

Abstract
Polarised targeting of diverse mRNAs to cellular protrusions is a hallmark of cell migration. Although a widespread phenomenon, definitive functions for endogenous targeted mRNAs and their relevance to modulation of in vivo tissue dynamics remain elusive. Here, using single‐molecule analysis, gene editing and zebrafish live‐cell imaging, we report that mRNA polarisation acts as a molecular compass that orients motile cell polarity and spatially directs tissue movement. Clustering of protrusion‐derived RNAseq datasets defined a core 192‐nt localisation element underpinning precise mRNA targeting to sites of filopodia formation. Such targeting of the small GTPase RAB13 generated tight spatial coupling of mRNA localisation, translation and protein activity, achieving precise subcellular compartmentalisation of RAB13 protein function to create a polarised domain of filopodia extension. Consequently, genomic excision of this localisation element and perturbation of RAB13 mRNA targeting—but not translation—depolarised filopodia dynamics in motile endothelial cells and induced mispatterning of blood vessels in zebrafish. Hence, mRNA polarisation, not expression, is the primary determinant of the site of RAB13 action, preventing ectopic functionality at inappropriate subcellular loci and orienting tissue morphogenesis.
Keywords: angiogenesis, endothelial cell, filopodia, mRNA targeting, zebrafish
Subject Categories: Development & Differentiation, Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control
A localisation element in its 3′UTR targets RAB13 mRNA to nascent filopodia in migrating cells, directing local mRNA translation, filopodia extension, and tissue morphogenesis in vivo.
Introduction
Dynamic subcellular polarisation of a myriad of proteins fundamentally shapes the front‐rear orientation and directed movement of motile cells during tissue formation (reviewed in Mayor & Etienne‐Manneville, 2016). In parallel, cell migration is associated with subcellular polarisation of numerous mRNAs (reviewed in Herbert & Costa, 2010), but whether this phenomenon is also relevant to the modulation of tissue dynamics remains an open question. However, in many other biological contexts, mRNA localisation and local translation are well‐established as key determinants of cell polarity (Buxbaum et al, 2015). This mode of spatial control of gene expression contributes to polarised cellular responses in broad contexts, ranging from axon growth (Leung et al, 2006; Yao et al, 2006) and synaptic function (e.g. Kang & Schuman, 1996; Lyles et al, 2006; Younts et al, 2016) to epithelial polarity (Nagaoka et al, 2012; Moor et al, 2017). Moreover, there is a wealth of data in diverse cell types demonstrating that large numbers of mRNAs are co‐distributed together at distinct subcellular sites, which has led to the idea that such mRNA polarisation functions to generate local transcriptomes (reviewed in Engel et al, 2020). This suggests that clusters of mRNAs encoding proteins belonging to common complexes and biological pathways co‐localise to participate in local processes (e.g. Mingle et al, 2005; Hotz & Nelson, 2017). Indeed, it has been proposed that such co‐distribution of mRNAs also ensures the fidelity of interactions between locally produced proteins in rapidly changing cell environments (Weatheritt et al, 2014). Nevertheless, during cell migration, the impact of mRNA polarisation on the control of translated protein function, local assembly of the migratory machinery and motile cell polarity remain poorly understood, as does the in vivo relevance of this phenomenon.
The polarised localisation of mRNAs is driven by cis‐localisation elements (LEs) that are commonly found in 3′ untranslated regions (UTRs) (Andreassi & Riccio, 2009; Mayr, 2016). Indeed, alternative 3′UTRs have been shown to control the spatial localisation of mRNAs (Taliaferro et al, 2016; Tushev et al, 2018) and modulate protein distribution (An et al, 2008; Ciolli Mattioli et al, 2019), suggesting that tight control of LE usage may underpin spatial regulation of gene expression. Although the complex sequence and structural composition of LEs render the identification of conserved RNA motifs within large groups of co‐localised mRNAs a challenging task, several individual LEs have been characterised in detail using diverse model organisms (reviewed in Jambhekar & Derisi, 2007). In vertebrate cells, LEs ranging from just a few to hundreds of nucleotides in length have been identified within 3′UTRs (e.g. Mowry & Melton, 1992; Kislauskis et al, 1994; Ainger et al, 1997). However, considering the difficulties in manipulating endogenous transcripts, our understanding of LE function during complex tissue formation in vertebrate organisms remains relatively poor.
Here, using novel reporter transgenics in zebrafish embryos and targeted gene editing, we shed light on the function of LE‐mediated mRNA polarisation in the control of tissue formation in vivo. Following clustering analysis of transcriptome‐wide data, we define a core group of 5 mRNAs that are universally targeted to the leading edge of migratory cells in vitro. Moreover, we identify a conserved RNA motif within the 3′UTRs of these genes and a 192‐nt LE containing four of these motifs that is sufficient to target transcripts to polarised sites of filopodia remodelling. Excision of this LE in transcripts encoding the small GTPase RAB13 perturbs mRNA localisation (but not translation) and was sufficient to depolarise RAB13‐mediated filopodia dynamics in motile endothelial cells in vitro. Hence, RAB13 mRNA polarisation achieves precise spatial compartmentalisation of RAB13 protein activity to the front of migrating cells and blocks ectopic protein action at inappropriate subcellular loci. Consequently, excision of the rab13 LE in zebrafish embryos also perturbed mRNA polarisation and induced mispatterning of nascent blood vessels. Altogether, our findings show that mRNA polarisation spatially restricts protein activity to precisely orient motile cell polarity and tissue movement in vivo.
Results
Clustering of RNAseq datasets identifies mRNAs exhibiting universal targeting to protrusions
To explore the ability of targeted mRNAs to direct tissue formation, we first aimed to define mRNA localisation motifs driving transcript polarisation in motile endothelial cells (ECs), as an initial step towards probing their function in coordinating blood vessel morphogenesis in vivo. As a starting point, we identified 233 transcripts enriched in fractionated cellular protrusions of migrating primary human umbilical vein ECs (HUVECs) in vitro (Fig 1A and B; Table EV1). ECs were seeded on Transwells in low serum and induced to migrate upon addition of VEGF‐A to the lower chamber (Fig 1A). Consequently, the motile protrusions and trailing cell bodies of ECs were separated and protrusion‐enriched transcripts identified by RNAseq (Fig 1B; Table EV1). k‐means clustering analysis of these data alongside RNAseq datasets from unrelated cell types (NIH/3T3 fibroblasts (Wang et al, 2017), MDA‐MB231 metastatic breast cancer cells (Mardakheh et al, 2015), induced neuronal cells (Zappulo et al, 2017)) revealed unexpected cell type‐specific diversity to transcript polarisation, with only five mRNAs exhibiting universal targeting to protrusions in all cell types tested (cluster k 5; RAB13, TRAK2, RASSF3, NET1, KIF1C; Figs 1B and C, and EV1A). Strikingly, cluster k 5 mRNAs shared near‐identical spatial distributions by single‐molecule FISH (smFISH) (Raj et al, 2008; Tsanov et al, 2016), being highly polarised to cellular protrusions relative to a control transcript, GAPDH (Fig 1D–G). Indeed, all cluster k 5 mRNAs exhibited a significantly higher Polarisation Index (PI) than GAPDH when co‐detected in migrating ECs (Fig 1E) and the ratio of k 5 mRNA PI to GAPDH PI was consistently greater than one in individual cells (Fig 1F). Moreover, k 5 transcripts were highly spatially distinct from other clusters, such as mRNAs of cluster k 7 that exhibited less‐polarised perinuclear targeting (Figs 1G and H, and EV1B and D). Likewise, protrusion localisation of the well‐established polarised mRNA ACTB (Condeelis & Singer, 2005) was significantly more diffuse than k 5 mRNAs, as were other cluster k 2 members such as PPDPF (Figs 1G and I, and EV1B and C). Finally, protrusion‐enriched mRNAs were also tightly clustered according to protein function (Table EV2), with k 5 transcripts specifically encoding cell periphery‐associated modulators of vesicle trafficking and membrane remodelling (Tommasi et al, 2002; Brickley et al, 2005; Kopp et al, 2006; Srougi & Burridge, 2011; Wu et al, 2011). Hence, tight coupling of distinct mRNA spatial distributions with discrete protein functionalities likely indicates that the universal polarisation of k 5 mRNAs is a key functional requirement in processes common to all motile cells.
Figure 1. Clustering of RNAseq datasets identifies mRNAs exhibiting universal targeting to protrusions in diverse cell types.
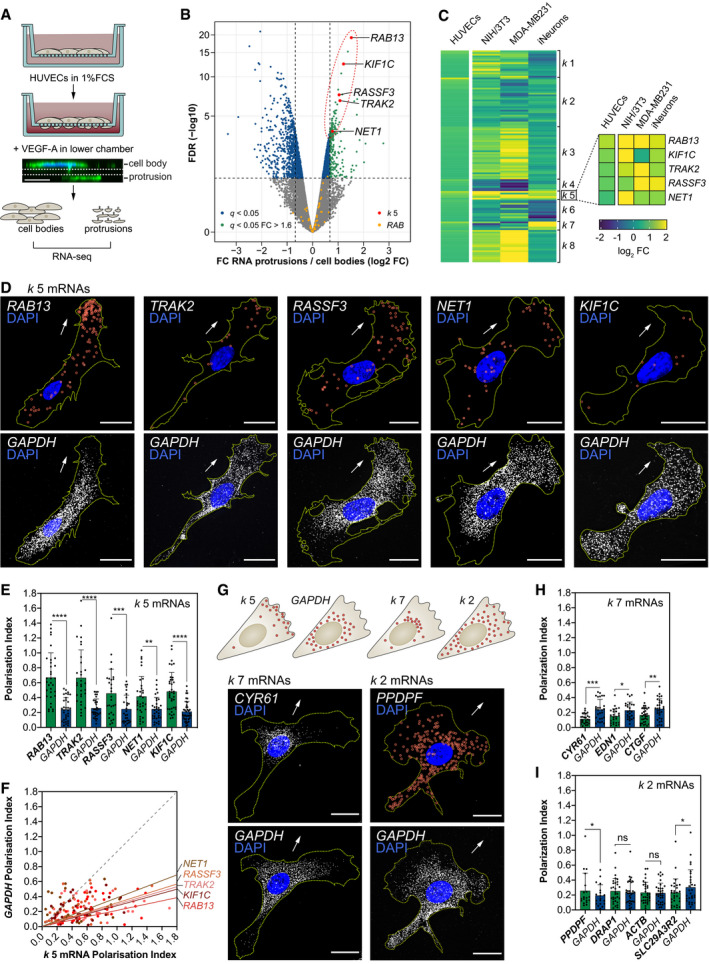
- Strategy used to screen for mRNAs enriched in motile protrusions of HUVECs migrating through Transwell membranes.
- RNAseq data are plotted in log2 fold change (FC) levels of protrusions over cell bodies against adjusted −log10 false discovery rate (FDR) (n = 2 replicates, average FC values are represented). The horizontal dashed line marks the FDR (q) = 0.05 threshold; vertical dashed lines mark the FC = 0.625 (left) or FC = 1.6 (right) thresholds.
- Heat map represents the k‐means clustering of transcript log2 FC levels (protrusions over cell bodies) extracted from RNAseq datasets published elsewhere. The corresponding HUVEC FC levels are shown in parallel.
- smFISH co‐detection of k 5 mRNAs and GAPDH in subconfluent motile HUVECs.
- Polarisation Index (PI) of k 5 mRNAs and GAPDH in co‐detected in HUVECs (n ≥ 28 cells; **P < 0.01, ***P < 0.001, ****P < 0.0001; Wilcoxon test).
- k 5 mRNA PIs plotted against respective GAPDH PIs. The slope of the coloured lines represents the average k 5 mRNA/GAPDH PI ratio; the dashed grey line represents a 1:1 ratio (n ≥ 28 cells).
- Top: distribution pattern of mRNAs clustered in k 2, k 5, k 7 and GAPDH. Bottom: smFISH co‐detection of exemplar k 7/k 2 mRNAs and GAPDH in subconfluent motile HUVECs.
- PIs of k 7 mRNAs and GAPDH co‐detected in HUVECs (n ≥ 25 cells; *P < 0.05, **P < 0.01, ***P < 0.001; paired t test).
- PIs of k 2 mRNAs and GAPDH co‐detected in HUVECs (n ≥ 19 cells; *P < 0.05; Wilcoxon test).
Figure EV1. Clustering of RNAseq datasets defines unexpected cell type‐specific diversity to mRNA polarisation.
