,4-benzoquinone as an oxidant gave 1c in 10% yield (entry 2). When o-chloranil was used in this reaction, only a trace amount of 1c was obtained. These results indicate that molecular oxygen is the optimal oxidant. The addition of bases to the oxidative coupling with molecular oxygen was also examined (entries 4–6). The yield of 1c was decreased by addition of Na2CO3 or K2CO3 (entries 4 and 5, respectively) but slightly increased by addition of Cs2CO3 (entry 6). Other reaction conditions including the base, solvent, and temperature were also investigated but the yield of 1c was not improved. Thus, the entry 6 gave the optimal conditions.
Table 1. Oxidative Condensation of 4-Chloro-1,2,3-benzenetriol (9) with N-Methylbenzene-1,2-diamine (4)a.

| entry | oxidant | additive | yield (%)b |
|---|---|---|---|
| 1c | O2 | 12 | |
| 2d | 1,4-benzoquinone | 10 | |
| 3d | o-chloranil | trace | |
| 4e | O2 | Na2CO3 | 5 |
| 5e | O2 | K2CO3 | 10 |
| 6e | O2 | Cs2CO3 | 17 |
Unless otherwise noted, reactions were performed using 4 (0.246 mmol) and 9 (1.0 equiv) in 2-propanol (2.0 mL).
Isolated yield.
The reaction was performed at room temperature (rt) under an oxygen atmosphere.
The reaction was performed at 0 °C under an argon atmosphere using the oxidant (3.0 equiv) indicated.
The reaction was performed at rt under an oxygen atmosphere using the additive (1.0 equiv) indicated.
Figure 3.
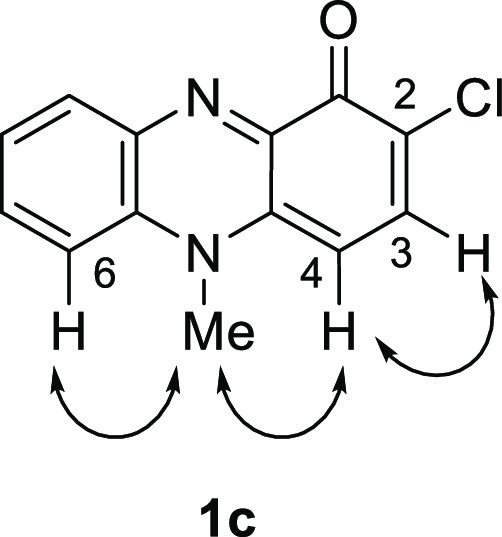
Selected NOESY correlations observed in 1c.
Using optimal conditions, several N-alkyl-2-halophenazin-1-ones were prepared (Table 2). The first synthesis of WS-5659 B (2c) was achieved by coupling between 5 and 9 (entry 1). The 1H and 13C NMR spectroscopic data were identical to those reported for natural 2c.12 Oxidative coupling of 9 with 6 gave 3c in 21% yield (entry 2). The desired oxidative coupling also proceeded using 4-bromo-1,2,3-benzenetriol (10)26 as a coupling partner to give N-alkyl-2-bromophenazin-1-ones. The coupling between 4 and 10 afforded 2-bromopyocyanin (1b) in 18% yield (entry 3). Marinocyanins A (2b) and B (3b) were synthesized by oxidative coupling of 10 with 5 and 6, respectively (entries 4 and 5).
Table 2. Oxidative Condensation of 4-Halo-1,2,3-benzenetriols with N-Alkylbenzene-1,2-diaminea.

| entry | 1,2-diamine | 4-halo-1,2,3-benzenetriols | product | yield (%)b |
|---|---|---|---|---|
| 1c | 5 | 9 | 2c | 20 |
| 2d | 6 | 9 | 3c | 21 |
| 3d | 4 | 10 | 1b | 18 |
| 4c | 5 | 10 | 2b | 10 |
| 5d | 6 | 10 | 3b | 15 |
Unless otherwise noted, reactions were performed at rt, with O2 bubbling or under an oxygen atmosphere, using N-alkylbenzene-1,2-diamine (30.0 mg) and 4-halo-1,2,3-benzenetriols (1.0 equiv) in the presence of Cs2CO3 (1.0 equiv).
Isolated yield.
The reaction was performed with oxygen bubbling.
The reaction was performed under an oxygen atmosphere.
