

Abstract
In the current study, virtual screening of a small library of 1302 pyrrolizines bearing urea/thiourea moieties was performed. The top-scoring hits were synthesised and evaluated for their cytotoxicity against three cancer (MCF-7, A2780, and HT29) and one normal (MRC-5) cell lines. The results of the MTT assay revealed potent cytotoxic activities for most of the new compounds (IC50 = 0.16–34.13 μM). The drug-likeness study revealed that all the new compounds conform to Lipinski’s rule. Mechanistic studies of compounds 18 b, 19a, and 20a revealed the induction of apoptosis and cell cycle arrest at the G1 phase in MCF-7 cells. The three compounds also displayed potent inhibitory activity against CDK-2 (IC50 = 25.53–115.30 nM). Moreover, the docking study revealed a nice fitting of compound 19a into the active sites of CDK-2/6/9. These preliminary results suggested that compound 19a could serve as a promising scaffold in the discovery of new potent anticancer agents.
Keywords: Pyrrolizine, urea derivatives, cytotoxicity, apoptosis, cell cycle, CDK-2
Graphical Abstract

Highlights
Virtual screening of 1302 pyrrolizines was done using the pharmacophore model of the multi-CDKI 3.
The top-scoring hits were synthesised and evaluated for their cytotoxic activities.
Compound 19a showed potent in vitro cytotoxic activity against CDK-2.
Compound 19a induced apoptosis and cell cycle arrest at the G1 phase in MCF-7 cells.
The docking study revealed nice fitting of compound 19a into CDK-2/6/9 with high binding affinities.
1. Introduction
Targeting the oncogenic protein kinases has emerged as a promising strategy in the development of new anticancer agents in the last three decades1,2. Currently, more than 40 kinase inhibitors were approved by the FDA for the treatment of different types of cancers3–5. Among these inhibitors, the cyclin-dependent kinase inhibitors (CDKIs) attracted much attention which could be due to the important role of CDKs in cell division and differentiation6,7. Among the CDK family, CDK-2 plays an important role in the progression of cells from G1 to S cell cycle phases6,7. The overexpression of CDK-2 was also reported in several solid tumours such as breast8, colon9, and ovarian cancers10. In addition, the increase in CDK-2 expression was also associated with the induction of the radio-resistance in glioblastoma cells11,12, and metastasis in prostate cancer13. CDK-2 also has a crucial role in DNA replication and apoptosis in different types of cancer6,12.
These findings highlighted the importance of CDK-2 as a potential target in cancer chemotherapy. Several small molecule inhibitors of CDK-2 have proved potent anticancer activities6,14. However, many of these inhibitors have also displayed pan-CDKs inhibitory activity which could be attributed to the high sequence similarity between different members of the CDK family6. Some of these inhibitors (roscovitine, dinaciclib, and Ro-3306) have succeeded to reach phase I/II clinical trials6,14.
Our literature review16–18 revealed several CDKIs bearing similar pharmacophoric groups which include two aryl/heterocyclic rings, two carbonyl groups, and a five-membered pyrazole/imidazole core, Figure 1. Among these inhibitors, compound 1 exhibited moderate inhibitory activity against CDK-2 with an IC50 value of 0.324 μM15. Compound 1 also exhibited inhibitory activity against CDK-1/4. The study of structure–activity relationship of compound 1 revealed improvement in CDK-2 inhibitory activity on replacement of the ketone oxygen by sulfur15.
Figure 1.

Multi-CDK inhibitors 1–5 bearing similar pharmacophoric features.
Compound 2 was reported among a series of pyrazole derivatives 2–4 with CDK-1/2 inhibitory activities16. Compound 2 displayed inhibitory activity against CDK-2 with an IC50 value of 140 nM. Substitution on the benzoyl ring in compound 2 with 2,6-difluoro groups afforded compound 3 with a 46-fold increase in CDK-2 inhibitory activity, Figure 1.
Moreover, compound 4 (AT7519) was obtained in an attempt to optimise the anticancer activity compound 3. However, compound 4 exhibited weaker inhibitory activity against CDK-2 (IC50 = 47 nM) compared to the parent compound 3 which indicates that the aromatic 4-fluorophenyl moiety is favoured for CDKs inhibition16, Figure 1. Mechanistic study of compound 4 also revealed high inhibitory activity against CDK-9, while weaker activity was observed against CDKs 1, 3, 4, and 617. Moreover, replacement of the 4-piperidinyl ring in compound 4 by the N-4-((2-aminophenyl)carbamoyl)benzyl moiety afforded compound 5 with higher inhibitory activities against CDK-1/218.
In addition, several CDKIs were designed bearing substituted urea moiety19–22. The importance of this moiety in the formation of hydrogen bonding network with the kinase domain which improves inhibitory activities against CDKs was discussed in several reports19,21,22. Honma et al. investigated the inhibitory activities of several diaryl urea derivatives against CDK-419. The results revealed higher kinase inhibitory activity for the derivatives with the bulky aryl moiety when substituted with H-bond donner (HBD)/acceptor (HBA) groups. Among these derivatives, compound 6 with the bulky 7-hydroxynaphthalen-1-yl moiety inhibited CDK-4 with an IC50 value of 7.6 μM, Figure 2. Compound 7 (CDKi 277, Figure 2) is also a thiazolyl urea derivative which exhibited potent ATP competitive inhibitory activity against CDKs 1, 2, and 5 with IC50 values of 8, 4, and 5 nM, respectively20.
Figure 2.

Diary urea derivatives 6–9 with their inhibitory activities against CDKs.
On the other hand, compound 8 was reported with moderate inhibitory activity against CDK-2 (IC50 = 14.3 μM)21. However, extending the chemical structure of the diaryl urea 9 with methyl piperazine and aminopyrimidine moieties allowed the new compound to occupy a larger volume of the active site of CDK-2, Figure 2. Accordingly, compound 9 exhibited higher inhibitory activity against CDK-2 than compound 822.
1.1. Rationale and design
Previously, we reported compound 10 among a series of pyrrolizine-5-carboxamide derivatives with potent cytotoxic activity against MCF-7 cells23. Although compound 10 was able to activate caspase 3/7 in MCF-7 cells, but the exact mechanism of action of this compound was not investigated. Recently, we reported different pyrrolizine derivatives with weak to moderate inhibitory activities against 20 oncogenic kinases24,25. Among these kinases, CDK-2 was the most affected one by all the tested compounds.
Based on the above-mentioned data, the current study was performed to optimise the cytotoxic potential, study the structure-activity relationship (SAR), and investigate the mechanism of action of the pyrrolizine-5-carboxamide 10. In this study, scaffold A (Figure 3) was designed bearing the pharmacophoric groups of the lead compound 10 and the multi-CDKIs 317. In addition, a small library of 1302 urea/thiourea derivatives was generated through different structural modifications in scaffold A, Supplementary data (Figures S1–S42).
Figure 3.

Rational design of scaffold A and general structure of the compound library.
In the last two decades, more than 500 crystal structure of CDKs were released in the protein data bank26,27. Among these, a large number of CDK-2 crystal structures bound to inhibitors of diverse chemical scaffolds were reported27. The use of the in silico studies (virtual screening, docking studies and molecular dynamics) in the discovery and identification of new potent CDKs inhibitors was previously discussed in several reports28–30. Among these techniques, virtual screening was used in several studies to design/identify novel CDK-2 inhibitors31,32. Poulsen et al. reported a nitrogen-linked macrocyclic CDK-2 inhibitor using structure-based design and docking studies33.
Moreover, the pharmacophore-based virtual screening was used widely in different steps of the drug discovery process. This technique depends on the generation of a 3D pharmacophore model based on a set of active ligands, a target–ligand complex or the apo target. The generated pharmacophore can then be used in screening virtual libraries of molecules to select/optimise the lead compounds34. The application of virtual screening was also succeeded in the identification of potent CDK-2 inhibitors of diverse chemical nature33,35.
In the current work, a pharmacophore-based virtual screening of the compound library was performed using the 3 D pharmacophore model of the multi-CDKI 3. The virtual screening was done using Pharmit (http://pharmit.csb.pitt.edu/)36. First, the pdb file of compound 3 (LZ9) bound to CDK-2 (pdb: 2VTP) was selected in the user interface. After loading the protein file, the program identified all the pharmacophoric features in the ligand (LZ9) based on the recorded ligand-protein interactions. This features included the aromatic, hydrophobic, hydrogen bond donor/acceptor, and positively/negatively charged moieties, Figure 4. In the current study, five of the pharmacophoric features in compound 3 which participate in the interactions with amino acids in the active site of CDK-2 were selected to generate the pharmacophoric model. These features included the aromatic phenyl, pyrazole ring (hydrophobic), and imidazole N2 (HBA1), the anilide NH (HBD), and the carbonyl oxygen (HBA2), Figure 4.
Figure 4.

Compound 3 (LZ9) bound to CDK-2 (pdb 2VTP); (A) pharmacophoric features of compound 3; (B) pharmacophoric features selected to generate the 3D pharmacophore model used in the virtual screening.
In the virtual screening, the maximum number of rotatable bonds was set to 9, while the radius of the pharmacophoric feature were set to the default values. Compounds with molecular weights > 500 were excluded to avoid violations from Lipinski’s rule. The top-scoring hits were ranked based on RMSD in Table 1.
Table 1.
The top-scoring hits of the compound library based on RMSD.
| Rank | Hita | RMSD | Rotatable bond |
|---|---|---|---|
| 1 | Naphthalen-1-yl-ureido | 0.463 | 8 |
| 2 | Naphthalen-1-yl-ureido | 0.516 | 8 |
| 3 | 3-Fluorophenyl-ureido | 0.522 | 9 |
| 4 | 4-Methoxyphenyl-ureido | 0.522 | 9 |
| 5 | Cyclohexyl-ureido | 0.522 | 9 |
| 6 | 4-Fluorophenyl-ureido | 0.522 | 9 |
| 7 | Naphthalen-2-yl-ureido | 0.522 | 9 |
| 8 | 4-Bromophenyl-ureido | 0.522 | 9 |
| 9 | Naphthalen-1-yl-ureido | 0.522 | 9 |
| 10 | Tert-butyl-ureido | 0.522 | 9 |
| 11 | Naphthalen-1-yl-thioureido | 0.522 | 9 |
| 12 | Cyclopentyl-ureido | 0.522 | 9 |
| 13 | Cycloheptyl-ureido | 0.522 | 9 |
| 14 | 4-Methoxyphenyl-ureido | 0.522 | 9 |
| 15 | 3-Bromophenyl-ureido | 0.522 | 9 |
aHits named according to their aryl urea/thiourea fragment.
The results of the virtual screening of the compound library revealed the best score for the naphthalen-1-yl bearing hits (R1 = H, CH3). The top 15-scoring-hits also included the naphthalen-1-yl-thioureido, two 4-methoxyphenyl-ureido and two fluorophenyl-ureido hits with the same RMSD, Table 1.
Based on the above results, the SAR study of scaffold A was achieved through the following modifications in compound 10 (Figure 5): (i) replacement of the 4-chlorophenyl moiety by naphthalen-1-yl moiety; (ii) replacement of the urea moiety in the naphthalenyl derivatives by thiourea; (iii) replacement of the chloro group in compound 10 with electron donating (R2 = 4-OCH3) or the electron withdrawing (R2 = 4-F) groups to study the impact of electronic effect of these substituents on cytotoxicity of the new compounds; (iv) replacement of the 4-chlorophenyl in compound 10 moiety by the aliphatic hexyl group to compare the impact of the aliphatic versus aromatic (phenyl/naphthalenyl) moieties on cytotoxic activity of the new compounds; (v) substitution on ring A with electron donating (R1 = 4-CH3) or with electron withdrawing (R1 = 4-Cl) to study the impact of these substituents on activity.
Figure 5.

