

Abstract
Eleven new acylphloroglucinols, including six new formylated phloroglucinol-monoterpene meroterpenoids, eucalyprobusals A–F (1–6), one monomeric acylphloroglucinol, eucalyprobusone B (7), and four dimeric acylphloroglucinols, eucalyprobusones C–F (8–11) were purified from the fruits of Eucalyptus robusta. The establishment of the structures of 1–11 was achieved by a combination of NMR and HRESIMS data analyses, electron circular dichroism (ECD), and single-crystal X-ray diffraction. Compounds 6, 8, and an inseparable mixture of 10 and 11 were found to be potent AChE inhibitors with IC50 values of 3.22 ± 0.36, 3.82 ± 0.22, and 2.55 ± 0.28 μM, respectively. Possible interaction sites of 6, 8, 10, and 11 with AChE were investigated by means of molecular docking studies, and the results revealed that AChE residues Asn87, Ser125, Thr83, Tyr133, Tyr124, Tyr337, and Tyr341 played crucial roles in the observed activity of the aforementioned compounds.
Keywords: Eucalyptus robusta, Acylphloroglucinols, Acetylcholinesterase inhibitory effect, Molecular docking
1. Introduction
Plants of Eucalyptus genus (Myrtaceae) are a prolific resource of structurally intriguing phloroglucinol derivatives, especially formylated phloroglucinol meroterpenoids (FPMs) [1–4]. These Eucalyptus secondary metabolites not only possess multifarious bioactive properties, including protein tyrosine phosphatase 1B inhibitory [2], immunosuppressive [3], antimicrobial [4,5], antiviral [6], anticancer [7–9], AChE inhibitory [10], and anti-leishmanial [11] effects, but also have attracted significant attention from the synthetic organic chemistry community [12–18]. Eucalyptus robusta, a tall arbor indigenous to Australia, is widely cultivated in south China. Its leaves have been traditionally used as a Chinese folk medicine to treat dysentery, malaria, and bacterial diseases [19], whereas its fruits are usually used for the main treatment of malaria.
Alzheimer’s disease (AD), a neurodegenerative disorder associated with memory and other cognitive functions, has been commonly known as one of the most burdensome threats to increasingly elderly people [20,21]. Currently, the causative factors of AD are not fully understood, pathophysiological brain hallmarks mainly include low levels of acetylcholine (ACh), amyloid-β (Aβ) deposits, and neurofibrillary tangles. Despite decades of studies for the basic biology of AD and significant pharmaceutical efforts to develop viable therapies, no effective therapy is available to totally cure AD or to significantly inhibit the progression of AD symptoms. Pharmacologically, three marketed acetylcholinesterase inhibitors (AChEIs) that are approved by U.S. FDA, named donepezil, rivas-tigmine, and galantamine [22], are only relevant medicines for the treatment of ameliorating the symptoms of AD patients. All these AChEIs acting on central nervous system (CNS) cholinergic pathways are now approved for mild to severe dementia, although they are widely used for patients in earlier predementia stages associated with significant progressive memory impairment. Therefore, it would be of great significance to hunt for potent AChEIs from medicinal plant resources. The PE (petroleum ether)–EtOAc (ethyl acetate) extract of E. robusta fruits displayed an AChE inhibitory rate of 68% at the concentration of 500 μg/mL, which prompted further phytochemical investigation with the aim at clarifying its bioactive constituents. As a result, six new FPMs, eucalyprobusals A–F (1–6), one monomeric acylphloroglucinol, eucalyprobusone B (7), and four acylphloroglucinol dimers, eucalyprobusones C–F (8–11) were isolated and structurally characterized (Fig. 1). AChE inhibitory assays of 1–11 were performed, and the possible action sites of 6, 8, 10, and 11 with AChE were also accomplished via molecular docking methods.
Fig. 1.
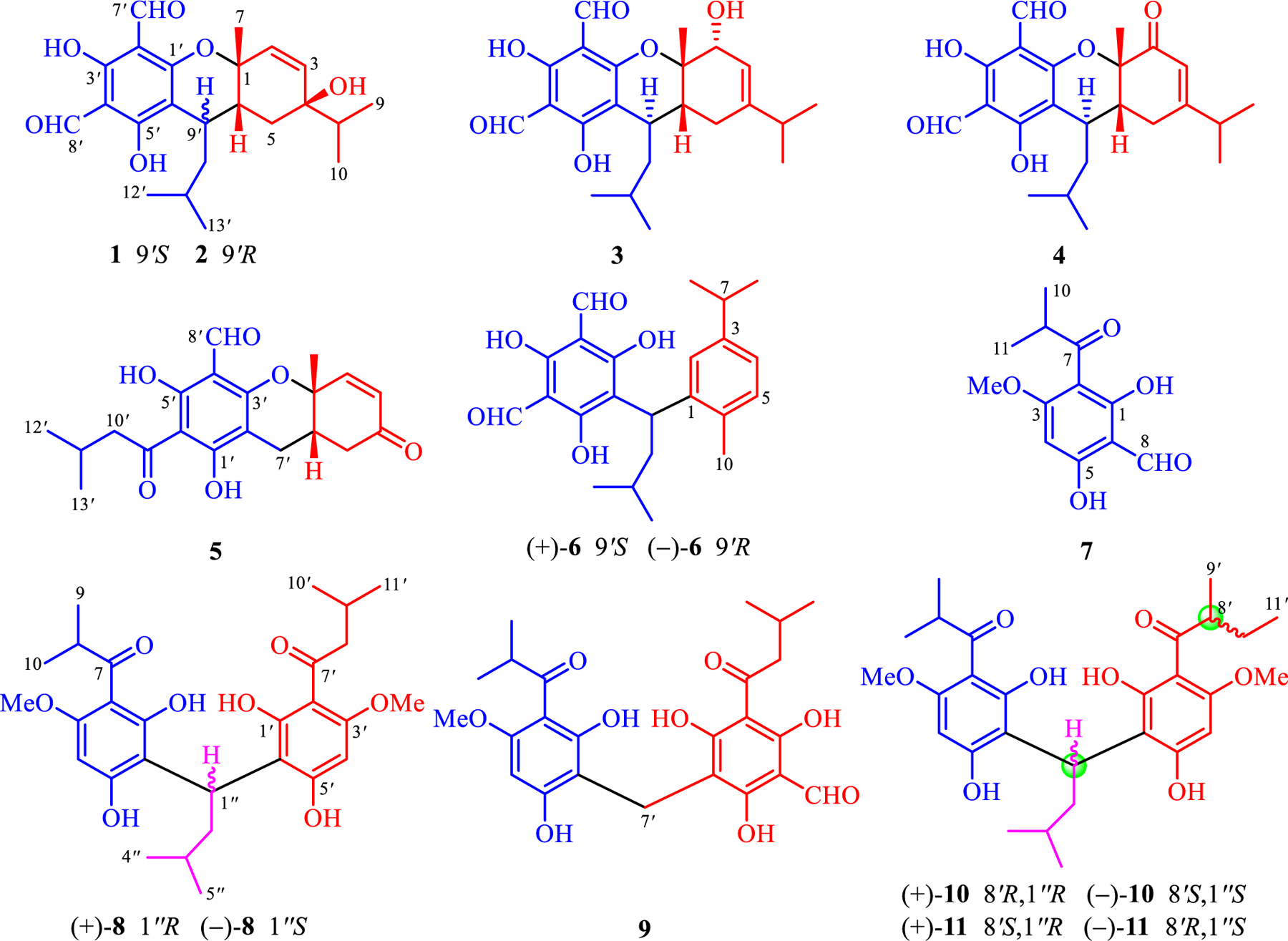
Structures of 1–11 isolated from E. robusta.
2. Experimental
2.1. General experimental procedures
Optical rotation and UV spectra were measured on a AUTOPOL VI automatic and a SHIMADZU UV-2700 UV–VIS instruments, respectively. CD data were recorded on an Applied Photophysics spectro-polarimeter. A Bruker FT-IR Tensor-27 infrared spectrophotometer was utilized for measuring the IR spectra (KBr disks). NMR spectra were collected on Bruker Ascend 500, 600, and 800 MHz instruments with various solvent (including CDCl3, methanol-d4, acetone-d6, and pyridine-d5) signals as referenced internal standards. An Agilent 1290 UPLC/6540 Q-TOF system was used for HRESIMS data. Crystallographic data of 1 and a mixture of 10 and 11 were obtained using a Bruker D8 QUEST diffractometer (λ = 1.54178 Å) with Cu Kα radiation. Silica gel, Sephadex LH-20, and MCI were applied as the packing materials for CC (column chromatography). Chiral analysis was performed on an Agilent 1100 instrument with a CHIRALPAK IC column (4.6 × 250 mm, 5 μm). A Hanbon Newstyle preparative HPLC instrument equipped with a SunFire Prep C18 column (10 × 250 mm, 5 μm) was used to purify compounds.
2.2. Plant material
The E. robusta fruits authenticated by Dr. Rong Li (Kunming Institute of Botany, CAS) were collected from Kunming, Yunnan province, People’s Republic of China. A voucher specimen (HY0032) is deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
2.3. Extraction and isolation
The dried E. robusta fruits (5.0 kg) were powdered and extracted with PE–EtOAc (1:1 v/v, 15 L × 3, each 24 h) to afford an inquinate residue. This crude extract (230.0 g) was subjected to silica gel CC eluting with PE–EtOAc (100:1→ 1:1, v/v) to afford six fractions (Fr. A–Fr. F) as monitored based on TLC by spraying with 10% FeCl3-EtOH. Fr. D (20.5 g) was separated on an RP-18 column (MeOH–H2O, 60:40→100:1, v/v, 1‰ FA in H2O) and was further purified with a Sephadex LH-20 column (MeOH) and semipreparative HPLC (MeCN–H2O, 80:20 v/v, 1‰ FA in H2O) to yield 7 (22.2 mg), 8 (10.2 mg), 9 (6.8 mg), and a mixture of 10 and 11 (31.1 mg). Fr. E (10.5 g) was fractionated by an RP-18 column (MeOH–H2O, 50:50→100:1, v/v, 1‰ FA in H2O) and further purified via semipreparative HPLC (MeOH–H2O, 98:2 v/v, 1‰ FA in H2O) to give 4 (2.4 mg), 5 (3.3 mg), and 6 (54.3 mg). Likewise, Fr. F (16.0 g) was separated on an RP-18 column (MeOH–H2O, 50:50→100:1, v/v, 1‰ FA in H2O) and followed by semipreparative HPLC (MeCN–H2O, 90:10 v/v, 1‰ FA in H2O) to afford 1 (12.1 mg), 2 (8.8 mg), and 3 (1.4 mg).
2.3.1. Eucalyprobusal A (1)
Yellowish crystals (methanol-acetone, 1:1 v/v); + 91.8 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 207 (4.26), 281 (4.47), 368 (3.57) nm; IR (KBr) νmax 3440, 2954, 1641, 1180, 781 cm−1; 1H (500 MHz, CDCl3) and 13C (125 MHz, CDCl3) NMR spectral data, see Table 1; (+)-HRESIMS m/z 425.1939 [M+Na]+ (calcd for C23H30O6Na, 425.1935).
Table 1.