Evaluation of Cytotoxic Activity against Human Cancer Cells and Normal Cells
Initially, the cytotoxic activity of 1a, 1b, and 1c against human promyelocytic leukemia HL-60 cells, human lung adenocarcinoma A549 cells, and human normal lung fibroblast MRC-5cells was evaluated using the WST-8 method.27 The concentrations of the compound required to reduce cell viability by 50% (IC50) are summarized in Table 3. Table 3 also shows the values of the selectivity index (SI),28−30 which is defined as the ratio of the IC50 value against normal cells to the corresponding IC50 value against cancer cells of similar tissue. Because both A549 and MRC-5 are derived from the lung, the SI value in Table 3 is defined as the IC50 value against normal MRC-5 cells divided by the IC50 value against A549 cells. Generally, the compounds with a SI value higher than 2 are considered to give selective cytotoxicity against cancer cells.28 SN-38, the active metabolite of irinotecan, was used as a positive control.31 The cytotoxicity against normal MRC-5 cells of 1a was less-potent than that against leukemia HL-60 cells and cancer A549 cells. The SI value of 1a (SI = 3.95) indicates a significant selectivity toward cancer cells. However, the cytotoxicity of 1b against normal MRC-5 cells was almost the same as that against leukemia HL-60 cells and cancer A549 cells. Among compounds 1a–c, the compound 1c exhibited the most potent and selective cytotoxicity against cancer A549 cells. At least 5.8-fold and 4.6-fold higher concentrations of 1c are required to induce cytotoxicity against MRC-5 cells, as compared to those required against A549 and HL-60 cells, respectively. The differential cytotoxicity of 1c was highly significant (SI = 5.80), suggesting that 1c is considered as a candidate for the development of chemotherapeutic agents. Pyocyanin (1a) has been reported to diffuse through the cell membrane and increase the intracellular ROS and Ca2+ levels.32 The presence of the chlorine atom at the 2-position in 1a may enhance the generation of ROSs or increase the chemical reactivity or binding affinity with the target molecules in A549 cells.
Table 3. Cytotoxic Activity of 1a–1c against Human Cancer Cells and Normal Cells (IC50, μM) and Their Selective Indexesa.
| IC50 (μM) |
||||
|---|---|---|---|---|
| compounds | HL-60 | A549 | MRC-5 | SIc |
| 1a | 5.82 ± 0.133 | 5.52 ± 0.280 | 21.8 ± 2.550 | 3.95 |
| 1b | 3.07 ± 0.080 | 2.59 ± 0.056 | 3.36 ± 0.204 | 1.30 |
| 1c | 3.56 ± 0.052 | 0.76 ± 0.065 | 4.41 ± 0.069 | 5.80 |
| SN-38b | <0.05 | 0.038 ± 0.003 | <0.1 | |
The concentration of the compound needed to reduce cell viability by 50% (IC50) was determined using the WST-8 assay. IC50 values are expressed as the mean ± SD of triplicate experiments.
SN-38 was used as a positive control.
The SI is defined as the IC50 value against normal MRC-5 cells divided by the IC50 value against A549 cells.
Next, the cytotoxic activity of 2a–c and 3a–c against HL-60, A549, and MRC-5 cells was evaluated (Table 4). Although these compounds showed cytotoxicity against HL-60 cells, compounds 3a and 3c displayed weaker activity than the other compounds. Compounds 2a–c and 3a–c exhibited cytotoxicity against A549 and MRC-5 cells at concentrations lower than 1 μM. Unfortunately, no significant difference in the IC50 values between these compounds was observed. The SI values of 2a–c and 3a–c are lower than 2. Selectivity against A549 cells was not observed. The displacement of the methyl group with the longer alkyl groups at the nitrogen atom in 1a and 1c decreased the selective cytotoxicity toward A549 cells.
Table 4. Cytotoxic Activity of 2 and 3 against Human Cancer Cells and Normal Cells (IC50, μM) and Their Selective Indexesa.
| IC50 (μM) |
||||
|---|---|---|---|---|
| compounds | HL-60 | A549 | MRC-5 | SIc |
| 2a | 0.36 ± 0.023 | 0.38 ± 0.003 | 0.68 ± 0.013 | 1.79 |
| 2b | 0.27 ± 0.017 | 0.73 ± 0.015 | 0.60 ± 0.008 | 0.82 |
| 2c | 0.23 ± 0.013 | 0.75 ± 0.006 | 0.37 ± 0.019 | 0.49 |
| 3a | 2.59 ± 0.195 | 0.88 ± 0.023 | 0.87 ± 0.063 | 0.99 |
| 3b | 0.67 ± 0.038 | 0.33 ± 0.010 | 0.54 ± 0.009 | 1.64 |
| 3c | 2.20 ± 0.070 | 0.66 ± 0.016 | 0.66 ± 0.041 | 1.00 |
| SN-38b | 0.056 ± 0.002 | 0.052 ± 0.003 | 0.092 ± 0.001 | 1.77 |
The concentration of the compound needed to reduce cell viability by 50% (IC50) was determined using the WST-8 assay. IC50 values are expressed as the mean ± SD of triplicate experiments.
SN-38 was used as a positive control.
The SI is defined as the IC50 value against normal MRC-5 cells divided by the IC50 value against A549 cells.
Conclusions
In this study, the synthesis of N-alkyl-2-halophenazin-1-one was established. Six N-alkyl-2-halophenazin-1-ones, including WS-9659 B (2c) and marinocyanins A (2b) and B (3b), were synthesized by the oxidative condensation of 4-halo-1,2,3-benzenetriols with N-alkylbenzene-1,2-diamines. The present strategy enables the highly convergent and selective synthesis of N-alkyl-2-halophenazin-1-ones. In addition, the cytotoxicity of compounds 1a–c, 2a–c, and 3a–c against human leukemia HL-60 cells, cancer A549 cells, and normal MRC-5 cells was evaluated. Among the compounds tested in this study, 2-chloropyocyanin (1c) exhibited the most selective cytotoxicity against A549 cells. The high SI value of 1c (SI = 5.80) suggests that 1c is considered as a potential candidate for the development of chemotherapeutic agents. Halogenated phenazines have recently attracted increasing attention as a promising class of biofilm-eradicating agents that effectively target multiple persistent bacterial phenotypes.33,34 Thus, halogenation of phenazines and phenazinones is one of the valuable approaches for improving their biological activities.35 The present study will be invaluable not only for analyzing the molecular and cellular biological activities of N-alkylphenazin-1-ones and their halogenated derivatives but also in the design of more potent and selective cytotoxic derivatives against cancer cells. Additional biological studies to investigate the mechanism of action of these compounds are currently underway.
