

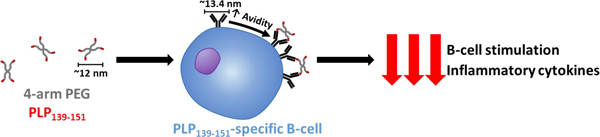
Abstract
Many autoimmune therapies focus on immune suppression to reduce symptom severity and halt disease progression; however, currently approved treatments lack specificity for the autoantigen and rely on more global immune suppression. Multivalent antigen arrays can disarm pathogenic autoimmune B cell populations that specifically recognize the antigen of interest via their B cell receptor (BCR). Disarmament may be achieved by BCR engagement, crosslinking, and sustained receptor occupancy as a result of multivalent, high avidity BCR binding. To engage and explore this mechanism, a tetramer display of the encephalogenic proteolipid peptide (PLP139–151), referred to as 4-arm PLP139–151, was synthesized by copper-catalyzed azide-alkyne cycloaddition chemistry. Subcutaneous administration of 4-arm PLP139–151 completely ameliorated symptoms of paralysis in a mouse model of multiple sclerosis known as experimental autoimmune encephalomyelitis. Competitive binding of 4-arm PLP139–151 to PLP139–151-specific IgG in mouse serum demonstrated the enhanced avidity associated with the multivalent array compared to free peptide. Furthermore, key PLP139–151-reactive B cells were depleted following 4-arm PLP139–151 treatment, resulting in significant reduction of pro-inflammatory cytokines. Together, these data demonstrate the potential of 4-arm PLP139–151 to silence autoreactive B cell populations and limit downstream activation of effector cells.
Keywords: Tolerance, autoimmunity, antigen specificity, multivalent, B cell receptor, EAE
Graphical Abstract
1. Introduction
Autoimmunity is characterized by a loss of immune tolerance toward self-antigens resulting in aberrant inflammatory events.1–2 Typical treatments for autoimmune diseases lack specificity for the offending, autoreactive T cells and B cells, which can induce a wide range of adverse side effects. Many such autoimmune therapies induce global immune suppression, hindering the immune system’s ability to eliminate pathogens.3 This immune suppression may relieve autoimmune symptoms, but it also leaves the patient susceptible to opportunistic infections. There is a crucial need for antigen-specific immunotherapies (ASITs) capable of inducing immune tolerance toward an autoantigen without compromising immune function.
In multiple sclerosis (MS), loss of immune tolerance toward myelin sheath autoantigens results in abnormal inflammatory responses in the central nervous system facilitated by both autoreactive T cells and B cells.4–5 The result of this inflammation is demyelination and neuronal degradation leading to neurological dysfunction.6 The role of B cells in MS pathology is still unclear, however, the success of B cell depletion in clinical studies utilizing Rituximab, an anti-CD20 mAb, suggests autoreactive B cells function beyond autoantibody secretion.7–10 MS patients who received Rituximab exhibited improvement in disease progression, even though they maintained high serum levels of autoantibodies against myelin antigens. These results indicated antibody-independent mechanisms for B cell involvement in MS, including antigen presentation to T cells and B cell cytokine and chemokine production.7 The successes of B cell depletion therapies in MS patients implicate autoreactive B cells as an excellent target for tolerance induction through ASITs. Autoreactive B cells may be targetable through their high-affinity membrane bound B cell receptor (BCR) specific for MS autoantigens.
Polymers with grafted antigens have been shown to specifically target and disarm B cells driving autoimmunity. Beginning in the 1970s, Dintzis and colleagues investigated polymers grafted with haptens (small molecule antigens) to induce antigen-specific B cell responses. The solubility, molecular weight of the polymer backbone, polymer type, and antigen valency were systematically varied and B cell responses observed.11 Interestingly, their findings revealed that large (>100 kDa) polymer backbones with increased antigen valency elicited an immunogenic response; however, smaller antigen arrays (<100 kDa) and lower valency arrays invoked long-term immune tolerance to the hapten (antigen). Additionally, based on their greater molecular weight in comparison to peptide antigen, multivalent arrays may also impart pharmacokinetic advantages regarding biotherapeutic distribution. Namely, following subcutaneous administration, drug absorption into the lymphatic system is increased with increasing molecular weight for therapeutics greater than 16 kDa and less than 150 kDa; a feature which may improve the exposure of multivalent arrays to target B cell populations in lymph nodes as compared to free peptide antigens.12–13 In more recent work, Dintzis and others demonstrated the potential for antigen arrays <100 kDa to selectively eliminate high affinity B cells specific for the antigen of interest.14 Our lab has investigated this phenomenon and determined BCR crosslinking and sustained receptor occupancy induced by antigen arrays may selectively target and disarm autoreactive B cells.15–17
Building upon these findings, our lab has developed a framework for multivalent antigen presentation known as soluble antigen arrays (SAgAs) and investigated their tolerogenic effects in a mouse model of MS known as experimental autoimmune encephalomyelitis (EAE).18 SAgAs are a multivalent array of MS autoantigen proteolipid protein (PLP139–151) with or without a cell adhesion inhibitor peptide (LABL) presented on a polymeric backbone of hyaluronic acid. SAgAs were shown to act in an antigen-specific manner and facilitate BCR crosslinking in B cells.19 These studies demonstrated potential therapeutic efficacy in the treatment of MS, prompting further exploration of PLP-specific multivalent array structures capable of engaging BCRs on the surface of B cells.
Herein, we outline the synthesis and characterization of a novel polymeric array of PLP139–151 for use in the EAE model of MS. This multivalent antigen array, referred to as 4-arm PLP139–151, consists of a 20 kDa polyethylene glycol (PEG) tetramer backbone with the PLP139–151 peptide epitope conjugated to each terminus. This structure offers control over the number and positioning of reactive sites, a simple synthetic process, and homogenous product. The efficacy of 4-arm PLP139–151 was demonstrated in vivo through a 25-day EAE study. Additionally, the immune mechanisms associated with 4-arm PLP139–151 treatment in splenocytes were explored through cell phenotyping using flow cytometry, co-stimulatory marker expression, and downstream effector cell responses gauged by quantification of inflammatory cytokine expression following treatment.
2. Experimental Section
Materials
20 kDa 4-arm PEG-azide was purchased from JenKem Technology USA (Beijing, China). Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) and sodium ascorbate (NaAsc) were purchased from Sigma-Aldrich (St. Louis, MO). Copper (II) sulfate pentahydrate (CuSO4 • 5 H2O) was purchased from Acros Organics (Geel, Belgium). Alkyne functionalized peptide bearing an N-terminal 4-pentynoic acid (homopropargyl, hp) modification, hpPLP139–151 (hp-HSLGKWLGHPDKF-OH), was obtained from Biomatik, USA, LLC (Wilmington, DE). All reagents were used as received without further purification. For in vitro cell assays and in vivo studies, female 4–6-week-old SJL/J (H-2) mice were purchased from Envigo Laboratories (Indianapolis, IN). For EAE induction, incomplete Freund’s adjuvant (IFA) and heat-killed mycobacterium tuberculosis H37RA were purchased from Difco (Sparks, MD). Additionally, pertussis toxin was purchased from List Biological Laboratories (Campbell, CA). For use in flow cytometry, TruStain fcX (anti-mouse CD16/32), R-phycoerythrin (PE)/Cy7-conjugated anti-mouse CD3, PE-conjugated anti-mouse CD86, FITC-conjugated anti-mouse CD80, AlexaFluor647-conjugated anti-mouse CD19, and BV421-conjugated anti-mouse CD11c were purchased from BioLegend (San Diego, CA).
Synthesis of 4-arm PLP139–151
4-arm PLP139–151 was synthesized by copper-catalyzed azide-alkyne cycloaddition (CuAAC) chemistry, as shown in Scheme 1. First, hpPLP139–151 (43 µmol) was added to a 20 mL scintillation vial with a stir bar. The powder was then dissolved in 5 mL of 50 mM phosphate buffer (pH 7.4) at room temperature. A 10 mM solution of 20 kDa 4-arm PEG-azide (10 µmol) in DMSO was then added to the solution, followed by a 120 mM solution of CuSO4• 5H2O (120 µmol) in 50 mM phosphate buffer (pH 7.4). Then, THPTA (600 umol) was added as a 600 mM solution in 50 mM phosphate buffer (pH 7.4). A 100 µL aliquot was removed for HPLC analysis before the addition of a 1 M solution of NaAsc (2.4 mmol) in 50 mM phosphate buffer (pH 7.4). The reaction was stirred at room temperature and the extent of conjugation was monitored by HPLC at various times. Upon completion of the reaction at 24 hrs, the solution was purified by semi-preparative HPLC utilizing a linear elution gradient of acetonitrile in water (constant 0.05% trifluoroacetic acid) over 20 min, with a Waters XBridge BEH C18, 5µm, 130 Å stationary phase (19 × 250 mm), with a 14.0 mL/min flow rate. The purified fraction was then concentrated under vacuum, transferred to vials, frozen, and lyophilized.
Scheme 1.

Reaction scheme for the synthesis of 4-arm PLP139–151.
