

Abstract
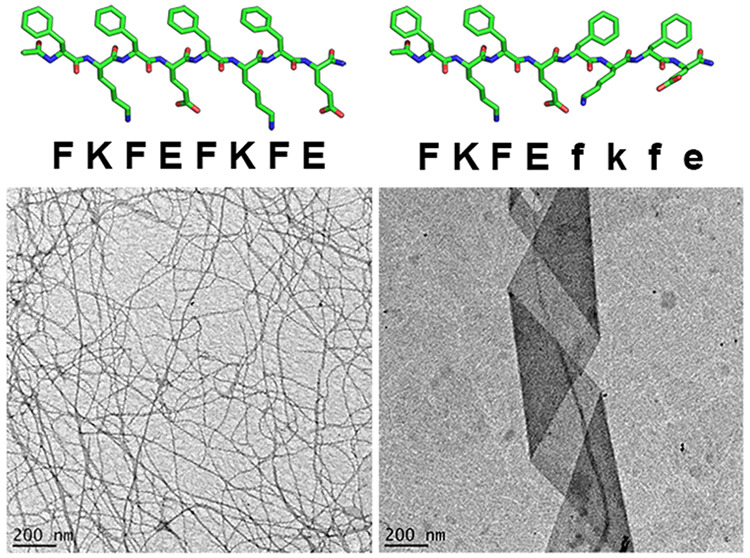
Patterned substitution of d-amino acids into the primary sequences of self-assembling peptides influences molecular-level packing and supramolecular morphology. We report that block heterochiral analogs of the model amphipathic peptide KFE8 (Ac-FKFEFKFE-NH2), composed of two FKFE repeat motifs with opposite chirality, assemble into helical tapes with dimensions greatly exceeding those of their fibrillar homochiral counterparts. At sufficient concentrations, these tapes form hydrogels with reduced storage moduli but retain the shear-thinning behavior and consistent mechanical recovery of the homochiral analogs. Varying the identity of charged residues (FRFEFRFE and FRFDFRFD) produced similarly sized nonhelical tapes, while a peptide with nonenantiomeric l- and d-blocks (FKFEFRFD) formed helical tapes closely resembling those of the heterochiral KFE8 analogs. A proposed energy-minimized model suggests that a kink at the interface between l- and d-blocks leads to the assembly of flat monolayers with nonidentical surfaces that display alternating stacks of hydrophobic and charged groups.
Self-assembling oligopeptides with alternating polar and nonpolar residues are attractive minimal systems that assemble into highly soluble fibrillar scaffolds with diverse applications in engineering and medicine.1,2 KFE8 (Ac-FKFEFKFE-NH2) has been extensively used to understand the roles of hydrophobicity, charge, ionic strength, sequence length, and residue patterning on molecular self-assembly and bulk material properties.2−6
An established method of controlling scaffold degradation is to incorporate peptides composed of non-natural d-amino acids.7,8 Studies using homochiral (all-l or all-d) enantiomers of self-assembling peptides have demonstrated differences in fibril handedness, proteolytic susceptibility, and bioactivity.9−12 Further, rippled β-sheets consisting of alternating l- and d-peptides have been shown to form hydrogels with enhanced viscoelasticity, while studies comparing homochiral and heterochiral combinations of syndiotactic and isotactic peptides found that the heterochiral combinations exhibited faster gelation kinetics and reduced viscoelasticity.13−15 As opposed to complete substitution, selective incorporation of d-amino acids is a facile approach to modulate the physicochemical and biological properties of peptide-based hydrogels.16−18 Recent findings by Marchesan and co-workers have revealed a conceptual framework describing the assembly behaviors of homochiral and heterochiral tripeptides composed of hydrophobic amino acids.16,17,19 These studies, among others, have laid the foundation for further exploration of chirality as a design tool to control emergent properties of peptide biomaterials.
In this work, we investigate the impact of block chirality patterns on peptide self-assembly and hydrogel rheological properties by substituting (d-Phe)-(d-Lys)-(d-Phe)-(d-Glu) (“fkfe”) for one or both FKFE repeats in KFE8. We synthesized the homochiral and block heterochiral analogs ll (FKFEFKFE), dd (fkfefkfe), ld (FKFEfkfe), and dl (fkfeFKFE) (Figure 1) and report that altering the chirality of a single repeat motif (ld or dl) significantly impacts peptide packing, supramolecular morphology, and hydrogel viscoelasticity.
Figure 1.
