

Abstract
Antiretroviral therapy has revolutionized the treatment of AIDS, turning a deadly disease into a manageable chronic condition. Life-long treatment is required because existing drugs do not eradicate HIV-infected cells. The emergence of drug-resistant viral strains and uncertain vaccine prospects highlight the pressing need for new therapeutic approaches with the potential to clear the virus. The HIV-1 accessory protein Nef is essential for viral pathogenesis, making it a promising target for antiretroviral drug discovery. Nef enhances viral replication and promotes immune escape of HIV-infected cells but lacks intrinsic enzymatic activity. Instead, Nef works through diverse interactions with host cell proteins primarily related to kinase signaling pathways and endosomal trafficking. This review emphasizes the structure, function, and biological relevance of Nef interactions with host cell protein-tyrosine kinases in the broader context of Nef functions related to enhancement of the viral life cycle and immune escape. Drug discovery targeting Nef-mediated kinase activation has allowed identification of promising inhibitors of multiple Nef functions. Pharmacological inhibitors of Nef-induced MHC-I down-regulation restore the adaptive immune response to HIV-infected cells in vitro and have the potential to enhance immune recognition of latent viral reservoirs as part of a strategy for HIV clearance.
Keywords: human immunodeficiency virus (HIV), AIDS, HIV-1 Nef, Src-family kinases, Tec-family kinases, Hck, Itk, Btk, dimerization, endocytosis, MHC-I, CD4, protein-protein interaction, SH3 domain, SH2 domain, infectious disease, bimolecular fluorescence complementation (BiFC), Src homology 3 domain (SH3 domain), Src homology 2 domain (SH2 domain), major histocompatibility complex (MHC)
The specter of the HIV pandemic first appeared in the United States Centers for Disease Control Morbidity and Mortality Weekly Report on June 5, 1981. A brief in that report recounted the appearance of Pneumocystis carinii pneumonia cases in the Los Angeles area, a diagnosis typically associated with severely immunocompromised patient populations but unexpected in young men (1). This report unknowingly provided the first description of what would become HIV/AIDS in the medical literature. Today, the AIDS pandemic persists as a global public health crisis, with 38 million people currently living with HIV worldwide. More than 1.7 million new infections and nearly 700,000 HIV-related deaths occurred in 2019, mainly in southern and eastern Africa. Over 30 million people have died from AIDS and related illnesses since the start of the pandemic (UNAIDS 2020 Fact Sheet).
Antiretroviral drug therapy for HIV/AIDS and barriers to cure
The first antiretroviral drug developed for HIV-1 was the nucleoside reverse transcriptase inhibitor (NRTI), zidovudine, which was approved by the United States Food and Drug Administration in 1987 (2). Subsequent development of additional NRTIs facilitated combination therapy that markedly delayed AIDS progression compared with zidovudine alone (3). However, AIDS-related fatalities continued to climb until nonnucleoside reverse transcriptase inhibitors and protease inhibitors were approved for clinical use (4, 5). Introduction of these new classes of inhibitors enabled a triple-drug regimen that resulted in dramatic suppression of viral loads and restored CD4+ T cell counts with a lower incidence of viral resistance. Combination antiretroviral therapy (cART) has continued to improve over the last 25 years, with more than 30 antiretroviral drugs now available across six classes of inhibitor types, including integrase inhibitors, viral fusion inhibitors, and coreceptor antagonists (6). Whereas cART has been extraordinarily successful for long-term management of HIV-1 infection, no existing drugs clear latent viral reservoirs. Thus, life-long drug administration is required.
Because HIV-1 is an integrating lentivirus, identification of curative strategies for HIV-1 infection has proven to be an even more difficult challenge than developing drugs to control infection. Clinical trials of HIV-1 vaccine candidates have been largely unsuccessful, with just one vaccine producing partial efficacy to date (7). In addition to vaccines, substantial research effort has addressed therapeutic cures for HIV-1 infection. One well-known cure strategy is based on the concept of “shock and kill,” in which latency reversal agents (LRAs) are used to reactivate the integrated provirus in latent reservoirs in the presence of antiretroviral drugs. Induction of viral protein expression may then promote immune recognition and clearance of latently infected cells (8). Recent examples of LRAs that induce latency reversal in animal models in the presence of antiretroviral drugs include engineered agonists for the interleukin-15 receptor (9) and a small molecule activator of the noncanonical NF-κB pathway (10), although neither treatment alone resulted in clearance of viral reservoirs.
The observations that LRAs can induce viral gene expression in the presence of antiretroviral drugs, but fail to prevent viral rebound following antiretroviral drug withdrawal, indicate that additional modalities are required to enhance the host immune response to the reactivated HIV-1 producer cell population. Inhibition of the HIV-1 Nef accessory protein, which plays a key role in immune escape of HIV-infected cells, may provide the additional immune system boost required for reduction of the latent reservoir.
HIV-1 Nef and viral pathogenesis
HIV-1 Nef is a relatively small (27–34 kDa, depending on the lentivirus), nonenzymatic accessory protein expressed early and throughout the viral life cycle. Nef was initially named on the basis of early evidence that it was a “negative factor” for HIV-1 replication (11). On the contrary, Nef plays prominent roles in HIV-1 pathogenesis by promoting viral replication and spread in vivo and by enabling immune escape of HIV-infected cells. Patients infected with HIV-1 harboring mutant or defective nef genes fail to progress to AIDS (12, 13). Similarly, rhesus macaques infected with SIV that does not express Nef develop low viral loads and fail to progress to simian AIDS (14). Nef-defective HIV-1 replicates poorly in humanized mice and does not induce significant CD4 T cell loss (15, 16). Finally, and remarkably, CD4 promoter–directed expression of HIV-1 Nef alone induces an AIDS-like syndrome in transgenic mice, suggesting a direct role for Nef in HIV-1 pathogenesis (17, 18).
Nef is comprised of two main parts: the N-terminal anchor region and a folded core (Fig. 1). The N-terminal 60 amino acids that comprise the anchor region are largely unstructured and bear a signal for myristoylation at the N-terminal glycine. NMR studies identified an amphipathic helix within the N-terminal anchor region (20), which together with the N-myristoyl group serves to localize Nef to host cell membranes (21). Following the anchor region is a folded core domain (∼105 residues) with a large flexible internal loop (∼25 residues). Because the anchor region is flexible, the folded core can move off the membrane (22, 23) to accommodate interactions with diverse host cell proteins, many of which are also associated with the membrane (Fig. 1). Nef does not exhibit any known enzymatic or biochemical activities, functioning instead through a diverse array of protein-protein interactions (24). Examples of the best-characterized Nef functions and attendant binding partners are summarized below. These include endocytic trafficking molecules followed by protein kinases, which are the primary focus of this review.
Figure 1.

Structural model of HIV-1 Nef and major classes of membrane-associated host cell partners. The overall structure of Nef is modeled in the center with major structural features indicated. The myristoylated (Myr) N-terminal anchor domain, together with a patch of basic amino acids from the amphipathic helix, tether Nef to the plasma membrane. The membrane anchor connects to the folded core, which encompasses multiple motifs essential for host cell protein recruitment. Highlighted is the conserved PxxPxR motif, which is essential for both SH3 domain interaction and binding to the AP-1 endocytic adaptor. Also shown is the flexible internal loop, which engages AP-2. Major classes of membrane-associated partner proteins include nonreceptor tyrosine kinases of the Src and Tec families (left), which Nef activates to enhance the viral life cycle. Nef also interacts with the endocytic adaptor proteins AP-1 and AP-2 to prevent cell-surface expression of multiple proteins, including MHC-I, CD4, and SERINC5, to promote viral infectivity and immune escape. This model of full-length Nef at the membrane is adapted from Geyer and Peterlin (19).
Nef alters membrane protein trafficking to facilitate HIV-1 infectivity and immune escape
Nef is remarkably well-adapted to hijack the intracellular trafficking machinery to modulate cell-surface protein expression. Two well-characterized Nef functions in this regard are down-regulation of CD4 and major histocompatibility complex I (MHC-I) proteins, which are orchestrated by Nef via the endocytic adaptor protein complexes, AP-1 and AP-2. Nef also drives down-regulation of the SERINC family of host cell restriction factors via AP-2 to enhance viral infectivity. Structural features and other aspects of these interactions are covered in several recent reviews (25, 26) and are not discussed in detail here.
CD4 down-regulation
Down-regulation of CD4 by Nef is conserved across virtually all HIV-1 subtypes (27). Whereas this function may seem counterintuitive at first, given the essential role of CD4 in viral entry, it is in full alignment with the role of Nef in cell survival and immune escape. Persistence of CD4 on the surface of infected cells may lead to cytotoxic superinfection, whereas interaction between CD4 and Env has been shown to trigger antibody-dependent cell-mediated cytotoxicity (28, 29). CD4 down-regulation is normally regulated through phosphorylation-dependent engagement of the cytosolic tail of CD4 by AP-2, which initiates clathrin-mediated endocytosis of CD4 into the endolysosomal pathway (30). Nef accelerates this endocytic process by engaging both the cytosolic tail of CD4 and the α and σ2 subunits of AP-2 (31–33). Notably, CD4 down-regulation is also triggered by the HIV-1 accessory protein Vpu. However, Vpu is expressed later in the viral life cycle and targets CD4 through a distinct ubiquitination-dependent process (34).
HIV-1 infectivity
Early work demonstrated that disruption of Nef expression impairs HIV-1 infectivity (35), implying a positive contribution of Nef to the infectivity of viral particles. Subsequent work established that infectivity enhancement requires Nef myristoylation, AP-2 association, and clathrin-mediated endocytosis (36, 37). Despite these advances, one key mechanism behind Nef infectivity enhancement remained elusive until 2015, when a novel host cell restriction factor known as SERINC5 was identified (38, 39). SERINC5 is a multipass transmembrane protein present on the surface of HIV-1 producer cells that is incorporated into the membrane of newly synthesized virions (38–40). SERINC5 disrupts viral fusion with host cells and delivery of the viral core through an Env-dependent mechanism (40). Nef counters the SERINC5 host defense mechanism by down-regulating it from the cell membrane through an AP-2–dependent pathway, thereby preventing incorporation into budding virions (39). Following down-regulation by Nef, internalized SERINC5 is targeted for degradation via the endolysosomal pathway (41).