Analytical Characterization of 4-arm PLP139–151
RP-HPLC analysis was conducted using a Waters Alliance HPLC system equipped with a dual wavelength UV/vis detector. Chromatographic conditions utilized a linear gradient from 5–95% acetonitrile in water (constant 0.05% trifluoroacetic acid) over 20 min, with a Waters XBridge C18, 5µm, 130 Å stationary phase (4.6 × 250 mm) with a 1.0 mL/min flow rate and detection at 214 nm. The followed equation was used to quantitate conjugation of PLP139–151
where Ncon = number of conjugated PLP139–151 molecules per backbone, nPLP139–151 = moles of PLP139–151 used in the reaction, n4-arm PEG-azide = number of moles of 20 kDa 4-arm PEG-azide used in the reaction, Vpre = total reaction volume before NaAsc is added, Vsam = volume of “pre-NaAsc” aliquot removed from the reaction mixture, PAt = the measure peak area of PLP139–151 at time t, and PAstart = the measure peak area of PLP139–151 before NaAsc is added to the reaction.
Nuclear magnetic resonance imaging (NMR) spectra were collected on a Bruker Avance AVIII 500 MHz spectrometer equipped with a dual carbon/proton cryoprobe, and all samples were dissolved in 600 µL D2O for analysis. MestReNova 12.0 was used for NMR data analysis.
Size exclusion chromatography (SEC) was conducted using a Waters Alliance HPLC system equipped with a dual wavelength UV/vis detector and an RI detector. Chromatographs were collected using a Waters XBridge® BEH 125 SEC, 2.5 µm, 125 Å stationary phase (7.8 × 300 mm) column at a flow rate of 0.5 ml/min over 60 minutes using saline as a mobile phase. The PEG standards were detected by the RI detector and 4-arm PEG-N3 and 4-arm PLP139–151 was analyzed at 214 nm.
Dynamic Light Scattering (DLS) was performed using a DynaPro Plate Reader (Wyatt Technology, Santa Barbara, CA). Incident light was detected in a backscattering configuration and analyzed with an autocorrelator. Sample (20 μL), in DI water, was placed in a clear-bottomed 384-well plate and read at 20 °C. Samples were measured 5 times with a 15 s acquisition time. Autocorrelation functions were fit using cumulant analysis, and intensity averaged values are reported. Errors are reported as standard deviation of 3 replicates.
Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS) was completed by spotting samples with α-Cyano-4-hydroxycinnamic acid matrix solution prior to analysis on a Bruker Autoflex MAX MALDI mass spectrometer. FlexControl 3.4 was utilized to analyze the data.
Induction of EAE
In vivo efficacy and in vitro cell assays were performed through induction of EAE in female 4–6-week-old SJL/J (H-2) mice. All protocols were approved through the University’s Institutional Animal Care and Use Committee with animals housed in pathogen-free conditions. Induction of EAE was carried out using an emulsion of 200 µg free PLP139–151 in PBS emulsified with Complete Freund’s Adjuvant (CFA) containing 4 mg/mL heat-killed M. Tuberculosis strain H37RA. This emulsion was administered subcutaneously to mice on day 0 in 50 µL injections above each shoulder and hind flank for a total injection volume of 200 µL per mouse. At this time, 100 µL intraperitoneal injections of pertussis toxin at 100 ng/mL in PBS were administered to the mice. The administration of pertussis toxin was also repeated on day 2. Beginning on day 7, severity of disease symptoms was recorded daily using the following clinical scoring system: 0, no clinical disease symptoms; 1, weakness or limpness of the tail; 2, weakness or partial paralysis of one or two hind limbs (paraparesis); 3, full paralysis of both hind limbs (paraplegia); 4, paraplegia plus weakness or paralysis of forelimbs; 5, moribund (euthanasia necessary). Mouse body weight was recorded daily throughout the entire study.
Competitive PLP139–151 ELISA
Competitive binding of 4-arm PLP139–151 to EAE serum antibodies was detected using an indirect ELISA. Initially, 100 µL of 1 mg/mL free PLP139–151 at pH 9.5 (50 mM sodium bicarbonate buffer) was incubated overnight in an Immulon 2HB plate at 4°C. Wash buffer consisting of 1x Phosphate Buffered Saline (PBS) with 0.5% Tween 20 was used to wash the plate 3 times with 300 µL/well. Block buffer was prepared using 0.2 µm filtered PBS containing 1% Bovine Serum Albumin (BSA). The plate was blocked with 250 µL of block buffer incubated at 37°C for 1 hour. Serum samples obtained from EAE mice at peak of disease severity were thawed and pre-incubated with one of the following treatments: 4-arm PLP139–151, 20 kDa 4-arm PEG-azide, free PLP139–151, or PBS. These treatments were maintained at 25 µM PLP139–151 basis and were incubated with the serum for 1 hour. Once again, the plate was washed 3 times with wash buffer, as described previously. Treated serum samples were added to the plate and serially diluted in reagent diluent consisting of 1% BSA and 0.5% Tween 20 in 1x PBS. Samples were incubated in the plate for 1 hour at 37°C. The plate was washed 3 times with wash buffer, as described previously. Secondary antibody solution was prepared using anti-mouse IgG diluted in reagent diluent. Each well was incubated with 100 µL of secondary antibody solution for 1 hour at 37°C. The plate was washed 3 times with wash buffer and 100 µL of TMB solution was added to each well. The plate was shaken at 200 rpm for 15 minutes prior to the addition of 100 µL/well of 2N sulfuric acid to quench the reaction. Lastly, the absorbance at 450/540 nm was read on a Spectramax M5 (Molecular Devices) plate reader.
In Vivo Efficacy
Treatment efficacy in vivo was determined using 6 mice per group. All mice were euthanized after 25 days. Treatments were subcutaneously administered on the back of the neck in 100 µL of 40 mg/mL. Each group was dosed on a 200 nmol PLP139–151 basis, with doses administered on days 4, 7, and 10 following EAE induction. Mouse weights were recorded daily beginning on day 0 and clinical scores were recorded daily beginning on day 7.
Splenocyte Isolation
Spleens were isolated from EAE mice at peak of disease for in vitro studies. Spleens were placed on ice in 5 mL of RPMI 1640 containing L-glutamine and 1% Penicillin-Streptomycin. Cellular extract was collected by pressing the spleens into a wire mesh using the plunger of a 1 mL syringe. The extract was centrifuged at 1100 xg for 5 minutes and resuspended in red blood cell lysis solution at room temperature for 5 minutes. The lysis solution was quenched through the addition of cold RPMI 1640 supplemented with L-glutamine, 1% Penicillin-Streptomycin, and 10% fetal bovine serum (FBS) (complete RPMI, cRPMI). The cells were then centrifuged at 1100 xg for 5 minutes and resuspended in cRPMI for counting in 0.04% trypan blue. Cells were counted using a Nexcelom Bioscience Cellometer Auto T4. In vitro cell incubation was carried out at 37°C and 5% CO2 for 72 hours following addition of treatment.
Cell Staining for Flow Cytometry
EAE splenocytes were incubated at 5 x 106 cells/well in a 24 well plate at a final volume of 1 mL. Treatments were added on a 25 µM PLP139–151 basis, and PLP139–151 rechallenge consisted of the addition of 25 µM free PLP139–151 to the well. Following 72 hours incubation with treatment, a portion of cell supernatant was removed for future processing and the remaining cells were washed with RPMI + 5% FBS and centrifuged. Cells were resuspended in 50 µL of 20 µg/mL TruStain fcX and placed on ice for 30 minutes. Next, 50 µL of antibody solution was added, according to manufacturer recommendations, and the solution was kept on ice for 1 hour. Antibody stains included: R-phycoerythrin (PE)/Cy7-conjugated anti-mouse CD3, PE-conjugated anti-mouse CD86, FITC-conjugated anti-mouse CD80, AlexaFluor647-conjugated anti-mouse CD19, and BV421-conjugated anti-mouse CD11c. Sample data was collected using a BD FACSFusion Cytometer with 30,000 events per sample. Sample data was analyzed using Kaluza and GraphPad Prism.
Cytokine Measurement
Prior to sample processing for flow cytometry, cell supernatant was removed from the samples. This supernatant was frozen at −80°C until cytokine analysis could be carried out. The secretion of IFN-γ, TNF-α, GM-CSF, IL-2, IL-6, IL-10, IL-12p70, IL-15, IL-17A, and IL-23 was detected using a U-Plex kit while following manufacturer instructions (Meso Scale Discovery).
Statistical Analysis
Statistical analysis in all assays was carried out using two-way analysis of variance (ANOVA) and Tukey multiple comparisons tests. Criteria for significance in cytokine analysis are as follows: * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. For cytometry samples * represents significance (p<0.05 or better) from the 4-arm PLP139–151 treatment group with 25 µM PLP139–151 rechallenge. Additionally, for flow cytometry samples # represents significance (p<0.05 or better) from the 4-arm PLP139–151 treatment group with no PLP139–151 rechallenge. All statistical analysis was performed using GraphPad Prism.
