

ABSTRACT
Clinical and experimental evidences indicate that epigenetic modifications induced by the prenatal environment are related to metabolic and reproductive derangements in polycystic ovary syndrome (PCOS). Alterations in the leptin and adiponectin systems, androgen signalling and antimüllerian hormone (AMH) levels have been observed in PCOS women and in their offspring. Using a targeted Next-Generation Sequencing (NGS), we studied DNA methylation in promoter regions of the leptin (LEP), leptin receptor (LEPR), adiponectin (ADIPOQ), adiponectin receptor 1 and 2 (ADIPOR1 and ADIPOR2), AMH and androgen receptor (AR) genes in 24 sons and daughters of women with PCOS (12 treated with metformin during pregnancy) and 24 children born to non-PCOS women during early infancy (2–3 months of age). Genomic DNA was extracted from whole blood, bisulphite converted and sequenced by NGS. Girls showed differences between groups in 1 CpG site of LEPR, 2 of LEP, 1 of ADIPOR2 and 2 of AR. Boys showed differences in 5 CpG sites of LEP, 3 of AMH and 9 of AR. Maternal metformin treatment prevented some of these changes in LEP, ADIPOR2 and partially in AR in girls, and in LEP and AMH in boys. Maternal BMI at early pregnancy was inversely correlated with the methylation levels of the ChrX-67544981 site in the whole group of girls (r = −0.530, p = 0.008) and with the global Z-score in all boys (r = −0.539, p = 0.007). These data indicate that the intrauterine PCOS environment predisposes the offspring to acquire certain sex-dependent DNA methylation patterns in the promoter regions of metabolic and reproductive genes.
KEYWORDS: Epigenetic, polycystic ovary syndrome (PCOS), androgen receptor, leptin, offspring
Introduction
Polycystic ovary syndrome (PCOS) is a highly prevalent and heterogenic disorder in women of reproductive age, characterized by hyperandrogenism and chronic anovulation, and closely associated with insulin resistance and obesity [1]. We and other authors have demonstrated that androgen excess and metabolic disturbances persist in PCOS women during pregnancy, predisposing to complications such as gestational diabetes (GDM) and pregnancy-induced hypertension. Moreover, this adverse maternal environment has also been associated with deleterious consequences for the foetus such as alterations in birth weight and development of metabolic and cardiovascular diseases during adult life [2,3]. Some of these disturbances are sex specific indicating that the maternal environment affects the male and female foetuses differently.
In previous studies we have observed higher leptin concentrations in cord blood of PCOS newborns compared to controls [4], a marker that has been associated with disturbances in BMI and insulin levels in PCOS women [5]. Daughters of PCOS women have higher antimüllerian hormone (AMH) concentrations since early infancy to puberty, evidencing an increased follicular mass [6,7] and an increased ovarian volume and hyperandrogenism during early and late puberty, respectively [8]. Regarding metabolic alterations, hypoadiponectinemia is present before the onset of puberty and hyperinsulinemia, reflecting insulin resistance, since prepuberty and later on [8,9]. On the other hand, sons of PCOS mothers show higher AMH concentrations from early infancy to prepuberty, suggesting an increase in the Sertoli cell number or function [10]. Moreover, these boys usually exhibit higher body weight since early infancy and insulin resistance when they get older [11]. Therefore, it seems that altered levels of leptin, adiponectin and AMH along with an abnormal androgen action are common features that appear since early infancy in both daughters and sons born to PCOS mothers. It is known that the androgen receptor modulates androgen action and several studies have supported that a higher activity in the AR is a determining factor in PCOS development [12–14].
During the last years, metformin has been used to treat the maternal complications associated with GDM and PCOS. Moreover, we have previously demonstrated that metformin can improve the altered endocrine-metabolic environment of PCOS mothers reducing AMH levels in their daughters, which might be associated with a decrease in their follicular mass [15]. Nevertheless, its long-term effects in the offspring could be debatable and need more research to be established.
It is now known that multiple mechanisms contribute to foetal programming in PCOS including genetic, epigenetic and environmental factors [16–19]. Epigenetics is a molecular phenomenon that regulates gene expression without changes in the DNA sequence modulating tissue-specific gene expression, genomic imprinting and X-chromosome inactivation [20]. Epigenetic modifications consist mainly of DNA methylation, histone modifications, chromatin reconstruction, and expression of non-coding RNA [21]. DNA methylation is the most stable and best understood epigenetic mechanism [22,23], which consists in the addition of methyl (-CH3) groups to the 5-carbon of cytosine mainly in CpG-dinucleotides (CpGs) regulating transcriptional expression of specific genes [24]. It has also been proposed that exposure to androgen excess during prenatal life may induce epigenetic changes inducing long-term modifications in the offspring [25]. Moreover, in PCOS women, specific genes have been demonstrated to be associated with aberrant DNA methylation in tissues and pathways associated to PCOS dysfunction [26–31]. In this context, it has been proposed that differences in the androgen receptor DNA methylation pattern could be associated to hyperandrogenism [32]. Recent studies suggest that adipokines are involved in the foetal metabolic health programming through epigenetic adaptations [33]. Finally, recent results showed a decreased methylation level of the AMH gene associated with an increase in AMH follicular levels in PCOS women [34]. Then, alterations observed in children born to PCOS women could be attributed to these modifications. Until now, only one study has approached this issue showing a differential DNA methylation pattern in umbilical cord blood from children born to PCOS women [35].
In the present study we hypothesized that, depending on their sex, children born to PCOS women could have a particular methylation pattern in the promoter region of key reproductive and metabolic genes, which may be modulated by the intrauterine environment. Therefore, our aim was to evaluate, in genomic DNA from whole blood, the methylation pattern of promoter regions of reproductive and metabolic genes in the offspring of PCOS women during early infancy (2–3 months of age) and compare it with controls and with children form PCOS women treated with metformin during pregnancy. Based on previous observations, we focused our analysis on the promoter regions of leptin (LEP), leptin receptor (LEPR), adiponectin (ADIPOQ), adiponectin receptor 1 and 2 (ADIPOR1, ADIPOR2), antimüllerian hormone (AMH), and the androgen receptor (AR) genes.
Subjects and methods
Subjects
Twenty-four Chilean infants born to PCOS women (PCOS) and 24 born to non-PCOS women (control) were included in the study. The PCOS daughters and sons groups included 12 female and 12 male infants (2–3 months old), born to PCOS mothers. The control daughters and sons groups included 12 female and 12 male infants born to mothers with regular menses and without hyperandrogenism. None of the subjects included in the study were genetically related with each other. In the PCOS group, 12 women were treated with metformin during the whole period of pregnancy (PCOS+M). PCOS and control infants were born from spontaneous singleton pregnancies. All infants were studied during early infancy (2–3 months of age). Most of these infants were included in previous studies carried out by our group [10,11,15]. Inclusion criteria for PCOS mothers and control mothers were similar to those previously reported [8,15].
The protocol was approved by the Institutional Review Board of the Faculty of Medicine University of Chile (Approval of Research Project Nº032-2015). All parents signed an informed consent before entering the study.
Pregnant women study protocol
PCOS mothers were recruited from patients attending the Unit of Endocrinology and Reproductive Medicine at the University of Chile who had desired fertility. Diagnosis of PCOS was made according to the diagnostic criteria for PCOS of the National Institutes of Health (NIH) consensus [36]. As part of their initial evaluation, all the patients underwent a lifestyle assessment and were placed on a diet and exercise treatment programme as previously described [15]. In addition, most of them received 1,500–2,000 mg metformin in standard formulation based on their weight, medication tolerance, and insulin levels. PCOS patients were instructed to stop metformin treatment upon a positive pregnancy test and those that required it continued with the medication during the whole pregnancy.
No medications to induce ovulation, such as clomiphene citrate or exogenous gonadotropins, were used. We excluded patients with hyperprolactinaemia, androgen-secreting neoplasm, Cushing syndrome, late-onset 21-hydroxylase deficiency, or thyroid disease.
As a control group, we selected pregnant women of similar age and socioeconomic level as the PCOS patients. These pregnant women had a history of regular 28- to 32-day menstrual cycles, absence of hirsutism and other clinical manifestations of hyperandrogenism, infertility, pregnancy complications, galactorrhoea, and thyroid dysfunction. All were healthy and were not receiving any drug therapy. These women were recruited from the antenatal care unit of San Juan de Dios hospital (Santiago, Chile) from the 12th week of gestation.
