

Abstract
A consensus virtual screening protocol has been applied to ca. 2000 approved drugs to seek inhibitors of the main protease (Mpro) of SARS-CoV-2, the virus responsible for COVID-19. 42 drugs emerged as top candidates, and after visual analyses of the predicted structures of their complexes with Mpro, 17 were chosen for evaluation in a kinetic assay for Mpro inhibition. Remarkably 14 of the compounds at 100-μM concentration were found to reduce the enzymatic activity and 5 provided IC50 values below 40 μM: manidipine (4.8 μM), boceprevir (5.4 μM), lercanidipine (16.2 μM), bedaquiline (18.7 μM), and efonidipine (38.5 μM). Structural analyses reveal a common cloverleaf pattern for the binding of the active compounds to the P1, P1′, and P2 pockets of Mpro. Further study of the most active compounds in the context of COVID-19 therapy is warranted, while all of the active compounds may provide a foundation for lead optimization to deliver valuable chemotherapeutics to combat the pandemic.
Keywords: SARS-CoV-2, virtual screening, drug repurposing, protease inhibitors
SARS-CoV-2, the cause of the COVID-19 pandemic,1 is a coronavirus (CoV) from the Coronaviridae family. Its RNA genome is ∼82% identical to that of SARS-CoV,2 which was responsible for the severe acute respiratory syndrome (SARS) pandemic in 2003.3 SARS-CoV-2 encodes two cysteine proteases: the chymotrypsin-like cysteine or main protease, known as 3CLpro or Mpro, and the papain-like cysteine protease, PLpro. They catalyze the proteolysis of polyproteins translated from the viral genome into nonstructural proteins essential for packaging the nascent virion and viral replication.4 Therefore, inhibiting the activity of these proteases would impede the replication of the virus. Mpro processes the polyprotein 1ab at multiple cleavage sites. It hydrolyzes the Gln-Ser peptide bond in the Leu-Gln-Ser-Ala-Gly recognition sequence. This cleavage site in the substrate is distinct from the peptide sequence recognized by other human cysteine proteases known to date.5 Thus, Mpro is viewed as a promising target for anti SARS-CoV-2 drug design; it has been the focus of several studies since the pandemic has emerged.2,4−7
An X-ray crystal structure of Mpro reveals that it forms a homodimer with a 2-fold crystallographic symmetry axis.2,5 Each protomer, with a length of 306 residues, is made of three domains (I–III). Domains II and I fold into a six-stranded β-barrel that harbors the active site.2,4,5 Domain III forms a cluster of five antiparallel α-helices that regulates the dimerization of the protease. A flexible loop connects domain II to domain III. The Mpro active site contains a Cys-His catalytic dyad and canonical binding pockets that are denoted P1, P1′, P2, P3, and P4.2 The amino acid sequence of the active site is highly conserved among coronaviruses.8 The catalytic dyad residues are His41 and Cys145, and residues involved in the binding of substrates include Phe140, His163, Met165, Glu166, and Gln189 (Figure 1). These residues have been found to interact with the ligands cocrystallized with Mpro in different studies.2,4,5 Crystallographic data also suggested that Ser1 of one protomer interacts with Phe140 and Glu166 of the other as the result of dimerization.2,4 These interactions stabilize the P1 binding pocket; thereby, dimerization of the main protease is likely for its catalytic activity.2,4
Figure 1.

Rendering of the residues near the catalytic site of MPro from a crystal structure at 1.31-Å resolution (PDB ID: 5R82). The catalytic residues are His41 and Cys145.
Drug repurposing is an important strategy for immediate response to the COVID-19 pandemic.9 In this approach, the main goal of computational and experimental studies has been to find existing drugs that might be effective against SARS-CoV-2. For instance, a molecular docking study suggested remdesivir as a potential therapeutic that could be used against SARS-CoV-2,10 which was supported experimentally by an EC50 value of 23 μM in an infected-cell assay.11 However, a clinical trial showed no statistically significant clinical benefits of remdesivir on adult patients hospitalized for severe COVID-19.12 Nonetheless, patients who were administered remdesivir in the same trial showed a faster time to clinical improvement in comparison to the placebo-control group.12 An EC50 value of 27 μM was also reported for lopinavir,11 suggesting it may have beneficial activity against SARS-CoV-2. However, neither lopinavir nor the lopinavir/ritonavir combination has thus far shown any significant benefits against COVID-19 in clinical trials. Chloroquine, hydroxychloroquine, and favipiravir have also been explored for repurposing against COVID-19; however, clinical studies with them have been controversial.13−16 These studies reflect the urgent need for systematic drug discovery efforts for therapies effective against SARS-CoV-2.