3. Results
Synthesis and Analytical Characterization of 4-arm PLP139–151
The 4-arm PLP139–151, with an average of 3.3 PLP139–151 molecules per polymer, was synthesized by conjugating multiple modified autoantigen (hpPLP139–151) molecules to 20 kDa 4-arm PEG-azide. Both qualitative and quantitative analytical techniques were used to characterize 4-arm PLP139–151 and its components. 1H/13C Heteronuclear Single Quantum Coherence (HSQC) NMR spectroscopy was used to confirm successful conjugation. The spectra of 20 kDa 4-arm PEG-azide showed one major resonance corresponding to the PEG backbone (Figure 1A). The unique resonance signal for the terminal alkyne of the homopropargyl linker on hpPLP139–151 (δ(1H) ≈ 2.6 ppm, δ(13C) ≈ 70 ppm) was clearly evident (Figure 1B). However, following conjugation, this resonance signal was absent (Figure 1C), indicating that 4-arm PLP139–151 contained only the conjugated peptide. Further, the incorporation of both components into the final product was observed by comparing the individual resonances from 20 kDa 4-arm PEG-azide (Figure 1A) and hpPLP139–151 (Figure 1B), to the HSQC spectrum of 4-arm PLP139–151 (Figure 1C).
Figure 1.
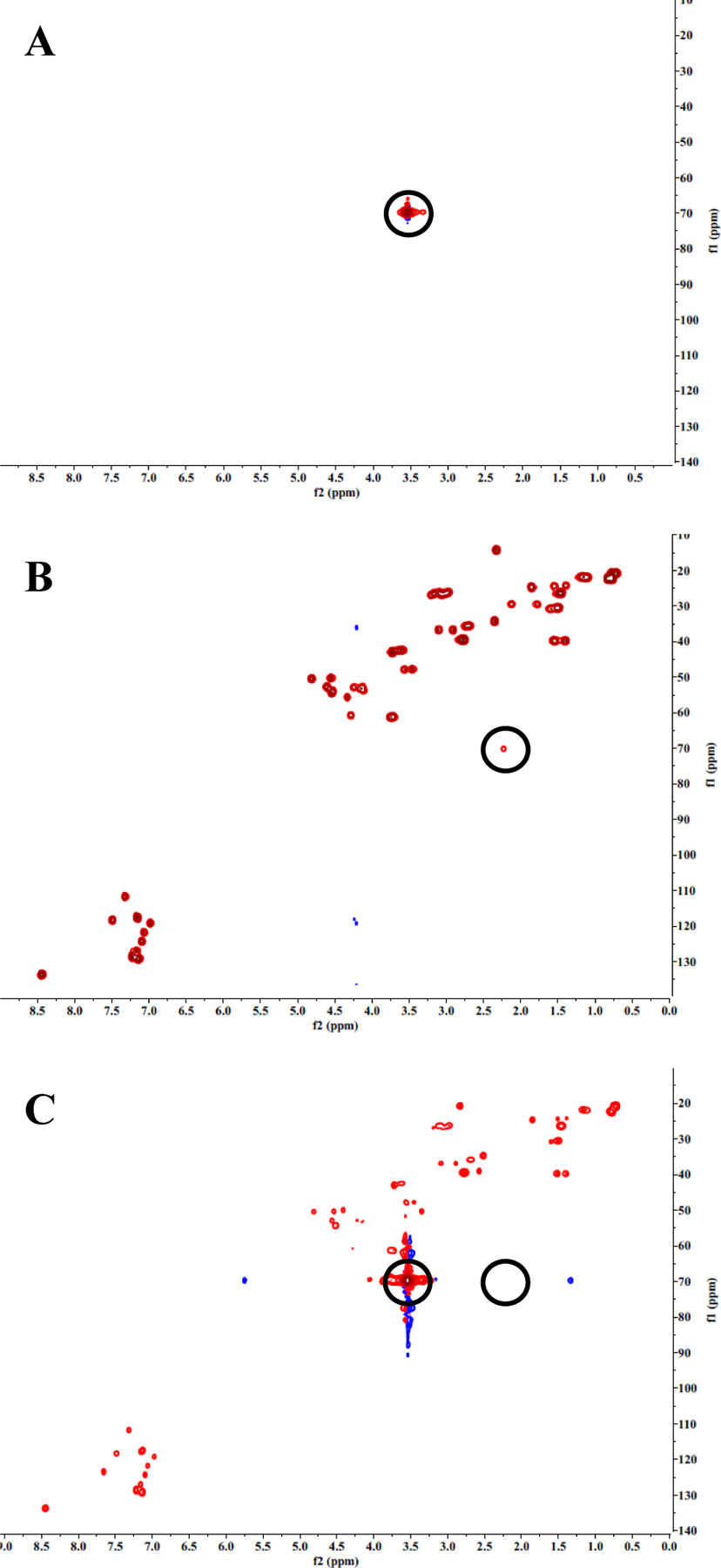
HSQC NMR spectra of 20 kDa 4-arm PEG-azide (A), hpPLP139–151 (B), and 4-arm PLP139–151 (C). hpPLP139–151 (B) shows a unique resonance from the alkyne peak. 4-arm PLP139–151 (C) does not show the resonance from the alkyne peak, indicating that no residual unconjugated PLP139–151 is present in the final product.
In order to gain information regarding the size of the tetramer, SEC, DLS, and MALDI-TOF data were collected. Results from SEC revealed molecular weights of 26,940 Da and 28,940 Da for 4-arm PEG-N3 and 4-arm PLP139–151, respectively. DLS data estimated the hydrodynamic radius of 1.67 nm, and 1.75 nm for 20 kDa 4-arm PEG-N3, and 4-arm PLP139–151, respectively. Finally, MALDI-TOF MS provided molecular weight determinations for the tetramer. Results showed that the average molecular weight of 20 kDa 4-arm PEG-N3 and 4-arm PLP139–151 were 20,367.485 Da and 23,758.924 Da, respectively. This change in molecular weight corresponds to the addition of 2.2 PLP139–151 molecules on the PEG backbone. The disagreement between the number of PLP molecules on the backbone likely results from the difficulty in accurately determining molecular weights of heterogeneous samples.
Competitive ELISA Demonstrating PLP139–151 Specificity
Serum from EAE mice was pre-incubated with 4-arm PLP139–151, 20 kDa 4-arm PEG-azide, or free PLP139–151 prior to addition to a PLP139–151-coated ELISA plate. The result of this pre-incubation was the loss of signal due to PLP139–151-specific antibodies binding during pre-incubation. As seen in Figure 2, pre-incubation with 4-arm PLP139–151 greatly reduced the signal in the competitive ELISA assay compared to pre-incubation with PBS, indicating that PLP139–151-specific antibody binding sites were occupied following 4-arm PLP139–151 pre-incubation. The 4-arm PLP139–151 was more effective at occupying the binding sites of PLP139–151-specific antibodies from EAE serum, compared to equimolar concentrations of free PLP139–151. Additionally, pre-incubation with 20 kDa 4-arm PEG-azide showed some reduction of the ELISA signal, suggesting free azides may introduce some degree of non-specific binding to serum antibodies (Figure S2).
Figure 2.
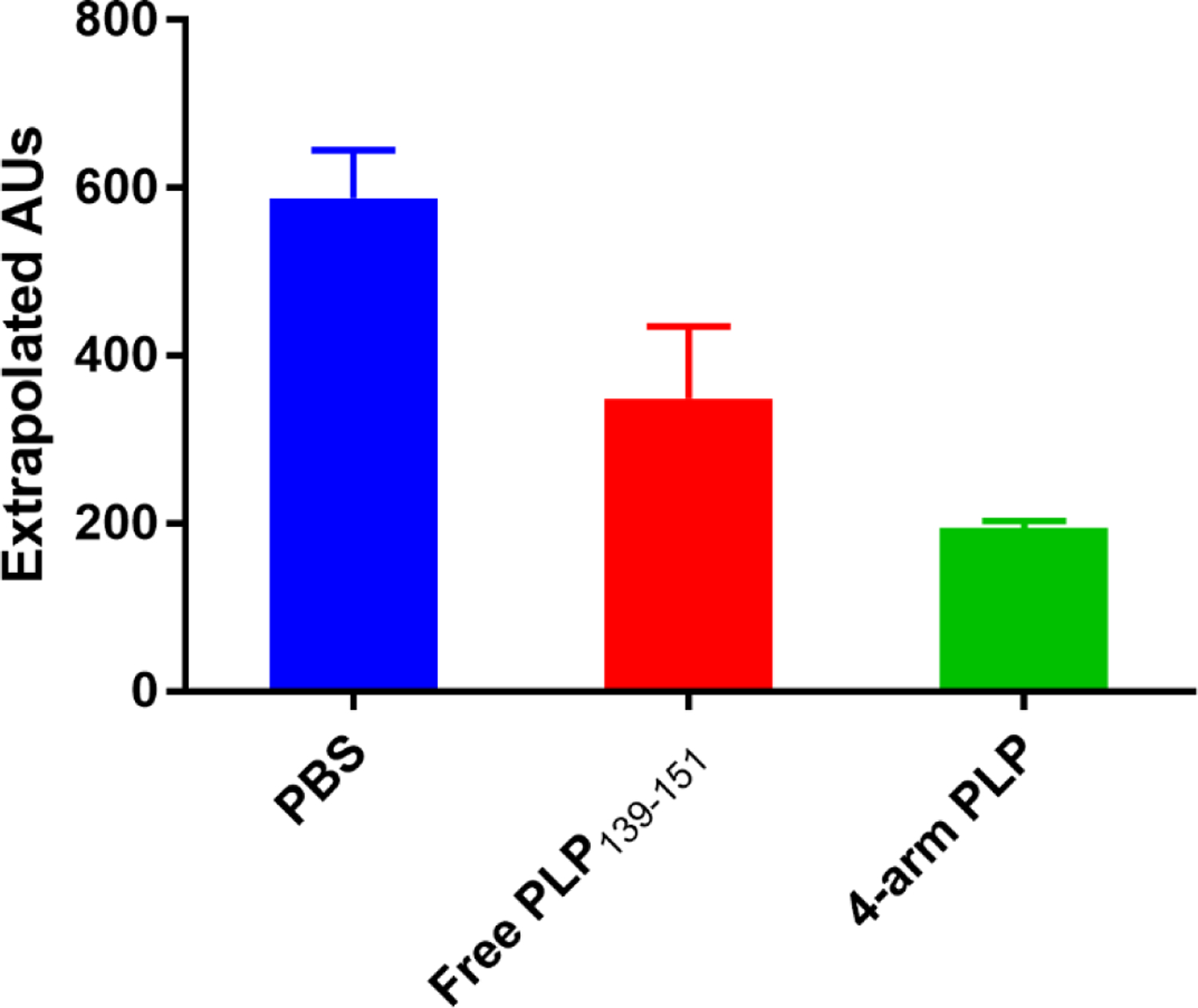
PLP139–151-specific IgG antibody titers in EAE mouse serum detected by ELISA following one hour serum incubation with (A) PBS vehicle control (B) Free PLP139–151 and (C) 4-arm PLP139–151.
