

ABSTRACT
Proteasome inhibition (PSMI) is known to activate macroautophagy (autophagy hereafter), but the underlying mechanisms remain to be fully delineated. Here we discuss our recent work identifying an important PPP3/calcineurin-TFEB-SQSTM1/p62 pathway in mediating activation of autophagy by PSMI, a compensatory process for the heart with proteasome malfunction. Through increasing PPP3/calcineurin activity and inhibiting MTOR signaling, PSMI promotes the dephosphorylation and thereby nuclear translocation of TFEB, resulting in transactivation of genes in the autophagic-lysosomal pathway (ALP) such as Mcoln1 and Sqstm1. We have discovered that SQSTM1 is required for not only induction of autophagy but also cardiac activation of TFEB by PSMI, unveiling a novel feedforward role for SQSTM1 in TFEB activation.
KEYWORDS: macroautophagy, PPP3/calcineurin, PSMC2, SQSTM1/p62, TFEB
The ubiquitin-proteasome system (UPS) and the ALP are pivotal to proteostasis. While the two pathways seem to function in parallel at basal conditions, increasing evidence suggests a strong interplay between them, especially when one of them is compromised. Chemical or genetic PSMI can induce autophagy in a variety of cell types and organs. However, molecular pathways that link proteasome malfunction to ALP activation were not well elucidated. Using mouse models and cultured cardiomyocytes with genetic and pharmacological interrogations, we have recently delineated a novel PPP3/calcineurin-TFEB- SQSTM1/p62 pathway in mediating autophagy activation by PSMI in cardiomyocytes and myocardium [1].
The Psmc1 gene encodes a stoichiometric subunit of the 19S proteasome. In characterizing a mouse model of perinatal cardiomyocyte-restricted knockout of Psmc1 (psmc1-CKO), we found marked increases in autophagosomes in cardiomyocytes. Attempts to measure myocardial autophagic flux in these mice failed because all the psmc1-CKO mice die by postnatal day 10 and cannot tolerate bafilomycin A1 treatment that was utilized to inhibit lysosomes. Hence, we had to turn to cell cultures for autophagic flux assays which confirmed that the increase of autophagosomes by genetic PSMI is due to increased autophagosome formation and indicative of autophagic activation.
TFEB (transcription factor EB) is a master transcription factor that regulates autophagy and lysosome biogenesis upon nutrient deprivation. At baseline condition, TFEB is phosphorylated by MTOR and thereby sequestered in the cytoplasm. As with starvation, PSMI diminishes TFEB phosphorylation and increases its nuclear translocation and its target gene expression in cultured cardiomyocytes and mouse hearts. Moreover, silencing of TFEB abolishes PSMI-induced autophagy gene expression, identifying TFEB as a major mediator for autophagy activation by PSMI.
PPP3/calcineurin is the main phosphatase for TFEB dephosphorylation and activation during starvation; thus, in search for the mechanism by which PSMI activates TFEB, we immediately thought of PPP3/calcineurin because we had reported that PSMI activates the myocardial PPP3/calcineurin-NFAT (nuclear factor of activated T cells) pathway. In this study, both genetic and pharmacological inhibition of PPP3/calcineurin abolishes the PSMI-induced TFEB activation in cardiomyocytes. Moreover, silencing of MCOLN1, a lysosomal calcium channel protein that mediates lysosomal calcium release into the cytosol and is required for PPP3/calcineurin activation, also attenuates PSMI-induced TFEB activation. Hence, the MCOLN1- PPP3/calcineurin-TFEB axis plays a key role in activation of autophagy by PSMI. MCOLN1 as a TFEB target gene plays a feedforward role in TFEB activation, but increasing PPP3/calcineurin activity by PSMI also may be attributable to TFEB protein stabilization, as PPP3/calcineurin is degraded by the UPS. Meanwhile, we observed that MTOR signaling is suppressed by PSMI; hence, deactivation of MTOR certainly also contributes to TFEB activation by PSMI.
Both mRNA and protein levels of SQSTM1 in mouse hearts and cultured cardiomyocytes are markedly upregulated by PSMI; this upregulation is significantly attenuated but not completely abolished by TFEB inactivation, indicating that TFEB-independent mechanisms may also be involved in SQSTM1 upregulation by PSMI. It was previously shown that SQSTM1 accumulation by autophagy deficiency is detrimental to the liver but compensatory to the brain. Our study demonstrates for the first time that SQSTM1 upregulation by PSMI is cardioprotective, as sqstm1-null mice are more susceptible to PSMI-induced cardiac diastolic malfunction. Mechanistically, SQSTM1 is required for the compensatory autophagic degradation of protein substrates that would be degraded by proteasomes. SQSTM1 recognizes ubiquitinated proteins via its UBA domain and targets them to phagophores via its LC3-interacting region/LIR. Our data from both mouse hearts and cultured cardiomyocytes showed that deletion of Sqstm1 abolishes PSMI-induced increases in autophagy flux and autophagic removal of ubiquitinated proteins. Also, unexpectedly, SQSTM1 deficiency prevents PSMI from inducing TFEB nuclear translocation, and diminishes TFEB target gene expression in mouse hearts, unveiling a novel feedforward role for SQSTM1 in TFEB activation. The underlying mechanism for this new role is unclear, but we found that MTOR frequently colocalizes with SQSTM1-positive protein aggregates in cardiomyocytes upon PSMI, prompting us to speculate that SQSTM1 senses proteotoxic stress and sequesters MTOR into protein aggregates, thereby preventing MTOR from interacting with and phosphorylating TFEB. Normally, SQSTM1 is an integral part of the MTOR complex and essential for MTOR activation at the lysosome membrane when nutrients are abundant. Notably, increases of SQSTM1 by autophagic impairment hijack ubiquitinated proteins from the proteasome and thereby impair UPS-mediated proteolysis. Hence, SQSTM1 exerts opposing effects on the cell during UPS and ALP malfunctioning.
