

Abstract
As one of the widely occurring RNA modifications, 5-methyluridine (m5U) has recently been shown to play critical roles in various biological functions and disease pathogenesis, such as under stress response and during breast cancer development. Precise identification of m5U sites on RNA is vital for the understanding of the regulatory mechanisms of RNA life. We present here m5UPred, the first web server for in silico identification of m5U sites from the primary sequences of RNA. Built upon the support vector machine (SVM) algorithm and the biochemical encoding scheme, m5UPred achieved reasonable prediction performance with the area under the receiver operating characteristic curve (AUC) greater than 0.954 by 5-fold cross-validation and independent testing datasets. To critically test and validate the performance of our newly proposed predictor, the experimentally validated m5U sites were further separated by high-throughput sequencing techniques (miCLIP-Seq and FICC-Seq) and cell types (HEK293 and HAP1). When tested on cross-technique and cross-cell-type validation using independent datasets, m5UPred achieved an average AUC of 0.922 and 0.926 under mature mRNA mode, respectively, showing reasonable accuracy and reliability. The m5UPred web server is freely accessible now and it should make a useful tool for the researchers who are interested in m5U RNA modification.
Keywords: 5-methyluridine, m5U, sequence-derived features, support vector machine, SVM
Graphical Abstract
Jie Jiang et al. present m5UPred, the first web server for identification of m5U sites from the primary sequences of RNA by advanced computational method in this study. It can help to accurately predict the m5U sites from RNA sequences, which will assist researchers to understand the RNA methylation mechanism and its biological functions.
Introduction
The development of high-resolution transcriptome mapping and quantification technologies has made epigenetic modifications of RNA one of the fastest-growing fields in biological research in the past several years. Over 170 post-transcriptional modifications have been identified, with the majority of them occurred in tRNAs and rRNAs.1 RNA modifications showed imperative roles in varied biological functions (e.g., embryonic stem cell development,2 cancer cell survival, migration,3 and response to environmental exposures4). Over 100 RNA modification enzyme mutations have been associated with human diseases.5 Besides the well-known role of fine-tuning RNA structures and functions to regulate gene expression and protein synthesis by RNA modifications,6 there are many other functions associated with RNA modifications. Some post-transcriptional RNA modifications have shown to be dynamic processes that have regulatory roles similar to post-translational protein modifications in controlling cell-type-specific functions.7
However, as one of the most abundant RNA modifications, the identification and functional characterization of 5-methyluridine (m5U) remain extremely limited in the literature. As a pyrimidine modification, m5U involves methylation at the 5-carbon position of uridine, which may be the first pyrimidine methyltransferases evolved to catalyze the pyrimidine-C5 methylation.8 The enzymes catalyzing the modification of m5U are TrmA in Escherichia coli,9,10 Trm2p in Saccharomyces cerevisiae,11 and TRMT2A and TRMT2B in mammals.12,13 m5U has been reported to participate in the development of breast cancer,5 systemic lupus erythematosus,14 and regulation of stress response and development in plants.15 Accurate identification of m5U sites is crucial to understanding fundamental biological processes and functions in all species. Some wet-lab experimental methods, such as miCLIP-Seq, iCLIP, and FICC-seq, have been developed to identify m5U sites.16 However, RNA sequencing could be a high-cost and time-consuming process, and the specificity of antibodies used for immunoprecipitation restricts the delivery of accurate sequencing results. Thus, only very limited data have been generated for m5U sites so far. In this study, we would like to propose an in silico technique for the identification of m5U sites based on sequence-derived information using a support vector machine (SVM) algorithm.
A number of computational methods have been developed to predict epigenetic modifications of RNA, including m6A site predictors WHISTLE,17 SRAMP,18 Gene2vec,19 iRNA-Methyl,20 and M6AMRFS;21 m5C site predictors RNAm5Cfinder,22 iRNA-m5C,23 and M5C-HPCR;24 pseudouridine site predictors iRNA-PseU,25 PseUI,26 PPUS,27 and PIANO;28 and one-stop platform iMRM for simultaneously identifying various RNA modifications in multiple species.29 However, to the best of our knowledge, there is no m5U site predictor available so far. Thus, in this study we would like to propose the first prediction framework, m5UPred, which can be utilized for high-accuracy identification of m5U site from RNA sequences. A user-friendly web server has also been developed and made free access publicly at https://www.xjtlu.edu.cn/biologicalsciences/m5u. We anticipate that our newly proposed model, m5UPred, could make the best use of limited experimentally detected data and facilitate the research of m5U site modification by providing an alternative computational approach.