In Vivo Efficacy of 4-arm PLP139–151
In vivo studies of 4-arm PLP139–151 in the EAE mouse model were conducted using subcutaneous injection of treatments in 40 mg/mL mannitol at equimolar PLP139–151 concentrations of 200 nmol. Treatments were administered on days 4, 7, and 10 with mice being observed for 25 days. Clinical scores were assigned based on disease severity, with higher scores representing more severe symptoms. Treatment with 4-arm PLP139–151 proved most effective at eliminating EAE symptoms, as mice treated with 4-arm PLP139–151 displayed no signs of paralysis (Figure 3). Treatment with 4-arm PLP139–151 presented a drastic change from the control treatments, which both developed symptoms of paralysis. Furthermore, the mouse weight data (Figure 4) paralleled the clinical score data, with the 4-arm PLP139–151 treated mice maintaining a healthy and stable weight gain throughout the study. This contrasted with the control groups, which displayed a rapid loss of weight beginning at approximately day 10.
Figure 3.
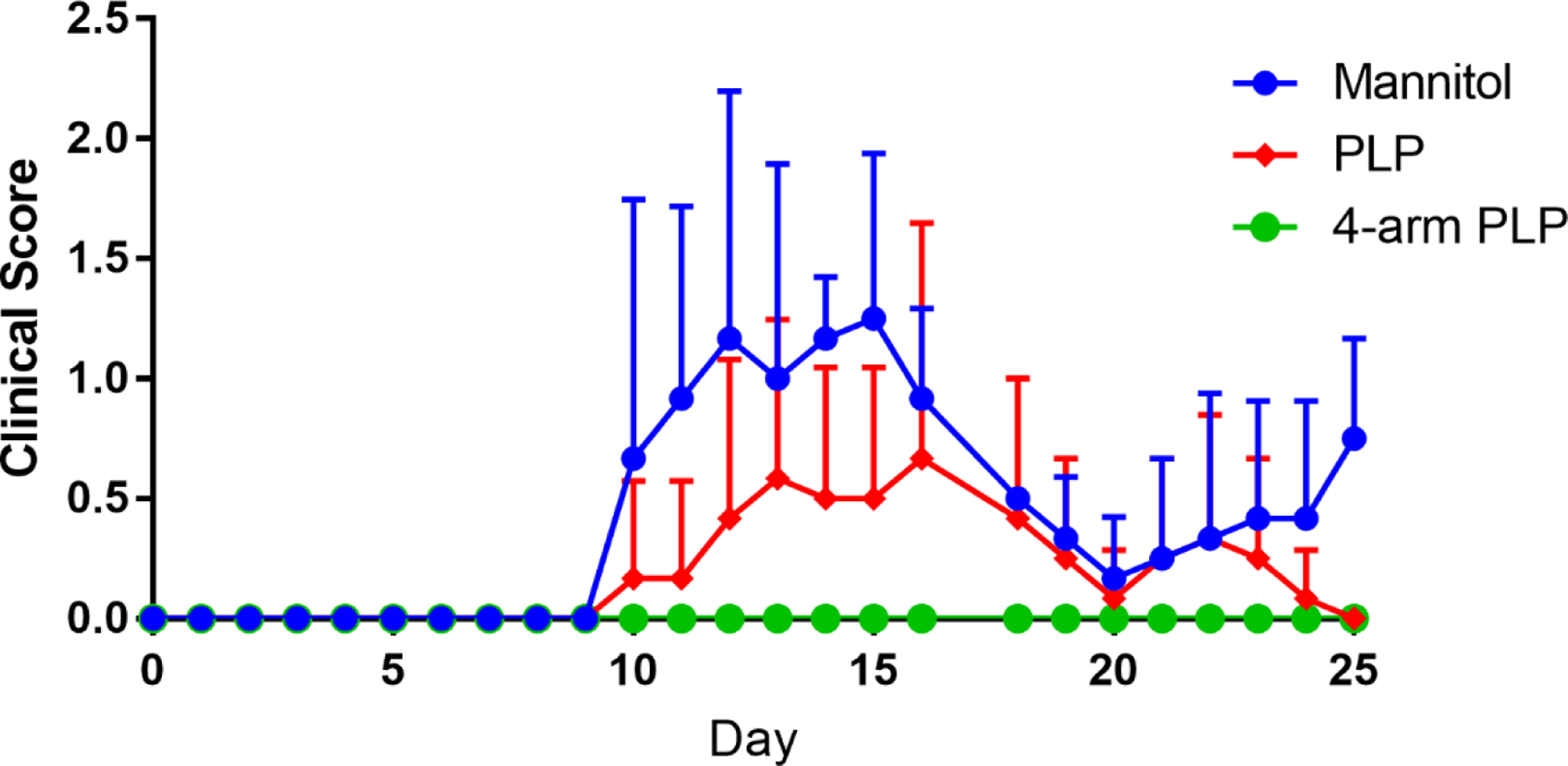
Clinical scores of EAE mice treated in vivo with (A) free PLP139–151 and (B) 4-arm PLP139–151. Treatments were administered subcutaneously on days 4, 7, and 10 with 40 mg/mL mannitol as vehicle. All doses were based on 200 nmol PLP139–151 basis. N=6 for all groups. Data is presented as mean ± SD.
Figure 4.
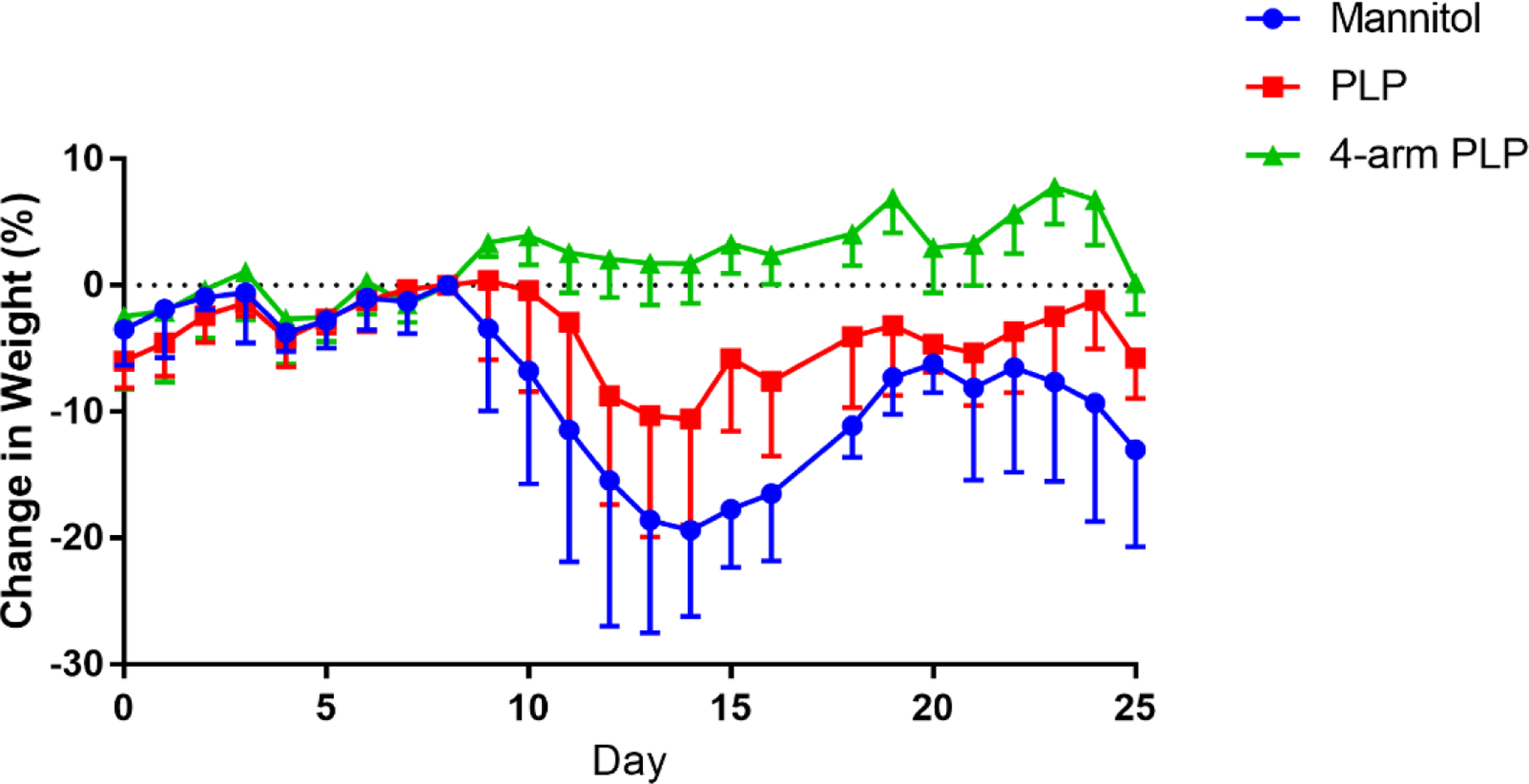
Animal weight data of EAE mice treated in vivo with (A) free PLP139–151 and (B) 4-arm PLP139–151. Treatments were administered subcutaneously on days 4, 7, and 10 with 40 mg/mL mannitol as vehicle. All doses were based on 200 nmol PLP139–151 basis. N=6 for all groups. Data is presented as mean ± SD of % change in weight normalized to mouse weight at day 8.
