

Abstract
Chemotherapy is central to oncology, perceived to operate only on prolific cancerous tissue. Yet many non-neoplastic tissues are more prolific than typical tumors. Chemotherapies achieve sufficient therapeutic windows to exert anti-neoplastic activity because they are pro-drugs that are bioactivated in cancer-specific environments. The advent of precision medicine has obscured this concept, favoring the development of high-potency kinase inhibitors. Inhibitors of essential mitotic kinases exemplify this paradigm shift, but intolerable on-target toxicities in more prolific normal tissues have led to repeated failures in the clinic. Proliferation rates alone cannot be used to achieve cancer specificity. Here, we discuss integrating the cancer-specificity of pro-drugs from classical chemotherapeutics and the potency of mitotic kinase inhibitors to generate a class of high-precision cancer therapeutics.
Keywords: cell cycle, kinesin, cyclin-dependent kinases, chemotherapy, targeted therapy, pro-drug, precision oncology
Genotoxic chemotherapies have high impact in the clinic, but research efforts now focus on molecular targeted therapies.
Chemotherapies inducing DNA damage remain the mainstay of cancer treatment. Such damage leads to double-stranded breaks, which triggers apoptosis during cell division [1–3]. It sometimes not fully appreciated how clinically impactful and transformative chemotherapeutic agents truly are, especially when used in combination. For example, the combination of cyclophosphamide with rituximab, vincristine, doxorubicin, and prednisone (R-CHOP) yields long-term remissions and even complete cures in certain cancers [4–8]. For glioblastoma (GBM), before the advent of temozolomide (TMZ) long-term survivorship was unheard off; now it is possible, even if only for a minority of patients [9].
A common misconception about chemotherapies is that they selectively kill cancer cells simply because they replicate more quickly than normal tissue [10–12]. Such pervasive misconceptions are perpetuated by popular, so-called “expert” [13,14] sources and fail to account for the many tissues in the human body that exhibit cell cycling rates higher than those found in tumors [15–20]. For instance, prolific cells, such as those in the bone marrow, can exhibit doubling times in as little as 17 hours [21], yet even the fastest growing tumors only double every 3 weeks [22]. Juxtaposing these rates evidences and dooms therapies that operate purely on the premise of fast proliferation as a point for differential sensitivity [23–31] (Table 1). In reality, most chemotherapeutics achieve cancer-selectivity because they are administered as pro-drugs that are preferentially biotransformed in cancer cells rather than prolific, non-neoplastic tissues such as hemopoietic tissues, gastrointestinal tract, mucus membranes, skin and hair follicles [32–37] (Table 2).
Table 1.
High potency mitotic kinase inhibitors stalled in late-stage clinical trials due to significant adverse events.
| Drug | Structure | Target (Ki) | Farthest clinical stage | Dose limiting toxicity | Best clinical response rate | Reference |
|---|---|---|---|---|---|---|
| Alisertib | 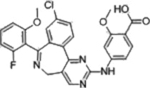 |
AURKA (1.2 nM) | Phase III (2012–2015) NCT01482962 |
Febrile neutropenia | 17% in aggressive B-cell Lymphoma | [23] |
| Ispinesib |  |
Eg5/KIF11 (1.7 nM) | Phase II (2004–2006) NCT00089973 |
Febrile neutropenia, vomiting | 0% in Malignant melanoma | [24–26] |
| Dinaciclib | 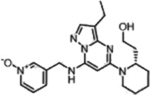 |
CDK2 (1 nM) CDK5 (1 nM) CDK1 (3 nM) CDK9 (4 nM) |
Phase I (2016–2018) NCT02684617 NCT03484520 |
Neutropenia, Pneumocytis | 11% PR in multiple myeloma | [27,28] |
| GSK461364 | 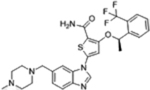 |
PLK1 (2.2 nM) | Phase I (2007–2009) NCT00536835 |
Neutropenia, thrombocytopenia, thrombic eboli, myelosuppression | 0% in solid tumors | [29–30] |
| BAY1217389 | 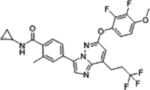 |
TTK (0.63 nM) | Phase I (2015–2019) NCT02366949 |
Hematologic toxicity, neutropenia, | N/A | [31] |
An overview of structures, corresponding target affinities, and farthest progression in clinical trials for several potent mitotic kinase inhibitors is shown. Even with single-digit nanomolar Kis, major dose-limiting toxicities are still observed, stalling these trials. According to RECIST criteria, none of these included drugs achieved a complete response (CR) at toxicity-limiting doses. The best clinical response rates (objective, PR) are less than 20%.
Table 2.
Many clinically impactful chemotherapeutic agents are pro-drugs and rely on cancer-specific biotransformation for specific killing.
| Representative example (class) | Structure of Administered Drug (pro-drug) | Active Agent (bio-transformed) | Mode of action | Mechanism of selectivity | Reference |
|---|---|---|---|---|---|
| Cyclophosphamide (Nitrogen mustards) | 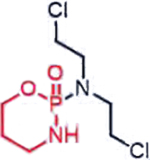 |
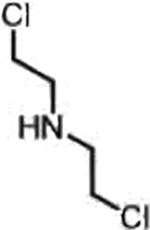 |
DNA damage through crosslinking | Loss of ALDH1A1 in de-differentiated cancer cells, basic tumor intracellular | [32] |
| Temozolomide (Triazenes) | 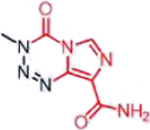 |
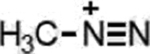 |
DNA damage through methylation | MGMT silencing, cytosolic alkaline pH in GBM | [33] |
| 5-fluorouracil (Nucleotide analogue) | 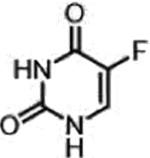 |
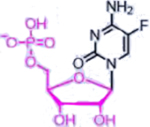 |
DNA, RNA damage, Inhibition of TS | Overexpression of enzymes of thymidine phosphate synthesis | [34] |
| Irinotecan (Topoisomerase Inhibitors) | 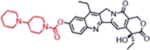 |
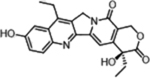 |
Topoisomerase inhibition leads to DNA breakage | Butyrylcholine Esterase activity, overexpressed in cancer cells | [35] |
| Cisplatin (Platinum agents) | 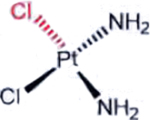 |
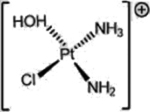 |
DNA damage: crosslinking | Aquation due to more alkaline intracellular and lower intracellular chloride | [36] |
| Methotrexate (Anti-folates) | 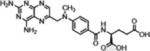 |
 |
Anti-folate: inhibition of nucleotide synthesis | Increased import via overexpressed folate receptor and trapping by polyglutamylation in cancer | [37] |
A representative panel of clinically useful conventional chemotherapies is shown. Combined treatment with one or more agents shown is responsible for curing a substantial number of cancer patients. Pro-drug moieties which undergo biological removal are indicated in red; those which undergo biological addition are indicated in magenta. Mechanisms outlined for each example are applicable for the broader class that it represents; for example, all nucleotide analogues require biotransformation to their monophosphate forms for bioactivity.
The development of mitotic kinase inhibitors came as an outgrowth of studies on the molecular mechanisms of the cell cycle in the 1990s [38–40], which promised the development of precise inhibitors of mitosis without the toxic sequelae of classical chemotherapeutics. Yet for all the initial excitement, mitotic kinase inhibitors have now been relegated to the drug development graveyard as they suffered from a lack of specificity between cancer cells and prolific, non-malignant cells. Multiple clinical trials have shown that they lack a therapeutic window and are dose-limited by toxicities to tissues such as the bone marrow and gut (Table 1). But given their high affinities and potencies, we contend that mitotic kinase inhibitors may find clinical utility if refashioned with cancer-specific pro-drug moieties. A detailed survey of the literature indicates that most classic chemotherapy drugs owe their therapeutic windows to specific pro-drug activation and de-activation pathways exclusive to cancer cells. Here, we will describe the utility of mitotic kinase inhibitors, review the bioactivation mechanisms of various chemotherapeutics, then synthesize these concepts to discuss the application of cancer-specific pro-drugs onto mitotic kinase inhibitors.
Mitotic kinases inhibitors exhibit highly selective toxicity to proliferating cells
The late 1980s and early 1990s saw pioneering studies of the eukaryotic cell cycle machinery in yeast [40–42]. From these initial studies, mitotic kinase inhibitors emerged as the next transformative hope in cancer medicine (Figure 1, Supplementary Tables S1, S2) [43–47], as they would target proteins involved in the cell cycle of proliferating, but not quiescent cells [48]. The majority of human tissues would thus be spared from potentially toxic side effects. Examples of such inhibitors and their corresponding targets include Dinaciclib [49] (pan-CDK), Ispinesib [24] (KIF11/Eg5), Alisertib [23] (AURAK) and Tozasertib [50] (pan-aurora kinase), and BI-2536 [51] (PLK1). A more exhaustive list can be found in Supplementary Tables S1, S2.
Figure 1. Essential Enzymes of the mitotic expression module are ideal targets for highly specific mitotic inhibitors.
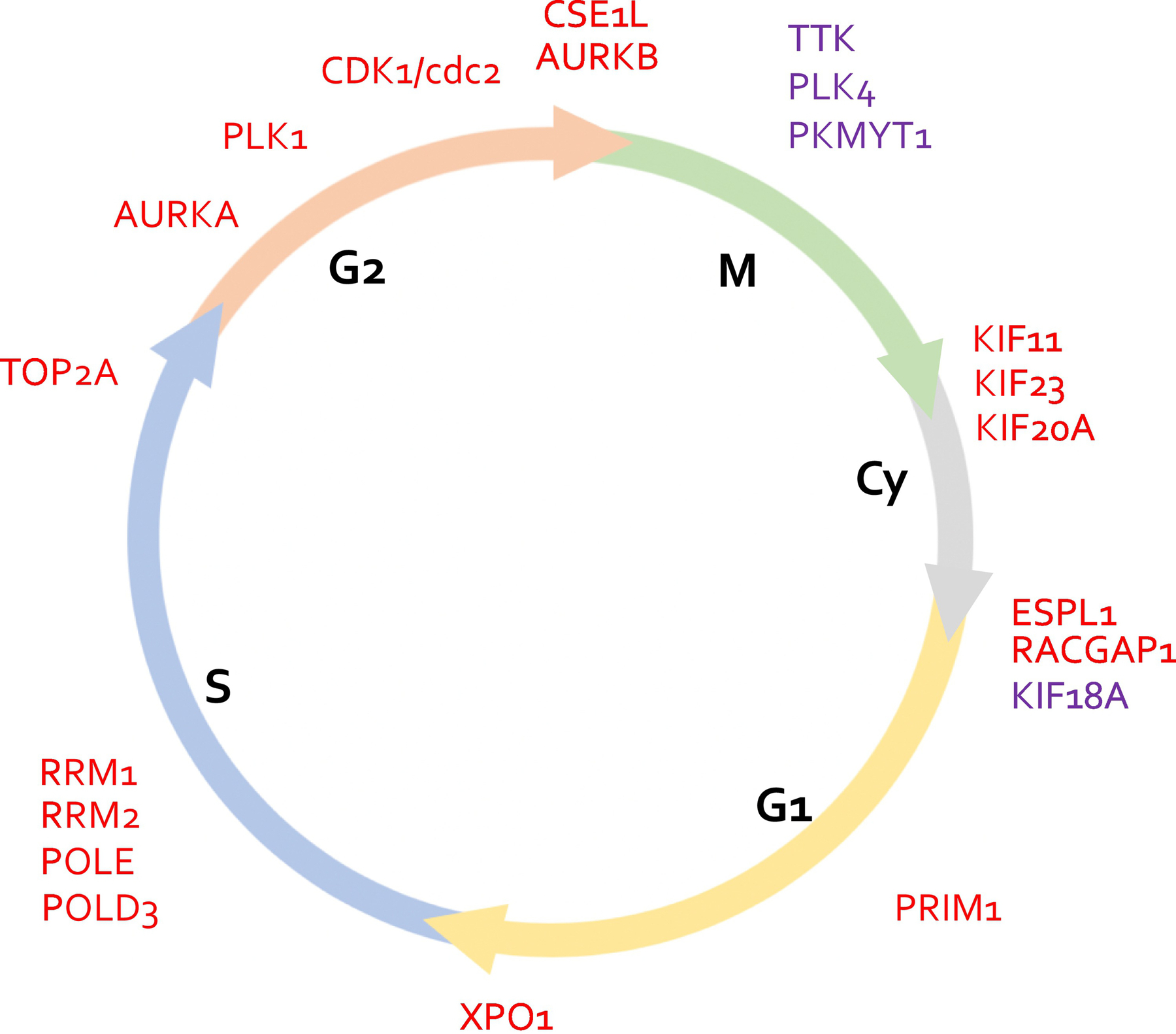
Systematic interrogation of the genes associated with the mitotic expression module [48] via CRISPR and RNAi drop-out screening via DepMap reveals essential cell cycle genes. Only proteins which were determined to be druggable are included. Proteins found to be common essential in 100% of the cancer cell lines included in DepMap are indicated in red while those found to be common essential in >95% of cancer cell lines are indicated in purple. An extended list of cell cycle-specific genes with essentiality scores can be found in Supplementary Tables I and II. Note that these lists include the accepted HUGO gene names used at the time of publication.