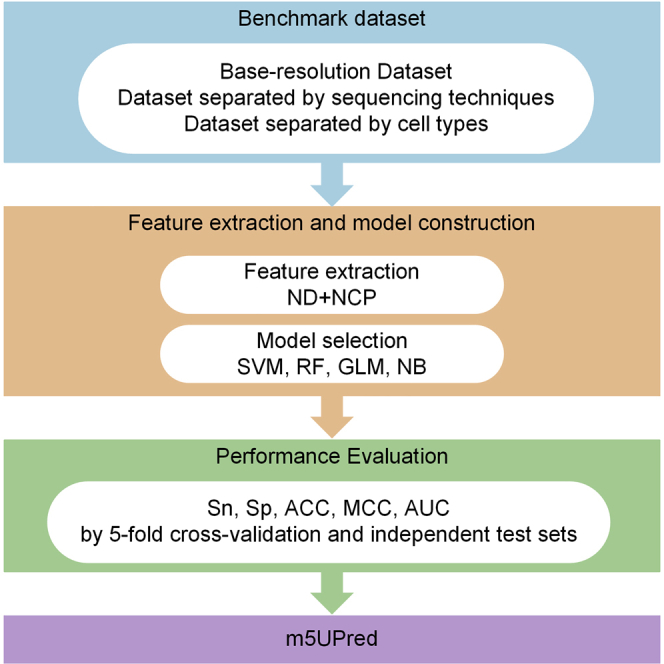
Results
Determine the Best Machine Learning Algorithm Used for m5U Site Prediction
In order to determine the best classifier for constructing m5UPred, the performances of different classifiers were tested on the independent dataset (see Table 1). SVM achieved area under the receiver operating characteristic curve (AUC) of 0.933 for full transcript mode and 0.951 for mature mRNA mode, which was the best among all classifiers and used to build m5UPred.
Table 1.
Performance Evaluation of m5UPred by Independent Testing Dataset
| Mode | Algorithms | Sn (%) | Sp (%) | ACC (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| Full transcript | SVM | 86.06 | 84.72 | 85.39 | 0.708 | 0.933a |
| RF | 85.18 | 83.44 | 84.31 | 0.687 | 0.929 | |
| NB | 86.06 | 61.53 | 73.80 | 0.491 | 0.845 | |
| GLM | 83.03 | 80.45 | 81.74 | 0.635 | 0.897 | |
| Mature mRNA | SVM | 86.99 | 89.07 | 88.03 | 0.761 | 0.951a |
| RF | 81.30 | 97.56 | 89.43 | 0.799 | 0.951 | |
| NB | 92.36 | 46.91 | 69.63 | 0.441 | 0.862 | |
| GLM | 85.57 | 81.54 | 83.56 | 0.672 | 0.915 |
We randomly selected 80% of experimentally validated m5U sites as training dataset and the performance of predictors was evaluated by the remaining 20% of m5U sites as independent testing data.
SVM achieved best performance among all classifier with AUC of 0.933 and 0.951 for full transcript mode and mature mRNA mode.
Performance Evaluation of m5UPred by Benchmark and Independent Testing Dataset
The prediction performance of m5UPred was evaluated by 5-fold cross-validation and an independent testing dataset, respectively (see Table 2). Using SVM as the final classifier, m5UPred was developed and tested using the same datasets that were previously applied for algorithm selection. In addition, the tune length of SVM in caret package was set to 9, with a random grid search for optimization. When evaluated by the independent testing data, m5UPred showed good generalization capability and achieved 0.767 Matthews correlation coefficient (MCC) with 88.35% accuracy and 0.956 AUC for full transcript mode and 0.795 MCC with 89.70% accuracy and 0.954 AUC for mature mRNA mode.
Table 2.