13C and 1H NMR data for eucalyprobusals A–E (1–5) in CDCl3
| Eucalyprobusal A (1)a | Eucalyprobusal B (2)a | Eucalyprobusal C (3)b | Eucalyprobusal D (4)a | Eucalyprobusal E (5)a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) |
| 1 | 76.5 | 75.9 | 72.7 | 82.4 | 76.3 | |||||
| 2 | 132.6 | 5.88 d (9.8) | 133.4 | 5.79 d (10.0) | 80.2 | 4.49 d (5.3) | 195.5 | 150.0 | 6.81 d (10.1) | |
| 3 | 135.9 | 5.83 dd (9.8, 1.0) | 135.3 | 5.83 d (10.0) | 111.6 | 5.10 d (5.3) | 121.9 | 5.85 s | 130.6 | 6.05 d (10.1) |
| 4 | 72.8 | 72.6 | 155.2 | 168.7 | 196.9 | |||||
| 5α | 27.0 | 1.54 dd (13.4, 2.4) | 35.4 | 1.51 brd (13.2) | 32.4 | 2.62 dd (18.8, 6.3) | 32.9 | 2.73 ddd (19.4, 5.6) | 40.4 | 2.64 dd (20.1, 7.5) |
| 5β | 1.38t (13.4) | 1.41 dd (13.2, 2.5) | 1.86 brd (18.8) | 2.46 brd (3.7) | 2.52t (7.5) | |||||
| 6 | 33.1 | 2.26 ddd (13.4, 6.2, 3.0) | 33.5 | 2.36 brdd (13.2, 3.2) | 40.2 | 2.23 brt (4.8) | 44.0 | 2.49 m | 35.0 | 2.54 m |
| 7 | 23.7 | 1.39 s | 27.2 | 1.55 s | 26.4 | 1.65 s | 22.5 | 1.63 s | 25.4 | 1.67 s |
| 8 | 37.8 | 1.76 sept (6.8) | 37.6 | 1.71 sept (6.8) | 34.5 | 2.06 sept. (6.8) | 35.6 | 2.43 sept. (6.8) | ||
| 9 | 16.2 | 0.93 d (6.8) | 16.2 | 0.92 d (6.8) | 21.1 | 0.74 d (6.8) | 20.5 | 1.13 d (6.8) | ||
| 10 | 17.3 | 0.87 d (6.8) | 17.3 | 0.83 d (6.8) | 20.9 | 0.72 d (6.8) | 20.7 | 1.12 d (6.8) | ||
| 1′ | 163.5 | 161.6 | 165.0 | 163.9 | 171.7 | |||||
| 2′ | 104.2 | 104.0 | 109.1 | 104.6 | 99.3 | |||||
| 3′ | 168.0 | 168.1 | 167.3 | 167.6 | 160.3 | |||||
| 4′ | 104.2 | 104.1 | 105.6 | 104.8 | 103.7 | |||||
| 5′ | 171.2 | 170.1 | 169.1 | 169.1 | 168.1 | |||||
| 6′ | 103.1 | 103.8 | 117.4 | 106.2 | 103.6 | |||||
| 7′a | 192.3 | 9.96 s | 192.3 | 9.99 s | 193.7 | 10.10 s | 192.6 | 10.14 s | 21.5 | 2.41 dd (16.9, 6.0) |
| 7′b | 2.81 dd (16.9, 6.0) | |||||||||
| 8′ | 191.8 | 10.14 s | 191.7 | 10.16 s | 192.3 | 10.21 s | 192.2 | 10.15 s | 191.4 | 10.04 s |
| 9′ | 28.4 | 3.21 ddd (10.9, 6.2, 3.9) | 34.0 | 2.64 brdd (10.2, 3.2) | 35.9 | 3.52 dt (8.2, 4.9) | 31.2 | 2.66 ddd (9.8, 6.3, 3.5) | 206.6 | |
| 10′a | 35.7 | 2.52 ddd (14.5, 10.9, 3.9) | 43.3 | 1.80 ddd (13.1, 10.2, 3.2) | 46.7 | 1.42 brdd (13.5, 6.8) | 43.6 | 1.61 2H m | 52.7 | 2.98 2H d (6.7) |
| 10′b | 1.26 ddd (14.2, 10.9, 3.9) | 1.58 ddd (14.2, 10.2, 3.2) | 1.27 brdd (13.5, 7.7) | |||||||
| 11′ | 24.5 | 1.75 m | 26.6 | 1.87 m | 25.6 | 1.47 m | 25.6 | 1.67 m | 27.5 | 1.92 m |
| 12′ | 20.9 | 1.00 d (6.5) | 21.6 | 1.01 d (6.5) | 22.3 | 0.93 d (6.3) | 22.2 | 0.97 d (6.4) | 22.7 | 0.98 d (6.7) |
| 13′ | 24.3 | 0.96 d (6.5) | 23.6 | 0.96 d (6.5) | 22.8 | 0.85 d (6.3) | 24.0 | 0.85 d (6.4) | 22.7 | 0.98 d (6.7) |
| OH-3′ | 13.43 s | 13.44 s | 13.29 s | 13.35 s | OH-1′ | 15.48 s | ||||
| OH-5′ | 13.82 s | 13.48 s | 13.31 s | 13.38 s | OH-5′ | 14.45 s | ||||
Data were recorded at 500 MHz.
Data were recorded at 800 MHz.
2.3.2. Eucalyprobusal B (2)
Yellowish amorphous powder; −31.4 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 206 (4.22), 281.5 (4.38), 373 (3.55) nm; ECD (MeOH, Δε) 204 (+3.53), 213 (+1.20), 226 (+3.73), 246 (+0.42), 274 (+10.22), 306 (−6.48) nm; IR (KBr) νmax 3441, 2952, 1641, 1179, 780 cm−1; 1H (500 MHz, CDCl3) and 13C (125 MHz, CDCl3) NMR spectral data, see Table 1; (+)-HRESIMS m/z 425.1942 [M+Na]+ (calcd for C23H30O6Na, 425.1935).
2.3.3. Eucalyprobusal C (3)
Yellowish amorphous powder; −254.3 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 206 (4.28), 273 (4.38), 343 (3.71), 380 (3.53) nm; ECD (MeOH, Δε) 221 + 27.39), 267 (−1.44), 290 (+2.04), 343 (−4.56) nm; IR (KBr) νmax 3439, 2943, 1632, 1430, 1057 cm−1; 1H (800 MHz, CDCl3) and 13C (200 MHz, CDCl3) NMR spectral data, see Table 1; (−)-HRESIMS m/z 401.1978 [M−H]− (calcd for C23H29O6, 401.1970).
2.3.4. Eucalyprobusal D (4)
Yellowish amorphous powder; −307.3 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 206 (3.15), 236 (3.09), 279 (3.23) nm; ECD (MeOH, Δε) 206 (−5.91), 242 (+9.73), 269 (−20.5), 316 (−0.59), 343 (−3.69) nm; IR (KBr) νmax 3436, 2937, 1721, 1629, 1468, 1024 cm−1; 1H (500 MHz, CDCl3) and 13C (125 MHz, CDCl3) NMR spectral data, see Table 1; (+)-HRESIMS m/z 423.1772 [M+Na]+ (calcd for C23H28O6Na, 423.1778).
2.3.5. Eucalyprobusal E (5)
Yellowish amorphous powder; −53.7 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 206 (4.00), 276 (4.09), 335 (3.39), 366 (3.20) nm; ECD (MeOH, Δε) 215 (+2.07), 239 (−0.61), 263 (+1.27), 292 (−0.58), 354 (+0.03) nm; 1H (500 MHz, methanol-d4) and 13C (125 MHz, methanol-d4) NMR spectral data, see Table 1; (+)-HRESIMS m/z 397.1051 [M+K]+ (calcd for C20H22O6K, 397.1048).
2.3.6. (±)-Eucalyprobusal F (6)
Yellowish gum; + 86.8 (c 0.10, MeOH) for (+)-6; −86.2 (c 0.10, MeOH) for (−)-6; UV (MeOH) λmax (log ε) 202 (4.38), 213 (4.37), 291 (4.27), 388 (3.76) nm; ECD (MeOH, Δε) 204 (+14.46), 227 (+1.06), 246 (+21.48), 264 (+1.75), 274 (+3.08), 322 (−0.21) nm for (+)-6; ECD (MeOH, Δε) 204 (−13.89), 227 (−1.03), 246 (−20.39), 264 (−1.68), 274 (−2.97), 322 (+0.20) for (−)-6; 1H (methanol-d4, 500 MHz) NMR δ 0.88 (3H, d, J = 6.6 Hz, H3-13′), 0.94 (3H, d, J = 6.6 Hz, H3-12′), 1.17 × 2 (6H, d, J = 7.0 Hz, H3-8/H3-9), 1.49 (1H, m, H-11′), 1.80 (1H, ddd, J = 14.5, 8.0, 6.6 Hz, H-10′b), 2.19 (1H, ddd, J = 14.5, 8.4, 5.2 Hz, H-10′a), 2.22 (3H, s, H3-10), 2.78 (1H, sept., J = 7.0 Hz, H-7), 4.65 (1H, dd, J = 9.6, 6.6 Hz, H-9′), 6.85 (1H, dd, J = 7.8, 1.5 Hz, H-4), 6.91 (1H, d, J = 7.8 Hz, H-5), 7.45 (1H, d, J = 1.5 Hz, H-2), 10.05 (2H, s, H-7′/H-8′); 13C (methanol-d4,125 MHz) NMR δ 19.5 (C-10), 22.9 (C-12′), 23.8 (C-13′), 24.6 (C-8), 24.7 (C-9), 27.4 (C-11′), 35.0 (C-9′), 35.2 (C-7), 42.9 (C-10′), 106.3 × 2 (C-2′/C-4′), 111.3 (C-6′), 124.6 (C-4), 127.7 (C-2), 131.0 (C-5), 134.7 (C-6), 142.9 (C-1), 146.8 (C-3), 169.1 (C-3′), 169.9 × 2 (C-1′/C-5′), 193.1 × 2 (C-7′/C-8′); (−)-HRESIMS m/z 383.1872 [M−H]− (calcd for C23H27O5, 383.1864).
2.3.7. Eucalyprobusone B (7)
Yellowish gum; UV (MeOH) λmax (log ε) 205 (3.87), 272 (4.22), 321 (3.59) nm; 1H NMR (CDCl3, 500 MHz) δ 1.16 × 2 (6H, d, J = 6.8 Hz, H 3–10/H3-11), 3.68 (1H, sept., J = 6.8 Hz, H-9), 3.95 (3H, s, OMe-3), 5.91 (1H, s, H-4), 10.20 (1H, s, H-8), 12.99 (1H, s, OH-5), 15.50 (1H, s, OH-1); 13C NMR (CDCl3, 125 MHz) δ 19.1 × 2 (C-10/C-11), 39.6 (C-9), 56.2 (OMe-3), 90.8 (C-4), 103.4 (C-2), 105.3 (C-6), 168.1 (C-3), 169.9 (C-5), 171.8 (C-1), 192.7 (C-8), 210.5 (C-7); (+)-HRESIMS m/z 261.0735 [M+Na]+ (calcd for C12H14O5Na, 261.0733).
2.3.8. ( ± )-Eucalyprobusone C (8)
Yellowish gum; + 72.5 (c 0.10, MeOH) for (+)-8; −72.4 (c 0.10, MeOH) for (−)-8; UV (MeOH) λmax (log ε) 207 (4.48), 302 (4.43) nm; ECD (MeOH, Δε) 213 (−3.46), 234 (+13.65), 264 (−0.15), 308 (+4.11) nm for (+)-8; ECD (MeOH, Δε) 213 (+3.40), 235 (−13.39), 264 (+0.15), 308 (−4.04) for (−)-8; 1H (600 MHz, acetone-d6) and 13C (150 MHz, acetone-d6) NMR spectral data, see Table 2; (+)-HRESIMS m/z 503.2645 [M+H]+ (calcd for C28H39O8, 503.2639).
Table 2.
1H and 13C NMR Data for Eucalyprobusones C (8) and D (9).
| Eucalyprobusone C (8)a | Eucalyprobusone D (9)b | |||
|---|---|---|---|---|
| No. | δC | δH (J in Hz) | δC | δH (J in Hz) |
| 1 | 165.8 | 163.6 | ||
| 2 | 104.6 | 104.0 | ||
| 3 | 162.5 | 162.0 | ||
| 4 | 93.6 | 6.10 s | 92.8 | 6.07 s |
| 5 | 164.4 | 162.8 | ||
| 6 | 110.3 | 106.0 | ||
| 7 | 211.3 | 211.1 | ||
| 8 | 39.9 | 3.82 sept. (6.7) | 39.3 | 3.79 sept. (6.8) |
| 9 | 19.6 | 1.13 d (6.7) | 19.2 | 1.17 d (6.8) |
| 10 | 19.7 | 1.12 d (6.7) | 19.2 | 1.17 d (6.8) |
| 1′ | 165.8 | 169.9 | ||
| 2′ | 104.6 | 105.6 | ||
| 3′ | 162.5 | 165.3 | ||
| 4′ | 93.6 | 6.10 s | 104.8 | |
| 5′ | 164.4 | 168.4 | ||
| 6′ | 110.3 | 103.5 | ||
| 7′ | 211.6 | 15.1 | 3.74 2H s | |
| 8′ | 27.8 | a 1.79 m, b 1.35 m | 193.2 | 10.15 s |
| 9′ | 46.7 | 3.69 sext. (6.7) | 207.2 | |
| 10′ | 16.8 | 1.11 d (6.7) | 52.2 | 3.00 2H d (6.7) |
| 11′ | 16.9 | 1.11 d (6.7) | 25.2 | 2.44 brsept. (6.7) |
| 12′ | 22.7 | 0.99 d (6.7) | ||
| 13′ | 22.7 | 0.99 d (6.7) | ||
| 1″ | 28.2 | 5.01 t (8.1) | ||
| 2″ | 40.8 | 2.08 2H m | ||
| 3″ | 27.4 | 1.44 brsept. (6.5) | ||
| 4″ | 22.8 | 0.88 d (6.5) | ||
| 5″ | 22.8 | 0.88 d (6.5) | ||
| OH-1 | 9.50 s | 16.76 s | ||
| OH-1′ | 9.50 s | 17.23 s | ||
| OH-3′ | 10.31 s | |||
| OH-5 | 8.93 s | |||
| OH-5′ | 14.48 s | |||
| OMe-3 | 56.2 | 3.90 s | 55.8 | 3.86 s |
| OMe-3′ | 56.2 | 3.89 s | ||
Data were recorded at 600 MHz in acetone-d6.
Data were recorded at 500 MHz in CDCl3.
2.3.9. Eucalyprobusone D (9)
Yellowish gum; UV (MeOH) λmax (log ε) 234 (4.42), 301 (3.44) nm; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR spectral data, see Table 2; (−)-HRESIMS m/z 499.1345 [M+K]+ (calcd for C24H28O9K, 499.1365).