Virtual screening and structural modifications of scaffold A.
2. Material and methods
2.1. Chemistry
Chemicals and reagents were purchased from Sigma-Aldrich, Darmstadt, Germany. All solvents were analytical grade and were used without purification. Melting points (m.p.) were determined by IA 9100MK digital melting point apparatus. TLC was used to check the purity of the new compounds. the IR spectra were recorded by BRUKER TENSOR 37 spectrophotometer using KBr disc. The IR spectra were expressed in wavenumber (cm−1). The 1H-NMR, 13 C-NMR, and DEPT C135 spectra were recorded in the Faculty of Pharmacy, Umm Al-Qura University, KSA using BRUKER AVANCE III at 500 and 125 MHz, respectively. The J constant was given in Hz. Shimadzu GCMS QP5050A spectrometer (EI, 70 eV, regional centre for mycology and biotechnology, Al-Azhar University, Cairo, Egypt) was used to perform the mass analyses of the new compounds. The quantitative elemental analyses were done in the microanalytical centre, Cairo University. Compounds 1237, 14a–c38–40, 15a–c41 were prepared according to the previous reports. Copies of spectral data including IR, 1H-NMR, 13 C-NMR, 13 C-NMR, DEPT C135, and mass spectra of the new compounds are provided in supplementary (Figures S43–S137). To facilitate the identification of different protons/carbons and their chemical shifts in each of the new compounds, the numbering of the atoms was illustrated in scaffold A, Figure 5.
2.1.1. General procedure for the preparation of compounds 16–20a–c
A mixture of the staring material (2 mmol), appropriate isocyanate/isothiocyanate (2.2 mmol), TEA (0.5 ml) in DCM (30 ml) was stirred at rt for 12 h. The solvent was evaporated, and the solid product was dissolved in chloroform-acetone (1:1). The formed precipitate was filtered, washed with acetone, and dried to give white amorphous solid product.
2.1.1.1. 7-Cyano-6-(3-hexylureido)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide (16a)
The title compound was prepared from the reaction of compound 15a (0.53 g, 2 mmol) and hexyl isocyanate (0.28 g, 2.2 mmol) according to the general procedure A. Compound 16a was obtained as white amorphous solid product, m.p. 217–19 °C, yield 57%. IRʋmax/cm−1 3351 (NHs), 3057 (aromatic C–H), 2934, 2872 (aliphatic C–H), 2225 (CN), 1708, 1646 (COs), 1599, 1538 (C = C, C = N), 1489, 1379, 1318, 1229 (C–N, C–O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 0.84 (t, 3H, J = 5.4 Hz, hexyl CH3), 1.29–1.23 (m, 6H, hexyl CH2-3 + CH2-4 + CH2-5), 1.44–1.40 (m, 2H, hexyl CH2-2), 2.47–2.42 (m, 2H, pyrrolizine CH2-2), 2.97 (t, 2H, J = 7.3 Hz, pyrrolizine CH2-1), 3.13 (q, 2H, J = 5.9 Hz, hexyl CH2-1), 4.27 (t, 2H, J = 6.6 Hz, pyrrolizine CH2-3), 6.77 (s, 1H, NHCH2-), 7.08 (t, 1H, 6.7 Hz, Ph CH-4), 7.33 (t, 2H, J = 7.6 Hz, Ph CH-3 + CH-5), 7.56 (d, 2H, J = 7.1 Hz, Ph CH-2 + CH-6), 8.36 (s, 1H, pyrrolizine-NHCO), 10.42 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 14.4 (CH3), 22.5 (hexyl CH2-5), 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 26.4 (hexyl CH2-3), 30.2 (hexyl CH2-2), 31.5 (hexyl CH2-4), 40.1 (hexyl CH2-1), 49.7 (pyrrolizine CH2-3), 84.3 (pyrrolizine C-7), 115.4 (CN), 119.3 (pyrrolizine C-5), 119.4 (Ph CH-2 + CH-6), 124.0 (Ph CH-4), 128.7 (pyrrolizine C-7a), 129.4 (Ph CH-3 + CH-5), 139.1 (pyrrolizine C-6), 146.0 (Ph C-1), 157.6 (NHCONH), 157.9 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 14.4 (CH3), 22.5 (hexyl CH2-5), 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 26.4 (hexyl CH2-3), 30.2 (hexyl CH2-2), 31.5 (hexyl CH2-4), 40.1 (hexyl CH2-1), 49.7 (pyrrolizine CH2-3), 119.4 (Ph CH-2 + CH-6), 124.0 (Ph CH-4), 129.4 (Ph CH-3 + CH-5). MS (EI): m/z (%) 395 ([M + 2]+, 5), 394 ([M + 1]+, 33), 393 ([M]+, 100), 392 ([M-1]+, 2), 292 (6), 266 (5), 117 (2), 91 (7), 77 (3). Anal. Calcd. for C22H27N5O2 (393.48): C, 67.15; H, 6.92; N, 17.80. Found: C, 66.81; H, 7.34; N, 17.93.
2.1.1.2. 7-Cyano-6-(3-hexylureido)-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (16b)
The title compound was prepared from the reaction of compound 15b (0.56 g, 2 mmol) and hexyl isocyanate (0.28 g, 2.2 mmol) according to the general procedure A. Compound 16 was obtained as white amorphous solid product, m.p. 231–33 °C, yield 51%. IRʋmax/cm−1 3314 (NHs), 2954, 2926, 2857 (aliphatic C–H), 2224 (CN), 1708, 1669, 1643 (COs), 1600, 1538 (C = C, C = N), 1433, 1318, 1243 (C–N, C–O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 0.84 (t, 3H, J = 5.3 Hz, hexyl CH3), 1.25–1.21 (m, 6H, hexyl CH2-3 + CH2-4 + CH2-5), 1.47–1.38 (m, 2H, hexyl CH2-2), 2.26 (s, 3H, tolyl CH3), 2.48–2.40 (m, 2H, pyrrolizine CH2-2), 2.97 (t, 2H, J = 6.0 Hz, pyrrolizine CH2-1), 3.13 (q, 2H, J = 6.7 Hz, hexyl CH2-1), 4.26 (t, 2H, J = 7.6 Hz, pyrrolizine CH2-3), 6.76 (s, 1H, NHCH2-), 7.13 (d, 2H, J = 7.1 Hz, Ph CH-3 + CH-5), 7.45 (d, 2H, J = 7.0 Hz, Ph CH-2 + CH-6), 8.34 (s, 1H, pyrrolizine-NHCO), 10.32 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 14.4 (hexyl CH3), 20.9 (tolyl CH3), 22.5 (hexyl CH2-5), 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 26.4 (hexyl CH2-3), 30.2 (hexyl CH2-2), 31.5 (hexyl CH2-4), 40.1 (hexyl CH2-1), 49.7 (pyrrolizine CH2-3), 84.3 (pyrrolizine C-7), 115.4 (CN), 119.4 (Ph CH-2 + CH-6), 119.7 (pyrrolizine C-5), 128.5 (pyrrolizine C-7a), 129.7 (Ph CH-3 + CH-5), 133.0 (Ph C-1), 136.6 (pyrrolizine C-6), 145.9 (Ph C-4), 157.6 (NHCONH), 157.7 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 14.4 (hexyl CH3), 20.9 (tolyl CH3), 22.5 (hexyl CH2-5), 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 26.4 (hexyl CH2-3), 30.2 (hexyl CH2-2), 31.5 (hexyl CH2-4), 40.1 (hexyl CH2-1), 49.7 (pyrrolizine CH2-3), 119.4 (Ph CH-2 + CH-6), 129.7 (Ph CH-3 + CH-5). MS (EI): m/z (%) 409 ([M + 2]+, 9), 408 ([M + 1]+, 35), 407 ([M]+, 100), 406 ([M-1]+, 4), 405 ([M-2]+, 1), 395 (2), 368 (3), 306 (25), 280 (3), 276 (3), 173 (3), 117 (12), 106 (3), 104 (12), 91 (9), 77 (25). Anal. Calcd. for C23H29N5O2 (407.51): C, 67.79; H, 7.17; N, 17.19. Found: C, 68.14; H, 6.86; N, 16.86.
2.1.1.3. N-(4-Chlorophenyl)-7-cyano-6-(3-hexylureido)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (16c)
The title compound was prepared from the reaction of compound 15c (0.60 g, 2 mmol) and hexyl isocyanate (0.28 g, 2.2 mmol) according to the general procedure A. Compound 16c was obtained as white amorphous solid product, m.p. 240–2 °C, yield 54%. IRʋmax/cm−1 3265 (NHs), 2954, 2923, 2857 (aliphatic C–H), 2222 (CN), 1672, 1641 (COs), 1597, 1567 (C = C, C = N), 1492, 1429, 1315, 1241 (C–N, C–O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 0.83 (t, 3H, J = 5.7 Hz, hexyl CH3), 1.23–1.19 (m, 6H, hexyl CH2-3 + CH2-4 + CH2-5), 1.44–1.36 (m, 2H, hexyl CH2-2), 2.47–2.42 (m, 2H, pyrrolizine CH2-2), 2.98 (t, 2H, J = 6.4 Hz, pyrrolizine CH2-1), 3.12 (q, 2H, J = 5.9 Hz, hexyl CH2-1), 4.25 (t, 2H, J = 6.8 Hz, pyrrolizine CH2-3), 6.77 (s, 1H, NHCH2-), 7.40 (d, 2H, J = 7.8 Hz, Ph CH-3 + CH-5), 7.58 (d, 2H, J = 7.6 Hz, Ph CH-2 + CH-6), 8.37 (s, 1H, pyrrolizine-NHCO), 10.55 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 14.4 (hexyl CH3), 22.5 (hexyl CH2-5), 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 26.4 (hexyl CH2-3), 30.1 (hexyl CH2-2), 31.5 (hexyl CH2-4), 40.1 (hexyl CH2-1), 49.7 (pyrrolizine CH2-3), 84.3 (pyrrolizine C-7), 115.3 (CN), 118.9 (Ph C-4), 121.0 (Ph CH-2 + CH-6), 127.5 (pyrrolizine C-5), 129.0 (pyrrolizine C-7a), 129.3 (Ph CH-3 + CH-5), 138.1 (pyrrolizine C-6), 146.2 (Ph C-1), 157.5 (NHCONH), 158.0 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 14.4 (hexyl CH3), 22.5 (hexyl CH2-5), 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 26.4 (hexyl CH2-3), 30.2 (hexyl CH2-2), 31.5 (hexyl CH2-4), 40.1 (hexyl CH2-1), 49.7 (pyrrolizine CH2-3), 121.0 (Ph CH-2 + CH-6), 129.3 (Ph CH-3 + CH-5). MS (EI): m/z (%) 430 ([M + 3]+, 9), 429 ([M + 2]+, 30), 428 ([M + 1]+, 33), 427 ([M]+, 100), 426 ([M-1]+, 1), 401 (2), 327 (8), 326 (17), 300 (4), 271 (2), 125 (2), 117 (3), 90 (4), 77 (2). Anal. Calcd. for C22H26ClN5O2 (427.93): C, 61.75; H, 6.12; N, 16.37. Found: C, 62.08; H, 6.43; N, 16.78.
2.1.1.4. 7-Cyano-6-(3-(4-methoxyphenyl)ureido)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide (17a)
The title compound was prepared from the reaction of compound 15a (0.53 g, 2 mmol) and 4-methoxyphenyl isocyanate (0.33 g, 2.2 mmol) according to the general procedure A. Compound 17a was obtained as white amorphous solid product, m.p. 226–8 °C, yield 62%. IRʋmax/cm−1 3412, 3309, 3262 (NHs), 2836 (aliphatic C–H), 2229 (CN), 1670, 1643 (COs), 1600, 1559 (C = C, C = N), 1445, 1322, 1248 (C–N, C–O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.48–2.44 (m, 2H, pyrrolizine CH2-2), 3.00 (t, 2H, J = 7.1 Hz, pyrrolizine CH2-1), 3.71 (s, 3H, OCH3), 4.29 (t, 2H, J = 7.4 Hz, pyrrolizine CH2-3), 6.86 (d, 2H, J = 8.6 Hz, MeO-Ph CH-3 + CH-5), 7.08 (t, 1H, J = 6.7 Hz, Ph CH-4), 7.37–7.34 (m, 4H, Ph CH-3 + CH-5+ MeO-Ph CH-2 + CH-6), 7.58 (d, 2H, J = 7.5 Hz, Ph CH-2 + CH-6), 8.49 (s, 1H, MeO-Ph-NH), 9.06 (s, 1H, pyrrolizine-NHCO), 9.94 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 55.6 (OCH3), 84.5 (pyrrolizine C-7), 114.4 (MeO-Ph CH-3+CH-5), 118.1 (CN), 119.9 (MeO-Ph CH-2 + CH-6), 121.0 (Ph CH-2 + CH-6), 124.2 (Ph CH-4), 129.4 (Ph CH-3 + CH-5), 132.8 (MeO-Ph C-1), 133.4 (pyrrolizine C-5), 139.0 (pyrrolizine C-7a), 146.3 (pyrrolizine C-6), 153.4 (Ph C-1), 154.5 (NHCONH), 155.3 (MeO-Ph C-4), 158.3 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 55.6 (OCH3), 114.4 (MeO-Ph CH-3+CH-5), 119.9 (MeO-Ph CH-2 + CH-6), 121.0 (Ph CH-2 + CH-6), 124.2 (Ph CH-4), 129.4 (Ph CH-3 + CH-5). MS (EI): m/z (%) 294 ([M-123 (4-methoxyaniline)]+, 2), 293 ([M-124]+, 6), 266 (1), 264 (2), 200 (6), 173 (7), 145 (3), 123 (17), 122 (5), 121 (3), 117 (29), 108 (41), 91 (29), 77 (28), 65 (100). Anal. Calcd. for C23H21N5O3 (415.44): C, 66.49; H, 5.09; N, 16.86. Found: C, 66.73; H, 4.77; N, 17.11.
2.1.1.5. 7-Cyano-6-(3-(4-methoxyphenyl)ureido)-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (17b)
The title compound was prepared from the reaction of compound 15 b (0.56 g, 2 mmol) and 4-methoxyphenyl isocyanate (0.33 g, 2.2 mmol) according to the general procedure A. Compound 17 b was obtained as white amorphous solid product, m.p. 234–6 °C, yield 63%. IRʋmax/cm−1 3401, 3275 (NHs), 3000 (aromatic C–H), 2954, 2857, 2835 (aliphatic C–H), 2222 (CN), 1670, 1644 (COs), 1600, 1563 (C = C, C = N), 1430, 1319, 1247 (C-N, C-O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.26 (s, 3H, CH3), 2.48–2.43 (m, 2H, pyrrolizine CH2-2), 3.00 (t, 2H, J = 6.4 Hz, pyrrolizine CH2-1), 3.72 (s, 3H, OCH3), 4.29 (t, 2H, J = 7.0 Hz, pyrrolizine CH2-3), 6.89 (d, 2H, J = 7.4 Hz, MeO-Ph CH-3 + CH-5), 7.14 (d, 2H, J = 7.3 Hz, tolyl CH-3 + CH-5), 7.37 (d, 2H, J = 7.3 Hz, MeO-Ph CH-2 + CH-6), 7.47 (d, 2H, J = 7.1 Hz, tolyl CH-2 + CH-6), 8.47 (s, 1H, MeO-Ph-NHCO), 9.05 (s, 1H, pyrrolizine-NHCO), 9.86 (s, 1H, tolyl-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 20.9 (CH3), 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.6 (pyrrolizine CH2-3), 55.6 (OCH3), 84.5 (pyrrolizine C-7), 114.5 (MeO-Ph CH-3+CH-5), 115.5 (CN), 118.2 (MeO-Ph C-1), 119.9 (MeO-Ph CH-2 + CH-6), 121.0 (tolyl CH-2 + CH-6), 128.9 (pyrrolizine C-5), 129.7 (tolyl CH-3+CH-5), 132.8 (pyrrolizine C-7a), 133.2 (tolyl C-1), 136.5 (pyrrolizine C-6), 146.2 (tolyl C-4), 154.5 (NHCONH), 155.3 (MeO-Ph C-4), 158.1 (tolyl-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 20.9 (CH3), 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 55.6 (OCH3), 114.5 (MeO-Ph CH-3+CH-5), 119.9 (MeO-Ph CH-2 + CH-6), 121.0 (tolyl CH-2 + CH-6), 129.7 (tolyl CH-3+CH-5). MS (EI): m/z (%) 431 ([M + 2]+, 3), 429 ([M]+, 13), 392 (3), 322 (23), 307 (23), 306 ([M-123 (4-methoxyaniline)]+, 23), 305 (41), 304 (9), 280 (7), 278 (37), 266 (5), 250 (8), 200 (7), 172 (10), 132 (13), 123 (2), 108 (8), 92 (17), 77 (46), 65 (100). Anal. Calcd. for C24H23N5O3 (429.47): C, 67.12; H, 5.40; N, 16.31. Found: C, 66.75; H, 4.89; N, 16.78.
2.1.1.6. N-(4-Chlorophenyl)-7-cyano-6-(3-(4-methoxyphenyl)ureido)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (17c)