Prediction Performance Using Cross Validation and Independent Testing Dataset
| Dataset | Testing Method | Sn (%) | Sp (%) | ACC (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| Full transcript | cross validation | 87.59 | 89.04 | 88.32 | 0.767 | 0.956 |
| independent test set | 87.90 | 88.80 | 88.35 | 0.767 | 0.956 | |
| Mature mRNA | cross validation | 88.64 | 91.18 | 89.91 | 0.798 | 0.956 |
| independent test set | 87.44 | 91.95 | 89.70 | 0.795 | 0.954 |
80% of experimentally validated m5U sites were used for training, while its performance was evaluated by the remaining 20% as independent testing data.
Performance Evaluation of m5UPred by Cross-Technique and Cross-Cell-Type Validation
It was shown previously that positive samples of RNA modification captured by different techniques may have different overall patterns.30 To test the performance and robustness of our newly proposed predictor in finding m5U sites generated from different techniques and cell types, we further evaluated the performance of m5UPred by cross-technique and cross-cell-type validation (Table 3). When tested by the independent dataset generated from another technique or cell type, m5UPred achieved AUC of 0.882 and 0.922 for cross-technique validation, and AUC of 0.899 and 0.926 for cross-cell-type validation, under full transcript mode and mature mRNA mode, respectively. It shows robustness and reliability of m5UPred.
Table 3.
Cross-Technique and Cross-Cell-Type Validation
| Mode | Testing Method | Evaluation Metric | Cross-Technique Validation |
Cross-Cell-Type Validation |
||||
|---|---|---|---|---|---|---|---|---|
| miCLIP-Seq | FICC-Seq | Average | HEK293 | HAP1 | Average | |||
| Full transcript | cross validation | Sn (%) | 86.70 | 89.80 | 88.25 | 86.26 | 89.67 | 87.96 |
| Sp (%) | 86.83 | 91.37 | 89.10 | 87.19 | 90.48 | 88.84 | ||
| ACC (%) | 86.76 | 90.58 | 88.67 | 86.72 | 80.15 | 83.44 | ||
| MCC | 0.735 | 0.812 | 0.773 | 0.735 | 0.901 | 0.818 | ||
| AUC | 0.946 | 0.966 | 0.956 | 0.942 | 0.969 | 0.955 | ||
| independent dataset | Sn (%) | 75.36 | 56.48 | 65.92 | 82.79 | 57.77 | 70.28 | |
| Sp (%) | 89.23 | 90.10 | 89.67 | 89.62 | 90.21 | 89.92 | ||
| ACC (%) | 82.29 | 73.29 | 77.79 | 86.20 | 73.99 | 80.10 | ||
| MCC | 0.652 | 0.495 | 0.574 | 0.726 | 0.507 | 0.617 | ||
| AUC | 0.910 | 0.853 | 0.882 | 0.941 | 0.857 | 0.899 | ||
| Mature mRNA | cross validation | Sn (%) | 88.34 | 94.14 | 91.24 | 89.86 | 95.32 | 92.59 |
| Sp (%) | 90.52 | 98.04 | 94.28 | 91.13 | 96.71 | 93.92 | ||
| ACC (%) | 89.43 | 96.09 | 92.76 | 90.50 | 96.02 | 93.26 | ||
| MCC | 0.789 | 0.922 | 0.856 | 0.810 | 0.920 | 0.865 | ||
| AUC | 0.962 | 0.992 | 0.977 | 0.964 | 0.987 | 0.975 | ||
| independent dataset | Sn (%) | 90.07 | 38.81 | 64.44 | 95.41 | 39.01 | 67.21 | |
| Sp (%) | 90.86 | 97.51 | 94.19 | 88.93 | 98.12 | 93.53 | ||
| ACC (%) | 90.46 | 68.16 | 79.31 | 92.17 | 68.57 | 80.37 | ||
| MCC | 0.809 | 0.449 | 0.629 | 0.845 | 0.461 | 0.653 | ||
| AUC | 0.970 | 0.873 | 0.922 | 0.981 | 0.871 | 0.926 | ||
As previously shown in Table 2, the experimentally validated m5U sites were further separated by high-throughput sequencing techniques and cell types, under full transcript and mature mRNA mode, respectively. Independent tests by the other technique or cell type are employed to evaluate the performance additional to 5-fold cross validation. When dataset miCLIP_F was used for training, its performance was tested by 5-fold cross-validation and an independent dataset of FICC_F. Similarly, while dataset FICC_F was used for training, miCLIP_F served as an independent test set. The same testing scheme was used for datasets separated by cell types as well. The performance of different sequencing techniques and cell types are then averaged. The performance evaluation of m5UPred by cross-technique and cross-cell-type validation using different machine learning classifiers is listed in Table S1.