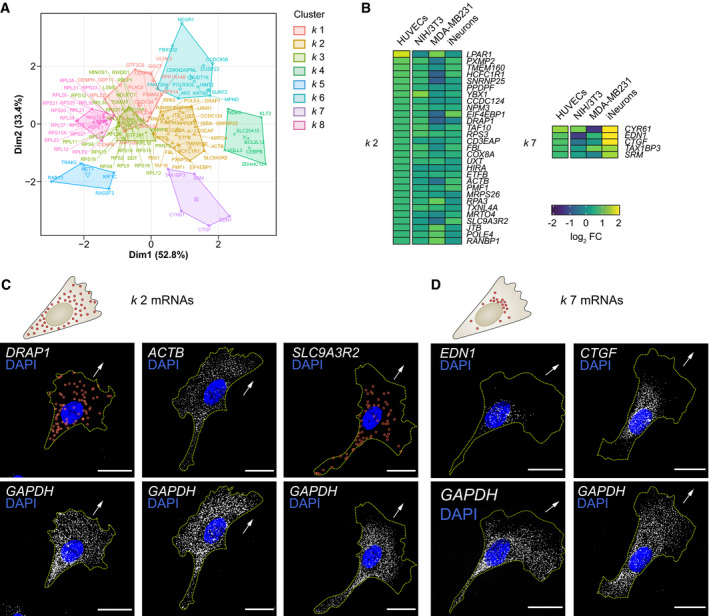
- Principal component plot depicting the k‐means clustering analysis of mRNAs enriched across cell protrusion types.
- Detail of the heat map shown in Fig 1C representing log2 fold change (FC) levels (protrusions over cell bodies) of mRNAs present in clusters k 2 and k 7. The corresponding HUVEC log2 FC levels are shown in parallel.
- Top: distribution pattern of mRNAs clustered in k 2. Bottom: smFISH co‐detection of k 2 mRNAs and GAPDH in HUVECs.
- Top: distribution pattern of mRNAs clustered in k 7. Bottom: smFISH co‐detection of k 7 mRNAs and GAPDH in HUVECs.
Clustering of RNAseq datasets defines an RNA motif enriched in 3′UTR sequences that target k 5 mRNAs to protrusions
Considering that the polarisation of cluster k 5 mRNAs was particularly acute, highly stereotyped and uniquely conserved amongst cell types (Fig 1C–G), we hypothesised that these transcripts employed common targeting mechanisms. Indeed, using the MEME Suite (Bailey & Elkan, 1994) we detected consistent repeat use of a conserved sequence motif in the 3′UTRs of all k 5 transcripts (Fig 2A and B, and Table EV3). This motif distinguished the k 5 mRNAs from other identified mRNA clusters, which contained the motif at much lower frequency (Fig 2C). Moreover, this motif was striking in its clustering as five repeats within a short 3′UTR region of RAB13, a known polarised mRNA (Mili et al, 2008; Jakobsen et al, 2013; Moissoglu et al, 2019) (Fig 2A). To interrogate its function, we tagged the non‐localising human HBB coding sequence with both the RAB13 3′UTR and the reporter MS2 hairpin repeats (Bertrand et al, 1998; Mili et al, 2008; Fig 2D). Following co‐expression with the MS2 capping protein (MCP)‐GFPnls that is usually confined to the nucleus (Fusco et al, 2003), the localisation of MS2‐tagged mRNAs can be monitored through changes to the spatial distribution of the GFP signal (Fig 2D). Potent localisation properties of the motif‐containing RAB13 3′UTR were confirmed upon expression of the MS2 reporter system in ECs (Fig 2E). Using this approach in combination with truncations or deletions of the RAB13 3′UTR, we identified a minimal 192‐nt LE encompassing four motif repeats that was both necessary and sufficient to exclusively polarise mRNA at motile EC protrusions (region 90–282 in Fig 2E). Furthermore, similar truncation and deletion analysis of the remaining k 5 mRNAs confirmed that these transcripts employ common cis‐regulatory mechanisms, as mRNA targeting ability was consistently reliant on motif‐containing 3′UTR regions (Fig 2F). Indeed, precise deletion of a single motif in TRAK2 was sufficient to entirely block mRNA targeting (Fig 2F). Of note, unlike TRAK2, more than two functional motifs were required to drive RAB13 polarisation, as deletions of individual motifs were tolerated and constructs containing two motifs were insufficient to drive RAB13 mRNA localisation (Fig 2E). Hence, distinct cluster k 5 mRNAs may exhibit different minimal requirements for the number of motifs needed to drive targeting.
Figure 2. Clustering of RNAseq datasets defines an RNA motif enriched in 3′UTR sequences that target k 5 mRNAs to protrusions.
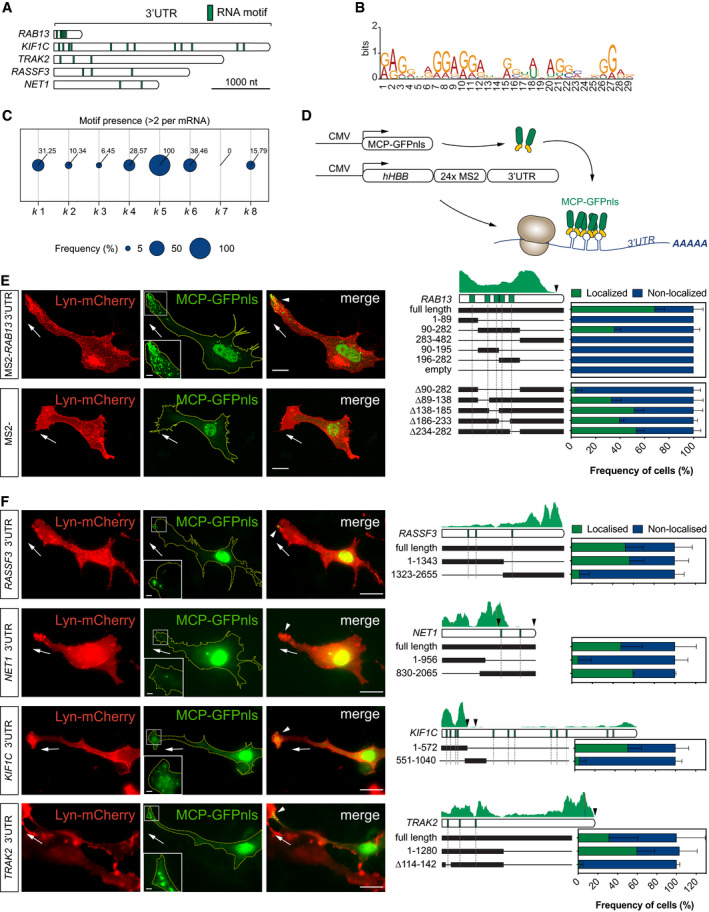
- Diagram of k 5 mRNA 3′UTRs and relative positions of the RNA motif shared between transcripts.
- RNA motif over‐represented in k 5 mRNA 3′UTRs.
- Frequency of mRNAs within each k‐means cluster containing at least 2 of the RNA motif over‐represented in k 5 mRNAs.
- Scheme depicts the in vitro MS2 system strategy. CMV promoter‐driven expression of MCP‐GFPnls and hHBB‐24xMS2‐tagged RAB13 3′UTR. The visualisation of MCP‐GFPnls bound to 24xMS2 allows the identification of the minimal region in the 3′UTR of RAB13 necessary for its localisation.
- Left: representative subconfluent motile cells co‐transfected with plasmids expressing Lyn‐mCherry, MCP‐GFPnls and 24xMS2‐RAB13 3′UTR or 24xMS2. Right: percentage of cells with MCP‐GFPnls localised to protrusions when co‐transfected with full length or deletion versions of RAB13 3′UTR (n ≥ 3 experiments).
- Left: representative cells co‐transfected with plasmids expressing Lyn‐mCherry, MCP‐GFPnls and 24xMS2‐k 5 3′UTRs. Right: percentage of cells with MCP‐GFPnls localised to protrusions when co‐transfected with full length or deletion versions of k 5 3′UTR (n ≥ 3 experiments).
CRISPR‐Cas9 excision of the 3′UTR localisation element of RAB13 disrupts mRNA targeting
As RAB13 was the only identified RAB small GTPase to exhibit such mRNA polarisation (Fig 1B), we hypothesised that the identified LE and targeting of the transcript were critical for RAB13 protein function in motile cells. However, studies probing the precise function of endogenous polarised mRNAs in motile cells are currently lacking, predominantly due to difficulties identifying targeting motifs and the potential propensity for genomic manipulation to perturb transcript stability and/or translation. However, precise genomic excision of the RAB13 minimal LE in ECs using CRISPR‐Cas9 tools did not perturb RAB13 mRNA or protein expression (Fig 3A–G). ECs were transfected with both a GFP‐expressing plasmid and CRISPR‐Cas9 ribonucleoprotein complexes targeting the 90–282‐nt LE in exon 8 of RAB13, prior to expansion of GFP‐expressing clones (Fig 3A). Clones were then selected for either biallelic deletion of the LE (∆LE) or presence of the full‐length RAB13 3′UTR (Wt) (Fig 3B) and were sequenced to confirm specific deletion of the LE in mutated clones (Figs 3C and EV2A). Moreover, RNAseq analysis verified that overall splicing of RAB13 mRNA was unaffected by genomic excision of the LE (Fig 3D) and confirmed the high specificity of these CRISPR‐Cas9 tools, as no nucleotide mismatches were observed at any putative low‐frequency off‐target sites (Fig EV2B). Importantly, excision of the LE did not perturb RAB13 mRNA levels or protein expression (Fig 3E–G), but did eradicate the polarised spatial pattern of RAB13 localisation, such that the transcript became diffusely distributed in ECs similar to GAPDH (Fig 3H). In particular, quantification of the PI of co‐detected RAB13 and GAPDH mRNAs revealed that loss of the LE consistently reduced RAB13 polarisation to levels equivalent to GAPDH controls (Fig 3I and J). Finally, correlation of RAB13 mRNA spot count versus RAB13 mRNA PI revealed that mRNA polarisation is actually independent of total mRNA levels and further confirmed that it is perturbed upon excision of the LE (Fig 3F). Hence, genomic excision of the 3′UTR LE specifically perturbs RAB13 mRNA polarisation.
Figure 3. CRISPR‐Cas9 excision of the 3′UTR localisation element of RAB13 disrupts mRNA targeting.
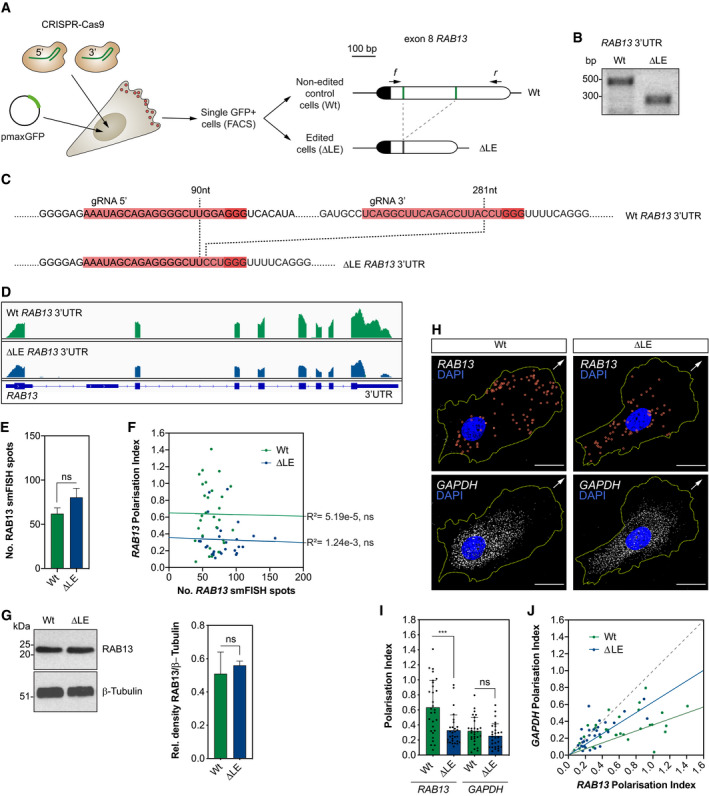
- CRISPR‐Cas9 strategy to derive HUVECs with an excision of the LE in the RAB13 3′UTR (∆LE) and parallel generation of wild‐type (Wt) control cells. The Wt RAB13 exon 8 is represented with its coding sequence in dark and the 3′UTR in clear boxes. The 5′ and 3′ gRNA‐targeted regions are represented with green lines. Arrows: relative positions of the forward (f) and reverse (r) PCR primers used to identify HUVECs with CRISPR‐Cas9-mediated excision of the LE.
- Representative genotyping PCR demonstrates the band size shift in ∆LE HUVECs.
- Detailed DNA sequence depicting nucleotide positions within the RAB13 3′UTR of Wt and ∆LE HUVECs.
- Wt and ∆LE HUVEC RNAseq mapped reads depicting RAB13 exon usage.
- Quantification of RAB13 mRNA smFISH spot number in Wt and ∆LE HUVECs (n = 3 experiments; ns: not significant; unpaired t test).
- Number of RAB13 mRNA smFISH spots plotted against the respective Polarisation Index (PI) (n = 29 cells; ns: not significant; linear regression).