MHC-I down-regulation
Antigen presentation by MHC-I is another critical target for Nef-mediated down-regulation. Antigenic peptides derived from proteolytically digested viral proteins are loaded onto MHC-I molecules within the endoplasmic reticulum and presented on the surface of infected cells, triggering recognition and killing of the infected cell by CD8 cytotoxic T cells. In contrast to CD4 down-regulation, Nef-mediated MHC-I down-regulation occurs via AP-1.
Two temporally distinct models of Nef-mediated antagonism of MHC-I have been reported. In the first model, also known as the “signaling mode,” Nef is recruited by the phosphofurin acidic cluster 2 (PACS-2) adaptor protein to the trans-Golgi network (TGN), where it drives the activation of Src-family kinases specific to the host cell lineage (Hck in macrophages; Lyn in T cells). Src-family kinase activity initiates a signaling cascade that ultimately increases levels of membrane phosphatidylinositol (3,4,5)-trisphosphate via phosphoinositide 3-kinase, causing activation of the small GTPases Arf1 and Arf6 and endocytosis of cell-surface MHC-I (42, 43). Internalized MHC-I is trapped in vesicles in complex with Nef and AP-1 and prevented from recycling back to the plasma membrane. In the second model, known as the “stoichiometric mode,” Nef associates with AP-1 and Arf1 to trap newly synthesized MHC-I molecules within the TGN, thereby preventing anterograde trafficking toward the plasma membrane (44). A temporal sequence for these two mechanisms has been proposed (42), with the signaling mode occurring earlier in the infection cycle. Both models require the association of Nef with AP-1 and the cytoplasmic tail of MHC-I, for which a crystal structure has been reported (45). Additional details of the endosomal trafficking pathways controlling MHC-I down-regulation by Nef are described in several other reviews (26, 46).
Nef and Src-family kinases
Recruitment and activation of Src-family kinases by Nef has been the focus of many structural, cellular, and in vitro studies. Early work demonstrated that of the eight mammalian Src-family members, Nef preferentially binds to Hck and Lyn via their SH3 domains (47, 48). Nef binding displaces the SH3 domain from its regulatory position on the back of the Hck kinase domain, resulting in constitutive kinase activation both in vitro and in cell-based systems (49–51). Nef-dependent Hck activation is a conserved function of all HIV-1 Nef M-group subtypes (52). Expression of a dominant-negative Hck mutant as well as knockdown of Hck expression both compromise HIV-1 transcription and viral replication in macrophages, a dominant site of Hck expression (53, 54). Compared with Hck, Lyn, and to some extent Src, interaction of other Src-family members with Nef is less definitive. One example is Lck, a critical component of the T cell receptor activation pathway (55). Whereas expression of Nef in T cell lines results in intracellular relocalization of Lck, it is less clear whether Nef binds directly to Lck within cells (56). When Nef and Lck are ectopically expressed in defined cellular systems, no change in Lck activity is observed, suggesting that interaction in T cell systems may be indirect (57–59).
A conserved PxxPxR motif is essential for SH3 domain binding and Src-family kinase activation by Nef
HIV-1 Nef contains a highly conserved PxxPxR motif, which forms a polyproline type II (PPII) helix that serves as the docking site for recruitment of Src-family kinases and other proteins with SH3 domains (60). This motif is required for activation of Hck and other Src-family members, as mutation of the core prolines in the PPII helix prevents Nef interaction and kinase activation (49, 50). Disruption of the PxxPxR motif also impacts Nef down-regulation of MHC-I (61) as well as enhancement of viral replication and pathogenicity in murine and primate models of AIDS (62, 63). However, down-regulation of CD4 by Nef is not affected by mutation of the PxxPxR motif (61), arguing against a role for Src-family kinase signaling or other SH3-binding proteins in this Nef function.
Structural basis of Src-family kinase activation by Nef
The first X-ray crystal structure of Nef (all structures described in this review are listed in Table 1) was reported in complex with the SH3 domain from the Src-family kinase, Fyn (64). The overall structure forms a 2:2 dimer of Nef·SH3 complexes, with Nef forming the dimer interface (Fig. 2A). This structure revealed that the PPII helix formed by the Nef PxxPxR motif engages hydrophobic grooves on the SH3 domain surface, with the arginine of the motif making an electrostatic contact with an aspartate residue in the SH3 domain RT loop (Fig. 2B).
Table 1.
X-ray crystal structures and NMR studies referenced in this review
| Protein or complex | PDB code | Method | Reference |
|---|---|---|---|
| Nef anchor region | 1QA4, 1QA5 | NMR | 20 |
| Nef·Fyn SH3 (R96I) | 1EFN | X-ray | 64 |
| Nef·Fyn SH3 | 1AVZ | X-ray | 65 |
| Nef | 2NEF | NMR | 66 |
| Nef·Hck SH3-SH2 | 4U5W | X-ray | 67 |
| Hck, near-full-length | 1QCF | X-ray | 68 |
| Nef·MHC-I·AP-1 µ1 | 4EN2 | X-ray | 45 |
| Nef·AP-2 α/σ2 | 4NEE | X-ray | 69 |
| Nef·CD4·AP-2 | 6URI | X-ray | 33 |
Figure 2.

X-ray crystal structures of HIV-1 Nef in complex with the SH3 domain of the Src-family kinase, Fyn. A, overall structure of the Nef core in complex with the Fyn SH3 R96I mutant. Nef·SH3 complexes form a 2:2 dimer, with Nef forming the dimer interface. The Nef monomers are modeled in blue (NefA) and green (NefB), respectively, with the SH3 domains in red (SH3A) and pink (SH3B). B, SH3 domain surface residues Tyr-91, Trp-119, and Tyr-137 form hydrophobic grooves that contact the Nef PxxPxR motif (orange). This interaction is oriented and stabilized by a polar contact between SH3 Asp-100 and Arg-77 from the Nef PxxPxR motif. C, high-affinity Nef·SH3 interaction requires Ile-96 from the SH3 RT loop (red), which accesses a hydrophobic pocket created by Nef residues Phe-90, Trp-113, and Tyr-120 (cyan). Structural details of the Nef homodimer interface from this complex are shown in Fig. 5. Models were produced with PyMOL using the crystal coordinates of the HIV-1 Nef core in complex with the Fyn R96I mutant SH3 domain (PDB code 1EFN).
Nef displays at least 10-fold higher affinity for the Hck SH3 domain relative to the Fyn SH3, a difference attributable to a single residue within the RT loop of the SH3 domain (48). The Hck SH3 domain has an isoleucine residue at position 963 within the RT loop, which engages a hydrophobic pocket in the folded core of Nef (70). The Fyn SH3 domain has an arginine at position 96, which is suboptimal for interaction with this Nef pocket, as revealed in a subsequent crystal structure (65) (PDB code 1AVZ). Replacement of Arg-96 in the Fyn SH3 domain with isoleucine restores higher affinity Nef binding, illustrating the importance of this residue (48). In fact, this modified Fyn SH3 domain (R96I) was used for the original crystal structure with Nef (PDB code 1EFN) to promote interaction with the hydrophobic pocket (Fig. 2C). Among the eight Src-family members, only Hck and Lyn have isoleucine at this SH3 position, thus explaining their unique sensitivity to activation following Nef engagement (59).
Other structures of SH3-bound HIV-1 Nef have used an artificially engineered SH3 domain in which the RT loop sequence was altered to enhance Nef binding. Replacement with five alternative RT loop residues increased Hck SH3 affinity for Nef by nearly 40-fold (71, 72). This optimized SH3 domain was subsequently combined with a single-chain anti-Nef antibody to create protein-based inhibitors of multiple Nef functions (73, 74). Introduction of the optimized RT loop sequence into full-length Hck enhanced interaction with Nef in vitro and induced allosteric changes in the Hck active site following Nef binding (75).
A more recent structure of the HIV-1 Nef core with the SH3-SH2 regulatory region of Hck (PDB code 4U5W) revealed additional contacts that may contribute to kinase activation (67). This newer structure also reveals a 2:2 dimer of Nef·SH3-SH2 complexes (Fig. 3). The Nef PxxPxR motif binds the SH3 surface in a manner identical to earlier structures, including interaction of the SH3 domain RT loop isoleucine residue with the Nef hydrophobic pocket. Unique to the Nef·SH3-SH2 structure, however, are reciprocal contacts between Glu-94 from each SH3 RT loop and Nef Arg-105 from the opposite complex, reorienting Nef Asp-123 to the surface. Overall, interaction of Nef with SH3-SH2 results in a much more compact structure compared with earlier Nef·SH3 complexes. Substitution of SH3 Glu-94 in the context of full-length Hck interferes with Nef interaction and kinase activation in cell-based assays, supporting a functional role for this intercomplex contact in kinase activation by Nef (67).
Figure 3.

X-ray crystal structure of the HIV-1 Nef core in complex with the Hck SH3-SH2 regulatory region. The overall structure is shown on the left, which crystallized as a dimer of Nef·SH3-SH2 complexes. The surfaces of the Nef core monomers are rendered in blue (NefA) and green (NefB), respectively. The SH3-SH2 subunits associated with each Nef monomer are shown as ribbons, with SH3-SH2B in the foreground (SH2 in blue, SH3 in red). The SH3-SH2A subunit is in the background (SH2 in light blue, SH3 in pink) with the second SH3 domain partially hidden. A close-up view of one Nef interface is shown on the right, with the Nef PxxPxR motif (orange) contacting the SH3 surface via Pro-72 and Pro-75, with Arg-77 making an ionic contact with SH3 RT loop Asp-100. Unique to this complex is a second polar contact between SH3 RT loop Glu-94 and Arg-105 from the opposing Nef monomer. Details of the unique Nef homodimer interface found in this structure are highlighted in Fig. 6. Modeling was performed with PyMOL using the crystal coordinates of the HIV-1 Nef core in complex with the Hck SH3-SH2 domain (PDB code 4U5W).