Thus, we decided to pursue discovery of small-molecule inhibitors of Mpro. The aim of this initial work was 2-fold: to identify known drugs that may be inhibitors, but also to identify structurally promising, synthetically accessible substructures suitable for subsequent lead optimization. Our expectation was that existing drugs may show activity but not at the low-nanomolar levels that are typical of effective therapies.17 This report provides results for the first goal. The work began by designing and executing a consensus molecular docking protocol to virtually screen ∼2000 approved drugs. The predicted structures (poses) of the complexes for the top-scoring 42 drugs received extensive scrutiny including consideration of intermolecular contacts, conformation, stability in molecular dynamics (MD) simulations, and potential for synthetic modification to arrive at 17 drugs, which were purchased and assayed for inhibition of Mpro. The outcome was strikingly successful with 14 of the 17 compounds showing some reduction of Mpro activity at 100 μM concentration, and with 5 compounds yielding IC50 values below 40 μM.
To begin, our analyses of more than 50 crystal structures of SARS-CoV-2 main protease in apo and holo forms showed small structural variations in the active site region. The overall root-mean-square deviation (RMSD) of all structures was ∼0.8 Å for Cα atoms. The presence of a ligand in the crystal structure likely places the side chains of the active site residues in positions that are more suitable for performing molecular docking compared to the apo form of the enzyme. Thus, we chose to use a high-resolution (1.31 Å) structure of Mpro cocrystallized with a noncovalent small fragment hit (PDB ID 5R82)18 for docking the approved drugs after removal of the fragment (Figure 1). The program Reduce19 was run on the structure for allowing side-chain flips, optimizing hydrogen bonds, and adding/removing hydrogen atoms. The pKa values of the ionizable residues of Mpro were predicted using the PROPKA320 and the H++ servers.21,22 Accordingly, lysines and arginines were positively charged, aspartic and glutamic acids were negatively charged, and all histidines were neutral. All histidines were built with the proton on Nε except for His80, which was protonated at Nδ. The resulting Mpro structure has a net charge of −4 e. Extensive visual inspection was carried out using UCSF Chimera.23
The next step was to pursue virtual screening by docking. Most docking programs apply methods to generate an initial set of conformations and tautomeric and protonation states for each ligand. This is followed by application of search algorithms and scoring functions to generate and score the poses of the ligand in the binding site of a protein. Scoring functions have been trained to reproduce a finite set of experimental ligand-binding affinities that are generally a mix of activity data converted to a free-energy scale. Therefore, the accuracy of the scores is dependent on multiple factors including the compounds that were part of the training set. To mitigate the biases, we performed four independent runs of protein–ligand docking with an in-house library of ca. 2000 approved, oral drugs using Glide SP, AutoDock Vina, and two protocols with AutoDock 4.2. The results were compiled, and further consideration focused on those compounds that ranked among the top 10% percent in at least 3 out of the 4 runs. The details of the docking protocols, correlations of docking scores, and the names and scores for all docked compounds are provided in the Supporting Information.
Molecular dynamics simulations were also performed starting from the Glide docking poses for the complexes of 14 high-interest compounds using the GROMACS software, version 2018a.24 The protonated Mpro dimer, with a net charge of −8 e, was represented by the OPLS-AA/M force field.25 TIP4P water was used as the solvent.26 Sodium counterions were added to neutralize the net charge of each system. The selected ligand candidates were represented by the OPLS/CM1A force field,27 as assigned by the BOSS software28 (version 4.9) and the LigParGen Python code.29 For neutral ligands, the CM1A partial atomic charges were scaled by a factor of 1.14.27 Each Mpro–ligand complex was placed at the center of a triclinic simulation box with 10-Å padding. For each complex, several ns of equilibration were followed by a 70 ns unrestrained MD run in the NPT ensemble at 310 K and 1 atm. Further details are provided in the Supporting Information.
On the experimental side, expression and purification of SARS-CoV-2 Mpro used a PGEX-6p-1 vector containing the gene for SARS-CoV-2 Mpro harboring a His6 tag followed by a modified PreScission cleavage site to produce recombinant protein, as previously described.5 Kinetic assays of SARS-CoV-2 Mpro activity were performed with compounds obtained from commercial sources except cinnoxicam, which had to be synthesized, and had purity >95% based on HPLC analysis. The kinetic measurements followed known procedures;5,7 100 nM Mpro in reaction buffer (20 mM Tris, 100 mM NaCl, 1 mM DTT, pH 7.3) was incubated with or without compound in DMSO at varying concentrations to a final DMSO concentration of 6% for 15 min with shaking at room temperature. The reaction was initiated by addition of substrate (Dabcyl-KTSAVLQ↓SGFRKM-E(Edans-NH2); GL Biochem) in reaction buffer, which is cleaved by Mpro, generating a product containing a free Edans group. Fluorescence was monitored at an excitation wavelength of 360 nm and emission wavelength of 460 nm. Intrinsic fluorescence of the FRET substrate in reaction buffer alone or in the presence of each compound was monitored simultaneously and subtracted from the kinetic measurements of Mpro-mediated substrate cleavage. Measurements from control wells subtracted from those of experimental wells, measuring cleavage of substrate by Mpro in the absence of any compound, contained 50 μM substrate with 6% DMSO in reaction buffer. For cleavage of substrate by Mpro in the presence of compound, the wells contained 50 μM substrate and compound at a concentration corresponding to a final DMSO concentration of 6% in reaction buffer. Baseline subtraction controlled for intrinsic fluorescence of each compound as well as intrinsic fluorescence of the uncleaved FRET substrate. All measurements were performed in triplicate and averaged.