In all pregnant women, duration of gestation, initial and final body mass index (BMI), and weight gain during pregnancy were recorded. During gestational weeks 22–28, all women were classified as having gestational diabetes mellitus or pregnancy-induced hypertension in accordance with the World Health Organization criteria (fasting glucose values >105 mg/dL; 2-hour glucose postload >140 mg/dL), pregnancy-induced hypertension (blood pressure ≥140/90 mm Hg without proteinuria at a gestational age > 20 weeks on two or more occasions) or preeclampsia (blood pressure ≥ 140/90 mm Hg with proteinuria > 0.3 g/24 h after 20 weeks’ gestation).
Offspring study protocol
All infants were examined twice, once during the first 3 days of life and again at 2–3 months of age. On both occasions the physical examination included weight and length, following the scheme described in previous studies in which most of these children participated [7,10,11,15]. Gestational age and the type of feeding was registered (exclusive breastfeeding, formula or mixed). In all infants, a blood sample was taken and stored at −80°C for DNA analysis.
Methylation analysis
DNA isolation
Genomic DNA was extracted from peripheral blood leukocytes in all infants using the E.Z.N.A.® Blood DNA Midi Kit (Omega Bio-tek, Inc.Qiagen, Hilden, Germany) following instructions provided by the manufacturer. The concentration and purity of DNA was determined using a Nanodrop spectrophotometer (Tecan Infinite 200 PRO).
Assay design, sample preparation, and multiplex targeted amplification
We selected the promoter regions of seven genes that, according to our previous observations and the literature, could be involved in the metabolic and endocrine changes found in the offspring of PCOS women, including leptin (LEP), leptin receptor (LEPR), adiponectin (ADIPOQ), adiponectin receptor 1 (ADIPOR1), adiponectin receptor 2 (ADIPOR2), anti-Müllerian hormone (AMH), and androgen receptor (AR) (Supplementary Table 1). Genomic DNA was bisulphite modified and then sequenced by the Zymo Research Corporation (Irvine, CA, USA). Assays were designed targeting CpG sites in the specified regions of interest (ROI) using primers created with Rosefinch, Zymo Research’s proprietary sodium bisulphite converted DNA-specific primer design tool (Supplementary Table 2). DNA samples were bisulphite converted using the EZ DNA Methylation-Lightning TM Kit (D5030, Zymo Research) according to the manufacturer’s instructions. The following processes included a targeted sequencing for DNA methylation analysis at multiple loci using a multiplex PCR strategy in combination with Next-Generation Sequencing (NGS) (MiSeq, Illumina, Inc., San Diego, CA). Multiplex amplification of all samples using ROI specific primer pairs and the Fluidigm Access ArrayTM System was performed according to the manufacturer’s instructions. The resulting amplicons were pooled for harvesting and subsequent barcoding according to the Fluidigm instrument’s guidelines. After barcoding, samples were purified using ZR-96 DNA Clean & Concentrator™ (D4023, Zymo Research), and then prepared for massively parallel sequencing using a MiSeq V2 300bp Reagent Kit and paired-end sequencing protocol according to the manufacturer’s guidelines.
Targeted sequence alignments and data analysis
Sequence reads were identified using standard Illumina base-calling software and then analysed using a Zymo Research proprietary analysis pipeline, which is written in Python. Low quality nucleotides and adapter sequences were trimmed off during analysis QC. Sequence reads were aligned back to the reference genome using Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/), an aligner optimized for bisulphite sequence data and methylation calling [37]. Paired-end alignment was used as default thus requiring both read 1 and read 2 to be aligned within a certain distance; otherwise both read 1 and read 2 were discarded. Index files were constructed using the bismark_genome_preparation command and the entire reference genome (GRCh38/hg38). The non-directional parameter was applied while running Bismark. All other parameters were set to default. Nucleotides in primers were trimmed off from amplicons during methylation calling.
Data analysis
The methylation level (β-value) of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T. Differential methylation was assessed per CpG site including samples with at least 10 reads. Moreover, Z-score value to every CpG site of each gene promoter region was calculated according the method proposed by Hertzberg [38]. For the calculation of the z-score of each promoter region, the sum of each individual z-score value was considered. Finally, we calculated a global z-score value, with the sum of each site that was statistically different between the PCOS and control group.
Transcription factors analysis
In order to identify potential transcription factor binding sites in a promoter sequence, computational identification was performed using TFSITESCAN tools and database (http://www.ifti.org/cgi-bin/ifti/Tfsitescan.pl). Only transcription factors binding on or closely near the CpGs were further studied.
Statistical evaluation
Data are expressed as median and range for anthropometric and biochemical variables, and as mean and standard deviation for DNA methylation. Normal distribution was assessed by the Kolmogorov-Smirnov test. The β-value and z-scores comparisons were performed according to the sex of the infants. Differences between groups were calculated through one-way ANOVA followed by Bonferroni post hoc test when data were normally distributed or Kruskal Wallis followed by Dunn test for skewed data. Categorical data were analysed using χ2 or Fisher’s exact test. Spearman correlations were used to evaluate the relationship between the variables of interest. Statistical analysis was performed using SPSS 23.0 package. A p-value of less than 0.05 was considered to be statistically significant.
Results
Clinical maternal data
Clinical characteristics of pregnant control and PCOS women (treated and non-treated with metformin during pregnancy) are shown in Table 1. PCOS women treated with metformin were older than control women (P= 0.010). Moreover, pre-pregnancy body weight, BMI and the prevalence of overweight/obesity were significantly higher in those PCOS women that required metformin during pregnancy compared to control women (P= 0.002, P < 0.001 and P = 0.004, respectively). At the beginning of pregnancy, both PCOS women treated and non-treated with metformin showed higher BMI compared to control women (P= 0.002 and P< 0.002, respectively). At the third trimester, BMI was higher in PCOS women compared to control women (P= 0.029). In turn, gestational weight gain (GWG) was lower in PCOS treated with metformin compared to controls and non-treated PCOS (P= 0.001 and P= 0.035, respectively). No differences were observed in the prevalence of pregnancy-induced hypertension but gestational diabetes mellitus (GDM) was more prevalent in both groups of PCOS women compared to controls. The clinical characteristics of PCOS women at diagnosis are shown in supplementary table 3.
Table 1.
Clinical and biochemical characteristics of control and PCOS pregnant women with (PCOS+M) and without metformin (PCOS)
| Control (n = 24) |
PCOS (n = 12) |
PCOS+M (n = 12) |
P-values | |
|---|---|---|---|---|
| Pre-pregnancy | ||||
| Age (years) | 23.5 (20.2–30.7) | 29.5 (26.0–31.0) | 30.5 (30.0–35.2) b | 0.013 |
| Weight (kg) | 59.5 (52.8–66.0) | 69.0 (56.0–80.0) | 72.0 (68.0–76.8)b | 0.002 |
| BMI (Kg/m2) | 22.9 (21.1–26.0) | 26.1 (23.7–29.1) | 29.3 (27.1–30.9) b | <0.001 |
| Overweight/obesity (%) | 29.2 | 58.3 | 83.3b | 0.007 |
| At beginning | ||||
| Weight (kg) | 59.0 (52.5–66.0) | 69.0 (57.0–80.0) | 70.8 (68.0–77.0)b | 0.002 |
| BMI (Kg/m2) | 23.7 (21.1–25.7) | 27.6 (24.4–29.9)a | 28.8 (27.1–30.9) b | <0.001 |
| At third trimester | ||||
| Weight (Kg) | 71.0 (65.8–77.8) | 78.0 (69.0–90.0)a | 78.8 (70.9–81.5) | 0.096 |
| BMI | 29.1 (26.7 – 31.3) | 30.2 (28.5 – 36.1)a | 31.9 (28.2 – 32.4) | 0.026 |
| SBP (mm Hg) | 120 (110–120) | 110 (105–110) | 120 (110–120) | 0.087 |
| DBP (mm Hg) | 70 (60–77) | 62 (60–70) | 70 (70 – 70) | 0.448 |
| GWG (Kg) | 12.9 (8.2–16.5) | 11.0 (6.5–15.8) | 5.0 (2.8–7.9)b, c | 0.001 |
| PIH (%) (n) | 0 | 8.3 (1/12) | 0 (0/12) | 0.216 |
| GDM (%) (n) | 0 | 16.7 (2/12)a | 66.7 (8/12) b,c | <0.001 |
BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; GWG: gestational weight gain; PIH: pregnancy induced-hypertension; GDM: gestational diabetes mellitus. Data are expressed as median and interquartile range for continuous variables and percentage for categorical variables. For continuous variables differences were calculated by one way ANOVA test followed by Bonferroni test or Kruskal Wallis followed by Dunn tests according to the normal distribution of data. Categorical variables were analysed by chi-square test. aP < 0.05 between control and PCOS; bP < 0.05 between control and PCOS+M; cP < 0.05 between PCOS and PCOS+M.
