

Abstract
Rationale:
Post-operative atrial fibrillation (POAF) is a common and troublesome complication of cardiac surgery. POAF is generally believed to occur when post-operative triggers act on a pre-existing vulnerable substrate, but the underlying cellular and molecular mechanisms are largely unknown.
Objective:
To identify cellular POAF-mechanisms in right-atrial samples from patients without a history of atrial fibrillation undergoing open-heart surgery.
Methods and Results:
Multicellular action potentials, membrane ion-currents (perforated patch-clamp) or simultaneous membrane-current (ruptured patch-clamp) and [Ca2+]i-recordings in atrial cardiomyocytes, along with protein-expression levels in tissue homogenates or cardiomyocytes, were assessed in 265 atrial samples from patients without or with POAF. No indices of electrical, profibrotic, or connexin remodeling were noted in POAF, but Ca2+-transient amplitude was smaller while spontaneous sarcoplasmic-reticulum (SR) Ca2+-release events and L-type Ca2+-current alternans occurred more frequently. Ca2+/calmodulin-dependent protein kinase-II (CaMKII) protein-expression, CaMKII-dependent phosphorylation of the cardiac ryanodine-receptor channel type-2 (RyR2) and RyR2 single-channel open-probability were significantly increased in POAF. SR Ca2+-content was unchanged in POAF despite greater SR Ca2+-leak, with a trend towards increased SR Ca2+-ATPase activity. POAF patients also showed stronger expression of activated components of the NLRP3-inflammasome system in atrial whole-tissue homogenates and cardiomyocytes. Acute application of interleukin-1β caused NLRP3-signaling activation and CaMKII-dependent RyR2/phospholamban hyperphosphorylation in HL-1-cardiomyocytes and enhanced spontaneous SR Ca2+-release events in both POAF-cardiomyocytes and HL-1-cardiomyocytes. Computational modeling showed that RyR2-dysfunction and increased SR Ca2+-uptake are sufficient to reproduce the Ca2+-handling phenotype and indicated an increased risk of proarrhythmic delayed afterdepolarizations in POAF-subjects in response to interleukin-1β.
Conclusions:
Pre-existing Ca2+-handling abnormalities and activation of NLRP3-inflammasome/CaMKII signaling are evident in atrial cardiomyocytes from patients who subsequently develop POAF. These molecular substrates sensitize cardiomyocytes to spontaneous Ca2+-releases and arrhythmogenic afterdepolarizations, particularly upon exposure to inflammatory mediators. Our data reveal a potential cellular and molecular substrate for this important clinical problem.
Subject Terms: Arrhythmias, Calcium Cycling/Excitation-Contraction Coupling, Computational Biology, Inflammation
Keywords: Post-operative atrial fibrillation, calcium-handling, action potentials, RyRs, NLRP3, ryanodine receptor, calcium-calmodulin-dependent protein kinase II
Graphical Abstract
POAF is a common complication of cardiac surgery and is associated with increased cost and longer hospital stays. Several preventive approaches have been proposed, but POAF-management remains challenging, partly due to incomplete understanding of underlying mechanisms. Although there is clinical evidence pointing to the presence of a pre-surgical vulnerable substrate contributing to POAF development, its nature and role in POAF development were unknown. Here, we identified a pre-existing substrate characterized by CaMKII-dependent calcium-handling abnormalities related to atrial cardiomyocyte NLRP3-inflammasome activation. This subclinical atrial cardiomyopathy is itself insufficient to cause AF, but determines which atria will cross the AF threshold in the presence of post-operative inflammatory triggers. Inflammation exacerbates pre-existing calcium-handling abnormalities in predisposed individuals to promote the formation of proarrhythmic delayed afterdepolarizations. Our study establishes a novel unifying paradigm, by suggesting that POAF patients share underlying atrial cardiomyocyte NLRP3-signaling activation and calcium-handling abnormalities with paroxysmal and persistent AF patients, predisposing them to POAF caused by transient surgery-induced inflammation (explaining the usually self-limited nature of POAF) and to long-term paroxysmal and persistent AF (explaining the predilection for recurrence). We propose that inhibition of CaMKII- and/or NLRP3-inflammasome-related signaling, for which therapeutic approaches exist, might constitute a novel pharmacological strategy to prevent POAF.
INTRODUCTION
Post-operative atrial fibrillation (POAF) is a common complication after surgery, affecting ~30% of patients undergoing open-heart surgery.1 Although generally a transient phenomenon with peak incidence around post-operative days 2–3, POAF is associated with increased short- and long-term morbidity and mortality.2–4 Several preventive approaches have been proposed, but POAF management remains challenging, partly due to incomplete understanding of underlying mechanisms.4,5
POAF is believed to result from transient peri-operative triggers interacting with a pre-existing arrhythmogenic substrate.4 The pre-existing POAF substrate has been studied in human atrial-samples obtained during cardiac surgery. While pre-operative left-atrial (LA) fibrosis has been noted in some studies,6,7 atrial fibrosis has not been consistently observed in right-atrial (RA) samples.4,6–8 There is no evidence for pre-existing electrical remodeling in human atrial cardiomyocytes (HAMs) of POAF-patients.6,9 Ca2+-handling abnormalities, a potential trigger of arrhythmogenesis, are present in both paroxysmal and long-standing persistent (‘chronic’) AF (pAF and cAF, respectively),10,11 as well as in systolic heart failure patients with increased AF-susceptibility,12 but have not yet been studied in sinus-rhythm patients who subsequently develop POAF.
Acute inflammation is a major transient post-operative factor that may trigger POAF. The time-course of AF post-surgery roughly parallels changes in inflammatory markers13–15 and a few studies identified associations between POAF-occurrence and systemic levels of pre-operative inflammatory biomarkers, notably interleukin (IL)-2 and IL-6.13,16 However, other studies found no correlation between POAF and pre- or post-operative C-reactive protein (CRP), IL-2, or IL-6.4,8,17 We recently identified abnormal NACHT, LRR, and PYD domains-containing protein-3 (NLRP3) inflammasome signaling within HAMs and showed a role in pAF and cAF, partly by altering Ca2+-handling.18 Abnormal Ca2+-signaling can, in turn, cause cardiomyocyte NLRP3-inflammasome activation.19,20 The arrhythmogenic HAM inflammasome-component activation and Ca2+-signaling abnormalities implicated in pAF and cAF have so far not been evaluated in POAF; we aimed to assess them in the present study.
We performed a comprehensive assessment of HAM-properties in RA-tissue samples from patients with sinus rhythm at the time of surgery who did not (Ctl) and who did subsequently manifest POAF to test the hypothesis that a pre-surgical cellular/tissue substrate exists and is a key requirement for POAF-development. The results identify a discrete pre-existing cellular substrate in patients who develop POAF, including increased priming and triggering of the NLRP3-inflammasome in HAMs and Ca2+-handling abnormalities characterized by reduced Ca2+-transient (CaT) amplitude, ryanodine-receptor channel type-2 (RyR2) dysfunction with enhanced sarcoplasmic-reticulum (SR) Ca2+-leak and spontaneous SR Ca2+-release events (SCaEs), along with increased sensitivity to inflammatory mediators. A computational HAM model incorporating the changes observed reproduces experimental findings and indicates that acute stimulation of the pre-existing substrate with inflammatory stimuli, as typically occur during the post-operative period, results in proarrhythmic delayed afterdepolarizations (DADs).
METHODS
A detailed description of all methods is provided in the online-only Data Supplement and the Major Resources Table in the Supplemental Materials. Key aspects are summarized below. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study design and human atrial samples.
Patients (>18 years) undergoing open-heart surgery without a previous history of AF and in normal sinus-rhythm at the time of surgery were included. All available RA-appendages were used consecutively; RA-appendages were not available from the following types of procedures: emergency surgery, off-pump and re-do procedures, or cases for which the surgeon judged that RA-sampling would excessively complicate the surgical procedure. Of the 241 RA-samples obtained, 92 were used for biochemical experiments, 140 for cardiomyocyte isolation and 9 for both (Online Figure I). 71 out of 149 RA-appendages (48%) directed to cardiomyocyte isolation provided Ca2+-tolerant HAMs of adequate quality for cellular electrophysiology (Online Figure I). As such, 71 RA-samples were included for patch-clamp/Ca2+-imaging and 101 for biochemical experiments (with tissue from 5 patients used for both). In addition, we included 48 consecutive patients for validation of patch-clamp/Ca2+-imaging experiments from a separate patient cohort and an independent cohort of 50 consecutive successful multicellular action potential (AP) recordings, resulting in a total of 265 studied patients. Experimental protocols were approved by ethical review boards of University Hospital Essen (#12–5268-BO), University Medical Center Hamburg-Eppendorf (WF-088/18), and Dresden University of Technology (EK114082002), respectively, and were conducted in accordance with the Declaration of Helsinki. Each patient provided written informed consent. All RA-samples were collected just prior to atrial cannulation for extracorporeal circulatory bypass, thus providing information on the potential pre-existing POAF-promoting substrate.
The POAF-occurrence was monitored during routine clinical care, including routine ECG-monitoring or assessment of symptoms, as per hospital standards. The absence (Ctl) or presence (POAF) of POAF diagnosis was employed to select samples for Western-blot experiments. All functional experiments (AP-recordings, patch-clamp±Ca2+-imaging experiments), which have to be performed within hours of tissue procurement, were performed without information about the subsequent post-operative evolution of the patients and were studied blinded to subsequent AF-occurrence that directed classification into Ctl or POAF. Patient characteristics are provided in Online Tables I-IV for all samples, samples used for patch-clamp/Ca2+-imaging experiments, AP-recordings, and biochemical/biophysical studies, respectively.
Atrial trabeculae/cardiomyocyte isolation.
Atrial trabeculae were isolated from RA-appendages.21 Cardiomyocyte-isolation was based on previously described enzymatic-digestion protocols.22–24 HAMs were suspended in EGTA-free storage-solution.
Sharp-electrode AP-recordings.
APs were recorded at 1-Hz in RA-trabeculae as described.21 Glass-microelectrodes filled with 2.5-mmol/L KCl had tip-resistances of 10–25 MΩ.
Perforated patch-clamp experiments.
HAM membrane-currents (corrected for membrane capacitance, expressed as pA/pF) were measured using whole-cell perforated-patch configuration (using 250-mg/mL amphotericin-B) at room-temperature.22 ICa,L was measured at 0.5-Hz during a 200-ms depolarizing pulse to 0-mV (additional ICa,L-protocols are in online-only Data Supplement). SR Ca2+-content was measured as integrated Na+/Ca2+-exchanger (NCX)-mediated current (INCX) during caffeine application (10-mmol/L) to empty the SR; SCaEs were quantified as frequency of transient-inward INCX at −50-mV and −80-mV.12,22
Simultaneous [Ca2+]i/membrane-current recording.
HAM membrane-currents were recorded with whole-cell ruptured-patch voltage-clamp configuration with simultaneous [Ca2+]i-measurements and expressed as current-densities (pA/pF). [Ca2+]i was measured in HAMs and in an immortalized mouse atrial-cardiomyocyte cell-line (HL-1 cells, Merck Millipore) and quantified using fluo-3-acetoxymethyl ester (Fluo-3) in bath and pipette solutions. Fluorescence was excited at 488-nm and emitted light (>510-nm) converted to [Ca2+]i using the formula [Ca2+]i=kd*F / (Fmax–F), where kd is the Fluo-3 dissociation constant (864-nmol/L), F is baseline-corrected Fluo-3 fluorescence, and Fmax is baseline-corrected Ca2+-saturated fluorescence obtained at the end of each experiment.10,11 In some experiments, results are presented as relative fluorescence changes (F/F0). ICa,L and corresponding CaTs were elicited using 100-ms depolarizing pulses to +10-mV (0.5-Hz).10,11 SR Ca2+-leak was measured in HAMs with 1-mmol/L tetracaine in the absence of extracellular Na+ and Ca2+, and SR Ca2+-content was determined as integrated INCX in response to 10-mmol/L caffeine, as described.10,11 SCaEs were measured prior to tetracaine application in HAMs and during acute IL-1β (40-ng/mL) application in HAMs and HL-1-cells.
Immunoblot.
Proteins were isolated from atrial-tissue homogenates, HAMs or HL-1-cells and protein-levels were determined using Western blot according to standard protocols.11,18,25,26 Antibodies are listed in Online Table V.
RyR2 single-channel recordings.
RyR2 single-channel recordings were performed as described10,27 using SR-membrane preparations incorporated into lipid-bilayer membranes formed across a 150-μm aperture of a polystyrene cuvette at 150-nmol/L free cytosolic [Ca2+].
Computational modeling.
We updated our recent computational HAM model with spatial atrial Ca2+-handling11,28 to reproduce experimentally-observed POAF-associated Ca2+-handling properties (Table VI and ‘Computational Modeling’ section in online-only Data Supplement). The model code is freely available on the authors’ website. For all simulations, experimental conditions (voltage-clamp protocol, pipette and bath solutions) were reproduced in the model. The effects of tetracaine and caffeine were simulated by blocking RyR2 by more than 99% and setting RyR2 open-probability to 1.0, respectively.11 Acute IL-1β-application was simulated as increased RyR2 open-probability developing during a 10-second period (details in Data Supplement).29,30
Statistics.