Web Implementation
To facilitate the access of our model by experimental researchers, a web server has been developed using Hyper Text Markup Language (HTML), Cascading Style Sheets (CSS), and Hypertext Preprocessor (PHP) and is accessible at https://www.xjtlu.edu.cn/biologicalsciences/m5u. It allows users to submit query RNA sequences in FASTA format with over 41 nt in length and a uridine in the center for analysis. The web server will evaluate the possibility of m5U modification in the given sequences, which return all putative m5U sites with download function (Figure 1).
Figure 1.

Input and Output of m5UPred Web Server
The input of m5UPred is FASTA sequences. The user can paste the sequences into a text box or provide a FASTA file. m5UPred will predict the m5U site possibility using our m5UPred model and returns predicted m5U sites. Results can be downloaded in table format.
Discussion
In this study, we extracted m5U modification sites from a data source generated by two sequencing methods and two cell types. A high-accuracy predictor was built by using a SVM model to predict the m5U modification sites. Satisfactory prediction performance was observed, with an average AUC of 0.956 for full transcript and 0.954 for mature mRNA when it was evaluated by an independent test set with 20% randomly selected sites from the dataset. The model was further tested by cross-technique and cross-cell-type validation. The results show that the positive m5U sites generated from different techniques and cell types share some common features, and these features can be captured by our m5UPred by extracting sequence-derived information. To help understand the sequence composition, we analyzed full transcript sequences from different cell types and sequencing methods by DREME31 motif discovery tool. The motifs identified are provided in Figure S1 and are consistent with the tRNA T-loop motif of GTTCG/AA proposed by the data source paper.16 A web server was developed based on our model and made available to the public to assist the prediction of m5U sites by other researchers.
It is worth noting that although m5U sites were generated under different conditions, they are still from a single source; this may pose an over-fitting problem, and the performance of the model may be over-estimated. We calculated the false discovery rate (FDR) and false omission rate (FOR) at different thresholds and the true positive rate (TPR). The result confirmed our previous statement about over-estimation, and the result is provided in the Supplemental Information. We will keep updating our model when more data are available. When comparing the crosslink peaks from two sequencing methods of the raw data, only around 35% of sites with over 5 crosslink peaks are identified by both methods;16 therefore, the true-positive m5U sites will need to be further confirmed by more experimental data. We noticed that tRNA modifications take the dominant position in the dataset we used for training, which was also confirmed by the data source paper. Considering the biological structures and functional difference of varied RNA molecules, further study will need to be performed to optimize the model using optimized window size, secondary structures, modification motif, and genome information to further improve the model robustness and generalization ability.
Materials and Methods
Training and Testing Datasets
Benchmark and Independent Testing Dataset Used for m5U Site Prediction
The positive samples (m5U sites) were obtained from the recently published single-nucleotide resolution m5U sequencing data,16 and the sequencing results were generated by FICC-seq and miCLIP-seq technologies on two cell lines, HEK293 and HAP1 (Table 4). Data were downloaded from Gene Expression Omnibus (GEO), with the GEO accession number GEO: GSE109183. Previous studies32,33 showed RNA sequence 41 nt in length with an RNA modification site in middle provided the most promising prediction result. Thus, we adopt this formula and designed our positive dataset by generating 41 nt sequences with experimental identified U sites in the center. Unmodified uridine sites located on the same transcripts of the positive m5U sites were randomly selected, and 10 negative datasets were generated. By combining each of these 10 negative sets with the positive data, 10 separate datasets were constructed with a 1:1 positive-to-negative ratio. Their prediction performances were averaged during the evaluation to reduce batch variance.
Table 4.