The results from the virtual screening were the predicted poses for the complexes and docking scores (in kcal/mol) that ranged from −10.85 to −0.59 for Glide, from −12.33 to −2.30 for AutoDock run 1, from −10.74 to −0.40 for AutoDock run 2, and from −8.50 to −2.10 for AutoDock Vina. As expected, the range of scores is wide and it is different from one docking program to another. The complete list of compounds and scores is provided in the SI. Compounds were ranked based on their docking scores, and the top 200 hits from the four docking runs were compared. As the result, 42 compounds with a consensus count of 4 or 3 were selected. This means that these compounds were among the top-200 ranked compounds in all 4 or at least 3 out of the 4 docking runs. The indications and mechanisms of action for the 42 drugs are shown in Table 1, and the structures of some of the ones that turned out to be most interesting are shown in Figure 2. The primary indications include bacterial and viral infections, hypertension, psychosis, inflammation, and cancer. Their mechanisms of action are also broad ranging from kinase and protease inhibitors to dopamine receptors agonists/antagonists and calcium channel blockers. It is not surprising that peptidic protease inhibitors are well-represented in view of the peptide substrate and prior discovery of peptidic inhibitors for Mpro and its SARS-CoV relative.7,30,31
Table 1. Consensus Count (CC), Indication and Mechanism of Action of the Top 42 Drugs Selected from Virtual Screeninga.
| Compound | CC | Indication | Mechanism of Action |
|---|---|---|---|
| avatrombopag maleate | 3 | Thrombocytopenia | Thrombopoietin receptor agonist |
| azelastine | 4 | Allergic rhinitis | Histamine H1-receptors antagonist |
| azilsartan Medoxomil | 4 | Hypertension | Angiotensin II receptor antagonist |
| bedaquiline | 3 | Tuberculosis | ATP synthase inhibitor |
| benzquercin | 4 | Inflammation | Flavonoid drug |
| boceprevir | 3 | Hepatitis C | Protease inhibitor |
| bromocriptine | 4 | Hyperprolactinemic disorders | Dopamine D2 receptor agonist |
| cabergoline | 4 | Hyperprolactinemic disorders | Dopamine D2 receptor agonist |
| carindacillin | 4 | Bacterial infection | Penicillin-binding protein |
| cinnoxicam | 4 | Inflammation | Prostaglandin synthesis inhibitor |
| clofazimine | 4 | Lepromatous leprosy | Destabilizing bacterial membrane |
| dexetimide | 3 | Neuroleptic parkinsonism | Muscarinic antagonist |
| dihydroergocristine | 4 | Peripheral vascular disease | Serotonin receptors antagonist |
| dihydroergocryptine | 4 | Parkinson’s disease | Dopamine receptor agonist |
| efonidipine | 4 | Hypertension | Calcium channel blocker |
| elbasvir | 3 | Hepatitis C | Protein 5A inhibitor |
| idarubicin | 4 | Acute myeloid leukemia | Topoisomerase II inhibitor |
| indinavir | 3 | HIV infection | Protease inhibitor |
| ketoconazole | 3 | Fungal infection | 14-α-sterol demethylase inhibitor |
| lapatinib | 4 | Breast and lung cancer | Kinase inhibitor |
| lercanidipine | 4 | Hypertension | Calcium channel blocker |
| lomitapide | 3 | Hypercholesterolemia | Triglyceride transfer inhibitor |
| lurasidone | 4 | Schizophrenia | Dopamine D2 receptor antagonist |
| macimorelin | 3 | Adult growth hormone deficiency | Ghrelin receptor agonist |
| manidipine | 3 | Hypertension | Calcium channel blocker |
| metergoline | 4 | Psychosis | Dopamine agonist |
| methoserpidine | 3 | Hypertension | Monoamine transport inhibitor |
| naldemedine | 3 | Opioid induced constipation | Opioid receptor antagonist |
| nelfinavir | 3 | HIV infection | Protease inhibitor |
| nicomol | 3 | Hyperlipidemia | - |
| nicomorphine | 4 | Analgesic | Opioid agonist |
| nilotinib | 4 | Chronic myeloid leukemia | Kinase inhibitor |
| perampanel | 4 | Partial-onset seizures | Glutamate receptor antagonist |
| periciazine | 3 | Psychosis | Dopamine D1 receptor antagonist |
| pipamazine | 3 | Psychosis | Dopamine receptor antagonist |
| saquinavir | 4 | HIV infection | Protease inhibitor |
| simvastatin | 3 | Hyperlipidemia | HMG-CoA reductase inhibitor |
| talampicillin | 3 | Antibacterial | Cell-wall synthesis inhibitor |
| telaprevir | 3 | Hepatitis C | Protease inhibitor |
| tipranavir | 3 | HIV infection | Protease inhibitor |
| tropesin | 3 | Inflammation | Prostaglandin synthesis inhibitor |
| zafirlukast | 4 | Asthma | Leukotriene receptor antagonist |
Assayed compounds are in bold.