Whereas the structural basis of Nef interaction with the Src-family kinase regulatory domains is well-known, no structures of Nef bound to full-length Hck or other Src-family members have been reported. However, the use of hydrogen-deuterium exchange MS (HX MS) has provided important clues to the overall structure of the active Nef·Hck complex. HX MS is based on the principle that exposure of a folded protein to D2O-based solvent results in exchange of labile hydrogen atoms for deuterium (76). Because deuteration causes the protein to gain mass (+1 Da per exchange event), the rate of hydrogen exchange can be monitored by MS (77). In folded proteins, hydrogens in unstructured or dynamic regions exposed to solvent undergo rapid deuteration, whereas buried hydrophobic regions or secondary structural elements stabilized by hydrogen bonding display slower exchange kinetics. Spatial resolution of deuterium incorporation at the peptide level is achieved by pepsin digestion after quenching the exchange reaction prior to chromatography and MS.
HX MS is particularly powerful when used to compare two states of a given protein (e.g. Hck in the presence and absence of Nef). Wales et al. (78) performed this comparison using recombinant near-full-length Hck (SH3-SH2-kinase-tail) and full-length Nef. The crystal structure of the identical near-full-length Hck protein has been reported (79) and shows that it adopts an assembled, inactive conformation driven by two primary intramolecular interactions: the SH3 domain binds to the SH2-kinase linker (which adopts a PPII helical conformation), whereas the SH2 domain engages the tyrosine-phosphorylated tail. Disruption of either interaction by mutagenesis leads to activation of Hck in cells (80), raising the question of whether Nef selectively disrupts SH3-mediated regulation of Hck or causes a more global disruption of the down-regulated state. Comparison HX MS experiments (78) of Hck alone (where the SH2-kinase linker is bound to the SH3 domain as in down-regulated Hck) versus Hck in complex with Nef (where the Nef PxxPxR motif is bound to the SH3 domain as required for Hck activation) demonstrated remarkably few changes to the rest of Hck upon Nef interaction. Very subtle changes in deuterium uptake by Hck were found in only a single peptic peptide that encompasses the C-terminal end of the SH2-kinase linker where it joins the N-lobe of the kinase domain. No changes in hydrogen exchange occurred in the SH2 domain or C-terminal tail, indicating that this regulatory interaction remains intact in the active Hck·Nef complex. This result indicates that Nef binding induces subtle perturbations in the conformation of Hck to induce its activation and is consistent with other work showing that short peptides or small molecules that disrupt the SH3-linker interface also stimulate Hck activity in vitro (81, 82).
In addition to Src-family kinase activation, the Nef PxxPxR motif also makes a structural contribution to MHC-I down-regulation. Selective activation of Hck in macrophages or Lyn in T cells is an essential step for the signaling mode of MHC-I down-regulation by Nef described above. In addition, an X-ray crystal structure of Nef in complex with the cytoplasmic tail of MHC-I and the μ1 subunit of AP-1 illuminates an SH3-independent role for the Nef PxxP motif (PDB code 4EN2) (45). In this complex, the Nef PxxPxR motif interacts directly with AP-1 μ1, helping to clamp the MHC-I tail into a binding groove within the heterocomplex. Thus, the Nef PxxPxR motif may provide a mechanism not only for Src-family kinase engagement and activation necessary for MHC-I down-regulation, but also for direct interaction with AP-1.
Nef and Tec-family kinases
A growing body of evidence has linked HIV-1 infection and Nef to activation of select members of the Tec kinase family that are expressed in HIV host cells. Tec-family kinases play essential roles in antigen receptor signaling in both T and B cells, as well as in lymphocyte development (83, 84). Readinger et al. (85) first reported a connection between the HIV-1 life cycle and Itk, a Tec-family member expressed in CD4 T cells that is essential for T cell receptor signaling. Using Itk-directed siRNA knockdown, a dominant-negative Itk mutant, and pharmacological Itk inhibitors, they demonstrated that loss of Itk activity reduced p24 capsid levels and virus spread in cell culture. Itk activity was also required for efficient transcription from the integrated provirus, an effect that was enhanced by overexpression of Itk. Whereas this study clearly demonstrated a role for Itk activity in the viral life cycle, it did not address how HIV-1 infection influenced Itk activity.
Subsequent work established that HIV-1 Nef selectively interacts with Tec-family kinases and that Nef-mediated kinase activation is essential for viral replication (86). Nef interaction with Tec-family members was explored using a cell-based bimolecular fluorescence complementation assay (BiFC), a technique widely used to assess protein-protein interactions in cells (87). In this approach, Nef and the presumptive partner kinase are fused to nonfluorescent N- and C-terminal fragments of Venus, a bright variant of YFP (88, 89). When co-expressed in cells, Nef-partner protein interaction juxtaposes the YFP fragments, resulting in complementation of the YFP fluorophore and a bright fluorescent signal that also reports subcellular localization.
Using BiFC, Tarafdar et al. (86) showed that Nef interacts with the Tec-family members Itk, Btk, and Bmx, but not Tec or Txk, with interaction localized almost exclusively to the plasma membrane. Variants of Nef representative of all major HIV-1 subtypes interacted strongly with Itk, supporting the conserved nature of this interaction. BiFC experiments with truncated forms of Itk and Btk, consisting only of the N-terminal PH domain and adjacent SH3 domain, retained interaction with Nef as well, suggestive of an SH3 domain–dependent interaction like Hck. However, unlike Hck, recombinant SH3 domains of Btk and Itk failed to interact with Nef by surface plasmon resonance in vitro,4 suggesting a more complex mechanism of interaction. A potent Itk inhibitor blocked WT but not Nef-defective HIV-1 infectivity and replication, supporting a role for Nef-dependent Itk activation in the viral life cycle (86).
More recent studies combined BiFC with anti-phosphotyrosine immunofluorescence microscopy to demonstrate that Nef recruits both Itk and Btk to the cell membrane, resulting in sustained kinase autophosphorylation (90). A myristoylation-defective Nef mutant failed to recruit the kinases to the membrane, providing evidence that Nef also induces kinase relocalization. Mutants of Nef defective for homodimer formation failed to induce Itk and Btk activation while retaining kinase interaction by BiFC, suggesting that Nef homodimers may recruit two kinase molecules to promote kinase activation via trans autophosphorylation (Fig. 4A). Importantly, HIV-1 infection increased endogenous Itk activity in T cell lines and donor-derived peripheral blood mononuclear cells (PBMCs), whereas HIV-1 expressing Nef dimerization-defective mutants was significantly attenuated for both Itk activation and viral replication.
Figure 4.

Nef-mediated activation of Itk requires Nef homodimers. A, Nef recruits Itk to the cytoplasmic face of the plasma membrane and induces 2:2 Nef·Itk complex formation. Regulatory domain displacement and kinase domain juxtaposition induces constitutive kinase activation through trans-autophosphorylation. Dimerization-defective Nef mutants still interact with Itk at the membrane but form inactive 1:1 complexes. 3, Src homology 3 domain; 2, Src homology 2 domain; K, kinase domain; P, activation loop phosphorylation. B, activation of the T-cell receptor complex normally requires antigen-loaded MHC molecules from an antigen-presenting cell. The MHC-bound receptor then activates the Src-family kinase Lck, which is associated with the cytosolic tail of CD4. Lck phosphorylates and activates Itk, which in turn activates phospholipase Cγ (PLCγ) by direct phosphorylation. Phospholipase Cγ generates diacylglycerol (DAG) and inositol triphosphate (IP3) via hydrolysis of membrane phosphatidylinositol 4,5-bisphosphate (PIP2), leading to activation of protein kinase Cθ (PKCθ) and the calcium-dependent protein-serine/threonine phosphatase, calcineurin (Cal). Protein kinase Cθ promotes activation of NF-κB via the CARMA1/BCL10/MALT1 complex (not shown), whereas calcineurin dephosphorylates NFAT to drive nuclear localization. The NF-κB and NFAT transcription factors both enhance transcription of the integrated HIV-1 provirus early in the viral life cycle (91). Direct activation of Itk by Nef at the membrane downstream of the TCR may promote viral transcription through this pathway (see “Nef and Tec-family kinases”). LTR, long terminal repeat.
Using the same approach, Li et al. (90) also established that SIV Nef (mac239 allele) interacted strongly with Itk and Btk at the plasma membrane and induced constitutive kinase autophosphorylation. This observation is significant because SIV Nef does not bind to the SH3 domain of Hck (92), providing further evidence that the mechanism of Tec-family kinase interaction with Nef may involve contacts in addition to the SH3 domain (86). Clarification of the mechanism awaits determination of a high-resolution crystal structure of Nef in complex with a Tec-family member or its regulatory region. Activation of Itk and Btk by Nef at the membrane in HIV-infected cells may circumvent immune receptor control of Tec-family kinase activity to enhance HIV-1 replication. For example, T cell receptor activation normally triggers activation of multiple nonreceptor tyrosine kinases, including Lck and ZAP-70 in addition to Itk (55). The presence of Nef in HIV-infected cells directly activates Itk, short-circuiting this normal control mechanism to promote viral transcription downstream. A model illustrating the possible relationship of Nef-mediated Itk activation to transcriptional activation of the integrated HIV-1 LTR is shown in Fig. 4B.
Structural basis and functional relevance of Nef homodimers
Early characterization of recombinant, purified Nef proteins revealed the presence of homodimers and higher-order oligomers in solution (93, 94). NMR spectroscopy demonstrated that Nef dimerization occurred between well-folded monomers, arguing against artifactual interaction of denatured protein subunits (93). Additional biophysical and biochemical analyses of recombinant Nef confirmed a reversible and concentration-dependent equilibrium between monomeric and dimeric forms of Nef in solution (66, 95), providing the first evidence that Nef has a natural tendency to oligomerize.
Crystal structures of HIV-1 Nef in complexes with Src-family kinase SH3 domains were the first to capture Nef in a dimeric state (64, 65). Within these structures, dimerization is mediated primarily by the αB helix of each Nef monomer with the dimer interface formed by a cluster of hydrophobic residues including Leu-112, Tyr-115, and Phe-121 (Fig. 5). The hydrophobic interface is flanked by reciprocal electrostatic interactions between Arg-105 and Asp-123, which appear to stabilize the dimer interface in this complex. Thus, the resulting structure is a dimer of Nef·SH3 complexes in which Nef is solely responsible for the intercomplex contacts. Nef residues that contribute to homodimer formation are highly conserved across HIV-1 M-group subtypes (52), with many conserved or homologous amino acids in the corresponding positions of Nef from HIV-2 and SIV Nef (95).
Figure 5.