Base-Resolution Dataset Used for m5U Site Prediction
| GEO Accession | Technique | Cell Line | No. of Sites |
Source | |
|---|---|---|---|---|---|
| Full | Mature | ||||
| GSE109183 | miCLIP-Seq | HEK293 | 3,696 | 1,232 | 15 |
| FICC-Seq | |||||
| HAP1 | |||||
80% of the data were randomly selected for training, while the remaining 20% were retained as an independent testing dataset for performance evaluation. All positive and negative sequences used in this project can be freely downloaded from https://www.xjtlu.edu.cn/biologicalsciences/m5u. and are provided in the Supplemental Information.
The performance of our predictor was evaluated under two modes, namely full transcript mode and mature mRNA mode. For the full transcript mode, the positive and negative m5U sites located in both exonic and intronic regions are all considered to generate the data, whereas, in the mature mRNA mode, only positive and negative m5U sites located on mature mRNA transcripts are employed. Positive m5U sites in different RNA families are listed in the Supplemental Information. For performance evaluation, 80% of the dataset was randomly selected as a benchmark training dataset, while the remaining 20% was used as independent testing data.
Dataset Separated by High-Throughput Sequencing Techniques and Cell Types
Besides randomly selected training and testing data from experimentally validated m5U sites, we applied two more strictly evaluation methods to validate the performance of the proposed predictor. The experimentally validated m5U sites were further divided by high-throughput sequencing technique and cell type (Table 5), from which the cross-technique and cross-cell-type testing were applied for performance evaluation under full transcript and mature mRNA modes, respectively. The performance was also evaluated by 5-fold cross-validation and an independent test set. But rather than randomly selecting 20% of the total data as a test set, we generated test sets by different techniques and cell types. Concretely, for cross-technique validation, when dataset miCLIP_F or miCLIP_M were used for training, their performance was tested by an independent dataset of FICC_F or FICC_M. Then, we switched the train and test set and used dataset FICC_F or FICC_M for training and miCLIP_F or miCLIP_M for independent testing. The validation scheme was applied for the dataset separated by cell types as well.
Table 5.
Dataset Separated by Sequencing Techniques and Cell Types
| Separation Method | Mode | Condition | Site No. | Dataset | Total No. |
|---|---|---|---|---|---|
| Technique | full | miCLIP-Seq | 2,225 | miCLIP_F | 3,696 |
| FICC-Seq | 1,471 | FICC_F | |||
| mature | miCLIP-Seq | 823 | miCLIP_M | 1,232 | |
| FICC-Seq | 409 | FICC_M | |||
| Cell type | full | HEK293 | 2,467 | HEK293_F | 3,696 |
| HAP1 | 1,229 | HAP1_F | |||
| mature | HEK293 | 868 | HEK293_M | 1,232 | |
| HAP1 | 364 | HAP1_M |
Feature Extraction
High classification accuracy of many machine learning algorithms largely relies on sequence encoding strategies for feature extraction of RNA sequences. To achieve the best performance, the chemical properties of nucleotides and their distribution information are employed for sequence feature extraction in this study. This strategy was adapted from previous work by Bari et al.,34 which was originally used for splicing site prediction from DNA sequence and lately is being widely used in encoding RNA sequences for RNA modification prediction.35, 36, 37
Nucleotide Density
The nucleotide density represents the distribution and frequency information of nucleotides at each position. The density () of a nucleotide at position can be calculated by the number () of occurred before position divided by . So, for a sequence “AUAGUCAUAA,” the density of A is 1, 0.67, 0.43, 0.44, and 0.50 at the 1st, 3rd, 7th, 9th, and 10th positions, respectively. Similarly, U is 0.50, 0.40, and 0.38 at positions 2, 5, and 8; C is 0.17 at position 6; and G is 0.25 at position 4 (Table 6).
Table 6.