2.3.10. (±)-Eucalyprobusones E (10) and F (11)
Colorless crystals (methanol-acetone, 1:1 v/v); [α] −0.67 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 207 (4.43), 302 (4.38) nm; 1H (600 MHz, pyridine-d5) and 13C (150 MHz, pyridine-d5) NMR spectral data, see Table 3; (+)-HRESIMS m/z 525.2467 [M+Na]+ (calcd for C28H38O8Na, 525.2459).
Table 3.
13C (150 MHz) and 1H (600 MHz) NMR data for eucalyprobusones E (10) and F (11) in pyridine-d5.
| Eucalyprobusone E (10) | Eucalyprobusone F (11) | |||
|---|---|---|---|---|
| No. | δC | δH (J in Hz) | δC | δH (J in Hz) |
| 1 | 166.6 | 165.6 | ||
| 2 | 104.6 | 105.1 | ||
| 3 | 161.4 | 161.5 | ||
| 4 | 92.6 | 6.27 s | 92.6 | 6.28 s |
| 5 | 164.4 | 164.5 | ||
| 6 | 111.1 | 111.1 | ||
| 7 | 210.0 | 210.2 | ||
| 8 | 39.5 | 3.75 sept. (6.7) | 39.5 | 3.75 sept. (6.7) |
| 9 | 19.4 | 1.13 d (6.7) | 19.4 | 1.13 d (6.7) |
| 10 | 19.5 | 1.10 d (6.7) | 19.5 | 1.10 d (6.7) |
| 1′ | 166.6 | 165.6 | ||
| 2′ | 104.6 | 105.1 | ||
| 3′ | 161.4 | 161.5 | ||
| 4′ | 92.6 | 6.28 s | 92.6 | 6.28 s |
| 5′ | 164.4 | 164.5 | ||
| 6′ | 111.1 | 111.1 | ||
| 7′ | 210.2 | 210.0 | ||
| 8′ | 46.2 | 3.65 m | 46.2 | 3.65 m |
| 9′ | 16.6 | 1.10 d (6.7) | 16.7 | 1.14 d (6.7) |
| 10′a | 27.3 | 1.80 m | 27.4 | 1.82 m |
| 10′b | 1.32 m | 1.34 m | ||
| 11′ | 12.0 | 0.80 t (7.4) | 12.1 | 0.84 t (7.4) |
| 1″ | 27.9 | 6.03 t (8.3) | 27.9 | 6.03 t (8.3) |
| 2″ | 41.8 | 2.47 2H t (7.5) | 41.8 | 2.47 2H t (7.5) |
| 3″ | 26.9 | 1.92, brsept. (6.6) | 26.9 | 1.92 brsept. (6.6) |
| 4″ | 22.8 | 0.90 d (6.6) | 23.0 | 1.09 d (6.6) |
| 5″ | 23.0 | 1.08 d (6.6) | 23.0 | 1.09 d (6.5) |
| OMe-3/3′ | 55.3 | 3.64 s | 55.3 | 3.65 s |
2.3.11. Crystallographic data for eucalyprobusal A (1)
C 23H30O6, M = 402.47, a = 10.3730(6) Å, b = 13.0331(8) Å, c = 31.5028(18) Å, α = 90°, β = 90°, γ = 90°, V = 4258.9(4) Å3, T = 100.(2) K, wavelength 1.54178 Å, orthorhombic crystal system, space group P212121, Z = 8, absorption coefficient 0.735 mm−1, μ(Cu Kα) = 0.735 mm−1, F(000) = 1728, crystal size 0.260 × 0.200 × 0.140 mm3, θ range for data collection 3.67 – 72.32°, index ranges −12 ≤ h ≤ 12, −13 ≤ k ≤ 16, −38 ≤ l ≤ 38, 35,845 reflections collected, 8336 independent reflections (Rint = 0.0249), completeness to θ (72.32°) 99.4%, data/restraints/parameters 8336/0/553, largest diff. peak and hole 0.205 and −0.154 e.Å−3. The final R1 values were 0.0247 [I > 2σ(I)]. The final wR(F2) values were 0.0647 [I > 2σ(I)]. The final R1 values were 0.0250 (all data). The final wR(F2) values were 0.0649 (all data). The goodness of fit on F2 was 1.042. Flack parameter = –0.01(2). Crystallographic data for 1 is deposited at CCDC (Cambridge Crystallographic Data Center) with a number of CCDC 2003650.
2.3.12. Crystallographic data for (±)-eucalyprobusones E (10) and F (11)
C 28H38O8, M = 502.58, a = 11.2889(2) Å, b = 11.5007(2) Å, c = 11.7348(2) Å, α = 81.6690(10)°, β = 78.5010(10)°, γ = 63.8270(10)°, V = 1337.06(4) Å3, T = 100.(2) K, wavelength 1.54178 Å, triclinic crystal system, space group P-1, Z = 2, absorption coefficient 0.744 mm−1, μ(Cu Kα) = 0.744 mm−1, F(000) = 540, crystal size 0.630 × 0.480 × 0.270 mm3, θ range for data collection 4.29–72.38°, index ranges −13 ≤ h ≤ 13, −14 ≤ k ≤ 14, −14 ≤ l ≤ 14, 42,727 reflections measured, 5244 independent reflections (Rint = 0.0421) completeness to θ (72.38°) 99.3%, data/restraints/parameters 5244/255/425, largest diff. peak and hole 1.093 and –0.430 e.Å−3. The final R1 values were 0.0707 [I > 2σ(I)]. The final wR(F2) values were 0.1933 [I > 2σ(I)]. The final R1 values were 0.0712 (all data). The final wR(F2) values were 0.1936 (all data). The goodness of fit on F2 was 1.111. Crystallographic data for 10 and 11 is deposited at CCDC (Cambridge Crystallographic Data Center) with a number of CCDC 2003659.
2.4. ECD computational methods
The ECD calculations of 2–6 and 8 were carried out using Gaussian 16 [23]. Conformational analysis of 2–6 and 8 was carried out by CONFLEX 8B software (CONFLEX Corporation, Tokyo, Japan) using MMFF94s molecular force field with a search limit of 1.0 kcal/mol to yield six, six, three, four, two, and 10 conformers, respectively. These initial structures were optimized via the Density Functional Theory (DFT) at the B3LYP/6–31+G(d) level in gas phase. The optimized conformations were used for ECD calculations by the Time Dependent DFT (TDDFT) at the B3LYP/6–311++G (2d, p) level.
2.5. AChE inhibitory assay
AChE inhibitory effects of all the isolated phloroglucinols were carried out on the basis of the spectrophotometric method in 96-well microplates with slightly modification [24]. Each well was filled with human acetylcholinesterase (0.02 U/mL, Sigma-Aldrich Corp., USA), phosphate buffer (pH = 8.0), and tested phloroglucinols (100, 50.0, 30.0, 10.0, 3.0, 1.0, and 0.2 μM) in DMSO and then incubated for 20 min at 37 °C. These reactions were initiated by the addition of 40 μL of solution containing Ellman’s reagent (DTNB, 0.625 mM of 5,5′-di-thiobis-2-nitrobenzoic acid) and acetylthiocholine iodide (0.625 mM) for AChE inhibitory assays, respectively. The results of acetylthiocholine hydrolysis were monitored at 405 nm for 1.0 h (30 s interval readings). DMSO and galanthamine were selected as the negative and positive controls, respectively. The percentage inhibition was calculated as follows:
(E and S are the average absorption values for the enzyme activities treated without and with tested compounds, respectively).
2.5.1. Molecular modeling
Discovery Studio was used to carry out molecular docking studies using recently published methods [25].
3. Results and discussion
3.1. Structural elucidation
Dried and powdered fruits of E. robusta were extracted three times by PE–EtOAc at room temperature. The obtained extract was separated using silica gel chromatrography to give six fractions (Fr. A–Fr. F). Fractions D, E, and F were repeatedly chromatographed on silica gel, Sephadex LH-20, and RP-18 columns as well as using semipreparative HPLC to yield 11 new acylphloroglucinols (1–11). The structures of 1–11 were elucidated employing a combination of NMR and HRMS data analyses; the absolute configurations of 1–6 and 8 were established based on X-ray diffraction or ECD calculations. 1–6 are phloroglucinol–monoterpene conjugates, whereas 7 and 8–11 are mono- and dimeric-acylphloroglucinols (Fig. 1), respectively. Among them, 10 and 11 were found to be an inseparable mixture of two pairs of enantiomers.
Eucalyprobusal A (1), a yellowish crystal, had a molecular formula of C23H30O6 as determined by the observed sodium adduct ion at m/z 425.1939 [M+Na]+ (calcd for C23H30O6Na, 425.1935) in the HRESIMS spectrum. The IR spectrum showed absorptions at 3440 and 1641 cm−1 which indicated the existence of hydroxy and carbonyl functionalities, respectively. The 1H NMR spectral data (Table 1) disclosed resonances for four secondary methyls (δH 0.87, d, J = 6.8 Hz, H 3–10; 0.93, d, J = 6.8 Hz, H3-9; 0.96, d, J = 6.5 Hz, H3-13′; 1.00, d, J = 6.5 Hz, H3-12′), a tertiary methyl (δH 1.39, s, H3-7), two olefinic protons (δH 5.83, dd, J = 9.8, 1.0 Hz, H-3; 5.88, d, J = 9.8 Hz, H-2), two aldehyde protons (δH 9.96, s, H-7′; 10.14, s, H-8′), and two hydroxy protons (δH 13.43, s, OH-3′; 13.82, s, OH-5′). Besides the characteristic signals for a diformylated phloroglucinol (DFPG) scaffold (δC 103.1, C-6′; 104.2 × 2, C-2′/C-4′; 163.5, C-1′; 168.0, C-3′; 171.2, C-5′; 191.8, C-8′; 192.3, C-7′), 13C NMR spectral data (Table 1) showed 23 carbon resonances ascribed to five methyls (δC 16.2, C-9; 17.3, C-10; 20.9, C-12′; 23.7, C-7; 24.3, C-13′), two methylenes (δC 27.0, C-5; 35.7, C-10′), four methines (δC 24.5, C-11′; 28.4, C-9′; 33.1, C-6; 37.8, C-8), an endocyclic double bond (δC 132.6, C-2; 135.9, C-3), and two oxygen-bearing quaternary carbons (δC 72.8, C-4; 76.5, C-1). The aforementioned NMR signals of 1 closely resembled those of eucalyptin D [6] recently obtained from E. globulus fruits, the difference being the configuration of the C-4 hydroxy group. Combined with three spin systems (Fig. 2) as furnished by the 1H−1H COSY experiment, HMBC correlations from H3-7 (δH 1.39) to C-6 (δC 33.1)/C-1 (δC 76.5)/C-2 (δC 132.6), from H3-10 (δH 0.87)/H3-9 (δH 0.93)/H2-5 (δH 1.54, 1.38)/H-2 (δH 5.88) to C-4 (δC 72.8), from OH-3′ (δH 13.43) to C-2′ (δC 104.2)/C-4′ (δC 104.2)/C-3′ (δC 168.0), from OH-5′ (δH 13.82) to C-6′ (δC 103.1)/C-4′ (δC 104.2)/C-5′ (δC 171.2), from H-7′ (δH 9.96) to C-2′ (δC 104.2), from H-8′ (δH 10.14) to C-4′ (δC 104.2), and from H-9′ (δH 3.21) to C-6′ (δC 103.1)/C-1′ (δC 163.5) indicated that 1 was a phloroglucinol-monoterpene conjugate. Although the observed ROESY correlations of both H-6 (δH 2.26) and H-9′ (δH 3.21) with H3-7 (δH 1.39) (Fig. S1, Supporting Information) revealed that these protons occupied the same side of the molecule and were stochastically assigned as β-oriented, no ROESY evidence was used to establish the configuration of the C-4 hydroxy group. Fortunately, needlelike crystals of 1 were obtained from a mixed solution of acetone and methanol (1:1, v/v). Single-crystal X-ray diffraction analysis with Cu Kα radiation (Fig. 3) of 1 not only resolved the configuration of C-4 hydroxyl, but also unequivocally established its absolute configuration (1R,4R,6R,9′S).
Fig. 2.