Results are presented as scatter-plots and mean±standard deviation (SD) for normally-distributed data or median and interquartile ranges for non-normally-distributed data. Normality was tested using D’Agostino&Pearson omnibus testing. Homogeneity of variance between Ctl and POAF was verified using the F-test in Prism-7 (GraphPad Software, San Diego, CA). P<0.05 was considered statistically significant. All comparisons were between two groups only: Ctl versus POAF, HL-1-cells± IL-1β, or HL-1-cells with IL-1β+KN-92 versus IL-1β+KN-93; in no cases were multiple comparisons made.
For patch-clamp/Ca2+-imaging and RyR2 single-channel recordings, in which each patient may contribute multiple data points, intra-class correlation coefficients were determined (Online Table VII) and multilevel mixed-effect models were employed, as previously described.31 Data were defined (1) by the individual patient ID and (2) the ID of the cardiomyocyte/channel within each patient. When multiple recordings per subject are available, sample-sizes are given as n/N, where n=cardiomyocytes and N=patients. The random effect within the model was the intercept to account for non-independent measurements in multiple cells from individual patients. Multilevel models were implemented in RStudio (Boston, MA) using lme4 with P-values derived using the Kenward-Roger approximation or nlme.32 Non-normally distributed data were log-transformed. For zero-value cases, all values were shifted by 0.01.33
For multicellular AP-recordings, biochemical experiments, and clinical parameters, for which each patient contributed a single data-point, unpaired two-tailed Students’ t-tests were used to compare means of normally-distributed continuous data. In case of heterogeneous variances, the Student’s t-test with Welch’s correction was employed. Non-normally-distributed continuous data or data for which normality could not be assessed were compared using Mann-Whitney tests. Categorical data were analyzed using Fisher’s exact or Chi-squared tests, as indicated. To investigate potential influences of individual clinical variables on specific parameters, two-way ANOVA sub-analyses with factors post-operative rhythm and either age, sex, left-atrial diameter, degree of mitral/tricuspid/aortic valve-insufficiency, use of beta-blockers or statins were performed (Online Table VIII).
RESULTS
Patients.
POAF-patients were on average older and had slightly lower estimated glomerular-filtration rate (Online Tables I-IV), but there were no statistically-significant differences in sex, body-mass index, indication for cardiac surgery, comorbidities, LA-size, degree of valvular regurgitation, left-ventricular ejection fraction, and pre- or post-operative CRP-levels between groups. Controlling for age, LA-size or valvular regurgitation did not alter the results (Online Table VIII). There was a trend towards increased use of dihydropyridine Ca2+-channel blockers and reduced use of lipid-lowering drugs and beta-blockers in POAF-samples (Online Table I-V), but this had minimal impact on the results (Online Table VIII). Other medication was not different between both groups.
Electrical and Ca2+-handling remodeling.
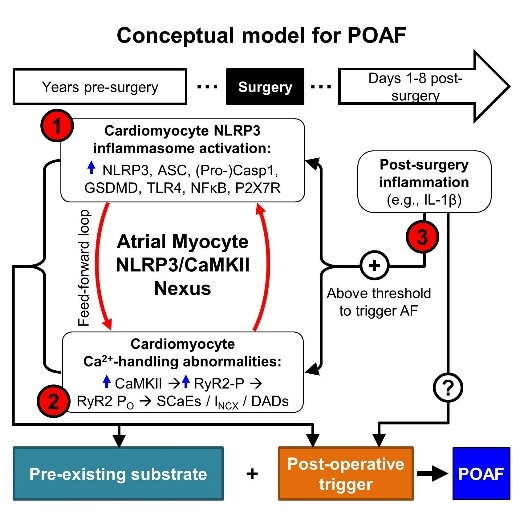
We simultaneously recorded depolarization-induced membrane currents and [Ca2+]i in the whole-cell ruptured-patch configuration (Figure 1A). Although peak and integrated ICa,L, overall diastolic and systolic [Ca2+]i-levels, CaT time-to-peak and time-constants of CaT-decay were similar in both groups (Figure 1B,D, Online Figure II), CaT-amplitude was 34% lower in POAF (Figure 1C), pointing to potentially-relevant Ca2+-handling abnormalities.
Figure 1. Ca2+-handling remodeling in Ctl and POAF patients.
A, Membrane current (IM, ruptured-patch) and Ca2+ transient (CaT) recorded in parallel under voltage-clamp conditions in response to a 100-ms depolarizing voltage step to +10 mV (inset) in Ctl or POAF atrial cardiomyocytes. B, L-type Ca2+-current (ICa,L) amplitude. C, ICa,L-triggered CaT-amplitude. D, time-constant (τ) of CaT-decay. N-numbers indicate numbers of cardiomyocytes/patients. *P<0.05 vs. Ctl based on multilevel mixed models with log-transformed data (B,D), or untransformed data (C) to account for non-independent measurements in multiple cells from individual patients.
ICa,L-amplitude, peak current-voltage relationship and steady-state inactivation (Online Figure IIIA-C) were also similar in both groups in perforated-patch recordings (which preserve the physiological intracellular milieu better than ruptured-patch experiments), whereas both inactivation and recovery from inactivation were significantly faster in POAF (Online Figure IIID-F). The proportion of HAMs with alternating or irregular ICa,L-patterns, known to be associated with repolarization alternans and reentrant arrhythmias,23,34 was significantly higher in POAF (Online Figure IV). No significant differences were observed in resting membrane-potential, AP-amplitude, maximum upstroke-velocity, conduction-time, plateau-potential or AP-duration (APD) at 20%, 50%, or 90% of repolarization (Online Figure V).
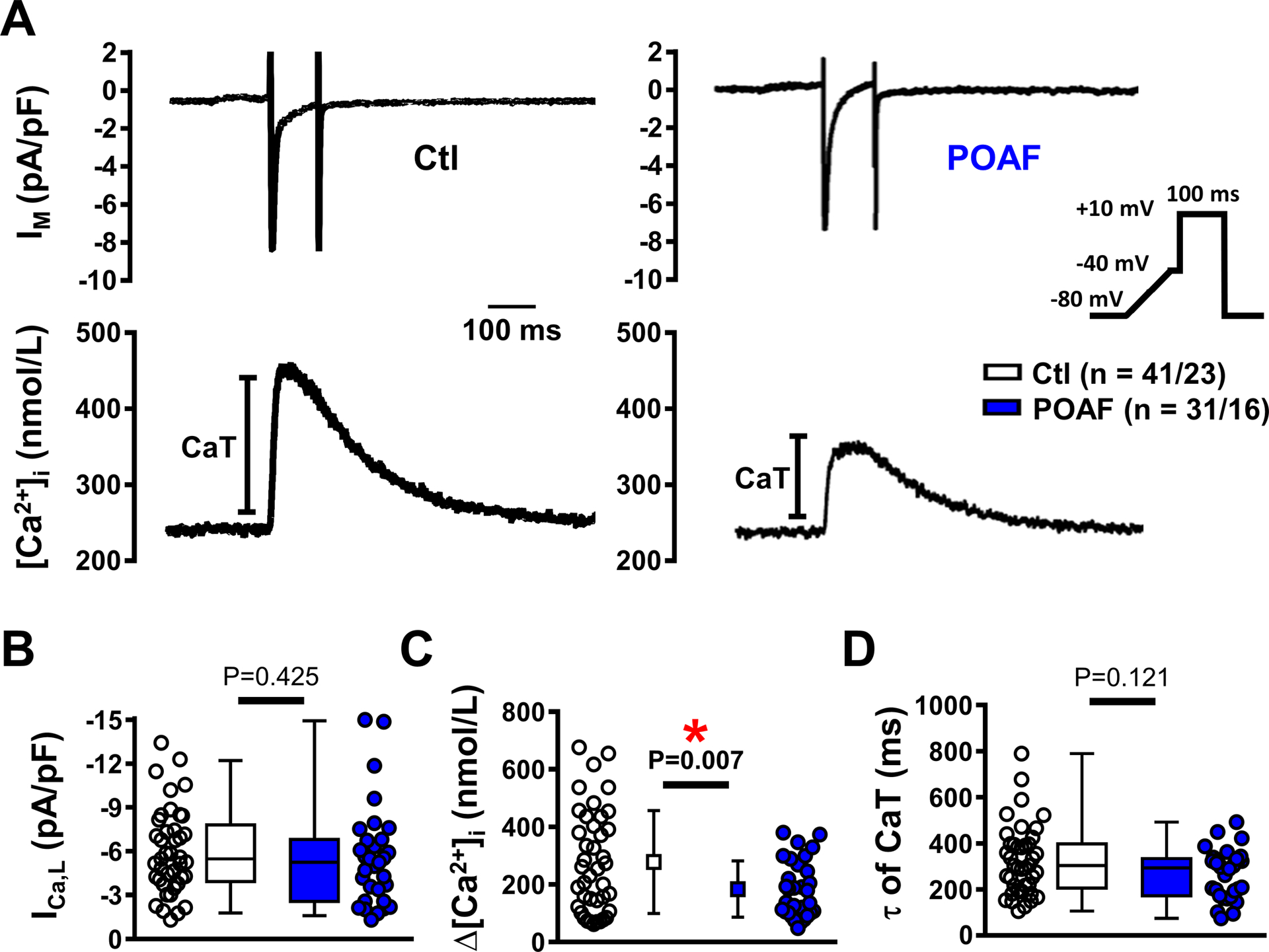
CaT-amplitude strongly depends on SR Ca2+-content. Therefore, we measured the amplitude of the caffeine (10-mmol/L)-induced CaT and corresponding INCX (Figure 2A),10,11 which were comparable between Ctl and POAF (Figure 2B). Peak INCX and its Ca2+-dependence (quantified as the slope of the INCX vs. [Ca2+]i-curve) were also similar in both groups (Figure 2C), indicating unchanged NCX-function.
Figure 2. Sarcoplasmic reticulum (SR) Ca2+-cycling and Na+/Ca2+-exchanger (NCX) function.
A, Caffeine-induced Ca2+ transient (cCaT, top) and corresponding inward current (bottom, largely mediated by NCX) at −80 mV in atrial cardiomyocytes from Ctl and POAF. B, Quantification of SR Ca2+-content as amplitude of cCaT (left) or integrated NCX-current (INCX, right). C, Peak INCX (left) and slope of its Ca2+-dependence (right). D, SR Ca2+-ATPase (SERCA) function determined by subtracting the rate-constants of cCaT-decay (kcaff, mediated by NCX and plasmalemmal Ca2+-ATPase, PMCA) from the rate-constants of systolic CaT-decay (ksyst, mediated by SERCA, NCX and PMCA) obtained during the same experiment. N-numbers indicate numbers of cardiomyocytes/patients. *P<0.05 vs. Ctl based on multilevel models (B,D), or multilevel models with log-transformed data (C).
The unaltered SR Ca2+-content in POAF was confirmed in independent perforated-patch experiments, excluding potential bias by Ca2+-indicator-mediated buffering (Online Figure VIA).
Previous work identified unaltered SR Ca2+-ATPase (SERCA)-2a expression, but reduced protein-levels of the SERCA2a-inhibitor sarcolipin, in atria of POAF-patients.35 Therefore, we assessed SERCA-function by subtracting the rate-constant of caffeine-induced CaT-decay (mediated by NCX and the plasmalemmal Ca2+-ATPase) from the rate-constant of systolic CaT-decay (mediated by SERCA, NCX and the plasmalemmal Ca2+-ATPase)10,11 Both rate constants had a significant but modest correlation (Pearson’s R2=0.34; P<0.01) and their subtraction revealed a trend towards increased SERCA-function in POAF (Figure 2D). We confirmed the unaltered protein expression of SERCA2a and phospholamban, and identified unchanged fractional phosphorylation-levels of phospholamban at Ser16 and Thr17 in POAF (Online Figure VIIA). Protein-levels of NCX1 were also comparable between Ctl and POAF (Online Figure VIIB), consistent with our functional data (Figure 2C).
SR Ca2+-leak and RyR2-dysfunction.