Infant clinical data
Clinical and anthropometric characteristics of infants born to control and PCOS women are shown in Table 2. At birth, daughters born to PCOS+M showed lower gestational age than controls (P = 0.004), whereas birth length tended to be lower in this group of girls compared to controls (P = 0.074). On the other hand, in sons, there were no differences between groups regarding clinical and anthropometric variables. The prevalence of small (SGA) and large (LGA) for gestational age was similar between groups in both daughters and sons.
Table 2.
Clinical characteristics of control and PCOS infants (daughters and sons) born to control and PCOS mothers treated (PCOS+M) and non-treated (PCOS) with metformin during pregnancy
| Daughters |
Sons |
|||||||
|---|---|---|---|---|---|---|---|---|
| Control (n = 12) |
PCOS (n = 6) |
PCOS+M (n = 6) |
P-values | Control (n = 12) |
PCOS (n = 6) |
PCOS+M (n = 6) |
P-values | |
| At birth | ||||||||
| Gestational age (wk) | 39.0 (39.0–40.0) | 39.0 (38.0–40.0) | 38.0 (36.0–38.0)b | 0.005 | 39.5 (38.2–40.0) | 39.5 (38.7–41.0) | 39.0 (37.7–39.5) | 0.474 |
| Weight (g) | 3545 (3257–3750) | 3275 (2690–3905) | 3285 (2895–3515) | 0.346 | 3575 (3402–4010) | 3588 (2965–3875) | 3417 (3015–3692) | 0.534 |
| Z-score weight | 0.76 (0.15–1.06) | 0.04 (−1.26–1.87) | 0.34 (−0.25–0.93) | 0.601 | 1.11 (0.55–1.93) | 1.03 (−0.73–1.58) | 0.57 (−0.25–1.34) | 0.631 |
| Length (cm) | 50.0 (49.0–51.0) | 47.2 (44.7–50.4) | 47.7 (45.9–49.2) | 0.046 | 50.0 (49.2–51.7) | 50.0 (49.2–51.0) | 49.5 (48.2–51.4) | 0.659 |
| Z-score length | 0.22 (−0.27–0.68) | −0.39 (−1.38–0.60) | −0.12 (−0.83–0.17) | 0.381 | 0.22 (0.06–0.85) | 0.30 -(0.25–0.54) | 0.13 -(0.70–0.88) | 0.775 |
| SGA (%) (n) | 0 (0/12) | 16.7 (1/6) | 0 (0/6) | 0.209 | 8.3 (1/12) | 16.7 (1/6) | 0 (0/6) | 0.579 |
| LGA (%) (n) | 16.7 (2/12) | 33.3 (2/6) | 0 (0/6) | 0.301 | 33.3 (4/12) | 33.3 (2/6) | 16.7 (1/6) | 0.739 |
| At study | ||||||||
| Age (years) | 0.17 (0.17–0.23) | 0.25 (0.17–0.25) | 0.25 (0.23–0.25)b | 0.006 | 0.17 (0.17–0.25) | 0.21 (0.17–0.27) | 0.25 (0.17–0.33) | 0.077 |
| Weight (g) | 6115 (5327–6650) | 6335 (5425–6605) | 5175 (4747–6337) | 0.336 | 5600 (5400–8160) | 6400 (6050–7540) | 6375 (5587–7012) | 0.040 |
| Z-score weight | 0.94 (0.18–1.48) | 0.73 (−0.04–1.28) | −1.02 (−1.52–0.42)b | 0.032 | −0.29 (−0.95–0.48) | 0.49 (−0.04–1.79) | 0.68 (−0.79–2.06) | 0.080 |
| Length (cm) | 58.0 (57.0–60.7) | 57.5 (55.5–59.0) | 57.5 (55.2–60.1) | 0.419 | 59.0 (58.0–60.9) | 60.0 (59.0–62.5) | 57.5 (57.0–61.0) | 0.267 |
| Z-score length | −0.13 (−0.59–1.45) | −0.73 (−1.56 – −0.08) | −1.30 (−2.17 – −0.20)b | 0.008 | −0.24 (−0.71–0.62) | 0.03 (−0.57–1.43) | −0.33 (−1.71–1.36) | 0.789 |
| Weight gain (g) | 2735 (1820–3212) | 2620 (2390–3170) | 2485 (1480–3327) | 0.886 | 2145 (1652–2732) | 2967 (2270–4147) | 3615 (2881–4510)b | 0.020 |
| EMB (%) (n) | 80 (8/10) | 83.3 (5/6) | 50.0 (3/6) | 0.338 | 77.8 (7/9) | 25.0 (1/4) | 66.7 (4/6) | 0.186 |
Results are expressed as median and interquartile range. SGA: small for gestational age; LGA: large for gestational age and EMB: exclusive maternal breastfeeding. Data are expressed as median and interquartile range for continuous variables and percentage for categorical variables. For continuous variables differences were calculated by one way ANOVA test followed by Bonferroni test or Kruskal Wallis followed by Dunn tests according to the normal distribution of data. Categorical variables were analysed by chi-square test. aP < 0.05 between control and PCOS; bP < 0.05 between control and PCOS+M; cP < 0.05 between PCOS and PCOS+M.
At the time of study, there were no differences in age between groups both in daughters and sons. Z-score of weight and length were lower in daughters of PCOS women treated with metformin during pregnancy compared to girls born to control women (P = 0.034 and P = 0.010). In turn, sons of PCOS+M tended to be heavier and gained more weight from birth to study time (P = 0.060 and P = 0.037, respectively). By the time of the study, the prevalence of exclusive maternal breastfeeding was similar between groups both in daughters and sons.
Methylation analysis in promoter region of candidate genes
In total, the methylation levels in 368 CpG sites distributed among the promoter regions of 7 genes (LEP, ADIPOQ, AMH, LEPR, ADIPOR1, ADIPOR2 and AR) were analysed.
Daughters showed differences in 1 CpG site located in the promoter region of LEPR, 2 in LEP, 1 in ADIPOR2 and 2 in AR (Figure 1(a–d)). In the Chr1-65419664 site of the LEPR promoter, the proportion of methylation was higher in infants born to PCOS and PCOS+M compared to those born to control women (P = 0.016 and P = 0.037, respectively) (Figure 1(a)). Moreover, the Chr7-128240906 and Chr7-128241078 sites in the promoter of LEP exhibited higher methylation levels in PCOS+M infants compared to PCOS (P = 0.007 and P = 0.033, respectively). A trend to higher methylation in the Chr7-128240906 site was observed in control infants compared to PCOS (P = 0.072) (Figure 1(b)). In the Chr12-1690290 site of the ADIPOR2 promoter, increased methylation levels were observed in infants born to PCOS mothers compared to controls and PCOS+M. (P = 0.022 and P = 0.019, respectively) (Figure 1(c)). Moreover, the ChrX-67543969 and ChrX-67544981 sites of the AR promoter were less methylated in PCOS compared to controls (P = 0.005 and P = 0.049, respectively) (Figure 1(d)).
Figure 1.