In summary, our study has delineated the MCOLN1-PPP3/calcineurin-TFEB-SQSTM1/p62 feedforward loop in the compensatory activation of the ALP by PSMI (Figure 1). This is highly significant, given that proteasome malfunction is increasingly recognized as a major driver for cardiac pathology, and therapeutic PSMI in chemotherapy induces cardiotoxicity. It is conceivable that suppressing any components of this pathway would be detrimental to a proteasome-impaired heart. Indeed, recent reports showed that heart-transplant patients receiving a rapamycin analog (MTOR inhibitor) as their immunosuppressant display better cardiac diastolic function than those using cyclosporin (a PPP3/calcineurin inhibitor) as the immunosuppressant.
Figure 1.
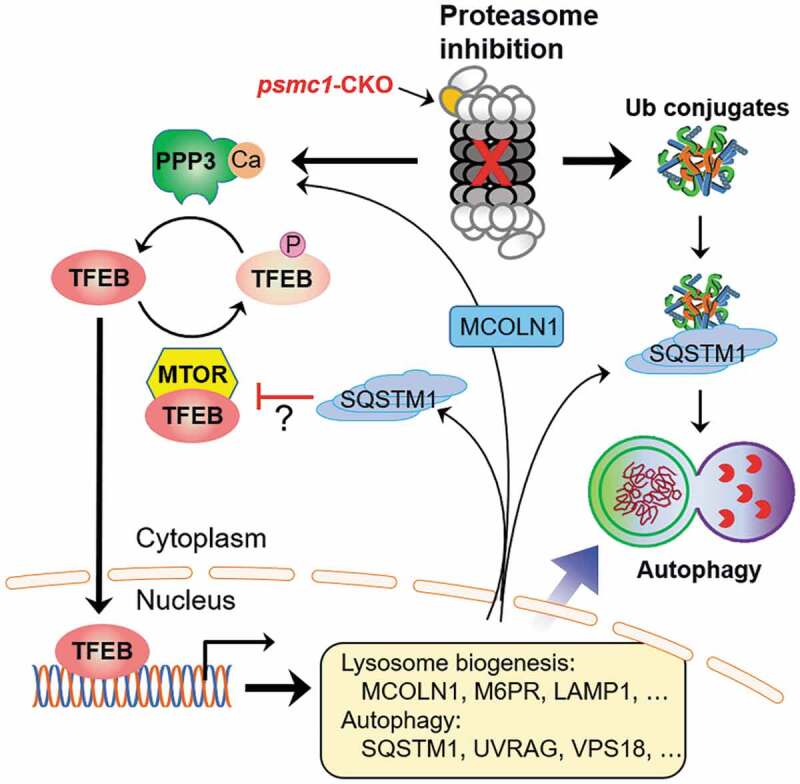
A MCOLN1-PPP3/calcineurin-TFEB-SQSTM1/p62 feed-forward loop activates autophagy following proteasome impairment. Proteasome malfunction promotes dephosphorylation and nuclear translocation of TFEB through stabilizing and activating PPP3/calcineurin and suppressing MTOR signaling. Activated TFEB induces the expression of genes required for autophagy and lysosome biogenesis, including those encoding MCOLN1 and SQSTM1. MCOLN1 in turn mediates lysosomal calcium release to further promote PPP3/calcineurin activation, whereas SQSTM1 appears to suppress MTOR, thereby leading to a persistent activation of TFEB and autophagy. Meanwhile, the upregulated SQSTM1 also promotes aggregation of ubiquitinated proteins and is required for targeting at least a subset of ubiquitinated proteins for autophagic degradation to compensate for proteasome malfunction. Adopted from Pan [1] with permission
Funding Statement
This work was supported by the American Heart Association [AHA00182]; National Heart, Lung, and Blood Institute [HL072166]; National Heart, Lung, and Blood Institute [HL131667]; National Heart, Lung, and Blood Institute [HL153614]; National Heart, Lung, and Blood Institute [HL124248]; National Heart, Lung, and Blood Institute [HL085629].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Pan B, Li J, Parajuli N, et al. The calcineurin-TFEB-p62 pathway mediates the activation of cardiac macroautophagy by proteasomal malfunction. Circ Res. 2020;127:502–518. [DOI] [PMC free article] [PubMed] [Google Scholar]