- Left: representative Western blotting (WB) of Wt and ∆LE HUVECs. Right: densitometry analysis of WB data (n = 3 samples; ns: not significant; unpaired t test).
- smFISH co‐detection of RAB13 and control GAPDH in Wt and ∆LE motile HUVECs cultured under subconfluent conditions.
- PI of RAB13 and GAPDH co‐detected in Wt and ∆LE HUVECs (n = 29 cells; ***P < 0.001, ns: not significant; Mann–Whitney test).
- RAB13 PI plotted against respective GAPDH PI. The slope of the coloured lines represents the average RAB13/GAPDH PI ratio; the dashed grey line represents a 1:1 ratio (n = 29 cells).
Figure EV2. CRISPR‐Cas9 editing of the RAB 3′UTR in vitro and in vivo does not generate off‐target mutations.
- Chromatogram confirming the excision of the LE within RAB13 3′UTR in HUVECs.
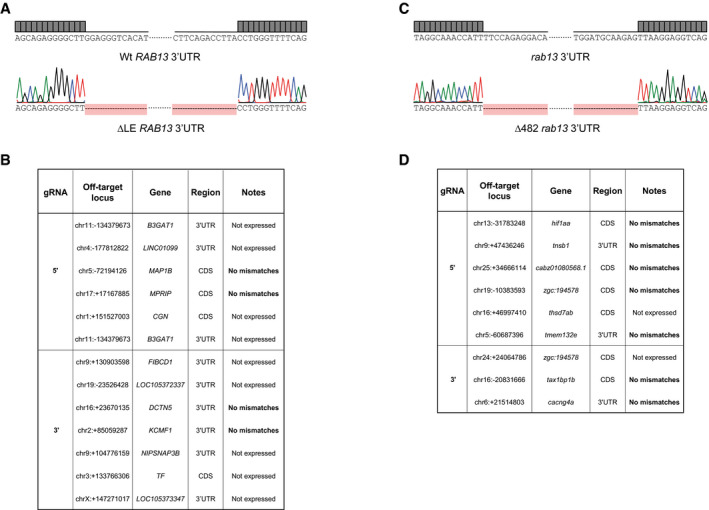
- List of predicted CRISPR‐Cas9 off‐target genes and RNAseq mismatch detection in CRISPR‐Cas9-derived HUVEC clones (n = 1 each genotype).
- Chromatogram confirming the CRISPR‐Cas9-mediated excision of 482‐nt within the rab13 3′UTR in zebrafish embryos.
- List of predicted CRISPR‐Cas9 off‐target genes and RNAseq mismatch detection in Tg(kdrl:EGFP) rab13 +/+ and rab13 ∆3′UTR/∆3′UTR embryos (n = 2 each genotype).
RAB13 mRNA polarisation spatially orients filopodia dynamics
RAB13 is an established modulator of cortical F‐actin crosslinking and cytoskeletal remodelling at leading front of migrating cells, via interaction with its effector protein, MICAL‐L2 (Sakane et al, 2012, 2013; Ioannou et al, 2015). Consistent with this function, live‐cell imaging of RAB13 3′UTR dynamics employing MS2 reporter constructs revealed enriched targeting of mRNA to sites of incipient filopodia formation in cell protrusions, suggesting a tight spatial coupling between RAB13 mRNA localisation and RAB13 protein activity (Fig 4A–C and Movie EV1). Furthermore, quantification revealed that filopodia preferentially emerged in close proximity to GFP particles transported by the RAB13 3′UTR reporter, but not with random cytoplasmic spot positions (Fig 4B). In addition, there was a putative causal relationship between RAB13 mRNA proximity and increased filopodia stability (Fig 4C). Likewise, induction of cell migration drove a significant increase in the levels and polarisation of RAB13 mRNA in vitro (Fig EV3), further indicating a dynamic involvement in leading‐edge remodelling and establishment of cell polarity. Hence, these data revealed that polarisation of RAB13 mRNA may spatially compartmentalise RAB13‐mediated F‐actin remodelling to orient motile cell polarity. Consistent with this hypothesis, loss of RAB13 mRNA polarisation—but not loss of expression—was indeed sufficient to depolarise filopodia dynamics in motile ECs (Fig 4D–G). When co‐cultured with fibroblasts to mimic polarised blood vessel sprouting (Hetheridge et al, 2019), Wt ECs exhibited highly polarised filopodia extensions biased towards the leading edge of motile cells (Fig 4D and F). In contrast, filopodia in ECs lacking the RAB13 LE (∆LE) were no longer spatially compartmentalised and became ectopically homogenously distributed along the distal–proximal cell axis (Fig 4D and F). Importantly, these observations were consistent between individual Wt and ∆LE CRISPR‐Cas9 clones (Fig 4G). Consequently, mutant ECs exhibited a significant increase in overall filopodia frequency (Fig 4E). Hence, these data indicate that tight control of RAB13 mRNA localisation spatially specifies a polarised domain of filopodia extension in motile cells (Fig 4H).
Figure 4. RAB13 mRNA polarisation spatially orients filopodia dynamics.
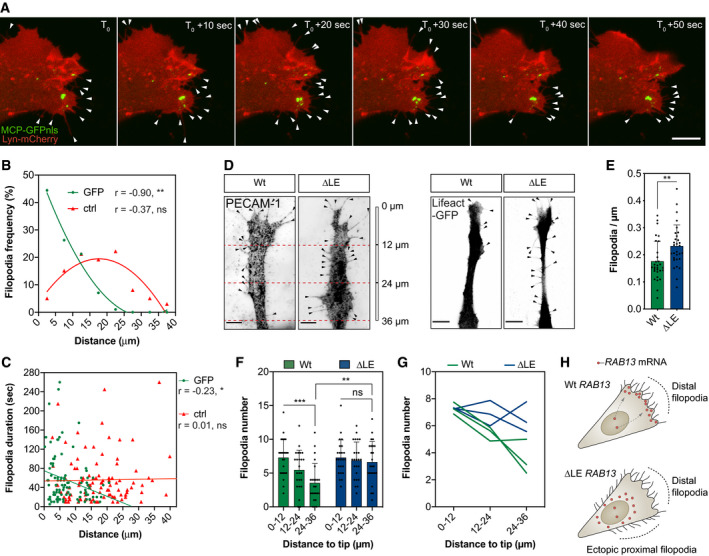
- Representative time‐lapse microscopy of a bEnd.3 cell co‐transfected with plasmids expressing Lyn‐mCherry, MCP‐GFPnls and 24xMS2‐RAB13 3′UTR.
- Frequency of newly formed filopodia formed within 5‐μm intervals relative to the nearest MCP‐GFPnls particle or a randomised (ctrl) position (n = 99 filopodia; **P < 0.01, ns: not significant; Pearson's r correlation).
- Distance of newly formed filopodia to MCP‐GFPnls or a ctrl position plotted against filopodia duration (n = 99 filopodia; *P < 0.05, ns: not significant; Spearman's r correlation).
- Wt and ∆LE HUVECs co‐cultured on fibroblast monolayers. Endothelial cells were identified either with an antibody against the endothelial cell marker PECAM‐1 (left) or through expression of a nucleofected plasmid encoding the cytoskeletal marker Lifeact‐GFP (right).
- Number of filopodia detected in co‐cultured HUVECs (n = 30 cells; **P < 0.01; unpaired t test).
- Number of filopodia detected in co‐cultured HUVECs within 12‐μm intervals relative to cell distal tip (n = 30 cells; **P < 0.01, ***P < 0.001, ns: not significant; one‐way ANOVA with Bonferroni's correction).
- Number of filopodia detected in individual clones of co‐cultured HUVECs within 12‐μm intervals relative to cell distal tip.
- Illustration of the spatial relationship between RAB13 mRNA localisation and sites of filopodia production.
Figure EV3. Induction of endothelial cell collective migration drives RAB13 mRNA polarisation in leader cells.
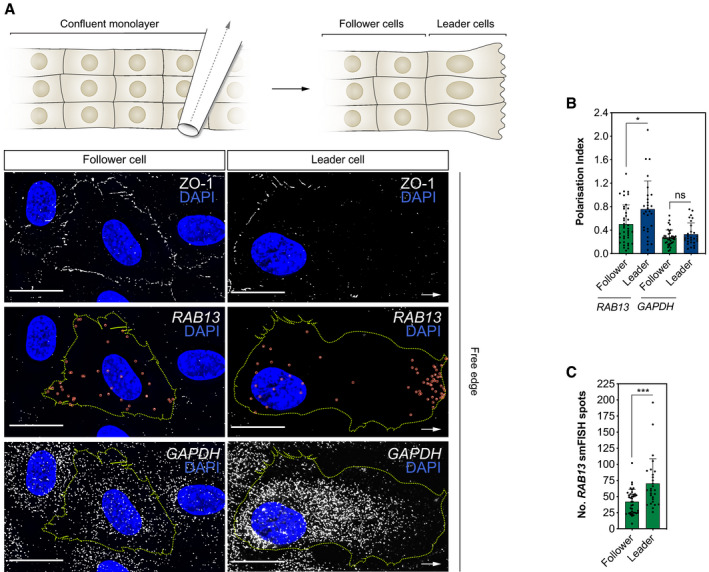
- Top: scratch wound assay generates a free edge on a confluent monolayer of HUVECs and encourages cell migration. Bottom: smFISH co‐detection of RAB13 mRNA and GAPDH mRNA in representative HUVECs migrating in a scratch wound assay. ZO‐1 immunolabelling defines cell boundaries.
- Polarisation Index of RAB13 and GAPDH co‐detected in HUVECs cultured in scratch wound assays (n ≥ 28 cells; *P < 0.05, ns: not significant; Mann–Whitney test).
- Quantification of the number of RAB13 mRNA smFISH spots per cell (n ≥ 28 cells; ***P < 0.001; Mann–Whitney test). Leader: cells identified at the edge of the scratch; follower: cells identified in confluent regions adjacent to leader cells.
mRNA polarisation achieves spatial compartmentalisation of RAB13 translation and protein function
These striking observations suggested that RAB13 mRNA polarisation acts to exclusively spatially compartmentalise RAB13‐mediated filopodia extension at distal sites. As such, targeting of RAB13 mRNA and local translation could effectively block ectopic protein function at inappropriate subcellular loci to orient motile cells. However, this could only be achieved if the sites of RAB13 mRNA localisation, translation and protein function were all tightly spatially coupled. Indeed, such coupling may be consistent with long‐standing proposals that newly translated RABs form a discrete protein pool from mature RABs, potentially with distinct interaction partners (e.g. specific RAB escorting proteins and GDP dissociation inhibitors) and separate biological functions (Pfeffer et al, 1995; Shen & Seabra, 1996; Seabra et al, 2002). Hence, local translation of polarised RAB13 transcript may generate nascent protein with distinct functional roles to mature RAB13 at specific subcellular sites, thus achieving tight spatial compartmentalisation of RAB13‐mediated membrane remodelling. As predicted, such coupling of polarised RAB13 mRNA localisation with local translation was confirmed in EC protrusions upon detection of nascent protein using puromycinilation‐proximity ligation assays (Puro‐PLA) (tom Dieck et al, 2015). ECs were cultured on Transwells and cell bodies removed prior to pulse labelling with puromycin to exclude detection of nascent proteins transported from the cell body to protrusions (Fig 5A). Isolated EC protrusions readily incorporated puromycin, which could be blocked upon pre‐incubation with the translation inhibitor anisomycin (Fig EV4), indicating active protein translation at the leading edge of migrating ECs. Importantly, Puro‐PLA on isolated EC protrusions using antibodies recognising puromycin and RAB13 revealed numerous distinct punctae corresponding to newly synthesised RAB13, unlike anisomycin pre‐treated and antibody‐free controls (Fig 5B and C). Hence, polarised targeting of RAB13 mRNA to motile cell protrusions drives local RAB13 translation. Moreover, spatial control of mRNA polarisation and local translation was coupled to regional compartmentalisation of RAB13 protein function, as loss of endogenous RAB13 specifically disrupted filopodia dynamics only at distal sites of mRNA targeting (Fig 5E–H). The siRNA‐mediated knockdown of RAB13 expression (Fig 5E) did not perturb RAB13‐independent filopodia at proximal regions in ECs (Fig 5H), but significantly depleted filopodia numbers at distal sites, as is particularly obvious in Fig 5F. Consequently, ECs exhibited overall reduced numbers of filopodia upon RAB13 knockdown (Fig 5G). This was not simply a consequence of spatial targeting of protein to the leading edge, as immunofluorescence assays revealed that RAB13 was homogeneously distributed throughout migrating cells (Fig 5D), indicating that in contrast to the mRNA encoding it, RAB13 steady‐state protein is not polarised. Alternatively, it was the location of RAB13 mRNA itself that defined the domain of RAB13‐dependent filopodia dynamics, as excision of the LE and diffuse mislocalisation of RAB13 mRNA was sufficient to drive ectopic depolarised filopodia (Fig 4D–G). Hence, the site of RAB13 mRNA localisation, translation and protein function appears to be tightly spatially coupled in migrating cells. Consequently, polarisation of RAB13 transcript forms a molecular compass that achieves precise subcellular compartmentalisation of protein function, defines a polarised domain of filopodia extension and ultimately orients motile cell polarity (Fig 5I).