Mitotic kinase inhibitors provide three key advantages over their chemotherapy predecessors. First, by only targeting cycling cells, they minimize toxicity to post-mitotic tissues such as the heart and liver. Second, they forgo DNA damage thus nullifying the risk of inducing treatment-related secondary cancers [52–54]. Finally, the resistance mechanisms derived from repeated DNA damage [55,56] cease to be an issue, making them a viable option even for tumors that have acquired complete resistance to cytotoxic chemotherapies. Though other mechanisms of resistance, such as active site mutations, may arise [57–60], they would be distinct from DNA-damage bypass-inviting synergistic combinations with cytotoxic chemotherapies. The pharmaceutical industry has already proven its consistent ability to counter binding site mutations by modifying the peripheral structures of a particular kinase inhibitor [61,62], overriding these concerns on resistance.
CDK1 is an excellent example of a target that encompasses the many advantages described above, as it is the only CDK uniformly essential for the execution of the cell cycle [63]. In mice, Cdk1-knockouts die at the early embryo stage. In contrast, Cdk2/4/5/6-knocouts are viable with growth stunted phenotypes [63]. Cdk7/8/9-knockout mice, while embryonic lethal and thus essential, are not exclusively mitotic kinases and their expression is not restricted to cell cycling [48]. CDK1 is an ideal cancer drug target due to its dispensability in post-mitotic tissues [64]. CRISPR and RNAi data via DepMap corroborates early molecular biology on CDK1-supporting its essentiality in all cancer cell lines profiled to-date. Other CDKs-such as CDK4/6-are only essential in some cell lines (Supplementary Tables S1, S2), underscoring their promotive but non-essential role in the cell cycle [63]. Unlike non-essential CDKs, CDK1 expression is strongly correlated with survival in many cancers in a manner similar to that of Ki67 and PCNA (Supplementary Tables S1, S2) [65–68]. Such properties coincide with the exclusivity of CDK1 expression in dividing cells, making it impossible for a highly specific CDK1 inhibitor to exhibit (on-target) side-effects to non-proliferative tissue. Following the rigorous candidate validation modeled for CDK1, we contend that other cell cycle-essential proteins would have similar proliferative-cell dependencies as CDK1 (Figure 1, Supplementary Tables S1, S2), though we note that the extensive bioinformatics data has yet to be corroborated with the full complement of biological validation, including mouse knockout phenotypes.
Inhibitors of essential cell cycle proteins lack a therapeutic window in the clinic due to high proliferation rates of critical non-neoplastic tissues.
The discrepancy between pre-clinical success and clinical failure of mitotic kinase inhibitors (Table I) can be explained by two reasons: 1.) interspecies differences between mice and humans and 2.) high proliferation index of critical non-neoplastic tissues versus tumors. Pre-clinical xenografted mouse tumors have significantly higher mitotic rates compared to those in actual human tumors [16,17,69,70]. Mitotic kinase inhibitors will thus inevitably perform better in murine models than in patients. Another shortcoming of pre-clinical mouse models is their maintenance in near aseptic specific pathogen free colony conditions. Within this environment, myelosuppression is of minor consequence, which sharply contrasts the reality for cancer patients.
The second, and most significant explanation for the clinical failure of mitotic kinase inhibitors lies in the on-target toxicity to prolific, non-malignant tissues which have higher proliferation indices than most cancers (Figure 2, Supplementary Figures S1 and S2). Two types of tissue are susceptible to mitotic kinase inhibition: cancerous and prolific, non-neoplastic tissues. Though these inhibitors were designed to act on the former, on-target toxicities to the latter limit the tolerated dose and ultimately, their therapeutic effects. Contrary to popular perception, some prolific, non-neoplastic tissue have much higher cell cycling rates compared to many cancers. Even for pre-clinical researchers, Timothy J. Mitchinson once noted that “…many basic cell biologists have a naïve view of human cancers as proliferating as fast as HeLa cells in a dish, as [he] did until recently” [10].
Figure 2: The mitotic rates of fast-growing normal tissues are higher than that of most tumors.
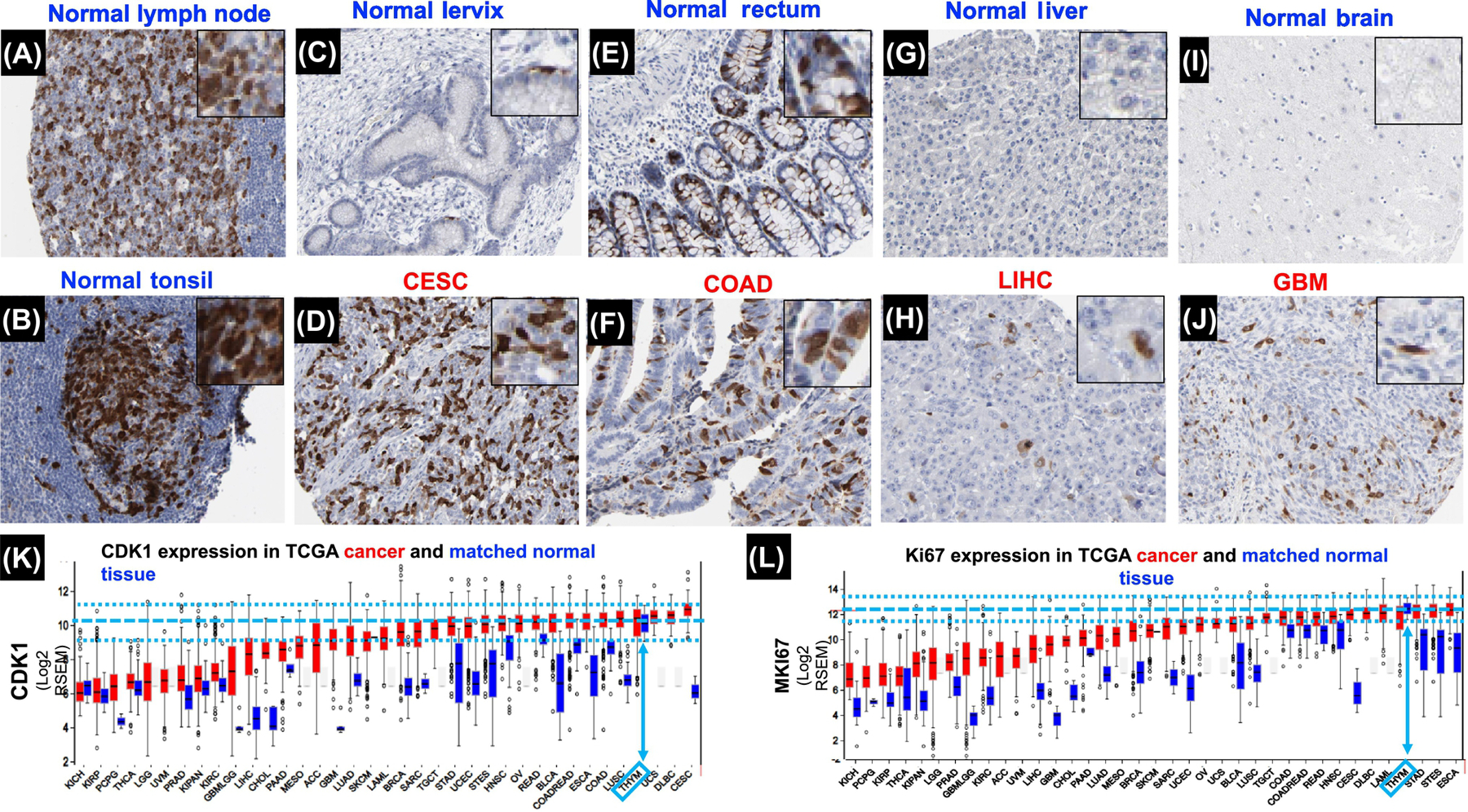
Immunohistochemistry (IHC) images compiled from the Human Protein Atlas show various normal and cancerous tissue were DAB-stained for CDK 1 [72] (antibody CAB003799) and counterstained with hematoxylin (blue, nuclei). CDK1 is only expressed during the G2/M and M phases and thus provides a simple representation of mitotic cells (Wolowiec et al. Int. J. Cancer 1995, Xi et al. Tumor Biol. 2015). Positively stained nuclei (brown) indicate a mitotic cell while non-stained cells (blue) are non-mitotic. A magnified view of strongly stained regions can be found in the top right corner of each image. A: Normal lymph node, germinal center (Patient ID: 1636), B: Normal tonsil (Patient ID: 2511); C: Normal Cervix (Patient ID: 2005), D: CESC, cervical squamous carcinoma, (Patient ID: 1751), E Normal rectum, (Patient ID: 2001); F, COAD, Colorectal adenocarcinoma (Patient ID: 2096); G: Normal liver (Patient ID: 1899); H: LIHC, liver hepatocellular carcinoma (Patient ID: 2325); I: Normal brain (Patient ID: 2521); J: GBM, glioblastoma (Patient ID: 1572).
Note the near absence of CDK1-postive in most normal tissues (C, G, I) and the high expression of CDK1 and Ki67 in tumors compared to matched normal tissues (K, L, red versus blue bars). In fact, strong positive staining is common in the germinal centers of hemopoietic tissues (A, B) and in the colorectal epithelium (E) compared to the cancerous tissues shown here (D, F, H, J). It is important to recall that highly proliferative cancers are not necessarily synonymous with the deadliest cancers. For example, CESC has a much higher proliferative index than LIHC or GBM yet the prognosis of the former is much better than in the latter.
K. RNA-Seq data for CDK1 from the TCGA Firebrowse concur with the qualitative mitotic indices obtained from the IHC images shown. Here, mRNA expression serves as a good approximation of proliferation, as CDK1 is only expressed during the M-phase. Bars (red, tumor; blue, normal control) represent mRNA expression (median, first and third quartiles, and 95% CI) of primary human tumors sequenced from diverse cancers in the TCGA.
mRNA expression of the thymus (bone marrow was not available), a representative hemopoietic tissue, is highlighted in light blue arrows, with the median and 95% CI indicated as light blue dashed lines. Overall, most cancers have lower expression of CDK1 compared to normal thymus, which concurs with the IHC data. L. RNA-Seq data for Ki67 from the TCGA Firebrowse follows a pattern similar to CDK1, with the majority of tumors having a much lower expression compared to normal hemopoietic tissues such as the thymus, indicating slower proliferation.