Figure 2.
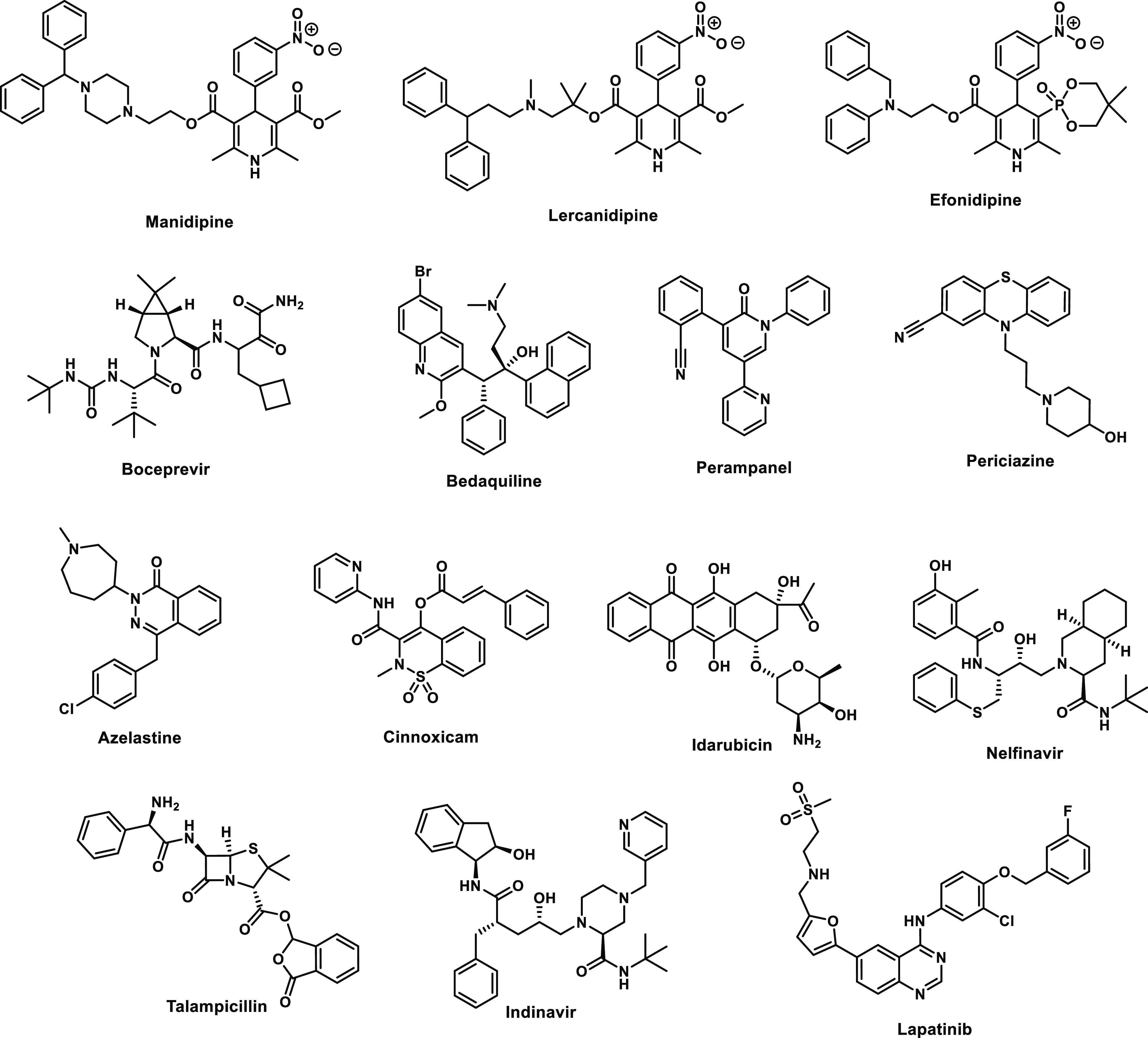
Selected high-scoring compounds from the consensus docking.