Experimental Section
General Information
All reactions sensitive to air or moisture were carried out under an argon atmosphere under anhydrous conditions, unless otherwise noted. Solvents and reagents were used without further purification unless otherwise noted. Analytical thin-layer chromatography (TLC) was performed using silica gel 60 F254 plates (0.25 mm, normal phase, Merck) and silica gel 60 RP-18 F254S plates (0.25 mm, reversed phase, Merck). Normal-phase flash column chromatography was performed using silica gel 60 (particle size 40–63 μm; 230–400 mesh ASTM; SilicaFlash F60, SiliCycle Inc.). Reversed-phase flash column chromatography was performed using octadecylsilyl silica gel (C18, particle size 15–30 μm, FUJIFILM Wako Pure Chemical Co.). Melting point (Mp) data were determined using a Shimadzu MM-2 instrument and were uncorrected. IR spectra were recorded on a Horiba FT-720 spectrometer, using KBr pellets (solid). 1H and proton-decoupled 13C (13C{1H}) NMR spectra were recorded on a Bruker Avance 400 spectrometer (400 and 100 MHz, respectively), using chloroform-d (CDCl3), dimethyl sulfoxide-d6 (DMSO-d6), and methanol-d4 (CD3OD) as the solvent. Chemical shift values are expressed in δ (ppm) relative to tetramethylsilane (TMS, δ 0.00 ppm) or the residual solvent resonance (CDCl3, δ 77.0 ppm for 13C NMR; DMSO-d6, δ 2.49 ppm for 1H NMR and δ 39.7 ppm for 13C NMR; and CD3OD, δ 3.30 ppm for 1H NMR and δ 49.0 ppm for 13C NMR). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, and m = multiplet), coupling constants (J; Hz), and integration. Mass spectra were obtained using a Sciex X500R quadrupole time-of-flight (QTOF) high-resolution mass spectrometer using electrospray ionization (ESI). High purity levels of all synthetic compounds were verified using 1H NMR spectra (see the Supporting Information). A549 cells were purchased from the Riken Cell Bank. HL-60 and MRC-5 cells were purchased from the National Institute of Biomedical Innovation, Health and Nutrition.
2-Bromo-5-methylphenazin-1(5H)-one (1b)
NBS (9.3 mg, 52.3 μmol) was added to a solution of 1a(22) (10.6 mg, 50.4 μmol) in CH2Cl2 (5.0 mL). The mixture was stirred at 0 °C for 1 h. The reaction was quenched by the addition of a saturated aqueous NaHCO3 solution. The mixture was diluted with CHCl3. After the layers were separated, the organic layer was washed with water and brine, dried over Na2SO4, and concentrated to give a residue. The residue was purified by reversed-phase flash chromatography (MeOH/H2O = 4/1) to give 1b (7.5 mg, 51%) as a dark-blue solid. Reversed-phase TLC (MeOH/H2O = 4/1) Rf 0.48; mp 129–130 °C; IR (KBr) νmax: 2958, 2914, 2848, 1724, 1620, 1560, 1493 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.24 (d, J = 8.1 Hz, 1H), 8.13–8.10 (m, 2H), 8.00 (t, J = 7.6 Hz, 1H), 7.67 (t, J = 7.6 Hz, 1H), 6.25 (d, J = 8.6 Hz, 1H), 4.04 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.0, 145.4, 144.5, 136.4, 136.0, 135.2, 133.0, 132.7, 125.8, 116.0, 108.3, 91.9, 35.7; HRMS (ESI/QTOF) m/z: [M + H]+ calcd for C13H1079BrN2O, 288.9971; found, 288.9969.
4-Chloro-5-methylphenazin-1(5H)-one (7)
NCS (7.1 mg, 53.2 μmol) was added to a solution of 1a(22) (10.6 mg, 50.4 μmol) in CH2Cl2 (5.0 mL). The mixture was stirred at 0 °C for 2 h. The reaction was quenched by the addition of a saturated aqueous NaHCO3 solution. The mixture was diluted with CHCl3. After the layers were separated, the organic layer was washed with water and brine, dried over Na2SO4, and concentrated to give a residue. The residue was purified by reversed-phase flash chromatography (MeOH/H2O = 4/1) to give 7 (8.0 mg, 65%) as a dark-blue solid. TLC (CHCl3/MeOH = 9/1) Rf 0.66; mp 125–127 °C; IR (KBr) νmax: 3035, 2920, 1728, 1637, 1574, 1514, 1493 cm–1; 1H NMR (400 MHz, CDCl3, TMS): δ 8.17 (dd, J = 8.1, 1.1 Hz, 1H), 7.74–7.70 (m, 1H), 7.44 (d, J = 10.0 Hz, 1H), 7.44–7.40 (m, 1H), 7.40 (d, J = 8.6 Hz, 1H), 6.47 (d, J = 10.0 Hz, 1H), 4.07 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 178.7, 146.9, 145.9, 136.0, 135.5, 134.6, 132.8, 132.7, 124.6, 119.1, 113.6, 100.3, 41.3; HRMS (ESI/QTOF) m/z: [M + H]+ calcd for C13H1035ClN2O, 245.0476; found, 245.0478.