The title compound was prepared from the reaction of compound 15c (0.60 g, 2 mmol) with 4-methoxyphenyl isocyanate (0.33 g, 2.2 mmol) according to the general procedure A. Compound 17c was obtained as white amorphous solid product, m.p. 245–7 °C, yield 69%. IRʋmax/cm−1 3294, 3163 (NHs), 3088 (aromatic C–H), 2212 (CN), 1663 (COs), 1631, 1605 (C = C, C = N), 1489, 1403, 1322, 1210 (C–N, C–O), 790 (C–Cl). 1H-NMR (CDCl3, 500 MHz, ppm) δ: δ 2.56–2.50 (m, 2H, pyrrolizine CH2-2), 2.98 (t, 2H, J = 7.5 Hz, pyrrolizine CH2-1), 3.80 (s, 3H, 4-OCH3), 4.39 (t, 2H, J = 7.2 Hz, pyrrolizine CH2-3), 6.20 (s, 1H, NH), 6.88 (d, 2H, J = 8,9 Hz, MeO-Ph CH-3 + CH-5), 7.31–7.22 (m, 5H, NH, Cl-Ph CH-3 + CH-5, MeO-Ph CH-2 + CH-6 + pyrrolizine-NHCO), 7.53 (d, 2H, J -= 8.8 Hz, Cl-Ph CH-2 + CH-6), 9.62 (s, 1H, Cl-Ph-NH). 13 C-NMR (CDCl3, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 55.7 (OCH3), 84.6 (pyrrolizine C-7), 114.6 (MeO-Ph CH-3+CH-5), 115.6 (CN), 117.0 (MeO-Ph C-1), 120.4 (MeO-Ph CH-2 + CH-6), 120.9 (Cl-Ph CH-2 + CH-6), 129.1 (4-Cl-Ph C-4), 129.8 (4-Cl-Ph CH-3+CH-5), 131.6 (pyrrolizine C-5), 133.0 (pyrrolizine C-7a), 136.7 (pyrrolizine C-6), 146.6 (4-Cl-Ph), 153.6 (NHCONH), 155.9 (MeO-Ph C-4), 159.0 (4-Cl-Ph-NHCO). DEPT C135 (CDCl3, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 55.7 (OCH3), 114.6 (MeO-Ph CH-3+CH-5), 120.4 (MeO-Ph CH-2 + CH-6), 120.9 (Cl-Ph CH-2 + CH-6), 129.8 (4-Cl-Ph CH-3+CH-5). MS (EI): m/z (%) 451 ([M + 2]+, 4), 450 ([M + 1]+, 5), 447 ([M-2]+, 2), 445 (4), 434 (5), 406 (3), 395 (8), 368 (10), 352 (6), 301 (6), 273 (44), 272 (100), 271 (14), 248 (3), 134 (6), 123 (62), 122 (7), 108 (78), 95 (17), 91 (4), 77 (20). Anal. Calcd. for C23H20ClN5O3 (449.89): C, 61.40; H, 4.48; N, 15.57. Found: C, 60.96; H, 4.81; N, 15.78.
2.1.1.7. 7-Cyano-6-(3-(4-fluorophenyl)ureido)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide (18a)
The title compound was prepared from the reaction of compound 15a (0.53 g, 2 mmol) and 4-fluorophenyl isocyanate (0.3 g, 2.2 mmol) according to the general procedure A. Compound 18a was obtained as white amorphous solid product, m.p. 242–4 °C, yield 65%. IRʋmax/cm−1 3402, 3366, 3336, 3280 (NHs), 3058, 3043 (aromatic C–H), 2969, 2895 (aliphatic C–H), 2221 (CN), 1655 (COs), 1601, 1549 (C = C, C = N), 1448, 1321, 1249 (C–N, C–O, C–F). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.48–2.44 (m, 2H, pyrrolizine CH2-2), 3.01 (t, 2H, J = 7.1 Hz, pyrrolizine CH2-1), 4.30 (t, 2H, J = 6.8 Hz, pyrrolizine CH2-3), 7.08 (t, 1H, J = 6.9 Hz, Ph CH-4), 7.14 (t, 2H, J = 8.2 Hz, F-Ph CH-2 + CH-6), 7.33 (t, 2H, J = 7.1 Hz, Ph CH-3 + CH-5), 7.47 (d, 2H, J = 8.5 Hz, F-Ph CH-3 + CH-5), 7.59 (d, 2H, J = 7.6 Hz, Ph CH-2 + CH-6), 8.55 (s, 1H, pyrrolizine-NHCO), 9.29 (s, 1H, F-Ph-NHCO), 9.84 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 84.6 (pyrrolizine C-7), 115.4 (CN), 115.8 (d, J = 22.3 Hz, F-Ph CH-3+CH-5), 117.8 (pyrrolizine C-5), 120.0 (Ph CH-2 + CH-6), 120.8 (d, J = 7.7 Hz, F-Ph CH-2 + CH-6), 124.2 (Ph CH-4), 129.1 (pyrrolizine C-7a), 129.3 (Ph CH-3 + CH-5), 136.2 (d, J = 2.7 Hz, F-Ph C-1), 139.0 (Ph CH-1), 146.3 (pyrrolizine C-6), 154.2 (NHCONH), 158.0 (d, J = 238.4 Hz, F-Ph C-4), 158.3 (PhNHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 115.8 (d, J = 22.3 Hz, F-Ph CH-3+CH-5), 120.0 (Ph CH-2 + CH-6), 120.8 (d, J = 7.7 Hz, F-Ph CH-2 + CH-6), 124.2 (Ph CH-4), 129.3 (Ph CH-3 + CH-5). MS (EI): m/z (%) 402 ([M-1]+, 2), 377 (4), 376 (17), 375 (50), 374 (17), 340 (4), 316 (32), 315 (22), 314 (100), 267 (2), 187 (4), 156 (2), 79 (5), 77 (4). Anal. Calcd. for C22H18FN5O2 (403.41): C, 65.50; H, 4.50; N, 17.36. Found: C, 65.83; H, 4.91; N, 17.62.
2.1.1.8. 7-Cyano-6-(3-(4-fluorophenyl)ureido)-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (18b)
The title compound was prepared from the reaction of compound 15b (0.56 g, 2 mmol) with 4-fluorophenyl isocyanate (0.3 g, 2.2 mmol) according to the general procedure A. Compound 18b was obtained as white amorphous solid product, m.p. 247–9 °C, yield 72%. IRʋmax/cm−1 3405, 3329, 3292 (NHs), 3077, 3037, 3001 (aromatic C-H), 2967, 2919, 2868 (aliphatic C–H), 2222 (CN), 1652 (COs), 1600, 1547 (C = C, C = N), 1435, 1320, 1210 (C–N, C–O, C–F). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.26 (s, 3H, CH3), 2.48–2.44 (m, 2H, pyrrolizine CH2-2), 3.00 (t, 2H, J = 6.4 Hz, pyrrolizine CH2-1), 4.29 (t, 2H, J = 7.3 Hz, pyrrolizine CH2-3), 7.15–7.12 (m, 4H, tolyl CH-3 + CH-5 and F-Ph CH-3 + CH-5), 7.48–7.46 (m, 4H, tolyl CH-2 + CH-6 and F-Ph CH-2 + CH-6), 8.57 (s, 1H, F-Ph-NHCO), 9.29 (s, 1H, pyrrolizine-NHCO), 9.74 (s, 1H, tolyl-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 20.9 (CH3), 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 84.6 (pyrrolizine C-7), 115.4 (CN), 115.8 (d, J = 22.3 Hz, F-Ph CH-3+CH-5), 118.0 (pyrrolizine C-5), 120.0 (tolyl CH-2 + CH-6), 120.8 (d, J = 7.7 Hz, F-Ph CH-2 + CH-6), 128.9 (pyrrolizine C-7a), 129.7 (tolyl CH-3 + CH-5), 133.2 (tolyl C-1), 136.2 (d, J = 2.1 Hz, F-Ph C-1), 136.4 (pyrrolizine C-6), 146.2 (tolyl C-4), 154.2 (NHCONH), 158.0 (d, J = 238.9 Hz, F-Ph C-4), 158.1 (tolyl-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 20.9 (CH3), 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 115.8 (d, J = 22.3 Hz, F-Ph CH-3+CH-5), 120.0 (tolyl CH-2 + CH-6), 120.7 (d, J = 7.7 Hz, F-Ph CH-2 + CH-6), 129.7 (tolyl CH-3 + CH-5). MS (EI): m/z (%) 418 ([M + 1]+, 2), 417 ([M]+, 6), 308 (4), 307 (20), 306 (82), 305 (9), 292 (6), 280 (8), 200 (16), 173 (11), 132 (9), 111 (54), 91 (43), 77 (92), 65 (100). Anal. Calcd. for C23H20FN5O2 (417.44): C, 66.18; H, 4.83; N, 16.78. Found: C, 65.72; H, 4.43; N, 16.65.
2.1.1.9. N-(4-Chlorophenyl)-7-cyano-6-(3-(4-fluorophenyl)ureido)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (18c)
The title compound was prepared from the reaction of compound 15c (0.60 g, 2 mmol) with 4-fluorophenyl isocyanate (0.3 g, 2.2 mmol) according to the general procedure A. Compound 18c was obtained as white amorphous solid product, m.p. 261–3 °C, yield 75%. IRʋmax/cm−1 3403, 3325, 3278 (NHs), 3062, 3046 (aromatic C–H), 2993, 2970 (aliphatic C–H), 2224 (CN), 1666 (COs), 1598, 1551 (C = C, C = N), 1431, 1315, 1233 (C–N, C–O, C–F), 774 (C–Cl). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.48–2.44 (m, 2H, pyrrolizine CH2-2), 3.01 (t, 2H, J = 6.7 Hz, pyrrolizine CH2-1), 4.29 (t, 2H, J = 6.9 Hz, pyrrolizine CH2-3), 7.13 (t, 2H, J = 7.8 Hz, F-Ph CH-3 + CH-5), 7.40 (d, 2H, J = 7.5 Hz, Cl-Ph CH-3 + CH-5), 7.48–7.43 (m, 2H, F-Ph CH-2 + CH-6), 7.62 (d, 2H, J = 7.4 Hz, Cl-Ph CH-2 + CH-6), 8.56 (s, 1H, F-PH-NHCO), 9.27 (s, 1H, pyrrolizine-NHCO), 9.94 (s, 1H, Cl-Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 84.5 (pyrrolizine C-7), 115.4 (CN), 115.8 (d, J = 22.3 Hz, F-Ph CH-3+CH-5), 117.3 (Cl-Ph C-4), 120.7 (d, J = 7.7 Hz, F-Ph CH-2 + CH-6), 121.7 (Cl-Ph CH-2 + CH-6), 127.7 (pyrrolizine C-5), 129.2 (Cl-Ph CH-3 + CH-5), 129.5 (pyrrolizine C-7a), 136.2 (d, J = 2.4 Hz, F-Ph C-1), 138.0 (pyrrolizine C-6), 146.5 (Cl-Ph C-1), 153.9 (NHCONH), 158.0 (d, J = 238.0 Hz, F-Ph C-4), 158.4 (Cl-Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 115.8 (d, J = 22.3 Hz, F-Ph CH-3+CH-5), 120.7 (d, J = 7.7 Hz, F-Ph CH-2 + CH-6), 121.7 (Cl-Ph CH-2 + CH-6), 129.2 (Cl-Ph CH-3 + CH-5). MS (EI): m/z (%) 438 ([M + 1]+, 5), 437 ([M]+, 15), 436 ([M-1]+, 2), 404 (6), 403 (14), 402 (2), 293 (14), 292 (82), 291 (7), 262 (2), 173 (5), 145 (3), 119 (15), 91 (43), 77 (37), 64 (100). Anal. Calcd. for C22H17ClFN5O2 (437.85): C, 60.35; H, 3.91; N, 15.99. Found: C, 59.92; H, 4.27; N, 16.22.
2.1.1.10. 7-Cyano-6-(3-(naphthalen-1-yl)ureido)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide (19a)
The title compound was prepared from the reaction of compound 15a (0.53 g, 2 mmol) with 1-naphthyl isocyanate (0.37 g, 2.2 mmol) according to the general procedure A. Compound 19a was obtained as white amorphous solid product, m.p. 258–61 °C, yield 74%. IRʋmax/cm−1 3393, 3262 (NHs), 3047 (aromatic C–H), 2987, 2951 (aliphatic C–H), 2223 (CN), 1671, 1640 (COs), 1553, 1528 (C = C, C = N), 1444, 1319, 1246 (C–N, C–O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.48–2.44 (m, 2H, pyrrolizine CH2-2), 3.02 (t, 2H, J = 6.5 Hz, pyrrolizine CH2-1), 4.30 (t, 2H, J = 7.5 Hz, pyrrolizine CH2-3), 7.08 (t, 1H, J = 7.3 Hz, Ph CH-4), 7.32 (t, 2H, J = 6.4 Hz, Ph CH-3 + CH-5), 7.53-7.48 (m, 2H, naphthalenyl CH-2 + pyrrolizine NH), 7.61 (d, 2H, J = 7.1 Hz, Ph CH-2 + CH-6), 7.66 (t, 1H, J = 8.8 Hz, naphthalenyl CH-3), 7.71 (d, 1H, J = 8.1 Hz, naphthalenyl CH-4), 7.87 (d, 1H, J = 7.7 Hz, naphthalenyl CH-5), 7.96 (t, 1H, J = 8.6 Hz, naphthalenyl CH-6), 8.11 (t, 1H, J = 7.5 Hz, naphthalenyl CH-7), 8.26 (d, 1H, J = 6.8 Hz, naphthalenyl CH-8), 8.89 (s, 1H, naphthalenyl-NHCO), 9.96 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 84.5 (pyrrolizine C-7), 117.9 (naphthalenyl CH-2), 118.1 (CN), 120.0 (Ph CH-2 + CH-6), 121.9 (naphthalenyl CH-4), 122.3 (naphthalenyl CH-8), 124.1 (naphthalenyl CH-7), 124.5 (naphthalenyl CH-6), 126.3 (naphthalenyl CH-3), 126.5 (Ph CH-4), 127.3 (naphthalenyl C-8a), 128.8 (naphthalenyl CH-5), 129.0 (pyrrolizine C-5), 129.3 (Ph CH-3 + CH-5), 134.2 (naphthalenyl C-4a), 134.5 (pyrrolizine C-7a), 134.8 (pyrrolizine C-6), 139.0 (Ph C-1), 146.2 (naphthalenyl C-1), 154.9 (NHCONH), 158.3 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 117.9 (naphthalenyl CH-2), 120.0 (Ph CH-2 + CH-6), 121.9 (naphthalenyl CH-4), 122.3 (naphthalenyl CH-8), 124.1 (naphthalenyl CH-7), 124.5 (naphthalenyl CH-6), 126.3 (naphthalenyl CH-3), 126.5 (Ph CH-4), 128.8 (naphthalenyl CH-5), 129.3 (Ph CH-3 + CH-5). MS (EI): m/z (%) 436 ([M + 1]+, 7), 432 ([M-3]+, 1), 424 (2), 412 (2), 331 (3), 312 (8), 287 (4), 189 (2), 178 (3), 169 (6), 143 (51), 116 (16), 115 (100), 114 (16), 91 (4), 90 (6), 75 (5). Anal. Calcd. for C26H21N5O2 (435.48): C, 71.71; H, 4.86; N, 16.08. Found: C, 72.12; H, 4.54; N, 16.25.
2.1.1.11. 7-Cyano-6-(3-(naphthalen-1-yl)ureido)-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (19b)
The title compound was prepared from the reaction of compound 15b (0.56 g, 2 mmol) with 1-naphthyl isocyanate (0.37 g, 2.2 mmol) according to the general procedure A. Compound 19 b was obtained as white amorphous solid product, m.p. 267–9 °C, yield 76%. IRʋmax/cm−1 3406, 3281 (NHs), 3050 (aromatic C–H), 2919, 2867 (aliphatic C–H), 2224 (CN), 1671, 1642 (COs), 1553, 1528 (C = C, C = N), 1433, 1390, 1298 (C–N, C–O). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.26 (s, 3H, CH3), 2.48–2.44 (m, 2H, pyrrolizine CH2-2), 3.01 (t, 2H, J = 6.4 Hz, pyrrolizine H2-1), 4.29 (t, 2H, J = 7.6 Hz, pyrrolizine CH2-3), 7.12 (d, 2H, J = 7.7 Hz, Ph CH-3 + CH-5), 7.49 (d, 2H, J = 8.1 Hz, Ph CH-2 + CH-6), 7.60–7.55 (m, 2H, naphthalenyl CH-2 + pyrrolizine-NH), 7.66 (t, 1H, J = 7.7 Hz, naphthalenyl CH-3), 7.71 (d, 1H, J = 8.0 Hz, naphthalenyl CH-4), 7.86 (d, 1H, J = 7.3 Hz, naphthalenyl CH-6), 7.96 (t, 1H, J = 7.9 Hz, naphthalenyl CH-7), 8.12–8.09 (m, naphthalenyl CH-5), 8.26 (d, 1H, J = 8.5 Hz, naphthalenyl CH-8), 8.88 (s, 1H, naphthalenyl-NHCO), 9.87 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 20.9 (CH3), 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 84.5 (pyrrolizine C-7), 115.5 (CN), 117.9 (naphthalenyl CH-2), 120.0 (Ph CH-2 + CH-6), 121.9 (naphthalenyl CH-4), 123.4 (naphthalenyl CH-8), 124.4 (naphthalenyl CH-7), 126.2 (naphthalenyl C-8a), 126.3 (naphthalenyl CH-6), 126.4 (naphthalenyl CH-3), 128.8 (pyrrolizine C-5), 129.0 (naphthalenyl CH-5), 129.7 (Ph CH-3 + CH-5), 133.2 (naphthalenyl C-4a), 134.2 (pyrrolizine C-7a), 134.5 (Ph C-1), 134.8 (pyrrolizine C-6), 136.5 (Ph C-4), 146.1 (naphthalenyl C-1), 155.0 (NHCONH), 158.1 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 20.9 (CH3), 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 117.9 (naphthalenyl CH-2), 120.0 (Ph CH-2 + CH-6), 121.9 (naphthalenyl CH-4), 123.4 (naphthalenyl CH-8), 124.4 (naphthalenyl CH-7), 126.3 (naphthalenyl CH-6), 126.4 (naphthalenyl CH-3), 129.0 (naphthalenyl CH-5), 129.7 (Ph CH-3 + CH-5). MS (EI): m/z (%) 450 ([M + 1]+, 5), 449 ([M]+, 8), 448 ([M-1]+, 2), 439 (5), 407 (3), 375 (3), 316 (8), 306 (49), 276 (3), 198 (3), 172 (4), 143 (16), 132 (16), 117 (33), 104 (17), 91 (17), 77 (88), 65 (100). Anal. Calcd. for C27H23N5O2 (449.50): C, 72.14; H, 5.16; N, 15.58. Found: C, 71.72; H, 4.86; N, 16.05.
2.1.1.12. N-(4-Chlorophenyl)-7-cyano-6-(3-(naphthalen-1-yl)ureido)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (19c)