Calculation of Nucleotide Density for a Sample Sequence
| Sequence | A | U | A | G | U | C | A | U | A | A |
|---|---|---|---|---|---|---|---|---|---|---|
| No. of same nucleotide accumulated | 1 | 1 | 2 | 1 | 2 | 1 | 3 | 3 | 4 | 5 |
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Nucleotide density | 1 | 0.5 | 0.67 | 0.25 | 0.4 | 0.17 | 0.43 | 0.38 | 0.44 | 0.5 |
Nucleotide Chemical Property
The encoding scheme of nucleotide chemical property was designed based on different chemical structures of four RNA nucleotides. Four building blocks of RNA, namely adenosine (A), uridine (U), guanosine (G), and cytosine (C), are categorized into different groups depending on the number of ring structures (two for A and G and one for C and T), existence of an amino group (A and C) or keto group (G and T), and strong (C and G) or weak (A and T) hydrogen bonds. Based on this information, together with the nucleotide density information, nucleotide at position from sequence (with length can be represented by the formula which satisfies the following equations:
| (1) |
Concretely, A, C, G, and U can be encoded as vectors (1,1,1, ), (0,1,0,), (1,0,0, ), and (0,0,1, ), respectively. So, each of the nucleotides in the RNA sequences will be transferred into four numeric values; thus, each of our 41 nt RNA sequences will be encoded into a 164-dimension vector.
Evaluation Matric
Five metrics were employed to evaluate the performance our model, namely Sn (Sensitivity), Sp (Specificity), MCC, ACC (overall accuracy), and AUC, with the equations below:
| (2) |
| (3) |
| (4) |
| (5) |
in which TP, TN, FP, and FN represent the number of true positives, true negatives, false positives, and false negatives, respectively. A threshold of 0.5 was applied to calculate Sn and Sp. R package ROCR38 was used to evaluate the performance of the model.
Choice of Machine Learning Classifier
SVM, random forest (RF), generalized linear model (GLM), and Naive Bayes (NB) are the most popular machine learning classifiers in RNA modification prediction and have been widely used for different site predictions.24,27,35 We evaluated the performance of these algorithms by an independent testing dataset, since the evaluation by cross-validation may over-estimate the performance of models.39 The R package caret was used to construct machine learning models, and all parameters were set by default for primitive evaluation. The results are shown in Table 1. SVM achieved the best results among all four algorithms, which was selected as our final algorithm to build our m5U site prediction model.
SVM
SVM is one of the most powerful yet flexible algorithms in bioinformatics research and has been used for many applications (e.g., mammalian microRNA target prediction,40 predicting the subcellular location of proteins,41 and RNA modification site prediction17,42). The final decision of SVM is determined by a representation of different classes in a hyperplane, which can be used for varied classification and regression tasks. In this study, we used the R package caret to construct our SVM model with a non-linear radial basis function. The prediction performance was evaluated by 5-fold cross-validation and an independent test set. Initially, all parameters were set by default to test the performance of the model, and then tune length was set to 9, with a random grid search for optimization. Random grid search has been proved by a previous study with better efficiency than manually set grid searches.43
Author Contributions
B.S. conceived the idea; K.C. and B.S. collected and processed the data; J.J. and B.S. constructed the predictive model and designed the experiments; Y.T. built the website; and J.J. and B.S. drafted the manuscript. All authors read, critically revised and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31671373) and the XJTLU Key Program Special Fund (KSF-E-51). This work is partially supported by the AI University Research Centre through XJTLU Key Programme Special Fund (KSF-P-02).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.09.031.
Supplemental Information
References
- 1.Boccaletto P., Machnicka M.A., Purta E., Piatkowski P., Baginski B., Wirecki T.K., de Crécy-Lagard V., Ross R., Limbach P.A., Kotter A. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–D307. doi: 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batista P.J., Molinie B., Wang J., Qu K., Zhang J., Li L., Bouley D.M., Lujan E., Haddad B., Daneshvar K. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delaunay S., Frye M. RNA modifications regulating cell fate in cancer. Nat. Cell Biol. 2019;21:552–559. doi: 10.1038/s41556-019-0319-0. [DOI] [PubMed] [Google Scholar]