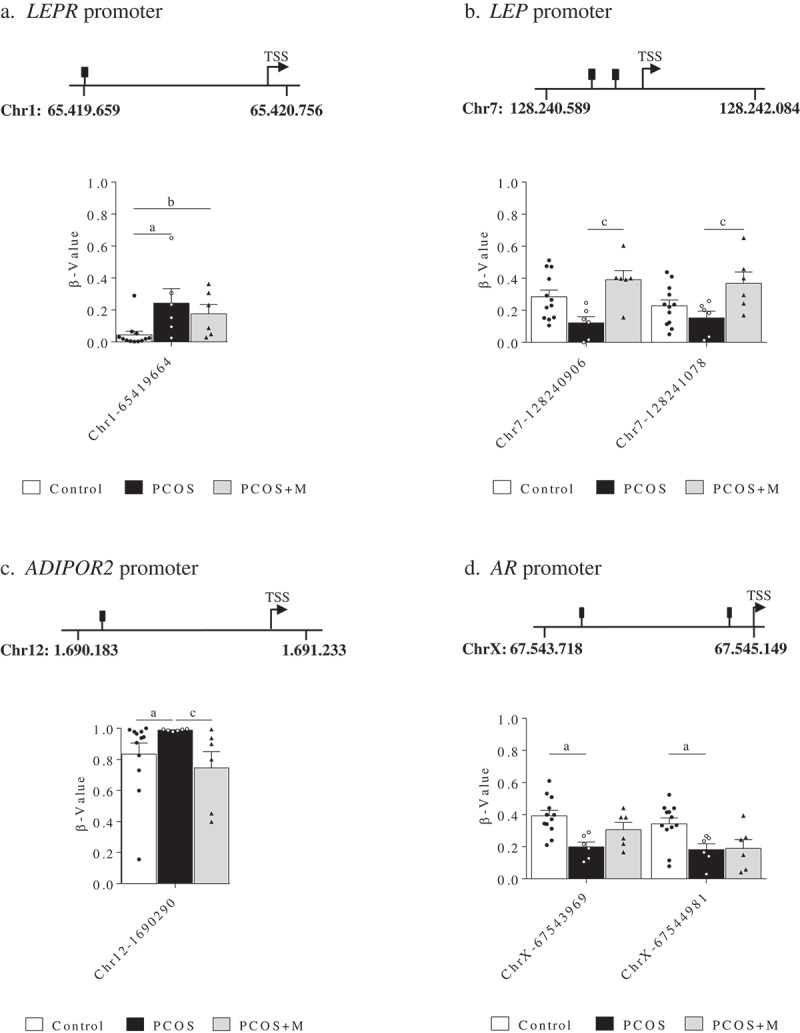
Methylation levels (β-value) in the promoter regions of the leptin receptor (LEPR) (a), leptin (LEP) (b), adiponectin receptor 2 (ADIPOR2) (c) and androgen receptor (AR) genes (d) in daughters of control (control, n = 12), PCOS women (PCOS, n = 6) and PCOS women treated with metformin during pregnancy (PCOS+M, n = 6). Data are shown as median ± SEM. Dots indicate the cases in each group. Differences were calculated by one-way ANOVA followed by Bonferroni test or Kruskal-Wallis test followed by Dunn test. aP < 0.05 between control and PCOS; bP < 0.05 between control and PCOS+M; cP < 0.05 between PCOS and PCOS+M
In sons, 5 CpG sites in the promoter region of LEP, 3 in AMH, and 9 in AR showed differences in methylation levels between groups (Figure 2(a–c)). In the promoter region of LEP (Figure 2(a)), the Chr7-128240873 site had lower methylation levels in PCOS compared to controls (P = 0.037), whereas in Chr7-128241155 methylation was lower in PCOS than in PCOS+M (P = 0.034). On the other hand, methylation in Chr7-128241074 was higher in PCOS compared to controls and PCOS+M (P = 0.008 and P = 0.012, respectively). In turn, methylation levels at the Chr7-128241028 site were lower (P = 0.028), whereas in Chr7-128241387 were higher in PCOS+M compared to controls (P = 0.028). In the promoter region of AMH (Figure 2(b)), methylation levels in Chr19-2248956 and Chr19-2249331 were lower in PCOS than in controls (P = 0.030 and P = 0.010, respectively) and the last site was also lower in PCOS compared to PCOS+M (P = 0.010). At Chr19-2249336, sons born to PCOS+M mothers had higher methylation levels than controls (P = 0.046). Finally, the AR promoter showed higher methylation levels in PCOS+M sons compared to controls and PCOS at the ChrX-67543762 site (P < 0.0001 and P < 0.0001, respectively) and lower methylation levels at ChrX-67544032 compared to controls and PCOS (P < 0.0001 and P = 0.048, respectively). In the other CpG sites, lower methylation levels were observed in sons born to PCOS+M compared to controls (ChrX-67543849 (P = 0.006), ChrX-67543889 (P = 0.043), ChrX-67543895 (P = 0.015), ChrX-67543899 (P = 0.017), ChrX-67544040 (P = 0.006), ChrX-67544221 (P = 0.041) and ChrX-67545002 (P = 0.022)) (Figure 2(c)).
Figure 2.
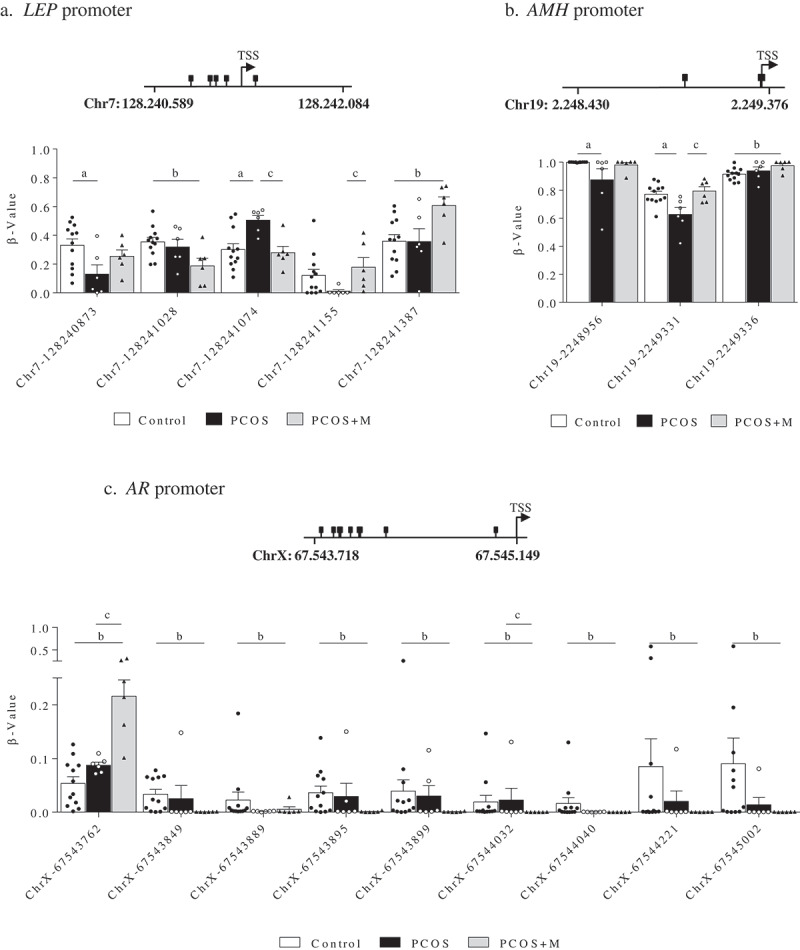
Methylation levels (β-value) in the promoter regions of the leptin (LEP) (a), antimüllerian hormone (AMH) (b) and androgen receptor (AR) genes (c) in sons of control (control, n = 12), PCOS women (PCOS, n = 6) and PCOS women treated with metformin during pregnancy (PCOS+M, n = 6). Data are shown as median ± SEM. Dots indicate the cases in each group. Differences were calculated by one-way ANOVA followed by Bonferroni test or Kruskal-Wallis test followed by Dunn test. aP < 0.05 between control and PCOS; bP < 0.05 between control and PCOS+M; cP < 0.05 between PCOS and PCOS+M
Methylation Z-score
The Z-scores for the promoter regions of LEP, LEPR, ADIPOQ, ADIPOR1, ADIPOR2 AMH and AR were comparable between the groups in both daughters and sons. The same was observed for the global Z-score in daughters (Figure 3(a)). However, the global Z-score was higher in control sons compared to PCOS and PCOS+M (P = 0.019 and P = 0.019, respectively) (Figure 3(b)).
Figure 3.
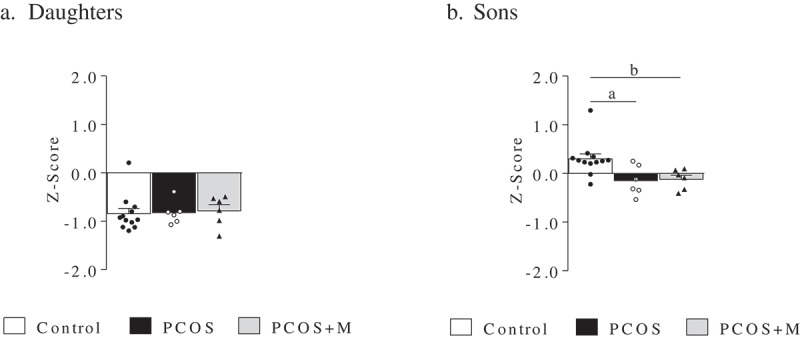
Z-Score of the promoter regions of LEP, LEPR, ADIPOQ, ADIPOR1, ADIPOR2, AMH and AR in daughters and sons of control (control, n = 12/12), PCOS women (PCOS, n = 6/6) and PCOS women treated with metformin during pregnancy (PCOS+M, n = 6/6). Data are shown as median ± SEM. Dots indicate the cases in each group. Differences were calculated by Kruskal-Wallis test followed by Dunn test. aP < 0.05 between control and PCOS; bP < 0.05 between control and PCOS+M
Correlation analysis
In daughters, the methylation levels of the chrX-67544981 site (AR) were significantly and inversely correlated with maternal BMI at early pregnancy (Figure 4(a)). On the other hand, in sons, the methylation levels in chr7-128241028 (LEP) and chrX-67543762 (AR) were significantly and positively correlated with the Z-Score of length and with the postnatal weight gain between birth and time of study, respectively (Figure 4(b–c)). Moreover, in sons, the global Z-Score of methylation was negatively associated with maternal BMI at early pregnancy and with postnatal weight gain (Figure 4(d–e)).