Figure 5. mRNA polarisation achieves spatial compartmentalisation of RAB13 translation and protein function.
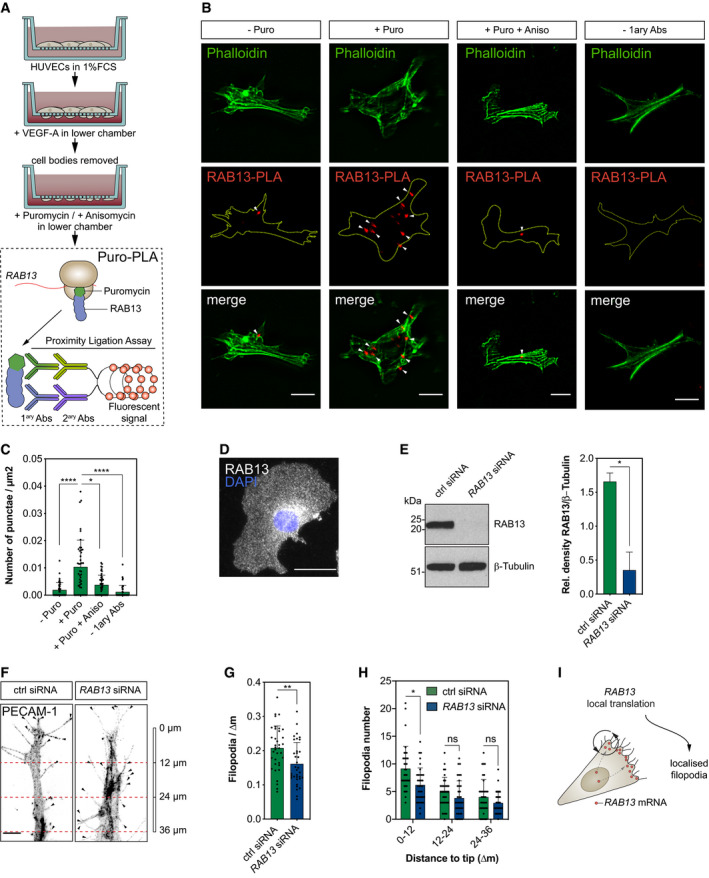
- Strategy used to detect local protein synthesis in protrusions formed by HUVECs migrating through Transwell membranes.
- Representative Puro‐PLA experiments detecting newly synthesised RAB13 in HUVEC protrusions present in the lower side of Transwell membranes. Puro: puromycin; Aniso: anisomycin; 1ary Abs: primary antibodies.
- Quantification of RAB13 Puro‐PLA punctae normalised to protrusion area (n ≥ 40 protrusions; *P < 0.05, ****P < 0.0001; Kruskal–Wallis test with Dunn's correction).
- Representative RAB13 IF assay on migrating HUVECs.
- Left: representative Western blotting (WB) of siRNA‐transfected HUVECs. Right: densitometry analysis of WB data (n = 3 samples; *P < 0.05; paired t test).
- Control (ctrl) and RAB13 siRNA‐treated HUVECs co‐cultured on fibroblast monolayers. Endothelial cells were identified with an antibody against the endothelial cell marker PECAM‐1.
- Number of filopodia detected in co‐cultured HUVECs (n ≥ 35 cells; **P < 0.01; unpaired t test).
- Number of filopodia detected in co‐cultured HUVECs within 12‐μm intervals relative to cell distal tip (n ≥ 35 cells; *P < 0.05, ns: not significant; Kruskal–Wallis test with Dunn's correction).
- Illustration of the spatial relationship between the sites of RAB13 mRNA localisation, local translation and RAB13 protein‐mediated filopodia distribution.
Figure EV4. Protein translation in endothelial cell protrusions.

Immunofluorescence analysis of HUVEC protrusions generated on the underside of Transwell membranes and exposed to puromycin after cell body removal. Yellow dashed lines outline protrusion borders; scale bars = 10 μm.
The 3′UTR of rab13 targets mRNA to endothelial cell protrusions in vivo
Although a widespread phenomenon, the functional role for localised mRNAs in tissue migration and vertebrate morphogenesis remains unexplored. Hence, having defined a key role for mRNA polarisation in the spatial control of EC behaviour in vitro, we then sought to define the broader relevance of this phenomenon to modulation of tissue dynamics in vivo. The production of polarised filopodia protrusions is a characteristic hallmark of motile endothelial tip cells, which lead new blood vessel branches during angiogenesis (Gerhardt et al, 2003; Isogai et al, 2003). As such, using live‐cell imaging approaches in the zebrafish model system, we probed the function of rab13 mRNA polarisation in the control of tip cell behaviour and angiogenesis in vivo. Firstly, we generated a novel vascular‐specific MCP‐GFPnls transgenic strain, Tg(fli1ep:MCP‐GFPnls), and monitored the targeting dynamics of a MS2‐tagged rab13 3′UTR reporter during intersegmental vessel (ISV) angiogenesis (Isogai et al, 2003; Fig 6A). Dynamic accumulation of MCP‐GFPnls adjacent to or within filopodia at the leading edge of ISV tip cells revealed that rab13 mRNA localisation in vivo closely mirrored the 3′UTR‐driven polarisation of human RAB13 mRNA in vitro (Fig 6B and C; Movies EV2 and EV3). Thus, the targeting function of the RAB13/rab13 3′UTR is highly conserved, despite rather low sequence conservation (Fig EV5B and C). Importantly, polarised localisation of MCP‐GFPnls was not observed in the absence of the MS2‐tagged rab13 3′UTR (Fig EV5A), confirming the presence of LEs within this region. Moreover, the polarised targeting of rab13 mRNA was retained in less‐motile ISV stalk cells, which trail tip cells, although at less dynamic and more discrete foci (Fig 6D; Movie EV4). Hence, the polarised targeting of RAB13/rab13 mRNA in motile cells is highly conserved between species and in tissues.
Figure 6. The 3′UTR of rab13 targets mRNA to endothelial cell protrusions in vivo .
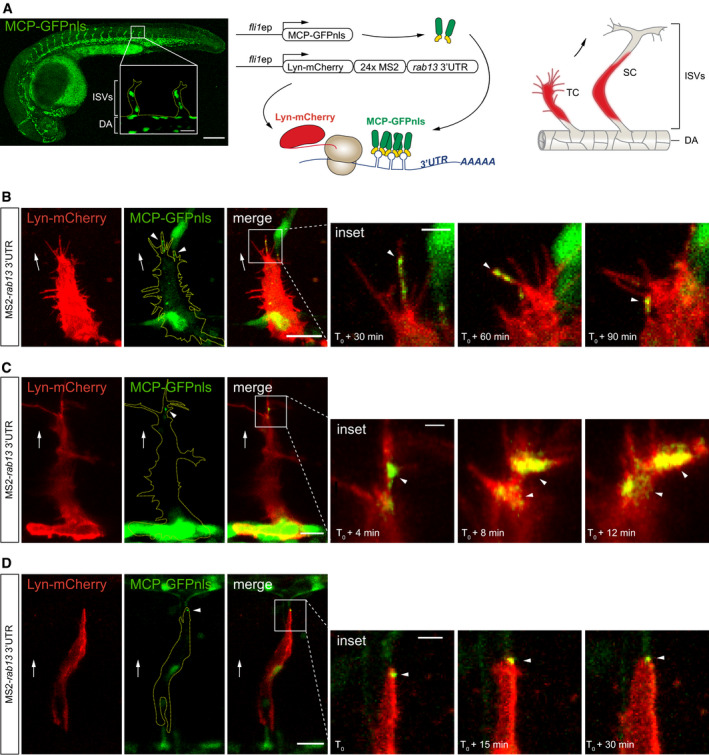
-
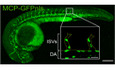
ALeft: Tg(fli1ep:MCP‐GFPnls) zebrafish embryo at 26 h post‐fertilisation (hpf) displaying vascular‐specific expression of MCP‐GFPnls. Inset shows the nuclear expression of MCP‐GFPnls in the intersomitic vessels (ISVs) sprouting from the dorsal aorta (DA). Middle: scheme depicts the in vivo MS2 system strategy with fli1 enhancer/promoter (fli1ep)‐driven expression of reporter constructs, simultaneous translation of Lyn‐mCherry reporter and binding of MCP‐GFPnls to 24xMS2‐rab13 3′UTR. Right: scheme illustrates ISV cells expressing Lyn‐mCherry imaged in panels B–D. TC: tip cell; SC: stalk cell.
-
B–DTime‐lapse microscopy of Tg(fli1ep:MCP‐GFPnls) tip and stalk cells displaying mosaic expression of Lyn‐mCherry‐24xMS2‐rab13 3′UTR in ISV cells.
Figure EV5. The 3′UTR of rab13 shows low conservation across species whilst retaining mRNA localisation potential.
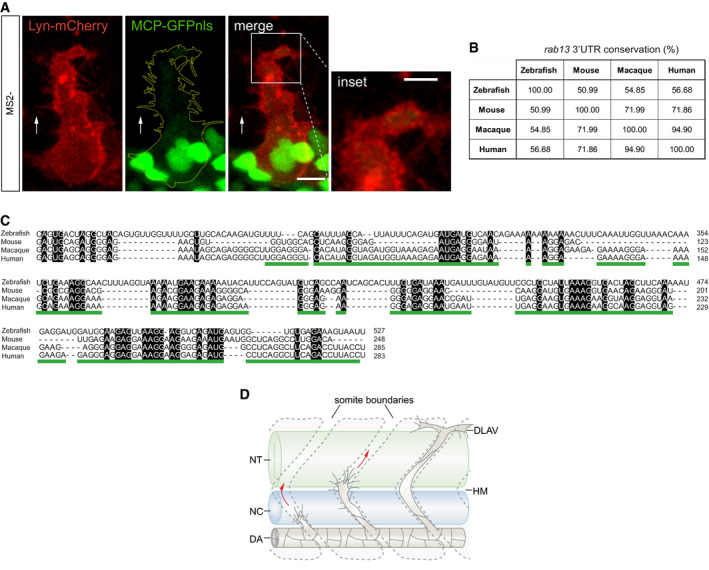
- Tg(fli1ep:MCP‐GFPnls) tip cell expressing a control Lyn‐mCherry‐24xMS2 construct.
- Percentage identity matrix of rab13 3′UTR orthologue sequences.
- Multiple sequence alignment between rab13 3′UTR orthologues. Black boxes indicate absolute nucleotide similarity. The human RAB13 3′UTR localisation element is underlined in green.
- Scheme depicts stages of zebrafish ISV sprouting. DA: dorsal aorta; DLAV: dorsal longitudinal anastomotic vessel; HM: horizontal myoseptum; NC: notochord; NT: neural tube.
CRISPR‐Cas9 editing of the zebrafish rab13 3′UTR perturbs mRNA polarisation
Next, we sought to determine whether rab13 mRNA localisation in vivo is functionally implicated in blood vessel spouting. Similar to in vitro experiments, we performed CRISPR‐Cas9‐mediated excision of a fragment within the rab13 3′UTR locus (∆3′UTR) to further confirm the presence of LEs and their potential role in rab13 mRNA polarisation in vivo. Microinjection of zebrafish embryos with CRISPR‐Cas9 ribonucleoprotein complexes targeting exon 8 of rab13 was sufficient to generate germline rab13 ∆3′UTR/∆3′UTR mutants lacking 482‐nt of the rab13 3′UTR, as confirmed by sequencing (Figs 7A–C and EV2C). Additional RNAseq analysis of mutant embryos verified that overall splicing of rab13 mRNA was unaffected by genomic excision of the LE (Fig 7D) and confirmed the high specificity of CRISPR‐mediated excision, as no nucleotide mismatches were observed at any putative low‐frequency off‐target sites (Fig EV2D). Moreover, genomic excision of the rab13 LE had no effect on mRNA levels (Fig 7E), although protein levels could not be tested due to a lack of good antibodies against zebrafish Rab13. Importantly, smFISH applied to explanted endothelial cells from dissociated rab13 ∆3′UTR/∆3′UTR mutant embryos confirmed that rab13 mRNAs lacking these LEs were more diffusely distributed (Fig 7F–H), similar to observations in human ECs (Fig 3H–J). In contrast, control kdr mRNAs displayed unperturbed PI measurements in rab13 mutant versus Wt ECs (Fig 7F and G). Hence, we reveal a previously unappreciated and conserved role for 3′UTR LEs in the dynamic polarisation of rab13 mRNA during cell migration.