Though not commonly known today, multiple studies since the 1960s have repeatedly shown that most tumors have lower proliferative indices than specific prolific, non-neoplastic tissues [16,18,19,71]. Typical tumors even of highly malignant cancers will have a Ki67 labeling index of around 15%, which pales in comparison to the greater-than-90% in germinal centers of lymph-nodes (Supplementary Figure S2). Comparative measurements of proliferation indices have since been performed on primary human tumors and have been deposited in public domain databases such as the Human Protein Atlas [72]. For example, the sparse immunohistochemistry (IHC) staining of Ki67 in the even most difficult to treat cancers (e.g. GBM, HCC) pales in comparison to the near complete labeling in lymph nodes, intestinal crypts, and skin (Supplementary Figure S2). Consistently high patterns of expression of other proliferation markers can also be observed for other prolific, non-neoplastic tissue such as hemopoietic tissues, gastrointestinal tract, mucus membranes, skin and hair follicles [73–75]. The fastest growing tumor cases in high frequency cancer types (breast, colon, lung, melanoma) human tumors double in as quickly as 20 days (50–80 days is more typical) [22]. This starkly contrasts the bone marrow, which has a doubling time of 24 hours, and the gut, which has a turnover time of 3 days. Histopathological determinants of proliferation evidencing the high proliferation indices in many non-neoplastic tissues are complemented by in vivo imaging techniques. Non-invasive positron emission tomography markers such as 18F-fluorothymidine have failed to see widespread clinical utility in the way 18F-fluorodeoxyglucose (18F-FDG) has due to the strong signal from the normal hemopoietic tissues such as the bone marrow [76,77] (Compare Figure 1A vs 1B in ref. [78]). These corroborative histopathological and imaging data epitomize the stark proliferation indices between prolific, non-neoplastic tissue and even the most fatal cancers. It is thus no surprise that drugs operating exclusively on the basis of proliferation rates fail to yield a therapeutic window due to toxicities to prolific, non-neoplastic tissues. In 2011, Komlodi-Pasztor et al. predicted that inhibitors of essential cell cycle kinases advancing to clinical trials at the time would ultimately fail from intolerable adverse events to normal prolific tissues [16,17]. Despite receiving much backlash for this stance at the time [79–81], Komlodi-Pasztor et al. proved correct, as inhibitors of essential mitotic kinases have failed to advance beyond phase 3 clinical trials as of 2020 (Table I). Seeing that no mitosis-targeting drug inherently possess a therapeutic window begs the question as to how any chemotherapeutic has efficacy at all. Investigation into their mechanisms of cancer selectivity may provide insight into designing a therapeutic window in mitotic kinase inhibitors.
Many chemotherapeutics achieve a therapeutic window through differential drug metabolism in tumor versus normal tissue.
Similar to mitotic kinase inhibitors, toxicity to prolific, non-neoplastic tissue such as bone marrow suppression, hair loss, stomatitis, diarrhea, and infertility are examples of common side effects associated with conventional cytotoxic chemotherapies. However, where mitotic kinase inhibitors fail to achieve a sufficient therapeutic window, conventional cytotoxic chemotherapies succeed well enough to impart clinically meaningful and, at times, truly transformative results [82,83]. Combinations of various chemotherapies have been central to cancer treatment, with a non-negligible number of patients across a range of cancers being effectively cured by such combinations [84,85]. While this is not well-appreciated in the cancer research community, many key chemotherapeutic drugs from diverse classes are pro-drugs; their therapeutic windows arise from preferential bioactivation in cancerous versus normal tissues (Table 2, Figures 3–4). A brief overview of functionally diverse, clinically impactful conventional chemotherapies makes the prevalence of the pro-drug theme quite apparent.
Figure 3. Mechanisms of action for common chemotherapeutics versus stalled clinical trial-stage mitotic kinase inhibitors.
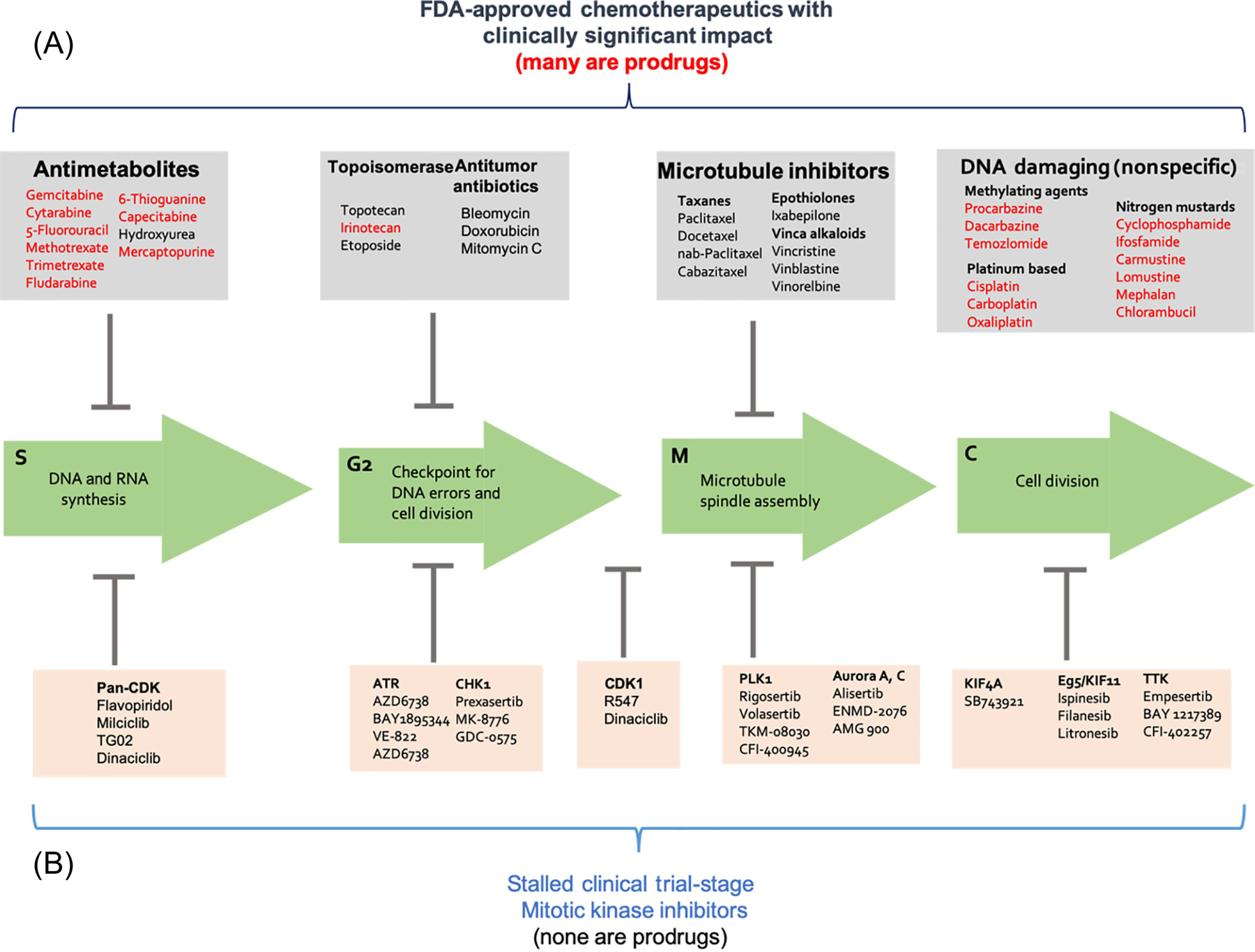
Top: Commonly prescribed conventional chemotherapies operate at various stages of the cell cycle and have made a significant impact in the clinic against a range of cancers. Bottom: Stalled, late-stage clinical trial mitotic kinase inhibitors. Many prevalent, FDA-approved chemotherapies used to treat a variety of cancers are administered as pro-drugs (indicated in red), which enables their specificity towards cancer cells over rapidly proliferating non-malignant cells. In sharp contrast, none of the modern mitotic kinase inhibitors are pro-drugs; all are administered as active compounds and experience little-to-no biotransformation.
Figure 4: Pro-drugging of nitrogen mustard has allowed the harnessing of an exceptionally toxic chemical into a diverse, clinically significant family of chemotherapeutics.
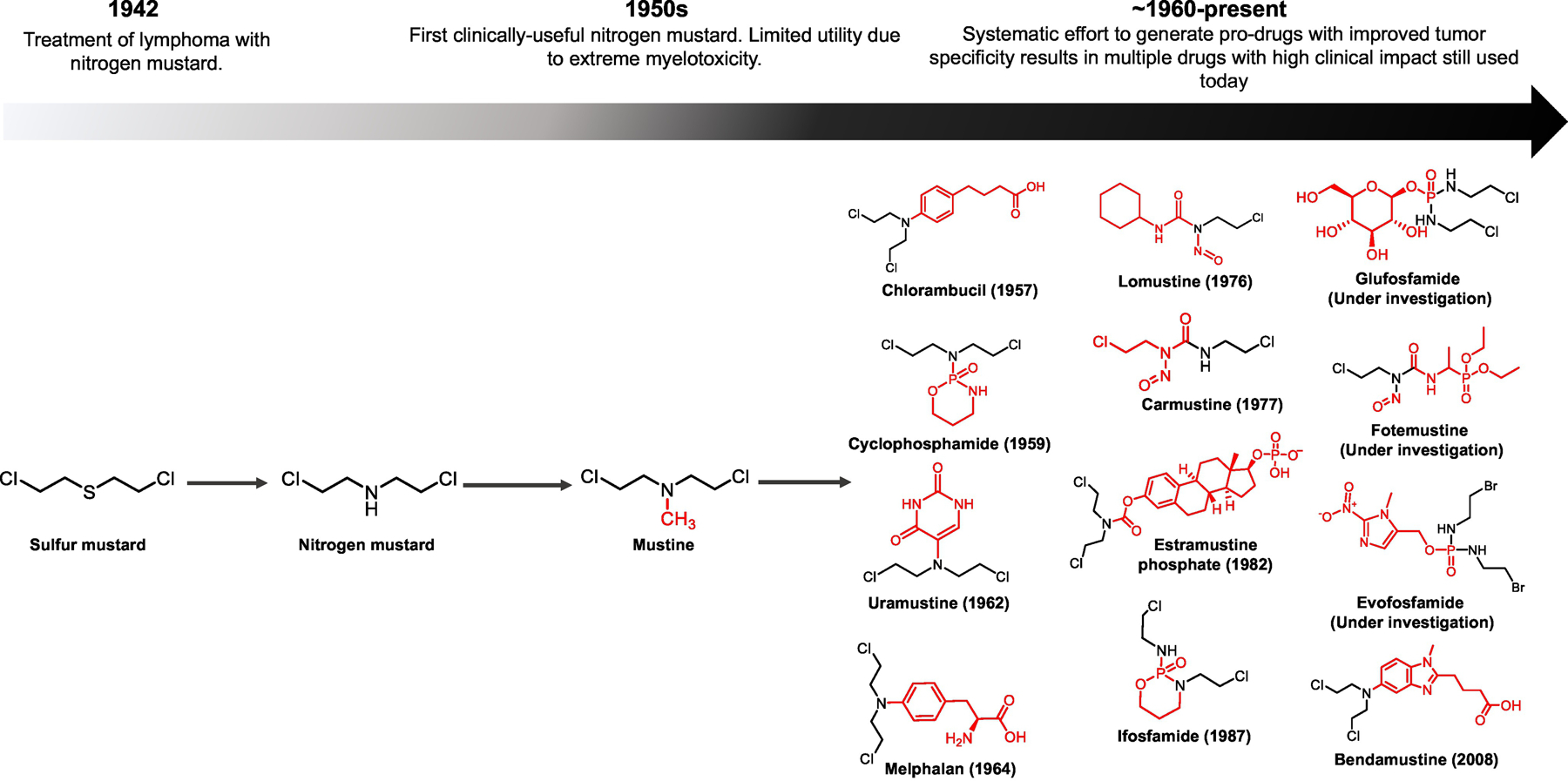
Initial observations of lymphopenia in survivors of chemical weapons exposure and animal experiments during WWII led to first clinical trials with nitrogen mustards [87]. A derivative of nitrogen mustard, mustine, received FDA approval for the treatment of leukemia and lymphoma. However, its utility was limited due to severe bone marrow suppression (though less than nitrogen mustard or sulfur mustard). A deliberate effort to generate pro-drugs that would result in more selective delivery of nitrogen mustard to cancer versus normal haemopoietic sites has been largely successful. This has resulted in numerous chemotherapeutic drugs that have made a tremendous clinical impact and that form the first line treatment of many cancers to this day. Research efforts in this area were most intense in 1960s but have continued in niche applications to this day, as exemplified by glufosfamide [118] and evofosfamide [131]. The latter, which is hypoxia-bioactivated, remains under active investigation. Another newer alkylating agent, fotemustine, is approved in Europe and is being investigated in the U.S. (NCT02460068).