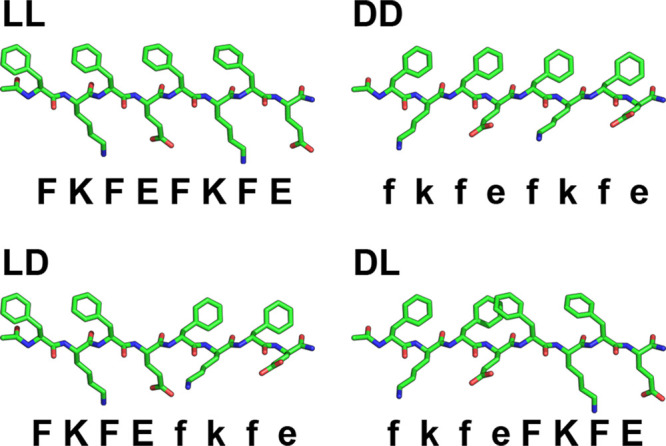
Structures of homochiral (ll and dd) and block heterochiral (ld and dl) KFE8.
Various assembly morphologies have been reported (e.g., fibrils, tapes, and ribbons) with diameters typically ranging from 3 to 20 nm and lengths on or near the micron scale.2−4,18,20−22 Transmission electron microscopy (TEM) images indicated that the homochiral ll and dd peptides reliably form nanofibers with dimensions in agreement with published values (ll: ∼8.1 ± 0.3 nm width, ∼19 nm pitch; dd: ∼7.9 ± 0.3 nm width, ∼20 nm pitch) (Figure 2A,B).4,13 In contrast, the ld and dl peptides assembled into supramolecular helices (Figure 2C,D) often appearing to possess pitch values of ∼900–1200 nm (Figure S1). A broad width distribution was observed (∼108 ± 55 nm); however, ∼90% of tapes were 50–150 nm wide with an average of ∼92 ± 22 nm (Figure S2). The helical morphology and approximate dimensions of the heterochiral assemblies were confirmed using two independent synthesis batches (Figure S3: additional TEM magnifications).
Figure 2.

Negative-stain TEM images for the ll (A), dd (B), ld (C), and dl (D) peptides at 0.25 mM in water.
Circular dichroism (CD) spectroscopy was used to determine the secondary structures of the analogs (Figure 3). The members of each enantiomeric pair produced mirror image spectra, with the chirality of the C-terminal blocks dictating their signs. Minima at ∼204 nm and ∼214 nm confirm the expected β-sheet structure of the homochiral peptide fibrils, while the ld and dl signatures are indicative of β-sheet and random coil morphologies. Second derivative FT-IR spectra of all samples showed major absorbances near 1630 and 1695 cm–1, indicative of β-sheet secondary structure and the presence of antiparallel β-strands (Figure S4).
Figure 3.
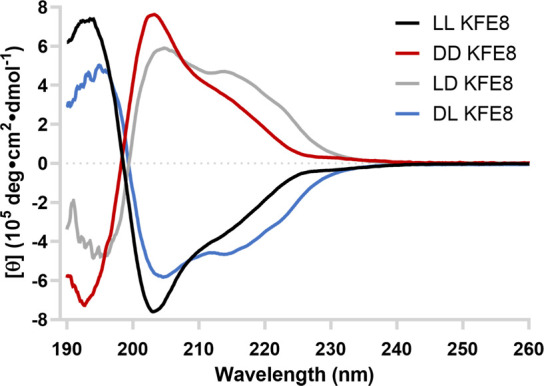
CD spectroscopy data for the peptides at 0.25 mM in water at room temperature.
Intrigued by how a small heterochiral peptide with dimensions on the order of a few nanometers or less can assemble into comparatively enormous supramolecular helical structures, we modeled supramolecular helical tapes to estimate the bend and twist angles that would yield structures of the specified pitch and radius (p S7). Based on this modeling, assembly of heterochiral peptides into supramolecular helices with a pitch of ∼1 μm and a radius of 100 nm is achieved when the bend (γν) is 0.077° and the twist (γθ) is 0.124° (Figure S5A). The corresponding values for all observed radii are shown in Figure S6. Alternatively, assembly of homochiral peptides into tighter helices with a pitch of 20 nm and a radius of 7 nm is achieved with increased bend and twist of 3.24° and 1.5°, respectively (Figure S5B).
Helical structures cannot form without both bend and twist. Twist in the absence of bend results in a nonhelical twisted fiber. The absence of bend is the result of a lack of anisotropy between the faces of a fiber, most commonly achieved in fibers that are two β-strands thick (i.e., a “ribbon”), such that the hydrophilic faces are exposed to solvent and hydrophobic faces are oriented toward the center of the fiber. Conversely, the presence of bend is typically the result of anisotropy between the faces of a fiber, most commonly achieved in fibers that are a single β-strand thick (i.e., a “tape”), such that one face is more hydrophilic and the other is more hydrophobic.