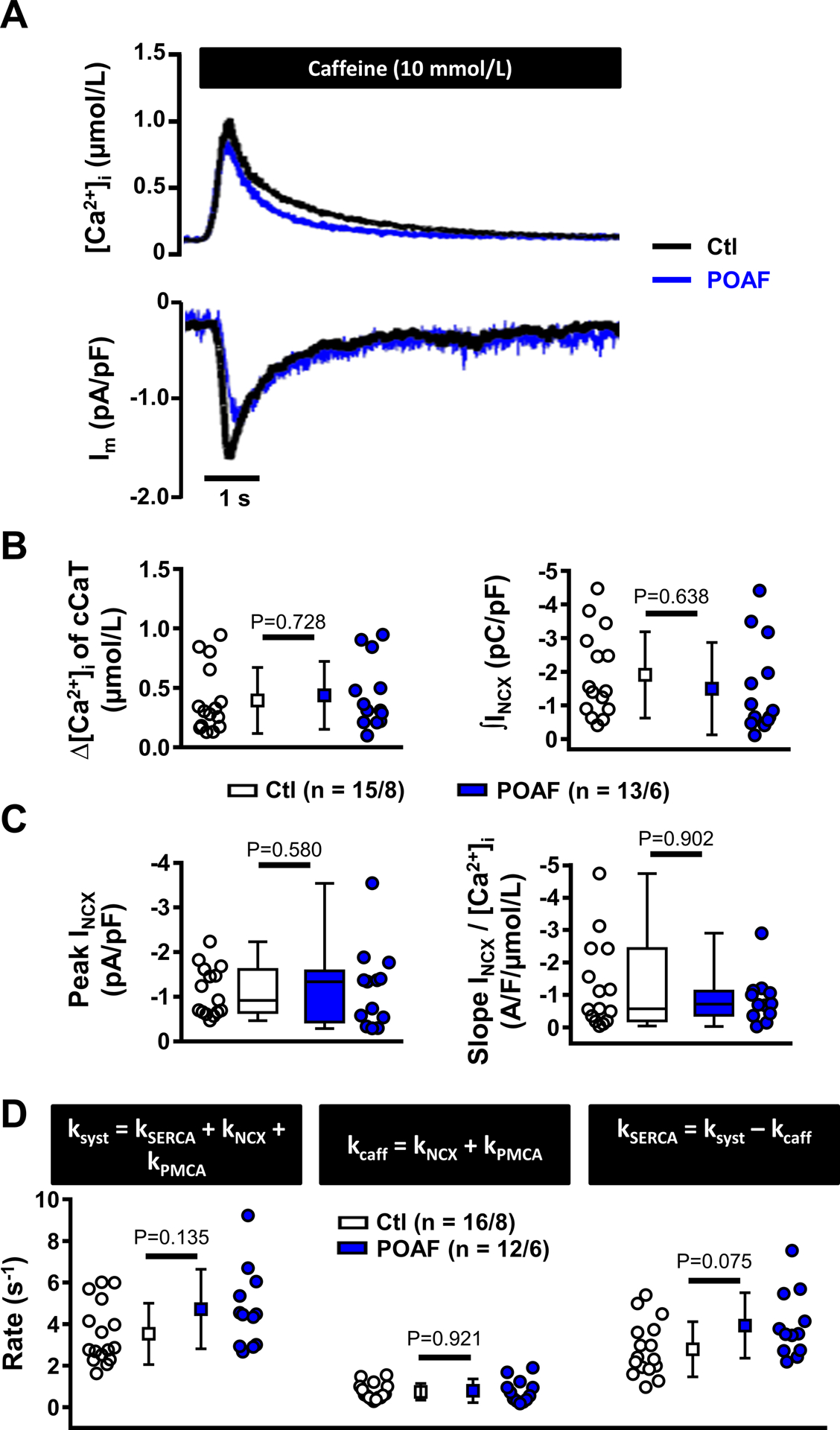
The quantitatively increased SERCA-function, along with unaltered SR Ca2+-content and reduced CaT-amplitude, suggest RyR2-dysfunction and increased SR Ca2+-leak. We measured the tetracaine-induced decrease in diastolic [Ca2+]i in the absence of extracellular Na+/Ca2+ (Figure 3A, left), which is proportional to the amount of RyR2-mediated SR Ca2+-leak.36 SR Ca2+-content was subsequently assessed using caffeine (10-mmol/L) in the same cell to determine leak/load ratios. SR Ca2+-leak/load ratio was significantly larger in POAF (Figure 3A, right), resulting from increased SR Ca2+-leak, along with a reduced caffeine-induced CaT-amplitude (Online Figure VIIIA-C). To assess whether the reduction of caffeine-induced CaT-amplitude under these specific (0-mmol/L extracellular Na+/Ca2+) conditions could be explained by increased expression of Ca2+-buffering proteins, we quantified the protein-levels of major Ca2+-buffers. Expression-levels of cardiac myosin-binding protein-C, troponin-I, troponin-C and calmodulin were similar between Ctl and POAF (Online Figure VIIID).
Figure 3. Sarcoplasmic reticulum (SR) Ca2+ leak and spontaneous SR Ca2+-release events (SCaEs).
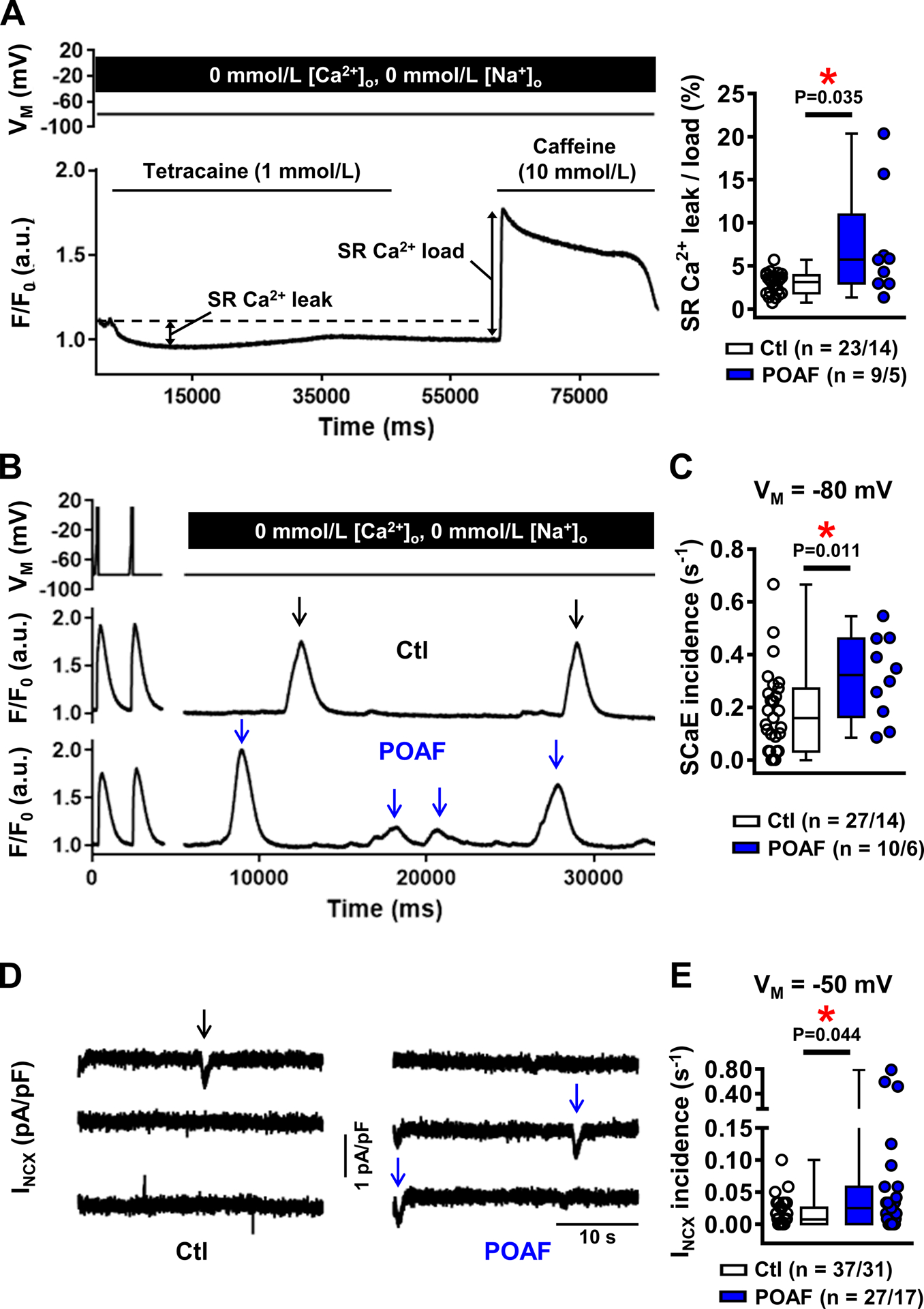
A, Experimental protocol employing tetracaine (1-mmol/L) to block RyR2 under 0-mmol/L extracellular Na+/Ca2+ to block sarcolemmal Ca2+-fluxes (left). The decrease in fluorescence in response to tetracaine shown in the example is proportional to SR Ca2+-leak.36 Subsequently, the amplitude of the caffeine-induced Ca2+-transient was used to assess SR Ca2+-load. Right panel shows SR Ca2+-leak/SR Ca2+-load ratio in Ctl and POAF. Individual leak and load components are shown in Online Figure VIII. B-C, Examples (B) and quantification (C) of SCaEs occurring prior to tetracaine application in the absence of sarcolemmal Ca2+-fluxes at −80 mV in Ctl and POAF. D-E, Examples (D) and quantification (E) of spontaneous INCX resulting from SCaEs (at −50 mV) in perforated-patch experiments in an independent cohort of Ctl and POAF cardiomyocytes. N-numbers indicate numbers of cardiomyocytes/patients. *P<0.05 vs. Ctl based on multilevel models with log-transformed data.
During the 0-mmol/L Na+/Ca2+ experiments, we observed more SCaEs, an indicator of RyR2-dysfunction, in POAF than Ctl (Figure 3B–C). We validated these findings and assessed their electrophysiological consequences by measuring spontaneous transient-inward INCX in perforated-patch experiments. Although the INCX-incidence at −80-mV was similar in Ctl and POAF (Online Figure VIB), when NCX-mediated Ca2+-extrusion was reduced by depolarizing the holding potential to −50-mV to challenge the system, POAF-HAMs had a statistically-significant greater transient INCX-incidence than Ctl-HAMs (Figure 3D–E).
Molecular basis of RyR2-dysfunction
To directly study RyR2-function we performed single-channel RyR2-recordings. RyR2 open-probability was significantly larger in POAF, due to reduced mean closed-times in the presence of normal open-times (Figure 4A). Immunoblots showed that total RyR2-protein-levels (Figure 4B), RyR2-phosphorylation at protein kinase-A site Ser2808 (Online Figure IXA), and protein-levels of the SR Ca2+-buffer calsequestrin (Online Figure IXB) were similar in Ctl and POAF. However, fractional RyR2-phosphorylation at the Ca2+/calmodulin-dependent protein kinase-II (CaMKII) site Ser2814 was 31% greater in POAF (Figure 4B–C), consistent with the significant increase in total protein-levels of the cytosolic CaMKIIδC (with preserved Thr287-autophosphorylation; Figure 4C). Total protein-levels or Thr287-autophosphorylation of the nuclear CaMKIIδB+δ9 were unchanged in POAF (Online Figure X). Thus, POAF patients have a pre-existing substrate characterized by Ca2+-handling abnormalities.
Figure 4. Ryanodine receptor channel type-2 (RyR2) dysfunction.