4-Chloro-5-{(2,4,4-trimethylcyclohex-1-en-1-yl)methyl}phenazin-1(5H)-one (8)
NCS (4.5 mg, 33.7 μmol) was added to a solution of 2a(22) (10.6 mg, 31.9 μmol) in CH2Cl2 (3.0 mL). The mixture was stirred at 0 °C for 1.5 h. The reaction was quenched by the addition of a saturated aqueous NaHCO3 solution. The mixture was diluted with CHCl3. After the layers were separated, the organic layer was washed with water and brine, dried over Na2SO4, and concentrated to give a residue. The residue was purified by reversed-phase flash chromatography (MeOH/H2O = 4/1) to give 8 (5.6 mg, 48%) as a dark-blue solid. TLC (CHCl3/MeOH = 9/1) Rf 0.47; mp 114–116 °C; IR (KBr) νmax: 2954, 2920, 2850, 1724, 1716, 1643, 1603, 1514, 1469 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.18 (dd, J = 8.0, 1.4 Hz, 1H), 7.65–7.61 (m, 1H), 7.46 (d, J = 10.1 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.27–7.25* (m, 1H), 6.49 (d, J = 10.1 Hz, 1H), 5.29 (s, 2H), 1.81 (br s, 5H), 1.48 (br s, 2H), 1.16 (t, J = 6.4 Hz, 2H), 0.73 (s, 6H), (*this proton signal was assigned based on the HMQC spectrum.); 13C{1H} NMR (100 MHz, CDCl3): δ 178.8, 146.9, 146.4, 136.1, 134.7, 134.0, 133.0 (2C), 130.3, 124.6, 123.2, 118.9, 114.5, 100.4, 54.2, 46.2, 35.0, 28.9, 27.9 (2C), 23.2, 19.3; HRMS (ESI/QTOF) m/z: [M + H]+ calcd for C22H2435ClN2O, 367.1572; found, 367.1579.
General Procedure for the Coupling between 4 and 9 under an Oxygen Atmosphere (Entries 1, 4, 5, and 6 in Table 1)
A solution of 4 (30.0 mg, 245.6 μmol) and 9 (1.0 equiv) in the absence or presence of the additive (1.0 equiv) indicated in Table 1 in 2-propanol (2.0 mL) was stirred under an O2 atmosphere at rt in the dark until no further TLC changes were observed. The reaction was quenched by the addition of 1 M aqueous HCl solution. The mixture was diluted with CHCl3. After the layers were separated, the aqueous layer was neutralized with 4 M aqueous NaOH solution. The aqueous layer was extracted with CHCl3 twice. The combined organic layer was dried over Na2SO4 and concentrated to give a residue. The residue was purified by silica gel column chromatography and reversed-phase flash chromatography.
Following the general procedure, the reaction was performed using 4 (30.0 mg, 245.6 μmol) and 9 (39.8 mg, 247.9 μmol) in the presence of Cs2CO3 (81.0 mg, 248.6 μmol) for 4 days to give 1c (10.3 mg, 17%) as a dark-blue solid (entry 6). Reversed-phase TLC (MeOH/H2O = 4/1) Rf 0.39; mp 124–125 °C; IR (KBr) νmax: 3043, 2924, 1716, 1622, 1560 cm–1; H NMR (400 MHz, DMSO-d6): δ 8.24 (dd, J = 8.2, 1.2 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 8.02–7.98 (m, 1H), 7.96 (d, J = 8.7 Hz, 1H), 7.66 (td, J = 8.2, 1.2 Hz, 1H), 6.27 (d, J = 8.7 Hz, 1H), 4.04 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6): δ 170.8, 145.9, 141.2, 136.2, 136.0, 134.6, 133.0, 132.7, 125.8, 118.0, 115.9, 90.7, 35.6; HRMS (ESI/QTOF) m/z: [M + H]+ calcd for C13H1035ClN2O, 245.0476; found, 245.0476.
General Procedure for the Coupling between 4 and 9 Using the Oxidant (Entries 2 and 3 in Table 1)
A solution of 4 (245.6 μmol) and 9 (1.0 equiv) in the presence of the oxidant (3.0 equiv) indicated in Table 1 in 2-propanol (2.0 mL) was stirred at 0 °C until no further TLC changes were observed. The reaction was quenched by the addition of 1 M aqueous HCl solution. The mixture was diluted with CHCl3. After the layers were separated, the aqueous layer was neutralized with 4 M aqueous NaOH solution. The aqueous layer was extracted with CHCl3 twice. The combined organic layer was dried over Na2SO4 and concentrated to give a residue. The residue was purified by silica gel column chromatography and reversed-phase flash chromatography to yield 1c.