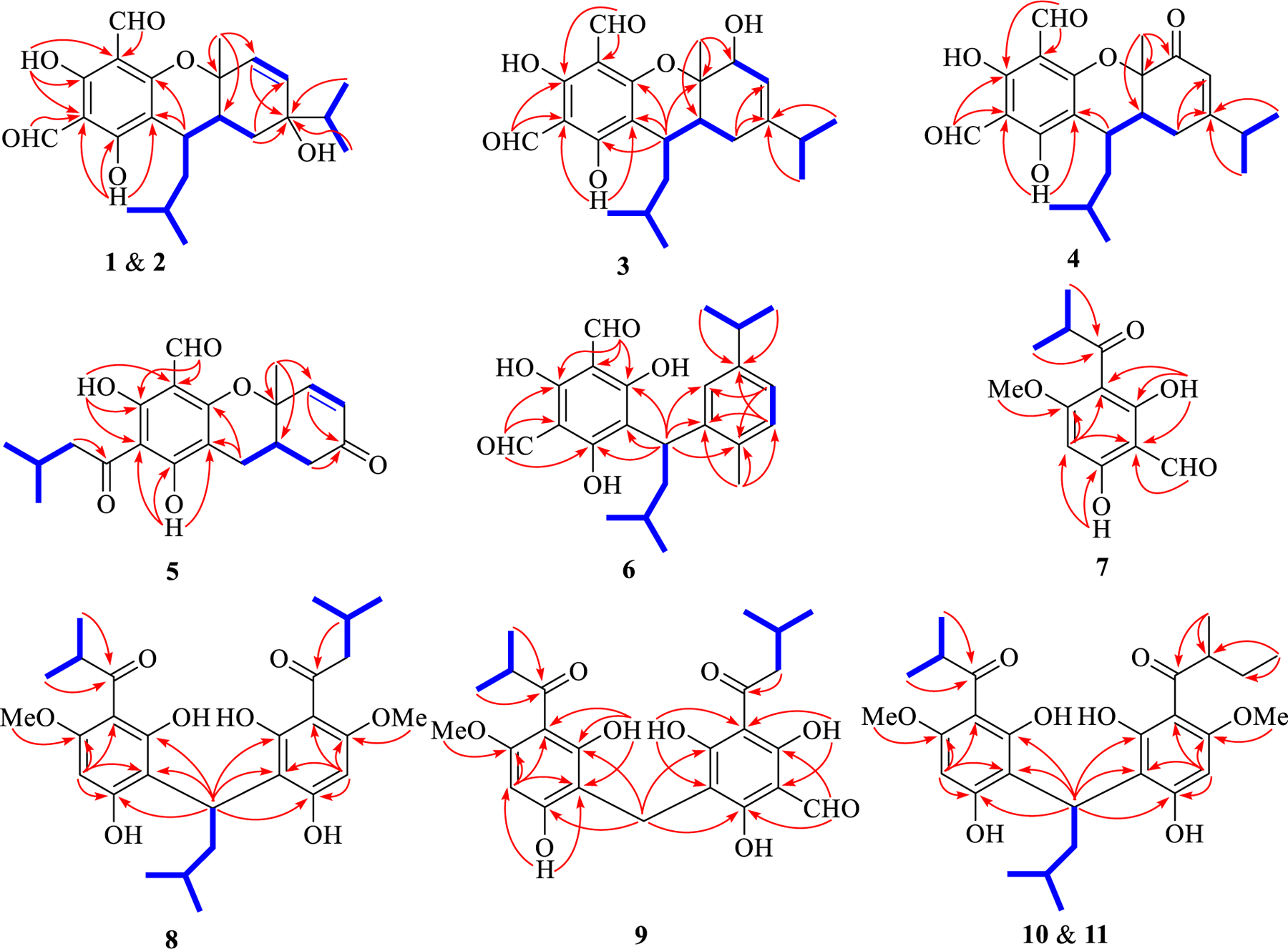
Selected 1H–1H COSY (blue bold line) and HMBC (red arrow) correlations of 1–11.
Fig. 3.
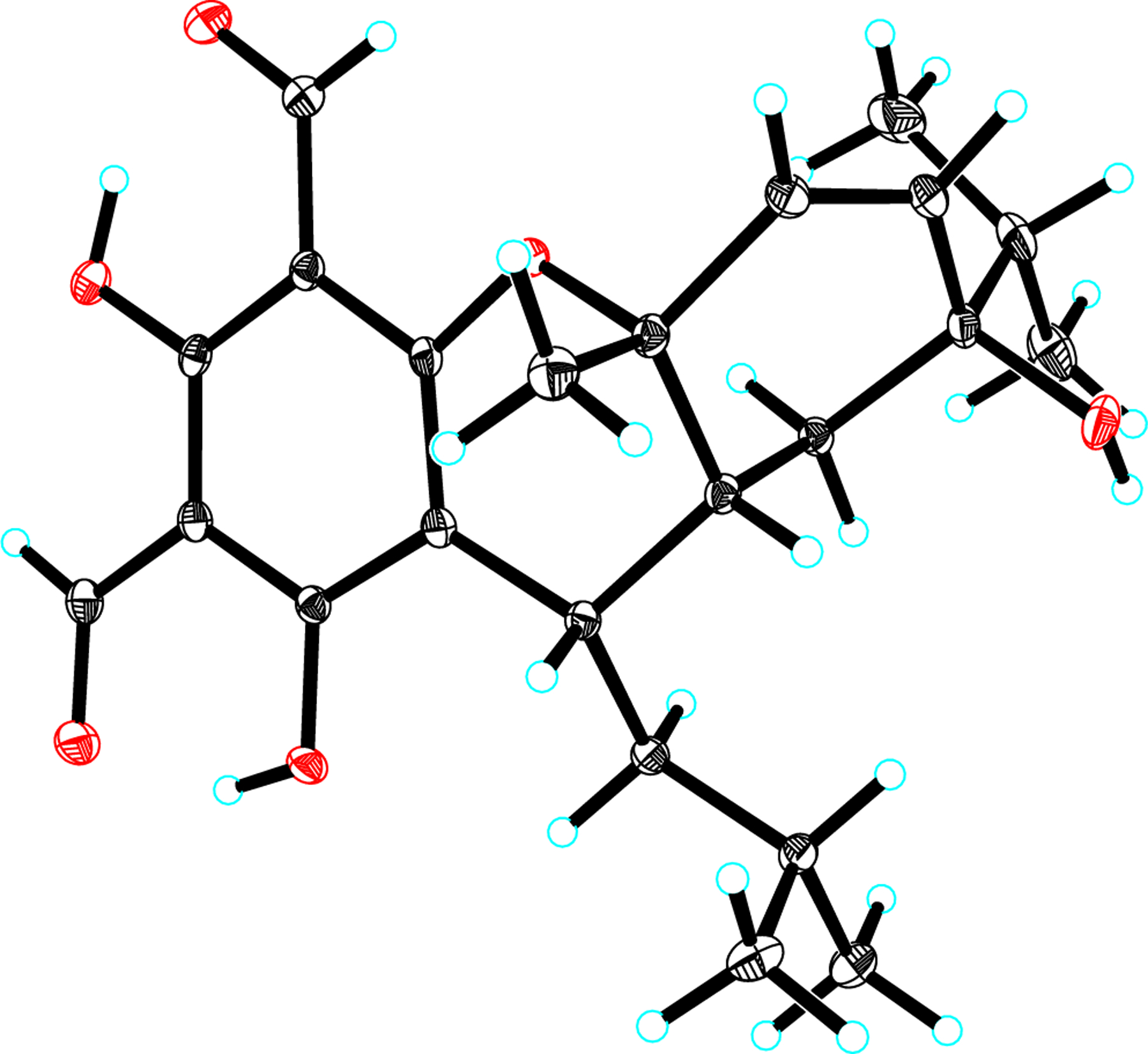
ORTEP drawing of 1.
Eucalyprobusal B (2) was assigned to have the same molecular formula (C23H30O6) according to its HRESIMS ion at m/z 425.1942 [M +Na]+ (calcd for C20H30O6Na, 425.1935). The 1D NMR data (Table 1) of 2 were highly similar to those of 1, and they shared the same planar architecture (Fig. 2) after detailed analysis of their 1H–1H COSY, HMBC, and HSQC data. Examination of the NMR data revealed that C-9’, C-10’, and C-11’ were significantly deshielded by ΔδC + 5.6, +7.6, and +2.1, respectively, indicating that 2 should be a C-9′ epimer of 1. This assumption was supported by the ROESY correlations (Fig. S1, Supporting Information) of both H-10′a (δH 1.80) and H-6 (δH 2.36) with H3-7 (δH 1.55). The absolute configuration (1R,4R,6R,9′R) of 2 was substantiated by a comparison of its calculated and experimental ECD spectra (Fig. 4).
Fig. 4.
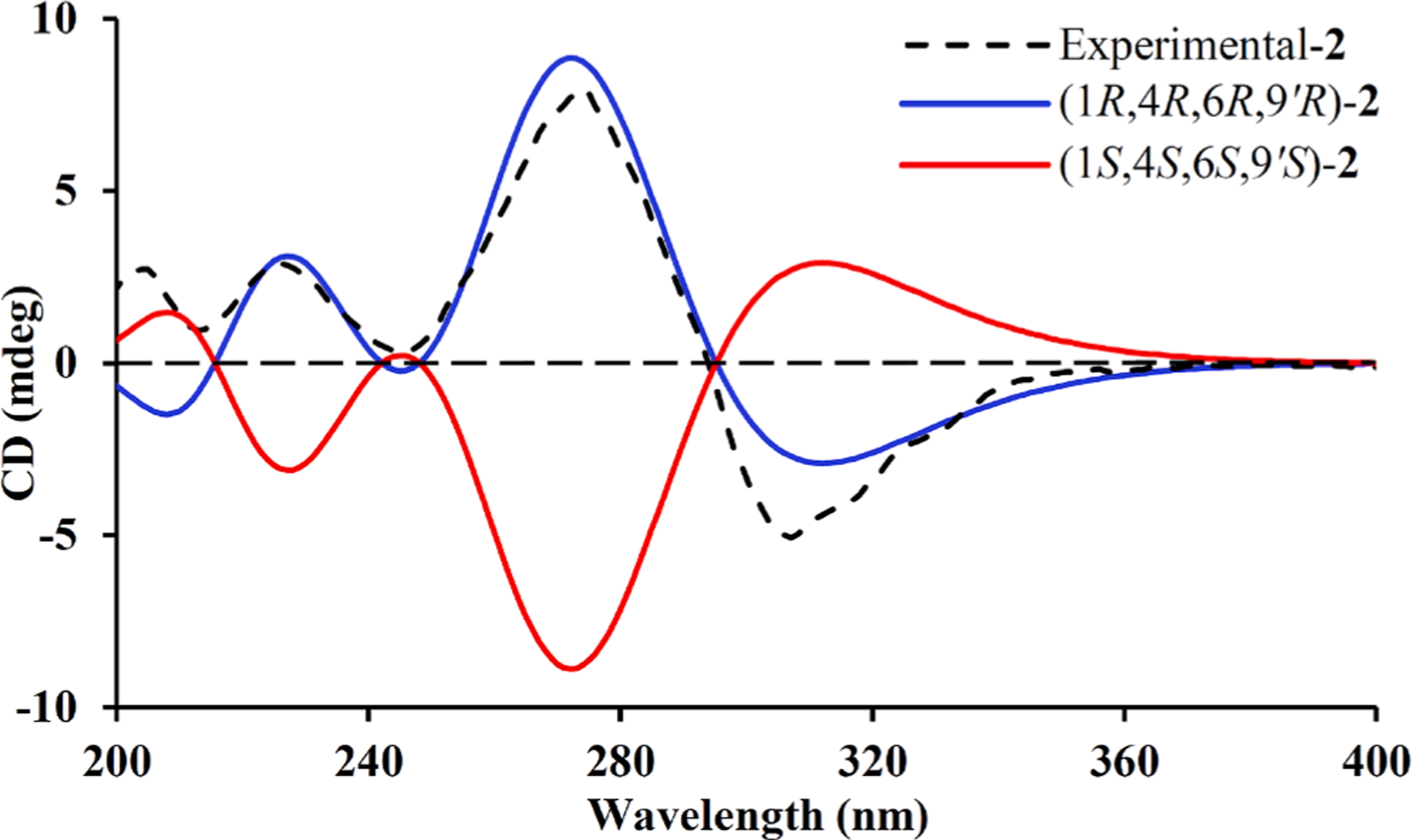
Calculated and experimental ECD spectra for 2.
Eucalyprobusal C (3) was determined to have the same molecular formula (C20H30O6) as those of 1 and 2 based on its HRESIMS ion at m/z 401.1978 [M−H]− (calcd for C20H29O6, 401.1970). Inspection of the 1H and 13C NMR data of 3 (Table 1) suggested that it was also a phloroglucinol–monoterpene adduct, of which the latter unit is similar to that of euglobal G6 [26]. The 1H–1HCOSY spectrum (Fig. 1) revealed the presence of three structural fragments, H-2–H-3, H3-9–H-8–H3-10, and H2-5–H-6–H-9′–H2-10′–H-11′–H3-12′ (H3-13′), for the monoterpene scaffold. In the HMBC spectrum, the observed correlations from H 3–7 (δH 1.65) to C-6 (δC 40.2)/C-1 (δC 72.7)/C-2 (δC 80.2), from H2-5 (δH 2.62, 1.86) to C-3 (δC 111.6)/C-4 (δC 155.2), and from H3-10 (δH 0.72)/Me-9 (δH 0.74) to C-4 validated the existence of a γ-terpinene derivative with a C-2 hydroxy group in 3. Compared with the remarkably different 13C NMR data for 1 and 2, the downfield chemical shifts of C-9’ (δC 35.9) and C-10’ (δC 46.7) indicated an α-oriented configuration for the H-9’ in 3. The ROESY correlations (Fig. S1, Supporting Information) of H-10′a (δH 1.42)/H-6 (δH 2.23)/H-2 (δH 4.49) with H3-7 (δH 1.65) proved that these protons were all β-oriented. The experimental ECD spectrum with two positive Cotton effects at 221 (+27.39) and 290 (+2.04) nm as well as two negative Cotton effects at 267 (−1.44) and 343 (−4.56) nm (Fig. 5) of 3 defined its absolute configuration (1S,2R,6R,9′R).