The title compound was prepared from the reaction of compound 15c (0.6 g, 2 mmol) with 1-naphthyl isocyanate (0.37 g, 2.2 mmol) according to the general procedure A. Compound 19c was obtained as white amorphous solid product, m.p. 274–6 °C, yield 72%. IRʋmax/cm−1 3396, 3282 (NHs), 3059, 3008 (aromatic C–H), 2225 (CN), 1672, 1641 (COs), 1597, 1556 (C = C, C = N), 1492, 1386, 1245 (C–N, C–O), 791 (C–Cl). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.47–2.44 (m, 2H, pyrrolizine CH2-2), 3.02 (t, 2H, J = 6.1 Hz, pyrrolizine CH2-1), 4.29 (t, 2H, J = 6.8 Hz, pyrrolizine CH2-3), 7.38 (d, 2H, J = 6.8 Hz, Ph CH-3 + CH-5), 7.55–7.47 (m, 3H, naphthalenyl CH-2 + CH-3 + CH-6), 7.63 (d, 2H, J = 6.8 Hz, Ph CH-2 + CH-6), 7.70 (d, 1H, J = 8.1 Hz, naphthalenyl CH-4), 7.84 (d, 1H, J = 6.8 Hz, naphthalenyl CH-5), 7.96 (t, 1H, J = 8.2 Hz, naphthalenyl CH-7), 8.11 (d, 1H, J = 6.7 Hz, naphthalenyl CH-8), 8.89 (s, 1H, pyrrolizine-NHCO), 9.22 (s, 1H, naphthalenyl-NHCO), 10.07 (s, 1H, Ph-NHCO). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 84.4 (pyrrolizine C-7), 115.5 (CN), 117.6 (naphthalenyl C-8a), 117.9 (naphthalenyl CH-2), 119.4 (naphthalenyl CH-4), 121.6 (Ph CH-2 + CH-6), 122.2 (naphthalenyl CH-8), 124.4 (naphthalenyl CH-7), 126.3 (naphthalenyl CH-6), 126.5 (naphthalenyl CH-3), 127.2 (Ph C-4), 127.65 (pyrrolizine C-5), 128.8 (naphthalenyl CH-5), 129.2 (Ph CH-3 + CH-5), 129.4 (naphthalenyl C-4a), 134.2 (pyrrolizine C-7a), 134.5 (pyrrolizine C-6), 138.1 (Ph C-1), 146.4 (naphthalenyl C-1), 154.7 (NHCONH), 158.4 (Ph-NHCO). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 117.9 (naphthalenyl CH-2), 119.4 (naphthalenyl CH-4), 121.6 (Ph CH-2 + CH-6), 122.2 (naphthalenyl CH-8), 124.4 (naphthalenyl CH-7), 126.3 (naphthalenyl CH-6), 126.5 (naphthalenyl CH-3), 128.8 (naphthalenyl CH-5), 129.2 (Ph CH-3 + CH-5). MS (EI): m/z (%) 471 ([M + 2]+, 1), 469 ([M]+, 3), 434 (2), 424 (2), 326 (3), 314 (6), 313 (21), 312 (100), 311 (19), 293 (2), 210 (2), 169 (2), 143 (10), 115 (54), 90 (6), 77 (25). Anal. Calcd. for C26H20ClN5O2 (469.92): C, 66.45; H, 4.29; N, 14.90. Found: C, 66.12; H, 4.67; N, 15.32.
2.1.1.13. 7-Cyano-6-(3-(naphthalen-1-yl)thioureido)-N-phenyl-2,3-dihydro-1H-pyrrolizine-5-carboxamide (20a)
The title compound was prepared from the reaction of compound 15a (0.53 g, 2 mmol) with 1-naphthyl isothiocyanate (0.41 g, 2.2 mmol) according to the general procedure A. Compound 20a was obtained as white amorphous solid product, m.p. 264–6 °C, yield 69%. IRʋmax/cm−1 3339, 3168 (NHs), 3054 (aromatic C-H), 2964, 2919 (aliphatic C–H), 2229 (CN), 1694 (COs), 1596, 1523 (C = C, C = N), 1488, 1398, 1331, 1216, 1189 (C–N, C–O, C = S). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.47–2.39 (m, 2H, pyrrolizine CH2-2), 3.07 (t, 2H, J = 6.8 Hz, pyrrolizine CH2-1), 4.21 (t, 2H, J = 7.0 Hz, pyrrolizine CH2-3), 7.20 (d, 1H, J = 7.3 Hz, naphthalenyl CH), 7.32 (t, 1H, J = 7.5 Hz, naphthalenyl CH), 7.41 (d, 1H, J = 7.6 Hz, naphthalenyl CH), 7.47 (t, 2H, J = 7.30 Hz, Ph CH-3 + CH-5), 7.63–7.52 (m, 6H, 5 aromatics Hs + pyrrolizine-NH), 7.87 (d, 1H, J = 8.0 Hz, naphthalenyl H), 7.97 (d, 1H, J = 7.9 Hz, aromatic CH), 8.04 (d, 1H, J = 8.3 Hz, naphthalenyl H), 9.85 (s, 1H, Ph-NH), 13.76 (s, 1H, naphthalenyl-NH). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.8 (pyrrolizine CH2-2), 25.3 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 77.4 (pyrrolizine C-7), 108.1 (CN), 110.4 (naphthalenyl C-8a), 120.4 (naphthalenyl CH-2), 123.7 (Ph CH-2 + CH-6), 126.1 (naphthalenyl CH-4), 126.3 (naphthalenyl CH-8), 126.6 (naphthalenyl CH-7), 126.6 (pyrrolizine C-5), 127.4 (naphthalenyl CH-6), 128.6 (Ph CH-3 + CH-5), 129.1 (naphthalenyl CH-3), 129.4 (Ph CH-4), 129.6 (naphthalenyl CH-5), 130.7 (naphthalenyl C-4a), 134.4 (pyrrolizine C-7a), 135.8 (pyrrolizine C-6), 139.5 (Ph C-1), 146.8 (naphthalenyl C-1), 159.9 (Ph-NHCO), 175.9 (NHCSNH). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 24.8 (pyrrolizine CH2-2), 25.3 (pyrrolizine CH2-1), 49.7 (pyrrolizine CH2-3), 120.4 (naphthalenyl CH-2), 123.7 (Ph CH-2 + CH-6), 126.1 (naphthalenyl CH-4), 126.3 (naphthalenyl CH-8), 126.6 (naphthalenyl CH-7), 127.4 (naphthalenyl CH-6), 128.6 (Ph CH-3 + CH-5), 129.1 (naphthalenyl CH-3), 129.4 (Ph CH-4), 129.6 (naphthalenyl CH-5). MS (EI): m/z (%) 452 ([M + 1]+, 1), 436 ([M-15]+, 2), 434 (7), 432 (13), 395 (2), 375 (4), 314 (100), 296 (4), 285 (5), 268 (2), 251 (2), 188 (6), 91 (6), 77 (11). Anal. Calcd. for C26H21N5OS (451.54): C, 69.16; H, 4.69; N, 15.51. Found: C, 69.46; H, 4.82; N, 15.21.
2.1.1.14. 7-Cyano-6-(3-(naphthalen-1-yl)thioureido)-N-(p-tolyl)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (20b)
The title compound was prepared from the reaction of compound 15b (0.56 g, 2 mmol) with 1-naphthyl isothiocyanate (0.41 g, 2.2 mmol) according to the general procedure A. Compound 20 b was obtained as white amorphous solid product, m.p. 279–81 °C, yield 72%. IRʋmax/cm−1 3379, 3281, 3132 (NHs), 3059 (aromatic C-H), 2929, 2837 (aliphatic C–H), 2225 (CN), 1664, (C = O), 1599, 1567, 1539 (C = C, C = N), 1459, 1376, 1237, 1151 (C–N, C–O, C = S). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.36 (s, 3H, CH3), 2.52–2.49 (m, 2H, pyrrolizine CH2-2), 3.07 (t, 2H, J = 6.8 Hz, pyrrolizine CH2-1), 4.21 (t, 2H, J = 6.8 Hz, pyrrolizine CH2-3), 7.05 (d, 1H, J = 7.1 Hz, naphthalenyl CH), 7.26 (d, 1H, J = 7.4 Hz, naphthalenyl CH), 7.45 (broad s, 1H, naphthalenyl CH), 7.61–7.51 (m, 4H, aromatic Hs), 7.86 (d, 1H, J = 7.8 Hz, naphthalenyl CH), 7.96 (d, 2H, J = 8.4 Hz, tolyl CH-2 + CH-6), 8.06–8.02 (m, 2H, aromatic Hs), 9.83 (s, 1H, pyrrolizine-NH), 10.56 (s, 1H, tolyl-NH), 13.71 (broad s, 1H, naphthalenyl-NH). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 21.3 (CH3), 25.1 (pyrrolizine CH2-2), 26.2 (pyrrolizine CH2-1), 48.8 (pyrrolizine CH2-3), 109.1 (pyrrolizine C-7), 110.4 (CN), 118.8 (naphthalenyl CH-2), 123.7 (tolyl CH-2 + CH-6), 126.1 (naphthalenyl CH-4), 126.3 (naphthalenyl CH-8), 126.6 (naphthalenyl C-8a), 126.6 (naphthalenyl CH-7), 127.0 (pyrrolizine C-5), 127.3 (naphthalenyl CH-6), 128.6 (tolyl CH-3 + CH-5), 129.3 (naphthalenyl CH-3), 129.9 (naphthalenyl CH-5), 130.7 (naphthalenyl C-5a), 131.9 ((pyrrolizine C-7a), 134.4 (tolyl C-1), 135.8 (pyrrolizine C-6), 136.1 (tolyl C-4), 145.6 (naphthalenyl C-1), 158.3 (tolyl-NHCO), 176.0 (NHCSNH). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 21.3 (CH3), 25.1 (pyrrolizine CH2-2), 26.2 (pyrrolizine CH2-1), 48.8 (pyrrolizine CH2-3), 118.8 (naphthalenyl CH-2), 123.7 (tolyl CH-2 + CH-6), 126.1 (naphthalenyl CH-4), 126.3 (naphthalenyl CH-8), 126.6 (naphthalenyl CH-7), 127.3 (naphthalenyl CH-6), 128.6 (tolyl CH-3 + CH-5), 129.3 (naphthalenyl CH-3), 129.9 (naphthalenyl CH-5). MS (EI): m/z (%) 466 ([M + 1]+, 28), 465 ([M]+, 16), 462 ([M-3]+, 18), 446 (15), 424 (17), 404 (37), 382 (57), 369 (42), 359 (22), 347 (15), 266 (46), 236 (27), 218 (18), 185 (16), 174 (25), 160 (19), 143 (4), 119 (22), 92 (100), 77 (29). Anal. Calcd. for C27H23N5OS (465.57): C, 69.65; H, 4.98; N, 15.04. Found: C, 69.54; H, 4.72; N, 14.87.
2.1.1.15. N-(4-chlorophenyl)-7-cyano-6-(3-(naphthalen-1-yl)thioureido)-2,3-dihydro-1H-pyrrolizine-5-carboxamide (20c)
The title compound was prepared from the reaction of compound 15c (0.6 g, 2 mmol) with 1-naphthyl isothiocyanate (0.41 g, 2.2 mmol) according to the general procedure A. Compound 20c was obtained as white amorphous solid product, m.p. 287–9 °C, yield 68%. IRʋmax/cm−1 3279, 3244, 3179 (NHs), 3053 (aromatic C–H), 2929 (aliphatic C–H), 2224 (CN), 1662 (C = O), 1600, 1567, 1542 (C = C, C = N), 1491, 1332, 1230, 1149 (C–N, C–O, C = S), 792 (C–Cl). 1H-NMR (DMSO-d6, 500 MHz, δ ppm): δ 2.49–2.41 (m, 2H, pyrrolizine CH2-2), 3.07 (t, 2H, J = 6.1 Hz, pyrrolizine CH2-1), 4.31 (t, 2H, J = 7.8 Hz, pyrrolizine CH2-3), 7.31–7.17 (m, 2H), 7.60–7.49 (m, 5H), 7.90 (d, 1H, J = 7.9 Hz, naphthalenyl CH-4), 8.00 (d, 1H, J = 7.0 Hz, naphthalenyl CH-5), 8.05 (d, 1H, J = 7.7 Hz, naphthalenyl CH-8), 10.12 (s, 1H, pyrrolizine-NH), 10.69 (s, 1H, Cl-Ph-NH), 11.55 (s, 1H, naphthalenyl-NH). 13 C-NMR (DMSO, 125 MHz, δ ppm): δ 24.9 (pyrrolizine CH2-2), 25.6 (pyrrolizine CH2-1), 49.6 (pyrrolizine CH2-3), 84.7 (pyrrolizine C-7), 108.7 (CN), 111.0 (naphthalenyl C-8a), 120.4 (Cl-Ph CH-2 + CH-6), 123.2 (naphthalenyl CH-2), 125.2 (naphthalenyl CH-4), 126.3 (naphthalenyl CH-8), 126.8 (Cl-Ph C-4), 127.1 (naphthalenyl CH-7), 127.2 (naphthalenyl CH-6), 128.0 (naphthalenyl CH-3), 128.8 (Cl-Ph CH-3 + CH-5), 129.3 (naphthalenyl CH-5), 134.4 (pyrrolizine C-5), 134.6 (naphthalenyl C-4a), 135.6 (pyrrolizine C-7a), 136.8 (pyrrolizine C-6), 138.4 (Cl-Ph C-1), 146.0 (naphthalenyl C-1), 158.4 (Cl-Ph-NHCO), 173.4 (NHCSNH). DEPT C135 (DMSO, 125 MHz, δ ppm): δ 25.0 (pyrrolizine CH2-2), 25.7 (pyrrolizine CH2-1), 49.6 (pyrrolizine CH2-3), 120.4 (Cl-Ph CH-2 + CH-6), 123.2 (naphthalenyl CH-2), 125.2 (naphthalenyl CH-4), 126.3 (naphthalenyl CH-8), 127.1 (naphthalenyl CH-7), 127.2 (naphthalenyl CH-6), 128.0 (naphthalenyl CH-3), 128.8 (Cl-Ph CH-3 + CH-5), 129.3 (naphthalenyl CH-5). MS (EI): m/z (%) 487 ([M + 2]+, 28), 486 ([M + 1]+, 47), 472 (25), 445 (52), 435 (85), 412 (58), 367 (74), 336 (93), 277 (100), 262 (37), 202 (52), 156 (59), 128 (24), 91 (23), 77 (4). Anal. Calcd. for C26H20ClN5OS (485.99): C, 64.26; H, 4.15; N, 14.41. Found: C, 64.56; H, 4.62; N, 14.12.
2.2. Biological evaluation
2.2.1. Cytotoxic activity
2.2.1.1. Cell culture
The cancer/normal cell lines used in this study were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The cell culture conditions were the same as in our previous reports25.
2.2.1.2. MTT assay
MTT assay was used to evaluate the cytotoxicity of the new compounds. The cancer/normal cells were incubated for 72 h with the test compounds (dissolved in DMSO). The cytotoxic activities (IC50 values) of the new compounds were determined according to the previous reports42.
2.2.2. Annexin V-FITC/PI staining assay
The apoptotic effect of compounds 18b, 19a, and 20a on MCF-7 cells was evaluated after treatment for 24 h at 0, 1, 5, and 10 µM. The assay was done using Annexin V-FITC/PI staining assay. Flow cytometry (Bechman Coulter, FC500, Brea, CA) was used differentiate apoptotic/necrotic cells from viable cells43.
2.2.3. Cell cycle analysis
The MCF-7 cells were incubated with compounds 18b, 19a, and 20a at 0, 1, 5, and 10 µM. After 24 h treatment. The PI-stained cells was used to analyse cells using flow cytometer (BC, FC500)44.
2.2.4. Cdk-2 inhibition assay
The CDK-2 inhibitory activities of compounds 18b, 19a, and 20a were determined in vitro using ADP-GloTM kinase assay (Promega, Madison, WI). The assay was done according to manufacturer’s instructions and as described in the previous report45. The results were represented as IC50 values, Table 4.
Table 4.
Inhibition of CDK-2 by compounds 18b, 19a, and 20a.
| Comp. no. | IC50 (nM)a ±SEM |
|---|---|
| 18b | 115.3 ± 3.20 |
| 19a | 25.53 ± 1.7 |
| 20a | 77.90 ± 2.2 |
| Staurosporine | 26.39 ± 0.9 |
IC50, concentration which decrease kinase activity to 50%.
aAverage of three determinations;.
2.3. Computational studies
2.3.1. Molecular docking studies
The docking studies were done using AutoDock 4.246. Preparation of ligands, proteins, grid, and docking parameter files was done following our previous reports47–49. The study was performed according to the previous report25. Discovery studio visualiser was used to visualise the binding interaction50. The pdb files of CDK-2 (pdb: 2VTP)16, CDCK-6 (pdb: 2EUF)51, and CDK-9 (pdb: 3TNH)52 were downloaded from protein data bank (http://www.rcsb.org/pdb). The results of the docking study of compounds 18b, 19a, and 20a were represented in Tables 5 and 6 and Figures 9–11. Moreover, the results of the docking study of the remaining compounds into CDKs are provided in supplementary data (Figures S138–S152).
Table 5.
Docking results of compounds 18b, 19a, 20a and LZ9 into CDK-2 (pdb: 2VTP)
| Ligand | ΔGba | Kib | HBsc | Atoms in H-bonding |
Length d(Å) | |
|---|---|---|---|---|---|---|
| In ligand | In protein | |||||
| 18 b | −9.16 | 193.5 nM | 3 | Urea NH | CO of Leu83 | 1.88 |
| CN | NH of Leu83 | 2.09 | ||||
| Ph-NH | CO of Gln131 | 2.06 | ||||
| 19a | −9.97 | 49.38 nM | 3 | CN | NH of Leu83 | 1.97 |
| Urea NH | CO of Leu83 | 1.80 | ||||
| Ph-NH | CO of Gln131 | 2.93 | ||||
| 20a | −9.54 | 101.28 nM | 4 | CN | NH of Leu83 | 1.90 |
| Thiourea S | CO of Asp86 | 2.07 | ||||
| Thiourea S | CO of Gln131 | 2.60 | ||||
| Ph-NH | CO of Gln131 | 2.76 | ||||
| LZ9 | −7.56 | 2.81 μM | 3 | Pyrazole-CO | NH of Glu81 | 2.21 |
| pyrazole NH | CO of Leu83 | 2.05 | ||||
| pyrazole NHe | CH of Phe82 | |||||
aBinding free energy (kcal/mol).
bInhibition constant (n/μM).
cHBs, number of hydrogen bonds.
dLength in angstrom (Å).
eCarbon hydrogen bond.
Table 6.
Docking results of compounds, 18b, 19a, and 20a into CDK-6 (pdb: 2EUF) and CDK-9 (pdb: 3TNH) in comparison to the native ligands, palbociclib (LQQ) and CAN508 (F18).
| Ligand | CDK-6 |
CDK-9 |
||||
|---|---|---|---|---|---|---|
| ΔGba | Kib | Fsc | ΔGba | Kib | Fsc | |
| 18b | −9.84 | 61.08 nM | 3.17 | −9.81 | 64.89 nM | 2.98 |
| 19a | −10.52 | 19.41 nM | 2.54 | −10.40 | 23.93 nM | 2.06 |
| 20a | −10.30 | 28.30 nM | 3.58 | −9.99 | 47.27 nM | 2.14 |
| LQQd | −11.54 | 3.48 nM | – | – | – | – |
| F18e | – | – | – | −6.29 | 24.67 μM | – |
aBinding free energy (kcal/mol).
bInhibition constant.
cFold selectivity towards CDK-6/9, calculated by dividing the Ki value against CDK-2 by the Ki value against CDK-6/9.
dLQQ, palbociclib/.
eF18, CAN508.
Figure 9.