Nitrogen mustards
The dismal clinical landscape of mitotic kinase inhibitors is eerily similar to that observed for nitrogen mustards in the early 1950s [86]: though there were hints of anti-neoplastic activity in model systems and in the clinic [87,88]. intolerable adverse effects [89,90] precluded further utility. To temper the indiscriminate toxicity of the aliphatic nitrogen mustard (Figure 4) [90–92], a concerted effort to reduce the electrophilicity of the mustard was made in the 1950s. It was then that the very first pro-drug iterations of the nitrogen mustard arose, with the FDA approving chlorambucil in 1957. More sophisticated efforts to modulate the reactivity of the mustard while increasing specificity for tumors followed with the conception of perhaps the most iconic nitrogen mustard pro-drug, cyclophosphamide. Cyclophosphamide is a DNA cross-linking agent that exhibits selectivity for cancer cells over prolific normal cells by exploiting differences in aldehyde dehydrogenase (ALDH) expression [93]. In normal tissue and especially stem cells, high expression of ALDH1A1 [94] prevents trapping of the phosphoramidate mustard intermediate in cells. However, in cancer cells, downregulation of ALDH1A1 [94–97] results in trapping of the aldophosphoramidate mustard, ultimately yielding the DNA-damaging agent nitrogen mustard.
Triazenes
Triazenes encompass a number of structurally complex alkylating agents, such as TMZ and dacarbazine. TMZ is DNA-methylating agent used primarily for the treatment of GBM [98]. While stable under acidic conditions (pH < 5), pro-drug hydrolysis increases exponentially above pH ~7 such at physiological pH 7.4, the compound is slowly hydrolyzed to the active agent, 5-(3- methyltriazen-1-yl) imidazole-4-carboxamide (MTIC) [99]. Subsequent proton transfers ultimately reveal the methyl diazonium ion, the active alkylating agent. TMZ is a particularly intriguing pro-drug that exploits the sharp pH gradient between the extracellular and intracellular environments of GBMs [100]. In contrast to normal tissue, tumors such as GBM have a uniquely acidic extracellular environment and a more alkaline intracellular environment [101]. Pro-drug specificity arises from exploiting the more Lewis basic, alkaline intracellular pH in gliomas compared to non-malignant tissue [102,103]; administration of the active metabolite, MTIC results in massive myelotoxicity and the absence of a therapeutic window [33]. This is supported by the vast discrepancy in potency against the GBM cells in vitro compared to that in vivo. While greater than 100 μM is typically required to kill even MGMT-negative GBM cells (e.g. U87), intratumoral concentrations of <1 μM are sufficient to elicit a marked tumor regression in U87-xenografts [104]. Though it has been established that a silenced MGMT promoter sensitizes GBM tumors to TMZ due to the inability to repair methylated DNA [98], this alone cannot explain why TMZ is the most clinically effective triazene against GBM, as many other agents methylate the same position on guanine. TMZ’s requirement for nucleophilic ring-opening under Lewis basic conditions [105] points to the more alkaline (or more Lewis basic) environment in tumors, compared to cultured cells, as a point for selectivity.
Nucleotide analogs
Nucleotide analogs are a diverse group of chemotherapeutics which require biotransformation into an active mono- or, for some, tri-phosphate nucleotide to disrupt various aspects of DNA replication. For example, the classic and successful chemotherapeutic drugs, 5-fluorouracil (5-FU), gemcitabine, and 6-mercaptopurine are all administered as pro-drugs (Table 2). None of the nucleotide analogs are administered in their phosphorylated forms: they are either given as nucleosides (e.g. gemcitabine) or nucleobase analogs (e.g. 5-FU, 6-mercaptopurine). Their therapeutic windows come from faster biotransformation into the active nucleotide analog due to higher expression of bio-transforming enzymes in cancer cells compared to prolific normal cells (Supplementary Figure S3). Biotransformation of 5-FU to the active 5-FU-deoxyribose phosphate can serve as an example. Selectivity arises from elevated expression of thymidine kinase (TK1) and thymidine phosphorylase (TYMP) and lower levels of the inactivating enzymes, dihydropyrimidine dehydrogenase (DYPD) and the drug target thymidylate synthase (TYMS) in cancer cells compared to their prolific, non-neoplastic counterparts (Supplementary Figure S3). While these enzymes are specific to selective bioactivation of 5-FU, the overall theme is the same for all nucleobase and nucleoside analogues [106,107]; differences arise from the identity of the drug as a purine or pyrimidine analog.
Efforts to the bypass these crucial biotransformation steps by generating cell-permeable, nucleotide monophosphate pro-drugs have been attempted using esterase-labile pro-drugs [108] or by using a ProTide approach [109]. A ProTide version of gemcitabine monophosphate (Acelarin) was recently being investigated in phase 3 clinical trials to determine whether it would show improved efficacy against metastatic pancreatic adenocarcinoma compared to gemcitabine (NCT03610100). However, the current phase 3 trial investigation has been suspended following a futility analysis [110]. Precise physiological reasons for the lack of efficacy were not disclosed, though based on enzyme expression from TCGA, pancreatic adenocarcinoma does not express particularly high levels of CTSA, one of the ProTide bioactivating enzymes, suggesting indiscriminate toxicity to prolific normal tissue. This reinforces the need for initial phosphorylation steps of nucleobase analogues to act as points of selectivity for pro-drug activation.
Anti-folates
Anti-folates consist of folic acid antagonists, such as methotrexate (MTX), pemetrexed, pralatrexate, and raltitrexed. Though these analogs may appear to be structurally inert, their identity as pro-drug mechanisms are quite intricate, as represented by the bioactivation of MTX. As an antimetabolite, MTX is substrate competitive with dihydrofolate and inhibits dihydrofolate reductase (DHFR). In many cancer cell lines [111–113], MTX achieves selectivity through polyglutamation [114]. The high affinity of MTX to DFHR combined with the formation of polyglutamated-MTX results in drug accumulation [114]. Selectivity is achieved by the ability for cancer cells to more readily convert MTX to its polyglutamated form [114]. Cell lines unable to form polyglutamates in less than 24 hours are insensitive to the drug because of their inability to generate a reserve of the polyglutamated-MTX [114].
The examples described above represent some of the most common classes of pro-drug chemotherapeutics prescribed in clinic but are not an exhaustive list. Toxicities observed when conventional chemotherapies override selective cancer-specific bioactivation pathways coincide with those observed for mitotic kinase inhibitors. Much in the way nitrogen mustards were decorated with pro-drug moieties to direct their toxic effects to cancerous tissue, we contend that the pro-drug strategy may also be applied to mitotic kinase inhibitors to enable their clinical utility.
Applying the pro-drug strategy to mitotic kinase inhibitors.
What medicinal chemists in the 1950s lacked in technology to target the active sites of specific mutated oncogenes or mitotic kinases, they compensated with the thoughtful assembly of tumor-specific chemical skeletons to harness the nonspecific potency of the nitrogen mustard. That the strategic attachment of chemical moieties can turn a chemical warfare agent engineered to kill into a therapeutic designed to cure testifies to the unique control endowed by rational pro-drug design. While the aforementioned properties do raise the possibility of using mitotic kinase inhibitors for antibody-drug conjugate purposes, the need for picomolar potency, a highly expressed surface marker antibody target, and on-going issues of premature linker cleavage make the application of this strategy problematic [115]. With these considerations in mind, we propound that strategic pro-drug attachment to mitotic kinase inhibitors leverages an opportune target with tumor specific bioactivation.
Mitotic kinase inhibitors are advantageous because, unlike cytotoxic DNA-damaging agents, they do not require a functional DNA repair or apoptotic machinery to convert DNA damage into cell death. Mitotic kinase inhibitors can also exert sustained effects: because they are not inactivated upon target interaction, they can be released by dying cells and permeate neighboring tumor cells. Their sub-nanomolar potency in cell-based assays also suggests that if even a fraction of administered pro-drug is bioactivated, cancer-specific killing could still be achieved. Finally, targeting cell cycle-essential proteins also focuses the scope of the possible target cells to those actively undergoing proliferation-leaving quiescent cells unaffected.
In addition to cancer-specific enzymatic or environmental activation, the ideal pro-drug inhibitor would also be orally bioavailable, with minimal metabolism in the extracellular space. The active pharmacophore itself should also be a “soft drug” [116] with a short systemic half-life to avoid toxicity against the proliferative tissue in the bone marrow. Following good pharmacokinetics and tissue penetration, the active agent should also have isolated efficacy against its target. The pro-drug activating enzyme should not be highly expressed in proliferative tissues such as the bone marrow. Where cell cycling rates are slower in other contexts, such as the liver, pro-drug enzyme expression levels are less of an issue, so long as the activating enzyme is expressed highly in the tumor.
It does not take an experienced chemist to see that all reported inhibitors are structurally inert compounds with few-if any-sites to attach metabolically labile pro-drug groups. Kinase inhibitors are ATP-competitive analogs and ATP is evidently a phosphate-containing, polyanionic molecule. However, kinase inhibitors are hard drugs: their design revolves around the adenine nucleobase while structural mimicry of the anionic phosphonate moiety has deliberately been engineered out of pharmacophore. Given the several advantages to harnessing tumor-specific environmental and metabolic aberrations, we contend that incorporating more sensitive moieties amenable to pro-drug attachment may actually prove to be an asset, rather than a vulnerability [117–119]. Large-scale genomic and proteomic datasets coupled with the advances in available bioinformatics tools [120,121] can vastly enrich the known scope of potential tumor-specific pro-drug-activating enzymes and, by extension, the possible pro-drug chemical moieties. In addition to the well-established bioactivation strategies illustrated earlier, the identification of tumor-specific enzymatic or environmental vulnerabilities can be used for pro-drug activation. Translating distinctive features of the tumor into a pro-drug moiety, indeed, requires a strong grasp of the relationship between chemical structure and its metabolic proclivities.
General biochemical characteristics common to tumors can be used for pro-drug design
Rational pro-drug design can be guided by either general or specific alterations that distinguish cancer cells from prolific, non-malignant cells. Where general characteristics often correlate with environmental factors, specific characteristics capitalize on individual alterations in enzymatic expression.
Example 1: pH-dependent pro-drugs.
Unlike other pro-drug strategies, pH-dependent protecting groups are the only class that relies solely on the inherent chemical properties of the molecule. Electrophilicity, inductive effects, and resonance are just a few key organic chemistry principles that should be taken into consideration when constructing viable, pH-dependent triggers. There are two main types of pH-dependent categories thus emerge: 1.) hydrolysis leaving groups and 2.) beta-elimination moieties (Supplementary Figure S4). One possible starting point to illustrate the concept of hydrolysis includes substituted phenols. The unusually low pKa of phenol (~10) can be modulated by making substitutions on the ring with either electron-donating or electron-withdrawing groups. The tunable nature of the phenol moiety is a good starting point for probing most ideal substitution pattern for controlled release when considering the range of possible pKas or Lewis basicity within the intratumoral space. Other good leave groups include well-known leaving groups such as mesylates and tosylates [122,123]. The second type of pH-dependent pro-drug include is beta-elimination moieties. Viable beta-elimination moieties are characterized by an electrophilic functionality two carbons away from releasable handle (Supplementary Figure S4). Electrophilic moieties include pseudohalogens such as the nitro or cyano group. Carbonyl groups may also be engineered for enhanced electrophilicity through halogen attachment at the alpha carbon. The identity of the electron-withdrawing group influences the pKa of the adjacent alpha proton. Under sufficiently alkaline conditions, Lewis base abstraction of the alpha proton results in elimination of the mitotic kinase inhibitor at the beta position.