Analysis of Key Immune Cell Populations
In vitro treatment of EAE splenocytes harvested at peak of disease with 4-arm PLP139–151 was assessed through flow cytometric analysis of key immune cell populations. These populations included CD11c+ antigen-presenting cells, CD19+ B cells, and CD3+ T cells. Additionally, the expression of two costimulatory markers, CD80 and CD86, was analyzed in these splenocytes. Treatment groups included media, 25 µM free PLP139–151, 4-arm PLP139–151, and a combination 4-arm PLP139–151 + 25 µM free PLP139–151. This strategy provided insight into the ability of these treatment options to suppress PLP139–151-specific cell expansion in the presence of free, cognate antigen. Additionally, 20 kDa 4-arm PEG-azide and 20 kDa 4-arm PEG-azide + 25 µM free PLP139–151 treatments were explored and these data are included in the supplement.
CD11c+ cells represented the smallest population observed in this study, and with the inclusion of 25 µM PLP139–151 rechallenge, the presence of this population was further reduced through treatment with 4-arm PLP139–151 (Figure 5). This reduction in CD11c+ cells was not observed in the presence of 20 kDa 4-arm PEG-azide without conjugation of PLP139–151. The total T cell populations remained relatively constant across all treatment options except for a small reduction in T cells when treated with 4-arm PLP139–151 alone, compared to media alone (Figure 6).
Figure 5.
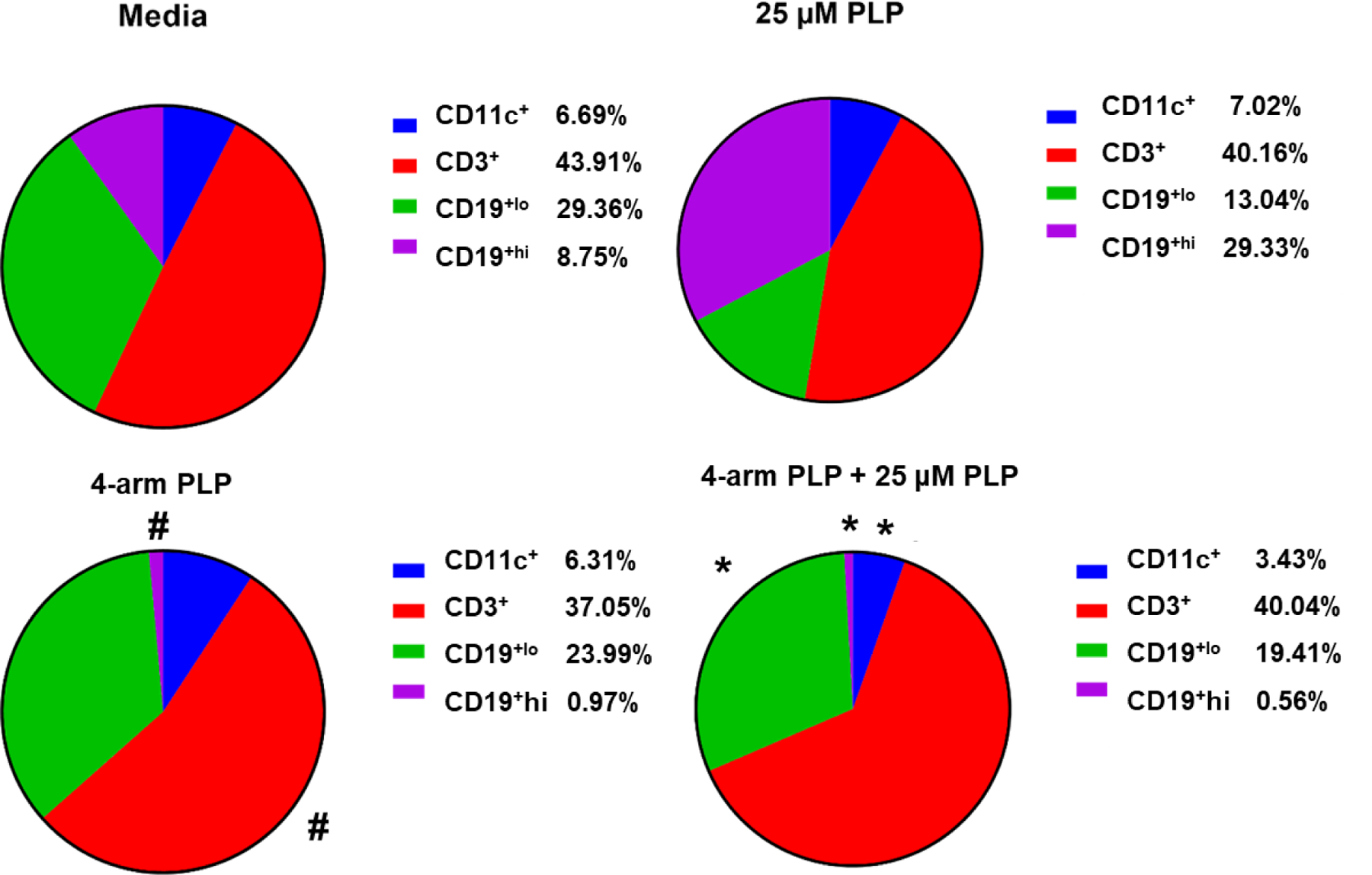
Percent of total singlet splenocytes expressing CD11c, CD3, CD19lo, and CD19hi following in vitro incubation for 72 hours with media, 25 µM free PLP139–151, 4-arm PLP139–151, and 4-arm PLP139–151 + 25 µM free PLP139–151. Data is presented as mean ± SD. N=3, * represents significance from 25 µM free PLP139–151. # represents significance from Media.
Figure 6.
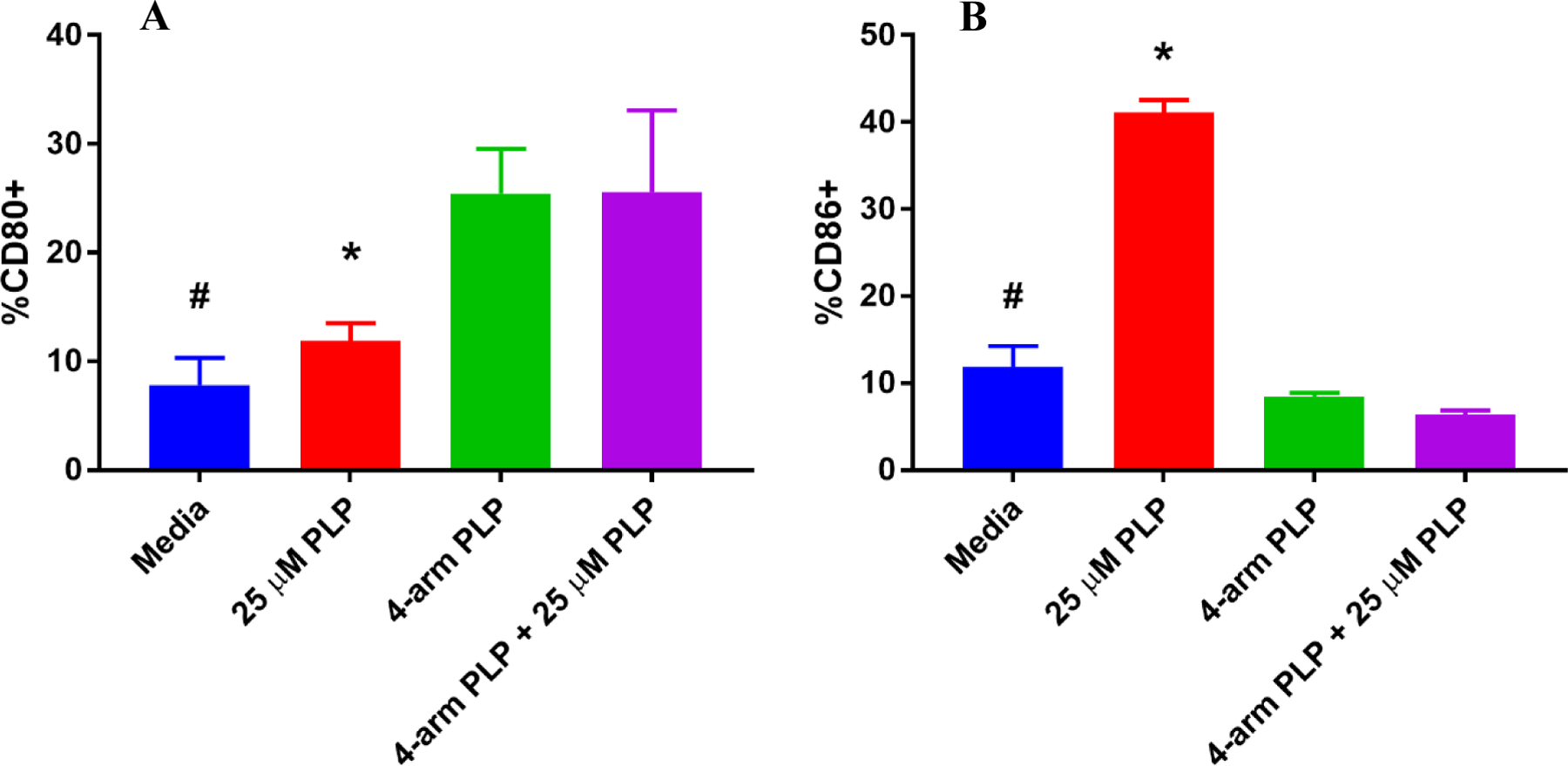
Percent of total singlet splenocytes expressing (A) CD80 and (B) CD86 following in vitro incubation for 72 hours with (I) media (II) 25 µM free PLP139–151 (III) 4-arm PLP139–151 and (IV) 4-arm PLP139–151 + 25 µM free PLP139–151. Data is presented as mean ± SD. N=3, * represents significance from 4-arm PLP139–151 + 25 µM free PLP139–151. # represents significance from 4-arm PLP139–151.