In almost all cases the predicted poses for the 42 compounds from the different docking programs agreed well. The poses from Glide were then subjected to extensive visual scrutiny to check for unsatisfied hydrogen-bonding sites, electrostatic mismatches, and unlikely conformation of the ligand. About half of the compounds were ruled out for further study due to the occurrence of such liabilities and the presence of multiple ester groups (e.g., methoserpidine and nicomol) or overall size and complexity (e.g., bromocriptine and benzquercin). The MD simulations were carried out for 14 compounds to check the stability of the docked structures; this contributed to ruling out metergoline and dihydroergocristine that showed above normal drift without return, and it raised suspicion about talampicillin.
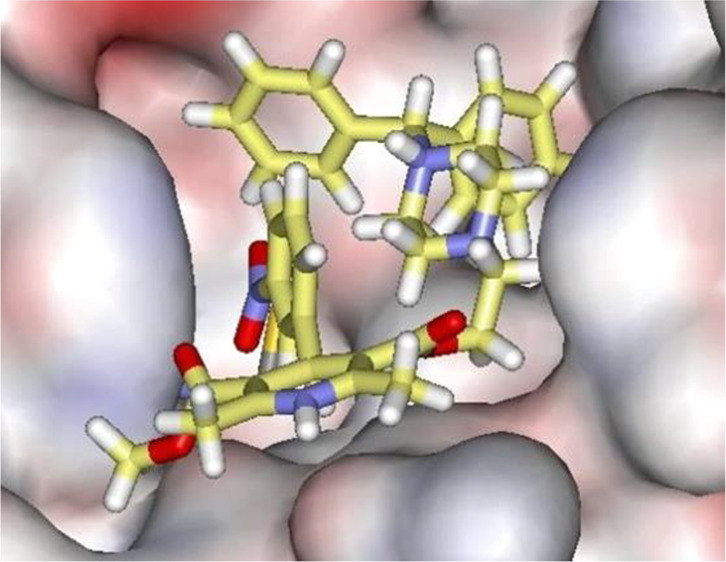
A repeated motif was apparent with high-scoring ligands having a cloverleaf pattern with occupancy of the P1, P1′, and P2 pockets, as illustrated, for example, in Figure 3 for the complex of azelastine. Other common elements are an edge-to-face aryl–aryl interaction with His41 and placement of a positively charged group in the P1 pocket in proximity to Glu166, e.g., the methylazepanium group of azelastine, the protonated trialkylamino group of bedaquiline, and the protonated piperazine of periciazine. However, Glu166 forms a salt-bridge with the terminal ammonium group Ser1B (Figure 1). The electrostatic balance seems unclear in this region, so our final selections included a mix of neutral and positively charged groups for the P1 site. The analysis of the high-scoring 42 compounds also considered structural variety and potential synthesis of analogs. In the end, we settled on 17 compounds, which are highlighted in Table 1, for purchase and assaying. Sixteen were commercially available, mostly from Sigma-Aldrich. The 17th, cinnoxicam, was not available, but it was prepared in a one-step synthesis from the commercially available ester components. It may be noted that three calcium channel blockers, efonidipine, lercanidipine, and manidipine, were purchased (Figure 2). This was not done owing to the characteristic dihydropyridine substructure, since this end of the molecule protrudes out of the P1′ site in the docked poses. It was for the variety in the left-sides of the molecules in Figure 2, which form the cloverleaf that binds in the P1, P1′, and P2 pockets, as illustrated in Figure 4 for manidipine. The steric fit in this region appears good with the two phenyl rings in the P1 and P2 sites, though the only potential hydrogen bond is between the nitro group and the catalytic Cys145.
Figure 3.

Glide docking pose for azelastine in surface (left; coloring by element) and stick (right) renderings. All illustrations are oriented with the P1 pocket to the left and P2 to the right, and all carbon atoms of ligands are in yellow.
Figure 4.

Glide docking pose for manidipine in surface (left) and stick (right) renderings.
The 17 known drugs were then evaluated using the FRET-based assay monitoring the fluorescence generated from the cleavage of a peptide substrate harboring an Edans–Dabcyl pair by recombinant SARS-CoV-2 Mpro. Remarkably, 14 of the drugs at 100 μM decreased Mpro activity (100 nM), as shown in Figure 5 and Table 2. Five drugs decreased Mpro activity to below 40%: manidipine, boceprevir, efonidipine, lercanidipine, and bedaquiline. Dose–response curves were obtained to determine IC50 values, when possible, as shown in Figure 6 for the five most potent inhibitors, with the raw data as a function of time and concentration given in Figure S3.
Figure 5.