Figure 7. CRISPR‐Cas9 editing of the zebrafish rab13 3′UTR perturbs mRNA polarisation.
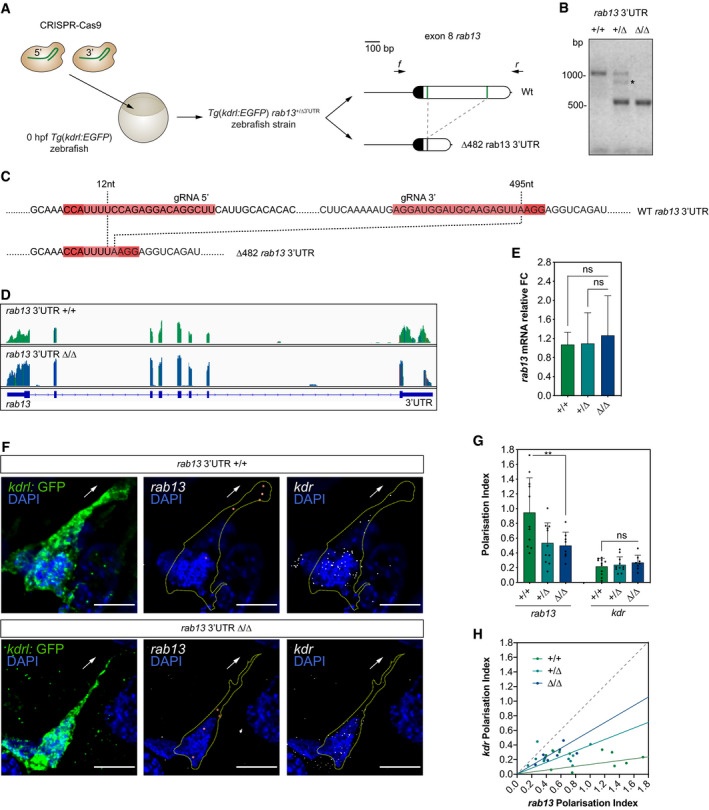
- CRISPR‐Cas9 strategy to generate the Tg(kdrl:EGFP) rab13 +/∆3′UTR zebrafish strain. The wild‐type (Wt) rab13 exon 8 is represented with its coding sequence in dark and the 3′UTR in clear boxes; the 5′ and 3′ gRNA‐targeted regions are represented with green lines. Arrows: relative positions of the forward (f) and reverse (r) PCR primers used to identify animals with CRISPR‐Cas9-mediated deletions (∆) in the rab13 3′UTR.
- Representative genotyping PCR demonstrates the band size shift in zebrafish harbouring a ∆482 rab13 3′UTR. Asterisk marks a heteroduplex formed between Wt and ∆482 rab13 3′UTR PCR amplicons.
- Detailed DNA sequence depicting nucleotide positions within the Wt and ∆482 rab13 3′UTR.
- RNAseq mapped reads depicting rab13 exon usage in Tg(kdrl:EGFP) rab13 +/+ and rab13 ∆3′UTR/∆3′UTR zebrafish embryos. Coloured lines indicate SNPs.
- qPCR analysis of rab13 mRNA levels in individual 26–28 hpf clutch‐matched sibling embryos (n ≥ 9 embryos; ns: not significant; Kruskal–Wallis test with Dunn's correction).
- smFISH detection of rab13 and kdr mRNA in cultured GFP‐expressing endothelial cells extracted from 48 hpf Tg(kdrl:EGFP) rab13 +/+ and rab13 ∆3′UTR/∆3′UTR zebrafish embryos.
- Polarisation Index (PI) of rab13 and kdr detected by smFISH in individual zebrafish cells (n ≥ 8 cells; **P < 0.01, ns: not significant; one‐way ANOVA with Bonferroni's correction).
- rab13 PI plotted against respective kdr PI. The slope of the coloured lines represents the average rab13/kdr PI ratio; the dashed grey line represents a 1:1 ratio (n ≥ 8 cells).
rab13 mRNA polarisation orients blood vessel morphogenesis
During ISV branching, migrating tip cells must make key directional decisions, particularly when negotiating the multi‐tissue junction of the horizontal myoseptum (Lu et al, 2004; Torres‐Vazquez et al, 2004; Lamont et al, 2009) (HM; Fig EV5D). Hence, we hypothesised that mRNA localisation‐mediated orientation of EC filopodia may indeed generate spatial cues that direct vascular tissue movement. Consistent with a key role for mRNA polarisation in the spatial coordination of vascular morphogenesis, live‐cell imaging of ISVs branching in Wt and rab13 ∆3′UTR/∆3′UTR embryos revealed that loss of rab13 polarisation severely perturbed tip cell path‐finding decisions (Fig 8A and B). Unlike ISVs in Wt and rab13 +/∆3′UTR embryos that efficiently negotiated their way past the HM position, ISVs in rab13 ∆3′UTR/∆3′UTR mutants struggled with this directional decision, resulting in a sevenfold increase in tip cells exhibiting ectopic misdirected branches (Fig 8A and B). Of interest, rab13 ∆3′UTR/∆3′UTR mutants were viable with no detectable gross defects in other embryonic or vascular tissues, indicating a highly specific ISV phenotype. More importantly, rab13 mRNA stability was unperturbed in rab13 ∆3′UTR/∆3′UTR mutant embryos (Fig 7E), indicating that observed defects were not due to decreased rab13 expression but a consequence of perturbed mRNA localisation. Thus, we provide the first in vivo evidence that spatial targeting of mRNAs and precise compartmentalisation of protein function generate key directional cues that orient motile cells during vertebrate tissue morphogenesis (Fig 8C).
Figure 8. rab13 mRNA polarisation orients blood vessel morphogenesis.
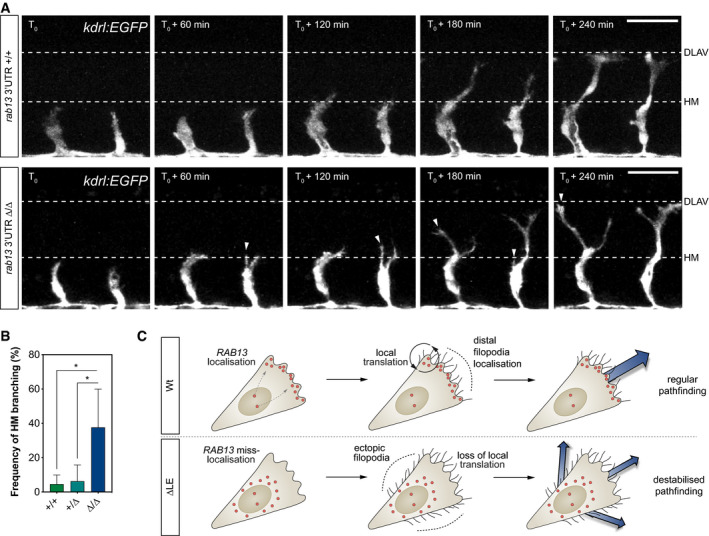
- Time‐lapse confocal microscopy of representative Tg(kdrl:EGFP) rab13 +/+ and rab13 ∆3′UTR/∆3′UTR embryos. DLAV: dorsal longitudinal anastomotic vessel; HM: horizontal myoseptum.
- Frequency of ISV ectopic branching occurring at the HM (n = 4 experiments; *P < 0.05; one‐way ANOVA with Bonferroni's correction).
- Illustration of the role for RAB13 mRNA localisation, local translation and compartmentalisation of RAB13 function in defining the orientation of EC filopodia dynamics, motile EC polarity and blood vessel pathfinding.
Discussion
Whilst it is well established that numerous transcripts are targeted to the leading protrusions of motile cells (Herbert & Costa, 2010), how this regulates translated protein function, its importance for cell migration and the in vivo relevance of this phenomenon are poorly understood. Here, using gene editing to modulate subcellular mRNA targeting, we reveal that tight spatial coupling of mRNA localisation, translation and protein function achieves precise subcellular compartmentalisation of protein action and prevents ectopic protein functionality at inappropriate subcellular loci. We find that such mRNA‐mediated spatial compartmentalisation of RAB13 activity serves to define a polarised domain of filopodia remodelling that orients motile cells. Moreover, using unique tissue‐specific reporters of mRNA localisation in intact vertebrates, alongside gene editing, we uncover a key role for rab13 mRNA localisation in the coordination of cell pathfinding during tissue morphogenesis. Hence, we define mRNA polarisation as a novel paradigm for the spatial control of motile cell polarity and oriented tissue movement in vivo.
Moreover, our findings lend weight to recent observations that newly synthesised protein can have a distinct functionality to pre‐existing protein (Kim et al, 2020). Considering that steady‐state RAB13 protein is homogeneously distributed in migrating ECs, our work indicates that nascent protein must have distinct functional properties to the mature protein pool that enables tight spatial coupling of RAB13 translation and local filopodia remodelling. Indeed, in a parallel study published back‐to‐back with our work, Moissoglu et al (2020) show that nascent RAB13 co‐translationally interacts with its exchange factor, RABIF, to drive local activation of newly synthesised protein in MDA‐MB‐231 cells. In their report, Moissoglu et al (2020) demonstrate that modulation of mRNA localisation using antisense oligonucleotides did not disrupt RAB13 expression, translation or steady‐state protein localisation, but shifted the site and levels of co‐translational RAB13 activation. Consequently, spatial manipulation of RAB13 mRNA targeting fundamentally defined the location of RAB13 protein action and perturbed cell protrusion in migration. Hence, the work by Moissoglu et al (2020) provides a convincing mechanistic basis for how RAB13 localisation ultimately orients motile cell polarity and tissue movement in vivo, via precise spatial control of co‐translational interactions with exchange factors that define the site of GTPase activation.
Considering that RAB13 is one of only five mRNAs exhibiting conserved polarisation in all cell types tested, the function of RAB13 mRNA targeting may be a universally conserved mechanism for spatial coordination of complex morphogenetic events. Moreover, it is striking that all five‐cluster k 5 mRNAs encode highly dynamic membrane trafficking and/or small GTPase‐regulating proteins, all known to modulate cell motility. Hence, for classes of proteins normally constant in motion, mRNA polarisation may be essential to spatially compartmentalise and precisely fix their site of function. It is tempting to speculate that cluster k 5 mRNA co‐targeting and localised translation may participate in a coordinated effort to modulate actin dynamics and/or membrane protrusion at the leading edge. As such, perturbation of the localisation mechanisms that transport RAB13 and other cluster k 5 mRNAs may be expected to generate a more acute phenotype than perturbation of RAB13 targeting alone. Indeed, other studies have provided evidence that mRNAs sharing subcellular compartments can encode subunits of common protein complexes involved in actin remodelling (Mingle et al, 2005) or components of related chemotaxis pathways (Hotz & Nelson, 2017). Nevertheless, the functional role of mRNA co‐localisation in the context of cell migration remains largely elusive. As ever‐increasing technological advances continue to unveil the nature of compartmentalised transcriptomes, this will shed further light on the patterns of co‐localised mRNAs (Eng et al, 2019; Xia et al, 2019) and protein synthesis (Chouaib et al, 2020) that likely underpin key events directing cell migration.
Whilst it is well established that polarised trafficking of RAB13 and other cluster k 5 mRNAs are attributed to their interaction with APC, a microtubule plus‐end associated protein that escorts mRNAs to the leading edge of motile cells (Mili et al, 2008; Wang et al, 2017), the function of APC itself in cell migration is unclear. Despite reports on the importance of APC for EC migration (Harris & Nelson, 2010), knockdown of APC does not impact protrusion formation during migration in all cell types (Mili et al, 2008). Yet, from our work and that of Moissoglu et al (2020), it is clear that the function of RAB13 mRNA polarisation in the coordination of cell movement is itself conserved between distinct cell types and organisms. As such, a full explanation for the context dependency of APC function has yet to be defined, but may indicate differences in the mode of motility employed by distinct cell types, the types of dynamic protrusions employed (be they RAB13‐dependent or not) or even hint at the use of currently unknown APC‐independent mRNA transport mechanisms in cell migration.
Finally, this work reveals an unexpected spatial diversity to identified clusters of polarised mRNAs. Considering our observations that the sites of mRNA targeting and protein function are tightly coupled, this raises the exciting possibility that other distinct mRNA distributions, such as the perinuclear localisation of cluster k 7, reflect even broader functionalities for compartmentalised gene expression/function in the coordination of diverse aspects of tissue development, health and disease.
Materials and Methods
Zebrafish husbandry
Zebrafish were grown and maintained according to UK Home Office regulation guidelines, and all studies were approved by the University of Manchester Ethical Review Board.