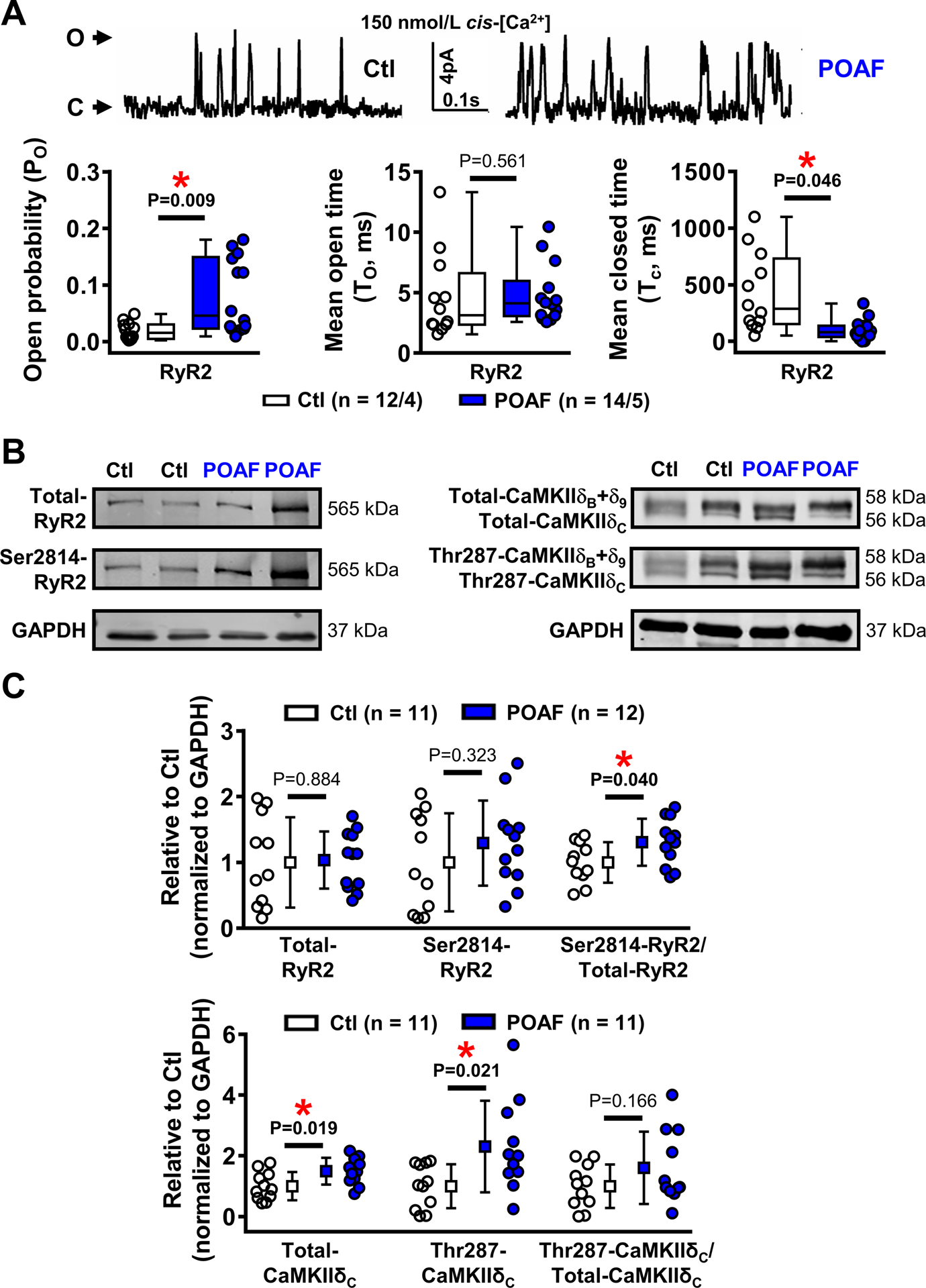
A, RyR2 single-channel recordings in planar lipid bilayers of Ctl and POAF patients (top) and quantification of RyR2 open-probability, mean open-time, and mean closed-time (bottom, left-to-right) at diastolic levels (150-nmol/L) of cytosolic (cis) Ca2+. B-C, Example Western blots (B) and protein-levels (C) of total and Ser2814-phosphorylated RyR2, as well as total and Thr287-autophosphorylated levels of nuclear and cytosolic Ca2+/calmodulin-dependent protein kinase-IIδ (CaMKIIδB+δ9 and CaMKIIδC, respectively) and their quantification (quantifications for CaMKIIδB+δ9 are shown in Online Figure X). N-numbers indicate numbers of channels/patients (panel A), or patients (panels B-C). *P<0.05 vs. Ctl based on multilevel models with log-transformed data (A) or unpaired Student’s t-test (B, C), with Welch’s correction for unequal variance for Thr287-CaMKIIδC.
Upregulation of NLRP3-inflammasome signaling
Given the contribution of persistently-activated cardiomyocyte NLRP3-inflammasome signaling to AF,18 and recent data supporting a direct link between CaMKIIδ and cardiomyocyte NLRP3-inflammasome activation,19,20 we hypothesized that POAF patients may have a pre-existing low-grade inflammatory-state characterized by enhanced atrial NLRP3-related signaling, which we evaluated by immunoblot. The specificity of antibodies against components of the NLRP3-inflammasome signaling cascade was validated with blocking peptides or short-hairpin RNA-mediated knockdown in atrial whole-tissue lysates, cardiomyocytes and fibroblasts from humans and canines, or HL-1-cells (Online Figures XI-XII). Protein-levels of NLRP3 (125 kDa)37 and pro-caspase-1 were increased in whole-tissue of POAF-patients (Figure 5A), with a trend towards increased protein-levels of the “apoptosis-associated speck-like protein containing a CARD” (ASC, +116%), indicating increased priming (transcription of components) of the NLRP3-inflammasome in POAF. Protein-levels of the inflammasome-dependent membrane-pore gasdermin-D were also significantly higher in POAF, whereas those of pro-IL-1β and the NLRP3-inflammasome products caspase-1-p20, gasdermin-D N-terminal fragment, and IL-1β were similar in Ctl and POAF (Figure 5A). The expression-levels of toll-like receptor-4 (TLR4) and nuclear-factor kappa-B (NFκB), two upstream regulators of NLRP3-complex priming, were also increased in POAF (+95% and +48%, respectively; Figure 5B). Protein-levels of the purinergic P2X7-receptor (P2X7R), which mediates K+-efflux and K+-depletion-induced triggering (assembly of components) of the NLRP3-complex, were 59% higher in POAF (Figure 5B), pointing to activation of atrial NLRP3-inflammasome through increased priming and triggering mechanisms.
Figure 5. Enhanced atrial NACHT, LRR, and PYD domains containing protein 3 (NLRP3)-inflammatory signaling in whole-tissue homogenates.
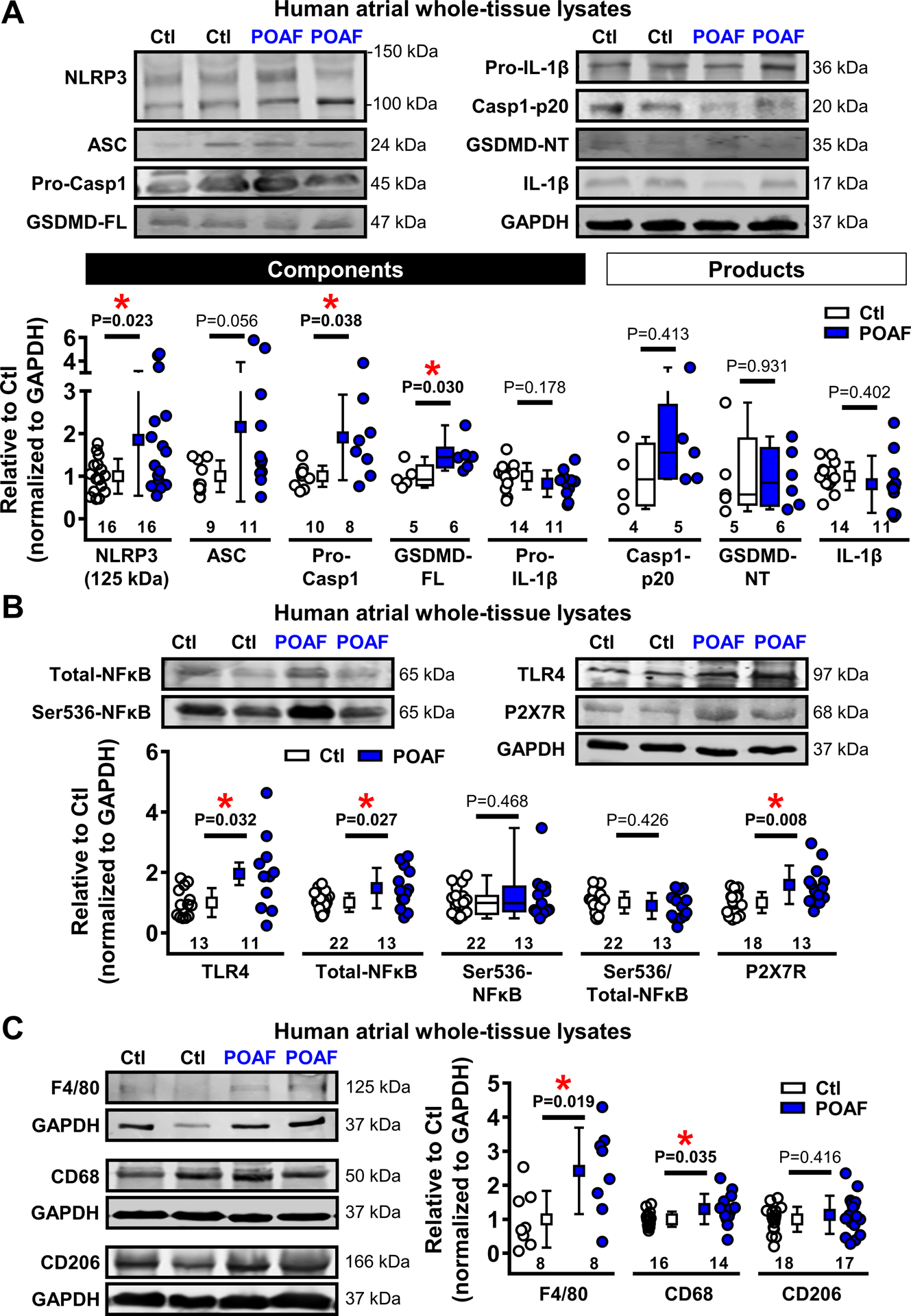
A, Western blots (top) and quantified protein-levels (bottom) of components and products of the atrial NLRP3-inflammasome in Ctl or POAF patients. ASC indicates apoptosis-associated speck-like protein containing a CARD; (Pro-)Casp1, (Pro-)caspase-1; CT, C-terminal; FL, full-length; GSDMD, gasdermin-D; (Pro-)IL-1β, (Pro-)interleukin-1β; NT, N-terminal; B, Western blots (top) and quantified protein-levels (bottom) of toll-like receptor-4 (TLR4), total and Ser536-phosphorylated nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) involved in priming, and purinergic P2X7 receptor (P2X7R), involved in triggering of the NLRP3-inflammasome. C, Western blots (left) and quantified protein-levels (right) of macrophage lineage markers F4/80, CD68 and CD206 in Ctl and POAF patients. Numbers below symbols indicate number of atrial samples. GAPDH was used as loading control. *P<0.05 vs. Ctl based on Mann-Whitney test (GSDM-FL, Casp1-p20, GSDMD-NT, and Ser536-NFκB), unpaired Student’s t-test (Pro- IL-1β, Ser536/Total-NFκB, F4/80, and CD206) or Student’s t-test with Welch’s correction for unequal variance (remaining proteins).
Results obtained in whole-tissue lysates can reflect changes in various cardiac cell-types. We assessed protein-levels of macrophage-markers (F4/80, CD68 and CD206) in whole-tissue and found increased F4/80- and CD68-levels in POAF (Figure 5C), pointing to a local inflammatory response. To investigate whether the NLRP3-inflammasome is present and remodeled in cardiomyocytes, we assessed its components in HAM fractions, which we have shown not to be contaminated by macrophages.18,26 Consistent with our whole-tissue findings, protein-levels of pro-caspase-1 and ASC were significantly increased in HAMs (Figure 6). Furthermore, active caspase-1-p10 and the cleaved N-terminal and C-terminal fragments of gasdermin-D, which are products of activated NLRP3-inflammasome, were also upregulated in POAF-HAMs. Consistent with the increased levels of the pore-forming N-terminal fragment of gasdermin-D, which propagates inflammatory signaling by secreting IL-1β out of the cell, intracellular HAM protein-levels of IL-1β were lower in POAF (Figure 6). Together, our data indicate enhanced NLRP3-inflammasone activation in POAF-HAMs, suggesting a low-grade atrial inflammatory-state, which might promote AF-initiation in the presence of appropriate post-operative AF-triggers.
Figure 6. Enhanced atrial NACHT, LRR, and PYD domains containing protein 3 (NLRP3)-inflammatory signaling in human atrial cardiomyocytes.