Nef homodimer interface from the X-ray crystal structure in complex with a Src-family kinase SH3 domain. Overview of the Nef dimer structure is shown in the top left, with the Nef monomers rendered in blue and green, respectively. The αB helices that form the dimer interface are highlighted (SH3 domains not shown for clarity). The αB helices are enlarged in the bottom left, illustrating the side chains of Leu-112, Tyr-115, and Phe-121 from each monomer, which form the hydrophobic core of the interface. Also shown are the reciprocal ionic contacts between Arg-105 and Asp-123. Both models are rotated 90° on the right. These models were produced with PyMOL using the crystal coordinates of the HIV-1 Nef core in complex with the Fyn SH3 domain R96I mutant (PDB code 1EFN). A model of the overall Nef·SH3 crystal structure is shown in Fig. 2A.
Subsequent studies validated the dimer seen in the Nef·SH3 crystal structures in terms of residues involved in Nef homodimer formation. Using the BiFC approach described above, Poe et al. (96) demonstrated that Nef forms homodimers in cells that localize to the plasma membrane and the TGN, two subcellular compartments essential for function. Mutagenesis of the hydrophobic core of the Nef dimer interface as well as Asp-123 and Arg-105 all reduced the BiFC signal for interaction, supporting a role for these residues in Nef homodimerization. Each mutation substantially attenuated Nef-dependent receptor down-regulation and viral replication, implying an important functional role for Nef dimers (96). In contrast, mutagenesis of the Nef myristoylation signal or the PxxPxR motif did not affect fluorescence complementation, indicating that membrane localization or SH3 domain engagement are not directly required for Nef dimerization in cells.
A more recent X-ray crystal structure (PDB code 4U5W) of HIV-1 Nef in complex with the complete regulatory region of Hck (tandem SH3-SH2 domains) demonstrates the plasticity of the Nef dimerization interface (67). Whereas the Nef αB helices still contribute to the dimer interface in the 4U5W structure, the orientation of the two Nef monomers is substantially altered relative to the Nef·SH3 complexes seen in 1EFN, and the orientation is stabilized by a much more extensive network of Nef·Nef contacts. Three interfaces stabilize the Nef homodimer observed in the crystal complex with the tandem Hck SH3-SH2 domains, which are modeled in Fig. 6. These remarkable changes in the overall homodimer orientation relative to the Nef·SH3 complex occur without changes to the overall structure of the Nef core.
Figure 6.

Homodimer interface from the X-ray crystal structure of Nef in complex with the Hck SH3-SH2 regulatory region. Three views of this Nef homodimer are shown on the left with the Nef monomers rendered in blue and green, respectively (SH3-SH2 proteins not shown for clarity). Three interfaces stabilize this homodimer, which are enlarged on the right. In the top view, the side chains of Leu-112, Tyr-115, Phe-121, and Pro-122 form a hydrophobic cup that interacts with Val-70 from the opposing monomer in reciprocal fashion. The side view shows the contributions of Ile-109, Leu-112, Trp-113, and His-116 to a hydrophobic interface between the αB helices. The bottom view shows reciprocal polar contacts formed by Ser-103 and Arg-106 with the main-chain carbonyls of Gly-95 and Gly-96; Leu-100 also makes a nonpolar contact in this interface. These models were produced with PyMOL using the crystal coordinates of the HIV-1 Nef core in complex with the Hck SH3-SH2 region (PDB code 4U5W). A model of the overall Nef·SH3-SH2 crystal structure is shown in Fig. 3A.
Reorientation of the Nef homodimer in the Hck SH3-SH2 complex results in movement of Nef Asp-123, which is buried in the Nef·SH3 structures, to a solvent-exposed position. The structure and position of the Nef loop displaying Asp-123 in the Nef·SH3-SH2 complex is nearly identical to the one observed in the crystal structure of the Nef·MHC-I·AP-1 complex (PDB code 4EN2), where it coordinates the association of Nef, the cytoplasmic tail of MHC-I, and the µ1 subunit of AP-1 (45, 67, 97). Mutagenesis of Asp-123 prevents Nef-mediated down-regulation of MHC-I, illustrating the functional importance of this residue to immune escape (45, 98). Whereas Nef crystallized as a monomer in the complex with MHC-I and AP-1 µ1, the possibility exists that interaction with Hck may promote the Nef structure required for MHC-I·AP-1 complex assembly as illustrated in Fig. 7. This possibility is consistent with the requirement for Src-family kinases in the early steps of MHC-I down-regulation as described above (42, 43).
Figure 7.
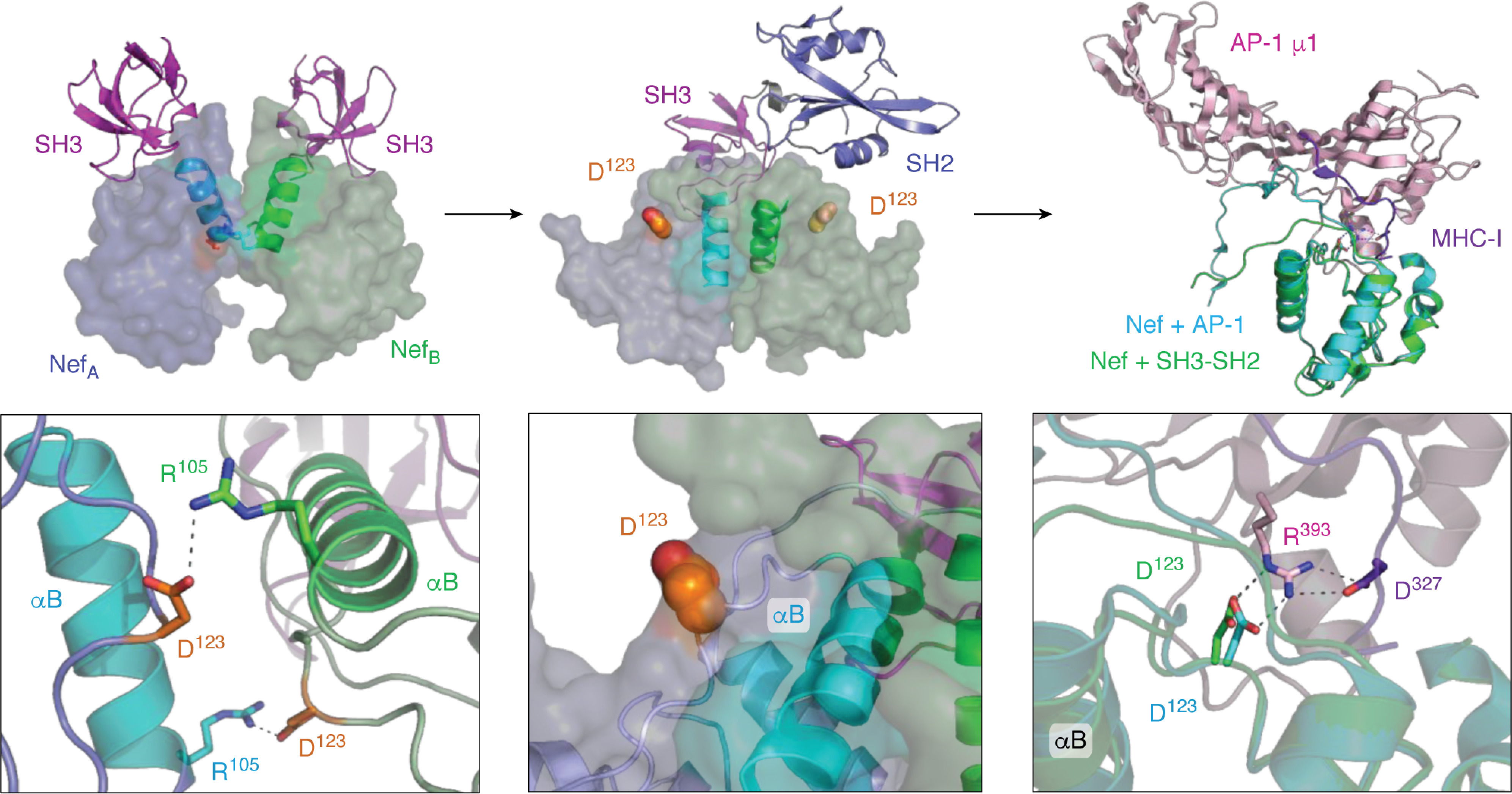
Interaction of HIV-1 Nef with the Src-family kinase Hck may induce conformational changes consistent with MHC-I/AP-1 recruitment. Left, crystal structure of the HIV-1 Nef core in complex with the SH3 domain of Fyn (high-affinity R96I mutant; PDB code 1EFN). Nef forms a dimer of Nef·SH3 complexes in this structure (NefA (blue) and NefB (green)). The dimer interface is formed by the Nef αB helices (highlighted helices). The SH3 domains are shown at the top in pink. The helical dimer interface is enlarged in the bottom panel and highlights the reciprocal polar contacts between Nef Asp-123 and Arg-105, which are buried in the core of this structure. The center, top panel shows the crystal structure of the Nef core in complex with the Hck SH3-SH2 region (PDB code 4U5W). The color scheme is as per the left panel with a single SH3-SH2 unit shown for clarity. Nef also crystallizes as a 2:2 complex of Nef·SH3-SH2 dimers in this structure, but the helical interface is completely reoriented relative to the Nef·SH3 complex such that the Nef Asp-123 side chain (orange) is now pointed toward the solvent. One of the Asp-123 residues is enlarged in the bottom panel. Right, structural alignment of one of the Nef core proteins (green ribbon) in the Nef·SH3-SH2 complex (PDB code 4U5W) with the Nef core (cyan ribbon) in complex with the AP-1/μ1 subunit (pink) and MHC-I tail peptide (purple) from PDB code 4EN2. The Nef core proteins from each complex adopt very similar conformations, with nearly identical positioning of Nef Asp-123 (enlarged in the bottom panel). Nef Asp-123 forms part of an ionic bond network, with Arg-393 from AP-1 and Asp-327 from MHC-I, that is required for Nef-mediated down-regulation of MHC-I and immune escape.