Fig. 5.
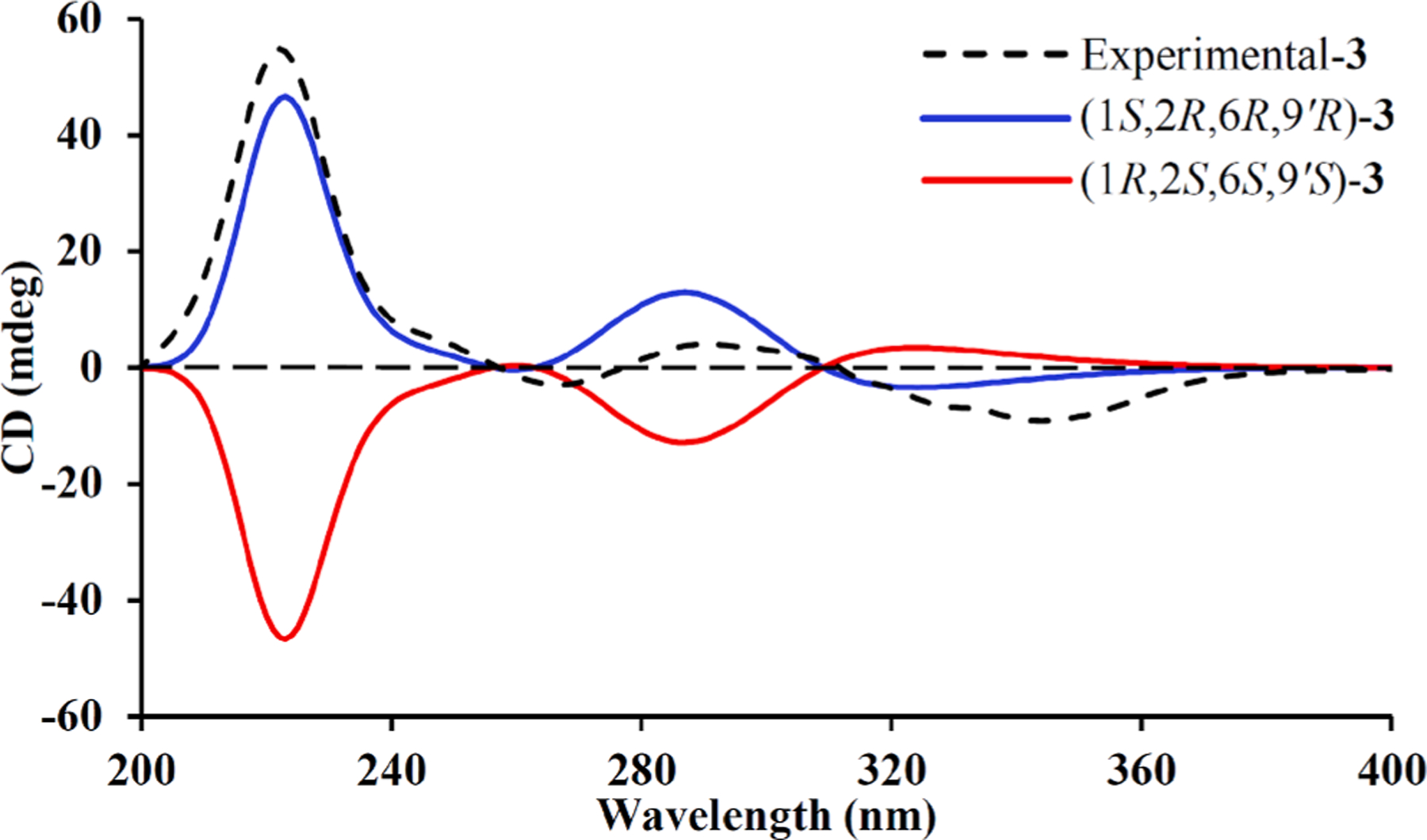
Calculated and experimental ECD spectra for 3.
Eucalyprobusal D (4) was shown to possess a molecular formula of C 20H30O6 due to its observed HRESIMS ion at m/z 423.1772 [M+Na]+ (calcd for C23H28O6Na, 423.1778). The NMR data (Table 1) of 4 highly resembled those of 3, with the exception for the presence of a ketone carbonyl group (δC 195.5, C-2) in 4, instead of an oxygenated methine (δC 80.2, C-2; δH 4.49, H-2) in 3. The placement of this ketone carbonyl carbon at C-2 was proved by the evidently observed HMBC correlations from H3-7 (δH 1.63) to C-6 (δC 44.0)/C-1 (δC 82.4)/C-2 (δC 195.5). Similarly, Me-7 in 4 was stochastically assigned a β-orientation, and the observed ROESY correlations (Fig. S1, Supporting Information) of H3-7 with both H-10′ (δH 1.61) and H-6 (δH 2.49) revealed the β-orientations for the C-9′ isopentyl group and H-6 (Fig. 1). The absolute configuration (1S,6R,9′R) of 4 was established by a comparison of its experimental and calculated ECD spectra (Fig. 6).
Fig. 6.
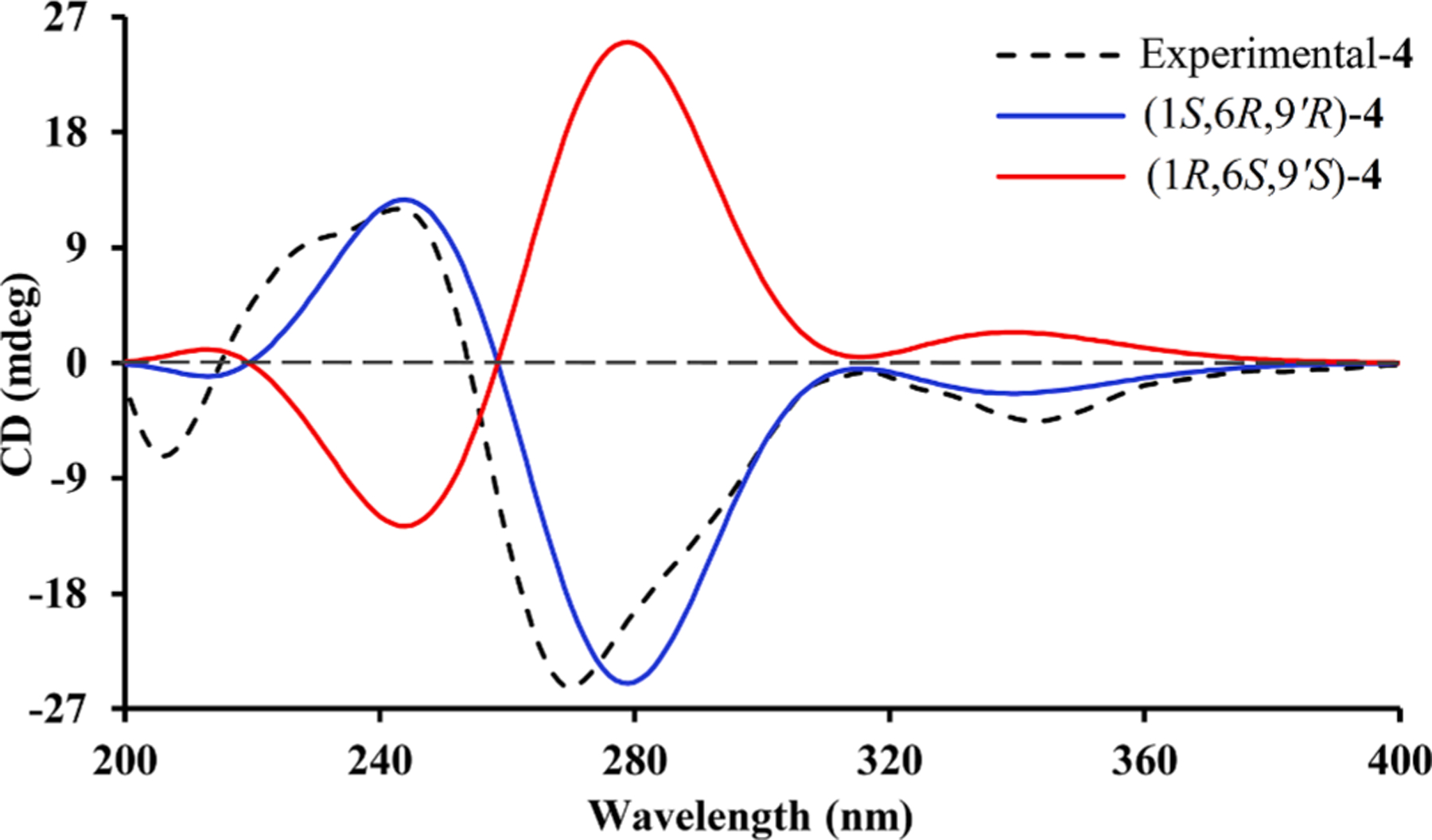
Calculated and experimental ECD spectra for 4.
Eucalyprobusal E (5) was proved to share a molecular formula of C 20H22O6 owing to its HRESIMS ion at m/z 397.1051 [M+K]+ (calcd for C20H22O6K, 397.1048). Its NMR data (Table 1) were highly similar to those of euglobal IIc [27], except for the presence of a ketone carbon (δC 196.9, C-4) and the disappearance of signals for the C-4 isopropyl functionality. Together with two fragments of H-2–H-3 and H2-5–H-6 revealed by the 1H–1H COSY spectrum, HMBC correlations from H3-7 (δH 1.67) to C-6 (δC 35.0)/C-1 (δC 76.3)/C-2 (δC 150.0) and from both H-2 (δH 6.81) and H2-5 (δH 2.64, 2.52) to C-4 (δC 196.9) indicated the existence of an α-phellandrene derivative with the loss of a C-4 isopropyl group (Fig. 1). In the ROESY spectrum, the key correlations of H 3–7 (δH 1.67) with H-6 (δH 2.54) suggested that they shared -β-configurations. The experimental ECD curve with three positive Cotton effects at 215 (+2.07), 263 (+1.27), and 354 (+0.03) nm as well as two negative Cotton effects at 239 (−0.61) and 292 (−0.58) nm (Fig. 7) defined the absolute configuration (1S,6S) of 5.
Fig. 7.
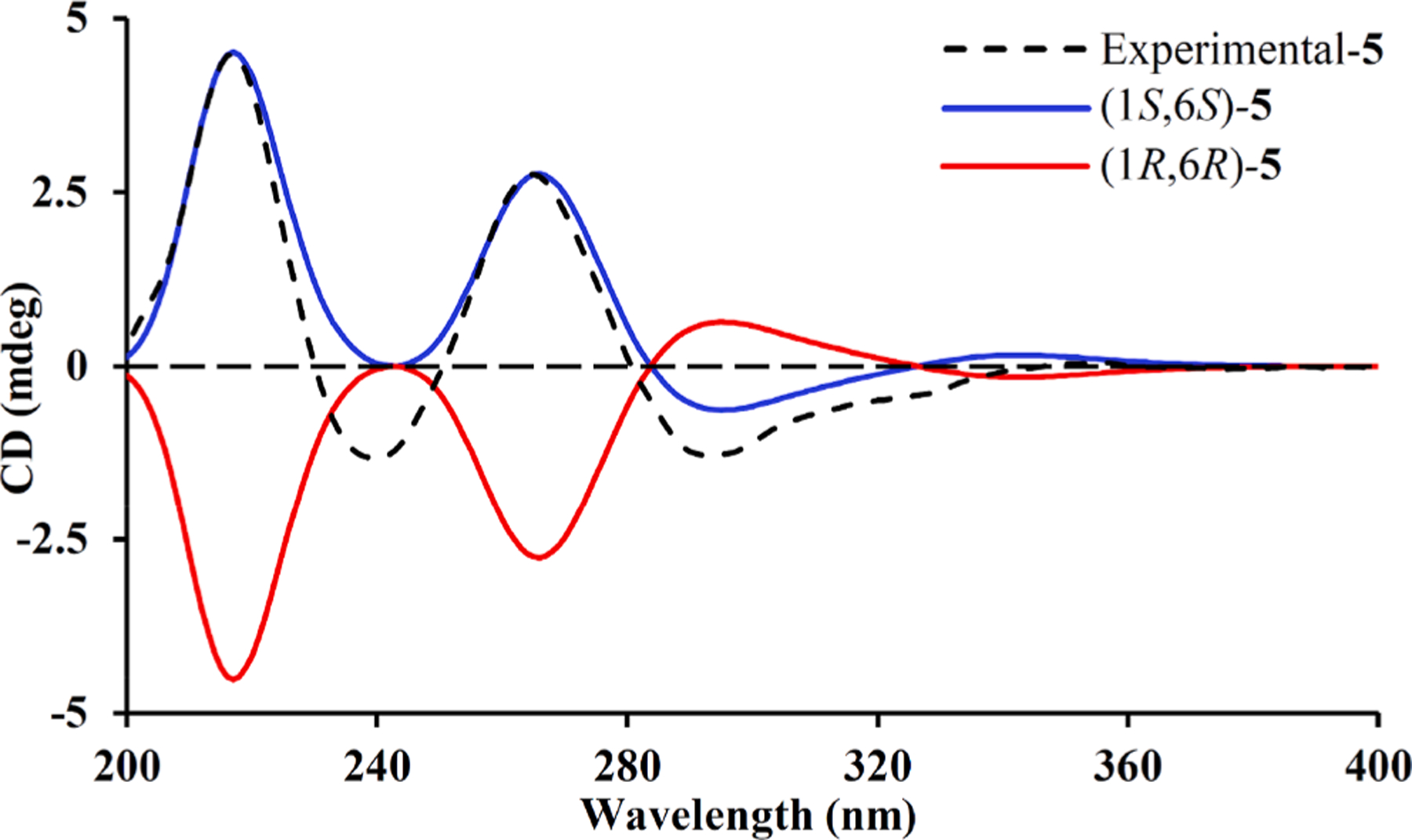
Calculated and experimental ECD spectra for 5.