Binding mode and interactions of compound 19a into CDK-2 (pdb: 2VTP)): (A) 3D binding mode of compound 19a into the active site of CDK-2, the native ligand LZ9 shown as yellow sticks, receptor shown as hydrogen-bond surface; (B) 2D binding mode of compound 19a into CDK-2 showing three conventional hydrogen bonds, one pi-cation, and several hydrophobic interactions, hydrogen atoms were omitted for clarity.
Figure 10.

Binding modes and interactions of compounds 18b/20a into CDK-2 (pdb: 2VTP)): A) 3 D binding mode of compound 18b into CDK-2, the native ligand LZ9 shown as yellow sticks, receptor shown as hydrogen-bond surface; B) 3 D binding mode of compound 20a into CDK-2, the native ligand LZ9 shown as yellow sticks; C) 2 D binding mode of compound 18b into CDK-2 showing three conventional hydrogen bonds and several hydrophobic interactions; (C) 2D binding mode of compound 20a into CDK-2 showing four conventional hydrogen bonds and one pi-anion, and several hydrophobic interactions, hydrogen atoms were omitted for clarity.
Figure 11.

3D binding modes, hydrogen bonding (shown as green dotted lines), and electrostatic interactions (shown as orang dotted lines) of compounds 18b, 19a, and 20a (shown as sticks) into CDK-6 (pdb: 2EUF) and CDK-9 (pdb: 3TNH): (A) compound 18b into CDK-6; (B) compound 19a into CDK-6; (C) compound 20a into CDK-6; (D) compound 18b into CDK-9; (E) compound 19a into CDK-9; (F) compound 20a into CDK-9, amino acid residues shown as lines coloured by element, hydrogen atoms were omitted for clarity.
2.3.2. Drug-likeness and ADME studies
SwissADME webserver (http://www.swissadme.ch/)53 was used to predict the physicochemical characters related to pharmacokinetic parameters of the new compounds. The study was performed following the previous report39. The molecular weights, ClogP, number of hydrogen bond donner (HBD), and hydrogen bond acceptor (HBA) of the new compounds were calculated checked for any violation from Lipinski’s rule54. The chemical structures were sketched and calculation start following submission of the chemical structures. MVs, TPSA, and DLSs were calculated using Molsoft webserver (http://molsoft.com/mprop/). The expected GIT absorption percent (%Abs) of the new compounds was also calculated according to the previous report55.
3. Results and discussion
3.1. Chemistry
Preparation of the starting compounds 1237 and 14a–c38–40 was performed according to the previous reports, Scheme 1. Briefly, the lactam derivative 11 was reacted with dimethyl sulphate followed by reaction with malononitrile to give compound 12. On the other hand, the anilides 14a–c were prepared form the reaction of (un)substituted-anilines with chloroacetyl chloride. The starting materials 15a–c were obtained from the reaction of compound 12 with the acetanilides 14a–c in refluxing acetone41.
Scheme 1.

Reagents and conditions: (a) (CH3)2SO4, benzene, CH2(CN)2; (b) ClCH2COCl, g. acetic acid, CH2COONa; (c) acetone, K2CO3, reflux, 24 h.
The target urea derivatives 16–20a–c (Scheme 2) were prepared from the reaction of compounds 15a–c with the appropriate isocyanate in DCM according to the previous report23.
Scheme 2.