Multiple lines of evidence support an essential role for Nef homodimer formation in nonreceptor tyrosine kinase activation. In an early study, Nef was fused to the estrogen receptor hormone-binding domain (Nef-ER fusion protein) to enable chemical control of Nef dimerization with 4-hydroxytamoxifen (4-HT) in cells (99). When Nef-ER and Hck were expressed together in rodent fibroblasts as a model system, 4-HT treatment induced Nef-ER dimer formation, Hck activation, and oncogenic transformation. Fibroblasts expressing Nef-ER and Hck in the presence of 4-HT produced a markedly enhanced transforming response relative to cells co-expressing WT Nef and Hck, indicating that enforced oligomerization may augment Hck activation by Nef. In complementary experiments, a Nef mutant defective for Hck SH3 domain binding (PxxPxR to AxxAxR mutant) suppressed signaling from the WT Nef·Hck complex, suggestive of a dominant-negative effect. Considering the Nef·SH3-SH2 complex structures discussed above, the NefAxxAxR mutant may form a mixed homodimer in cells (i.e. NefPxxPxR·NefAxxAxR) to which only one Hck molecule can bind, producing an inactive, dead-end complex. These studies provided the first clue that Hck SH3 domain displacement from the SH2-kinase linker may not be sufficient to fully activate the kinase. Rather, juxtaposition of two Hck molecules may be required, such that autophosphorylation can proceed via a trans mechanism.
Mutants of HIV-1 Nef that are defective for homodimer formation have been reanalyzed in recent work (90). These mutants involve three hydrophobic residues that contribute to the Nef dimer interface in the crystal complexes with both the SH3 domain alone and with the dual SH3-SH2 domain of Hck (Nef Leu-112, Tyr-115, and Phe-121; Figs. 5 and 6). Recombinant full-length Nef proteins with these mutations are predominantly (Y115D) or exclusively (L112D and F121A) monomers by analytical size exclusion chromatography and show reduced propensity to form homodimers in the cell-based BiFC assay (90). The observation that these mutations do not completely abolish Nef homodimer formation in the BiFC assay likely relates to enhanced local concentrations of Nef at the cell membrane plus the fact that the YFP fluorophore is irreversibly reconstituted once formed (87). Nevertheless, each of these mutants showed greatly diminished capacity to activate the Tec-family kinases Itk and Btk at the cell membrane (90). Similar results were shown with Hck using the same cell-based assay (100), suggesting that activation of both kinase families requires a Nef dimer. Remarkably, one of these mutants, Nef-F121A, suppressed both Itk and Btk autophosphorylation below background levels observed in the absence of Nef while retaining interaction with each kinase (90), suggesting the formation of nonproductive Nef-kinase complexes. This observation provides further support for the trans-autophosphorylation mechanism of kinase activation modeled for Itk in Fig. 4A.
Crystal structures of Nef in complexes with isolated SH3 domains versus the Hck SH3-SH2 domain provide snapshots of very different homodimerization interfaces. This raises the important question of whether distinct dimer conformations are possible in solution and ultimately in cells. Moroco et al. (97) explored this issue using HX MS to compare the solution conformation of Nef alone and in complexes with the Hck SH3 and dual SH3-SH2 domain proteins previously used for crystallography. HX MS showed that the Nef αB-helix is protected from deuterium uptake in Nef complexes with both SH3 and SH3-SH2 compared with Nef alone, consistent with a role for the Nef αB helix in dimer formation. This result also shows that complex formation with Hck or other Src-family kinases stabilizes the Nef homodimer in solution. Subsequent comparative HX MS analysis of a Nef-D123N mutant showed protection of the αB helix when bound to SH3-SH2 but not when bound to SH3 alone. This result is consistent with the crystal structures, where Asp-123 contributes to Nef dimer formation when bound to SH3 alone but is surface-exposed when bound to SH3-SH2 (Fig. 7). These results support the idea that alternative dimeric states of Nef exist in solution and by extension in cells, despite a common fold of the structured core. Furthermore, Src-family kinase binding to Nef not only induces kinase activation but also triggers dynamic changes in Nef essential for recruitment of other host cell effectors.
Harnessing kinase activation by Nef for anti-retroviral drug discovery
Nef is a challenging target for traditional drug discovery campaigns based on high-throughput chemical library screening because it lacks intrinsic enzymatic or biochemical activity (101). Instead, Nef functions via interactions with diverse host cell proteins to enhance HIV-1 infectivity, replication, and immune escape as described above. Structural studies have established that host effector protein binding involves distinct Nef surfaces, complicating rational design of a universal Nef inhibitor capable of blocking its pleiotropic actions (33, 45, 67, 69). These issues have been circumvented in part through the development of a high-throughput screening assay for inhibitors of Nef-dependent activation of Hck (102, 103). In this approach, the recombinant Nef·Hck complex was used for the primary chemical library screen with Hck serving as a “reporter” for Nef using a kinase assay designed for high-throughput screening. Subsequent counterscreens of the hit compounds against Hck alone allowed for identification of Nef-dependent inhibitors, which were subsequently shown to exhibit antiretroviral activity.
Two classes of small molecule inhibitors were identified through this screening strategy. The first class, characterized by a 4-amino-diphenylfurano-pyrimidine (DFP) scaffold, was discovered in a small library of kinase inhibitor–biased compounds (Fig. 8) (103). Whereas these compounds work directly via the Hck active site, they showed enhanced potency for Hck inhibition in the presence of Nef, suggesting that Nef binding may allosterically impact the Hck active site to enhance inhibitor binding. This conclusion is supported by subsequent HX MS studies of the Nef·Hck complex in the presence of a DFP-based compound with antiretroviral activity (78). DFP-based Hck inhibitors also blocked Nef-mediated enhancement of viral replication across a wide range of Nef subtypes, providing additional evidence that Src-family kinase signaling is important to viral replication (52).
Figure 8.

Examples of small molecule inhibitors discovered using a Nef-coupled Hck kinase activity assay. Combining recombinant Nef and Hck proteins in vitro enabled high-throughput screening for inhibitors of the active complex (illustrated at the right; 3, SH3 domain; 2, SH2 domain; K, kinase domain). Screening of a small kinase inhibitor–biased library identified diphenylfuranopyrimidine 4-amino propanol (DFP-4AP), which inhibits the Nef-Hck complex via the Hck kinase domain (103). Screening of a large, more diverse library identified the compound B9 (102), which binds directly to Nef and inhibits Hck by an allosteric mechanism that may be related to Nef homodimer formation (left). Medicinal chemistry optimization has led to more potent analogs, such as FC-8052, which shares a hydroxypyrazole core with B9 (red). FC-8052 binds to recombinant Nef in vitro with a KD value of ∼10 pm, compared with about 80 nm for B9, and inhibits Nef-dependent HIV-1 replication in PBMCs in the subnanomolar range (100).
A subsequent screen of a large, diverse chemical library using this kinase-coupled approach identified a unique Nef inhibitor based on a hydroxypyrazole scaffold (102). This compound, a diphenyl hydroxypyrazolodiazene known as B9, bound directly to recombinant Nef by surface plasmon resonance (Fig. 8). B9 demonstrated inhibitory activity against multiple Nef functions, including enhancement of viral infectivity, replication, and MHC-I down-regulation (102, 104). Synthesis and characterization of more than 200 analogs of B9 have been reported without the potentially carcinogenic diazene functionality (100, 105). Several of these analogs bound to recombinant Nef with KD values in the nanomolar to picomolar range and inhibited Nef-mediated enhancement of HIV-1 replication in donor PBMCs with low nanomolar potency. Inhibitors in this class also blocked Nef-dependent activation of Itk and Hck, raising the possibility that they may influence Nef dimerization as a potential mechanism of action. Importantly, inhibitor treatment of cells expressing the kinases alone did not affect basal kinase activity, supporting a Nef-dependent mechanism of action. Ongoing challenges for Nef inhibitor development include structural analysis of their binding mode within Nef, which will clarify their mechanism of action and guide medicinal chemistry optimization for in vivo testing.
As described above, Nef prevents cell-surface display of MHC-I complexes with HIV-1 antigenic peptides on infected cells, allowing escape from cytotoxic T lymphocytes. In this way, Nef inhibits clearance of the virus and may contribute to establishment and maintenance of the persistent viral reservoir (106). Mujib et al. (104) showed that the direct acting Nef inhibitor B9, as well as several first-generation B9 analogs (105), restored MHC-I to the surface of HIV-infected CD4 T cells. Moreover, when Nef inhibitor–treated cells were co-cultured with autologous CD8 T cells expanded in the presence of HIV-1 antigenic peptides, the CD8 T cells became activated and killed the infected CD4 target cells. These findings raise the exciting possibility that Nef inhibitors may enhance CTL-mediated responses to help clear latent viral reservoirs. Newer analogs of B9 with enhanced affinity for Nef also retain the ability to reverse Nef-dependent down-regulation of MHC-I (100).
Summary and prospects
The Nef proteins of HIV-1 and other primate lentiviruses have been the subject of intense research for more than 30 years. Nef interacts with a diverse array of host cell proteins to benefit the virus, only a small subset of which are discussed here. This review focused primarily on structural aspects of Nef-dependent protein-tyrosine kinase activation and the relationship of these interactions to the viral life cycle, intracellular trafficking pathways, and immune escape.
Recurrent themes in the biology of Nef relate to its dynamic nature, diverse signaling partners, and ability to form alternative homodimer conformations and possibly larger oligomeric complexes. These versatile properties allow Nef to assemble signaling complexes that bypass normal cellular control mechanisms. This concept is particularly clear for Nef-mediated activation of nonreceptor tyrosine kinases, which overrides normal kinase control mechanisms through recruitment and juxtaposition of kinase molecules in the membrane. In this way, Nef mimics well-known mechanisms of cytoplasmic kinase activation by receptor tyrosine kinases as well as antigen and cytokine receptors. Interestingly, mutants of Nef attenuated for homodimer formation not only fail to activate Src- and Tec-family kinases, but are also unable to down-regulate CD4 and MHC-I. Yet in crystal structures of Nef in complex with AP-1/MHC-I (PDB code 4EN2) as well as AP-2/CD4 (PDB code 6URI), Nef is present as a monomer. This observation suggests that dynamic monomer-dimer transitions may be important for some but not all Nef functions. In the case of MHC-I down-regulation, Nef-dependent Src-family kinase activation has been linked to this event. In addition, interaction with the Hck regulatory region induces a Nef conformation compatible with AP-1/MHC-I recruitment (Fig. 7), suggesting that Src-family kinase engagement may induce a dimer-monomer transition in the MHC-I down-regulation pathway. On the other hand, a recent structure of Nef in complex with AP-2 and the cytoplasmic tail of CD4 shows that residues involved in homodimer formation in complexes with Src kinases interact either directly with CD4 or internally with Nef (33). In the case of CD4, therefore, mutations that render Nef defective for homodimer formation may also prevent interaction with AP-2 and CD4 directly. Regardless of the mechanism, mutagenesis studies show that homodimerization is linked to most Nef functions, and pharmacological perturbation of Nef dimer formation may provide a new approach to antiretroviral therapy with the potential for viral clearance.