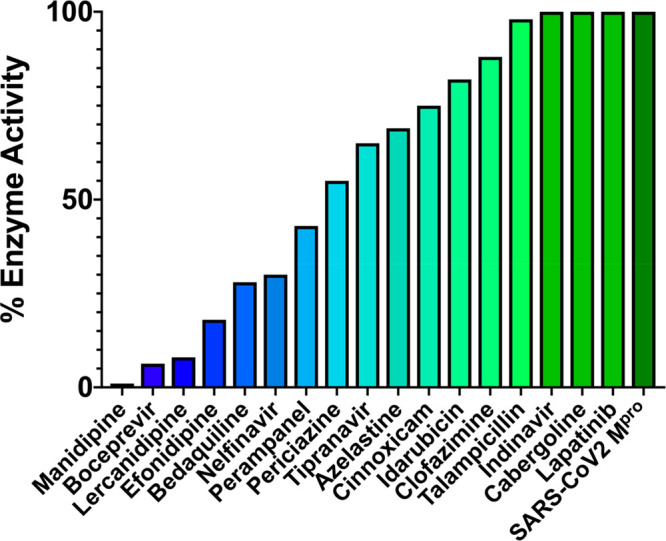
Ranking of the 17 compounds by percent residual enzyme activity monitored by cleavage product fluorescence following a 1 h incubation of 100 nM Mpro with 100 μM compound. Compounds are ranked from most (blue) to least (green) active.
Table 2. Measured Activities of the 17 Compounds Tested for Inhibition of Mpro.
| Compound | % Activity at 100 μM | IC50(μM) |
|---|---|---|
| manidipine | 1 | 4.81 ± 1.87 |
| boceprevir | 6 | 5.40 ± 1.53 |
| lercanidipine | 8 | 16.2 ± 2.94 |
| efonidipine | 18 | 38.5 ± 0.41 |
| bedaquiline | 28 | 18.7 ± 4.20 |
| perampanel | 43 | 100–250a,b |
| periciazine | 55 | 250a |
| nelfinavir | 64 | 250–600a |
| tipranavir | 65 | >600a |
| azelastine | 69 | 20–100a |
| cinnoxicam | 75 | >600a |
| idarubicin | 82 | 250–600a |
| clofamizine | 88 | >600a |
| talampicillin | 90 | 250–600a |
| indinavir | 100 | NA |
| cabergoline | 100 | NA |
| lapatinib | 100 | NA |
Estimate due to incomplete inhibition at 600 μM.
Fluorescence of compound interfered with assay.
Figure 6.

IC50 plots and values for the top five compounds active against SARS-CoV-2 Mpro from in vitro FRET-based assay. IC50 plots were generated from averaged kinetic data in triplicate for (A) manidipine, (B) boceprevir, (C) lercanidipine, (D) efonidipine, and (E) bedaquiline.
The calcium channel-blockers manidipine, lercanidipine, and efonidipine inhibit Mpro activity with IC50 values of 4.8 μM, 16.2 μM, and 38.5 μM, respectively. As suggested from Figure 4, the variation likely arises primarily from differences in binding of the left sides of the molecules (Figure 2) in the P1, P1′, and P2 pockets. It has previously been proposed that such compounds might be useful for treatment of SARS-CoV-2 infection for their role as calcium channel blockers, not as Mpro inhibitors.32 Boceprevir, a hepatitis C virus protease inhibitor, inhibits Mpro with an IC50 of 5.4 μM; its IC50 has been previously reported as 4.13 μM.7 Bedaquiline, approved for the treatment of multidrug-resistant tuberculosis, inhibits Mpro with an IC50 of 18.7 μM. The IC50 of nelfinavir, an HIV protease inhibitor, was estimated to be between 250 and 600 μM. Vatansever et al. have reported an IC50 for nelfinavir of 234 μM.33 The other 12 compounds have not been previously reported as Mpro inhibitors to our knowledge. Perampanel appears to be the sixth most active compound at 100 μM, though its IC50 could not be calculated reliably, as its intrinsic fluorescence interfered with the fluorescence measurements.