Reagents and conditions: (d) hexyl isocyanate, TEA, DCM, stir, rt, 12 h; (e) appropriate isocyanate, TEA, DCM, stir, rt, 12 h; (f) appropriate isocyanate/isothiocyanate, TEA, DCM, stir, rt, 12 h.
The crude white solid product was dissolved in chloroform/acetone and the white amorphous solid formed was collected. The chemical structural of the new compounds was confirmed by spectral and elemental analyses, Supplementary data (Figures S43–S137).
3.2. Biological evaluation
3.2.1. Cytotoxic activity
3.2.1.1. Evaluation of cytotoxic activity
All the new compounds were evaluated for their cytotoxicity against three cancer (MCF-7, A2780, and HT29) cell lines. Selection of these cell lines was based on previous findings which reported an overexpression of CDK-2 in breast, ovarian, and colon cancers8–10.
The investigation of cytotoxicity of the new compounds was performed using the MTT assay42. The results were expressed in IC50 values (the concentration required to inhibit the growth of cancer cells by 50% compared to the untreated control), Table 2.
Table 2.
Cytotoxic activity of compounds 10, 16–20a–c and lapatinib against MCF-7, A2780, and HT29 cell lines. 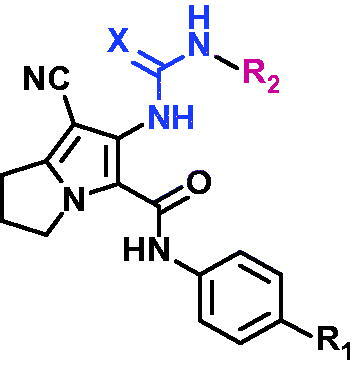
| Comp. no. | R1 | R2 | X | (IC50 ± SD μM)a |
Average μM | ||
|---|---|---|---|---|---|---|---|
| MCF-7 | A2780 | HT29 | |||||
| 16a | H | -C6H13 | O | 0.59 ± 0.01 | 0.35 ± 0.18 | 0.85 ± 0.07 | 0.60 |
| 16b | CH3 | -C6H13 | O | 1.17 ± 0.31 | 0.23 ± 0.04 | 1.56 ± 0.14 | 0.99 |
| 16c | Cl | -C6H13 | O | 34.13 ± 4.61 | 0.53 ± 0.10 | 3.10 ± 0.10 | 12.59 |
| 17a | H | 4-MeOPh | O | 0.90 ± 0.11 | 0.44 ± 0.03 | 5.29 ± 1.51 | 2.21 |
| 17b | CH3 | 4-MeOPh | O | 2.85 ± 0.31 | 0.42 ± 0.07 | 1.44 ± 0.17 | 1.57 |
| 17c | Cl | 4-MeOPh | O | 3.36 ± 0.83 | 0.54 ± 0.02 | 0.42 ± 0.16 | 1.44 |
| 18a | H | 4-F-Ph | O | 10.76 ± 0.07 | 10.58 ± 2.83 | 9.13 ± 0.85 | 10.16 |
| 18b | CH3 | 4-F-Ph | O | 0.19 ± 0.02 | 0.97 ± 0.14 | 9.71 ± 2.25 | 3.62 |
| 18c | Cl | 4-F-Ph | O | 0.29 ± 0.19 | 0.21 ± 0.10 | 1.38 ± 0.02 | 0.63 |
| 19a | H | 1-naphthalenyl | O | 0.40 ± 0.42 | 0.32 ± 0.17 | 0.20 ± 0.01 | 0.31 |
| 19b | CH3 | 1-naphthalenyl | O | 0.44 ± 0.09 | 5.39 ± 1.13 | 0.68 ± 0.14 | 2.17 |
| 19c | Cl | 1-naphthalenyl | O | 1.27 ± 0.01 | 3.07 ± 1.50 | 2.56 ± 0.70 | 2.30 |
| 20a | H | 1-naphthalenyl | S | 0.42 ± 0.15 | 0.16 ± 0.01 | 0.72 ± 0.12 | 0.43 |
| 20b | CH3 | 1-naphthalenyl | S | 4.97 ± 0.42 | 2.04 ± 0.95 | 3.83 ± 0.58 | 3.61 |
| 20c | Cl | 1-naphthalenyl | S | 1.76 ± 0.95 | 2.81 ± 0.98 | 3.67 ± 1.65 | 2.75 |
| 10 | 3,5-diMec | 4-Cl-Ph | O | 1.12 ± 0.10 | – | – | – |
| Lap. | – | – | – | 6.77 ± 1.16 | 10.52 ± 0.76 | 12.46 ± 1.27 | 9.92 |
aIC50 is the concentration of test compounds which reduce cellular growth to 50% after treatment for 72 h.
bResults represent mean IC50 value ± SD (n = 3); Lap, lapatinib.
cCI50 value quoted from our previous publication23.
The results revealed the ability of the new compounds to inhibit the growth of the selected cancer cell lines with IC50 values in the range of 0.16–34.13 μM. Among these derivatives, compound 18b was the most active against MCF-7 cells (IC50 = 0.19 μM), while compounds 19a and 20a were the most potent in inhibiting the growth of HT29 and A2780 cell line, respectively.
The hexyl urea derivatives 16a–c displayed weak to potent cytotoxic activity against the three cell lines (IC50 = 0.23–34.13 μM). Among the three derivatives, compound 16b was the most active against A2780, while compound 16a showed the highest cytotoxic activity against MCF-7 and HT29 cell lines, Table 2.
Furthermore, the 4-methoxyphenyl-urea derivatives 17a–c showed potent cytotoxic activity against the three cancer cell lines with IC50 values in the range of 0.42–5.29 μM. Compound 17c was more active against MCF-7 and HT29 cell lines compared to its hexyl urea 16c. However, compounds 17a,b were less active against MCF-7 and A2780 cell lines than their hexyl analogs 16a,b, Table 2.
The 4-fluorophenyl urea derivatives 18a–c showed moderate to potent cytotoxic activities against the three cancer cell lines (IC50 = 0.19–10.76 μM). Among these derivatives compound 18b was the most active against MCF-7 cells. Moreover, compound 18c exhibited higher cytotoxic activities towards MCF-7 and A2780 cell lines compared to the hexyl 16c and methoxyphenyl 17c analogues, Table 2.
The naphthalenyl urea derivatives 19a–c exhibited the highest cytotoxicity with IC50 in the range of 0.20–5.39 μM. Among these derivatives, compound 19a displayed the highest cytotoxic activity against HT29 cell line. On the other hand, the thiourea derivatives 20a–c exhibited higher cytotoxic activities against A2780 cells than their urea analogues 19a–c. However, they (20a–c) were less active against MCF-7 and HT29 cells compared to compounds 19a–c, Table 2.
In conclusion, the results of MTT assay of most of the new compounds revealed higher cytotoxicity compared to lapatinib (IC50 = 6.77–12.46 μM). In addition, most of the new compounds (16a, 17a, 18b,c, 19a–c) exhibited higher cytotoxic activities against MCF-7 than the parent compound 10, Table 2.
3.2.1.2. Evaluation of cytotoxic selectivity
Beside the cytotoxic activity, toxicity and selectivity towards normal cells are also critical factors in the discovery of new anticancer agents56. Accordingly, cytotoxicity of the new reported compounds (16–20) was also evaluated in the current study using the normal human foetal lung fibroblast (MRC-5). The investigation was performed using the MTT assay according to the previous reports25. The results were presented in Table 3.
Table 3.
Cytotoxicity of compounds 16–20a–c, and lapatinib against normal MRC-5 cells and their selectivity indices (SIs).
| Comp. no. | IC50 (µM ± SD)a MRC-5 | Selectivity indexb |
||
|---|---|---|---|---|
| MCF-7 | A2780 | HT29 | ||
| 16a | 11.86 ± 0.87 | 20.10 | 33.89 | 13.95 |
| 16b | 2.65 ± 0.29 | 2.26 | 11.52 | 1.70 |
| 16c | 0.75 ± 0.14 | 0.02 | 1.42 | 0.24 |
| 17a | 2.30 ± 0.15 | 2.56 | 5.23 | 0.43 |
| 17b | 6.20 ± 0.88 | 2.18 | 14.76 | 4.31 |
| 17c | 3.80 ± 0.25 | 1.13 | 7.04 | 9.05 |
| 18a | 10.70 ± 0.61 | 0.99 | 1.01 | 1.17 |
| 18b | 1.46 ± 0.42 | 7.68 | 1.51 | 0.15 |
| 18c | 0.10 ± 0.01 | 0.34 | 0.48 | 0.07 |
| 19a | 2.67 ± 0.37 | 6.68 | 8.34 | 13.35 |
| 19b | 1.54 ± 0.20 | 3.50 | 0.29 | 2.26 |
| 19c | 2.68 ± 0.37 | 2.11 | 0.87 | 1.05 |
| 20a | 0.43 ± 0.13 | 1.02 | 2.69 | 0.60 |
| 20b | 2.00 ± 0.75 | 0.40 | 0.98 | 0.52 |
| 20c | 2.92 ± 0.60 | 1.66 | 1.04 | 0.80 |
| Lapatinib | 6.74 ± 1.22 | 1.00 | 0.64 | 0.54 |
aIC50 against MRC-5 cells after 72 h treatment with the test compound, results represent mean IC50 ± SD (n = 3).
bSelectively index (SI) = IC50 value against normal MRC-5 cells/IC50 value against cancer cell line.
The results revealed IC50 values in the range of 0.10–11.86 µM. To evaluate the cytotoxic selectivity of the new compounds, the selectivity index (SI) for each compound was calculated. The SI was obtained by dividing its IC50 value of the compound against normal MRC-5 cells by its IC50 value against the corresponding cancer cell line, Table 3.
Among the new compounds, the hexyl urea derivatives 16a–c inhibited the growth of MRC-5 cells with IC50 values in the range of 0.75–11.86 µM. among these derivatives, compound 16a showed the highest selectivity towards the three cell lines with SIs in the range of 13.95–33.89.
Moreover, the 4-methoxyphenyl urea derivatives 17a–c exhibited higher selectivity (SI = 5.23–14.76) towards the ovarian cancer cell line (A2780) compared to their 4-fluorophenyl analogues 18a–c. However, compound 18b displayed more than seven times higher selectivity towards the cancerous MCF-7 cells compared to lapatinib. In addition, the naphthalenyl urea derivatives 19a–c displayed higher selectivity towards MCF-7 and HT29 cells compared to their thiourea derivatives 20a–c, Table 3.
3.2.1.3. Structure–activity relationship (SAR)
The relationship between the chemical structure of the new compounds and their cytotoxic activity/selectivity was presented in Figure 6. Compound 16a which exhibited potent submicromolar cytotoxicity (IC50 = 0.35–0.85 μM) and was the most selective towards three cancer cell lines.
Figure 6.

SAR of cytotoxicity and selectivity of the new compounds against MCF-7, A2780, and HT29 cells.
Substitution on the phenyl ring in compound 16a with 4-CH3 increased the cytotoxic activity against A2780 cells. On the other hand, significant decreases in cytotoxic activities of compound 16a against both MCF-7 and HT29 cell lines were observed on substitution with either 4-CH3/Cl groups on the phenyl ring. Moreover, substitution with CH3/Cl groups at the para-position of the phenyl ring in compound 16a was associated with decrease in selectivity towards the three cancer cell lines, Figure 6.
Replacement of the hexyl group in compound 16a by 4-methoxyphenyl afforded compound 17a with slight decrease in cytotoxicity against both MCF-7 and A2780 cells, while significant reduction in activity against HT29 cells was observed. Substitution on the phenyl ring (A) in compound 17a with 4-CH3/Cl resulted in increase in cytotoxic activity against HT29 cells, while cytotoxicity against MCF-7 was decreases, Figure 6.
In addition, replacement of the OCH3 group in compound 17a by F afforded compound 18a with sharp decrease in cytotoxicity against the three cancer cell lines. On the other hand, substitution on the phenyl ring of compound 18a with either the electron donating 4-CH3 or the electron withdrawing 4-Cl groups improved cytotoxicity against both MCF-7 and A2780 cell lines, Figure 6.
Replacement of the hexyl group in compound 16a by 1-naphthalenyl group afforded compound 19a which was more active as cytotoxic agent against the three cancer cell lines. Although cytotoxicity of compound 19a was decreased on substitution with 4-CH3/Cl groups on ring A, but the produced derivatives 19b,c were more active than their hexyl analogues 16b,c against MCF-7 and HT29 cells, Figure 6.
Replacement of urea oxygen in compound 19a by sulphur yielded compound 20a with higher cytotoxicity against A2780 cells but lower cytotoxic activities towards HT29 cells. Substitution on ring A of compound 20a with the 4-CH3/Cl groups resulted in weaker cytotoxic activities against the three cancer cell lines, Figure 6.
3.2.2. Annexin V-FITC/PI apoptosis assay
Previously, compound 10, the lead compound in this study was reported to induce apoptosis in MCF-7 cells through activation of caspase 3/723. Accordingly, the ability of the new compounds to induce apoptosis in the same cells (MCF-7) was investigated in the current study. Compounds 18 b, 19a, and 20a the most active against MCF-7 cells were selected for this study. The ability of these compounds to induce apoptosis was evaluated using annexin V fluorescein isothiocyanate (FITC)/Propidium Iodide (PI) staining assay according to the previous report43. MCF-7 cells were treated with each the test compounds for 24 h. The results were presented in Figure 7.
Figure 7.

Annexin V phases of MCF-7 treated with compound 18b, 19a, and 20a at 0, 1, 5, and 10 µM (24 h, x-axis: Annexin V; y-axis: PI). C1: necrotic cells; C2: late apoptosis; C3: live cells; C4: early apoptosis. Data shown is mean % cell number ± SD (n = 3). Experiment was repeated x3 times.
The results revealed a significant dose dependent increase of the apoptotic events by compound 18 b from 0% in the control, to 20%, 23%, and 37% at 1, 5, and 10 µM, respectively. Similarly, compound 20a caused increase of 38%, 39%, and 47% at 1, 5, and 10 µM, respectively. in addition, compound 19a caused the highest increase in apoptotic events with 68%, 88%, and 90% at 1, 5, and 10 µM, respectively, Figure 7.
3.2.4. Cell-cycle analysis
The effect of compounds 18 b, 19a, and 20a, on cell-cycle perturbation in MCF-7 cells was investigated based on previous report44. The MCF-7 cells were treated with each of the three compounds at 0, 1, 5, and 10 µM for 24 h. Following this treatment, the propidium iodide (PI)-stained cells were analysed. The results are outlined in Figure 8.
Figure 8.