Embryo micro‐injections and generation of zebrafish strains
To generate the transgenic zebrafish strain Tg(fli1ep:MCP‐GFPnls) using Tol2 transposon transgenesis, 32 pg of Cerulean‐H2B:basfli1ep:MCP‐GFPnls Tol2‐based plasmid was co‐injected with 32 pg Tol2 mRNA into one‐cell stage AB zebrafish embryos. The next day, embryos with mosaic GFP expression were selected, raised to adulthood and then outbred to AB zebrafish to identify founders with germline transmission of the transgene. Adult Tg(fli1ep:MCP‐GFPnls) were inbred, and one‐cell stage embryos were co‐injected with 32 pg of Cerulean‐H2B:basfli1ep:Lyn‐mCherry‐24xMS2‐rab13‐3′UTR Tol2‐based plasmid and 32 pg Tol2 mRNA for mosaic expression analysis.
The mutant rab13 ∆3′UTR strain was generated with CRISPR‐Cas9 tools. One‐cell stage Tg(kdrl:EGFP)s843 embryos (Jin et al, 2005) were injected with 150 pg of each in vitro transcribed gRNA and co‐injected with 150 pg Cas9 NLS nuclease (New England Biolabs). Embryos were raised to adulthood and outbred to AB zebrafish to identify founders with germline transmission deletions in the rab13 3′UTR. Heterozygous animals harbouring a 482‐nucleotide deletion in the rab13 3′UTR (Tg(kdrl:EGFP)s843 rab13 +/∆3′UTR) were in‐crossed, and the resulting embryos were used for live‐cell imaging analysis.
gRNA generation and in vitro transcription
The online CRISPRscan tool (Moreno‐Mateos et al, 2015) was used to design gRNAs targeting the zebrafish 3′UTR region in the rab13 locus (Table EV4) and to determine off‐target loci. Next, 0.3 μM oligonucleotides comprising the target sequences (flanked by the T7 promoter and the Tail annealing sequence) were mixed with 0.45 μM Tail primer (Table EV4) and PCR‐amplified with Platinum Pfx DNA Polymerase (Thermo Fisher Scientific) in a T100 thermal cycler (Bio‐Rad). The following cycling conditions were used: 1 cycle of initial denaturation at 94°C for 10 min, 30 cycles of denaturation at 94°C for 30 s, annealing at 45°C for 30 s, extension at 68°C for 30 s and a final extension cycle at 68°C for 7 min. Subsequently, 200 ng of PCR‐amplified templates was used to transcribe gRNAs using a MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific), following the manufacturer's recommendations.
To synthesise Tol2 mRNA, 1 μg NotI‐linearised pCS2‐TP plasmid was transcribed using a SP6 mMESSAGE mMACHINE kit (Thermo Fisher Scientific) according to the manufacturer's protocol.
Embryo genotyping
Genomic DNA was extracted by incubating either whole embryos or embryo heads in lysis buffer (10 mM Tris–HCl pH 8, 1 mM EDTA, 80 mM KCl, 0.3% NP40, 0.3% Tween) containing 0.5 μg/μl Proteinase K (Promega) at 55°C for 1–2 h, followed by a denaturation step at 95°C for 15 min in a T100 thermal cycler. Genotyping PCR was performed using 2 μl genomic DNA, 0.4 μM zebrafish genotyping primers (Fig 7A and Table EV4) and 1× MyTaq Red DNA Polymerase (Bioline) according to the manufacturer's protocol in a T100 thermal cycler. PCRs were resolved in 1% agarose (Bioline) gels containing 0.5 μg/ml ethidium bromide (Sigma) for analysis. PCR products were cloned into TOPO‐TA vectors (Thermo Fisher Scientific) according to the manufacturer's protocol and analysed via Sanger sequencing on an ABI 3730 device.
Cell culture, scratch wound and co‐culture angiogenesis assays
Trunks of 26–48 hpf embryos were incubated in trypsin–EDTA solution (Sigma) at 28°C for 15 min. Trypsinisation was quenched with complete L‐15 medium (Sigma) containing 10% foetal bovine serum (FBS, Sigma) and 10 U/ml–100 μg/ml penicillin–streptomycin (Sigma). Cells were pelleted at 376 g for 5 min at room temperature (RT), resuspended in complete ECGM2 (PromoCell) and cultured on fibroblast‐coated coverslips. Zebrafish cells were cultured in 24‐well plates and maintained at 28°C for 18 h.
HUVECs (PromoCell) were cultured in complete ECGM2 (PromoCell) in gelatin‐coated (Millipore) dishes. Human pulmonary fibroblasts (HPF; PromoCell) were cultured in M199 (Thermo Fisher Scientific) containing 10% FBS, 50 μg/ml gentamycin (Sigma) and 50 ng/ml amphotericin (Sigma). Brain endothelial cells (bEnd.3 and bEnd.5) were cultured in DMEM (Sigma) supplemented with 10% FBS, 10 ng/ml recombinant human VEGF‐A (PeproTech) and 10 U/ml–100 μg/ml penicillin–streptomycin.
For scratch wound assays, HUVECs cultured on gelatin‐coated coverslips were grown to confluence and used in scratch wound assays as described elsewhere (Liang et al, 2007).
Co‐cultures of HUVEC and HPF and the corresponding siRNA‐mediated knockdown experiments were performed as previously described by Hetheridge et al (2019).
CRISPR‐Cas9 cell editing and cell transfections
The online Alt‐R CRISPR‐Cas9 design tool (https://eu.idtdna.com) was used to design crRNAs targeting the RAB13 3′UTR locus and to determine off‐target loci. HUVECs were transfected with Alt‐R CRISPR‐Cas9 ribonucleoprotein complexes (Integrated DNA Technologies) targeting the 90–282‐nt localisation element within the 3′UTR. Briefly, each sequence‐specific crRNA (Table EV4) was mixed with tracrRNA at 1:1 50 μM, incubated at 95°C for 5 min in a T100 thermal cycler and allowed to cool to RT for 60 min. Next, 12 μM each crRNA:tracrRNA (gRNA) was incubated with 20 μM Alt‐R Cas9 nuclease in PBS (Sigma) at RT for 20 min to form ribonucleoprotein complexes and mixed with 500 × 103 HUVECs. Additionally, 2 μg pmaxGFP Vector (Lonza) was included in the HUVEC‐ribonucleoprotein mix to identify transfected cells. Transfections were performed in a Nucleofector 2b Device (Lonza), using a HUVEC Nucleofector Kit (Lonza) according to the manufacturer's instructions, and the cells were further cultured for 72 h. Afterwards, single GFP‐expressing cells were isolated in a FACSAria Fusion cell sorter (BD Biosciences) into gelatin‐coated 96‐well plates to grow individual clones. Genomic DNA was extracted from expanded HUVEC clones and PCR‐analysed with sequence‐specific primers (Fig 3A and Table EV4) as described for zebrafish embryo genotyping. Clones with either biallelic deletion of the localisation element (ΔLE) or with the full‐length RAB13 3′UTR (Wt) were maintained until passage 6, and three clones from each genotype were used for analysis.
Knockdown experiments were performed with ON‐TARGETplus Non‐targeting Control or RAB13 siRNAs (Horizon) using GeneFECTOR (VennNova) as previously described (Hetheridge et al, 2019).
For in vitro MS2 experiments, bEnd.3 or bEnd.5 cells were transfected with pcDNA3‐Lyn‐mCherry, pCS2‐MCP‐GFPnls and different versions of pcDNA3‐HBB‐24XMS2SL‐RAB13 3′UTR. Briefly, 100 × 103 cells/well cultured in 6‐well plates were transfected with 0.5–1 μg each plasmid DNA using Lipofectamine 2000 or Lipofectamine 3000 following the manufacturer's protocol (Thermo Fisher Scientific) and analysed 48 h later.
Transwell assays and cell body/protrusion fractionation
Transwell experiments to segregate cell bodies and protrusions were performed as described elsewhere (Mili et al, 2008), with the following modifications: 1.5 × 106 HUVECs were cultured for 2 h in 24‐mm Transwells (Costar), containing 3‐μm‐pore polycarbonate membranes, in M199 (Thermo Fisher Scientific) supplemented with 1% FBS. Subsequently, 25 ng/ml VEGF‐A was added to the lower chambers to promote cell migration over the next hour. Whilst only 1 Transwell was used for the cell body fraction, 2 Transwells were used to harvest each HUVEC protrusion sample.
RNA isolation, qPCR and RNAseq
Embryo and cell‐derived RNA was isolated using a RNAqueous‐Micro Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. For gene expression analysis, cDNA was synthesised with a High‐Capacity RNA‐to‐cDNA Kit (Thermo Fisher Scientific) following the manufacturer's protocol.
qPCR experiments were performed with 1–2 μl cDNA, 0.25 μM gene‐specific primers (Table EV4) and 1× Power SYBR Green Master Mix (Thermo Fisher Scientific) in a StepOne Real‐Time PCR System (Applied Biosystems). GAPDH expression was used to normalise gene expression levels, and the relative mRNA levels were analysed with the 2 −ΔΔCT method.
For RNAseq, quality and integrity of RNA samples obtained from HUVEC cell bodies and protrusions were assessed using a 2200 TapeStation (Agilent Technologies). Next, RNAseq libraries were generated using the TruSeq Stranded mRNA assay (Illumina) according to the manufacturer's protocol. Adapter indices were used to multiplex libraries, which were pooled prior to cluster generation using a cBot instrument. The loaded flow cell was then paired‐end‐sequenced (76 + 76 cycles, plus indices) on an Illumina HiSeq 4000 instrument, and the output data were demultiplexed (allowing one mismatch) and BCL‐to‐Fastq conversion performed using Illumina's bcl2fastq software, v2.17.1.14. Sequence adapters were removed, and reads were quality trimmed using Trimmomatic v0.36 (Bolger et al, 2014) (Transwell samples) or BBDuk (part of the BBMap suite; v36.32) (HUVEC and zebrafish CRISPR‐Cas9 experiments). Processed reads from the human‐derived samples were mapped against the reference human genome (hg38) using STAR v2.5.3/2.7.2b (Dobin et al, 2013), and counts per gene were calculated using annotation from GENCODE v30/32 (http://www.gencodegenes.org/). Zebrafish‐derived samples were mapped against the reference assembly GRCz11 and gene annotation from Ensembl v99. Normalisation and differential expression was calculated with Bioconductor package DESeq2 v1.24 (Transwell samples), and RNAseq mapped reads were visualised with Jalview v2.11.0 (Waterhouse et al, 2009) (HUVEC and zebrafish CRISPR‐Cas9 experiments).
smFISH
Zebrafish cells and HUVECs were fixed in methanol‐free 4% formaldehyde (Thermo Fisher Scientific) and used in smFISH assays. Briefly, cells were permeabilised with 70% ethanol at RT for 1 h or 4°C overnight, washed with smFISH wash buffer (2× SSC, 10% formamide) and incubated with smFISH probes (Table EV5) in smFISH hybridisation buffer (10% dextran sulphate, 2× SSC, 10% formamide) at 37°C overnight. Afterwards, cells were washed with smFISH wash buffer twice at 37°C for 30 min, washed once with 2× SSC for 10 min, counterstaining with 1 μg/ml DAPI (Sigma) and washed twice with PBS for 5 min at RT. Coverslips were air‐dried and mounted on microscope slides with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). All probes targeting protrusion‐enriched mRNAs were designed with Stellaris Probe Designer (LGC Biosearch Technologies), synthesised and labelled with Quasar 570 or Quasar 670 (LGC Biosearch Technologies). Alternatively, probes were synthesised with an upstream FLAP sequence (CCTCCTAAGTTTCGAGCTGGACTCAGTG) (Tsanov et al, 2016) and annealed to a complementary FLAP probe labelled with Alexa 594 (Integrated DNA Technologies). Co‐hybridisation experiments were carried out with predesigned GAPDH probes labelled with Quasar 670 (HUVECs) or kdr probes labelled with Quasar 570 (zebrafish cells) (LGC Biosearch Technologies).
Puro‐PLA and immunofluorescence (IF)
For Puro‐PLA, cell bodies of HUVECs cultured in Transwells were scraped off and remaining protrusions were exposed to 3 μM puromycin (Sigma) added to lower chambers for 6 min. In translation inhibition experiments, 40 μM anisomycin (Sigma) was added to the lower Transwell chamber 30 min before cell body removal and 6 min after cell body removal together with 3 μM puromycin. Subsequently, HUVEC protrusions grown in Transwell membranes were fixed in methanol‐free 4% formaldehyde, removed from the Transwell inserts and used in Puro‐PLA experiments as described elsewhere (tom Dieck et al, 2015). Following the Puro‐PLA protocol, Transwell membranes were incubated for 20 min with 1:40 Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific) in PBS, washed in Duolink wash buffer B (Sigma) and mounted on microscope slides with Duolink In Situ Mounting Medium containing DAPI (Sigma).