Example 2: Hypoxia activated pro-drugs.
Hypoxic regions within a tumor are another well-established, general feature amenable to pro-drug design. Under low oxygen conditions, a class of NADH dehydrogenases known as nitroreductases display heightened activity [124]. In some extreme cases, median tumoral oxygen levels can be less than 2% O2 [125,126], compared to roughly 7% O2 in normal tissues [127]. Hypoxia is associated with upregulation of enzymes known as nitroreductases and azoreductases (quinonereductases) [128], which reduce nitroaromatic moieties. Nitroaromatics have used in hypoxia-activated probes [129,130] and in pro-drug contexts, as seen for the bioreducible alkylating agent, TH-302 (evofosfamide) [131]. Popular pro-drug moieties include nitrofuran [132], nitrothiophene [133], and nitroimidazole [131]. Azoreductases have similar substrate scope as nitroreductases, though are also capable of reducing azo moieties, (R1-N=N-R2, where R1 and R2 are typically aryl groups) providing another option for potential pro-drug attachment [134,135].
Using tumor-type specific pro-drug activating enzyme expression for pro-drug design
Pro-drug strategies can also capitalize on unique enzyme elevations within certain cancers. Even before the ubiquity of bioinformatic data, chemists in the 1950s aimed to package alkylating agents, such as mustine, with chemical moieties that would be hydrolyzed by increased expression of phosphoramidases in cancer cells to release active agent [136]. This ultimately resulted in the synthesis of cyclophosphamide. Though it was later determined that cyclophosphamide was not bioactivated by phosphoramidases [96,137], the idea of harnessing elevated enzymatic activity in cancer cells for selective delivery is not new. But unlike early pro-drug endeavors, the wide availability of bio- and cheminformatic data enables precise identification of elevated enzyme isoforms within a cancer subtype for highly specific drug delivery.
Example 1: Tyrosinase is highly expressed in melanoma.
Human Protein Atlas and TCGA FireBrowse [138] data indicate that tyrosinase (TYR) is highly overexpressed in uveal and cutaneous melanoma [139] but is not remarkably elevated other non-neoplastic tissue. As its name suggests, the amino acid tyrosine is the natural substrate for TYR, which undergoes phenolic oxidation to form a quinone species that eventually results in melanin [140]. Uniquely high expression of TYR in melanoma compels the design and attachment of tyrosine-like prodrugs to mitotic kinase inhibitors. Though the concept of tyrosinase activated pro-drugs has permeated the drug delivery literature for quite some time [141–143], it has yet to be applied to the delivery of mitotic kinase inhibitors. There are two themes notable in the tyrosinase pro-drug development field: attachment to DNA-damaging agents [142,144] and dormant compounds that become cytotoxic upon oxidation by tyrosinase [145]. For reasons discussed previously (inappropriate pharmacophore), these approaches had shortcomings that preluded further clinical development. Despite the drawbacks in rationale, these applications show that rational design of a chemical moiety can be subject to tyrosinase activation [141–143]. Changing the pharmacophore to a mitotic kinase inhibitor inherently narrows the scope of possible targetable cells to those that are actively dividing. Adding a tyrosinase-activatable pro-drug onto a mitotic kinase inhibitor confers yet another layer of specificity, as only highly prolific cells expressing high levels of tyrosinase will be targeted. Among retinal, brain, and melanotic cells, only the latter encompasses both characteristics (Figure 5). Cancer cell specificity is thus conferred on the bases of proliferation index and tyrosinase expression, which vastly narrows the scope of targetable tissue.
Figure 5. Melanomas express high levels of TYR and CDK1.
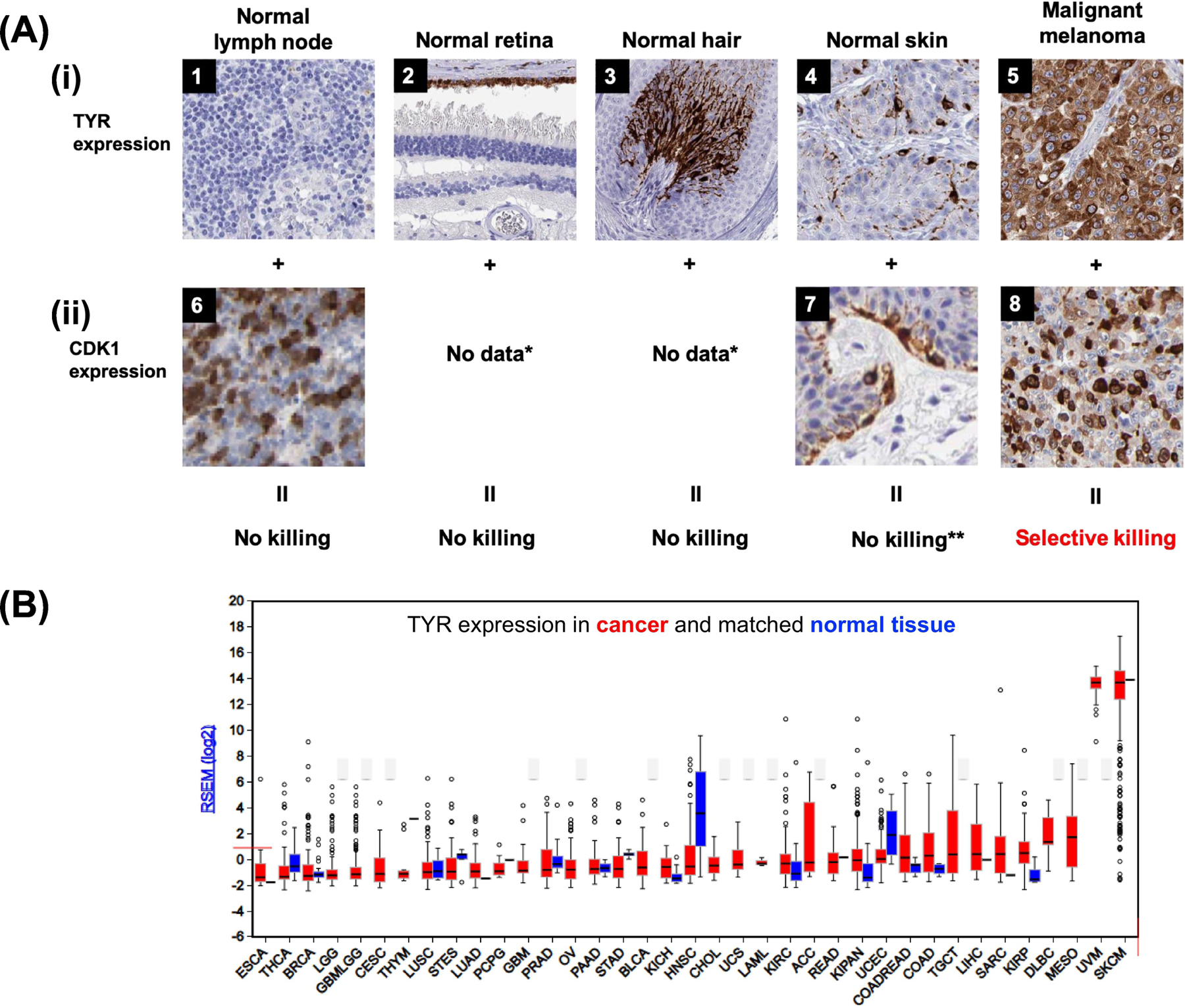
A: IHC images compiled from the Human Protein Atlas show normal and cancerous tissue DAB-stained for tyrosinase (antibody HPA064583) and CDK1 (antibody: CAB003799). Top row: Positively stained cells (brown) indicate the expression of TYR while negatively stained cells (blue) indicate the absence of TYR. Bottom row: Positively stained nuclei (brown) indicate the expression of CDK1 while negatively stained cells (blue) indicate the absence of CDK1. TYR is a pro-drug activating enzyme. In addition to melanotic cells (5, Patient ID: 5526), cells in the retina (2, Patient ID: N/A), hair (3, Patient ID: N/A), and skin (4, Patient ID: 4406) also express TYR to a much lesser degree. However, the retina, hair, and skin (7, Patient ID: 1876) do not have as high of a mitotic index compared to melanoma (8, Patient ID: 179). The normal lymph node is shown as an example of a proliferative non-neoplastic tissue (6, Patient ID; 1636) that has no expression of TYR (1, Patient ID: 1004). B: RNA-Seq data for TYR from the TCGA Firebrowse concur with the qualitative mitotic indices obtained from the IHC images shown. Bars (red, tumor; blue, normal control) represent mRNA expression (median, first and third quartiles, and 95% CI) of primary human tumors sequenced from diverse cancers in the TCGA.
*No IHC images available via Human Protein Atlas
**Though it may appear that certain regions (namely, melanocytes) of the skin show high expression of TYR, melanocytes are minimally proliferative and would thus experience minimal killing by a tyrosinase-activated mitotic kinase inhibitor.
Example 2: Paraoxonase 2 exhibits uniquely high expression in GBM.
The aforementioned workflow can be used to pinpoint enzymes with the elevated expression in other cancer subtypes. A survey of the Human Protein Atlas and TCGA FireBrowse reveals that paraoxonase 2 (PON2) expression is distinctly high in GBM but not in prolific, non-malignant tissue such as that in the lymph node (Figure 6). A mitotic kinase inhibitor protected with a PON2-specific pro-drug would exhibit singular activity against GBM. Though other tissues in the body have high expression of PON2, they have extremely low proliferation indices (Figure 6). GBM cells are the only cells that are actively dividing and expressing PON2, subjecting them to unique targeting by a PON2-activated mitotic kinase inhibitor. While non-malignant brain tissue has high expression of PON2 (Figure 6), the mitotic kinase inhibitor would exert selective toxicity to dividing GBM. Few studies have reported the synthesis of PON-activated pro-drugs [146]. Most of the biochemical data on PONs largely details the substrate proclivities of PON1 and, to a lesser extent, PON3. Though it appears that PON pro-drug design efforts are not intentionally isoform-specific, following the existing biochemical literature has inadvertently resulted in the design of lactone-containing pro-drugs more specific for PON1/3 [147,148]. Though there is considerable substrate overlap between PON1 and PON3, PON2 exhibits a comparatively unique substrate scope. Unlike PON1/3, PON2 is the only isoform able to hydrolyze N-acyl homoserine lactone derivatives [149], which serves as a helpful starting point for PON2-specific pro-drug design.
Figure 6. A PON2 bioactivated CDK1 inhibitor pro-drug only kills cells that express both enzymes.