Figure 4.
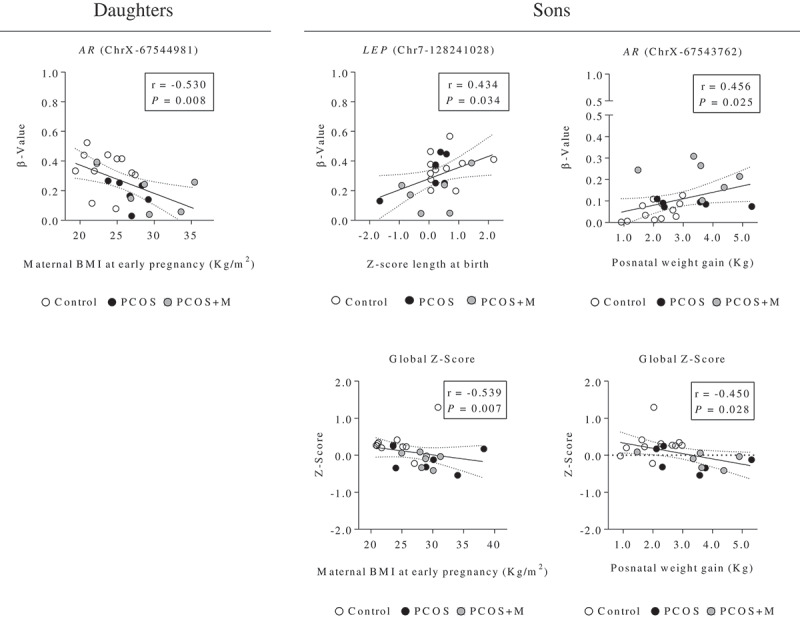
Correlation analysis between the methylation levels (β-value) of the CpG sites of the promoter regions of the AR (chromosome X, ChrX), and LEP (chromosome 7, Chr7) genes (and Z-Score of the analysed promoter regions), and the maternal characteristics and anthropometric parameters at birth and postnatal age in daughters and sons. Dots indicate the cases in each group. The associations between variables were calculated by Spearman’s rank correlation analysis. P < 0.05 was considered as significant level
Predicted transcription factor (TF) binding sites
In daughters, the differently methylated site, Chr7-128240906, in the LEP promoter has putative binding sites for the transcription factors SP1 (specificity protein 1) and GKLF (gut-enriched Krüppel:like factor). Moreover, the Chr1-65419662 site of the LEPR promoter binds SOX9 (SRY-Box 9 Protein), and the Chr12-1690290 site of the ADIPOR2 promoter binds E4 F1 (E4F transcription factor 1). In turn, the AR promoter has different methylation sites associated with EGR1 (early growth response 1), SET (Suppressor of variegation, Enhancer of Zeste, Trithorax) and MYND (myeloid:Nervy:DEAF1) in the ChrX-67543969 site, whereas the ChrX-67544580 site binds MED1 (mediator of RNA polymerase II transcription subunit 1).
In sons, CpG sites in the LEP promoter exhibit binding sites for AP2 (Activator protein 2) in Chr7-128241387 and GCF (GC factor) in Chr7-128241155. The Chr19-2249336 site of the AMH promoter can be regulated by CNRE (cAMP negative response element). In the AR promoter, we found 4 sites that bind transcription factors: the ChrX-67543762 and ChrX-67543895 sites bind E2 F factor, the ChrX-67543849 site binds XRE (xenobiotic responsive element), and EBS1 (Ets binding site 1) and EGR1 are very close to this site. Finally, AP2 alpha and gamma, and CAF1 (chromatin assembly factor complex) are transcription factors associated to the ChrX-67545002 site.
Discussion
Using a targeted NGS approach, we found that the offspring of women with PCOS, at early infancy, show a sex-specific DNA methylation pattern in the promoter regions of genes associated with reproductive and metabolic features of PCOS. Interestingly, metformin treatment during pregnancy in PCOS women normalized the methylation levels in some of these CpG sites, suggesting that the intrauterine environment of PCOS women may confer a different methylation pattern to their offspring compared to children born to women without PCOS.
Both sons and daughters of women with PCOS showed differences in the methylation levels of specific sites in the promoter regions of LEP and AR. We also observed that daughters of these women had changes in the methylation levels of LEPR and ADIPOR2. Previous data indicate that deregulation of the expression and secretion of leptin in metabolic diseases is associated with modifications in its promoter methylation [39]. Interestingly, it has been described that DNA methylation of the LEP and ADIPOR2 promoters in cord blood and placenta are associated with maternal and infant perinatal factors [40,41]. In this regard, in placenta of women with PCOS, a reduced gene expression of LEP and LEPR have been observed, whereas in cord blood, higher circulating levels of leptin have been reported in newborns of women with PCOS [4,42]. Similarly, prenatal androgenization in the sheep model, resembling PCOS pregnancy, produces an increase of the ADIPOR2 gene expression in fat, muscle and liver of the female offspring at the peripubertal period [43]. It has been observed that the expression of adipokines and their receptors is modulated by the promoter methylation status [44–47], moreover, methylation of these genes has been associated with metabolic alterations and BMI [47,48]. In the same line, we have described that the circulating leptin-adiponectin ratio is associated with metabolic abnormalities in daughters of women with PCOS during the pubertal transition [49]. Therefore, it is possible to suggest that alterations in the leptin-adiponectin system that have been frequently observed at different ages in the offspring of women with PCOS, could be determined by epigenetic modifications that occur during early life.
Same as with the LEP promoter, AR exhibited a differential pattern of methylation according to sex, indicating that epigenetic modifications could affect the expression of the androgen receptor, and hence, androgen action. As stated before, hyperandrogenism is central in the pathophysiology of PCOS. Therefore, our observations in peripheral leukocytes from PCOS daughters agree with this asseveration because a hypomethylation in the AR promoter suggests an increased expression of the AR. On the other hand, DNA methylation is also an important epigenetic mechanism involved in the X chromosome inactivation (XCI), where the AR gene is [50]. It has been proposed that a non-random XCI, may contribute significantly to the expression of PCOS [14]. Nevertheless, while this analysis was not considered in the present study, in a previous report we did not find significant differences in the pattern of XCI between daughters of PCOS and control women at 2–3 months of life [51], as has been reported in adult PCOS women [32,52,53]. In addition, in that study we showed that shorter CAG repeats in the AR, which favours its activity, are associated with abnormalities in the lipid profile of young daughters of PCOS women [51]. Consistently, an elevated protein and gene expression of the androgen receptor have been found in the ovaries and liver of the female offspring of prenatally androgenized sheep, indicating that the mechanisms that regulate the sensitivity to androgen action are determined during foetal life [54,55]. Moreover, the AR promoter methylation has been linked with BMI and fat mass according to gender [56–58].
One of the most constant features that we have observed in the offspring of PCOS women is high AMH serum concentrations. In the present study only sons of PCOS women showed lower methylation levels in CpG sites of the AMH promoter, which is in accordance with the higher serum AMH concentrations described in them during infancy and childhood [59]. On the other hand, we did not observe differences in AMH DNA methylation in girls, which could suggest that the increase in the follicular mass is responsible for this feature more than an increased gene expression. Thus, it seems that, opposite to what was observed in boys, apparently this gene is not modified by epigenetic regulation in girls.
It is currently accepted that an adverse in utero environment can influence the establishment of epigenetic marks during foetal development with consequences later in life [60,61]. In this regard, the negative correlation found between maternal BMI and the CpG methylation level in the AR promoter in girls and with the global z-score in boys, highlights the importance of the maternal metabolic condition for the acquisition of specific epigenetic marks in their offspring. On the other hand, the relationships found between the methylation levels of the LEP and AR sites and the global methylation z-score with postnatal anthropometric features in male newborns highlights the effect of these epigenetic mechanisms in postnatal life. Regarding these anthropometric parameters, only postnatal weight gain was different among sons, specifically sons of PCOS women who took metformin during pregnancy gained almost 1.5 kg more than sons born to control women, while the z-score for length at birth was comparable between groups.