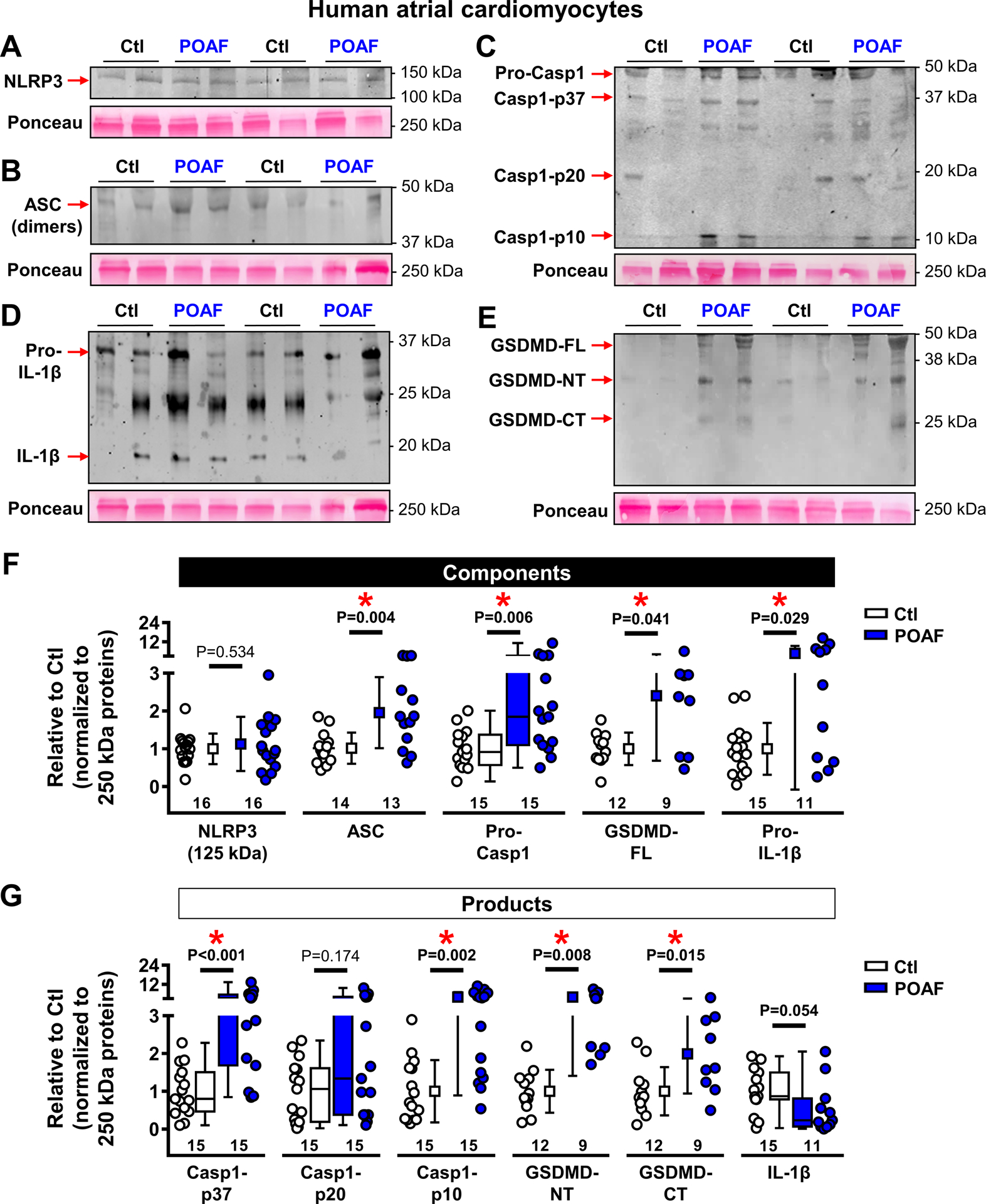
A-E, Western blots of NLRP3 (A), apoptosis-associated speck-like protein containing a CARD (ASC, B), (Pro-)caspase1 (Pro-Casp1, C), (Pro-)interleukin-1β (Pro-IL-1β, D) and full-length (FL), N-terminal fragment (NT) or C-terminal fragment (CT) of gasdermin-D (GSDMD, E). Please note the absence of a ~100 kDa NLRP3 band in HAMs vs whole-tissue homogenates (Figure 5).37 F-G, Quantification of components (F) and products (G) of the atrial NLRP3-inflammasome in atrial cardiomyocytes of Ctl or POAF patients. Total protein-levels at 250 kDa (Ponceau staining) were used as loading control. N-numbers indicate numbers of patients and kDa labels reflect values of molecular weight markers. *P<0.05 vs. Ctl based on Mann-Whitney test (Pro-Casp1, Casp1-p37, Casp1-p20, and IL-1β), unpaired Student’s t-test (GSDM-CT) or Student’s t-test with Welch’s correction for unequal variance (remaining proteins).
Indices of profibrotic and connexin remodeling.
Systemic inflammation has been associated with reentry-promoting profibrotic remodeling.4 We assessed established profibrotic markers in RA whole-tissue, but found no significant differences between groups (Online Figure XIII). Similarly, we found no significant differences in total connexin-40 or connexin-43, nor in Ser368-phosphorylated connexin-43 (Online Figure XIV). Thus, despite NLRP3-signaling consistent with an inflammatory state, we found no evidence that profibrotic signaling and connexin remodeling contribute to the pre-existing POAF-substrate.
Role of NLRP3-inflammasome signaling and its proarrhythmic consequences in the post-operative period.
To assess the timing of post-operative inflammation in relation to the first POAF-episode in our patients, we characterized the time-course of serum CRP-changes (Figure 7A). The peak incidence of the first POAF-episode was on days 2–3, coinciding with the maximal values of serum CRP-concentration. We therefore hypothesized that the production of inflammatory mediators in the post-operative period may act on the pre-existing atrial HAM-substrate (as revealed by our intra-operative tissue samples) to cause proarrhythmic DAD-mediated triggered activity. Because it is not possible to obtain human atrial tissue at the moment of POAF-development, we applied an acute stimulus with the major inflammatory-mediator IL-1β (40-ng/mL) to HAMs to mimic post-operative inflammation (Figure 7B). Acute IL-1β application induced SCaEs more frequently in POAF-HAMs and provoked a larger per-HAM frequency of SCaE-occurrence for POAF within HAMs showing SCaEs (Figure 7C–D), supporting the notion that post-operative inflammation might provide a sufficient trigger for POAF-initiation when superimposed on the pre-existing substrate.
Figure 7. Proarrhythmic effects of post-operative inflammation in patients with pre-existing substrates.
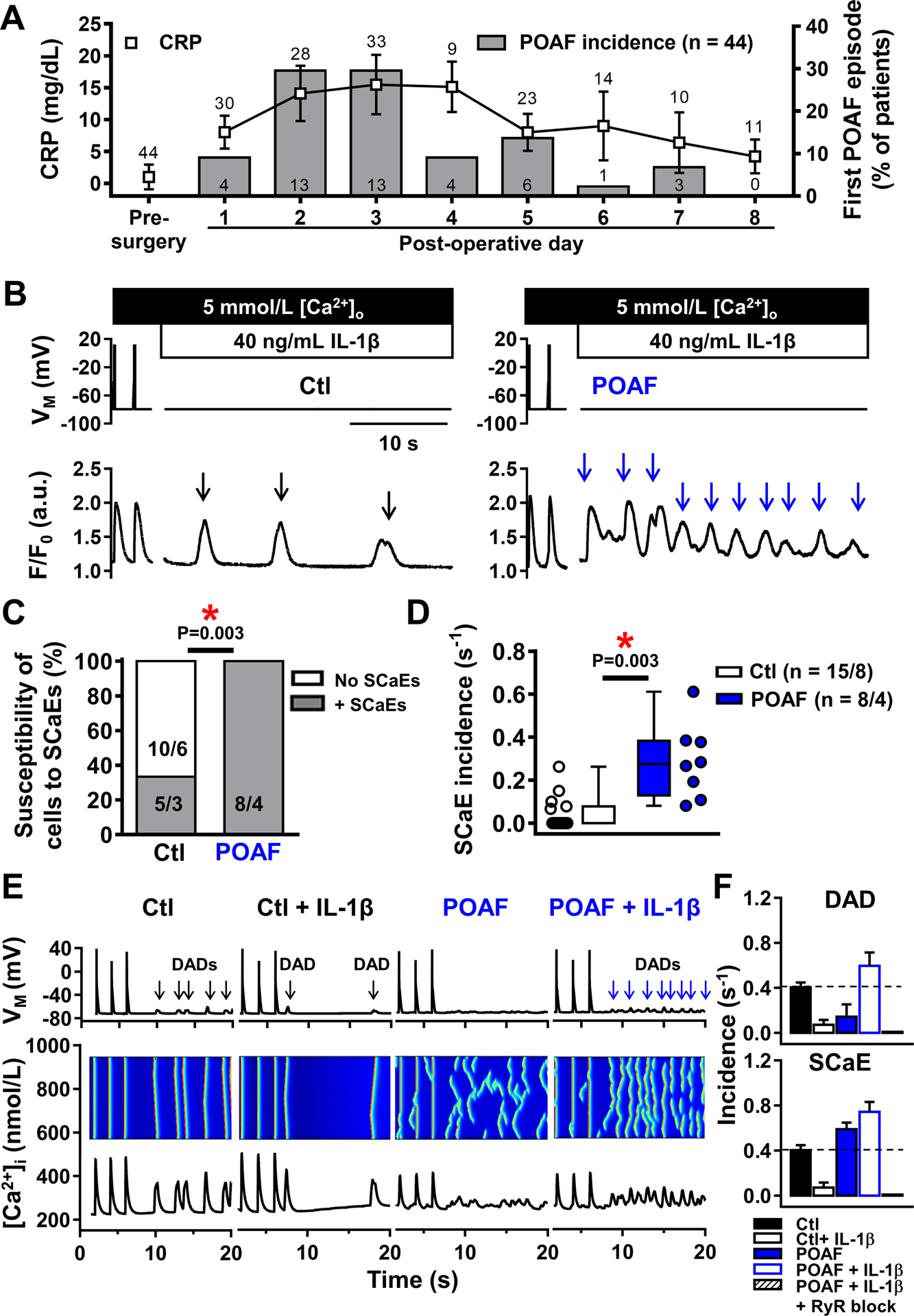
A, Coincidence of post-operative C-reactive-protein (CRP) increases (symbols) and POAF development (bars) in 44 patients with regular ECG- and CRP-measurements, showing peak incidence of POAF on days with highest CRP-levels (post-operative days 2–3). Numbers above symbols indicate number of available CRP-measurements; numbers in bars indicate absolute number of patients with a first POAF episode on a given post-operative day. B, Depolarization-induced Ca2+-transients and proarrhythmic spontaneous sarcoplasmic-reticulum Ca2+-release events (SCaEs; indicated by arrows) in a Ctl (left) and POAF (right) cardiomyocyte acutely exposed to 40-ng/mL IL-1β to mimic post-operative inflammation. C,D, Susceptibility of individual cardiomyocytes to (C) and incidence of (D) SCaEs in Ctl and POAF. One Ctl patient had one cardiomyocyte with SCaEs and one without and is therefore listed twice in panel C. E, Simulated action potentials, longitudinal line-scans and whole-cell CaTs (top-to-bottom) at 0.5-Hz and follow-up showing SCaEs and corresponding delayed afterdepolarizations (DADs) in Ctl, Ctl+IL-1β, POAF, and POAF+IL-1β models. The novel POAF human atrial cardiomyocyte model was developed by implementing the experimentally observed increases in RyR2 open-probability and SERCA function (Online Table VI). F, Quantification of SCaE (top) and DAD (bottom) incidence in 6 replications of the Ctl, Ctl+IL-1β, POAF, POAF+IL-1β and POAF+IL-1β+RyR2 block models with stochastic RyR2 gating. N-numbers indicate numbers of patients (A) or cardiomyocytes/patients (C,D). *P<0.05 vs. Ctl based on Fisher’s exact test (C) or multilevel models with log-transformed data (D).
We then applied computational modeling to: 1) determine whether the incorporation of our experimentally-observed altered RyR2-gating and trend towards increased SERCA-function in POAF into an established in silico HAM-model would result in relative AP- and Ca2+-handling properties similar to those we observed in Ctl versus POAF; and 2) examine the potential arrhythmogenic consequences (e.g., DADs) of POAF-associated remodeling. We adjusted our previously-developed HAM-model incorporating spatial Ca2+-handling11,28 to reproduce key Ca2+-handling properties observed in Ctl-HAMs (Online Figures XV-XVI). We then developed a POAF-version based on our experimental data, by incorporating altered RyR2-gating and increased SERCA-function. With this limited set of experimentally-guided changes, the POAF-model fully recapitulated the experimentally-observed POAF-associated reduction in CaT-amplitude, unaltered SR Ca2+-load and increased SR Ca2+-leak (Online Figure XVI-XVII). The POAF-model also reproduced the observed SCaE differences, with simulated IL-1β causing a large increase in SCaE-incidence in POAF versus Ctl (+509% versus +122%, with and without IL-1β-effects, respectively; Online Figure XVIII).
We then employed the POAF-model to evaluate the occurrence of arrhythmogenic DADs during current-clamp simulations. The Ctl-model showed a limited number of SCaEs with corresponding DADs, both with and without simulated IL-1β (Figure 7E–F, Online Figure XIX). Although the incidence of ScaEs was reduced in the Ctl-model following IL-1β stimulation, total SR Ca2+-leak was increased (Online Figure XX), suggesting that IL-1β-mediated SR Ca2+-leak does not translate into proarrhythmic Ca2+-waves without pre-existing RyR2-dysfunction. DADs, but not ScaEs, were abolished after removal of extracellular Ca2+ and Na+, whereas ScaEs disappeared during simulated RyR2-inhibition (Online Figure XIX), supporting the causal role of RyR2-mediated SR Ca2+-leak and the associated depolarizing INCX in the proarrhythmic response. The model with the pre-existing POAF-substrate also showed an increased ScaE-incidence (+45%), but in the absence of IL-1β the ScaEs were small and too dispersed to produce clear DADs (Figure 7E–F). With IL-1β stimulation, the POAF-model showed a large increase in ScaEs (+84%) and more large-amplitude DADs (+47%), which disappeared with simulated RyR2-block (Figure 7F). Overall, computational modeling supports the internal consistency of our findings and is compatible with the notion that post-operative inflammation acting on a pre-existing cellular substrate elicits proarrhythmic cellular responses contributing to POAF.