For IF experiments, cells and Transwell membranes containing protrusions were permeabilised in PBS containing 0.2–0.5% Triton X‐100 (Sigma), blocked in 4% goat serum (Sigma) for 15 min and incubated with primary antibodies in blocking solution at 4°C overnight. Next, cells were washed in PBS containing 0.2% Tween, incubated with secondary antibodies at RT for 1 h, counterstaining with 1 μg/ml DAPI and washed again. Transwell membranes were further incubated with 1:40 Phalloidin Alexa Fluor 488 (Thermo Fisher Scientific) in PBS at RT for 20 min before washing. Cells and Transwell membranes were mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific).
Western blotting
Proteins were extracted with RIPA buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP‐40, 1% sodium deoxycholate and 0.1% SDS) and quantified with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) following the supplier's recommendations. Samples were denatured with Laemmli buffer (250 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 0.0025% bromophenol blue, 2.5% β‐mercaptoethanol) at 95°C for 5 min, loaded on 10% Mini‐PROTEAN TGX precast protein gels (Bio‐Rad) and separated in a Mini‐PROTEAN Electrophoresis System (Bio‐Rad). Proteins were transferred onto nitrocellulose membranes using a Trans‐Blot Turbo Transfer System RTA Kit following the manufacturer's protocols (Bio‐Rad). Subsequently, membranes were blocked in 5% milk (Sigma) or 5% BSA (Sigma) in TBS containing 0.1% Tween at RT for 1 h and incubated with primary antibodies at 4°C overnight. The next day, membranes were washed with TBS containing 0.1% Tween, incubated with secondary antibodies at RT for 1 h and washed again. Signal detection was carried out with SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific) according to the supplier's recommendations.
Antibodies
Primary and secondary antibodies were used at the following concentrations: 1:1,600 mouse PECAM‐1 89C2 (Cell Signaling Technology), 1:100 rabbit RAB13 (Puro‐PLA, Millipore), 1:1,000 rabbit RAB13 (Western blotting and IF, Cambridge Bioscience), 1:3,500 mouse puromycin (Kerafast), 1:1,000 rabbit β‐tubulin 9F3 (Cell Signaling Technology), 1:200 mouse ZO‐1 1A12 (Thermo Fisher Scientific), 1:500 goat anti‐mouse Alexa Fluor 488 or Alexa Fluor 568 (Thermo Fisher Scientific), 1:500 goat anti‐rabbit Alexa Fluor 568 (Thermo Fisher Scientific), 1:5,000 goat anti‐mouse HRP‐linked (Cell Signaling Technology) and 1:5,000 goat anti‐rabbit HRP‐linked antibody (Cell Signaling Technology).
Plasmid construction
The pCS2‐MCP‐GFPnls plasmid used in in vitro MS2 system assays was generated excising a MCP‐GFPnls fragment with SpeI and KpnI from pMS2‐GFP, a gift from Robert Singer (Addgene plasmid # 27121) (Fusco et al, 2003), and subcloning it into a pCS2 + vector using the XbaI and KpnI sites.
To construct the Cerulean‐H2B:basfli1ep:MCP‐GFPnls Tol2‐based plasmid for in vivo studies, MCP‐GFPnls was amplified from pMS2‐GFP with sequence 0.3 μM specific primers (Table EV4) and Platinum Pfx DNA Polymerase in a T100 thermal cycler. Subsequently, the PCR product was cloned into a pDONR221 P3‐P2 using Gateway Technology (Thermo Fisher Scientific) according to the manufacturer's manual. The final Tol2‐based construct was assembled into the pTol2Dest(R1R2) (Addgene plasmid # 73484) (Villefranc et al, 2007) using Gateway 3‐fragment recombination with pE(L1L4)Cerulean‐H2B in the first position, pE(R4R3)basfli1ep (De Bock et al, 2013) in the second position and pE(L3L2)MCP‐GFPnls in the third position.
For in vitro MS2 system experiments, 3′UTRs were PCR‐amplified from human genomic DNA with 0.3 μM sequence‐specific primers (Table EV4) using Platinum Pfx DNA Polymerase or MyTaq Red DNA Polymerase (Bioline) in a T100 thermal cycler and the resulting PCR product was cloned using either Zero Blunt PCR or TOPO TA Cloning Kits (Thermo Fisher Scientific), following the manufacturer's manual. Next, the human HBB gene was PCR‐amplified using 0.3 μM sequence‐specific primers (Table EV4) and Platinum Pfx DNA Polymerase in a T100 thermal cycler and cloned into the NotI and BamHI sites of the pCR4‐24XMS2SL‐stable plasmid, a gift from Robert Singer (Addgene plasmid # 31865) (Bertrand et al, 1998). Subsequently, a multiple cloning site (MCS; Table EV4) was introduced into the BglII and SpeI sites of pCR4‐HBB‐24XMS2SL and the recombinant HBB‐24XMS2SL‐MCS sequence was subcloned into the pcDNA3 mammalian expression vector (Thermo Fisher Scientific) using the NotI and XbaI sites. Full length 3′UTRs were then subcloned into pcDNA3‐HBB‐24XMS2SL‐MCS using NheI and XhoI/ApaI sites. Alternatively, truncated and deletion versions of the 3′UTRs were generated by PCR using 0.3 μM sequence‐specific primers (Table EV4) and Platinum Pfx DNA Polymerase, Phusion High‐Fidelity polymerase (NEB) or using QuikChange II Site‐Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer's instructions and introduced into the pcDNA3‐HBB‐24XMS2SL‐MCS using the NheI and XhoI sites.
In order to generate the zebrafish MS2 system reporter construct, the 24XMS2SL cassette was firstly subcloned from pCR4‐24XMS2SL‐stable into a kdrl:Lyn‐mCherry Tol2‐based plasmid (Costa et al, 2016) using a BamHI site. Next, the zebrafish rab13 3′UTR was PCR‐amplified with 0.4 μM sequence‐specific primers (Table EV4) and MyTaq Red DNA Polymerase from zebrafish genomic DNA in a T100 thermal cycler and then subcloned into the Tol2 kdrl:Lyn‐mCherry‐24XMS2SL plasmid using NheI and BglII sites. The resulting Lyn‐mCherry‐24XMS2SL‐rab13 3′UTR recombinant sequence was amplified with 0.3 μM sequence‐specific primers (Table EV4) and Platinum Pfx DNA Polymerase in a T100 thermal cycler and subcloned into a pDONR221 P3‐P2 using Gateway Technology. Lastly, the final Tol2‐based construct was assembled into the pTol2Dest(R1R2) using Gateway 3‐fragment recombination with pE(L1L4)Cerulean‐H2B in the first position, pE(R4R3)basfli1ep in the second position and Lyn‐mCherry‐24XMS2SL‐rab13 3′UTR in the third position.
All plasmid maps and details are available upon request.
Microscopy
Confocal time‐lapse imaging of zebrafish embryos was carried out as previously described (Costa et al, 2016). MS2 system‐transfected cells were live‐imaged every 5 s in a Nikon A1R‐inverted confocal microscope equipped with an Okolab incubation chamber, using a 60× objective. Fixed images of cultured cells and Transwell membranes were acquired on an Olympus IX83‐inverted microscope using Lumencor LED excitation, either a 60×/1.42 PlanApo or a 100×/1.35 UplanApo objective and a Sedat QUAD (DAPI/FITC/TRITC/Cy5) filter set (Chroma 89000). The images were collected using a R6 (Qimaging) CCD camera with a Z optical spacing of 0.2 μm. Raw images were then deconvolved using the Huygens Pro software (SVI), and maximum intensity projections of these images were used for analysis.
smFISH spot quantification, Polarisation Index and filopodia analysis
Processed smFISH images were used to calculate mRNA polarisation with the PI metric developed by Park et al (2012) and to assess mRNA spot number with FISH‐quant (Mueller et al, 2013).
For the studies of filopodia distance to GFP signal in MS2 system movies, filopodia parameters (position, duration and frequency) of MS2 system‐transfected cells were determined using Filopodyan plugin for FIJI (Urbancic et al, 2017). Only filopodia that emerged and retracted through the duration of the movies were analysed. Subsequently, the coordinates of GFP particles were extracted with the TrackMate plugin for FIJI (Tinevez et al, 2017). In order to generate control coordinates in each movie, the Lyn‐mCherry channel was thresholded to generate regions of interest (ROI) and the FIJI built‐in macro function “random” was used within the ROI at each frame. The Euclidean distances between the base of newly formed filopodia and both the nearest GFP particle and the control randomised coordinate were calculated.
RNA motif enrichment analysis and Gene Ontology
Discovery of recurring motifs across the k 5 mRNA 3′UTRs was carried out using MEME (Bailey & Elkan, 1994) (settings set to: mode ‐ anr; nmotifs: 5; minw: 6; maxw: 50; objfun classic: markov_order 0). The only motif present in all 3′UTR was selected for downstream studies. In order to quantify the frequency mRNA within the remaining k‐means clusters containing at least two repeats of the studied RNA motif, 3′UTR sequences were scanned in FIMO (Grant et al, 2011) using the position‐specific probability matrix obtained in MEME (settings set to: match P‐value < 1E‐5).
Gene Ontology studies were performed using DAVID using the HUVEC‐enriched mRNAS as background (Huang da et al, 2009a,b).
Statistics and k‐means clustering
All data are represented as means ± standard deviation. Statistical analysis of the data was carried out using GraphPad Prism software and RStudio. D'Agostino–Pearson or Shapiro–Wilk normality tests were applied to smFISH, Puro‐PLA, filopodia, ISV branching data and RNA, protein levels to determine the appropriate statistical test. Statistical significance is reported for P < 0.05.
k‐means clustering was performed in RStudio. Briefly, mRNA fold changes (FC) between cell bodies and protrusions of the cell types mentioned in the main text were obtained from the respective publications—NIH/3T3 fibroblasts (Wang et al, 2017), MDA‐MB231 metastatic breast cancer cells (Mardakheh et al, 2015) and induced neuronal cells (Zappulo et al, 2017). mRNAs were included in the clustering analysis if they were enriched in HUVEC protrusions (FC > 1.6, FDR < 0.05) and expressed in all three cell types. The log2 FC values between cell fractions were extracted, scaled and centred, followed by k‐means clustering (k = 8). The number of clusters was defined by the number of cell types and by the possible transcript statuses (enriched or depleted in each of the three cell types) − 23 = 8. The output is represented in heat maps using the log2 FC data prior to scaling and centring.
Author contributions
GC and SPH conceptualised the study. GC contributed to methodology. GC involved in formal analysis. GC, JB and NT investigated the study. GC and SPH wrote the original draft of the manuscript. GC and SPH wrote, reviewed and edited the manuscript. GC and SPH supervised the study. GC and SPH acquired the funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Review Process File
Source Data for Figure 3
Source Data for Figure 5
Acknowledgements
We are grateful to E. Schuman and S. tom Dieck at the MPI for Brain Research, Frankfurt, Germany, for help and guidance setting up Puro‐PLA assays. Moreover, we thank Rob Wilkinson at the University of Nottingham, UK, for access to his preliminary results for CRISPRi‐mediated rab13 knockdown in zebrafish. We also thank N. Papalopulu and group members at the University of Manchester, UK, for critical feedback, reagents and materials. We wish to thank members of the University of Manchester Biological Services, Genomic Technologies, Bioimaging Facilities and Flow Cytometry Facilities, for technical support. This work was funded by the Wellcome Trust (095718/Z/11/Z and 219500/Z/19/Z to S.P.H.), Wellcome Institutional Strategic Support Fund (204796/Z/16/Z to G.C.) and the British Heart Foundation (PG/16/2/31863 to S.P.H.).
The EMBO Journal (2020) 39: e106003
Contributor Information
Guilherme Costa, Email: g.costa@qub.ac.uk.
Shane P Herbert, Email: shane.herbert@manchester.ac.uk.
Data availability
The RNAseq datasets presented in this study have been deposited to the Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE133055 and GSE155449.
RStudio scripts are available upon request.