Following the general procedure, the reaction was performed using 4 (30.0 mg, 245.6 μmol), 9 (40.0 mg, 249.1 μmol), and 1,4-benzoquinone (79.6 mg, 736.4 μmol) for 1 h to give 1c (6.1 mg, 10%) (entry 2).
General Procedure of N-{(2,4,4-Trimethylcyclohex-1-en-1-yl)methyl}benzene-1,2-diamine (5) with 4-Halo-1,2,3-benzenetriols (General Procedure A; Entries 1, 4 in Table 2)
A solution of N-alkylbenzene-1,2-diamine (30.0 mg) and 4-halo-1,2,3-benzenetriols (1.0 equiv) in the presence of the Cs2CO3 (1.0 equiv) in 2-propanol (1.0 mL) was stirred and bubbled with O2 from an O2 balloon at rt in the dark until no further TLC changes were observed. The reaction was quenched by the addition of water. The mixture was diluted with CHCl3. After the layers were separated, the organic layer was washed with water and brine, dried over Na2SO4, and concentrated to give a residue. The residue was purified by silica gel column chromatography and reversed-phase flash chromatography.
Marinocyanin A (2b)
Following the general procedure A, the reaction was performed using 5 (30.0 mg, 122.8 μmol) and 10 (25.2 mg, 122.9 μmol) in the presence of Cs2CO3 (40.0 mg, 122.8 μmol) in 2-propanol with O2 bubbling for 2 days. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 50/1) and reversed-phase flash chromatography (MeOH/H2O = 4/1) to give 2b (5.0 mg, 10%) as a dark-blue solid. The structure of 2b was confirmed by comparison of the 1H and 13C{1H} NMR spectra with those reported for 2b.22
WS-9659 B (2c)
Following the general procedure A, the reaction was performed using 5 (30.0 mg, 122.8 μmol) and 9 (19.9 mg, 123.9 μmol) in the presence of Cs2CO3 (40.2 mg, 123.4 μmol) in 2-propanol with O2 bubbling for 2 days. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 50/1) and reversed-phase flash chromatography (MeOH/H2O = 4/1) to give 2c (8.9 mg, 20%) as a dark-blue solid. Reversed-phase TLC (MeOH/H2O = 4/1) Rf 0.43; mp 154–157 °C (lit. 152–153 °C);12 IR (KBr) νmax: 2918, 2848, 2362, 1699, 1626, 1604, 1562, 1495 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.40 (d, J = 7.8 Hz, 1H), 7.82 (t, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.57–7.52 (m, 2H), 5.89 (d, J = 8.4 Hz, 1H), 5.10 (br s, 2H), 1.91 (s, 3H), 1.88 (br s, 2H), 1.53–1.45 (br s 2H), 1.20 (t, J = 6.3 Hz, 2H), 0.83 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3): δ 172.7, 146.5, 140.9, 136.2, 135.4, 134.1, 133.8, 133.2, 131.5, 125.1, 120.9, 120.3, 113.6, 90.4, 50.1, 46.2, 34.8, 28.9, 28.0 (2C), 22.7, 19.5; HRMS (ESI/QTOF) m/z: [M + H]+ calcd for C22H2435ClN2O, 367.1572; found, 367.1568.
General Procedure of N-Methylbenzene-1,2-diamine (4) or N-(3-Methylbut-2-en-1-yl)benzene-1,2-diamine (6) with 4-Halo-1,2,3-benzenetriols (General Procedure B; Entries 2, 3, and 5 in Table 2)
A solution of N-alkylbenzene-1,2-diamine (30.0 mg) and 4-halo-1,2,3-benzenetriols (1.0 equiv) in the presence of Cs2CO3 (1.0 equiv) in 2-propanol was stirred under an O2 atmosphere at rt in the dark until no further TLC changes were observed. The reaction was quenched by the addition of 1 M aqueous HCl solution. The mixture was diluted with CHCl3. After the layers were separated, the aqueous layer was neutralized with 4 M aqueous NaOH solution. The aqueous layer was extracted with CHCl3 twice. The combined organic layer was dried over Na2SO4 and concentrated to give a residue. The residue was purified by silica gel column chromatography or reversed-phase flash chromatography.
2-Bromo-5-methylphenazin-1(5H)-one (1b)
Following the general procedure B, the reaction was performed using 4 (30.0 mg, 245.6 μmol) and 10 (50.7 mg, 247.3 μmol) in the presence of Cs2CO3 (80.5 mg, 247.1 μmol) in 2-propanol (2.0 mL) under an O2 atmosphere for 3 days. The residue was purified by reversed-phase flash chromatography (MeOH/H2O = 2/1) to give 1b (12.7 mg, 18%) as a dark-blue solid.