Funding and additional information—Work in the authors' laboratories has been supported by National Institutes of Health Grants AI057083, AI102724, and AI126617 (to T. E. S.) and GM086507, GM101135, and CA233978 (to J. R. E.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
HIV-1 Nef amino acid numbering is based on the crystal structure of the Nef core in complex with the R96I mutant of the Fyn SH3 domain (PDB code 1EFN). Amino acid numbering for Hck and other Src-family kinases is based on the crystal structure of Src (PDB code 2SRC).
K. Whitlatch and T. E. Smithgall, unpublished data.
- NRTI
- nucleoside reverse transcriptase inhibitor
- cART
- combination antiretroviral therapy
- LRA
- latency reversal agent
- SIV
- simian immunodeficiency virus
- MHC
- major histocompatibility complex
- TGN
- trans-Golgi network
- PPII
- polyproline type II
- SH2
- Src homology 2
- SH3
- Src homology 3
- HX
- hydrogen-deuterium exchange
- BiFC
- bimolecular fluorescence complementation
- PBMC
- peripheral blood mononuclear cell
- ER
- endoplasmic reticulum
- 4-HT
- 4-hydroxytamoxifen
- DFP
- 4-amino-diphenylfurano-pyrimidine
- PDB
- Protein Data Bank
- YFP
- yellow fluorescent protein.
References
- 1. Jaffe H. W. (2008) The early days of the HIV-AIDS epidemic in the U.S.A. Nat. Immunol. 9, 1201–1203 10.1038/ni1108-1201 [DOI] [PubMed] [Google Scholar]
- 2. Marwick C. (1987) AZT (zidovudine) just a step away from FDA approval for AIDS therapy. JAMA 257, 1281–1282 10.1001/jama.1987.03390100015002 [DOI] [PubMed] [Google Scholar]
- 3. Hammer S. M., Katzenstein D. A., Hughes M. D., Gundacker H., Schooley R. T., Haubrich R. H., Henry W. K., Lederman M. M., Phair J. P., Niu M., Hirsch M. S., and Merigan T. C. (1996) A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N. Engl. J. Med. 335, 1081–1090 10.1056/NEJM199610103351501 [DOI] [PubMed] [Google Scholar]
- 4. Vella S., Schwartlander B., Sow S. P., Eholie S. P., and Murphy R. L. (2012) The history of antiretroviral therapy and of its implementation in resource-limited areas of the world. AIDS 26, 1231–1241 10.1097/QAD.0b013e32835521a3 [DOI] [PubMed] [Google Scholar]
- 5. Sepkowitz K. A. (2001) AIDS–the first 20 years. N. Engl. J. Med. 344, 1764–1772 10.1056/NEJM200106073442306 [DOI] [PubMed] [Google Scholar]
- 6. Dionne B. (2019) Key principles of antiretroviral pharmacology. Infect. Dis. Clin. North Am. 33, 787–805 10.1016/j.idc.2019.05.006 [DOI] [PubMed] [Google Scholar]
- 7. Rerks-Ngarm S., Pitisuttithum P., Nitayaphan S., Kaewkungwal J., Chiu J., Paris R., Premsri N., Namwat C., de Souza M., Adams E., Benenson M., Gurunathan S., Tartaglia J., McNeil J. G., Francis D. P., et al. (2009) Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361, 2209–2220 10.1056/NEJMoa0908492 [DOI] [PubMed] [Google Scholar]
- 8. Kim Y., Anderson J. L., and Lewin S. R. (2018) Getting the “kill” into “shock and kill”: strategies to eliminate latent HIV. Cell Host Microbe 23, 14–26 10.1016/j.chom.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McBrien J. B., Mavigner M., Franchitti L., Smith S. A., White E., Tharp G. K., Walum H., Busman-Sahay K., Aguilera-Sandoval C. R., Thayer W. O., Spagnuolo R. A., Kovarova M., Wahl A., Cervasi B., Margolis D. M., et al. (2020) Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 578, 154–159 10.1038/s41586-020-1946-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nixon C. C., Mavigner M., Sampey G. C., Brooks A. D., Spagnuolo R. A., Irlbeck D. M., Mattingly C., Ho P. T., Schoof N., Cammon C. G., Tharp G. K., Kanke M., Wang Z., Cleary R. A., Upadhyay A. A., et al. (2020) Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 578, 160–165 10.1038/s41586-020-1951-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahmad N., and Venkatesan S. (1988) Nef protein of HIV-1 is a transcriptional repressor of HIV-1 LTR. Science 241, 1481–1485 10.1126/science.3262235 [DOI] [PubMed] [Google Scholar]
- 12. Rhodes D. I., Ashton L., Solomon A., Carr A., Cooper D., Kaldor J., and Deacon N. (2000) Characterization of three nef-defective human immunodeficiency virus type 1 strains associated with long-term nonprogression. Australian Long-Term Nonprogressor Study Group. J. Virol. 74, 10581–10588 10.1128/jvi.74.22.10581-10588.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirchhoff F., Greenough T. C., Brettler D. B., Sullivan J. L., and Desrosiers R. C. (1995) Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332, 228–232 10.1056/NEJM199501263320405 [DOI] [PubMed] [Google Scholar]
- 14. Kestler H. W. 3rd, Ringler D. J., Mori K., Panicali D. L., Sehgal P. K., Daniel M. D., and Desrosiers R. C. (1991) Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65, 651–662 10.1016/0092-8674(91)90097-I [DOI] [PubMed] [Google Scholar]
- 15. Watkins R. L., Foster J. L., and Garcia J. V. (2015) In vivo analysis of Nef's role in HIV-1 replication, systemic T cell activation and CD4+ T cell loss. Retrovirology 12, 61 10.1186/s12977-015-0187-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou W., Denton P. W., Watkins R. L., Krisko J. F., Nochi T., Foster J. L., and Garcia J. V. (2012) Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology 9, 44 10.1186/1742-4690-9-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanna Z., Kay D. G., Cool M., Jothy S., Rebai N., and Jolicoeur P. (1998) Transgenic mice expressing human immunodeficiency virus type 1 in immune cells develop a severe AIDS-like disease. J. Virol. 72, 121–132 10.1128/JVI.72.1.121-132.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hanna Z., Kay D. G., Rebai N., Guimond A., Jothy S., and Jolicoeur P. (1998) Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 95, 163–175 10.1016/S0092-8674(00)81748-1 [DOI] [PubMed] [Google Scholar]
- 19. Geyer M., and Peterlin B. M. (2001) Domain assembly, surface accessibility and sequence conservation in full length HIV-1 Nef. FEBS Lett. 496, 91–95 10.1016/S0014-5793(01)02394-8 [DOI] [PubMed] [Google Scholar]
- 20. Geyer M., Munte C. E., Schorr J., Kellner R., and Kalbitzer H. R. (1999) Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J. Mol. Biol. 289, 123–138 10.1006/jmbi.1999.2740 [DOI] [PubMed] [Google Scholar]
- 21. Gerlach H., Laumann V., Martens S., Becker C. F., Goody R. S., and Geyer M. (2010) HIV-1 Nef membrane association depends on charge, curvature, composition and sequence. Nat. Chem. Biol. 6, 46–53 10.1038/nchembio.268 [DOI] [PubMed] [Google Scholar]
- 22. Akgun B., Satija S., Nanda H., Pirrone G. F., Shi X., Engen J. R., and Kent M. S. (2013) Conformational transition of membrane-associated terminally acylated HIV-1 Nef. Structure 21, 1822–1833 10.1016/j.str.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kent M. S., Murton J. K., Sasaki D. Y., Satija S., Akgun B., Nanda H., Curtis J. E., Majewski J., Morgan C. R., and Engen J. R. (2010) Neutron reflectometry study of the conformation of HIV Nef bound to lipid membranes. Biophys. J. 99, 1940–1948 10.1016/j.bpj.2010.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jäger S., Cimermancic P., Gulbahce N., Johnson J. R., McGovern K. E., Clarke S. C., Shales M., Mercenne G., Pache L., Li K., Hernandez H., Jang G. M., Roth S. L., Akiva E., Marlett J., et al. (2011) Global landscape of HIV-human protein complexes. Nature 481, 365–370 10.1038/nature10719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buffalo C. Z., Iwamoto Y., Hurley J. H., and Ren X. (2019) How HIV Nef proteins hijack membrane traffic to promote infection. J. Virol. 93, e01322 10.1128/JVI.01322-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pereira E. A., and daSilva L. L. (2016) HIV-1 Nef: taking control of protein trafficking. Traffic 17, 976–996 10.1111/tra.12412 [DOI] [PubMed] [Google Scholar]
- 27. Mariani R., and Skowronski J. (1993) CD4 down-regulation by nef alleles isolated from human immunodeficiency virus type 1-infected individuals. Proc. Natl. Acad. Sci. U. S. A. 90, 5549–5553 10.1073/pnas.90.12.5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lama J. (2003) The physiological relevance of CD4 receptor down-modulation during HIV infection. Curr. HIV Res. 1, 167–184 10.2174/1570162033485276 [DOI] [PubMed] [Google Scholar]
- 29. Pham T. N., Lukhele S., Hajjar F., Routy J. P., and Cohen E. A. (2014) HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 11, 15 10.1186/1742-4690-11-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pitcher C., Höning S., Fingerhut A., Bowers K., and Marsh M. (1999) Cluster of differentiation antigen 4 (CD4) endocytosis and adaptor complex binding require activation of the CD4 endocytosis signal by serine phosphorylation. Mol. Biol. Cell 10, 677–691 10.1091/mbc.10.3.677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaudhuri R., Lindwasser O. W., Smith W. J., Hurley J. H., and Bonifacino J. S. (2007) Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J. Virol. 81, 3877–3890 10.1128/JVI.02725-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garcia J. V., and Miller A. D. (1991) Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350, 508–511 10.1038/350508a0 [DOI] [PubMed] [Google Scholar]