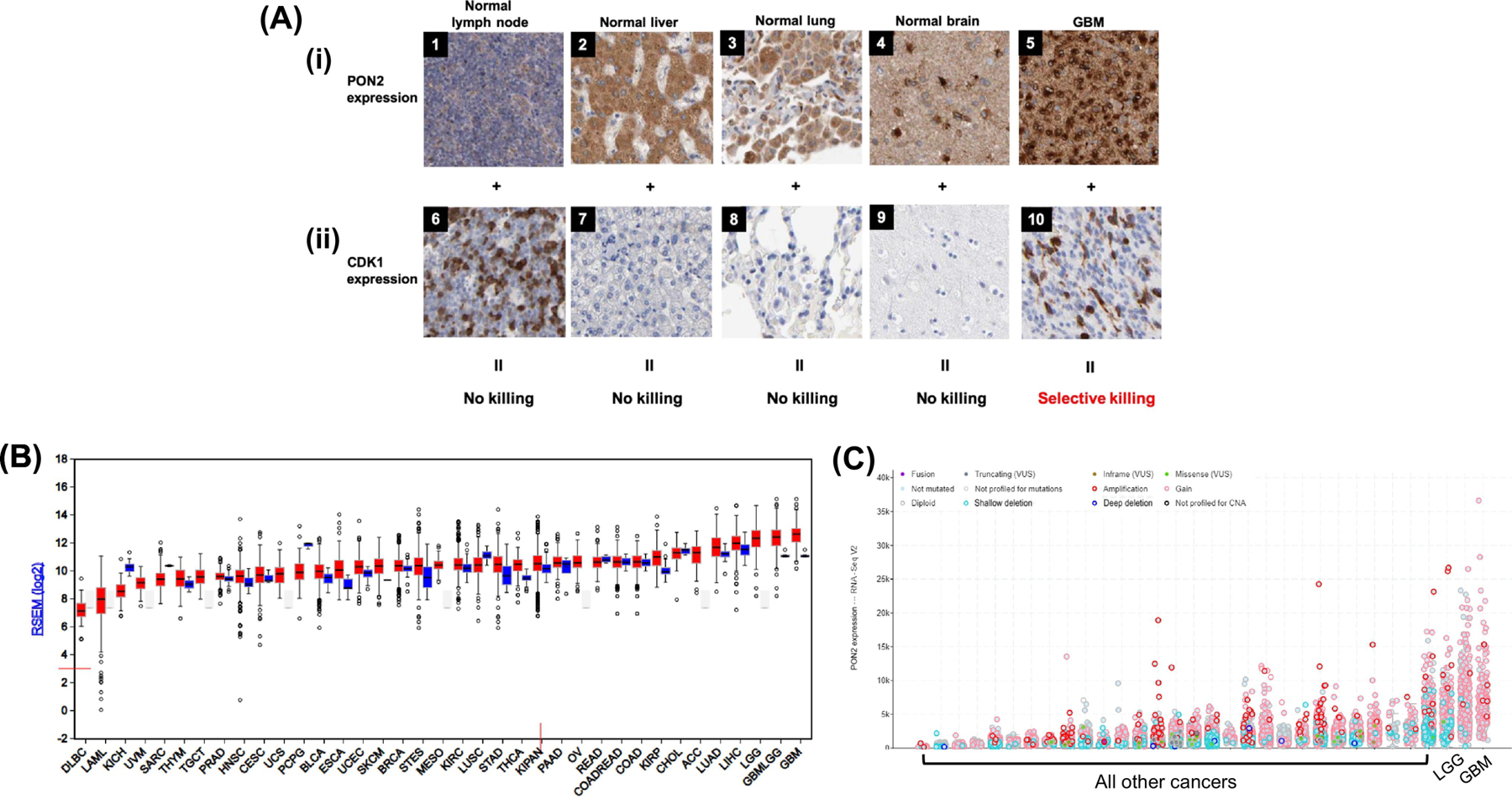
IHC images compiled from the Human Protein Atlas show normal and cancerous tissue DAB-stained for PON2 (antibody HPA029193) and CDK1 (antibody: CAB003799). Top row: Positively stained cells (brown) indicate the expression of PON2 while negatively stained cells (blue) indicate the absence of PON2. Bottom row: Positively stained nuclei (brown) indicate the expression of CDK1 while negatively stained nuclei (blue) indicate the absence of CDK1. Though PON2 expression is high in both normal brain tissue (4, Patient ID: 1609) and in GBM (5, Patient ID: 2843), CDK1 is only expressed in GBM (10, Patient ID: 1578) and not in the normal brain (9, Patient ID: 2521). Pro-drug activation by PON2 in non-malignant brain cells to release the mitotic kinase inhibitor is thus of no consequence to normal brain cells. Similarly, normal liver (2, Patient ID 3222) and lung cells (3, Patient ID 2268) exhibit high levels of PON2 but do not express CDK1 (7, 8, Patient IDs: 1846, 2417). For comparison, CDK1 is highly expressed in proliferative, normal cells such as in the lymph node (6, Patient ID: 1636) but PON2 is not expressed at notable levels (1, Patient ID: 1724). B. RNA-Seq data for PON2 from the TCGA Firebrowse concur with the qualitative mitotic indices obtained from the IHC images shown. Bars (red, tumor; blue, normal control) represent mRNA expression (median, first and third quartiles, and 95% CI) of primary human tumors sequenced from diverse cancers in the TCGA. C. RNA-Seq data with copy number analyses for PON2 across various cancers adapted from TCGA cBioPortal. Amplifications (red circles) and gain-of-function mutations (pink circles) in PON2 are only elevated in GBM and LGG.
Concluding Remarks
In the original paper detailing the structure of cyclophosphamide and its efficacy in rats, Arnold and colleagues reasoned that it would be “advantageous not to apply the substance in its active form, but by introducing suitable chemical groups to transform it into an inert transport substance which will be reconverted into the active form at the site of therapeutic action in the body” [136]. Some may contend that this drug delivery rationale was necessary then due to the nonspecific toxicity of the nitrogen mustard; equipped with technological advances in the post-genomic era, Arnold’s statement may seem archaic. But we contend that it is because of the growing body of public domain bioinformatic data that this strategy is more relevant today than ever before. The ubiquity of various omics data and complementary advances in molecular modelling and structure-based drug design today enables the design of any enzyme-specific pro-drug. Mitotic kinase inhibitors naturally emerge as ideal candidates for rational pro-drug design, as their targetable scope is inherently limited to cycling cells. Target specificity is thus achieved on the basis of two criteria: 1.) metabolic differences between prolific normal cells and cancer cells and 2.) exquisite affinity an inhibitor will have for its essential cell cycle target. Execution of this strategy fundamentally rests on a shift in medicinal chemistry from designing metabolically inert drug to incorporating chemical moieties amenable to pro-drug attachment (see Outstanding Questions). It is thus through the tools driving precision oncology endeavors today that the inadvertent wisdom of our predecessors is most resonant. A drug delivery approach which seemed so obvious in the 1950s can now be realized to its fullest.
Outstanding Questions.
How do biochemical/metabolic differences vary across cancer types and tumors at the individual patient level?
Can tumor biochemical microenviromental conditions be manipulated to increase pro-drug bioactivation?
What specific chemical moieties constitute pro-drug structures that are differentially metabolized by cancerous versus normal tissues?
What advances in synthetic chemistry are needed to enable the transition from designing metabolically inert drugs to incorporating more “vulnerable” chemical moieties?
How does the multidrug resistance phenotype affect sensitivity to mitotic kinase inhibitors?
To what extent do pro-drug bioactivation mechanisms associated with classical chemotherapies contribute to the mechanisms of cancer resistance and how does this affect the efficacy of (pro-drug) mitotic kinase inhibitors?
Supplementary Material
Highlights.
A common misconception is that all cancers proliferate more rapidly than normal tissue. However, many non-malignant tissues have proliferation indices higher than certain cancers.
Many successful DNA-damaging chemotherapies are pro-drugs and achieve sufficient therapeutic windows due to selective bioactivation in cancer cells over normal cells.
Despite strong pre-clinical promise, inhibitors of cell-essential mitotic kinases were relegated to the drug development graveyard due to severe off-target toxicities to rapidly proliferating normal tissues.
The utility of mitotic kinase inhibitors may by applying the pro-drug strategy, used successfully in classical chemotherapies, onto this class of inhibitors. Selectivity for cancer cells will then arise from proliferation rate and tumor-specific pro-drug bioactivation.
Acknowledgements.
This work was supported by the following grants to F.L.M.: NIH 1R21CA226301, American Cancer Society RSG-15-145-01-CDD, National Comprehensive Cancer Network YIA170032 and the Andrew Sabin Family Foundation Fellows Award. Funding from the University of Texas MD Anderson Cancer Center/Glioblastoma Moon Shot, CABI/GE In-Kind Research Grant (MI2), Brockman Medical Research Foundation and the SPORE in Brain Cancer (2P50CA127001) funds are gratefully acknowledged. We dedicate this work in the memory of Kenneth Scott, a brilliant cancer researcher who, at the prime of his life, succumbed to the very illness he was working to eradicate.
Glossary.
- Pro-drug
a drug that is inactive against its target as administered and must be metabolically converted to its active form
- Bioactivation
a metabolic series of enzymatic or environmental transformations made to a drug that changes its chemical structure
- Therapeutic window
amount of drug required to exert effects on the target population of cells without inducing toxicity to normal tissues
- Proliferation index
the number of cells in a tumor that are actively dividing
- Adverse events
unexpected medical events that occur during therapy; they can be classified as mild, moderate, or severe. Examples of severe adverse events include bone marrow suppression and neutropenia
- Hard drug
a metabolically inert drug that is active when administered
- Pharmacophore
portion of a molecular structure that has bioactivity against its target
- Lewis base
chemical entities that can donate an electron pair to an acceptor molecule
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest
References
- 1.DeVita VT and Chu E (2008) A History of Cancer Chemotherapy. Cancer Res. 68, 8643–8653 [DOI] [PubMed] [Google Scholar]
- 2.Roos WP and Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol. Med 12, 440–50 [DOI] [PubMed] [Google Scholar]
- 3.Norbury CJ and Zhivotovsky B (2004) DNA damage-induced apoptosis. Oncogene 23, 2797–808 [DOI] [PubMed] [Google Scholar]
- 4.Molyneux EM et al. (2012) Burkitt’s lymphoma. Lancet 379, 1234–44 [DOI] [PubMed] [Google Scholar]
- 5.DeVita VT (1975) Single Agent Versus Combination Chemotherapy. CA-A Cancer J. Clin 25, 152–158 [DOI] [PubMed] [Google Scholar]
- 6.Hoster E et al. (2014) The Addition of Rituximab to CHOP Improves Failure-Free and Overall Survival of Mantle-Cell Lymphoma Patients - a Pooled Trials Analysis of the German Low-Grade Lymphoma Study Group (GLSG). Blood 124, [Google Scholar]
- 7.Dann EJ et al. (2016) Addition of Intermediate-Dose Methotrexate Is Beneficial in Terms of Both Progression-Free Survival and Overall Survival for Patients with Diffuse Large B Cell Lymphoma Receiving Either R-CHOP or CHOP. Blood 128, [Google Scholar]
- 8.Fukuda M et al. (1999) Combination therapy for advanced breast cancer: cyclophosphamide, doxorubicin, UFT, and tamoxifen. Oncology (Williston Park). 13, 77–81 [PubMed] [Google Scholar]
- 9.Tykocki T and Eltayeb M (2018) Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci 54, 7–13 [DOI] [PubMed] [Google Scholar]
- 10.Mitchison TJ (2012) The proliferation rate paradox in antimitotic chemotherapy. Mol. Biol. Cell 23, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer PM and Gianella-Borradori A (2003) CDK inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 12, 955–970 [DOI] [PubMed] [Google Scholar]
- 12.Zaharevitz DW et al. (1999) Discovery and initial characterization of the paullones, a novel class of small-molecule inhibitors of cyclin-dependent kinases. Cancer Res. 59, 2566–9 [PubMed] [Google Scholar]
- 13.How chemotherapy works | Cancer in general | Cancer Research UK. . [Online] Available: https://www.cancerresearchuk.org/about-cancer/cancer-in-general/treatment/chemotherapy/how-chemotherapy-works. [Accessed: 11-Sep-2019]
- 14.How Chemotherapy Works | Breastcancer.org. . [Online] Available: https://www.breastcancer.org/treatment/chemotherapy/how_it_works. [Accessed: 11-Sep-2019]
- 15.Skarin AT et al. (2010) Atlas of diagnostic oncology, Mosby Elsevier. [Google Scholar]
- 16.Komlodi-Pasztor E et al. (2012) Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin. Cancer Res 18, 51–63 [DOI] [PubMed] [Google Scholar]
- 17.Komlodi-Pasztor E et al. (2011) Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol 8, 244–50 [DOI] [PubMed] [Google Scholar]
- 18.Skipper HE and Perry S (1970) Kinetics of normal and leukemic leukocyte populations and relevance to chemotherapy. Cancer Res. 30, 1883–97 [PubMed] [Google Scholar]
- 19.Skipper HE (1971) Kinetics of mammary tumor cell growth and implications for therapy. Cancer 28, 1479–1499 [DOI] [PubMed] [Google Scholar]
- 20.Buehring GC and Williams RR (1976) Growth rates of normal and abnormal human mammary epithelia in cell culture. Cancer Res. 36, 3742–7 [PubMed] [Google Scholar]
- 21.Boll IT and Fuchs G (1970) A kinetic model of granulocytopoiesis. Exp. Cell Res 61, 147–52 [DOI] [PubMed] [Google Scholar]
- 22.Tubiana M (1989) Tumor Cell Proliferation Kinetics and Tumor Growth Rate. Acta Oncol. (Madr) 28, 113–121 [DOI] [PubMed] [Google Scholar]
- 23.Manfredi MG et al. (2011) Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res 17, 7614–24 [DOI] [PubMed] [Google Scholar]