Previously, we reported that metformin treatment ameliorates the endocrine and metabolic alterations in women with PCOS during pregnancy resulting in the improvement of ovarian PCOS markers in their female offspring [15]. Along with this, a recent study demonstrated that short-term metformin administration, at therapeutic doses, has a rapid effect on epigenetic regulation in human white blood cells producing both hypo and hypermethylation in the promoters of different genes [62]. In the present study, we observed that metformin treatment during pregnancy reversed the effect of PCOS on the methylation patterns of some CpG sites of the LEP, ADIPOR2 and had a partial effect on the AR promoters in daughters, whereas in sons, it had an effect on the LEP and AMH promoters. Interestingly, in sons of women with PCOS, in the CpG sites of the AR gene, the methylation level was only modified in the metformin group, especially in ChrX-67543762 and ChrX-67544032, which seems to suggest that the PCOS effect was enhanced by metformin treatment. The impact of these findings is unclear as the long-term impact of intrauterine metformin exposure on childhood development is an unanswered question. Studies evaluating children of patients with gestational diabetes exposed to metformin vs. insulin show a neutral effect in body fat, visceral adipose tissue and intrahepatic fat [63]. We observed that sons of women with PCOS treated with metformin gained more weight from birth until 3 months old, which is consistent with previous observations that have shown an increase in BMI and in the prevalence of overweight or obesity in prepubertal PCOS sons exposed to metformin during pregnancy, (4–7 years old) [64,65]. Nevertheless, it is important to note that in those PCOS women who were medicated with metformin during pregnancy, a large percentage developed GDM probably due to their higher pre-pregnancy metabolic risk, which makes difficult to dissect the effect of metformin from the effect of maternal GDM. In this regard, several studies have established an association between GDM and an altered epigenetic profile in the offspring, finding both increases and decreases of methylation levels depending on the genes studied [66,67]. The significance of these findings in terms of long-term cardiovascular risk is uncertain.
DNA methylation is involved in essential processes that regulate gene expression such as the binding of transcription factors to regulatory elements or direct transcriptional inhibition as in the X chromosome inactivation [68]. DNA methylation has been generally related to gene inactivation and to repression of transcription factor binding ability [69–74]. Therefore, the final effect depends on both mechanisms [24]. The consequences of the inactivation of a gene are relative, since it depends on whether the expression of such a gene is favourable or deleterious for a particular condition. Unfortunately, we were unable to perform gene expression analysis (RNA from blood leucocytes) since the original study was not designed for this purpose. The LEP promoter can potentially bind the transcription factors SP1 and GKLF (KLF4), which are important regulators of the leptin gene, adipogenesis and oxidative stress [75–78]. Particularly KLF4 also promotes macrophage polarization towards an antiinflamatory phenotype (M2) [79]. A study with PCOS women showed activation of KLF-4 after treatment with electroacupunture, associated with epigenetic and transcriptional changes that elicit metabolic improvement [76]. On the other hand, SP1 may also function as a cellular glucose sensor and the effect of its regulation depends on the maturation of the adipocyte [75,78]. Many of these functions have to do with increased leptin transcription. In this context, if the leptin promoter is more methylated, this may indeed inhibit transcription factors binding, avoiding that they exert their action at the promoter level [77,78] and possibly reducing their positive metabolic effects. On the other hand, the transcription factors EGR, MED1, E2 F and XRE have been related to AR promoter regulation. In this regard, the overexpression of EGR-1 enhances AR translocation to the nucleus increasing its activity [80] and therefore, could contribute to the hyperandrogenic state; interestingly higher levels of this protein have been observed in obese women with PCOS [81]. In turn, MED1 and E2 F have been described as co-activators of the AR, participating in the regulation of the expression of cell cycle genes [82,83], which could be involved in the proliferation of ovarian granulosa cells, a phenomenon that has been observed in females born to prenatally androgenized sheep and in the offspring of women with PCOS [84,85]. Moreover, in humans, polymorphisms of the XRE gene have been associated with susceptibility to polycystic ovaries [86]. It is likely that lower DNA methylation of the AR promoter may favour DNA binding of EGR, MED1, E2 F and XRE increasing AR transcription, which could worsen hyperandrogenism and its consequences in the PCOS condition. Although, we cannot exclude that that the binding of some of these transcription factors, such as EGR1, to their target sequences may occur independently of the methylation status [87].
The differential methylation pattern observed in the offspring of PCOS women may reflect adaptive changes generated during pregnancy as a result of an altered intrauterine environment, they may be directly inherited from the mother, or result from a combination of both. Further modulation of these processes may occur during postnatal life through environmental exposure to other factors such as hormones, nutrients, lifestyle, etc. which may reverse or worsen these effects, making them susceptible for therapeutic interventions [88–90].
Although it would be ideal to evaluate the target tissue this is not always possible. Thus, peripheral blood leucocytes DNA is a good surrogate. In this regard, a genome-wide epigenetic study has reported high consistency between peripheral blood and ovarian tissue from PCOS women [29,91]. However, Sang et al did not found consistency between the methylation level of peripheral blood leukocytes and tissue regarding the follistatin promoter in endometrial tissue of PCOS women [92]. In turn, LEP and ADIPOQ, methylation in peripheral leukocytes is correlated with their expression in subcutaneous and visceral adipose tissue [93]. Therefore, whole blood could serve as a useful surrogate measure of the tissue status in terms of epigenetics in the PCOS scenario [94–96]. Another limitation of our study is the small simple size, mainly due to the age of the children studied, the careful selection of PCOS patients and the time required to follow the pregnancies. Finally, although it was not contemplated in this study, it would have been very interesting to have the mothers’ DNA to compare the methylation pattern with that of their children, since these data would have enriched the interpretation of the results.
In summary, we observed that both daughters and sons born to women with PCOS have sex-dependent differences in the methylation levels of CpG sites in the promoter regions of metabolic and reproductive genes such as LEP, LEPR, ADIPOR2, AMH and AR. The intrauterine environment at least in part, mediates these modifications, as treatment with metformin during pregnancy is able to change them. Therefore, our data support the concept that the maternal environment in women with PCOS may induce epigenetic modifications in the DNA methylation profile both in their sons and daughters, which can program the expression of future reproductive and metabolic derangements.
Supplementary Material
Funding Statement
This work was supported by the Fondo Nacional de Desarrollo Científico y Tecnológico (National Fund for Scientific and Technological Research) FONDECYT Grant [1151531]; [1181798] and VID research travelling support grant of the University of Chile [UCH-1566]. The authors express their gratitude to Mr Patricio Miranda and Miss Madián García for their commitment and dedication to the study.