- 33. Kwon Y., Kaake R. M., Echeverria I., Suarez M., Karimian Shamsabadi M., Stoneham C., Ramirez P. W., Kress J., Singh R., Sali A., Krogan N., Guatelli J., and Jia X. (2020) Structural basis of CD4 downregulation by HIV-1 Nef. Nat. Struct. Mol. Biol. 27, 822–828 10.1038/s41594-020-0463-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Binette J., Dubé M., Mercier J., Halawani D., Latterich M., and Cohen E. A. (2007) Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology 4, 75 10.1186/1742-4690-4-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chowers M. Y., Spina C. A., Kwoh T. J., Fitch N. J., Richman D. D., and Guatelli J. C. (1994) Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 68, 2906–2914 10.1128/JVI.68.5.2906-2914.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Münch J., Rajan D., Schindler M., Specht A., Rücker E., Novembre F. J., Nerrienet E., Müller-Trutwin M. C., Peeters M., Hahn B. H., and Kirchhoff F. (2007) Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J. Virol. 81, 13852–13864 10.1128/JVI.00904-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Basmaciogullari S., and Pizzato M. (2014) The activity of Nef on HIV-1 infectivity. Front. Microbiol. 5, 232 10.3389/fmicb.2014.00232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rosa A., Chande A., Ziglio S., De Sanctis V., Bertorelli R., Goh S. L., McCauley S. M., Nowosielska A., Antonarakis S. E., Luban J., Santoni F. A., and Pizzato M. (2015) HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526, 212–217 10.1038/nature15399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Usami Y., Wu Y., and Göttlinger H. G. (2015) SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526, 218–223 10.1038/nature15400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sood C., Marin M., Chande A., Pizzato M., and Melikyan G. B. (2017) SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 292, 6014–6026 10.1074/jbc.M117.777714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi J., Xiong R., Zhou T., Su P., Zhang X., Qiu X., Li H., Li S., Yu C., Wang B., Ding C., Smithgall T. E., and Zheng Y. H. (2018) HIV-1 Nef antagonizes SERINC5 restriction by downregulation of SERINC5 via the endosome/lysosome system. J. Virol. 92, e00196 10.1128/JVI.00196-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dikeakos J. D., Atkins K. M., Thomas L., Emert-Sedlak L., Byeon I. J., Jung J., Ahn J., Wortman M. D., Kukull B., Saito M., Koizumi H., Williamson D. M., Hiyoshi M., Barklis E., Takiguchi M., et al. (2010) Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol. Biol. Cell 21, 3279–3292 10.1091/mbc.E10-05-0470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hung C. H., Thomas L., Ruby C. E., Atkins K. M., Morris N. P., Knight Z. A., Scholz I., Barklis E., Weinberg A. D., Shokat K. M., and Thomas G. (2007) HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 1, 121–133 10.1016/j.chom.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 44. Kasper M. R., Roeth J. F., Williams M., Filzen T. M., Fleis R. I., and Collins K. L. (2005) HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J. Biol. Chem. 280, 12840–12848 10.1074/jbc.M413538200 [DOI] [PubMed] [Google Scholar]
- 45. Jia X., Singh R., Homann S., Yang H., Guatelli J., and Xiong Y. (2012) Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 19, 701–706 10.1038/nsmb.2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pawlak E. N., and Dikeakos J. D. (2015) HIV-1 Nef: a master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 1850, 733–741 10.1016/j.bbagen.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 47. Saksela K., Cheng G., and Baltimore D. (1995) Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 14, 484–491 10.1002/j.1460-2075.1995.tb07024.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee C.-H., Leung B., Lemmon M. A., Zheng J., Cowburn D., Kuriyan J., and Saksela K. (1995) A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 14, 5006–5015 10.1002/j.1460-2075.1995.tb00183.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moarefi I., LaFevre-Bernt M., Sicheri F., Huse M., Lee C.-H., Kuriyan J., and Miller W. T. (1997) Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650–653 10.1038/385650a0 [DOI] [PubMed] [Google Scholar]
- 50. Briggs S. D., Sharkey M., Stevenson M., and Smithgall T. E. (1997) SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 272, 17899–17902 10.1074/jbc.272.29.17899 [DOI] [PubMed] [Google Scholar]
- 51. Lerner E. C., and Smithgall T. E. (2002) SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat. Struct. Biol. 9, 365–369 10.1038/nsb782 [DOI] [PubMed] [Google Scholar]
- 52. Narute P. S., and Smithgall T. E. (2012) Nef alleles from all major HIV-1 clades activate Src-family kinases and enhance HIV-1 replication in an inhibitor-sensitive manner. PLoS One 7, e32561 10.1371/journal.pone.0032561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Komuro I., Yokota Y., Yasuda S., Iwamoto A., and Kagawa K. S. (2003) CSF-induced and HIV-1-mediated distinct regulation of Hck and C/EBPβ represent a heterogeneous susceptibility of monocyte-derived macrophages to M-tropic HIV-1 infection. J. Exp. Med. 198, 443–453 10.1084/jem.20022018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Biggs T. E., Cooke S. J., Barton C. H., Harris M. P., Saksela K., and Mann D. A. (1999) Induction of activator protein 1 (AP-1) in macrophages by human immunodeficiency virus type-1 NEF is a cell-type-specific response that requires both hck and MAPK signaling events. J. Mol. Biol. 290, 21–35 10.1006/jmbi.1999.2849 [DOI] [PubMed] [Google Scholar]
- 55. Courtney A. H., Lo W. L., and Weiss A. (2018) TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 10.1016/j.tibs.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Collette Y., Dutartre H., Benziane A., Ramos-Morales F., Benarous R., Harris M., and Olive D. (1996) Physical and functional interaction of Nef with Lck–HIV-1 Nef-induced T-cell signaling defects. J. Biol. Chem. 271, 6333–6341 10.1074/jbc.271.11.6333 [DOI] [PubMed] [Google Scholar]
- 57. Briggs S. D., Lerner E. C., and Smithgall T. E. (2000) Affinity of Src family kinase SH3 domains for HIV Nef in vitro does not predict kinase activation by Nef in vivo. Biochemistry 39, 489–495 10.1021/bi992504j [DOI] [PubMed] [Google Scholar]
- 58. Mitchell J. L., Trible R. P., Emert-Sedlak L. A., Weis D. D., Lerner E. C., Applen J. J., Sefton B. M., Smithgall T. E., and Engen J. R. (2007) Functional characterization and conformational analysis of the Herpesvirus saimiri Tip-C484 protein. J. Mol. Biol. 366, 1282–1293 10.1016/j.jmb.2006.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trible R. P., Emert-Sedlak L., and Smithgall T. E. (2006) HIV-1 Nef selectively activates SRC family kinases HCK, LYN, and c-SRC through direct SH3 domain interaction. J. Biol. Chem. 281, 27029–27038 10.1074/jbc.M601128200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Herna R. G., and Saksela K. (2000) Interactions of HIV-1 NEF with cellular signal transducing proteins. Front. Biosci. 5, D268–D283 [DOI] [PubMed] [Google Scholar]
- 61. Mangasarian A., Piguet V., Wang J. K., Chen Y. L., and Trono D. (1999) Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal α helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 73, 1964–1973 10.1128/JVI.73.3.1964-1973.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hanna Z., Weng X., Kay D. G., Poudrier J., Lowell C., and Jolicoeur P. (2001) The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J. Virol. 75, 9378–9392 10.1128/JVI.75.19.9378-9392.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Khan I. H., Sawai E. T., Antonio E., Weber C. J., Mandell C. P., Montbriand P., and Luciw P. A. (1998) Role of the SH3-ligand domain of simian immunodeficiency virus Nef in interaction with Nef-associated kinase and simian AIDS in rhesus macaques. J. Virol. 72, 5820–5830 10.1128/JVI.72.7.5820-5830.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee C.-H., Saksela K., Mirza U. A., Chait B. T., and Kuriyan J. (1996) Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 85, 931–942 10.1016/S0092-8674(00)81276-3 [DOI] [PubMed] [Google Scholar]
- 65. Arold S., Franken P., Strub M.-P., Hoh F., Benichou S., Benarous R., and Dumas C. (1997) The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure 5, 1361–1372 10.1016/S0969-2126(97)00286-4 [DOI] [PubMed] [Google Scholar]
- 66. Grzesiek S., Bax A., Clore G. M., Gronenborn A. M., Hu J. S., Kaufman J., Palmer I., Stahl S. J., and Wingfield P. T. (1996) The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat. Struct. Biol. 3, 340–345 10.1038/nsb0496-340 [DOI] [PubMed] [Google Scholar]
- 67. Alvarado J. J., Tarafdar S., Yeh J. I., and Smithgall T. E. (2014) Interaction with the Src homology (SH3-SH2) region of the Src-family kinase Hck structures the HIV-1 Nef dimer for kinase activation and effector recruitment. J. Biol. Chem. 289, 28539–28553 10.1074/jbc.M114.600031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sicheri F., Moarefi I., and Kuriyan J. (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385, 602–609 10.1038/385602a0 [DOI] [PubMed] [Google Scholar]
- 69. Ren X., Park S. Y., Bonifacino J. S., and Hurley J. H. (2014) How HIV-1 Nef hijacks the AP-2 clathrin adaptor to downregulate CD4. Elife 3, e01754 10.7554/eLife.01754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Choi H. J., and Smithgall T. E. (2004) Conserved residues in the HIV-1 Nef hydrophobic pocket are essential for recruitment and activation of the Hck tyrosine kinase. J. Mol. Biol. 343, 1255–1268 10.1016/j.jmb.2004.09.015 [DOI] [PubMed] [Google Scholar]
- 71. Hiipakka M., Poikonen K., and Saksela K. (1999) SH3 domains with high affinity and engineered ligand specificities targeted to HIV-1 Nef. J. Mol. Biol. 293, 1097–1106 10.1006/jmbi.1999.3225 [DOI] [PubMed] [Google Scholar]
- 72. Horenkamp F. A., Breuer S., Schulte A., Lülf S., Weyand M., Saksela K., and Geyer M. (2011) Conformation of the dileucine-based sorting motif in HIV-1 Nef revealed by intermolecular domain assembly. Traffic 12, 867–877 10.1111/j.1600-0854.2011.01205.x [DOI] [PubMed] [Google Scholar]