Marinocyanin B (3b)
Following the general procedure B, the reaction was performed using 6 (30.0 mg, 170.2 μmol) and 10 (35.8 mg, 174.6 μmol) in the presence of Cs2CO3 (53.5 mg, 164.2 μmol) in 2-propanol (1.0 mL) under an O2 atmosphere for 2 days. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 50/1) and reversed-phase flash chromatography (MeOH/H2O = 4/1) to give 3b (8.5 mg, 15%) as a dark-blue solid. The structure of 3b was confirmed by comparison of the 1H and 13C{1H} NMR spectra with those reported for 3b.22
2-Chloro-5-(3-methylbut-2-en-1-yl)phenazin-1(5H)-one (3c)
Following the general procedure B, the reaction was performed using 6 (30.0 mg, 170.2 μmol) and 9 (27.3 mg, 170.0 μmol) in the presence of Cs2CO3 (55.6 mg, 170.6 μmol) in 2-propanol (1.5 mL) under an O2 atmosphere for 2 days. The residue was purified by reversed-phase flash chromatography (MeOH/H2O = 2/1) to give 3c (10.6 mg, 21%) as a dark-blue solid. Reversed-phase TLC (MeOH/H2O = 4/1) Rf 0.38; mp 145–149 °C; IR (KBr) νmax: 3035, 2958, 2918, 2848, 1724, 1624, 1562, 1496 cm–1; 1H NMR (400 MHz, CD3OD): δ 8.31 (d, J = 7.8 Hz, 1H), 8.07–8.03 (brm, 1H), 7.99–7.93 (m, 2H), 7.72 (t, J = 7.8 Hz, 1H), 6.41 (d, J = 8.7 Hz, 1H), 5.37 (br s, 2H), 5.21–5.18 (br s, 1H), 2.03 (s, 3H), 1.80 (d, J = 0.9 Hz, 3H); 13C{1H} NMR (100 MHz, CD3OD): δ 171.4, 146.4, 144.3, 140.4, 138.9, 138.1, 134.5, 134.0, 133.5, 128.0, 119.5, 117.3, 116.8, 93.8, 48.7*, 25.6, 18.7 (*this carbon signal was assigned based on the HMQC spectrum.); HRMS (ESI/QTOF) m/z: [M + H]+ calcd for C17H1635ClN2O, 299.0946; found, 299.0942.
Cell Lines and Culturing
A549 cells were cultured at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HL-60 cells were cultured at 37 °C with 5% CO2 in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS. MRC-5 cells were cultured in Minimum Essential Medium Eagle-alpha modification (α-MEM) without ribonucleosides and deoxyribonucleosides supplemented with 10% FBS.
Cytotoxicity Evaluation
Cell growth was evaluated using cell counting kit-8 according to the manufacturer’s instructions based on the WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] assay.27 For the assay, the adherent A549 and MRC-5 cells were cultured in a 96-well plate with each well containing 2000 cells in a total volume of 100 μL, and HL-60 cells were cultured in a 96-well plate with each well containing 5000 cells in a total volume of 100 μL. The concentration of DMSO in the cell cultures was 0.1–0.2% (v/v). The plates also included blank wells (0 cells) and control wells (2000 cells/100 μL for the adherent cells and 5000 cells/100 μL for HL-60 cells). A549 and MRC-5 cells were preincubated for 24 h before exposure to the test compounds. The plates were incubated with various concentrations of each compound for 72 h. At the end of incubation, 10 μL of the solution provided with the cell counting kit was then added, and the resulting mixture was incubated for 2 h at 37 °C. The absorbance values were then measured at 450 nm with a 96-well plate reader. Cell growth inhibition was evaluated as the ratio of the absorbance of the sample to that of the control.
Acknowledgments
This study was partly supported by JSPS KAKENHI (no. 20K05868) to K.K.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04253.
1H, 13C{1H}, and 2D NMR spectra for synthetic compounds (PDF)
Author Present Address
† Shogo Kamo: Faculty of Pharmaceutical Sciences, Hoshi University, 2-4-41 Ebara, Shinagawa-ku, Tokyo 142-8501, Japan.
Author Present Address
‡ Shusuke Tomoshige: Graduate School of Life Sciences, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan.
The authors declare no competing financial interest.
Supplementary Material
References
- Cimmino A.; Culver C. A.; van Breemen R. B. Phenazines and Cancer. Nat. Prod. Rep. 2012, 29, 487–501. 10.1039/c2np00079b. [DOI] [PubMed] [Google Scholar]
- Pierson L. S. III Metabolism and Function of Phenazines in Bacteria: Impacts on the Behavior of Bacteria in the Environment and Biotechnological Process. Appl. Microbiol. Biotechnol. 2010, 86, 1659–1670. 10.1007/s00253-010-2509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen J. B.; Nielsen J. Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity. Chem. Rev. 2004, 104, 1663–1686. 10.1021/cr020473j. [DOI] [PubMed] [Google Scholar]