Eucalyprobusal F (6) had a molecular formula of C23H28O5 as deduced from its HRESIMS ion at m/z 383.1872 [M−H]− (calcd for C 20H27O5, 383.1864). The 1H NMR spectrum displayed resonances for four secondary methyls (δH 0.88, d, J = 6.6 Hz, H3-13′; 0.94, d, J = 6.6 Hz, H3-12′; 1.17 × 2, both d, J = 7.0 Hz, H3-8/H3-9), a tertiary methyl (δH 2.22, s, H3-10), three aromatic protons (δH 6.85, dd, J = 7.8, 1.5 Hz, H-4; 6.91, d, J = 7.8 Hz, H-5; 7.45, d, J = 1.5 Hz, H-2), and two aldehyde protons (δH 10.05 × 2, s, H-7′/H-8′). Apart from the readily discernable signals attributable for a diformylated phloroglucinol unit (δC 106.3 × 2, C-2′/C-4′; 111.3, C-6′; 169.1, C-3′; 169.9 × 2, C-1′/C-5′; 193.1 × 2, C-7′/C-8′), the 13C NMR data indicated the occurrence of five methyls (δC 19.5, C-10; 22.9, C-12′; 23.8, C-13′; 24.6, C-8; 24.7, C-9], one methylene (δC 42.9, C-10′), three methines (δC 27.4, C-11′; 35.0, C-9′; 35.2, C-7), and a trisubstituted benzene ring (δC 124.6, C-4; 127.7, C-2; 131.0, C-5; 134.7, C-6; 142.9, C-1; 146.8, C-3). Along with two spin systems in the 1H–1H COSY spectrum (Fig. 1), the observed HMBC correlations from H-4 (δH 6.85) to C-2 (δC 127.7)/C-6 (δC 134.7), from H-5 (δH 6.91) to C-1 (δC 142.9)/C-3 (δC 146.8), from H3-8/H3-9 (δH 1.17, both) to C-3 (δC 146.8), and from H3-10 (δH 2.22) to C-5 (δC 131.0)/C-6 (δC 134.7)/C-1 (δC 142.9) allowed the establishment of the monoterpene moiety as p-cymene.7 The linkage of monoterpene and phloroglucinol units via a C-1–C-9′ bond was determined by the key HMBC correlations (Fig. 1) from H-9′ (δH 4.65) to C-2 (δC 127.7)/C-6 (δC 134.7)/C-1 (δC 142.9)/C-6′ (δC 111.3)/C-1′ (δC 169.9)/C-5′ (δC 169.9). Meroterpenoid 6 was determined to be a racemic mixture by HPLC analysis using a CHIRALPAK IC column (Fig. S2, Supporting Information). Chiral separation followed by ECD calculations determined the absolute configurations (9′S) and (9′R) for (+)-6 and (−)-6, respectively (Fig. 8).
Fig. 8.
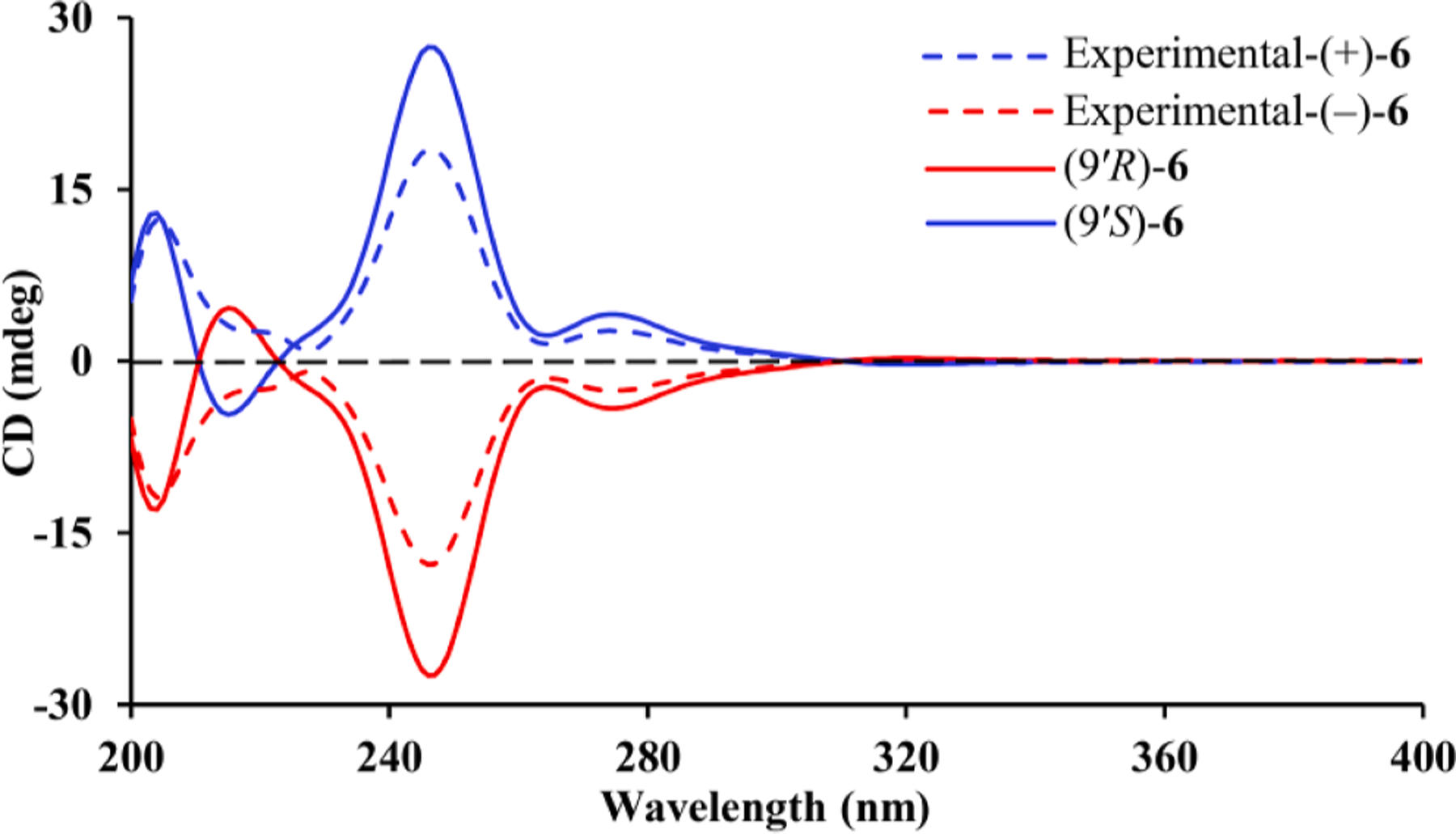
Calculated and experimental ECD spectra of (±)-6.
Eucalyprobusone B (7) possessed a molecular formula of C12H14O5 as revealed by an HRESIMS ion at m/z 261.0735 [M+Na]+ (calcd for C 12H14O5Na, 261.0733). With the assistance of HSQC spectrum, the 1H and 13C NMR data showed the characteristic resonances for an isopropenyl (δH 1.16 × 2, d, J = 6.8 Hz, H3-10/H3-11; δC 19.1 × 2, H3-10/H3-11; δH 3.68, sept., J = 6.8 Hz, H-9; δC 39.6, C-9), one methoxy group (δH 3.95, s, OMe-3; δC 56.2, OMe-3), one pentasubstituted aromatic ring (δH 5.91, s, H-4; δC 90.8, CH-4; δC 103.4, C-2; δC 105.3, C-6; δ C 168.1, C-3; δC 169.9, C-5; δC 171.8, C-1), an aldehyde group (δH 10.20, s, H-8; δC 192.7, CH-8), a ketone carbonyl (δC 210.5, C-7), and two hydroxy protons (δH 12.99, s, OH-5; δH 15.50, s, OH-1). The aforementioned data indicated that 7 was a monomeric formylated phloroglucinol similar to 1,5-dihydroxy-2-(2′-methylpropionyl)-3-methoxy-6-methylbenzene [28], except for the replacement of a C-8 methyl (δH 1.97, s; δC 7.4) in the former by a formyl group (δH 10.20, s; δ C 192.7) in 7. The HMBC correlations (Fig. 1) from H3-10 (δH 1.16)/H3-11 (δH 1.16) to C-7 (δC 210.5), from OMe-3 (δH 3.95) to C-3 (δC 168.1), from H-8 (δH 10.20) to C-6 (δC 105.3), from OH-5 (δH 12.99) to C-4 (δC 90.8)/C-5 (δC 169.9), and from OH-1 (δH 15.50) to C-2 (δC 103.4)/C-6 (δC 105.3)/C-1 (δC 171.8) established the structure of 7.
Eucalyprobusone C (8) was deduced to have a molecular formula of C28 H38O8 by its HRESIMS ion at m/z 503.2645 [M+H] + (calcd for C28H39O8, 503.2639). The 1D NMR spectral data of 8 (Table 2) indicated it was a dimeric resorcinol analogue. Similar to 7, the discernable signals for an isopropenyl (δH 1.12, d, J = 6.7 Hz, H3-10; δH 1.13, d, J = 6.7 Hz, H3-9; δH 3.82, sept., J = 6.7 Hz, H-8), an isobutyl (δH 1.11 × 2, d, J = 6.7 Hz, H3-10′/H3-11′; 1.79 and 1.35, both m, H2-8′; 3.69, sext., J = 6.7 Hz, H-9′), an isopentyl (δH 0.88 × 2, d, J = 6.5 Hz, H3-4′’/H3-5′’; 1.44, brsept., J = 6.5 Hz, H-3′’; 2.08, 2H, m, H 2–2′’; 5.01, t, J = 8.1 Hz, H-1′’), two methoxy groups (δH 3.89, s, OMe-3′; 3.90, s, OMe-3), two aromatic protons (δH 6.10 × 2, s H-4/H-4′), and two hydroxy protons (δH 9.50 × 2, s, OH-1/OH-1′) were readily recognized in the 1H NMR spectrum (Table 2) of 8. Together with three fragments in blue bold lines (Fig. 1) as revealed by the 1H–1H COSY spectrum, HMBC correlations from both H3-10 (δH 1.12) and H3-9 (δH 1.13) to C-7 (δC 211.3), from H-9′ (δH 3.69) to C-7′ (δC 211.6), from OMe-3′ (δH 3.89) to C-3′ (δC 162.5), from OMe-3 (δH 3.90) to C-3 (δC 162.5), from both H-4 (δH 6.10) and H-4′ (δH 6.10) to C-2 (δC 104.6)/C-2′ (δC 104.6)/C-6 (δC 110.3)/C-6′ (δC 110.3)/C-3 (δC 162.5)/C-3′ (δC 162.5)/C-5 (δC 164.4)/C-5′ (δC 164.4), and from H-1′’ (δH 5.01) to C-6 (δC 110.3)/C-6′ (δC 110.3)/C-3 (δC 162.5)/C-3′ (δC 162.5)/C-5 (δC 164.4)/C-5′ (δC 164.4)/C-1 (δC 165.8)/C-1′ (δC 165.8) not only verified the presence of two methoxy resorcinol moieties, but also substantiated that these two units were connected via a C-6–C-1′’–C-6′ bond. An HPLC analysis equipped with a CHIRALPAK IC column (Fig. S2, Supporting Information) indicated that 8 was a racemic mixture, and ECD calculations (Fig. 9) was used to establish the absolute configurations (1′’S) and (1′’R) for (+)-8 and (−)-8, respectively.
Fig. 9.