The computed structures for the complexes of bedaquiline and boceprevir are illustrated in Figure 7. For bedaquiline, the three pockets are occupied by the ammonium containing side chain, the phenyl group, and the naphthyl group, respectively, while the quinoline fragment extends toward the solvent, and there are no clear protein–ligand hydrogen bonds. The activity of this compound does suggest that positively charged groups may be acceptable in the P1 site. For boceprevir, the dimethylcyclopropyl subunit is predicted to sit in P1, the side chain with the cyclobutyl and terminal ketoamide groups is in P1′, the proximal tert-butyl group is in P2, the distal tert-butyl group is in the hydrophobic pocket at P4/P5, and there are hydrogen bonds with the NH of Gly143, the carbonyl oxygen of Thr26, and three for the urea group with the NH group of Glu166 and the side chain carbonyl of Gln166. All computed poses are for noncovalent docking; few known drugs have sufficiently electrophilic sites to serve as warheads for covalent docking, so this was not pursued. However, there is a crystal structure that has been deposited in the Protein Databank (ID 6WNP) for a boceprevir–Mpro complex covalently linked to Cys145 at the keto group adjacent to the terminal amide group.34 This anchoring causes the cyclobutylmethyl and dimethylcyclopropyl groups to sit in P1 and P2, respectively, and the urea-containing appendage with the two tert-butyl groups to lie in the P3–P5 region. The docked noncovalent structure is compelling, and it was the 10th best ranked compound with a Glide SP score of −9.5. The orientational difference with the crystal structure for the covalent complex is interesting and may represent a noncovalent binding mode on the pathway to the covalent attachment.
Figure 7.

Renderings of Glide docking poses for bedaquiline (left) and boceprevir (right).
In summary, the present virtual screening study was highly successful in identifying 14 known drugs as showing inhibitory effect on the main protease of SARS-CoV-2. The consensus scoring approach using three docking programs and four protocols was effective in narrowing down ca. 2000 candidate drugs to 42 of high interest. The final 17 compounds that were selected for assay did reflect additional human visualization and analyses. Five compounds were identified with IC50 values below 40 μM with manidipine, boceprevir, lercanidipine, and bedaquiline below 20 μM at 4.8, 5.4, 16.2, and 18.7 μM, respectively. Such potencies are very successful for a virtual screening exercise but likely insufficient for repurposing. However, all of the active compounds reported here may provide a foundation for lead optimization to deliver valuable chemotherapeutics to combat the COVID-19 pandemic.
Acknowledgments
Gratitude is expressed for support to the U.S. National Institutes of Health (GM32136) and to the Yale University School of Medicine for a CoReCT Pilot Grant. The Mpro plasmid was kindly provided by the Hilgenfeld lab.5
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00521.
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; Yuan M.-L.; Zhang Y.-L.; Dai F.-H.; Liu Y.; Wang Q.-M.; Zheng J.-J.; Xu L.; Holmes E. C.; Zhang Y.-Z. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Drosten C.; Günther S.; Preiser W.; van der Werf S.; Brodt H.-R.; Becker S.; Rabenau H.; Panning M.; Kolesnikova L.; Fouchier R. A. M.; Berger A.; Burguière A.-M.; Cinatl J.; Eickmann M.; Escriou N.; Grywna K.; Kramme S.; Manuguerra J.-C.; Müller S.; Rickerts V.; Stürmer M.; Vieth S.; Klenk H.-D.; Osterhaus A. D. M. E.; Schmitz H.; Doerr H. W. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Jin Z.; Zhao Y.; Sun Y.; Zhang B.; Wang H.; Wu Y.; Zhu Y.; Zhu C.; Hu T.; Du X.; Duan Y.; Yu J.; Yang X.; Yang X.; Yang K.; Liu X.; Guddat L. W.; Xiao G.; Zhang L.; Yang H.; Rao Z. Structural Basis for the Inhibition of SARS-CoV-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno A.; Mestres-Truyol J.; Ojeda-Montes M. J.; Macip G.; Saldivar-Espinoza B.; Cereto-Massagué A.; Pujadas G.; Garcia-Vallvé S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. 10.3390/ijms21113793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse J. S.; Lalonde T.; Xu S.; Liu W. R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushpakom S.; Iorio F.; Eyers P. A.; Escott K. J.; Hopper S.; Wells A.; Doig A.; Guilliams T.; Latimer J.; McNamee C.; Norris A.; Sanseau P.; Cavalla D.; Pirmohamed M. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discovery 2019, 18, 41–58. 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- Elfiky A.; Anti-HCV A. Nucleotide Inhibitors, Repurposing against COVID-19. Life Sci. 2020, 248, 117477. 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy K.-T.; Wong A. Y.-L.; Kaewpreedee P.; Sia S. F.; Chen D.; Hui K. P. Y.; Chu D. K. W.; Chan M. C. W.; Cheung P. P.-H.; Huang X.; Peiris M.; Yen H.