References
- Ainger K, Avossa D, Diana AS, Barry C, Barbarese E, Carson JH (1997) Transport and localization elements in myelin basic protein mRNA. J Cell Biol 138: 1077–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B et al (2008) Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134: 175–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassi C, Riccio A (2009) To localize or not to localize: mRNA fate is in 3′UTR ends. Trends Cell Biol 19: 465–474 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM (1998) Localization of ASH1 mRNA particles in living yeast. Mol Cell 2: 437–445 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley K, Smith MJ, Beck M, Stephenson FA (2005) GRIF‐1 and OIP106, members of a novel gene family of coiled‐coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem 280: 14723–14732 [DOI] [PubMed] [Google Scholar]
- Buxbaum AR, Haimovich G, Singer RH (2015) In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol 16: 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouaib R, Safieddine A, Pichon X, Imbert A, Kwon OS, Samacoits A, Traboulsi A‐M, Robert M‐C, Tsanov N, Coleno E et al (2020) A dual protein‐mrna localization screen reveals compartmentalized translation and widespread co‐translational RNA targeting. Dev Cell S1534‐5807(20)30584‐0 [DOI] [PubMed] [Google Scholar]
- Ciolli Mattioli C, Rom A, Franke V, Imami K, Arrey G, Terne M, Woehler A, Akalin A, Ulitsky I, Chekulaeva M (2019) Alternative 3′ UTRs direct localization of functionally diverse protein isoforms in neuronal compartments. Nucleic Acids Res 47: 2560–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J, Singer RH (2005) How and why does beta‐actin mRNA target? Biol Cell 97: 97–110 [DOI] [PubMed] [Google Scholar]
- Costa G, Harrington KI, Lovegrove HE, Page DJ, Chakravartula S, Bentley K, Herbert SP (2016) Asymmetric division coordinates collective cell migration in angiogenesis. Nat Cell Biol 18: 1292–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G et al (2013) Role of PFKFB3‐driven glycolysis in vessel sprouting. Cell 154: 651–663 [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Kochen L, Hanus C, Heumuller M, Bartnik I, Nassim‐Assir B, Merk K, Mosler T, Garg S, Bunse S et al (2015) Direct visualization of newly synthesized target proteins in situ . Nat Methods 12: 411–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC et al (2019) Transcriptome‐scale super‐resolved imaging in tissues by RNA seqFISH. Nature 568: 235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel KL, Arora A, Goering R, Lo HG, Taliaferro JM (2020) Mechanisms and consequences of subcellular RNA localization across diverse cell types. Traffic 21: 404–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, Bertrand E (2003) Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol 13: 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161: 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ES, Nelson WJ (2010) Adenomatous polyposis coli regulates endothelial cell migration independent of roles in beta‐catenin signaling and cell‐cell adhesion. Molecular Biology of the Cell 21: 2611–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert SP, Costa G (2019) Sending messages in moving cells: mRNA localization and the regulation of cell migration. Essays Biochem 63: 595–606 [DOI] [PubMed] [Google Scholar]
- Hetheridge C, Mavria G, Mellor H (2011) Uses of the in vitro endothelial‐fibroblast organotypic co‐culture assay in angiogenesis research. Biochem Soc Trans 39: 1597–1600 [DOI] [PubMed] [Google Scholar]
- Hotz M, Nelson WJ (2017) Pumilio‐dependent localization of mRNAs at the cell front coordinates multiple pathways required for chemotaxis. Nat Commun 8: 1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Ioannou MS, Bell ES, Girard M, Chaineau M, Hamlin JN, Daubaras M, Monast A, Park M, Hodgson L, McPherson PS (2015) DENND2B activates Rab13 at the leading edge of migrating cells and promotes metastatic behavior. J Cell Biol 208: 629–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S, Lawson ND, Torrealday S, Horiguchi M, Weinstein BM (2003) Angiogenic network formation in the developing vertebrate trunk. Development 130: 5281–5290 [DOI] [PubMed] [Google Scholar]
- Jakobsen KR, Sorensen E, Brondum KK, Daugaard TF, Thomsen R, Nielsen AL (2013) Direct RNA sequencing mediated identification of mRNA localized in protrusions of human MDA‐MB‐231 metastatic breast cancer cells. J Mol Signaling 8: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambhekar A, Derisi JL (2007) Cis‐acting determinants of asymmetric, cytoplasmic RNA transport. RNA 13: 625–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY (2005) Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132: 5199–5209 [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM (1996) A requirement for local protein synthesis in neurotrophin‐induced hippocampal synaptic plasticity. Science 273: 1402–1406 [DOI] [PubMed] [Google Scholar]
- Kim NY, Lee S, Yu J, Kim N, Won SS, Park H, Heo WD (2020) Optogenetic control of mRNA localization and translation in live cells. Nat Cell Biol 22: 341–352 [DOI] [PubMed] [Google Scholar]
- Kislauskis EH, Zhu X, Singer RH (1994) Sequences responsible for intracellular localization of beta‐actin messenger RNA also affect cell phenotype. J Cell Biol 127: 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp P, Lammers R, Aepfelbacher M, Woehlke G, Rudel T, Machuy N, Steffen W, Linder S (2006) The kinesin KIF1C and microtubule plus ends regulate podosome dynamics in macrophages. Mol Biol Cell 17: 2811–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RE, Lamont EJ, Childs SJ (2009) Antagonistic interactions among Plexins regulate the timing of intersegmental vessel formation. Dev Biol 331: 199–209 [DOI] [PubMed] [Google Scholar]
- Leung KM, van Horck FP, Lin AC, Allison R, Standart N, Holt CE (2006) Asymmetrical beta‐actin mRNA translation in growth cones mediates attractive turning to netrin‐1. Nat Neurosci 9: 1247–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Park AY, Guan JL (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro . Nat Protoc 2: 329–333 [DOI] [PubMed] [Google Scholar]
- Lu X, Le Noble F, Yuan L, Jiang Q, De Lafarge B, Sugiyama D, Breant C, Claes F, De Smet F, Thomas JL et al (2004) The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system. Nature 432: 179–186 [DOI] [PubMed] [Google Scholar]
- Lyles V, Zhao Y, Martin KC (2006) Synapse formation and mRNA localization in cultured Aplysia neurons. Neuron 49: 349–356 [DOI] [PubMed] [Google Scholar]
- Mardakheh FK, Paul A, Kumper S, Sadok A, Paterson H, McCarthy A, Yuan Y, Marshall CJ (2015) Global analysis of mRNA, translation, and protein localization: local translation is a key regulator of cell protrusions. Dev Cell 35: 344–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor R, Etienne‐Manneville S (2016) The front and rear of collective cell migration. Nat Rev Mol Cell Biol 17: 97–109 [DOI] [PubMed] [Google Scholar]
- Mayr C (2016) Evolution and biological roles of alternative 3′UTRs. Trends Cell Biol 26: 227–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S, Moissoglu K, Macara IG (2008) Genome‐wide screen reveals APC‐associated RNAs enriched in cell protrusions. Nature 453: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingle LA, Okuhama NN, Shi J, Singer RH, Condeelis J, Liu G (2005) Localization of all seven messenger RNAs for the actin‐polymerization nucleator Arp2/3 complex in the protrusions of fibroblasts. J Cell Sci 118: 2425–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissoglu K, Yasuda K, Wang T, Chrisafis G, Mili S (2019) Translational regulation of protrusion‐localized RNAs involves silencing and clustering after transport. Elife 8: e44752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissoglu K, Stueland M, Gasparski AN, Wang T, Jenkins LM, Hastings ML, Mili S (2020) RNA localization and co‐translational interactions control RAB13 GTPase function and cell migration. EMBO J 39: e104958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor AE, Golan M, Massasa EE, Lemze D, Weizman T, Shenhav R, Baydatch S, Mizrahi O, Winkler R, Golani O et al (2017) Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357: 1299–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ (2015) CRISPRscan: designing highly efficient sgRNAs for CRISPR‐Cas9 targeting in vivo . Nat Methods 12: 982–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowry KL, Melton DA (1992) Vegetal messenger RNA localization directed by a 340‐nt RNA sequence element in Xenopus oocytes. Science 255: 991–994 [DOI] [PubMed] [Google Scholar]
- Mueller F, Senecal A, Tantale K, Marie‐Nelly H, Ly N, Collin O, Basyuk E, Bertrand E, Darzacq X, Zimmer C (2013) FISH‐quant: automatic counting of transcripts in 3D FISH images. Nat Methods 10: 277–278 [DOI] [PubMed] [Google Scholar]
- Nagaoka K, Udagawa T, Richter JD (2012) CPEB‐mediated ZO‐1 mRNA localization is required for epithelial tight‐junction assembly and cell polarity. Nat Commun 3: 675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Trcek T, Wells AL, Chao JA, Singer RH (2012) An unbiased analysis method to quantify mRNA localization reveals its correlation with cell motility. Cell Rep 1: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR, Dirac‐Svejstrup AB, Soldati T (1995) Rab GDP dissociation inhibitor: putting rab GTPases in the right place. J Biol Chem 270: 17057–17059 [DOI] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane A, Abdallah AA, Nakano K, Honda K, Ikeda W, Nishikawa Y, Matsumoto M, Matsushita N, Kitamura T, Sasaki T (2012) Rab13 small G protein and junctional Rab13‐binding protein (JRAB) orchestrate actin cytoskeletal organization during epithelial junctional development. J Biol Chem 287: 42455–42468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane A, Alamir Mahmoud Abdallah A, Nakano K, Honda K, Kitamura T, Imoto I, Matsushita N, Sasaki T (2013) Junctional Rab13‐binding protein (JRAB) regulates cell spreading via filamins. Genes Cells 18: 810–822 [DOI] [PubMed] [Google Scholar]
- Seabra MC, Mules EH, Hume AN (2002) Rab GTPases, intracellular traffic and disease. Trends Mol Med 8: 23–30 [DOI] [PubMed] [Google Scholar]
- Shen F, Seabra MC (1996) Mechanism of digeranylgeranylation of Rab proteins. Formation of a complex between monogeranylgeranyl‐Rab and Rab escort protein. J Biol Chem 271: 3692–3698 [DOI] [PubMed] [Google Scholar]
- Srougi MC, Burridge K (2011) The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB‐mediated cell death after DNA damage. PLoS ONE 6: e17108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro JM, Vidaki M, Oliveira R, Olson S, Zhan L, Saxena T, Wang ET, Graveley BR, Gertler FB, Swanson MS et al (2016) Distal alternative last exons localize mRNAs to neural projections. Mol Cell 61: 821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: an open and extensible platform for single‐particle tracking. Methods 115: 80–90 [DOI] [PubMed] [Google Scholar]
- Tommasi S, Dammann R, Jin SG, Zhang XF, Avruch J, Pfeifer GP (2002) RASSF3 and NORE1: identification and cloning of two human homologues of the putative tumor suppressor gene RASSF1. Oncogene 21: 2713–2720 [DOI] [PubMed] [Google Scholar]
- Torres‐Vazquez J, Gitler AD, Fraser SD, Berk JD, Van NP, Fishman MC, Childs S, Epstein JA, Weinstein BM (2004) Semaphorin‐plexin signaling guides patterning of the developing vasculature. Dev Cell 7: 117–123 [DOI] [PubMed] [Google Scholar]
- Tsanov N, Samacoits A, Chouaib R, Traboulsi AM, Gostan T, Weber C, Zimmer C, Zibara K, Walter T, Peter M et al (2016) smiFISH and FISH‐quant – a flexible single RNA detection approach with super‐resolution capability. Nucleic Acids Res 44: e165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tushev G, Glock C, Heumuller M, Biever A, Jovanovic M, Schuman EM (2018) Alternative 3′ UTRs modify the localization, regulatory potential, stability, and plasticity of mrnas in neuronal compartments. Neuron 98: 495–511 e496 [DOI] [PubMed] [Google Scholar]
- Urbancic V, Butler R, Richier B, Peter M, Mason J, Livesey FJ, Holt CE, Gallop JL (2017) Filopodyan: an open‐source pipeline for the analysis of filopodia. J Cell Biol 216: 3405–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villefranc JA, Amigo J, Lawson ND (2007) Gateway compatible vectors for analysis of gene function in the zebrafish. Dev Dyn 236: 3077–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Hamilla S, Cam M, Aranda‐Espinoza H, Mili S (2017) Extracellular matrix stiffness and cell contractility control RNA localization to promote cell migration. Nat Commun 8: 896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatheritt RJ, Gibson TJ, Babu MM (2014) Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems. Nat Struct Mol Biol 21: 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Agrawal S, Vasanji A, Drazba J, Sarkaria S, Xie J, Welch CM, Liu M, Anand‐Apte B, Horowitz A (2011) Rab13‐dependent trafficking of RhoA is required for directional migration and angiogenesis. J Biol Chem 286: 23511–23520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, Zhuang X (2019) Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle‐dependent gene expression. Proc Natl Acad Sci USA 116: 19490–19499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Sasaki Y, Wen Z, Bassell GJ, Zheng JQ (2006) An essential role for beta‐actin mRNA localization and translation in Ca2 + ‐dependent growth cone guidance. Nat Neurosci 9: 1265–1273 [DOI] [PubMed] [Google Scholar]
- Younts TJ, Monday HR, Dudok B, Klein ME, Jordan BA, Katona I, Castillo PE (2016) Presynaptic protein synthesis is required for long‐term plasticity of GABA release. Neuron 92: 479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulo A, van den Bruck D, Ciolli Mattioli C, Franke V, Imami K, McShane E, Moreno‐Estelles M, Calviello L, Filipchyk A, Peguero‐Sanchez E et al (2017) RNA localization is a key determinant of neurite‐enriched proteome. Nat Commun 8: 583 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Review Process File
Source Data for Figure 3
Source Data for Figure 5
Data Availability Statement
The RNAseq datasets presented in this study have been deposited to the Gene Expression Omnibus repository (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE133055 and GSE155449.
RStudio scripts are available upon request.