- 4.Yang C. ToxPoint: Dissecting Functional RNA Modifications in Responses to Environmental Exposure-Mechanistic Toxicology Research Enters a New Era. Toxicol. Sci. 2020;174:1–2. doi: 10.1093/toxsci/kfz252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonkhout N., Tran J., Smith M.A., Schonrock N., Mattick J.S., Novoa E.M. The RNA modification landscape in human disease. RNA. 2017;23:1754–1769. doi: 10.1261/rna.063503.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y.T., Terns R.M., Terns M.P. Fine-Tuning of RNA Functions by Modification and Editing, H. Grosjean, ed. Top. Curr. Genet. Vol. 12. Springer; 2004. Mechanisms and functions of RNA-guided RNA modification; pp. 223–262. [Google Scholar]
- 7.He C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 2010;6:863–865. doi: 10.1038/nchembio.482. [DOI] [PubMed] [Google Scholar]
- 8.Bujnicki J.M., Feder M., Ayres C.L., Redman K.L. Sequence-structure-function studies of tRNA:m5C methyltransferase Trm4p and its relationship to DNA:m5C and RNA:m5U methyltransferases. Nucleic Acids Res. 2004;32:2453–2463. doi: 10.1093/nar/gkh564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ny T., Björk G.R. Cloning and restriction mapping of the trmA gene coding for transfer ribonucleic acid (5-methyluridine)-methyltransferase in Escherichia coli K-12. J. Bacteriol. 1980;142:371–379. doi: 10.1128/jb.142.2.371-379.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urbonavičius J., Jäger G., Björk G.R. Amino acid residues of the Escherichia coli tRNA(m5U54)methyltransferase (TrmA) critical for stability, covalent binding of tRNA and enzymatic activity. Nucleic Acids Res. 2007;35:3297–3305. doi: 10.1093/nar/gkm205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordlund M.E., Johansson J.O.M., von Pawel-Rammingen U., Byström A.S. Identification of the TRM2 gene encoding the tRNA(m5U54)methyltransferase of Saccharomyces cerevisiae. RNA. 2000;6:844–860. doi: 10.1017/s1355838200992422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell C.A., Minczuk M. TRMT2B is responsible for both tRNA and rRNA m5U-methylation in human mitochondria. RNA Biol. 2020;17:451–462. doi: 10.1080/15476286.2020.1712544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang Y.H., Nishimura S., Oishi H., Kelly V.P., Kuno A., Takahashi S. TRMT2A is a novel cell cycle regulator that suppresses cell proliferation. Biochem. Biophys. Res. Commun. 2019;508:410–415. doi: 10.1016/j.bbrc.2018.11.104. [DOI] [PubMed] [Google Scholar]
- 14.Guo G., Wang H., Shi X., Ye L., Yan K., Chen Z., Zhang H., Jin Z., Xue X. Disease Activity-Associated Alteration of mRNA m5 C Methylation in CD4+ T Cells of Systemic Lupus Erythematosus. Front. Cell Dev. Biol. 2020;8:430. doi: 10.3389/fcell.2020.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y., Pang C., Li X., Hu Z., Lv Z., Zheng B., Chen P. Identification of tRNA nucleoside modification genes critical for stress response and development in rice and Arabidopsis. BMC Plant Biol. 2017;17:261. doi: 10.1186/s12870-017-1206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter J.M., Emmett W., Mozos I.R., Kotter A., Helm M., Ule J., Hussain S. FICC-Seq: a method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res. 2019;47:e113. doi: 10.1093/nar/gkz658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen K., Wei Z., Zhang Q., Wu X., Rong R., Lu Z., Su J., de Magalhães J.P., Rigden D.J., Meng J. WHISTLE: a high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019;47:e41. doi: 10.1093/nar/gkz074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou Q., Xing P., Wei L., Liu B. Gene2vec: gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. RNA. 2019;25:205–218. doi: 10.1261/rna.069112.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W., Feng P., Ding H., Lin H., Chou K.C. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Qiang X., Chen H., Ye X., Su R., Wei L. M6AMRFS: Robust prediction of n6-methyladenosine sites with sequence-based features in multiple species. Front. Genet. 2018;9:495. doi: 10.3389/fgene.2018.00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J., Huang Y., Yang X., Zhou Y., Zhou Y. RNAm5Cfinder: A Web-server for Predicting RNA 5-methylcytosine (m5C) Sites Based on Random Forest. Sci. Rep. 2018;8:17299. doi: 10.1038/s41598-018-35502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lv H., Zhang Z.M., Li S.H., Tan J.X., Chen W., Lin H. Evaluation of different computational methods on 5-methylcytosine sites identification. Brief. Bioinform. 2020;21:982–995. doi: 10.1093/bib/bbz048. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M., Xu Y., Li L., Liu Z., Yang X., Yu D.J. Accurate RNA 5-methylcytosine site prediction based on heuristic physical-chemical properties reduction and classifier ensemble. Anal. Biochem. 2018;550:41–48. doi: 10.1016/j.ab.2018.03.027. [DOI] [PubMed] [Google Scholar]