Acute IL-1β drives a self-amplifying feed-forward loop via a NLRP3/CaMKII nexus
We observed increased SCaE-generation in POAF-HAMs acutely exposed to IL-1β (Figure 7B,C). To examine potential mechanisms of IL-1β-action, we examined its acute effects on SCaEs and the expression of relevant proteins in HL-1-cells. Consistent with our HAM and in silico data, acute IL-1β stimulation increased the number of SCaEs in HL-1-cells (Figure 8A). On Western blot, 5-min incubation with IL-1β significantly increased RyR2-phosphorylation at Ser2808 and Ser2814, and phospholamban phosphorylation at Thr17, but not Ser16 (Figure 8B,C). The IL-1β-mediated increases in phospholamban-Thr17 and RyR2-Ser2814 (but not RyR2-Ser2808) phosphorylation were prevented by pre-incubation with the CaMKII-inhibitor KN-93 (but not the negative-control analog KN-92) (Figure 8C, right), pointing towards acute CaMKII-mediated promotion of Ca2+-handling abnormalities and SR Ca2+-release events in response to acute IL-1β exposure. In addition, 5-min incubation with IL-1β increased the formation of NLRP3-inflammasome mediators, active caspase-1-p20 and the pore-forming gasdermin-D N-terminal fragments (Figure 8D,E), in a CaMKII-independent manner (Figure 8E, right), without changes in total NLRP3-expression.
Figure 8. Acute interleukin-1β (IL-1β) stimulation promotes ryanodine receptor type-2 (RyR2) channel hyperphosphorylation, spontaneous Ca2+-release events (SCaEs) and NACHT, LRR, and PYD domains containing protein 3 (NLRP3)-inflammatory signaling in HL-1 cells.
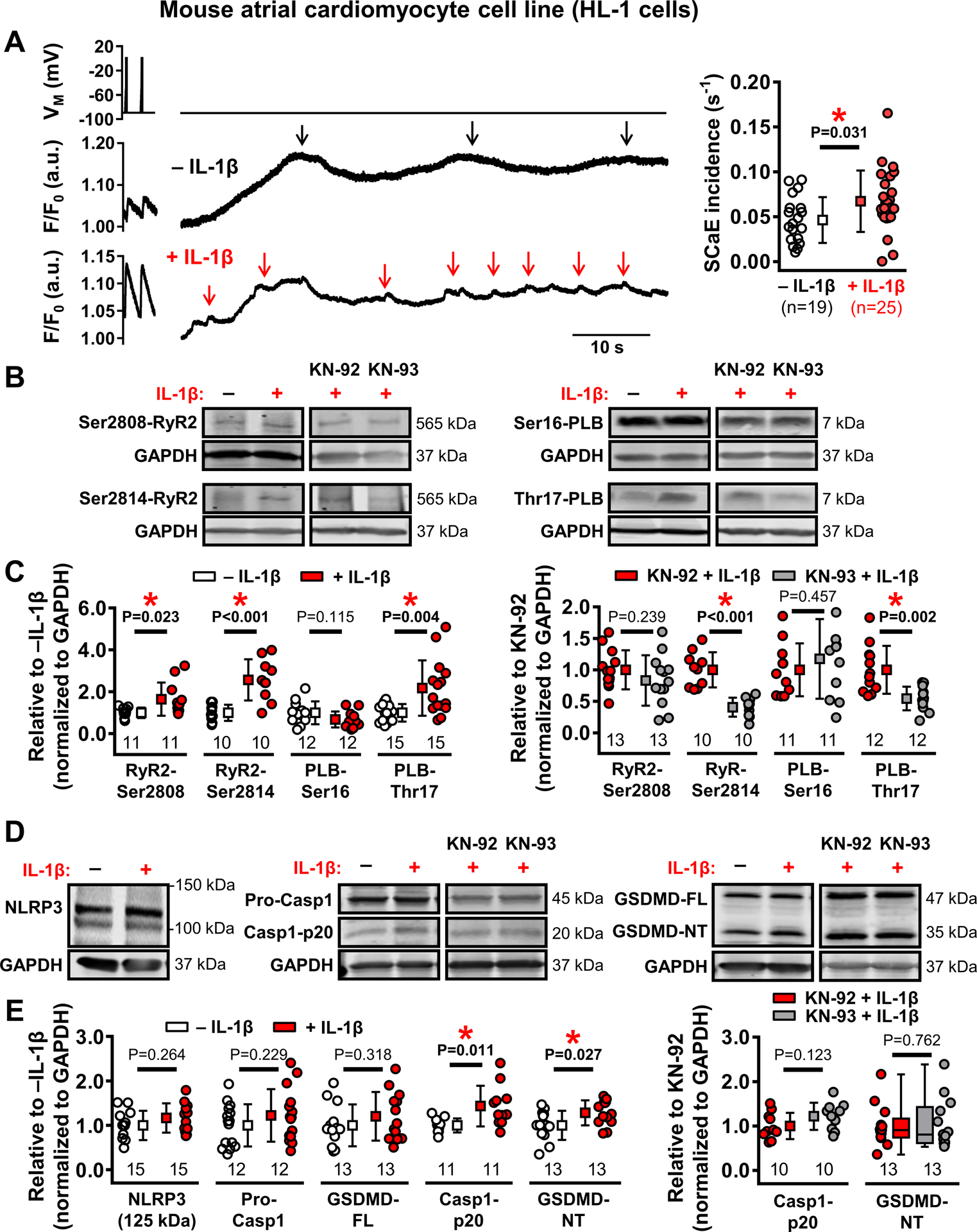
A, Examples of depolarization-induced Ca2+-transients and SCaEs (indicated by arrows) in HL-1 cells at baseline (top) and acutely exposed to 40-ng/mL IL-1β to mimic post-operative inflammation (bottom), as well as incidence of SCaEs in both groups (right). B-C, Western blots (B) and group data of RyR2 phosphorylation at Ser2808 and Ser2814, as well as phospholamban phosphorylation at Ser16 and Thr17 at baseline and after 5 minutes of IL-1β stimulation (C, left) and 5-min IL-1β-induced phosphorylation levels of RyR2 and phospholamban in the presence of the Ca2+/calmodulin-dependent protein kinase-II inhibitor KN-93 or its inactive control KN-92 (C, right). D-E, Similar to panels B-C for protein-levels of NLRP3-inflammatory signaling components. N-numbers indicate number of cells (A) or batches of cells (B-E). *P<0.05 vs. baseline (panels A, C, E) or vs. KN-92 (right panels of C,E) based on unpaired Student’s t-test (A-E), with Welch’s correction for unequal variance for RyR2-Ser2808, RyR2-Ser2814 (C, left), PLB-Thr17 (C), and Casp1-p20 (E, left); or Mann-Whitney test (GSDMD-NT in panel E, right).
DISCUSSION
Here, we have identified a predisposing cellular substrate for POAF consisting of (1) HAM Ca2+-handling abnormalities resulting from CaMKII-dependent RyR2-hyperphosphorylation and (2) NLRP3-inflammasome activation, without AF-promoting changes in AP-properties, connexin remodeling or active profibrotic signaling (Online Figure XXI). Despite the presence of abnormalities in patients destined to manifest POAF, none of these patients had manifested clinical AF, indicating that these abnormalities were insufficient in themselves to generate AF. Our data suggest that this substrate determines which atria will cross the AF-threshold, initiating POAF, when acted upon by post-operative triggers. In particular, post-operative inflammation can exacerbate pre-existing Ca2+-handling abnormalities, promoting the formation of DADs that can lead to POAF. Our observations provide novel insights into the potential cellular and molecular substrates underlying clinical POAF.
Mechanisms of POAF and comparison with previous work.
The vulnerable POAF-substrate is generally investigated by correlating the properties of atrial samples obtained during cardiac surgery with the occurrence of subsequent POAF. Prior to the present study, the data available were limited and inconclusive. Previous studies found similar mRNA-expression of all major ion-channel subunits in LA and RA of Ctl- and POAF-patients4 and unchanged K+-currents and APD in RA-cardiomyocytes from POAF-patients,9,38 consistent with our data. Both increased and unchanged ICa,L-amplitude have been reported in POAF;9,39 here, we found unchanged ICa,L-amplitude in POAF-patients with both ruptured and perforated patch-clamp methods. We did observe an increased susceptibility to beat-to-beat ICa,L-alternans in POAF, which may contribute to APD-alternans, a known reentry-promoting mechanism.34 Although the development of APD-alternans at faster rates was not evaluated in the human multicellular preparations studied here, we have recently shown that Ca2+-handling abnormalities like those observed in POAF can promote APD-alternans and AF in dogs.40 Thus, although classical indices of pre-existing electrical remodeling are absent in POAF, dynamic repolarization heterogeneities, potentially exacerbated by post-operative triggers, may contribute to POAF-initiation and -maintenance by favoring ectopic-activity-mediated induction of reentry and require further investigation.
Previously, we comprehensively analyzed HAM Ca2+-handling in cAF10 and pAF11 patients. In pAF,11 both SERCA-related SR Ca2+-uptake and RyR2 Ca2+-leak/single-channel open-probability are enhanced, as in POAF (present study). Like those destined to experience POAF, pAF-patients have an underlying abnormality in Ca2+-handling that predisposes to AF-generation, but that requires some additional condition to initiate AF. In pAF, the superimposed factor(s) that trigger(s) AF-episodes is unclear, whereas for POAF superimposed surgery-induced inflammation plays a key role. In cAF, SR Ca2+-leak, single RyR2 open-probability and DAD-susceptibility are also increased,10 although what proportion of these abnormalities contribute to AF-maintenance and what proportion are due to persistent AF is unclear. POAF and cAF share decreased CaT-amplitude and RyR2-mediated SR Ca2+-leak.10 In POAF, the non-significant reduction in SR Ca2+-load and steep dependence of CaT-amplitude on SR Ca2+-load41 likely contribute to the reduced CaT-amplitude. By contrast, reduced CaT-amplitude in cAF is largely due to decreased ICa,L and SERCA, along with increased NCX.10 Finally, we showed that Ca2+-handling abnormalities in POAF are not in themselves sufficient to trigger arrhythmogenic afterdepolarizations, which are only elicited in the presence of an inflammatory mediator (IL-1β). Thus, although all forms of AF exhibit Ca2+-handling abnormalities that can elicit arrhythmogenic afterdepolarizations, there are important mechanistic differences in the potential cause of RyR2 dysfunction and the role of SR Ca2+-load, which may be relevant for the development of future therapeutic strategies.
Previous work identified reduced mRNA and protein-levels of SERCA2a-inhibitory sarcolipin in POAF,35 pointing to altered SR Ca2+-uptake. In agreement, we noted a trend towards increased SERCA-function in POAF, and identified a novel critical role for CaMKII-dependent RyR2-leak, likely underlying the reduced CaT-amplitude and increased incidence of SCaEs and DADs.
Systemic inflammation is considered a major post-operative trigger for POAF.13–15 In dogs with pericardiotomy or right-atriotomy, post-operative inflammation correlates with conduction inhomogeneities and AF-inducibility localized around the RA incision-site, which are inhibited by anti-inflammatory treatment.42–45 These data implicate a susceptibility to reentry due to acute inflammation in a clinically-relevant animal model. However, spontaneous AF is not reported in this model, suggesting that additional factors (such as DAD-induced triggered activity) might be needed to act on the reentry substrate to induce spontaneous POAF. Some,13,16 but not all,8,17 clinical studies have associated POAF with elevated pre-operative inflammatory markers. However, prior to the present study it was unknown whether atria of patients with increased susceptibility for POAF have a pre-existing inflammatory or cellular electrophysiological state that supports AF-induction. We detected increased priming and triggering of the NLRP3-inflammasome and activated caspase-1 in POAF-HAMs at the onset of cardiac surgery. Protein-levels of IL-1β were lower in POAF, likely due to caspase-1-mediated increases in the pore-forming N-terminal fragment of gasdermin-D, which mediates the exit of IL-1β,46 thereby increasing the release of IL-1β from HAMs and promoting inflammatory signaling through autocrine and paracrine effects.
CaMKIIδ modulates the expression of inflammatory genes and NLRP3-inflammasome activation in mouse hearts with transverse-aortic banding or chronic angiotensin-II-treatment,19,20 suggesting that the pre-operative CaMKIIδ-activation that we identified in POAF may represent a critical nodal point for both RyR2-mediated SR Ca2+-leak with subsequent increases in SCaEs and NLRP3-inflammasome activation.47 Oxidative stress is increased in POAF-patients48 and may contribute to both CaMKIIδ and NLRP3-inflammasome activation.47,49 Our data indicate that acute IL-1β stimulation promotes CaMKII-dependent RyR2-hyperphosphorylation, exacerbates cardiomyocyte Ca2+-handling abnormalities and causes DAD-generation. In addition, acute IL-1β application promoted NLRP3-inflammasome activation, but this effect was not CaMKII-dependent, pointing to the involvement of alternative signaling pathways. Together with evidence for a role for inflammatory cells in lone AF,50 these findings suggest that pre-operative local inflammation may contribute to the observed cardiomyocyte NLRP3-inflammasome remodeling. Furthermore, our findings show that post-operative IL-1β releases from inflammatory cells or HAMs might create an additional self-amplifying feed-forward loop of NLRP3-inflammasome activation at the expense of promotion of CaMKII-dependent Ca2+-handling abnormalities, mechanistically linking IL-1β- and CaMKII-signaling pathways with NLRP3-inflammasome activation. Our data support a model in which the post-surgical IL-1β-induced increases in phosphorylation levels of Ca2+-handling proteins exacerbate the pre-existing SR-dysfunction, reaching a value above the threshold needed to cause potentially-proarrhythmic SCaEs and DADs that might predispose to POAF (Online Figure XXI).