Flow cytometry bar chart showing the effect of compounds 18b, 19a, and 20a at 0, 1, 5, and 10 μM on cell cycle distribution of MCF-7 cells after treatment for 24 h, values represent the mean ± SD (n = 3), experiment was repeated ×3.
The results of the cell cycle analysis revealed G1 -S cell cycle arrest by the three compounds (18b, 19a, and 20a). Compound 19a caused the highest increase in the number of cells in the G1 cell-cycle phase (70%) at the expense of the other phases, compared to 18b (53%) and 20a (57%), in Figure 8. These results agree with previous reports which indicate that CDK-2 inhibitors arrest cell cycle at the G1 phase6,7.
3.2.4. Cdk-2 inhibitory activity
Targeting CDKs is one of the promising strategies in cancer chemotherapy6. The success of this strategy was confirmed by many CDK inhibitors which reached phase I/II clinical trials as potential anticancer agents14. Previously, several compounds with pyrrolizine-5-carboxamide scaffold exhibited weak to moderate inhibitory activity against CDK-224,25.
In the current study, compounds 18b, 19a, and 20a, the most active in cytotoxic assay (Table 2) were selected to evaluate for their inhibitory activity against CDK-2 in vitro. The assay was performed using Adenosine diphosphate (ADP)-GloTM kinase assay kit (Promega, Madison, WI) following the previous report45. Briefly, the ability of CDK-2 to convert ATP to ADP was used as a measure of the kinase activity. The inhibition in activity of the kinase was determined after treatment with the test compounds relative to the control (DMSO, 5%). Staurosporine was used as positive control. The results of this assay are presented as IC50 in Table 4.
The results revealed high in vitro inhibitory activity against CDK-2 by the tested compounds with IC50 in the nanomolar range. Compound 19a exhibited the highest inhibitory activity against CDK-2, which was comparable to that of the positive control (staurosporine). The inhibitory activity of compound 19a was nearly three-fold higher than its thiourea analogue 20a. Moreover, the inhibitory activities of the two naphthalenyl derivatives 19a and 20a against CDK-2 was higher than that of the 4-methoxyphenyl urea 18b, Table 4.
3.3. Computational studies
3.3.1. Molecular docking studies
To identify the potential mechanism of action and understand the results of the CDK-2 inhibitory assay (Table 4), a molecular docking study was performed into the active site of CDK-2 protein. The study was performed to evaluate the binding free energies (affinities) and inhibition constants of the new compounds into CDK-2. The crystal structure of CDK-2 protein (pdb: 2VTP)16 was obtained from protein data bank (https://www.rcsb.org/structure/3TNW). AutoDock 4.2 was used to carry out the docking study46. The docking procedures were performed according to the previous report25. The grid and docking parameter files were prepared according to the previous reports47–49. Visualisation of the binding interactions of the new compounds in the active sites of CDK-2 was performed by Discovery studio visualizer50.
3.3.1.1. Docking study into CDK-2
Initially, the validation of the docking procedures into the active site of CDK-2 was performed. The native ligand, compound 3 (LZ9) was re-docked into the active site of CDK-2. The results revealed superposition of the re-docked ligand 3 over the position of the co-crystallized ligand with RMSD of 0.75 Å (Supplementary data, Figure S138). The results of the docking study of compounds 18b, 19a, and 20a revealed nice fitting into CDK-2 with binding free energy in the range of −9.16 to −9.77 kcal/mol. The binding affinities of the three compounds towards CDK-2 was also higher than that of the native ligand LZ9, Table 5. The results of the docking study of the remaining derivatives are provided in supplementary data (Table S1 and Figures S139–S150).
Among the three derivatives, compound 19a showed higher binding affinity towards CDK-2 compared to compounds 18b and 20a which was matched with their inhibitory activities against CDK-2, Table 5. In addition, the thiourea 20a exhibited also higher affinity to CDK-2 compared to the 4-flourophenyl derivative 18b which was also matched with their IC50 values towards CDK-2, Table 4. To explain these results, the binding modes, orientations, and interactions of the three compounds (18b, 18a, and, 20a) were investigated in comparison to the native ligand to understand their inhibitory activities against CDK-2.
Investigation of the binding mode of the best fit conformation of compound 19a revealed partial superposition of the pyrrole ring over the pyrazole ring in compound 3. Moreover, the naphthalenyl moiety in compound 19a superposed over the 4-fluorophenyl moiety of compound 3, Figure 9.
Compound 19a exhibited three conventional hydrogen bonds with Leu83 (2 HBs) and Gln131 with bond length in the range of 1.80–2.93 Å, Table 5. The pyrrolidine ring in compound 19a also formed one carbon hydrogen bond with Asp145. Compound 19a also showed one pi-anion interaction with Asp145 and a cluster of hydrophobic interactions with Val18, Ala31, Val64, Phe80, Leu134, and Ala144, Figure 9. These binding mode and interactions of compound 19a with CDK-2 could account for its high inhibitory activity against CDK-2.
Compound 20a also adopted binding orientations in which the naphthalenyl-thiourea side chain superpose the position of 4-fluorophenyl-carboxamide moiety in compound 3, Figure 10. Moreover, the phenyl-carboxamide side chain in compound 20a adopted an orientation superposing over the position of the difluorophenyl-carboxamide chain in LZ9, while the pyrrole ring in compound 20a overlaid partially over the pyrazole ring in LZ9.
The binding interactions of compound 20a with CDK-2 included four conventional hydrogen bonds with Leu83, Asp86, and Gln131 (2 HBs) amino acids. In addition, one pi-anion and several hydrophobic interactions were also formed between compound 20a and amino acids in the active site of CDK-2, Figure 10.
Although compound 20a showed higher number of hydrogen bonds with CDK-2, but it exhibited lower affinity than compound 19a towards CDK-2. This could be attributed to the stronger (shorter) hydrogen bonds formed by compound 19a with CDK-2 compared to compound 20a, Table 5.
On the other hand, compound 18b exhibited three conventional hydrogen bonds with Leu83 and Gln131 and one carbon hydrogen bond with Asp145. Several hydrophobic interactions of the alkyl and pi-alkyl types were also observed between compound 18b and amino acids in the active site of CDK-2, Figure 10.
The bulkier size of compounds 19a and 20a compared to compound 18b could account even partially to their higher affinities and higher inhibitory activities towards CDK-2. In addition, the higher binding affinities of compounds 19a and 20a compared to compound 18b could be due to higher number and/or stronger hydrogen bonding interactions compared to compound 18b. Moreover, each of the two compounds (19a and 20a) exhibited one electrostatic interaction of pi-anion type which was absent with compound 18b. these findings could account even partially for the higher inhibitory activity of compounds 19a and 20a compared to compound 18b.
Moreover, the results of the docking study into CDK-2 indicated the important role of the urea/thiourea moieties in the formation of hydrogen bonding interactions between the tested pyrrolizines (18b, 19a, and 20a) and amino acids in the active site of CDK-2. In addition, the pyrrolidine ring in the three compounds also contributed hydrophobically in the binding interactions into CDK-2 through the formation of a cluster of hydrophobic interactions with Ala31, Phe80, Leu134, and Ala144.
However, compounds 18b, 19a and 20a were designed based on the chemical structure of the multi-CDKI 3. Accordingly, a molecular docking study was performed to investigate other potential targets which could mediate their cytotoxic activities.
3.3.1.2. Docking into CDK-6/9
The multi-CDKIs 1–4 exhibited potent anticancer activities against breast, ovarian, and colon cancers which could be attributed to their pan-CDKs inhibitory activities15–18,57. The ability of these compounds to inhibit multi-CDKs could be attributed to the shared pharmacophoric groups between the four compounds, Figure 1. In addition, high sequence similarities were confirmed between different members in the CDK family. CDK-6 and CDK-9 have sequence similarity of 44% and 32% with CDK-2, respectively6. These similarities could also allow the CDK-2 inhibitors to target other members in the CDK family.
Based on these findings, compounds 18b, 19a, and 20a which were designed bearing the pharmacophoric features of the multi-CDKI 3 were docked into the active sites of CDK-6 (pdb: 2EUF)51 and CDK-9 (pdb: 3TNH)52. The aim of this study was to evaluate the ability of the three compounds to bind to and inhibit these two kinases. Selection of these three compounds was based on their high CDK-2 inhibitory activities, Table 4. In addition, the two CDKs 6/9 were selected based on the results of the cell cycle analysis which revealed G1 to S cell-cycle arrest by the three compounds (18b, 19a, and 20a), Figure 8.
The docking procedures into CDK-6/9 were essentially the same as those applied in the docking study into CDK-225. Validation of docking study into CDK-6 revealed superposition of the re-docked ligand, palbociclib over the co-crystallized ligand with RMSD of 0.61 Å, while the re-docked CAN508 into CDK-9 superposed the position of the native ligand with RMSD of 1.06 Å (Supplementary data, Figures S151 and S152). The docking results were presented in Table 6.
The results of the docking study into CDK-6 revealed good fitting of the three compounds with binding free energy in the range of −9.84 to −10.52 kcal/mol compared to −11.54 for palbociclib. Among the three pyrrolizines, the 4-fluorophenyl urea 19a displayed the highest affinity towards CDKs-6, while compound 18b showed the lowest binding free energy, Table 6.
Investigation of the binding interaction of compounds 18b, 19a, and 20a with CDK-6 revealed the formation of 1–3 conventional hydrogen bonds compared to one conventional hydrogen bond for palbociclib. The three compounds also exhibited several types of hydrophobic interactions with amino acids into the active site of CDK-6, Figure 11.
The tested compounds 18b, 19a, and 20a exhibited higher binding affinities (ΔGb = −9.81 to −10.40 kcal/mol) towards CDK-9 compared to the native ligand, CAN508 (ΔGb = −6.29 kcal/mol). The high affinities of the new compounds could be attributed to their bulkier size compared to CAN508 which allow them to occupy larger volume of the active site in CDK-9.
Compound 19a also showed the highest affinity towards CDK-9. Investigation of the binding interactions of compound 18b into CDK-9 revealed one conventional hydrogen bond with Ile25, while compound 19a showed three hydrogen conventional bonds Thr29, Lys48, and Ala153, Figure 11. The results of the docking study into CDKs-6/9 indicated that the three compounds (18b, 19a, and 20a) could have potential inhibitory activities against the two kinases.
In attempt to compare the binding affinities of the three compounds against the CDKs, fold selectivity was calculated, Table 6. The three compounds displayed higher affinities towards CDK-6/9 than CDK-2 with fold selectivity in the range of 2.06–3.58. These results suggest that the three compounds could have better inhibitory activities against CDK-6/9 than CDK-2.
3.3.2. In silico ADME and drug-likeness studies
One of the major problems in drug discovery and development is the poor pharmacokinetic profile of a drug candidate58,59. The success of a drug candidate to pass the clinical investigation depends mainly on its pharmacokinetic as well as pharmacodynamic profile. Accordingly, the in silico studies of the physicochemical properties related to the pharmacokinetics of drug candidates have attracted considerable attention in the last two decades60,61.
In the current study, the physicochemical properties related to the pharmacokinetic (PK) profiles of the new compounds 16–20a–c were evaluated using SwissADME (http://www.swissadme.ch/)53. The new compounds were also evaluated for their drug-likeness score using the online Molsoft tools (http://www.molsoft.com/mprop/). To predict the oral activity, the new compounds were also evaluated for any violation from Lipinski’s rule54. The calculated values of the new compounds were compared with those of the multi-CDKIs, flavopiridol, palbociclib, compounds 3 and 10. The results of this study are presented in Table 7.
Table 7.
Molecular properties related to PKs and DLS of the new compounds in comparison to compounds 3, 10, flavopiridol and palbociclib.
| Physicochemical properties |
Lipinski’s rule | %Abs a | BS | DLS b | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | MW | MV | TPSA | MlogP | RBs | HBA | HBD | ||||
| 16a | 393.48 | 431.15 | 73.39 | 1.97 | 11 | 3 | 3 | Yes | 83.68 | 0.55 | 0.24 |
| 16b | 407.51 | 452.09 | 73.39 | 2.18 | 11 | 3 | 3 | Yes | 83.68 | 0.55 | 0.35 |
| 16c | 427.93 | 448.35 | 73.39 | 2.45 | 11 | 3 | 3 | Yes | 83.68 | 0.55 | 0.84 |
| 17a | 415.44 | 427.60 | 79.50 | 1.71 | 8 | 4 | 3 | Yes | 81.57 | 0.55 | 0.45 |
| 17b | 429.47 | 448.54 | 79.50 | 1.92 | 8 | 4 | 3 | Yes | 81.57 | 0.55 | 0.48 |
| 17c | 449.89 | 444.79 | 79.50 | 2.19 | 8 | 4 | 3 | Yes | 81.57 | 0.55 | 1.01 |
| 18a | 403.41 | 401.67 | 71.96 | 2.39 | 7 | 4 | 3 | Yes | 84.17 | 0.55 | 0.65 |
| 18b | 417.44 | 422.61 | 71.96 | 2.61 | 7 | 4 | 3 | Yes | 84.17 | 0.55 | 0.71 |
| 18c | 437.85 | 418.86 | 71.96 | 2.87 | 7 | 4 | 3 | Yes | 84.17 | 0.55 | 0.86 |
| 19a | 435.48 | 446.29 | 70.99 | 2.68 | 7 | 3 | 3 | Yes | 84.51 | 0.55 | 0.14 |
| 19b | 449.50 | 467.23 | 70.99 | 2.88 | 7 | 3 | 3 | Yes | 84.51 | 0.55 | 0.30 |
| 19c | 469.92 | 463.49 | 70.99 | 3.15 | 7 | 3 | 3 | Yes | 84.51 | 0.55 | 0.71 |
| 20a | 451.54 | 460.28 | 57.51 | 2.67 | 7 | 2 | 3 | Yes | 89.16 | 0.55 | 0.11 |
| 20b | 465.57 | 481.22 | 57.51 | 2.87 | 7 | 2 | 3 | Yes | 89.16 | 0.55 | 0.28 |
| 20c | 485.99 | 477.48 | 57.51 | 3.14 | 7 | 2 | 3 | Yes | 89.16 | 0.55 | 0.69 |
| 10 | 447.92 | 469.04 | 58.48 | 2.92 | 7 | 2 | 3 | Yes | 88.82 | 0.55 | 0.71 |
| 3 | 360.29 | 306.76 | 70.18 | 2.64 | 6 | 6 | 3 | Yes | 84.79 | 0.55 | 0.98 |
| Flavopiridol | 401.8 | 382.75 | 71.93 | 1.75 | 2 | 5 | 3 | Yes | 84.18 | 0.55 | 1.48 |
| Palbociclib | 447.53 | 479.27 | 84.30 | 1.13 | 5 | 6 | 2 | Yes | 79.92 | 0.55 | 0.62 |
MW: molecular weight; MV: molecular volume (A3); TPSA: topological polar surface area (A2); MlogP: logP (Moriguchi etal.); RBs: number of rotatable bonds; HBA: number of hydrogen bond acceptors; HBD: number of hydrogen bond donors.
a%Abs (% absorbed orally) = 109−(0.345 × TPSA; BS: bioavailability score.
bDLS: drug-likeness score; Lipinski’s rule, yes mean no violation; MV, TPSA, and DLS were calculated using Molsoft (http://www.molsoft.com/mprop/); other parameters calculated using SwissADME (http://www.swissadme.ch/).