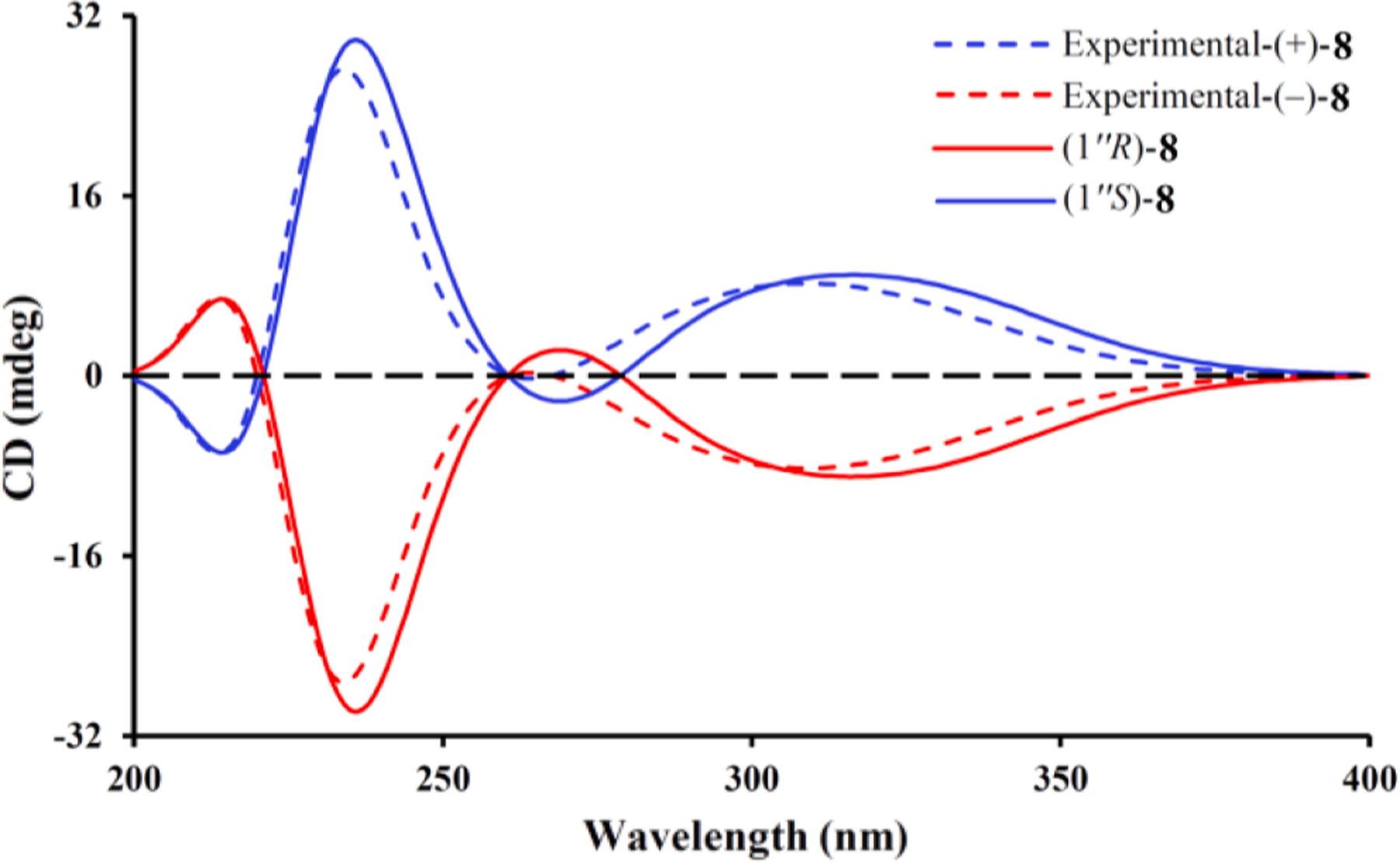
Calculated and experimental ECD spectra of (±)-8.
Eucalyprobusone D (9) possessed a molecular formula of C24H28O9 as inferred from an HRESIMS ion at m/z 499.1345 [M+K]+ (calcd for C24 H28 O9K, 499.1365). The 1H–1H COSY spectrum (Fig. 1) indicated two coupled systems of H3-9–H-8–H3-10 and H2-10′–H-11′–H3-12′(H3-13′). In the HMBC spectrum (Fig. 1), correlations from H3-9/H3-10 (both δH 1.17) to C-7 (δC 211.1), from OMe-3 (δH 3.86)/H-4 (δH 6.07) to C-3 (δC 162.0), from OH-1 (δH 16.76) to C-1 (δC 163.6)/C-2 (δC 104.0)/ C-6 (δC 106.0), and from OH-5 (δH 8.93) to C-4 (δC 92.8)/C-6 (δC 106.0) revealed the occurrence of an isobutyryl methoxy resorcinol moiety, whereas the observed correlations from H2-10′ (δH 3.00) to C-9’ (δC 207.2), from H-8′ (δH 10.15) to C-3′ (δC 165.3)/C-5′ (δC 168.4), from OH-5′ (δH 14.48) to C-6′ (δC 103.5)/C-4′ (δC 104.8), and from OH-1′ (δH 17.23) to C-6′ (δC 103.5)/C-2′ (δC 105.6) allowed the establishment of a mono-formylated isovaleryl phloroglucinol unit. The key HMBC correlations from H2-7′ (δH 3.74) to C-5 (δC 162.8)/C-1 (δC 163.6)/C-3′ (δC 165.3)/C-1′ (δC 169.9) unequivocally revealed that the two mono-phloroglucinol derivatives were connected by a C-7′–C-6 bond.
Eucalyprobusones E (10) and F (11) were isolated as two pairs of enantiomers with the same molecular formula (C28H38O8) as that of 9 by HRESIMS (m/z 525.2467 [M+Na]+, calcd for C28H38O8Na, 525.2459). A comparison of the 1D NMR data (Table 3) with those of 9 revealed that the isovaleryl in the latter was replaced by a sec-isovaleryl in the former ones. This was confirmed by the HMBC correlations from H 3–9′ (δH 1.10, J = 6.7 Hz for 10; 1.14, J = 6.7 Hz for 11) to C-8′ (δC 46.2 for both 10 and 11)/C-7′ (δC 210.2 for 10, 210.0 for 11), and from Me-11′ (δH 0.80, t, J = 6.7 Hz for 10; 0.84, t, J = 6.7 Hz for 11) to C-10′ (δC 27.3 for 10, 27.4 for 11)/C-8′ (δC 46.2 for both 10 and 11). Interestingly, an optical rotation value of 0.67 (c 0.15, MeOH) obtained for the mixture of 10 and 11 indicated that they should be racemic. Fortunately, a triclinic crystal obtained from a mixed solvent of acetone-MeOH (1:1, v/v) of 10 and 11 was selected for subsequent X-ray diffraction study. The results revealed that C-7′ sec-isobutyl and C-1′’ isobutyl units were both unordered, suggesting the occurrence of two pairs of enantiomers (Fig. 10). Nevertheless, it was not feasible to obtain (+)-10, (–)-10, (+)-11, and (–)-11 by a chiral column after several attempts (Fig. S2, Supporting Information). Taking the relationships between the specific rotation values and absolute configurations of (+)-8 and (–)-8 into consideration, the absolute configurations of (+)-10, (–)-10, (+)-11, and (–)-11 could be provisionally assigned as (8′R,1′’S), (8′R,1′’R), (8′S,1′’S), and (8′S,1′’R), respectively, owing to the fact that the existence of the C-7′ sec-isobutyl could not strikingly affect the holistic absolute configurations that were determined by the specific rotation values [29].
Fig. 10.
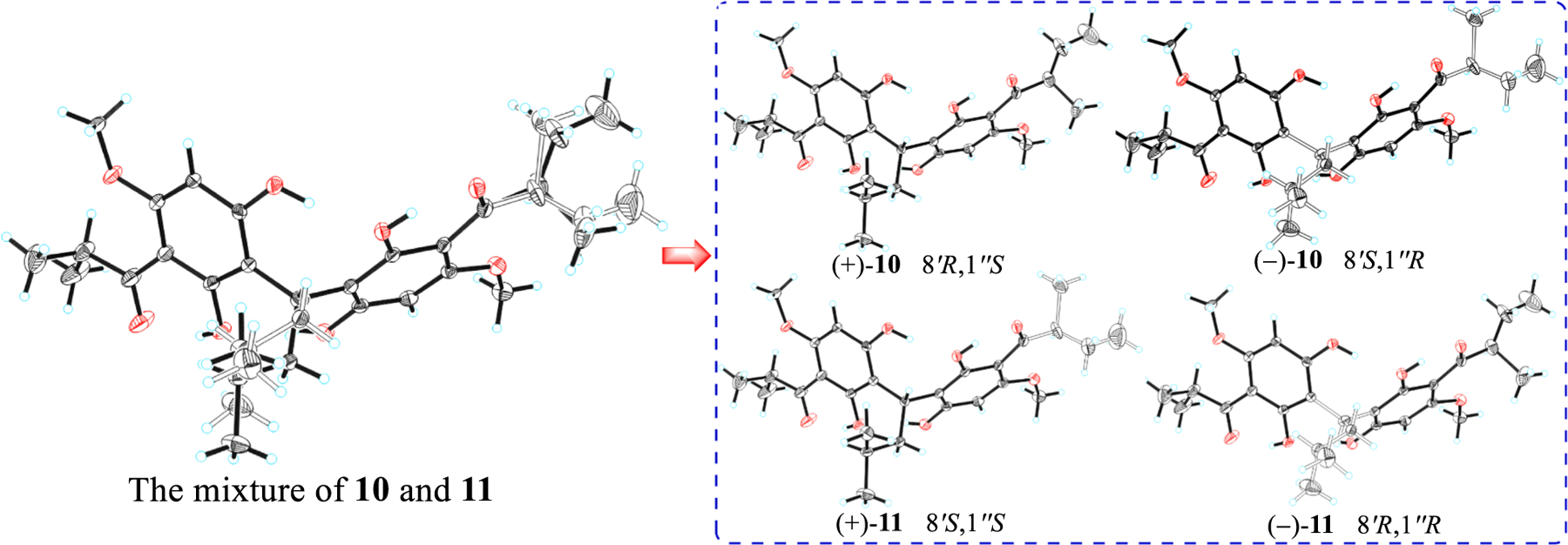
ORTEP drawing of (±)-10 and (±)-11.
3.2. AChE inhibitory effects
Given the PE–EtOAc extract of E. robusta fruits was AChE inhibitory (500 μg/mL, 68%), all the isolated acylphloroglucinols were screened for AChE inhibitory effects. At a concentration of 40.0 μM, only acylphloroglucinols 6 and 8–11 showed AChE inhibitory activities with inhibition rates ranging from 93.02 ± 0.71 to 71.97 ± 2.20%. Further studies indicated these compounds were AChE inhibitory with IC50 values ranging from 2.55 ± 0.28 to 36.22 ± 2.29 μM, with the mixture of 10 and 11 being the most effective possessing an IC50 value of 2.55 ± 0.28 μM (Table 4). Taking their structural characteristics and AChE inhibitory data into consideration, the observable structure–activity relationships can be summarized as follows (i) both FPMs featuring with a dihydropyran ring and acylphloroglucinol monomer were inactive; (ii) acylphloroglucinol dimers that be connected via an isopentyl moiety showed stronger AChE inhibitory effects than that of being linked by C-7′; (iii) the mixtures of (+)-6/(−)-6 and (+)-8/(−)-8 showed stronger AChE inhibitory activities than (+)-6, (−)-6, (+)-8, or (−)-8. Compared with structurally diverse acylphloroglucinol-like compounds reported from various plants [30–34], acylphloroglucinols 6, 8, and the mixture of 10 and 11 isolated from E.robusta fruits displayed more potential AChE inhibitory effects. With regard to acylphloroglucinol derivatives obtained from species of Myrtaceae, apart from polymethylated phloroglucinol meroterpenoids (PPMs) isolated from Rhodomyrtus tomentosa [35], the current findings indicated that FPM and acylphloroglucinol heterodimers [10] connected only by an isopentyl unit are more likely to be AChE inhibitors.
Table 4.
AChE inhibitory effects of acylphloroglucinols 1–11.
| Compound | IC50 ± SD(μM) | Compound | IC50 ± SD (μM) |
|---|---|---|---|
| 1 | > 40.0 | 7 | > 40.0 |
| 2 | > 40.0 | 8 | 3.82 ± 0.22 |
| 3 | > 40.0 | (+)-8 | 4.96 ± 0.68 |
| 4 | > 40.0 | (−)-8 | 6.02 ± 0.54 |
| 5 | > 40.0 | 9 | 36.22 ± 2.29 |
| 6 | 3.22 ± 0.36 | 10 + 11 | 2.55 ± 0.28 |
| (+)-6 | 4.79 ± 0.57 | ||
| (−)-6 | 5.85 ± 0.76 | Galantaminea | 1.05 ± 0.06 |
Positive drug.