- 24.Lad L et al. (2008) Mechanism of inhibition of human KSP by ispinesib. Biochemistry 47, 3576–85 [DOI] [PubMed] [Google Scholar]
- 25.Carol H et al. (2009) Initial testing (stage 1) of the kinesin spindle protein inhibitor ispinesib by the pediatric preclinical testing program. Pediatr. Blood Cancer 53, 1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee CW et al. (2008) A phase II study of ispinesib (SB-715992) in patients with metastatic or recurrent malignant melanoma: a National Cancer Institute of Canada Clinical Trials Group trial. Invest. New Drugs 26, 249–255 [DOI] [PubMed] [Google Scholar]
- 27.Parry D et al. (2010) Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther 9, 2344–2353 [DOI] [PubMed] [Google Scholar]
- 28.Kumar SK et al. (2015) Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 125, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olmos D et al. (2011) Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin. Cancer Res 17, 3420–30 [DOI] [PubMed] [Google Scholar]
- 30.Gilmartin AG et al. (2009) Distinct Concentration-Dependent Effects of the Polo-like Kinase 1-Specific Inhibitor GSK461364A, Including Differential Effect on Apoptosis. Cancer Res. 69, 6969–6977 [DOI] [PubMed] [Google Scholar]
- 31.Wengner AM et al. (2016) Novel Mps1 Kinase Inhibitors with Potent Antitumor Activity. Mol. Cancer Ther 15, 583–592 [DOI] [PubMed] [Google Scholar]
- 32.Russo JE and Hilton J (1988) Characterization of cytosolic aldehyde dehydrogenase from cyclophosphamide resistant L1210 cells. Cancer Res. 48, 2963–8 [PubMed] [Google Scholar]
- 33.Newlands ES et al. (1997) Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev 23, 35–61 [DOI] [PubMed] [Google Scholar]
- 34.van Triest B et al. (1999) Thymidylate synthase level as the main predictive parameter for sensitivity to 5-fluorouracil, but not for folate-based thymidylate synthase inhibitors, in 13 nonselected colon cancer cell lines. Clin. Cancer Res 5, 643–54 [PubMed] [Google Scholar]
- 35.Morton CL et al. (1999) The anticancer prodrug CPT-11 is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to SN-38 by butyrylcholinesterase. Cancer Res. 59, 1458–63 [PubMed] [Google Scholar]
- 36.Davies MS et al. (2000) Slowing of Cisplatin Aquation in the Presence of DNA but Not in the Presence of Phosphate: Improved Understanding of Sequence Selectivity and the Roles of Monoaquated and Diaquated Species in the Binding of Cisplatin to DNA. Inorg. Chem 39, 5603–5613 [DOI] [PubMed] [Google Scholar]
- 37.Rajagopalan PTR et al. (2002) Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci 99, 13481–13486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piwnica-Worms H (1996) Reversible phosphorylation and mitotic control. J. Lab. Clin. Med 128, 350–4 [DOI] [PubMed] [Google Scholar]
- 39.Hartwell L and Kastan M (1994) Cell cycle control and cancer. Science (80-.). 266, 1821–1828 [DOI] [PubMed] [Google Scholar]
- 40.Draetta G et al. (1988) Human cdc2 protein kinase is a major cell-cycle regulated tyrosine kinase substrate. Nature 336, 738–44 [DOI] [PubMed] [Google Scholar]
- 41.Draetta G (1990) Cell cycle control in eukaryotes: molecular mechanisms of cdc2 activation. Trends Biochem. Sci 15, 378–83 [DOI] [PubMed] [Google Scholar]
- 42.Hartwell LH and Kastan MB (1994) Cell cycle control and cancer. Science (80-.). 266, 1821–8 [DOI] [PubMed] [Google Scholar]
- 43.Sausville EA (2004) Aurora kinases dawn as cancer drug targets. Nat. Med 10, 234–235 [DOI] [PubMed] [Google Scholar]
- 44.Schmidt M and Bastians H (2007) Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist. Updat 10, 162–81 [DOI] [PubMed] [Google Scholar]
- 45.Gascoigne KE and Taylor SS (2009) How do anti-mitotic drugs kill cancer cells? J. Cell Sci 122, 2579–85 [DOI] [PubMed] [Google Scholar]
- 46.Miglarese MR and Carlson RO (2006) Development of new cancer therapeutic agents targeting mitosis. Expert Opin. Investig. Drugs 15, 1411–25 [DOI] [PubMed] [Google Scholar]
- 47.Wood KW et al. (2001) Past and future of the mitotic spindle as an oncology target. Curr. Opin. Pharmacol 1, 370–7 [DOI] [PubMed] [Google Scholar]
- 48.Bar-Joseph Z et al. (2008) Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc. Natl. Acad. Sci. U. S. A 105, 955–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parry D et al. (2010) Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther 9, 2344–53 [DOI] [PubMed] [Google Scholar]
- 50.Harrington EA et al. (2004) VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med 10, 262–7 [DOI] [PubMed] [Google Scholar]
- 51.Steegmaier M et al. (2007) BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol 17, 316–22 [DOI] [PubMed] [Google Scholar]
- 52.Neugut AI et al. Poor survival of treatment-related acute nonlymphocytic leukemia. JAMA 264, 1006–8 [PubMed] [Google Scholar]
- 53.PDQ Adult Treatment Editorial Board, P.A.T.E. (2002) Adult Acute Myeloid Leukemia Treatment (PDQ®): Health Professional Version, National Cancer Institute (US). [PubMed] [Google Scholar]
- 54.Getta BM et al. (2016) Treatment outcomes and secondary cancer incidence in young patients with hairy cell leukaemia. Br. J. Haematol 175, 402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith MA et al. (1999) Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J. Clin. Oncol 17, 569–77 [DOI] [PubMed] [Google Scholar]
- 56.Hawkins MM et al. (1992) Epipodophyllotoxins, alkylating agents, and radiation and risk of secondary leukaemia after childhood cancer. BMJ 304, 951–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi S et al. (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 352, 786–92 [DOI] [PubMed] [Google Scholar]
- 58.Awad MM et al. (2013) Acquired resistance to crizotinib from a mutation in CD74-ROS1. N. Engl. J. Med 368, 2395–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heidel F et al. (2006) Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 107, 293–300 [DOI] [PubMed] [Google Scholar]
- 60.Shah NP et al. (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–25 [DOI] [PubMed] [Google Scholar]
- 61.Dalle S et al. (2011) Vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med 365, 1448–9; author reply 1450 [DOI] [PubMed] [Google Scholar]
- 62.Robichaux JP et al. (2018) Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat. Med 24, 638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santamaría D et al. (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–5 [DOI] [PubMed] [Google Scholar]
- 64.Diril MK et al. (2012) Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. U. S. A 109, 3826–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piao J et al. (2019) High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 701, 15–22 [DOI] [PubMed] [Google Scholar]
- 66.Jayanthi VSPKSA et al. (2019) Grade-specific diagnostic and prognostic biomarkers in breast cancer. Genomics DOI: 10.1016/j.ygeno.2019.03.001 [DOI] [PubMed] [Google Scholar]
- 67.Pabla S et al. (2019) Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.A pathology atlas of the human cancer transcriptome: CDK1. , The Human Protein Atlas. [Online] Available: https://www.proteinatlas.org/ENSG00000170312-CDK1/pathology. [Accessed: 12-Sep-2019]
- 69.Livingston RB et al. (1974) In vitro determination of thymidine-3H labeling index in human solid tumors. Cancer Res. 34, 1376–80 [PubMed] [Google Scholar]
- 70.Weidner N et al. (1994) Correlation of Ki-67 antigen expression with mitotic figure index and tumor grade in breast carcinomas using the novel "paraffin"-reactive MIB1 antibody. Hum. Pathol 25, 337–42 [DOI] [PubMed] [Google Scholar]
- 71.Buehring GC and Williams RR (1976) Growth rates of normal and abnormal human mammary epithelia in cell culture. Cancer Res. 36, 3742–7 [PubMed] [Google Scholar]
- 72.Uhlén M et al. (2015) Proteomics. Tissue-based map of the human proteome. Science (80-.). 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 73.Cooper GM (2000) Cell Proliferation in Development and Differentiation In The Cell: A Molecular Approach. 2nd edition 2nd EditionSinaur Associates [Google Scholar]
- 74.Richardson RB et al. (2014) Greater organ involution in highly proliferative tissues associated with the early onset and acceleration of ageing in humans. Exp. Gerontol 55, 80–91 [DOI] [PubMed] [Google Scholar]
- 75.Szabo E (2001) Lung epithelial proliferation: a biomarker for chemoprevention trials? J. Natl. Cancer Inst 93, 1042–3 [DOI] [PubMed] [Google Scholar]
- 76.Been LB et al. (2004) [18F]FLT-PET in oncology: current status and opportunities. Eur. J. Nucl. Med. Mol. Imaging 31, 1659–72 [DOI] [PubMed] [Google Scholar]
- 77.Minamimoto R et al. (2012) 4’-[Methyl-11C]-Thiothymidine PET/CT for Proliferation Imaging in Non-Small Cell Lung Cancer. J. Nucl. Med 53, 199–206 [DOI] [PubMed] [Google Scholar]
- 78.Minamimoto R et al. (2012) 4’-[Methyl-11C]-thiothymidine PET/CT for proliferation imaging in non-small cell lung cancer. J. Nucl. Med 53, 199–206 [DOI] [PubMed] [Google Scholar]
- 79.Wissing MD et al. (2013) Tales of how great drugs were brought down by a flawed rationale--letter. Clin. Cancer Res 19, 1303. [DOI] [PubMed] [Google Scholar]
- 80.Tunquist BJ et al. (2013) Tales of How Great Drugs Were Brought Down by a Flawed Rationale--Letter. Clin. Cancer Res 19, 1302–1302 [DOI] [PubMed] [Google Scholar]
- 81.Kitagawa K (2011) Too early to say, "no targeting of mitosis!". Nat. Rev. Clin. Oncol 8, 444; author reply 444 [DOI] [PubMed] [Google Scholar]
- 82.Sullivan RD et al. (1959) Antimetabolite-metabolite combination cancer chemotherapy.Effects of intra-arterial methotrexate-intramuscular citrovorum factor therapy in human cancer. Cancer 12, 1248–1262 [DOI] [PubMed] [Google Scholar]
- 83.Nathanson L et al. (1969) Concurrent combination chemotherapy of human solid tumors: experience with a three-drug regimen and review of the literature. Cancer Res. 29, 419–25 [PubMed] [Google Scholar]
- 84.Ross MB et al. (1985) Improved survival of patients with metastatic breast cancer receiving combination chemotherapy. Comparison of consecutive series of patients in 1950s, 1960s, and 1970s. Cancer 55, 341–346 [DOI] [PubMed] [Google Scholar]
- 85.Frei E et al. (1973) Combination chemotherapy in advanced Hodgkin’s disease. Induction and maintenance of remission. Ann. Intern. Med 79, 376–82 [DOI] [PubMed] [Google Scholar]
- 86.Singh RK et al. (2018) Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem 151, 401–433 [DOI] [PubMed] [Google Scholar]