Two populations of CD19+ B cells were observed throughout this study. The first population, labeled CD19+lo exhibited a lower expression of CD19 on the cell surface (Figure S6). This population remained mostly constant across all treatment groups with the exception of a slight increase in CD19+lo cells in splenocytes treated with 4-arm PLP139–151 + 25 µM free PLP139–151 (Figure 5). Most interestingly, the CD19+hi B cell population was highly responsive to PLP139–151-rechallenge, as the inclusion of 25 µM free PLP139–151 resulted in marked expansion of this cell population (Figure 5). Furthermore, treatment with 4-arm PLP139–151 nearly completely eliminated CD19+hi B cells from these splenocytes, both in the presence of 25 µM free PLP139–151 and without rechallenge (Figure 5).
Costimulatory markers CD80 and CD86 were observed in EAE splenocytes following treatment, and treatment with 4-arm PLP139–151 also had striking results on the expression of these costimulatory molecules. CD80 expression was increased following treatment with 4-arm PLP139–151, a result that was maintained both with and without PLP139–151 rechallenge (Figure 6A). In contrast, the expression of CD86 was significantly reduced in splenocytes treated with 4-arm PLP139–151 (Figure 6B). The expression of CD86 was upregulated in the presence of 25 µM free PLP139–151 in the control treatment groups, but the inclusion of 4-arm PLP139–151 prevented this upregulation despite the presence of PLP139–151 rechallenge (Figure 6B).
Cytokine Expression
In addition to investigating changes in key immune cell population statistics, the treated samples were also tested for differences in cytokine expression. Cytokines investigated in this assay included IFN-γ, TNF-α, GM-CSF, IL-2, IL-6, IL-10, IL-12p70, IL-15, IL-17A, and IL-23. Samples were designed for analysis in triplicate, however, GM-CSF, IL-12p70, IL-15, and IL-23 each resulted in fewer than 3 samples within the detectable range. Of the remaining cytokines, the effects of 4-arm PLP139–151 were clear. General inflammatory cytokines such as IFN-γ (Figure 7A) and TNF-α (Figure 7B) were highly expressed in the presence of 25 µM free PLP139–151 for control treatment groups. Despite this increase due to the presence of free PLP139–151, 4-arm PLP139–151 treatment prevented the increased secretion of IFN-γ (Figure 7A) and TNF-α (Figure 7B) in the presence of PLP139–151 rechallenge. It should be noted that treatment with 20 kDa 4-arm PEG-azide in the presence of 25 µM free PLP139–151 significantly increased the secretion of TNF-α (Figure S5B). The anti-inflammatory effects of 4-arm PLP139–151 were also observed in expression of IL-2 (Figure 7C), where response to PLP139–151 rechallenge was not as pronounced. IL-17A (Figure 7D) and IL-6 (Figure 7E) expression was also increased in the presence of free PLP139–151, but treatment with 4-arm PLP139–151 significantly reduced the secretion of both cytokines in response to PLP139–151 rechallenge. Lastly, expression of IL-10 (Figure 7F) appeared to remain unchanged in response to the treatment groups explored in this study.
Figure 7.

Concentrations of (A) IFN-γ (B) TNF-α (C) IL-2 (D) IL-17A (E) IL-6 and (F) IL-10 following 72 hr in vitro incubation with (I) media (II) 25 µM free PLP139–151 (III) 4-arm PLP139–151 and (IV) 4-arm PLP139–151 + 25 µM free PLP139–151. Data is presented as mean ± SD. N=3, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
4. Discussion
Soluble multivalent antigen arrays have the potential to selectively disarm, tolerize, or delete self-reactive B cells at various stages of B cell maturity due to their high affinity interactions with the BCR, crosslinking of BCRs, and sustained receptor occupancy.14, 20–21 As discussed above, early works in the development of multivalent arrays for tolerance induction indicated that low molecular weight antigen arrays (<100 kDa) were most effective at deactivating antigen-specific B cells, suggesting enhanced suppressive activity compared to larger constructs.22 In studies reported here, tetramer antigen arrays (~25 kDa) displaying PLP139–151 were investigated as a multivalent, antigen-specific therapy for inhibiting EAE induced by the PLP139–151 epitope.
PLP139–151 induction of EAE is known to invoke antigen-specific B and T cells responses that drive disease.23 Adoptive transfer experiments and rechallenge studies using EAE immune cells (e.g. EAE splenocytes) have confirmed the specificity of the immune response to PLP139–151.24–25 Furthermore, EAE induced by PLP139–151 has not shown immune reactivity to null antigens.26–27 Thus, our research did not aim to reiterate the specificity of PLP139–151 immune responses. Instead, we designed and synthesized the 4-arm PLP139–151 for comparison to free (monovalent) PLP139–151, ultimately showing this format change yields opposing mechanisms of disarming autoimmune B cells versus autoimmune stimulation, respectively.
The multivalent nature of 4-arm PLP139–151 more effectively blocked PLP139–151-specific antibodies in solution due to higher avidity; a feature resulting from the molecular design of 4-arm PLP139–151. As seen in Figure 2, pre-incubation with 4-arm PLP139–151 resulted in the greatest reduction of absorbance in the competitive ELISA assay. Samples pre-incubated with 4-arm PLP139–151 displayed significantly reduced absorbance in comparison to samples pre-incubated with an equimolar amount of free PLP139–151. Notably, the theoretical span of the 5 kDa PEG arms in 20 kDa 4-arm PEG-azide is ~6 nm.28–29 As such, the distance between two PLP139–151 moieties in 4-arm PLP139–151 ranges from 8 nm to 12 nm, which is nearly identical to the spacing between the two arms of murine IgG antibody subclasses, which are reported to range from approximately 11.7 nm to 13.4 nm.30–31 Specifically, since the PLP139–151 multimer described herein has an average of 2.2 PLP139–151 molecules on the PEG backbone, the distance between epitopes should be around 12 nm; whereas if all arms were occupied, lateral distances could range from ~8.5 nm between adjacent peptides for a fully occupied 4-arm structure to a maximum of ~12 nm spacing when fully stretched. Further studies utilizing various sizes of PEG backbones will need to be conducted to fully elucidate the effects of autoantigen spacing on efficacy.
In vivo treatment of EAE mice through subcutaneous injection of 4-arm PLP139–151 prevented symptom onset in “at-risk” animals. As seen in Figure 3, subcutaneous administration of 4-arm PLP139–151 dosed at a 200 nmol PLP139–151 equivalent basis was capable of completely ameliorating EAE symptoms in mice, with none of the treated mice showing signs of paralysis throughout the 25-day study. This was in contrast to mice treated with mannitol or with free PLP139–151, both of which experienced symptoms of paralysis. Additionally, mice treated with 4-arm PLP139–151 also maintained a healthy weight throughout the study (Figure 4), further supporting the efficacy associated with multivalent display of antigen.