Disclosure statement
Authors have no conflict of interests
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14(5):270–284. [DOI] [PubMed] [Google Scholar]
- [2].Sir-Petermann T, Hitchsfeld C, Maliqueo M, et al. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum Reprod. 2005;20(8):2122–2126. [DOI] [PubMed] [Google Scholar]
- [3].Vanky E, Engen Hanem LG, Abbott DH.. Children born to women with polycystic ovary syndrome-short- and long-term impacts on health and development. Fertil Steril. 2019;111(6):1065–1075. [DOI] [PubMed] [Google Scholar]
- [4].Maliqueo M, Echiburu B, Crisosto N, et al. Metabolic parameters in cord blood of newborns of women with polycystic ovary syndrome. Fertil Steril. 2009;92(1):277–282. [DOI] [PubMed] [Google Scholar]
- [5].Chakrabarti J. Serum leptin level in women with polycystic ovary syndrome: correlation with adiposity, insulin, and circulating testosterone. Ann Med Health Sci Res. 2013;3(2):191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Crisosto N, Codner E, Maliqueo M, et al. Anti-Mullerian hormone levels in peripubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92(7):2739–2743. [DOI] [PubMed] [Google Scholar]
- [7].Sir-Petermann T, Codner E, Maliqueo M, et al. Increased anti-Mullerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(8):3105–3109. [DOI] [PubMed] [Google Scholar]
- [8].Sir-Petermann T, Codner E, Perez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94(6):1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sir-Petermann T, Maliqueo M, Codner E, et al. Early metabolic derangements in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92(12):4637–4642. [DOI] [PubMed] [Google Scholar]
- [10].Recabarren SE, Sir-Petermann T, Rios R, et al. Pituitary and testicular function in sons of women with polycystic ovary syndrome from infancy to adulthood. J Clin Endocrinol Metab. 2008;93(9):3318–3324. [DOI] [PubMed] [Google Scholar]
- [11].Recabarren SE, Smith R, Rios R, et al. Metabolic profile in sons of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(5):1820–1826. [DOI] [PubMed] [Google Scholar]
- [12].Schuring AN, Welp A, Gromoll J, et al. Role of the CAG repeat polymorphism of the androgen receptor gene in polycystic ovary syndrome (PCOS). Exp Clin Endocrinol Diabetes. 2012;120(2):73–79. [DOI] [PubMed] [Google Scholar]
- [13].Xita N, Georgiou I, Lazaros L, et al. The role of sex hormone-binding globulin and androgen receptor gene variants in the development of polycystic ovary syndrome. Hum Reprod. 2008;23(3):693–698. [DOI] [PubMed] [Google Scholar]
- [14].Shah NA, Antoine HJ, Pall M, et al. Association of androgen receptor CAG repeat polymorphism and polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(5):1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Crisosto N, Echiburu B, Maliqueo M, et al. Improvement of hyperandrogenism and hyperinsulinemia during pregnancy in women with polycystic ovary syndrome: possible effect in the ovarian follicular mass of their daughters. Fertil Steril. 2012;97(1):218–224. [DOI] [PubMed] [Google Scholar]
- [16].Luque-Ramírez M, San Millán JL, Escobar-Morreale HF. Genomic variants in polycystic ovary syndrome. Clin Chim Acta. 2006;366(1–2):14–26. [DOI] [PubMed] [Google Scholar]
- [17].Escobar-Morreale HF, Luque-Ramirez M, San Millan JL. The molecular-genetic basis of functional hyperandrogenism and the polycystic ovary syndrome. Endocr Rev. 2005;26(2):251–282. [DOI] [PubMed] [Google Scholar]
- [18].Hickey TE, Legro RS, Norman RJ. Epigenetic modification of the X chromosome influences susceptibility to polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(7):2789–2791. [DOI] [PubMed] [Google Scholar]
- [19].Gur EB, Karadeniz M, Turan GA. Fetal programming of polycystic ovary syndrome. World J Diabetes. 2015;6(7):936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Christensen BC, Marsit CJ. Epigenomics in environmental health. Front Genet. 2011;2:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Guerrero-Bosagna C, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol Cell Endocrinol. 2012;354(1–2):3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Meer A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. [DOI] [PubMed] [Google Scholar]
- [24].Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. [DOI] [PubMed] [Google Scholar]
- [25].Li Z, Huang H. Epigenetic abnormality: a possible mechanism underlying the fetal origin of polycystic ovary syndrome. Med Hypotheses. 2008;70(3):638–642. [DOI] [PubMed] [Google Scholar]
- [26].Wang P, Zhao H, Li T, et al. Hypomethylation of the LH/choriogonadotropin receptor promoter region is a potential mechanism underlying susceptibility to polycystic ovary syndrome. Endocrinology. 2014;155(4):1445–1452. [DOI] [PubMed] [Google Scholar]
- [27].Sang Q, Li X, Wang H, et al. Quantitative methylation level of the EPHX1 promoter in peripheral blood DNA is associated with polycystic ovary syndrome. PloS One. 2014;9(2):e88013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu YY, Sun CX, Liu YK, et al. Promoter methylation of CYP19A1 gene in Chinese polycystic ovary syndrome patients. Gynecol Obstet Invest. 2013;76(4):209–213. [DOI] [PubMed] [Google Scholar]
- [29].Wang XX, Wei JZ, Jiao J, et al. Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget. 2014;5(16):6603–6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Qu F, Wang FF, Yin R, et al. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: hyperandrogenism induces epigenetic alterations in the granulosa cells. J Mol Med. 2012;90(8):911–923. [DOI] [PubMed] [Google Scholar]
- [31].Pan JX, Tan YJ, Wang FF, et al. Aberrant expression and DNA methylation of lipid metabolism genes in PCOS: a new insight into its pathogenesis. Clin Epigenetics. 2018;10(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hickey T, Chandy A, Norman RJ. The androgen receptor CAG repeat polymorphism and X-chromosome inactivation in Australian Caucasian women with infertility related to polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87(1):161–165. [DOI] [PubMed] [Google Scholar]
- [33].Houde AA, Hivert MF, Bouchard L. Fetal epigenetic programming of adipokines. Adipocyte. 2013;2(1):41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wijaya AD, Febri RR, Desmawati, et al. DNA methylation analysis of anti-mullerian hormone gene in ovarian granulosa cells in PCOS patients. J Phys. 2018;1073:032077. [Google Scholar]
- [35].Lambertini L, Saul SR, Copperman AB, et al. Intrauterine reprogramming of the polycystic ovary syndrome: evidence from a pilot study of cord blood global methylation analysis. Front Endocrinol (Lausanne). 2017;8:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif AGJ, Haseltine FP, Merriam GR, editors. Polycystic ovary syndrome. Oxford, UK: Blackwel; 1992. p. 59–69. [Google Scholar]
- [37].Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics. 2011;27(11):1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hertzberg L, Izraeli S, Domany E. STOP: searching for transcription factor motifs using gene expression. Bioinformatics. 2007;23(14):1737–1743. [DOI] [PubMed] [Google Scholar]
- [39].Wroblewski A, Strycharz J, Swiderska E, et al. Molecular insight into the interaction between epigenetics and leptin in metabolic disorders. Nutrients. 2019;11(8):1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lesseur C, Armstrong DA, Paquette AG, et al. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol Cell Endocrinol. 2013;381(1–2):160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nogues P, Dos Santos E, Jammes H, et al. Maternal obesity influences expression and DNA methylation of the adiponectin and leptin systems in human third-trimester placenta. Clin Epigenetics. 2019;11(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maliqueo M, Sundstrom Poromaa I, Vanky E, et al. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum Reprod. 2015;30(3):692–700. [DOI] [PubMed] [Google Scholar]
- [43].Puttabyatappa M, Andriessen V, Mesquitta M, et al. Developmental programming: impact of gestational steroid and metabolic milieus on mediators of insulin sensitivity in prenatal testosterone-treated female sheep. Endocrinology. 2017;158(9):2783–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tobi EW, Lumey LH, Talens RP, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18(21):4046–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stoger R. In vivo methylation patterns of the leptin promoter in human and mouse. Epigenetics. 2006;1(4):155–162. [DOI] [PubMed] [Google Scholar]
- [46].Melzner I, Scott V, Dorsch K, et al. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem. 2002;277(47):45420–45427. [DOI] [PubMed] [Google Scholar]
- [47].Garcia-Cardona MC, Huang F, Garcia-Vivas JM, et al. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int J Obes (Lond). 2014;38(11):1457–1465. [DOI] [PubMed] [Google Scholar]
- [48].Yousefi M, Karmaus W, Zhang H, et al. The methylation of the LEPR/LEPROT genotype at the promoter and body regions influence concentrations of leptin in girls and BMI at age 18 years if their mother smoked during pregnancy. Int J Mol Epidemiol Genet. 2013;4(2):86–100. [PMC free article] [PubMed] [Google Scholar]
- [49].Maliqueo M, Galgani JE, Perez-Bravo F, et al. Relationship of serum adipocyte-derived proteins with insulin sensitivity and reproductive features in pre-pubertal and pubertal daughters of polycystic ovary syndrome women. Eur J Obstet Gynecol Reprod Biol. 2012;161(1):56–61. [DOI] [PubMed] [Google Scholar]
- [50].Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Annu Rev Genet. 1997;31:571–610. [DOI] [PubMed] [Google Scholar]
- [51].Echiburu B, Perez-Bravo F, Maliqueo M, et al. CAG repeat polymorphism of androgen receptor gene and X-chromosome inactivation in daughters of women with polycystic ovary syndrome (PCOS): relationship with endocrine and metabolic parameters. Gynecol Endocrinol. 2012;28(7):516–520. [DOI] [PubMed] [Google Scholar]