The molecular weights of the new compounds were in the range 393.48–485.99 Da which was less than 500. Moreover, the calculated molecular volumes of the new compounds were in the range of 401.67 − 481.22 A3, which was comparable to multi-CDKIs, flavopiridol and palbociclib. In addition, the new compounds showed topological polar surface areas (TPSA) in the range of 57.51–79.50 A2 compared to 71.93 and 84.30 A2 for flavopiridol and palbociclib, respectively, Table 7.
The Clog P (MlogP) values of the new compounds were in the range of 1.71–3.15, which was higher than those of flavopiridol and palbociclib but was comparable with that compound 3. Compounds 19c and 20c were the most lipophilic with ClogP values of 3.15 and 3.14, respectively. The chemical structure of the new compounds have 2–4 hydrogen bond acceptors (HBA <10) and 3 hydrogen bond donors (HBD < 5). These results indicated that all the new compounds conform to Lipinski’s rule. In addition, the new compounds showed drug-likeness score (DLS) in the range of 0.11–1.01, compared to 0.62 for palbociclib, Supplementary data (Table S2). Among the new compounds, compound 17c displayed the highest DLS.
The expected GIT absorption percent (%Abs) of the new compounds was also calculated according to the previous report55. The new compounds exhibited %Abs in the range of 81.57–89.16% compared to 84.79–84.18% for flavopiridol.
The study also revealed that compounds 17a,b, 18a,b, 19a, and 20a,b are expected to be substrates for P-glycoprotein and predicted to be effluated from the CNS by P-glycoprotein, Supplementary data (Table S3).
4. Conclusions
In the current study, a library of 1302 pyrrolizine derivatives was designed based on the pyrrolizine-5-carboxamide scaffold 10. The 3D pharmacophore model of the multi-CDKI 3 was used in the virtual screening of this library. The top-scoring hits were synthesised and evaluated for their cytotoxic activities against three cancer (MCF-7, A2780, and HT29) cell lines. The results revealed cytotoxic activities against the three cancer cell lines with IC50 values in the range of 0.16–34.13 μM. Among the new derivatives, compound 18b was the most active against MCF-7 cells (IC50 = 0.19 μM). In addition, compounds 19a and 20a were the most potent in inhibiting the growth of and HT29 and A2780 cell lines, respectively. The results of the MTT assay of the new compounds against normal MRC-5 cells also revealed IC50 values in the range of 0.10–11.86 µM. Compounds 18b, 19a, and 20a increased the apoptotic events and arrested MCF-7 cells at the G1 phase. Mechanistic study of compounds 18b, 19a, and 20a revealed potent in vitro inhibition in CDK-2 activity (IC50 = 25.53–115.3 nM). Among the three derivatives, compound 19a exhibited the highest inhibitory activity against CDK-2. In silico ADME study revealed that all new compounds conform to Lipinski’s rule. In addition, the molecular docking analyses indicated that compounds 18b, 19a, and 20a fit snugly into the active sites of CDK-2/6/9. Among the three derivatives, compound 19a also displayed the highest binding scores and exhibited several hydrogen bonding interactions with the three kinases. These preliminary results suggested that compound 19a could serve as a promising scaffold in the discovery and development of new potent anticancer agents.
Supplementary Material
Funding Statement
This work was funded by National Science, Technology and Innovation Plan (MAARIFAH), King Abdulaziz City for Science and Technology (KACST), Kingdom of Saudi Arabia, Grant no. 12-MED2304-10-R.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Ferguson FM, Gray NS.. Kinase inhibitors: the road ahead. Nat Rev Drug Discov 2018;17:353–77. [DOI] [PubMed] [Google Scholar]
- 2.Bhullar KS, Lagaron NO, McGowan EM, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018;17:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu P, Nielsen TE, Clausen MH.. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 2015;36:422–39. [DOI] [PubMed] [Google Scholar]
- 4.Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res 2020;152:104609. [DOI] [PubMed] [Google Scholar]
- 5.Jeong W, Doroshow JH, Kummar S.. United States Food and Drug Administration approved oral kinase inhibitors for the treatment of malignancies. Curr Probl Cancer 2013;37:110–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tadesse S, Caldon EC, Tilley W, Wang S.. Cyclin-dependent kinase 2 inhibitors in cancer therapy: an update. J Med Chem 2019;62:4233–51. [DOI] [PubMed] [Google Scholar]
- 7.Tsai LH, Lees E, Faha B, et al. The cdk2 kinase is required for the G1-to-S transition in mammalian cells. Oncogene 1993;8:1593–602. [PubMed] [Google Scholar]
- 8.Keyomarsi K, Pardee AB.. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A 1993;90:1112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Fang D, Chen H, et al. Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 2015;6:20801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto H, Monden T, Miyoshi H, et al. Cdk2/cdc2 expression in colon carcinogenesis and effects of cdk2/cdc2 inhibitor in colon cancer cells. Int J Oncol 1998;13:233–9. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Yang T, Xu G, et al. Cyclin-dependent kinase 2 promotes tumor proliferation and induces radio resistance in glioblastoma. Transl Oncol 2016;9:548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faber AC, Chiles TC.. Inhibition of cyclin-dependent kinase-2 induces apoptosis in human diffuse large B-cell lymphomas. Cell Cycle 2007;6:2982–9. [DOI] [PubMed] [Google Scholar]
- 13.Yin X, Yu J, Zhou Y, et al. Identification of CDK2 as a novel target in treatment of prostate cancer. Future Oncol 2018;14:709–18. [DOI] [PubMed] [Google Scholar]
- 14.Lin ZP, Zhu YL, Ratner ES.. Targeting cyclin-dependent kinases for treatment of gynecologic cancers. Front Oncol 2018;8:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamdouchi C, Keyser H, Collins E, et al. The discovery of a new structural class of cyclin-dependent kinase inhibitors, aminoimidazo[1,2-a]pyridines. Mol Cancer Ther 2004;3:1–9. [PubMed] [Google Scholar]
- 16.Wyatt PG, Woodhead AJ, Berdini V, et al. Identification of N-(4-piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a novel cyclin dependent kinase inhibitor using fragment-based X-ray crystallography and structure based drug design. J Med Chem 2008;51:4986–99. [DOI] [PubMed] [Google Scholar]
- 17.Seftel MD, Kuruvilla J, Kouroukis T, et al. The CDK inhibitor AT7519M in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and mantle cell lymphoma. A Phase II study of the Canadian Cancer Trials Group. Leuk Lymphoma 2017;58:1358–65. [DOI] [PubMed] [Google Scholar]
- 18.Cheng C, Yun F, Ullah S, Yuan Q.. Discovery of novel cyclin-dependent kinase (CDK) and histone deacetylase (HDAC) dual inhibitors with potent in vitro and in vivo anticancer activity. Eur J Med Chem 2020;189:112073. [DOI] [PubMed] [Google Scholar]
- 19.Honma T, Hayashi K, Aoyama T, et al. Structure-based generation of a new class of potent Cdk4 inhibitors: new de novo design strategy and library design. J Med Chem 2001;44:4615–27. [DOI] [PubMed] [Google Scholar]
- 20.Payton M, Chung G, Yakowec P, et al. Discovery and evaluation of dual CDK1 and CDK2 inhibitors. Cancer Res 2006;66:4299–308. [DOI] [PubMed] [Google Scholar]
- 21.Wróbel TM, Kiełbus M, Kaczor AA, et al. Discovery of nitroaryl urea derivatives with antiproliferative properties. J Enzyme Inhib Med Chem 2016;31:608–18. [DOI] [PubMed] [Google Scholar]
- 22.Alexander LT, Möbitz H, Drueckes P, et al. Type II inhibitors targeting CDK2. ACS Chem Biol 2015;10:2116–25. [DOI] [PubMed] [Google Scholar]
- 23.Gouda AM, Abdelazeem AH, Arafa e-SA, Abdellatif KR.. Design, synthesis and pharmacological evaluation of novel pyrrolizine derivatives as potential anticancer agents. Bioorg Chem 2014;53:1–7. [DOI] [PubMed] [Google Scholar]
- 24.Gouda AM, Abdelazeem AH, Omar HA, et al. Pyrrolizines: design, synthesis, anticancer evaluation and investigation of the potential mechanism of action. Bioorg Med Chem 2017;25:5637–51. [DOI] [PubMed] [Google Scholar]
- 25.Shawky AM, Abourehab MAS, Abdalla AN, Gouda AM.. Optimization of pyrrolizine-based Schiff bases with 4-thiazolidinone motif: design, synthesis and investigation of cytotoxicity and anti-inflammatory potency. Eur J Med Chem 2020;185:111780. [DOI] [PubMed] [Google Scholar]
- 26.Cheng W, Yang Z, Wang S, et al. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur J Med Chem 2019;164:615–39. [DOI] [PubMed] [Google Scholar]
- 27.Tutone M, Almerico AM.. Recent advances on CDK inhibitors: an insight by means of in silico methods. Eur J Med Chem 2017;142:300–15. [DOI] [PubMed] [Google Scholar]
- 28.Ece A, Sevin F.. The discovery of potential cyclin A/CDK2 inhibitors: a combination of 3D QSAR pharmacophore modeling, virtual screening, and molecular docking studies. Med Chem Res 2013;22:5832–43. [Google Scholar]
- 29.Mohammad T, Batra S, Dahiya R, et al. Identification of high-affinity inhibitors of cyclin-dependent kinase 2 towards anticancer therapy. Molecules 2019;24:4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakanishi I, Murata K, Nagata N, et al. Identification of protein kinase CK2 inhibitors using solvent dipole ordering virtual screening. Eur J Med Chem 2015;96:396–404. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Q, Muegge I.. Scaffold hopping through virtual screening using 2D and 3D similarity descriptors: ranking, voting, and consensus scoring. J Med Chem 2006;49:1536–48. [DOI] [PubMed] [Google Scholar]
- 32.Liang J-W, Wang M-Y, Wang S, et al. Identification of novel CDK2 inhibitors by a multistage virtual screening method based on SVM, pharmacophore and docking model. J Enzyme Inhib Med Chem 2020;35:235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulsen A, William A, Blanchard S, et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J Mol Model 2013;19:119–30. [DOI] [PubMed] [Google Scholar]
- 34.Horvath D. Pharmacophore-based virtual screening. Methods Mol Biol 2011;672:261–98. [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Li S, Liu F, et al. Discovery of novel nonpeptide allosteric inhibitors interrupting the interaction of CDK2/cyclin A3 by virtual screening and bioassays. Bioorg Med Chem Lett 2015;25:4069–73. [DOI] [PubMed] [Google Scholar]
- 36.Sunseri J, Koes DR.. Pharmit: interactive exploration of chemical space. Nucleic Acids Res 2016;44:W442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouda AM, Ali HI, Almalki WH, Azim MA, et al. Design, synthesis, and biological evaluation of some novel pyrrolizine derivatives as COX inhibitors with anti-inflammatory/analgesic activities and low ulcerogenic liability. Molecules 2016;21:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Attalah KM, Abdalla AN, Aslam A, Ahmed M, et al. Ethyl benzoate bearing pyrrolizine/indolizine moieties: design, synthesis and biological evaluation of anti-inflammatory and cytotoxic activities. Bioorg Chem 2020;94:103371. [DOI] [PubMed] [Google Scholar]
- 39.Gouda AM, Abdelazeem AH, Abdalla AN, Ahmed M.. Pyrrolizine-5-carboxamides: exploring the impact of various substituents on anti-inflammatory and anticancer activities. Acta Pharm 2018;68:251–73. [DOI] [PubMed] [Google Scholar]
- 40.Belal A, Gouda AM, Ahmed AS, Abdel Gawad NM.. Synthesis of novel indolizine, diazepinoindolizine and pyrimidoindolizine derivatives as potent and selective anticancer agents. Res Chem Intermed 2015;41:9687–701. [Google Scholar]
- 41.Elsaady MT, Gouda AM, Edrees FH, Gawad NMA.. Synthesis and biological evaluation of some novel Schiff base derivatives as potential anticancer agents. J Chem Pharm Res 2016;8:273–82. [Google Scholar]
- 42.Malki WH, Gouda AM, Ali HEA, et al. Structural-based design, synthesis, and antitumor activity of novel alloxazine analogues with potential selective kinase inhibition. Eur J Med Chem 2018;152:31–52. [DOI] [PubMed] [Google Scholar]
- 43.Alkahtani HM, Alanazi MM, Aleanizy FS, et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: molecular docking studies. Saudi Pharm J 2019;27:682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abdalla AN, Shaheen U, Abdallah QMA, et al. Proapoptotic activity of Achillea membranacea essential oil and its major constituent 1,8-cineole against A2780 ovarian cancer cells. Molecules 2020;25:1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Chen Y, Cheng X, et al. Design, synthesis and biological evaluation of pyrimidine derivatives as novel CDK2 inhibitors that induce apoptosis and cell cycle arrest in breast cancer cells. Bioorg Med Chem 2018;26:3491–501. [DOI] [PubMed] [Google Scholar]
- 46.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gouda AM, Almalki FA.. Carprofen: a theoretical mechanistic study to investigate the impact of hydrophobic interactions of alkyl groups on modulation of COX-1/2 binding selectivity. SN Appl Sci 2019;1:332. [Google Scholar]
- 48.Almalki FA, Gouda AM, Bin Ali MH, Almehmadi OM.. Profens: a comparative molecular docking study into cyclooxygenase-1/2. Drug Invent Today 2019;11:480–7. [Google Scholar]
- 49.Attallah KM, Gouda AM, Ibrahim IT, Abouzeid L.. Design, synthesis, 99mTc labeling, and biological evaluation of a novel pyrrolizine derivative as potential anti-inflammatory agent. Radiochemistry 2017;59:630–8. [Google Scholar]
- 50.Dassault Systems BIOVIA, Discovery studio visualizer, v16.1.0.15350. San Diego (CA): Dassault Systems; 2016. [Google Scholar]
- 51.Lu H, Schulze-Gahmen U.. Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J Med Chem 2006;49:3826–31. [DOI] [PubMed] [Google Scholar]
- 52.Baumli S, Hole AJ, Noble ME, Endicott JA.. The CDK9 C-helix exhibits conformational plasticity that may explain the selectivity of CAN508. ACS Chem Biol 2012;7:811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daina A, Michielin O, Zoete V.. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3–25. [DOI] [PubMed] [Google Scholar]
- 55.Zhao YH, Abraham MH, Le J, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res 2002;19:1446–57. [DOI] [PubMed] [Google Scholar]
- 56.Atkins JH, Gershell LJ.. Selective anticancer drugs. Nat Rev Drug Discov 2002;1:491–2. [DOI] [PubMed] [Google Scholar]
- 57.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES.. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015;14:130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Urbán L.. The impact of early ADME profiling on drug discovery and development strategy. DDW Drug Discov World 2004;5:73–86. [Google Scholar]
- 59.Li AP. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today 2001;6:357–66. [DOI] [PubMed] [Google Scholar]
- 60.Gleeson MP, Hersey A, Hannongbua S.. In-silico ADME models: a general assessment of their utility in drug discovery applications. Curr Top Med Chem 2011;11:358–81. [DOI] [PubMed] [Google Scholar]
- 61.Yan G, Wang X, Chen Z, et al. In-silico ADME studies for new drug discovery: from chemical compounds to Chinese herbal medicines. Curr Drug Metab 2017;18:535–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.