Investigations into the phenotypic changes associated with the amelioration of EAE symptoms at peak of disease supported the role of B cell interactions in the efficacy of 4-arm PLP139–151 treatment. Most notably, 72-hour in vitro treatment with 4-arm PLP139–151 resulted in nearly complete depletion of CD19+hi cells (Figure 5), a B cell population which was highly enhanced in response to treatment with 25 µM free PLP139–151. We postulate that these CD19+hi B cells are associated with EAE disease onset due to their prevalence following treatment with 25 µM free PLP139–151. This is supported by literature indicating overexpression of CD19 in transgenic mice results in enhanced proliferative response to antigens and may result in a breakdown of peripheral tolerance, allowing autoreactive B cells to overcome anergy.32–33 In addition, high avidity interactions between antigen and respective BCRs in the absence of secondary co-stimulatory signals are often associated with induction of B cell anergy, a state of immunological unresponsiveness.34–37 Therefore, depletion of CD19+hi B cells specific to PLP139–151 is likely a key mechanism by which 4-arm PLP139–151 is capable of ameliorating EAE symptoms.
In addition to the depletion of CD19+hi B cells, 4-arm PLP139–151 treatment also altered the expression of co-stimulatory markers CD80 and CD86. CD80 expression significantly increased following treatment with 4-arm PLP139–151 both with and without the addition of 25 µM free PLP139–151 (Figure 6A). Conversely, the expression of CD86 was significantly reduced when treated with 4-arm PLP139–151 + 25 µM free PLP139–151, in comparison to 25 µM free PLP139–151 alone (Figure 6B). In order to interpret these results, it is important to first understand the role these co-stimulatory markers play with regards to T cell activation. Primarily expressed on antigen-presenting cells, the ligands CD80 and CD86 modulate T cell responses through binding to one of two receptors: CD28 or cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). It has been demonstrated that the functions of these two ligands are distinct and opposing with regards to T cell activation.38–41 Manzotti and colleagues found that ligation of CTLA-4 with CD86 was associated with T cell proliferation, whereas ligation of CTLA-4 with CD80 resulted primarily in inhibition of T cell activation.38 Based on these data, a model was proposed in which CD80 preferentially interacts with CTLA-4 in the absence of inflammatory stimuli, restricting T cell activation.41 Therefore, the resulting shift in CD80/CD86 expression observed in EAE splenocytes following treatment with 4-arm PLP139–151 may inhibit the T cell response to PLP. A similar result has been observed in studies of soluble antigen arrays (SAgAs) at varying antigen valency, wherein the lowest valency SAgAs most strongly displayed an upregulation of CD80 and downregulation of CD86.16 This effect was less pronounced as SAgA antigen valency was increased, suggesting low valency antigen arrays (e.g. <4–6 antigens per backbone) may have the greatest immunosuppressive effects, a result in agreement with the studies performed here.
In order to determine the effect of 4-arm PLP139–151 treatment on T cell response, we quantified the expression of key inflammatory cytokines following in vitro treatment. Generally, only samples which included 25 µM free PLP139–151 resulted in significant changes in cytokines (Figure 7). EAE splenocytes treated with 25 µM free PLP139–151 alone resulted in increased secretion of inflammatory cytokines such as IFN-γ, TNF-α, IL-2, IL-17A, and IL-6, clearly showing immune stimulation associated with PLP139–151 rechallenge. Interestingly, splenocytes treated with 4-arm PLP139–151 + 25 µM free PLP139–151 expressed significantly lower levels of each of these inflammatory cytokines despite the inclusion of free, cognate antigen. In contrast, prior reports of multivalent SAgAs displaying PLP139–151 showed increases of IL-17, TNF-α, and IL-6 in the presence of free antigen.16 Thus, the 4-arm PLP139–151 may be a more potent inhibitor of effector T cell activation in the presence of antigen rechallenge as compared to SAgAs; however, further studies directly comparing the two multivalent arrays are necessary to determine precisely how this difference is propagated to effector T cells. The proinflammatory cytokines studied here have been implicated in the onset of EAE and inhibiting their secretion may represent a shift away from pathogenic T helper subsets; however, further studies focused on T-cell response to 4-arm PLP139–151 treatment will be necessary.42–46
5. Conclusions
The synthesis and characterization of a soluble tetrameric antigen display system was tested for the induction of antigen-specific tolerance. EAE was induced using PLP139–151 and subsequently treated using the very same PLP139–151 peptide or a ~25 kDa 4-arm PLP139–151 construct designed to target and disarm pathogenic B cells specific to this epitope. The simple change to a 4-arm PLP139–151 structure completely negated the development of EAE in contrast to treatment using the same dose of ‘free’ PLP139–151 peptide. In vitro studies of 4-arm PLP139–151 with EAE mouse splenocytes probed the mechanism and demonstrated that the therapeutic efficacy observed in vivo was likely the result of depletion of PLP139–151-specific CD19+hi B cells. Depletion of CD19+hi B cells is consistent with our hypothesized mechanism of avidity-induced BCR crosslinking and sustained receptor occupancy leading to B cell disarmament. Furthermore, the mechanism was affirmed in splenocyte cultures containing free PLP139–151, since the expected immune stimulation upon cognate antigen rechallenge was blocked by 4-arm PLP139–151. Additionally, the overall expression of CD80, associated with preferential inhibition of T cells, was increased, while the expression of CD86, associated with T cell proliferation, was decreased. This indicated the 4-arm PLP139–151 inhibited PLP139–151-specific inflammatory responses and reduced activation of effector T cells. A reduction in numerous inflammatory cytokines in response to free PLP139–151 further corroborated this trend. In summary, EAE was completely blocked in mice via antigen-specific disarmament of encephalogenic autoimmune cells when treated with a multivalent 4-arm PLP139–151 version of the normally encephalogenic PLP139–151 epitope.
Supplementary Material
Table 1.
Size information for hpPLP139–151, 20 kDa 4-Arm PEG-azide, and 4-arm PLP139–151 from SEC, DLS, and MALDI-TOF analysis.
| Size by SEC (Da) | Size by DLS (nm) | Size by MALDI-TOF (Da) | |
|---|---|---|---|
| hpPLP139–151 | n.d. | n.d. | 1,602 |
| 20 kDa 4-arm PEG-N3 | 26,940 | 1.7 ± 0.14 | 20,367 |
| 4-arm PLP139–151 | 28,940 | 1.8 ± 0.15 | 23,748 |
6. Acknowledgements
We gratefully acknowledge support from the National Institutes of Health Graduate Training Program in Dynamic Aspects of Chemical Biology Grant (T32 GM008545) from the National Institutes of General Medical Sciences (S.N.J.). Additionally, we would like to acknowledge support from the National Institutes of Health Biotechnology Training Grant (T32 GM008359) (M.A.C.). We would like to thank the PhRMA Foundation Postdoctoral Fellowship in Pharmaceutics and the Madison and Lila Self Graduate Fellowship for their support as well (J.D.G.). We would also like to thank the KU NMR facility and the KU Macromolecule and Vaccine Stabilization Center (MVSC) for collaboration and instrument use. Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grant # S10RR024664 and NSF Major Research Instrumentation Award # 1625923. Support for the MALDI instrumentation was provided by NIH grant P20GM103638 and NIGMS grant P20GM113117.
Footnotes
Supporting Information
Figure S1. SEC chromatograms of PEG standards. Figure S2. PLP139–151-specific IgG antibody titers in EAE mouse serum. Figure S3. Flow cytometric analysis of surface receptor expression in EAE splenocytes following in vitro treatment. Figure S4. Flow cytometric analysis of CD80 and CD86 expression in EAE splenocytes following in vitro treatment. Figure S5. ELISA analysis of cytokine expression of EAE splenocytes following in vitro treatment. Figure S6. Representative flow cytometric analysis displaying gating schemes for each fluorescent channel. Figure S7. Representative flow cytometric plots displaying changes in CD19 expression following treatment.