- 73. Breuer S., Schievink S. I., Schulte A., Blankenfeldt W., Fackler O. T., and Geyer M. (2011) Molecular design, functional characterization and structural basis of a protein inhibitor against the HIV-1 pathogenicity factor Nef. PLoS One 6, e20033 10.1371/journal.pone.0020033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lülf S., Matz J., Rouyez M. C., Järviluoma A., Saksela K., Benichou S., and Geyer M. (2014) Structural basis for the inhibition of HIV-1 Nef by a high-affinity binding single-domain antibody. Retrovirology 11, 24 10.1186/1742-4690-11-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pene-Dumitrescu T., Shu S. T., Wales T. E., Alvarado J. J., Shi H., Narute P., Moroco J. A., Yeh J. I., Engen J. R., and Smithgall T. E. (2012) HIV-1 Nef interaction influences the ATP-binding site of the Src-family kinase, Hck. BMC Chem. Biol 12, 1 10.1186/1472-6769-12-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Englander S. W., and Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655 10.1017/s0033583500005217 [DOI] [PubMed] [Google Scholar]
- 77. Zhang Z., and Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 10.1002/pro.5560020404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wales T. E., Hochrein J. M., Morgan C. R., Emert-Sedlak L. A., Smithgall T. E., and Engen J. R. (2015) Subtle dynamic changes accompany Hck activation by HIV-1 Nef and are reversed by an antiretroviral kinase inhibitor. Biochemistry 54, 6382–6391 10.1021/acs.biochem.5b00875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schindler T., Sicheri F., Pico A., Gazit A., Levitzki A., and Kuriyan J. (1999) Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol. Cell 3, 639–648 10.1016/S1097-2765(00)80357-3 [DOI] [PubMed] [Google Scholar]
- 80. Briggs S. D., and Smithgall T. E. (1999) SH2-kinase linker mutations release Hck tyrosine kinase and transforming activities in rat-2 fibroblasts. J. Biol. Chem. 274, 26579–26583 10.1074/jbc.274.37.26579 [DOI] [PubMed] [Google Scholar]
- 81. Dorman H. R., Close D., Wingert B. M., Camacho C. J., Johnston P. A., and Smithgall T. E. (2019) Discovery of non-peptide small molecule allosteric modulators of the Src-family kinase, Hck. Front. Chem. 7, 822 10.3389/fchem.2019.00822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moroco J. A., Craigo J. K., Iacob R. E., Wales T. E., Engen J. R., and Smithgall T. E. (2014) Differential sensitivity of Src-family kinases to activation by SH3 domain displacement. PLoS One 9, e105629 10.1371/journal.pone.0105629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Andreotti A. H., Schwartzberg P. L., Joseph R. E., and Berg L. J. (2010) T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol. 2, a002287 10.1101/cshperspect.a002287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hussain A., Yu L., Faryal R., Mohammad D. K., Mohamed A. J., and Smith C. I. (2011) TEC family kinases in health and disease—loss-of-function of BTK and ITK and the gain-of-function fusions ITK-SYK and BTK-SYK. FEBS J. 278, 2001–2010 10.1111/j.1742-4658.2011.08134.x [DOI] [PubMed] [Google Scholar]
- 85. Readinger J. A., Schiralli G. M., Jiang J. K., Thomas C. J., August A., Henderson A. J., and Schwartzberg P. L. (2008) Selective targeting of ITK blocks multiple steps of HIV replication. Proc. Natl. Acad. Sci. U. S. A. 105, 6684–6689 10.1073/pnas.0709659105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tarafdar S., Poe J. A., and Smithgall T. E. (2014) The accessory factor Nef links HIV-1 to Tec/Btk kinases in an Src homology 3 domain-dependent manner. J. Biol. Chem. 289, 15718–15728 10.1074/jbc.M114.572099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Romei M. G., and Boxer S. G. (2019) Split green fluorescent proteins: scope, limitations, and outlook. Annu. Rev. Biophys. 48, 19–44 10.1146/annurev-biophys-051013-022846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., and Miyawaki A. (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20, 87–90 10.1038/nbt0102-87 [DOI] [PubMed] [Google Scholar]
- 89. Rekas A., Alattia J. R., Nagai T., Miyawaki A., and Ikura M. (2002) Crystal structure of Venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J. Biol. Chem. 277, 50573–50578 10.1074/jbc.M209524200 [DOI] [PubMed] [Google Scholar]
- 90. Li W. F., Aryal M., Shu S. T., and Smithgall T. E. (2020) HIV-1 Nef dimers short-circuit immune receptor signaling by activating Tec-family kinases at the host cell membrane. J. Biol. Chem. 295, 5163–5174 10.1074/jbc.RA120.012536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Heusinger E., and Kirchhoff F. (2017) Primate lentiviruses modulate NF-κB activity by multiple mechanisms to fine-tune viral and cellular gene expression. Front. Microbiol. 8, 198 10.3389/fmicb.2017.00198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Collette Y., Arold S., Picard C., Janvier K., Benichou S., Benarous R., Olive D., and Dumas C. (2000) HIV-2 and SIV Nef proteins target different Src family SH3 domains than does HIV-1 Nef because of a triple amino acid substitution. J. Biol. Chem. 275, 4171–4176 10.1074/jbc.275.6.4171 [DOI] [PubMed] [Google Scholar]
- 93. Kienzle N., Freund J., Kalbitzer H. R., and Mueller-Lantzsch N. (1993) Oligomerization of the Nef protein from human immunodeficiency virus (HIV) type 1. Eur. J. Biochem. 214, 451–457 10.1111/j.1432-1033.1993.tb17941.x [DOI] [PubMed] [Google Scholar]
- 94. Azad A. A., Failla P., Lucantoni A., Bentley J., Mardon C., Wolfe A., Fuller K., Hewish D., Sengupta S., Sankovich S., Grgacic E., McPhee D., and Macreadie I. and (1994) Large-scale production and characterization of recombinant human immunodeficiency virus type 1 Nef. J. Gen. Virol. 75, 651–655 10.1099/0022-1317-75-3-651 [DOI] [PubMed] [Google Scholar]
- 95. Arold S., Hoh F., Domergue S., Birck C., Delsuc M. A., Jullien M., and Dumas C. (2000) Characterization and molecular basis of the oligomeric structure of HIV-1 Nef protein. Protein Sci. 9, 1137–1148 10.1110/ps.9.6.1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Poe J. A., and Smithgall T. E. (2009) HIV-1 Nef dimerization is required for Nef-mediated receptor downregulation and viral replication. J. Mol. Biol. 394, 329–342 10.1016/j.jmb.2009.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Moroco J. A., Alvarado J. J., Staudt R. P., Shi H., Wales T. E., Smithgall T. E., and Engen J. R. (2018) Remodeling of HIV-1 Nef structure by Src-family kinase binding. J. Mol. Biol. 430, 310–321 10.1016/j.jmb.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liu L. X., Heveker N., Fackler O. T., Arold S., Le Gall S., Janvier K., Peterlin B. M., Dumas C., Schwartz O., Benichou S., and Benarous R. (2000) Mutation of a conserved residue (D123) required for oligomerization of human immunodeficiency virus type 1 Nef protein abolishes interaction with human thioesterase and results in impairment of Nef biological functions. J. Virol. 74, 5310–5319 10.1128/.74.11.5310-5319.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ye H. H., Choi H. J., Poe J., and Smithgall T. E. (2004) Oligomerization is required for HIV-1 Nef-induced activation of the Src family protein-tyrosine kinase, Hck. Biochemistry 43, 15775–15784 10.1021/bi048712f [DOI] [PubMed] [Google Scholar]
- 100. Shi H., Tice C. M., Emert-Sedlak L., Chen L., Li W. F., Carlsen M., Wrobel J. E., Reitz A. B., and Smithgall T. E. (2020) Tight-binding hydroxypyrazole HIV-1 Nef inhibitors suppress viral replication in donor mononuclear cells and reverse Nef-mediated MHC-I downregulation. ACS Infect. Dis. 6, 302–312 10.1021/acsinfecdis.9b00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Smithgall T. E., and Thomas G. (2013) Small molecule inhibitors of the HIV-1 virulence factor, Nef. Drug Discov. Today Technol. 10, 523–529 10.1016/j.ddtec.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Emert-Sedlak L. A., Narute P., Shu S. T., Poe J. A., Shi H., Yanamala N., Alvarado J. J., Lazo J. S., Yeh J. I., Johnston P. A., and Smithgall T. E. (2013) Effector kinase coupling enables high-throughput screens for direct HIV-1 Nef antagonists with antiretroviral activity. Chem. Biol. 20, 82–91 10.1016/j.chembiol.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Emert-Sedlak L., Kodama T., Lerner E. C., Dai W., Foster C., Day B. W., Lazo J. S., and Smithgall T. E. (2009) Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem. Biol. 4, 939–947 10.1021/cb900195c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mujib S., Saiyed A., Fadel S., Bozorgzad A., Aidarus N., Yue F. Y., Benko E., Kovacs C., Emert-Sedlak L., Smithgall T. E., and Ostrowski M. A. (2017) Pharmacologic HIV-1 Nef blockade enhances the recognition and elimination of latently HIV-1 infected CD4 T cells by autologous CD8 T cells. J. Clin. Invest. Insight 2, e93684 10.1172/jci.insight.93684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Emert-Sedlak L. A., Loughran H. M., Shi H., Kulp J. L. 3rd, Shu S. T., Zhao J., Day B. W., Wrobel J. E., Reitz A. B., and Smithgall T. E. (2016) Synthesis and evaluation of orally active small molecule HIV-1 Nef antagonists. Bioorg. Med. Chem. Lett. 26, 1480–1484 10.1016/j.bmcl.2016.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Omondi F. H., Chandrarathna S., Mujib S., Brumme C. J., Jin S. W., Sudderuddin H., Miller R. L., Rahimi A., Laeyendecker O., Bonner P., Yue F. Y., Benko E., Kovacs C. M., Brockman M. A., Ostrowski M., et al. (2019) HIV subtype and Nef-mediated immune evasion function correlate with viral reservoir size in early-treated individuals. J. Virol. 93, e01832 10.1128/JVI.01832-18 [DOI] [PMC free article] [PubMed] [Google Scholar]