- Guttenberger N.; Blankenfeldt W.; Breinbauer R. Recent Developments in the Isolation, Biological Function, Biosynthesis, and Synthesis of Phenazine Natural Products. Bioorg. Med. Chem. 2017, 25, 6149–6166. 10.1016/j.bmc.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Jayaseelan S.; Ramaswamy D.; Dharmaraj S. Pyocyanin: Production, Applications, Challenges and New Insights. World J. Microbiol. Biotechnol. 2014, 30, 1159–1168. 10.1007/s11274-013-1552-5. [DOI] [PubMed] [Google Scholar]
- Lau G. W.; Hassett D. J.; Ran H.; Kong F. The Role of Pyocyanin in Pseudomonas aeruginosa Infection. Trends Mol. Med. 2004, 10, 599–606. 10.1016/j.molmed.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Price-Whelan A.; Dietrich L. E. P.; Newman D. K. Rethinking ‘Secondary’ Metabolism: Physiological Roles for Phenazine Antibiotics. Nat. Chem. Biol. 2006, 2, 71–78. 10.1038/nchembio764. [DOI] [PubMed] [Google Scholar]
- Meirelles L. A.; Newman D. K. Both Toxic and Beneficial Effects of Pyocyanin Contribute to the Lifecycle of Pseudomonas aeruginosa. Mol. Microbiol. 2018, 110, 995–1010. 10.1111/mmi.14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser N. R.; Saunders S. H.; Newman D. K. The Colorful World of Extracellular Electron Shuttles. Annu. Rev. Microbiol. 2017, 71, 731–751. 10.1146/annurev-micro-090816-093913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa K. C.; Glasser N. R.; Conway S. J.; Newman D. K. Pyocyanin Degradation by a Tautomerizing Demethylase Inhibits Pseudomonas aeruginosa Biofilms. Science 2017, 355, 170–173. 10.1126/science.aag3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S.; Furihata K.; Hayakawa Y.; Noguchi T.; Seto H. Lavanducyanin, a New Antitumor Substance Produced by Streptomyces sp. J. Antibiot. 1989, 42, 1196–1198. 10.7164/antibiotics.42.1196. [DOI] [PubMed] [Google Scholar]
- Nakayama O.; Yagi M.; Tanaka M.; Kiyoto S.; Okuhara M.; Kohsaka M. WS-9659 A and B, Novel Testosterone 5α-Reductase Inhibitors Isolated from a Streptomyces. I. Taxonomy, Fermentation, Isolation, Physico-chemical Characteristics. J. Antibiot. 1989, 42, 1221–1229. 10.7164/antibiotics.42.1221. [DOI] [PubMed] [Google Scholar]
- Nakayama O.; Shigematsu N.; Katayama A.; Takase S.; Kiyoto S.; Hashimoto M.; Kohsaka M. WS-9659 A and B, Novel Testosterone 5α-Reductase Inhibitors Isolated from a Streptomyces. II. Structural Elucidation of WS-9659 A and B. J. Antibiot. 1989, 42, 1230–1234. 10.7164/antibiotics.42.1230. [DOI] [PubMed] [Google Scholar]
- Nakayama O.; Arakawa H.; Yagi M.; Tanaka M.; Kiyoto S.; Okuhara M.; Kohsaka M. WS-9659 A and B, Novel Testosterone 5α-Reductase Inhibitors Isolated from a Streptomyces. III. Biological Characteristics and Pharmacological Characteristics. J. Antibiot. 1989, 42, 1235–1240. 10.7164/antibiotics.42.1235. [DOI] [PubMed] [Google Scholar]
- Kondratyuk T. P.; Park E.-J.; Yu R.; van Breemen R. B.; Asolkar R. N.; Murphy B. T.; Fenical W.; Pezzuto J. M. Novel Marine Phenazines as Potential Cancer Chemopreventive and Anti-inflammatory Agents. Mar. Drugs 2012, 10, 451–464. 10.3390/md10020451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura S.; Eda S.; Funayama S.; Komiyama K.; Takahashi Y.; Woodruff H. B. Studies on a Novel Antitumor Antibiotic, Phenazinomycin: Taxonomy, Fermentation, Isolation, and Physicochemical and Biological Characteristics. J. Antibiot. 1989, 42, 1037–1042. 10.7164/antibiotics.42.1037. [DOI] [PubMed] [Google Scholar]
- Funayama S.; Eda S.; Komiyama K.; O̅mura S.; Tokunaga T. Structure of Phenazinomycin, a Novel Antitumor Antibiotic. Tetrahedron Lett. 1989, 30, 3151–3154. 10.1016/s0040-4039(00)99188-3. [DOI] [Google Scholar]
- Li S.; Hu X.; Li L.; Hu X.; Wang J.; Hu X.; Liu H.; Yu L.; You X.; Jiang B.; Wu L. 1-Hydroxy-7-oxolavanducyanin and Δ7″,8″-6″-Hydroxynaphthomevalin from Streptomyces sp. CPCC 203577. J. Antibiot. 2020, 73, 324–328. 10.1038/s41429-020-0282-9. [DOI] [PubMed] [Google Scholar]