- [52].Rajender S, Carlus SJ, Bansal SK, et al. Androgen receptor CAG repeats length polymorphism and the risk of polycystic ovarian syndrome (PCOS). PLoS One. 2013;8(10):e75709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dasgupta S, Sirisha PV, Neelaveni K, et al. Androgen receptor CAG repeat polymorphism and epigenetic influence among the south Indian women with polycystic ovary syndrome. PLoS One. 2010;5(8):e12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ortega HH, Salvetti NR, Padmanabhan V. Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction. 2009;137(5):865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hogg K, Wood C, McNeilly AS, et al. The in utero programming effect of increased maternal androgens and a direct fetal intervention on liver and metabolic function in adult sheep. PloS One. 2011;6(9):e24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ammerpohl O, Bens S, Appari M, et al. Androgen receptor function links human sexual dimorphism to DNA methylation. PLoS One. 2013;8(9):e73288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Moverare-Skrtic S, Mellstrom D, Vandenput L, et al. Peripheral blood leukocyte distribution and body mass index are associated with the methylation pattern of the androgen receptor promoter. Endocrine. 2009;35(2):204–210. [DOI] [PubMed] [Google Scholar]
- [58].Vottero A, Capelletti M, Giuliodori S, et al. Decreased androgen receptor gene methylation in premature pubarche: a novel pathogenetic mechanism? J Clin Endocrinol Metab. 2006;91(3):968–972. [DOI] [PubMed] [Google Scholar]
- [59].Lukas-Croisier C, Lasala C, Nicaud J, et al. Follicle-stimulating hormone increases testicular anti-mullerian hormone (AMH) production through sertoli cell proliferation and a nonclassical cyclic adenosine 5ʹ-monophosphate-mediated activation of the AMH gene. Mol Endocrinol. 2003;17(4):550–561. [DOI] [PubMed] [Google Scholar]
- [60].Fleisch AF, Wright RO, Baccarelli AA. Environmental epigenetics: a role in endocrine disease? J Mol Endocrinol. 2012;49(2):R61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gluckman PD, Hanson MA. Developmental and epigenetic pathways to obesity: an evolutionary-developmental perspective. Int J Obesity. 2008;32(Suppl 7):S62–71. [DOI] [PubMed] [Google Scholar]
- [62].Elbere I, Silamikelis I, Ustinova M, et al. Significantly altered peripheral blood cell DNA methylation profile as a result of immediate effect of metformin use in healthy individuals. Clin Epigenetics. 2018;10(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rowan JA, Rush EC, Plank LD, et al. Metformin in gestational diabetes: the offspring follow-up (MiG TOFU): body composition and metabolic outcomes at 7–9 years of age. BMJ Open Diabetes Res Care. 2018;6(1):e000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hanem LGE, Stridsklev S, Juliusson PB, et al. Metformin use in PCOS pregnancies increases the risk of offspring overweight at 4 years of age: follow-up of two RCTs. J Clin Endocrinol Metab. 2018;103(4):1612–1621. [DOI] [PubMed] [Google Scholar]
- [65].Hanem LGE, Salvesen O, Juliusson PB, et al. Intrauterine metformin exposure and offspring cardiometabolic risk factors (PedMet study): a 5–10 year follow-up of the PregMet randomised controlled trial. Lancet Child Adolesc Health. 2019;3(3):166–174. [DOI] [PubMed] [Google Scholar]
- [66].Weng X, Liu F, Zhang H, et al. Genome-wide DNA methylation profiling in infants born to gestational diabetes mellitus. Diabetes Res Clin Pract. 2018;142:10–18. [DOI] [PubMed] [Google Scholar]
- [67].Houshmand-Oeregaard A, Hansen NS, Hjort L, et al. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin Epigenetics. 2017;9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Senner CE. The role of DNA methylation in mammalian development. Reprod Biomed Online. 2011;22(6):529–535. [DOI] [PubMed] [Google Scholar]
- [69].Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb). 2010;105(1):4–13. [DOI] [PubMed] [Google Scholar]
- [70].Campanero MR, Armstrong MI, Flemington EK. CpG methylation as a mechanism for the regulation of E2F activity. Proc Natl Acad Sci U S A. 2000;97(12):6481–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Comb M, Goodman HM. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990;18(13):3975–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mancini DN, Singh SM, Archer TK, et al. Site-specific DNA methylation in the neurofibromatosis (NF1) promoter interferes with binding of CREB and SP1 transcription factors. Oncogene. 1999;18(28):4108–4119. [DOI] [PubMed] [Google Scholar]
- [73].Yin H, Blanchard KL. DNA methylation represses the expression of the human erythropoietin gene by two different mechanisms. Blood. 2000;95(1):111–119. [PubMed] [Google Scholar]
- [74].Gu P, Le Menuet D, Chung AC, et al. Differential recruitment of methylated CpG binding domains by the orphan receptor GCNF initiates the repression and silencing of Oct4 expression. Mol Cell Biol. 2006;26(24):9471–9483. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [75].Chen J, Meng Y, Zhou J, et al. Identifying candidate genes for Type 2 Diabetes Mellitus and obesity through gene expression profiling in multiple tissues or cells. J Diabetes Res. 2013;2013:970435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kokosar M, Benrick A, Perfilyev A, et al. A single bout of electroacupuncture remodels epigenetic and transcriptional changes in adipose tissue in polycystic ovary syndrome. Sci Rep. 2018;8(1):1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Brey CW, Nelder MP, Hailemariam T, et al. Kruppel-like family of transcription factors: an emerging new frontier in fat biology. Int J Biol Sci. 2009;5(6):622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Roy D, Farabaugh KT, Wu J, et al. Coordinated transcriptional control of adipocyte triglyceride lipase (Atgl) by transcription factors Sp1 and peroxisome proliferator-activated receptor gamma (PPARgamma) during adipocyte differentiation. J Biol Chem. 2017;292(36):14827–14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liao X, Sharma N, Kapadia F, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121(7):2736–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yang SZ, Abdulkadir SA. Early growth response gene 1 modulates androgen receptor signaling in prostate carcinoma cells. J Biol Chem. 2003;278(41):39906–39911. [DOI] [PubMed] [Google Scholar]
- [81].Gonzalez F, Kirwan JP, Rote NS, et al. Glucose ingestion stimulates atherothrombotic inflammation in polycystic ovary syndrome. Am J Physiol Endocrinol Metab. 2013;304(4):E375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yin M, Wang X, Yao G, et al. Transactivation of micrornA-320 by microRNA-383 regulates granulosa cell functions by targeting E2F1 and SF-1 proteins. J Biol Chem. 2014;289(26):18239–18257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liu G, Sprenger C, Wu PJ, et al. MED1 mediates androgen receptor splice variant induced gene expression in the absence of ligand. Oncotarget. 2015;6(1):288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Das M, Djahanbakhch O, Hacihanefioglu B, et al. Granulosa cell survival and proliferation are altered in polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(3):881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Salvetti NR, Ortega HH, Veiga-Lopez A, et al. Developmental programming: impact of prenatal testosterone excess on ovarian cell proliferation and apoptotic factors in sheep1. Biol Reprod. 2012;87(1). DOI: 10.1095/biolreprod.112.100024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Babu KA, Rao KL, Kanakavalli MK, et al. CYP1A1, GSTM1 and GSTT1 genetic polymorphism is associated with susceptibility to polycystic ovaries in South Indian women. Reprod Biomed Online. 2004;9(2):194–200. [DOI] [PubMed] [Google Scholar]
- [87].Zandarashvili L, White MA, Esadze A, et al. Structural impact of complete CpG methylation within target DNA on specific complex formation of the inducible transcription factor Egr-1. FEBS Lett. 2015;589(15):1748–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cole E, Brown TA, Pinkerton KE, et al. Perinatal exposure to environmental tobacco smoke is associated with changes in DNA methylation that precede the adult onset of lung disease in a mouse model. Inhal Toxicol. 2017;29(10):435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol. 2015;1238:3–25. [DOI] [PubMed] [Google Scholar]
- [90].Teruel M, Sawalha AH. Epigenetic variability in systemic lupus erythematosus: what we learned from genome-wide DNA methylation studies. Curr Rheumatol Rep. 2017;19(6):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Li S, Zhu D, Duan H, et al. Differential DNA methylation patterns of polycystic ovarian syndrome in whole blood of Chinese women. Oncotarget. 2016;8(13):20656–20666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sang Q, Zhang S, Zou S, et al. Quantitative analysis of follistatin (FST) promoter methylation in peripheral blood of patients with polycystic ovary syndrome. Reprod Biomed Online. 2013;26(2):157–163. [DOI] [PubMed] [Google Scholar]
- [93].Houde -A-A, Légaré C, Hould F-S, et al. Cross-tissue comparisons of leptin and adiponectin: DNA methylation profiles. Adipocyte. 2014;3(2):132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].VA-m ER, Gomez-Viais YI, Garcia-Gomez E, et al. DNA methylation in the pathogenesis of polycystic ovary syndrome. Reproduction. 2019;158(1):R27–R40. [DOI] [PubMed] [Google Scholar]
- [95].Slieker RC, Bos SD, Goeman JJ, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ma B, Wilker EH, Willis-Owen SA, et al. Predicting DNA methylation level across human tissues. Nucleic Acids Res. 2014;42(6):3515–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.