Pre-operative structural remodeling might contribute to POAF and LA-fibrosis is a consistent finding.6,7 A greater degree of RA-fibrosis has also been reported in some,8,51 but not all studies.6,52 Although inflammation can activate profibrotic signaling, we did not find significant differences in expression of profibrotic proteins in POAF. Connexin remodeling can promote reentrant arrhythmias even in the absence of fibrosis, but in agreement with most published studies,4,53 our data argue against a major role for connexin remodeling in POAF.
Novelty and potential clinical implications.
CaMKII-dependent Ca2+-handling abnormalities are well established in patients with persistent AF and some animal models, promoting the progression of AF-related remodeling.10–12 Here, we provide the first evidence for a potential role of these phenomena in POAF. The identification of CaMKII-related abnormalities as part of the vulnerable substrate before the occurrence of atrial arrhythmias in the present work supports their potential causative role.
We recently identified abnormal NLRP3-inflammasome activation in pAF and cAF HAMs.18 Here, we established for the first time that pre-existing activation of the atrial cardiomyocyte NLRP3-inflammasome also contributes to the POAF-predisposing substrate. In a meta-analysis of 22 randomized-controlled trials, peri-operative statin use was associated with a decreased inflammatory response and lower POAF-risk.54 We similarly observed a tendency towards lower levels of lipid-lowering drugs in POAF-patients (Online Table I). Moreover, use of lipid-lowering drugs was associated with lower pre-operative NLRP3 protein-levels in atrial whole-tissue lysates and HAMs (Online Figure XXII). These results are consistent with a canine sterile pericarditis model showing that atorvastatin prevents maintenance of AF by inhibiting inflammation.44 This finding, while only hypothesis-generating, nonetheless provides interesting ideas for future work. The molecular signature of NLRP3-inflammasome activation is distinct in pAF (increase in NLRP3-inflammasome priming only) compared to cAF and POAF (increases in both priming and triggering of NLRP3-inflammasome).18 Nevertheless, all AF forms show evidence for atrial NLRP3-inflammasome activation.
We identified molecular evidence for the presence of a subclinical atrial cardiomyopathy, in terms of inflammatory and Ca2+-handling changes, that predisposes POAF-patients to AF-development. Indeed, there is clinical evidence pointing to the presence of a pre-existing atrial cardiomyopathy in patients who develop POAF.55 Our study provides the molecular links between the atrial cardiomyopathy and arrhythmogenic afterdepolarizations involved in AF-pathogenesis. Further work is needed to better define the factors leading to this AF-promoting atrial cardiomyopathy. Finally, recent work has emphasized the substantial risk for AF-recurrence in POAF-patients, and the many AF-associated risk factors that they display.4 Our findings provide a novel unifying paradigm, by suggesting that POAF-patients share underlying atrial-cardiomyocyte NLRP3-signaling activation with pAF- and cAF-individuals, predisposing POAF-patients to both POAF caused by transient surgery-induced inflammation (explaining the usually self-limited nature of POAF) and to long-term pAF and/or cAF (explaining the predilection for recurrence).
Potential limitations
Patient follow-up, including POAF-detection, was performed according to routine clinical practice and was based on ECG-monitoring and symptoms, in line with previous studies.48 However, brief asymptomatic POAF-episodes may have gone undetected in Ctl-patients. Also we cannot rule out that some patients may have had asymptomatic pAF prior to their surgery. Because it is not possible to study POAF-mechanisms at the time of the arrhythmia in patients, we mimicked post-operative inflammation with an acute application of a high concentration of IL-1β, but other inflammatory cytokines likely contribute and the post-operative inflammatory response develops slower in vivo (Figure 7A). These aspects warrant additional studies. In addition, the precise role of oxidative stress, consistently shown to be enhanced in POAF,48 was not addressed here and should be delineated in subsequent work. Although our experimental data and computer simulations suggest that acute IL-1β application is sufficient to produce cellular arrhythmogenic responses in predisposed cardiomyocytes, additional factors such as dysregulation of the autonomic nervous system may also contribute to POAF-initiation.56 How the autonomic nervous system contributes to the molecular pathophysiology of POAF is unknown and will require extensive experimental work, particularly in view of the evidence for substantial protective actions of β-adrenoceptor blockers4 and the known DAD-promoting effects of β-adrenoceptor activation.57 Because of limited accessibility to human atria, particularly the LA, our work was restricted to RA-appendages. Different atrial regions may undergo distinct remodeling and further work should be directed to assessing whether the RA changes we studied in detail also apply to LA. There is at present no direct evidence that DADs in the RA-appendage can lead to AF in patients.
Since this is the first study to identify pre-existing Ca2+-handling and NLRP3-inflammasome remodeling in patients that go on to develop POAF, it was not possible to perform a power calculation for predetermined outcome parameters. POAF-patients had similar clinical parameters to Ctl-patients except greater age and lower estimated glomerular filtration rate, consistent with previous studies.58 We also identified differences in the use of statins or dihydropyridine Ca2+-channel blockers for some subgroups (Online Tables I-IV). Atrial diameter was not different between Ctl and POAF-patients, but atrial volume index may be a more reliable indicator of atrial size. Since this parameter was not available for the majority of our patients, the impact of atrial volume on atrial cellular function needs further investigation. CaMKII-activation exhibited a weak dependence on age (Online Figure XXIII), consistent with previous work,59 and TLR4-levels also increased with advancing age (Online Figure XXIV). However, neither age nor dihydropyridine Ca2+-channel blocker therapy influenced any of the other findings. Thus, while the greater age of POAF-patients likely contributes to some components of POAF-pathogenesis, it fails to account for the other important aspects.
A large number of atrial samples were collected and studied, but each sample can only be used for a limited subset of experiments. Thus, of necessity different experimental series include experiments from different sets of patients and while we performed extensive analyses to assess the influence of factors like patient age, the influence of medications, atrial size, valvular regurgitation (Online Figures XXII-XXXII, Online Table X), we cannot fully exclude contributions from other, unrecognized factors. The clinical profiles of the patient subgroups used for patch-clamp/Ca2+-imaging and molecular-biology/biochemistry experiments were comparable. The patient cohort used for multicellular AP-recordings showed differences in distribution of some clinical parameters; this needs to be considered in relating the similar AP-properties that we noted in Ctl and POAF to the other findings in our paper. In addition, some individual experiments were inevitably unsuccessful (Online Figure I), potentially introducing bias. However, it is reassuring that POAF-incidence was similar in successful and unsuccessful experiments and that a re-analysis of Western blot experiments blinded to post-operative status produced similar results (Online Figure XXXIII).
Conclusions
We have performed the first detailed systematic analysis of the atrial cellular and molecular features associated with POAF in a substantial patient population. Our findings provide new insights into the underlying mechanism, identifying a novel pre-existing substrate characterized by CaMKII-dependent Ca2+-handling abnormalities and atrial-cardiomyocyte NLRP3-inflammasome activation. These observations provide a unifying model of AF that accounts for the high long-term AF recurrence-rate in POAF patients and the transient occurrence of POAF post-operatively. Novel strategies to inhibit CaMKII- and/or NLRP3-inflammasome-related signaling might provide new therapeutic approaches to POAF prevention and management.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is Known?
Post-operative atrial fibrillation (POAF) frequently occurs after cardiac surgery and while post-operative inflammation is considered a major trigger, the molecular mechanisms are poorly defined.
The NLRP3 (NACHT, LRR, and PYD domains containing protein-3) inflammasome contributes to paroxysmal and persistent AF, but has not been evaluated in POAF.
Calcium-handling abnormalities and CaMKII (calcium/calmodulin-dependent protein kinase-II) have been implicated in atrial arrhythmogenesis, but their role in POAF is unknown.
What New Information Does This Article Contribute?
We identify the presence of a subclinical pre-surgical atrial cardiomyopathy comprising NLRP3-inflammatory signaling and CaMKII-mediated calcium-handling changes that predispose POAF-patients to AF development.
Acute IL-1β stimulation promotes CaMKII-dependent RyR2 hyperphosphorylation, exacerbating proarrhythmic cardiomyocyte calcium-handling abnormalities and causes CaMKII-independent NLRP3-inflammasome activation.
These observations provide a unifying model of AF involving a vulnerable substrate and inflammatory triggering that accounts for the transient occurrence of POAF post-operatively along with the high long-term AF recurrence rate in POAF patients.
ACKNOWLEDGMENTS
The authors gratefully acknowledge expert technical assistance from Konstanze Fischer, Monika Hagedorn, Annette Kötting-Dorsch, Ramona Nagel, Simone Olesch, as well as extensive input from Anna Nozza of the Montreal Heart Institute Biostatistics Unit.
FUNDING SOURCES
The authors’ work was supported by the Netherlands Organization for Scientific Research (ZonMW Veni 91616057 to JH), the National Institutes of Health (R01-HL131517 to DD; R01-HL136389 to NL and DD; R01-HL089598 to XHTW and DD; and R01-HL-091947, R01-HL117641, and R01-HL134824 to XHTW), the German Research Foundation (DFG, Do 769/4-1 to DD), the Canadian Institutes of Health Research (1484011) and Heart and Stroke Foundation of Canada (18-22032) to SN. UR is a member of the SFB1425 (German Research Foundation).
Nonstandard Abbreviations and Acronyms:
- AP
Action potential
- APD
AP duration
- ASC
Apoptosis-associated speck-like protein containing a CARD
- cAF
Long-standing persistent (‘chronic’) atrial fibrillation
- CaMKII
Ca2+/calmodulin-dependent protein kinase-II
- (Pro-)Casp1
(Pro-)Caspase-1
- CaT
Ca2+-transient
- cCaT
Caffeine-induced Ca2+-transient
- CRP
C-reactive protein
- Ctl
Control patients without post-operative atrial fibrillation
- DAD
Delayed afterdepolarization
- GSDMD
Gasdermin-D
- HAM
Human atrial cardiomyocyte
- ICa,L
L-type Ca2+-current
- IL
Interleukin
- INCX
Na+/Ca2+-exchange current
- LA
Left atrial
- NCX
Na+/Ca2+-exchanger
- NFκB
Nuclear-factor kappa-B
- NLRP
NACHT, LRR, and PYD domains-containing protein-3
- P2X7R
P2X7-receptor
- pAF
Paroxysmal atrial fibrillation
- PMCA
Plasmalemmal Ca2+-ATPase
- POAF
Post-operative atrial fibrillation
- RA
Right atrial
- RyR2
Ryanodine-receptor channel type-2
- SERCA2a
SR Ca2+-ATPase-2a
- SCaEs
Spontaneous SR Ca2+-release events
- SD
Standard deviation
- SR
Sarcoplasmic reticulum
- TLR4
Toll-like receptor-4
Footnotes
DISCLOSURES
XHTW is a co-founder and Scientific Advisory Board member of Elex Biotech, a drug-development company focused on novel compounds for cardiac arrhythmia disorders and heart failure. DD is a member of the scientific advisory boards of OMEICOS Therapeutics GmbH and Acesion Pharma. Other authors have no relevant conflicts of interest to disclose.