- 25.Chen W., Tang H., Ye J., Lin H., Chou K.C. iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids. 2016;5:e332. doi: 10.1038/mtna.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He J., Fang T., Zhang Z., Huang B., Zhu X., Xiong Y. PseUI: Pseudouridine sites identification based on RNA sequence information. BMC Bioinformatics. 2018;19:306. doi: 10.1186/s12859-018-2321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y.H., Zhang G., Cui Q. PPUS: a web server to predict PUS-specific pseudouridine sites. Bioinformatics. 2015;31:3362–3364. doi: 10.1093/bioinformatics/btv366. [DOI] [PubMed] [Google Scholar]
- 28.Song B., Tang Y., Wei Z., Liu G., Su J., Meng J., Chen K. PIANO: A Web Server for Pseudouridine-Site (Ψ) Identification and Functional Annotation. Front. Genet. 2020;11:88. doi: 10.3389/fgene.2020.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu K., Chen W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics. 2020;36:3336–3342. doi: 10.1093/bioinformatics/btaa155. [DOI] [PubMed] [Google Scholar]
- 30.Song B., Tang Y., Chen K., Wei Z., Rong R., Lu Z., Su J., de Magalhães J.P., Rigden D.J., Meng J. m7GHub: deciphering the location, regulation and pathogenesis of internal mRNA N7-methylguanosine (m7G) sites in human. Bioinformatics. 2020;36:3528–3536. doi: 10.1093/bioinformatics/btaa178. [DOI] [PubMed] [Google Scholar]
- 31.Bailey T.L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng P., Ding H., Yang H., Chen W., Lin H., Chou K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids. 2017;7:155–163. doi: 10.1016/j.omtn.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng P., Ding H., Chen W., Lin H. Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Mol. Biosyst. 2016;12:3307–3311. doi: 10.1039/c6mb00471g. [DOI] [PubMed] [Google Scholar]
- 34.Bari A.T.M.G., Reaz M.R., Choi H.J.J.B. DNA Encoding for Splice Site Prediction in Large DNA Sequence. In: Hong B., Meng X., Chen L., Winiwarter W., Song W., editors. Database Systems for Advanced Applications. Lecture Notes in Computer Science. Springer; 2013. pp. 46–58. [Google Scholar]
- 35.Yang H., Lv H., Ding H., Chen W., Lin H. IRNA-2OM: A sequence-based predictor for identifying 2′-O-Methylation Sites in Homo sapiens. J. Comput. Biol. 2018;25:1266–1277. doi: 10.1089/cmb.2018.0004. [DOI] [PubMed] [Google Scholar]
- 36.Chen W., Feng P., Tang H., Ding H., Lin H. RAMPred: Identifying the N1-methyladenosine sites in eukaryotic transcriptomes. Sci. Rep. 2016;6:31080. doi: 10.1038/srep31080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen W., Tang H., Lin H. MethyRNA: a web server for identification of N6-methyladenosine sites. J. Biomol. Struct. Dyn. 2017;35:683–687. doi: 10.1080/07391102.2016.1157761. [DOI] [PubMed] [Google Scholar]
- 38.Sing T., Sander O., Beerenwinkel N., Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 39.Baron G. 2016. Comparison of Cross-Validation and Test Sets Approaches to Evaluation of Classifiers in Authorship Attribution Domain. ISCIS 2016; pp. 81–89. [Google Scholar]
- 40.Liu H., Yue D., Chen Y., Gao S.J., Huang Y. Improving performance of mammalian microRNA target prediction. BMC Bioinformatics. 2010;11:476. doi: 10.1186/1471-2105-11-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu C.-S., Lin C.-J., Hwang J.-K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004;13:1402–1406. doi: 10.1110/ps.03479604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y., He N., Chen Y., Chen Z., Li L. BERMP: a cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018;14:1669–1677. doi: 10.7150/ijbs.27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.James B., Yoshua B. Random Search for Hyper-Parameter Optimization. J. Mach. Learn. Res. 2012;13:281–305. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.