-L. Remdesivir, Lopinavir, Emetine, and Homoharringtonine Inhibit SARS-CoV-2 Replication in Vitro. Antiviral Res. 2020, 178, 104786. 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang D.; Du G.; Du R.; Zhao J.; Jin Y.; Fu S.; Gao L.; Cheng Z.; Lu Q.; Hu Y.; Luo G.; Wang K.; Lu Y.; Li H.; Wang S.; Ruan S.; Yang C.; Mei C.; Wang Y.; Ding D.; Wu F.; Tang X.; Ye X.; Ye Y.; Liu B.; Yang J.; Yin W.; Wang A.; Fan G.; Zhou F.; Liu Z.; Gu X.; Xu J.; Shang L.; Zhang Y.; Cao L.; Guo T.; Wan Y.; Qin H.; Jiang Y.; Jaki T.; Hayden F. G.; Horby P. W.; Cao B.; Wang C. Remdesivir in Adults with Severe COVID-19: A Randomised, Double-Blind, Placebo-Controlled, Multicentre Trial. Lancet 2020, 395, 1569–1578. 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M.; De Giglio M. A. R.; Roviello G. N. SARS CoV-2: Recent Reports on Antiviral Therapies Based on Lopinavir/Ritonavir, Darunavir/Umifenovir, Hydroxychloroquine, Remdesivir, Favipiravir and Other Drugs for the Treatment of the New Coronavirus. Curr. Med. Chem. 2020, 27, 4536–4541. 10.2174/0929867327666200416131117. [DOI] [PubMed] [Google Scholar]
- Singh A. K. Chloroquine and Hydroxychloroquine in the Treatment of COVID-19 with or without Diabetes: A Systematic Search and a Narrative Review with a Special Reference to India and Other Developing Countries. Diabetes Metab. Syndr. 2020, 14, 241–246. 10.1016/j.dsx.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Ye F.; Zhang M.; Cui C.; Huang B.; Niu P.; Liu X.; Zhao L.; Dong E.; Song C.; Zhan S.; Lu R.; Li H.; Tan W.; Liu D. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. 10.1093/cid/ciaa237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geleris J.; Sun Y.; Platt J.; Zucker J.; Baldwin M.; Hripcsak G.; Labella A.; Manson D. K.; Kubin C.; Barr R. G.; Sobieszczyk M. E.; Schluger N. W. Observational Study of Hydroxychloroquine in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2020, 382, 2411–2418. 10.1056/NEJMoa2012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann M. M. Molecular Obesity, Potency, and Other Addictions in Drug Discovery. MedChemComm 2011, 2, 349–355. 10.1039/C1MD00017A. [DOI] [Google Scholar]
- Fearon D.; Powell A. J.; Douangamath A.; Owen C. D.; Wild C.; Krojer T., Lukacik P.; Strain-Damerell C. M.; Walsh M. A.; von Delft F.. PanDDA analysis of COVID-19 main protease against the DSI-poised Fragment Library. PDB ID: 5R82.
- Word J. M.; Lovell S. C.; Richardson J. S.; Richardson D. C. Asparagine and Glutamine: Using Hydrogen Atom Contacts in the Choice of Side-Chain Amide Orientation. J. Mol. Biol. 1999, 285, 1735–1747. 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p Ka Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. H++: A Server for Estimating PKas and Adding Missing Hydrogens to Macromolecules. Nucleic Acids Res. 2005, 33, 368–371. 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandakrishnan R.; Aguilar B.; Onufriev A. V. H++ 3.0: Automating PK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012, 40, 537–541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera: A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M. J.; Tirado-Rives J.; Jorgensen W. L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. 10.1021/acs.jctc.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. Potential Energy Functions for Atomic-Level Simulations of Water and Organic and Biomolecular Systems. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 6665–6670. 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. Molecular Modeling of Organic and Biomolecular Systems Using BOSS and MCPRO. J. Comput. Chem. 2005, 26, 1689–1700. 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- Dodda L. S.; Cabeza de Vaca I.; Tirado-Rives J.; Jorgensen W. L. LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands. Nucleic Acids Res. 2017, 45, 331–336. 10.1093/nar/gkx312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; et al. Structure-based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T.; Manickam M.; Namasivayam V.; Hayashi Y.; Jung S.-H. An Overview of Severe Acute Respiratory Syndrome – Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danta C. C. Calcium Channel Blockers: A Possible Potential Therapeutic Strategy for the Treatment of Alzheimer’s Dementia Patients with SARS-CoV-2 Infection. ACS Chem. Neurosci. 2020, 11, 2145–2148. 10.1021/acschemneuro.0c00391. [DOI] [PubMed] [Google Scholar]
- Vatansever E. C.; Yang K.; Kratch K. C.; Drelich A.; Cho C. C.;, Mellot D. M.; Xu S.; Tseng C. K.; Liu W. R.. Targeting the SARS-CoV-2 Main Protease to Repurpose Drugs for COVID-19. bioRxiv 2020, May 23:2020.05.23.112235. 10.1101/2020.05.23.112235. [DOI] [Google Scholar]
- Anson B.; Mesecar A.. X-ray Structure of SARS-CoV-2 Main Protease Bound to Boceprivir at 1.45 Å. PDB ID: 6WNP. 10.2210/pdb6wnp/pdb. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.