- Asolkar R. N.; Singh A.; Jensen P. R.; Aalbersberg W.; Carté B. K.; Feussner K.-D.; Subramani R.; DiPasquale A.; Rheingold A. L.; Fenical W. Marinocyanins, Cytotoxic Bromo-phenazinone Meroterpenoids from a Marine Bacterium from the Streptomycete Clade MAR4. Tetrahedron 2017, 73, 2234–2241. 10.1016/j.tet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selected reports on the synthesis of pyocyanin:; a Surrey A. R. Pyocyanine. Org. Synth. 1946, 26, 86–90. 10.1002/0471264180.os026.25. [DOI] [PubMed] [Google Scholar]; b Surrey A. R. Pyocyanin. Org. Synth. Collect. 1955, 3, 753–756. [Google Scholar]; c McIlwain H. Phenazine series. VI. Reactions of Alkylphenazonium. Salts; the Phenazyls. J. Chem. Soc. 1937, 1704–1711. 10.1039/jr9370001704. [DOI] [Google Scholar]; d Mortzfeld F. B.; Pietruszka J.; Baxendale I. R. A Simple and Efficient Flow Preparation of Pyocyanin a Virulence Factor of Pseudomonas aeruginosa. Eur. J. Org. Chem. 2019, 2019, 5424–5433. 10.1002/ejoc.201900526. [DOI] [Google Scholar]
- A report on the synthesis of lavanducyanin:Kinoshita Y.; Watanabe H.; Kitahara T.; Mori K. Concise Construction of N-Alkylated Phenazinone Skeletons: Synthesis of Lavanducyanin (WS-9659A). Synlett 1995, 186–188. 10.1055/s-1995-4905. [DOI] [Google Scholar]
- Kohatsu H.; Kamo S.; Tomoshige S.; Kuramochi K. Total Syntheses of Pyocyanin, Lavanducyanin, and Marinocyanins A and B. Org. Lett. 2019, 21, 7311–7314. 10.1021/acs.orglett.9b02601. [DOI] [PubMed] [Google Scholar]
- Bondi A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Li J.-T.; Wang L.-X.; Wang D.-X.; Zhao L.; Wang M.-X. Synthesis, Resolution, Structure, and Racemization of Inherently Chiral 1,3-Alternate Azacalix[4]pyrimidines: Quantification of Conformation Mobility. J. Org. Chem. 2014, 79, 2178–2188. 10.1021/jo500054v. [DOI] [PubMed] [Google Scholar]
- Smith T. J.; Wearne R. H.; Wallis A. F. A. Preparation of Chlorosyringols and Chloropyrogallols-components of Pulp Bleaching Effluents. Chemosphere 1994, 28, 921–930. 10.1016/0045-6535(94)90009-4. [DOI] [Google Scholar]
- Pla D.; Albericio F.; Álvarez M. Regioselective Monobromination of Free and Protected Phenols. Eur. J. Org. Chem. 2007, 2007, 1921–1924. 10.1002/ejoc.200600971. [DOI] [Google Scholar]
- Ishiyama M.; Miyazono Y.; Sasamoto K.; Ohkura Y.; Ueno K. A Highly Water-soluble Disulfonated Tetrazolium Salt as a Chromogenic Indicator for NADH as well as Cell Viability. Talanta 1997, 44, 1299–1305. 10.1016/s0039-9140(97)00017-9. [DOI] [PubMed] [Google Scholar]
- Badisa R. B.; Darling-Reed S. F.; Joseph P.; Cooperwood J. S.; Latinwo L. M.; Goodman C. B. Selective Cytotoxic Activities of Two Novel Synthetic Drugs on Human Breast Carcinoma MCF-7 Cells. Anticancer Res. 2009, 29, 2993–2996. [PMC free article] [PubMed] [Google Scholar]
- Musa M. A.; Badisa V. L. D.; Latinwo L. M.; Waryoba C.; Ugochukwu N. In Vitro Cytotoxicity of Benzopyranone Derivatives with Basic Side Chain Against Human Lung Cell Lines. Anticancer Res. 2010, 30, 4613–4617. [PMC free article] [PubMed] [Google Scholar]
- Zainal-Abidin M. H.; Hayyan M.; Ngoh G. C.; Wong W. F. Doxorubicin Loading on Functional Graphene as a Promising Nanocarrier Using Ternary Deep Eutectic Solvent Systems. ACS Omega 2020, 5, 1656–1668. 10.1021/acsomega.9b03709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda N.; Nagata H.; Furuta T.; Yokokura T. Metabolism and Pharmacokinetics of the Camptothecin Analogue CPT-11 in the Mouse. Cancer Res. 1990, 50, 1715–1720. [PubMed] [Google Scholar]
- Liu K.; Wang X.; Sha K.; Zhang F.; Xiong F.; Wang X.; Chen J.; Li J.; Churilov L. P.; Chen S.; Wang Y.; Huang N. Nuclear Protein HMGN2 Attenuates Pyocyanin-induced Oxidative Stress via Nrf2 Signaling and Inhibits Pseudomonas aeruginosa Internalization in A549 Cells. Free Radic. Biol. Med. 2017, 108, 404–417. 10.1016/j.freeradbiomed.2017.04.007. [DOI] [PubMed] [Google Scholar]
- Garrison A. T.; Abouelhassan Y.; Kallifidas D.; Bai F.; Ukhanova M.; Mai V.; Jin S.; Luesch H.; Huigens R. W. III Halogenated Phenazines that Potently Eradicate Biofilms, MRSA Persister Cells in Non-Biofilm Cultures and Mycobacterium tuberculosis. Angew. Chem., Int. Ed. 2015, 54, 14819–14823. 10.1002/anie.201508155. [DOI] [PubMed] [Google Scholar]
- Garrison A. T.; Abouelhassan Y.; Norwood V. M.; Kallifidas D.; Bai F.; Nguyen M. T.; Rolfe M.; Burch G. M.; Jin S.; Luesch H.; Huigens R. W. III Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines that Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 3808–3825. 10.1021/acs.jmedchem.5b02004. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yang Z.; Liu Y.; Lu Y.; Chen K.; Zhu W. Halogen Bond: Its Role Beyond Drug-Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model. 2014, 54, 69–78. 10.1021/ci400539q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.