- 87.Goodman LS et al. (1984) Landmark article Sept. 21, 1946: Nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders By Goodman Louis S., Wintrobe Maxwell M., Dameshek William, Goodman Morton J., Gilman Alfred and McLennan Margaret T.. JAMA 251, 2255–61 [DOI] [PubMed] [Google Scholar]
- 88.GARDIKAS C and WILKINSON JF (1952) Alkylamines in the treatment of leukaemia. Lancet 2, 161–6 [DOI] [PubMed] [Google Scholar]
- 89.Sullivan RD et al. (1953) The effect of intra-arterial nitrogen-mustard. (Methylbis(2-chloroethyl)amine hydrochloride). Therapy on human skin. Cancer 6, 288–293 [DOI] [PubMed] [Google Scholar]
- 90.BATEMAN JC et al. (1951) Hematologic effects of regional nitrogen mustard therapy. Blood 6, 26–38 [PubMed] [Google Scholar]
- 91.DAMESHEK W et al. (1949) NITROGEN MUSTARD THERAPY IN HODGKIN’S DISEASE. Blood 4, [PubMed] [Google Scholar]
- 92.BARBERIO JR et al. (1951) Effects of intra-arterial administration of nitrogen mustard. Cancer 4, 1341–63 [DOI] [PubMed] [Google Scholar]
- 93.Hilton J (1984) Role of aldehyde dehydrogenase in cyclophosphamide-resistant L1210 leukemia. Cancer Res. 44, 5156–60 [PubMed] [Google Scholar]
- 94.Tomita H et al. (2016) Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Emadi A et al. (2009) Cyclophosphamide and cancer: golden anniversary. Nat. Rev. Clin. Oncol 6, 638–647 [DOI] [PubMed] [Google Scholar]
- 96.Sladek N (1973) Bioassay and relative cytotoxic potency of cyclophosphamide metabolites generated in vitro and in vivo. Cancer Res. 33, 1150–8 [PubMed] [Google Scholar]
- 97.Cox PJ et al. (1976) Studies on the selective action of cyclophosphamide (NSC-26271): Inactivation of the hydroxylated metabolite by tissue-soluble enzymes. Cancer Treat. Rep 60, 321–6 [PubMed] [Google Scholar]
- 98.Hegi ME et al. (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med 352, 997–1003 [DOI] [PubMed] [Google Scholar]
- 99.Saleem A et al. (2003) Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 63, 2409–15 [PubMed] [Google Scholar]
- 100.Friedman HS et al. (2000) Temozolomide and treatment of malignant glioma. Clin. Cancer Res 6, 2585–97 [PubMed] [Google Scholar]
- 101.Gerweck LE and Seetharaman K (1996) Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 56, 1194–8 [PubMed] [Google Scholar]
- 102.Hugg JW et al. (1992) Phosphorus-31 MR spectroscopic imaging (MRSI) of normal and pathological human brains. Magn. Reson. Imaging 10, 227–243 [DOI] [PubMed] [Google Scholar]
- 103.Reid J et al. (1997) Pharmacokinetics of 3-methyl-(triazen-1-yl)imidazole-4-carboximide following administration of temozolomide to patients with advanced cancer. Clin. Cancer Res 3, 2393–8 [PubMed] [Google Scholar]
- 104.Grossman R et al. (2013) Microdialysis measurement of intratumoral temozolomide concentration after cediranib, a pan-VEGF receptor tyrosine kinase inhibitor, in a U87 glioma model. Cancer Chemother. Pharmacol 72, 93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bonmassar L et al. (2013) Triazene compounds in the treatment of acute myeloid leukemia: a short review and a case report. Curr. Med. Chem 20, 2389–401 [DOI] [PubMed] [Google Scholar]
- 106.Nelson JA et al. (1975) Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res. 35, 2872–8 [PubMed] [Google Scholar]
- 107.Plunkett W et al. (1995) Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin. Oncol 22, 3–10 [PubMed] [Google Scholar]
- 108.Farquhar D et al. (1994) Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2’-deoxy-5-fluorouridine 5’-monophosphate (FdUMP): a strategy to introduce nucleotides into cells. J. Med. Chem 37, 3902–9 [DOI] [PubMed] [Google Scholar]
- 109.McGuigan C et al. (2011) Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem 54, 7247–58 [DOI] [PubMed] [Google Scholar]
- 110.20-Aug-(2019), Enrollment in the Independent Investigator-Sponsored Phase III Metastatic Pancreatic Study ACELARATE Has Been Suspended Following a Prespecified Futility Analysis. , Globe Newswire [Google Scholar]
- 111.Jolivet J et al. (1982) Synthesis, Retention, and Biological Activity of Methotrexate Polyglutamates in Cultured Human Breast Cancer Cells. J. Clin. Invest 70, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frei E et al. (1985) Alkylating agent resistance: in vitro studies with human cell lines. Proc. Natl. Acad. Sci. U. S. A 82, 2158–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koizumi S (1988) Impairment of methotrexate (MTX)-polyglutamate formation of MTX-resistant K562 cell lines. Jpn. J. Cancer Res 79, 1230–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chabner BA et al. (1985) Polyglutamation of methotrexate. Is methotrexate a prodrug? J. Clin. Invest 76, 907–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Diamantis N and Banerji U (2016) Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 114, 362–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bodor N and Buchwald P (2000) Soft drug design: general principles and recent applications. Med. Res. Rev 20, 58–101 [DOI] [PubMed] [Google Scholar]
- 117.Lin Y-H et al. (2018) Eradication of ENO1-deleted Glioblastoma through Collateral Lethality. bioRxiv DOI: 10.1101/331538 [DOI] [Google Scholar]
- 118.Ciuleanu TE et al. (2009) A randomised Phase III trial of glufosfamide compared with best supportive care in metastatic pancreatic adenocarcinoma previously treated with gemcitabine. Eur. J. Cancer 45, 1589–1596 [DOI] [PubMed] [Google Scholar]
- 119.Yan VC et al. (2020) Aliphatic Amines are Viable Pro-drug Moieties in Phosphonoamidate Drugs. bioRxiv DOI: 10.1101/2020.04.05.026583 [DOI] [PubMed] [Google Scholar]
- 120.Cerami E et al. (2012) The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li H et al. (2019) The landscape of cancer cell line metabolism. Nat. Med 25, 850–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hartley JA et al. (1999) DNA alkylation and interstrand cross-linking by treosulfan. Br. J. Cancer 79, 264–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tong WP and Ludlum DB (1980) Crosslinking of DNA by busulfan. Formation of diguanyl derivatives. Biochim. Biophys. Acta 608, 174–81 [DOI] [PubMed] [Google Scholar]
- 124.Hecht HJ et al. (1995) Crystal structure of NADH oxidase from Thermus thermophilus. Nat. Struct. Biol 2, 1109–14 [DOI] [PubMed] [Google Scholar]
- 125.Movsas B et al. (1999) Hypoxic regions exist in human prostate carcinoma. Urology 53, 11–18 [DOI] [PubMed] [Google Scholar]
- 126.Koong AC et al. (2000) Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys 48, 919–22 [DOI] [PubMed] [Google Scholar]
- 127.McKeown SR (2014) Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol 87, 20130676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ryan A et al. (2011) Activation of nitrofurazone by azoreductases: multiple activities in one enzyme. Sci. Rep 1, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gammon ST et al. (2019) Mechanism-Specific Pharmacodynamics of a Novel Complex-I Inhibitor Quantified by Imaging Reversal of Consumptive Hypoxia with [18F]FAZA PET In Vivo. Cells 8, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Varia MA et al. (1998) Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol. Oncol 71, 270–7 [DOI] [PubMed] [Google Scholar]
- 131.Laubach JP et al. (2019) A Phase I/II Study of Evofosfamide, A Hypoxia-activated Prodrug with or without Bortezomib in Subjects with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res 25, 478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yan VC et al. (2020) Bioreducible Pro-drug Inhibitors of Enolase. ChemRxiv DOI: 10.26434/CHEMRXIV.12033303.V1 [DOI] [Google Scholar]
- 133.Borch RF et al. (2000) Synthesis and Evaluation of Nitroheterocyclic Phosphoramidates as Hypoxia-Selective Alkylating Agents. J. Med. Chem 43, 2258–2265 [DOI] [PubMed] [Google Scholar]
- 134.Ryan A (2017) Azoreductases in drug metabolism. Br. J. Pharmacol 174, 2161–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.FOUTS JR et al. (1957) Enzymatic reduction of prontosil and other azo dyes. J. Pharmacol. Exp. Ther 120, 291–300 [PubMed] [Google Scholar]
- 136.ARNOLD H et al. (1958) Chemotherapeutic Action of a Cyclic Nitrogen Mustard Phosphamide Ester (B 518-ASTA) in Experimental Tumours of the Rat. Nature 181, 931–931 [DOI] [PubMed] [Google Scholar]
- 137.Brock N and Hohorst H-J (1967) Metabolism of cyclophosphamide. Cancer 20, 900–904 [DOI] [PubMed] [Google Scholar]
- 138.Broad Institute of MIT & Harvard (2015), FireBrowse: TYR differential plot. . [Online] Available: http://firebrowse.org/viewGene.html?gene=tyr. [Accessed: 12-Sep-2019]
- 139.Boyle J et al. (2002) Tyrosinase expression in malignant melanoma, desmoplastic melanoma, and peripheral nerve tumors. Arch Pathol. Lab Med 126, 816–22 [DOI] [PubMed] [Google Scholar]
- 140.Fitzpatrick TB et al. (1950) Tyrosinase in Human Skin: Demonstration of Its Presence and of Its Role in Human Melanin Formation. Science (80-.). 112, 223–225 [DOI] [PubMed] [Google Scholar]
- 141.Morrison ME et al. (1985) In vitro studies of 2,4-dihydroxyphenylalanine, a prodrug targeted against malignant melanoma cells. Proc. Natl. Acad. Sci. U. S. A 82, 2960–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jordan AM et al. (2001) Melanocyte-Directed enzyme prodrug therapy (MDEPT). Bioorg. Med. Chem 9, 1549–1558 [DOI] [PubMed] [Google Scholar]
- 143.Jimbow K et al. (1993) Exploitation of pigment biosynthesis pathway as a selective chemotherapeutic approach for malignant melanoma. J. Invest. Dermatol 100, 231S–238S [PubMed] [Google Scholar]
- 144.Marcella Gabrielle Mendes M et al. (2016) Targeted Prodrug Design for the Treatment of Malignant Melanoma. J. Dermatology Res. Ther 2, [Google Scholar]
- 145.Jawaid S et al. (2009) Tyrosinase activated melanoma prodrugs. Anticancer. Agents Med. Chem 9, 717–27 [DOI] [PubMed] [Google Scholar]
- 146.Furlong CE et al. (2016) Paraoxonases-1, −2 and −3: What are their functions? Chem. Biol. Interact 259, 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Draganov DI et al. (2005) Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J. Lipid Res 46, 1239–47 [DOI] [PubMed] [Google Scholar]
- 148.Ishizuka T et al. (2012) Paraoxonase 1 as a Major Bioactivating Hydrolase for Olmesartan Medoxomil in Human Blood Circulation: Molecular Identification and Contribution to Plasma Metabolism. Drug Metab. Dispos 40, 374–380 [DOI] [PubMed] [Google Scholar]
- 149.Billecke S et al. (2000) Human Serum Paraoxonase (pon1) Isozymes Q and R Hydrolyze Lactones and Cyclic Carbonate Esters. Drug Metab. Dispos 28, 1335–1342 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.