3.3. Molecular docking investigation
Considering acylphloroglucinols 6, 8, and the mixture of 10 and 11 displayed good AChE inhibitory properties, molecular modeling investigations were used to better understand their mechanism of action and the binding modes with AChE (Fig. 11). The results revealed that all these isolates could be buried into the hydrophobic pocket of AChE. More specifically, (i) the acylphloroglucinol unit of 6 appears to form hydrogen bonds with the Tyr337, Tyr341, Thr83, and Ser125 residues, the phenyl ring of the monoterpene moiety was bound to the Trp86 residue via the π–π stacking interactions, and the terminal methyl fragments of the isopentyl moiety formed π-σ stacking interactions with Phe297 and Tyr124 residues; (ii) both the C-5 and C-5′ hydroxy groups of 8 could form hydrogen bonds with only the Tyr124 residue and two phenyl rings showed π–π interactions with Tyr341 and Trp86 residues, respectively, and the terminal methyl fragments of isopentyl, isobutyl, and isopropyl showed π–σ stacking interactions with Tyr337, Trp286/Phe297, and Trp86/Tyr337 residues, respectively; (iii) the phloroglucinol units of both 10 and 11 could form hydrogen bonds with Ser125, Tyr124, Tyr133, Tyr337, and Asn87 residues, the phenyl rings bearing a sec-isovaleryl group displayed π–π interactions with Trp86 and Tyr341 residues; (iv) the phenyl rings bearing a sec-isovaleryl group of 10 also showed π–π interaction with Tyr337 residue; (v) the terminal methyl fragments of 10 exhibited π-σ stacking interactions with Phe295, Phe297, Tyr124, and Trp86 residues, whereas those of 11 displayed π-σ stacking interactions with only Tyr124 and Trp286 residues. Through docking analysis, the racemic acylphloroglucinols 10 and 11 shared more interaction sites with AChE than 6 and 9 did, which were also consistent with the results of their AChE inhibitory assay.
Fig. 11.
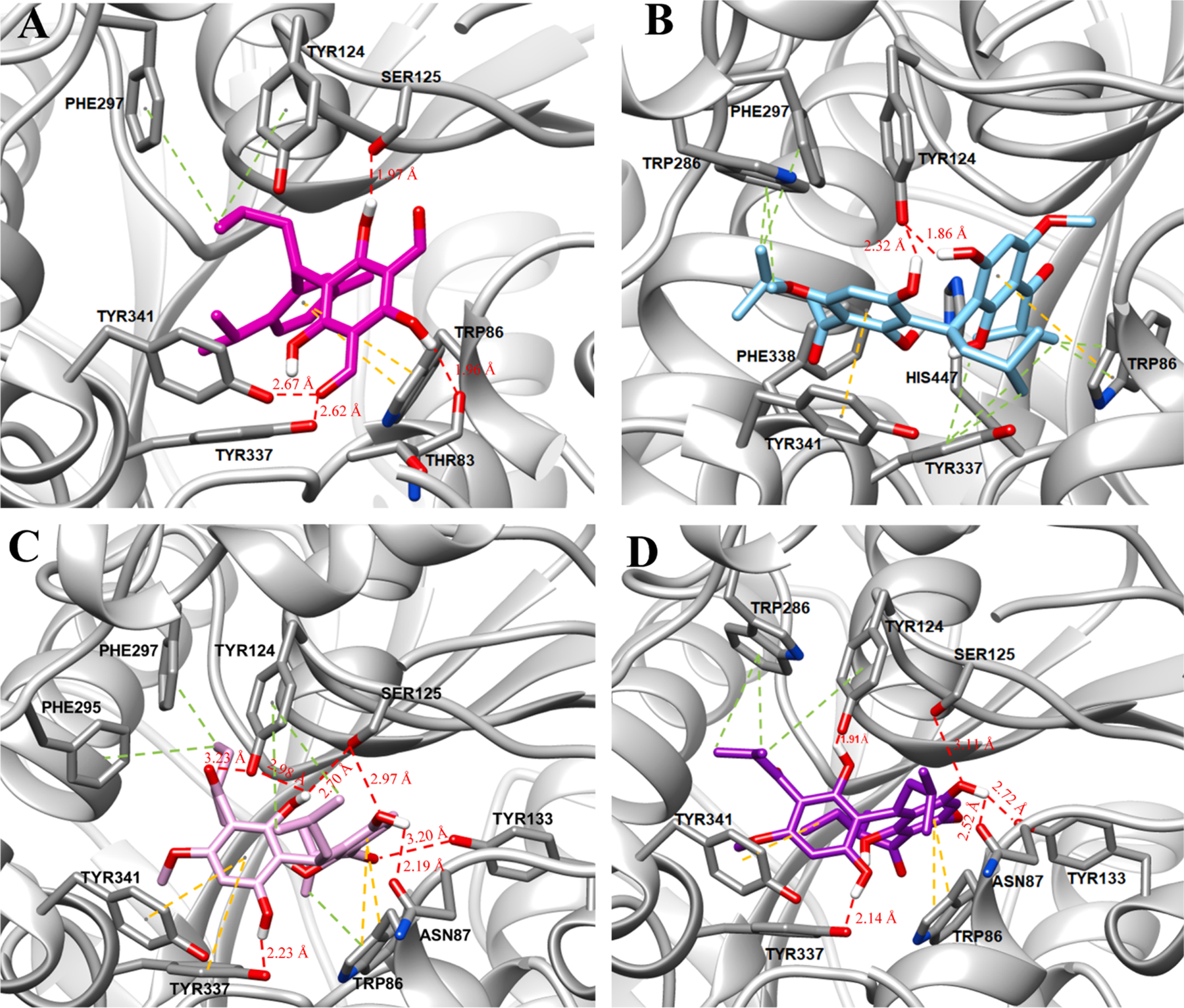
The binding modes of 6 (A), 8 (B), 10 (C), and 11 (D) with human AChE (PDB ID: 4M0F). Hydrogen bond interactions are depicted with red dashes, while π–π and π–σ stacking interactions are displayed with yellow and green dashes, respectively.
4. Conclusion
In summary, the systematically phytochemical investigation of E. robusta fruits resulted in the isolation of 11 new acylphloroglucinols, including six new formylated phloroglucinol-monoterpene meroterpenoids (1–6), one monomeric acylphloroglucinol (7), and four dimeric acylphloroglucinols (8–11). Although all attempts to separate the 10 and 11 mixture have failed, X-ray diffraction was crucial for confirming their structures and absolute configurations. Compounds 6, 8, and the mixture of 10 and 11 displayed significant AChE inhibitory effects, and the possible interaction sites of these four compounds with AChE were investigated by molecular docking, which could be recognized as lead compounds for treatment of Alzheimer’s disease.
Supplementary Material
Acknowledgments
The authors are grateful to the National Natural Science Foundation of China (31970377), the Yunnan Science Foundation for Excellent Young Scholars, the Youth Innovation Promotion Association CAS (2019381), the Ten Thousand Talents Plan of Yunnan Province for Industrial Technology Leading Talents, the Major Biomedical Project of Yunnan Province (2019ZF010), the State Key Laboratory of Phytochemistry and Plant Resources in West China (P2019-ZZ02), and the National Institutes of Health (J.A.P., Jr.) (R35 GM118173), and the State Key Laboratory of Functions and Applications of Medicinal Plants (FAMP201901K) for financial support.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2020.104127.
References
- [1].Ghisalberti EL, Phytochemistry 41 (1996) 7–22. [DOI] [PubMed] [Google Scholar]
- [2].Yu Y, Gan LS, Yang SP, Sheng L, Liu QF, Chen SN, Li J, Yue JM, J. Nat. Prod 79 (2016) 1365–1372. [DOI] [PubMed] [Google Scholar]
- [3].Pham TA, Hu XL, Huang XJ, Ma MX, Feng JH, Li JY, Hou JQ, Zhang PL, Nguyen VH, Nguyen MT, Xiong F, Fan CL, Zhang XQ, Ye WC, Wang H, J. Nat. Prod 82 (2019) 859–869. [DOI] [PubMed] [Google Scholar]
- [4].Shang ZC, Han C, Xu JL, Liu RH, Yin Y, Wang XB, Yang MH, Kong LY, Phytochemistry 163 (2019) 111–117. [DOI] [PubMed] [Google Scholar]
- [5].Li RH, Shang ZC, Li TX, Yang MH, Kong LY, Antimicrob. Agents Ch 61 (2017) e02702–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Umehara K, Singh IP, Etoh H, Takasaki M, Konoshima T, Phytochemistry 49 (1998) 1699–1704. [DOI] [PubMed] [Google Scholar]
- [7].Qin XJ, Jin LY, Yu Q, Liu H, Khan A, Hao XJ, An LK, Liu HY, J. Nat. Prod 81 (2018) 2638–2646. [DOI] [PubMed] [Google Scholar]
- [8].Liu H, Feng MY, Yu Q, Yan H, Zeng Y, Qin XJ, He L, Liu HY, Tetrahedron 74 (2018) 1540–1545. [Google Scholar]
- [9].Yin S, Xue JJ, Fan CQ, Miao ZH, Ding J, Yue JM, Org. Lett 9 (2007) 5549–5552. [DOI] [PubMed] [Google Scholar]
- [10].Qin XJ, Feng MY, Liu H, Ni W, Rauwolf T, Porco JA Jr., Yan H, He L, Liu HY, Org. Lett 20 (2018) 5066–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bharate SB, Singh IP, Bioorg. Med. Chem. Lett 21 (2011) 4310–4313. [DOI] [PubMed] [Google Scholar]
- [12].Lal K, Zarate EA, Youngs WJ, Salomom RG, J. Am. Chem. Soc 108 (1986) 1312–1314. [Google Scholar]
- [13].Salomon RG, Lal K, Mazza SM, Zarate EA, Youngs WJ, J. Am. Chem. Soc 110 (1988) 5213–5214. [Google Scholar]
- [14].Chiba K, Arakawa T, Tada M, Chem. Commun (1996) 1763–1764. [Google Scholar]
- [15].Khambay BPS, Beddie DG, Hooper AM, Simmonds MSJ, Tetrahedron 59 (2003) 7131–7133. [Google Scholar]
- [16].Bharate SB, Singh IP, Tetrahedron Lett. 47 (2006) 7021–7024. [Google Scholar]
- [17].Singh IP, Sidana J, Bharate SB, Foley WJ, Nat. Prod. Rep 27 (2010) 393–416. [DOI] [PubMed] [Google Scholar]
- [18].Alliot J, Gravel E, Larquetoux L, Nicolas M, Doris E, J. Nat. Prod 76 (2013) 2436–2349. [DOI] [PubMed] [Google Scholar]
- [19].Nanjing University of Chinese Medicine, In Dictionary of traditional Chinese medicine, Shanghai Science and Technology Press: Shanghai: 1 (2006) 184–185. [Google Scholar]
- [20].Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M, Lancet 336 (2005) 2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu J, Dumontet V, Simonin AL, Iorga BI, Guerineau V, Litaudon M, Nguyen VH, Gueritte F, J. Nat. Prod 74 (2011) 2081–2088. [DOI] [PubMed] [Google Scholar]
- [22].Zhang HY, Acta Pharmacol. Sin 33 (2012) 1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ, Gaussian 16 Revision C.01, Gaussian Inc, Wallingford CT, 2019. [Google Scholar]
- [24].Ellman GL, Courtney D, Andres V Jr., Featherstone RM, Biochem. Pharmacol 7 (1961) 88–95. [DOI] [PubMed] [Google Scholar]
- [25].Zhou Y, Sun W, Peng J, Yan H, Zhang L, Liu X, Zuo Z, Bioorg. Chem 93 (2019) 103322. [DOI] [PubMed] [Google Scholar]
- [26].Singh IP, Umehara K, Asai T, Ethoh H, Takasak M, Konoshima T, Phytochemistry 47 (1998) 1157–1159. [Google Scholar]
- [27].Chiba K, Arakawa T, Tada M, J. Chem. Soc. Perkin Trans 1 (1998) 2939–2942. [Google Scholar]
- [28].Shiu WKP, Gibbons S, Phytochemistry 67 (2006) 2568–2572. [DOI] [PubMed] [Google Scholar]
- [29].Qin XJ, Liu H, Yu Q, Yan H, Tang JF, An LK, Khan A, Chen QR, Hao XJ, Liu HY, Tetrahedron 73 (2017) 1803–1811. [Google Scholar]
- [30].Lou H, Yi P, Hu Z, Li Y, Zeng Y, Gu W, Huang L, Yuan C, Hao X, Fitoterapia 143 (2020) 104550. [DOI] [PubMed] [Google Scholar]
- [31].Orhan IE, Jedrejek D, Senol FS, Salmas RE, Durdagi S, Kowalska I, Pecio L, Oleszek W, Phytomedicine 42 (2018) 25–33. [DOI] [PubMed] [Google Scholar]
- [32].Yang XW, Li MM, Liu X, Ferreira D, Ding Y, Zhang JJ, Liao Y, Qin HB, Xu G, J. Nat. Prod 78 (2015) 885–895. [DOI] [PubMed] [Google Scholar]
- [33].Popoola OK, Marnewick JL, Rautenbach F, Iwuoha EI, Hussein AA, Molecules 20 (2015) 17309–17324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu X, Yang XW, Chen CQ, Wu CY, Zhang JJ, Ma JZ, Wang H, Yang LX, Xu G, J. Nat. Prod 76 (2013) 1612–1618. [DOI] [PubMed] [Google Scholar]
- [35].Qin XJ, Rauwolf TJ, Li PP, Liu H, McNeely J, Hua Y, Liu HY, Proco JA, Angew. Chem. Int. Ed 58 (2019) 4291–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.