REFERENCES
- 1.Filardo G, Damiano RJ Jr., Ailawadi G, Thourani VH, Pollock BD, Sass DM, Phan TK, Nguyen H, da Graca B. Epidemiology of new-onset atrial fibrillation following coronary artery bypass graft surgery. Heart. 2018;104:985–992 [DOI] [PubMed] [Google Scholar]
- 2.Ahlsson A, Fengsrud E, Bodin L, Englund A. Postoperative atrial fibrillation in patients undergoing aortocoronary bypass surgery carries an eightfold risk of future atrial fibrillation and a doubled cardiovascular mortality. Eur J Cardiothorac Surg. 2010;37:1353–1359 [DOI] [PubMed] [Google Scholar]
- 3.Melduni RM, Schaff HV, Bailey KR, Cha SS, Ammash NM, Seward JB, Gersh BJ. Implications of new-onset atrial fibrillation after cardiac surgery on long-term prognosis: a community-based study. Am Heart J. 2015;170:659–668 [DOI] [PubMed] [Google Scholar]
- 4.Dobrev D, Aguilar M, Heijman J, Guichard JB, Nattel S. Postoperative atrial fibrillation: mechanisms, manifestations and management. Nat Rev Cardiol. 2019;16:417–436 [DOI] [PubMed] [Google Scholar]
- 5.Ha AC, Mazer CD, Verma S, Yanagawa B, Verma A. Management of postoperative atrial fibrillation after cardiac surgery. Curr Opin Cardiol. 2016;31:183–190 [DOI] [PubMed] [Google Scholar]
- 6.Swartz MF, Fink GW, Lutz CJ, Taffet SM, Berenfeld O, Vikstrom KL, Kasprowicz K, Bhatta L, Puskas F, Kalifa J, et al. Left versus right atrial difference in dominant frequency, K+ channel transcripts, and fibrosis in patients developing atrial fibrillation after cardiac surgery. Heart Rhythm. 2009;6:1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swartz MF, Fink GW, Sarwar MF, Hicks GL, Yu Y, Hu R, Lutz CJ, Taffet SM, Jalife J. Elevated pre-operative serum peptides for collagen I and III synthesis result in post-surgical atrial fibrillation. J Am Coll Cardiol. 2012;60:1799–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goette A, Juenemann G, Peters B, Klein HU, Roessner A, Huth C, Rocken C. Determinants and consequences of atrial fibrosis in patients undergoing open heart surgery. Cardiovasc Res. 2002;54:390–396 [DOI] [PubMed] [Google Scholar]
- 9.Workman AJ, Pau D, Redpath CJ, Marshall GE, Russell JA, Kane KA, Norrie J, Rankin AC. Post-operative atrial fibrillation is influenced by beta-blocker therapy but not by pre-operative atrial cellular electrophysiology. J Cardiovasc Electrophysiol. 2006;17:1230–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molina CE, Abu-Taha IH, Wang Q, Rosello-Diez E, Kamler M, Nattel S, Ravens U, Wehrens XHT, Hove-Madsen L, Heijman J, et al. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients With and Without Atrial Fibrillation. Front Physiol. 2018;9:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ucar HI, Tok M, Atalar E, Dogan OF, Oc M, Farsak B, Guvener M, Yilmaz M, Dogan R, Demircin M, et al. Predictive significance of plasma levels of interleukin-6 and high-sensitivity C-reactive protein in atrial fibrillation after coronary artery bypass surgery. Heart Surg Forum. 2007;10:E131–135 [DOI] [PubMed] [Google Scholar]
- 14.Maesen B, Nijs J, Maessen J, Allessie M, Schotten U. Post-operative atrial fibrillation: a maze of mechanisms. Europace. 2012;14:159–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaireviciute D, Blann AD, Balakrishnan B, Lane DA, Patel JV, Uzdavinys G, Norkunas G, Kalinauskas G, Sirvydis V, Aidietis A, et al. Characterisation and validity of inflammatory biomarkers in the prediction of post-operative atrial fibrillation in coronary artery disease patients. Thromb Haemost. 2010;104:122–127 [DOI] [PubMed] [Google Scholar]
- 16.Pretorius M, Donahue BS, Yu C, Greelish JP, Roden DM, Brown NJ. Plasminogen activator inhibitor-1 as a predictor of postoperative atrial fibrillation after cardiopulmonary bypass. Circulation. 2007;116:I1–7 [DOI] [PubMed] [Google Scholar]
- 17.Jacob KA, Nathoe HM, Dieleman JM, van Osch D, Kluin J, van Dijk D. Inflammation in new-onset atrial fibrillation after cardiac surgery: a systematic review. Eur J Clin Invest. 2014;44:402–428 [DOI] [PubMed] [Google Scholar]
- 18.Yao C, Veleva T, Scott L Jr., Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha I, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138:2227–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight. 2018;3:97054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018;138:2530–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loose S, Mueller J, Wettwer E, Knaut M, Ford J, Milnes J, Ravens U. Effects of IKur blocker MK-0448 on human right atrial action potentials from patients in sinus rhythm and in permanent atrial fibrillation. Front Pharmacol. 2014;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llach A, Molina CE, Prat-Vidal C, Fernandes J, Casado V, Ciruela F, Lluis C, Franco R, Cinca J, Hove-Madsen L. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur Heart J. 2011;32:721–729 [DOI] [PubMed] [Google Scholar]
- 23.Molina CE, Llach A, Herraiz-Martinez A, Tarifa C, Barriga M, Wiegerinck RF, Fernandes J, Cabello N, Vallmitjana A, Benitez R, et al. Prevention of adenosine A2A receptor activation diminishes beat-to-beat alternation in human atrial myocytes. Basic Res Cardiol. 2016;111:5. [DOI] [PubMed] [Google Scholar]
- 24.Voigt N, Pearman CM, Dobrev D, Dibb KM. Methods for isolating atrial cells from large mammals and humans. J Mol Cell Cardiol. 2015;86:187–198 [DOI] [PubMed] [Google Scholar]
- 25.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–680 [DOI] [PubMed] [Google Scholar]
- 26.Graf EM, Bock M, Heubach JF, Zahanich I, Boxberger S, Richter W, Schultz JH, Ravens U. Tissue distribution of a human CaV1.2 alpha1 subunit splice variant with a 75 bp insertion. Cell Calcium. 2005;38:11–21 [DOI] [PubMed] [Google Scholar]
- 27.Wehrens XH, Lehnart SE., Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840 [DOI] [PubMed] [Google Scholar]
- 28.Sutanto H, van Sloun B, Schonleitner P, van Zandvoort M, Antoons G, Heijman J. The Subcellular Distribution of Ryanodine Receptors and L-Type Ca2+ Channels Modulates Ca2+-Transient Properties and Spontaneous Ca2+-Release Events in Atrial Cardiomyocytes. Front Physiol. 2018;9:1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao CM, Xia Q, Bruce IC, Shen YL, Ye ZG, Lin GH, Chen JZ, Li GR. Influence of interleukin-2 on Ca2+ handling in rat ventricular myocytes. J Mol Cell Cardiol. 2003;35:1491–1503 [DOI] [PubMed] [Google Scholar]
- 30.Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-α and IL-1β increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47:378–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE, MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luke SG. Evaluating significance in linear mixed-effects models in R. Behav Res Methods. 2017;49:1494–1502 [DOI] [PubMed] [Google Scholar]
- 33.Feng C, Wang H, Lu N, Tu XM. Log transformation: application and interpretation in biomedical research. Stat Med. 2013;32:230–239 [DOI] [PubMed] [Google Scholar]
- 34.Franz MR, Jamal SM, Narayan SM. The role of action potential alternans in the initiation of atrial fibrillation in humans: a review and future directions. Europace. 2012;14 Suppl 5:v58–v64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaman JA, Harling L, Ashrafian H, Darzi A, Gooderham N, Athanasiou T, Peters NS. Post-operative atrial fibrillation is associated with a pre-existing structural and electrical substrate in human right atrial myocardium. Int J Cardiol. 2016;220:580–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600 [DOI] [PubMed] [Google Scholar]
- 37.Fender AC, Kleeschulte S, Stolte S, Leineweber K, Kamler M, Bode J, Li N, Dobrev D. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res Cardiol. 2020;115:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobrev D, Wettwer E, Kortner A, Knaut M, Schuler S, Ravens U. Human inward rectifier potassium channels in chronic and postoperative atrial fibrillation. Cardiovasc Res. 2002;54:397–404 [DOI] [PubMed] [Google Scholar]
- 39.Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM, Nerbonne JM. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–436 [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Xiong F, Qi XY, Xiao J, Villeneuve L, Abu-Taha I, Dobrev D, Huang C, Nattel S. Altered calcium handling produces reentry-promoting action potential alternans in atrial fibrillation-remodeled hearts. JCI Insight. 2020;5:e133754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522:259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishii Y, Schuessler RB, Gaynor SL, Yamada K, Fu AS, Boineau JP, Damiano RJ, Jr. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation. 2005;111:2881–2888 [DOI] [PubMed] [Google Scholar]
- 43.Ishii Y, Schuessler RB, Gaynor SL, Hames K, Damiano RJ Jr. Postoperative atrial fibrillation: The role of the inflammatory response. J Thorac Cardiovasc Surg. 2017;153:1357–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumagai K, Nakashima H, Saku K. The HMG-CoA reductase inhibitor atorvastatin prevents atrial fibrillation by inhibiting inflammation in a canine sterile pericarditis model. Cardiovasc Res. 2004;62:105–111 [DOI] [PubMed] [Google Scholar]
- 45.Goldstein RN, Ryu K, Khrestian C, van Wagoner DR, Waldo AL. Prednisone prevents inducible atrial flutter in the canine sterile pericarditis model. J Cardiovasc Electrophysiol. 2008;19:74–81 [DOI] [PubMed] [Google Scholar]
- 46.Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017;27:673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nattel S, Heijman J, Zhou L, Dobrev D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy. Circ Res. 2020;127:51–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoniades C, Demosthenous M, Reilly S, Margaritis M, Zhang MH, Antonopoulos A, Marinou K, Nahar K, Jayaram R, Tousoulis D, et al. Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. J Am Coll Cardiol. 2012;59:60–70 [DOI] [PubMed] [Google Scholar]
- 49.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, et al. Oxidized Ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation. 2013;128:1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chimenti C, Russo MA, Carpi A, Frustaci A. Histological substrate of human atrial fibrillation. Biomed Pharmacother. 2010;64:177–183 [DOI] [PubMed] [Google Scholar]
- 51.Li JY, Lai YJ, Yeh HI, Chen CL, Sun S, Wu SJ, Lin FY. Atrial gap junctions, NF-kappaB and fibrosis in patients undergoing coronary artery bypass surgery: the relationship with postoperative atrial fibrillation. Cardiology. 2009;112:81–88 [DOI] [PubMed] [Google Scholar]
- 52.Grammer JB, Bohm J, Dufour A, Benz M, Lange R, Bauernschmitt R. Atrial fibrosis in heart surgery patients Decreased collagen III/I ratio in postoperative atrial fibrillation. Basic Res Cardiol. 2005;100:288–294 [DOI] [PubMed] [Google Scholar]
- 53.Lezoualc’h F, Steplewski K, Sartiani L, Mugelli A, Fischmeister R, Bril A. Quantitative mRNA analysis of serotonin 5-HT4 receptor isoforms, calcium handling proteins and ion channels in human atrial fibrillation. Biochem Biophys Res Commun. 2007;357:218–224 [DOI] [PubMed] [Google Scholar]
- 54.Zhen-Han L, Rui S, Dan C, Xiao-Li Z, Qing-Chen W, Bo F. Perioperative statin administration with decreased risk of postoperative atrial fibrillation, but not acute kidney injury or myocardial infarction: A meta-analysis. Sci Rep. 2017;7:10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guenancia C, Sagnard A, Bouchot O, Lorgis L. Should we predict post-operative atrial fibrillation with atrial cardiomyopathy biomarkers? Int J Cardiol. 2020;307:71–72 [DOI] [PubMed] [Google Scholar]
- 56.Amar D, Zhang H, Miodownik S, Kadish AH. Competing autonomic mechanisms precede the onset of postoperative atrial fibrillation. J Am Coll Cardiol. 2003;42:1262–1268 [DOI] [PubMed] [Google Scholar]
- 57.Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. 2014;114:1500–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Limite LR, Magnoni M, Berteotti M, Peretto G, Durante A, Cristell N, Laricchia A, Camici PG, Alfieri O, Cianflone D. The predictive role of renal function and systemic inflammation on the onset of de novo atrial fibrillation after cardiac surgery. Eur J Prev Cardiol. 2016;23:206–213 [DOI] [PubMed] [Google Scholar]
- 59.Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, Chen B, Wu X, Ginsburg KS, Bakhos M, et al. Stress Signaling JNK2 Crosstalk With CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ Res. 2018;122:821–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A, Ravens U. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306 [DOI] [PubMed] [Google Scholar]
- 61.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363 [DOI] [PubMed] [Google Scholar]
- 62.Qi XY, Yeh Y-H, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJJM, Dobrev D, Nattel S. Cellular Signaling Underlying Atrial Tachycardia Remodeling of L-type Calcium Current. Circulation Research. 2008;103:845–854 [DOI] [PubMed] [Google Scholar]
- 63.Qi X-Y, Huang H, Ordog B, Luo X, Naud P, Sun Y, Wu C-T, Dawson K, Tadevosyan A, Chen Y, et al. Fibroblast Inward-Rectifier Potassium Current Upregulation in Profibrillatory Atrial Remodeling. Circulation Research. 2015;116:836–845 [DOI] [PubMed] [Google Scholar]
- 64.Sugishita K, Kinugawa K, Shimizu T, Harada K, Matsui H, Takahashi T, Serizawa T, Kohmoto O. Cellular basis for the acute inhibitory effects of IL-6 and TNF- alpha on excitation-contraction coupling. J Mol Cell Cardiol. 1999;31:1457–1467 [DOI] [PubMed] [Google Scholar]
- 65.Lee SH, Chen YC, Chen YJ, Chang SL, Tai CT, Wongcharoen W, Yeh HI, Lin CI, Chen SA. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 2007;80:1806–1815 [DOI] [PubMed] [Google Scholar]
- 66.Kao YH, Chen YC, Cheng CC, Lee TI, Chen YJ, Chen SA. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med. 2010;38:217–222 [DOI] [PubMed] [Google Scholar]
- 67.Hobai IA, Morse JC, Siwik DA, Colucci WS. Lipopolysaccharide and cytokines inhibit rat cardiomyocyte contractility in vitro. J Surg Res. 2015;193:888–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin-1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. 1997;81:493–503 [DOI] [PubMed] [Google Scholar]
- 69.Hafner-Bratkovic I, Susjan P, Lainscek D, Tapia-Abellan A, Cerovic K, Kadunc L, Angosto-Bazarra D, Pelegrin P, Jerala R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat Commun. 2018;9:5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.