References
- 1.Torkildsen O; Myhr KM; Bo L, Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur J Neurol 2016, 23 Suppl 1, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones JL; Coles AJ, New treatment strategies in multiple sclerosis. Exp Neurol 2010, 225 (1), 34–9. [DOI] [PubMed] [Google Scholar]
- 3.Boster A; Edan G; Frohman E; Javed A; Stuve O; Tselis A; Weiner H; Weinstock-Guttman B; Khan O, Intense immunosuppression in patients with rapidly worsening multiple sclerosis: treatment guidelines for the clinician. Lancet Neurol 2008, 7 (2), 173–83. [DOI] [PubMed] [Google Scholar]
- 4.Fletcher JM; Lalor SJ; Sweeney CM; Tubridy N; Mills KH, T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 2010, 162 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehmann-Horn K; Kronsbein HC; Weber MS, Targeting B cells in the treatment of multiple sclerosis: recent advances and remaining challenges. Ther Adv Neurol Disord 2013, 6 (3), 161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amor S; Puentes F; Baker D; van der Valk P, Inflammation in neurodegenerative diseases. Immunology 2010, 129 (2), 154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar-Or A; Calabresi PA; Arnold D; Markowitz C; Shafer S; Kasper LH; Waubant E; Gazda S; Fox RJ; Panzara M; Sarkar N; Agarwal S; Smith CH, Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol 2008, 63 (3), 395–400. [DOI] [PubMed] [Google Scholar]
- 8.Hauser SL; Waubant E; Arnold DL; Vollmer T; Antel J; Fox RJ; Bar-Or A; Panzara M; Sarkar N; Agarwal S; Langer-Gould A; Smith CH, B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008, 358 (7), 676–88. [DOI] [PubMed] [Google Scholar]
- 9.Kappos L; Li D; Calabresi PA; O’Connor P; Bar-Or A; Barkhof F; Yin M; Leppert D; Glanzman R; Tinbergen J; Hauser SL, Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378 (9805), 1779–87. [DOI] [PubMed] [Google Scholar]
- 10.Sorensen PS; Lisby S; Grove R; Derosier F; Shackelford S; Havrdova E; Drulovic J; Filippi M, Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology 2014, 82 (7), 573–81. [DOI] [PubMed] [Google Scholar]
- 11.Dintzis HM; Dintzis RZ; Vogelstein B, Molecular determinants of immunogenicity: the immunon model of immune response. Proceedings of the National Academy of Sciences 1976, 73 (10), 3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diao L; Meibohm B, Pharmacokinetics and Pharmacokinetic–Pharmacodynamic Correlations of Therapeutic Peptides. Clinical Pharmacokinetics 2013, 52 (10), 855–868. [DOI] [PubMed] [Google Scholar]
- 13.Richter WF; Bhansali SG; Morris ME, Mechanistic Determinants of Biotherapeutics Absorption Following SC Administration. The AAPS Journal 2012, 14 (3), 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reim JW; Symer DE; Watson DC; Dintzis RZ; Dintzis HM, Low molecular weight antigen arrays delete high affinity memory B cells without affecting specific T-cell help. Mol Immunol 1996, 33 (17–18), 1377–88. [DOI] [PubMed] [Google Scholar]
- 15.Hartwell BL; Pickens CJ; Leon M; Northrup L; Christopher MA; Griffin JD; Martinez-Becerra F; Berkland C, Soluble antigen arrays disarm antigen-specific B cells to promote lasting immune tolerance in experimental autoimmune encephalomyelitis. J Autoimmun 2018, 93, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin JD; Leon MA; Salash JR; Shao M; Hartwell BL; Pickens CJ; Sestak JO; Berkland C, Acute B-Cell Inhibition by Soluble Antigen Arrays Is Valency-Dependent and Predicts Immunomodulation in Splenocytes. Biomacromolecules 2019, 20 (5), 2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leon MA; Wemlinger SM; Larson NR; Ruffalo JK; Sestak JO; Middaugh CR; Cambier JC; Berkland C, Soluble Antigen Arrays for Selective Desensitization of Insulin-Reactive B Cells. Mol Pharm 2019, 16 (4), 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sestak J; Mullins M; Northrup L; Thati S; Forrest ML; Siahaan TJ; Berkland C, Single-step grafting of aminooxy-peptides to hyaluronan: a simple approach to multifunctional therapeutics for experimental autoimmune encephalomyelitis. J Control Release 2013, 168 (3), 334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartwell BL; Martinez-Becerra FJ; Chen J; Shinogle H; Sarnowski M; Moore DS; Berkland C, Antigen-Specific Binding of Multivalent Soluble Antigen Arrays Induces Receptor Clustering and Impedes B Cell Receptor Mediated Signaling. Biomacromolecules 2016, 17 (3), 710–22. [DOI] [PubMed] [Google Scholar]
- 20.Johnson JG; Jemmerson R, Tolerance induction in resting memory B cells specific for a protein antigen. The Journal of Immunology 1992, 148 (9), 2682–2689. [PubMed] [Google Scholar]
- 21.Linton PJ; Rudie A; Klinman NR, Tolerance susceptibility of newly generating memory B cells. The Journal of Immunology 1991, 146 (12), 4099–4104. [PubMed] [Google Scholar]
- 22.Symer DE; Reim J; Dintzis RZ; Voss EW; Dintzis HM, Durable elimination of high affinity, T cell-dependent antibodies by low molecular weight antigen arrays in vivo. The Journal of Immunology 1995, 155 (12), 5608–5616. [PubMed] [Google Scholar]
- 23.Miyagaki T; Fujimoto M; Sato S, Regulatory B cells in human inflammatory and autoimmune diseases: from mouse models to clinical research. Int Immunol 2015, 27 (10), 495–504. [DOI] [PubMed] [Google Scholar]
- 24.McRae BL; Miller SD, Fine specificity of CD4+ T cell responses to the dominant encephalitogenic PLP 139–151 peptide in SJL/J mice. Neurochem Res 1994, 19 (8), 997–1004. [DOI] [PubMed] [Google Scholar]
- 25.Luca ME; Kel JM; van Rijs W; Wouter Drijfhout J; Koning F; Nagelkerken L, Mannosylated PLP(139–151) induces peptide-specific tolerance to experimental autoimmune encephalomyelitis. J Neuroimmunol 2005, 160 (1–2), 178–87. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi N; Kobayashi H; Gu L; Malefyt T; Siahaan TJ, Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J Pharmacol Exp Ther 2007, 322 (2), 879–86. [DOI] [PubMed] [Google Scholar]
- 27.Wegmann KW; Wagner CR; Whitham RH; Hinrichs DJ, Synthetic Peptide dendrimers block the development and expression of experimental allergic encephalomyelitis. J Immunol 2008, 181 (5), 3301–9. [DOI] [PubMed] [Google Scholar]
- 28.Choi CHJ; Zuckerman JE; Webster P; Davis ME, Targeting kidney mesangium by nanoparticles of defined size. Proceedings of the National Academy of Sciences 2011, 108 (16), 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saffer EM; Lackey MA; Griffin DM; Kishore S; Tew GN; Bhatia SR, SANS study of highly resilient poly(ethylene glycol) hydrogels. Soft Matter 2014, 10 (12), 1905–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sosnick TR; Benjamin DC; Novotny J; Seeger PA; Trewhella J, Distances between the antigen-binding sites of three murine antibody subclasses measured using neutron and X-ray scattering. Biochemistry 1992, 31 (6), 1779–86. [DOI] [PubMed] [Google Scholar]
- 31.Reth M, Matching cellular dimensions with molecular sizes. Nat Immunol 2013, 14 (8), 765–7. [DOI] [PubMed] [Google Scholar]
- 32.Fujimoto M; Poe JC; Inaoki M; Tedder TF, CD19 regulates B lymphocyte responses to transmembrane signals. Semin Immunol 1998, 10 (4), 267–77. [DOI] [PubMed] [Google Scholar]
- 33.Engel P; Zhou L-J; Ord DC; Sato S; Koller B; Tedder TF, Abnormal B lymphocyte delevopment, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity 1995, 3 (1), 39–50. [DOI] [PubMed] [Google Scholar]
- 34.Parker DC, T Cell-Dependent B Cell Activation. Annual Review of Immunology 1993, 11 (1), 331–360. [DOI] [PubMed] [Google Scholar]
- 35.Gauld SB; Benschop RJ; Merrell KT; Cambier JC, Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol 2005, 6 (11), 1160–7. [DOI] [PubMed] [Google Scholar]
- 36.Hartwell BL; Pickens CJ; Leon M; Berkland C, Multivalent Soluble Antigen Arrays Exhibit High Avidity Binding and Modulation of B Cell Receptor-Mediated Signaling to Drive Efficacy against Experimental Autoimmune Encephalomyelitis. Biomacromolecules 2017, 18 (6), 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarkoni Y; Getahun A; Cambier JC, Molecular underpinning of B-cell anergy. Immunol Rev 2010, 237 (1), 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manzotti CN; Tipping H; Perry LC; Mead KI; Blair PJ; Zheng Y; Sansom DM, Inhibition of human T cell proliferation by CTLA-4 utilizes CD80 and requires CD25+ regulatory T cells. Eur J Immunol 2002, 32 (10), 2888–96. [DOI] [PubMed] [Google Scholar]
- 39.Zheng Y; Manzotti CN; Liu M; Burke F; Mead KI; Sansom DM, CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol 2004, 172 (5), 2778–84. [DOI] [PubMed] [Google Scholar]
- 40.Nova-Lamperti E; Fanelli G; Becker PD; Chana P; Elgueta R; Dodd PC; Lord GM; Lombardi G; Hernandez-Fuentes MP, IL-10-produced by human transitional B-cells down-regulates CD86 expression on B-cells leading to inhibition of CD4+T-cell responses. Sci Rep 2016, 6, 20044–20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sansom DM; Manzotti CN; Zheng Y, What’s the difference between CD80 and CD86? Trends in Immunology 2003, 24 (6), 313–318. [DOI] [PubMed] [Google Scholar]
- 42.Arellano G; Ottum PA; Reyes LI; Burgos PI; Naves R, Stage-Specific Role of Interferon-Gamma in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front Immunol 2015, 6, 492–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofman FM; Hinton DR; Johnson K; Merrill JE, Tumor necrosis factor identified in multiple sclerosis brain. The Journal of experimental medicine 1989, 170 (2), 607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruddle NH; Bergman CM; McGrath KM; Lingenheld EG; Grunnet ML; Padula SJ; Clark RB, An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. The Journal of experimental medicine 1990, 172 (4), 1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komiyama Y; Nakae S; Matsuki T; Nambu A; Ishigame H; Kakuta S; Sudo K; Iwakura Y, IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 2006, 177 (1), 566–73. [DOI] [PubMed] [Google Scholar]
- 46.Serada S; Fujimoto M; Mihara M; Koike N; Ohsugi Y; Nomura S; Yoshida H; Nishikawa T; Terabe F; Ohkawara T; Takahashi T; Ripley B; Kimura A; Kishimoto T; Naka T, IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences 2008, 105 (26), 9041–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.