Small- and wide-angle X-ray scattering (SAXS and WAXS) confirmed morphological differences between the homochiral and heterochiral peptides at both the molecular and supramolecular levels. The ll and dd peptides formed fibrils ∼9.8 Å thick, consistent with the reported β-sheet helix.8 In contrast, the ld and dl peptides formed tapes ∼6.7 Å thick, which corresponds with monolayer sheets. WAXS curves show both similarities and differences between the homochiral and block heterochiral analogs (Figure 4B, Table S2). The strongest common peak, at 4.74–4.78 Å (q = 13.2 nm–1), represents β-sheet spacing. The ld and dl peptides have unique peaks at 5.4 and 9.6 Å, the first of which consists of a close doublet at 5.5 and 5.3 Å that matches the 5.4 Å (q = 11.4 nm–1) reflection observed in Aβ(42) amyloid films. The second, seen as a kink in the slope of the curves at 9.6 Å, roughly corresponds to β-solenoid spacing typically observed at ∼9.4 Å (q = 6.6 nm–1).23 A simple antiparallel model (Figure 5) of the heterochiral peptide forms monolayer β-sheets with a characteristic 9.5 Å repeat that approximately matches that of a β-solenoid (Figure S8).24
Figure 4.
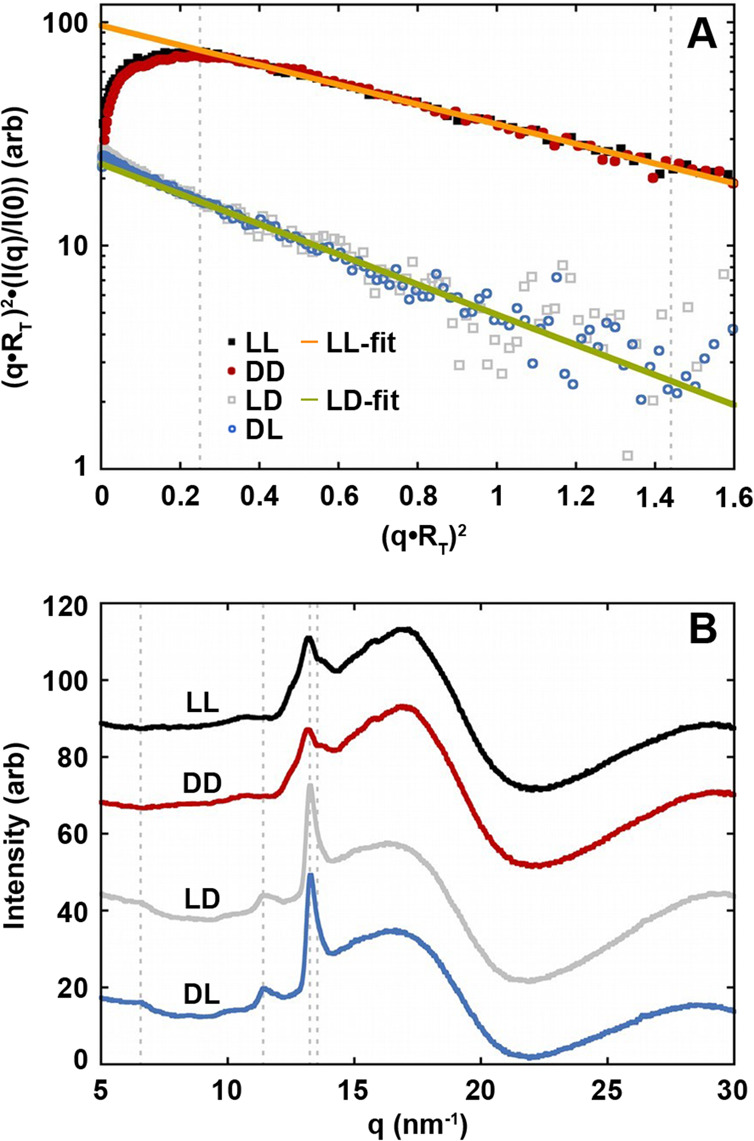
SAXS and WAXS data for the homochiral and block heterochiral peptides. (A) Kratky–Porod plots and Guinier radius of gyration of thickness fits to the ll/dd (Rt = 9.8 Å) and ld/dl (Rt = 6.7 Å) data. Guinier fits used data in the range (q·Rt)2 = 0.25 to 1.44 as marked by the vertical dashed lines. (B) WAXS plots. The dashed lines mark the published 9.4 Å β-solenoid (q = 6.6 nm–1), 5.4 Å Aβ(42)-amyloid (q = 11.4 nm–1), 4.76 Å β-sheet (q = 13.2 nm–1), and 4.64 Å β-mismatch (q = 13.5 nm–1) peaks.
Figure 5.
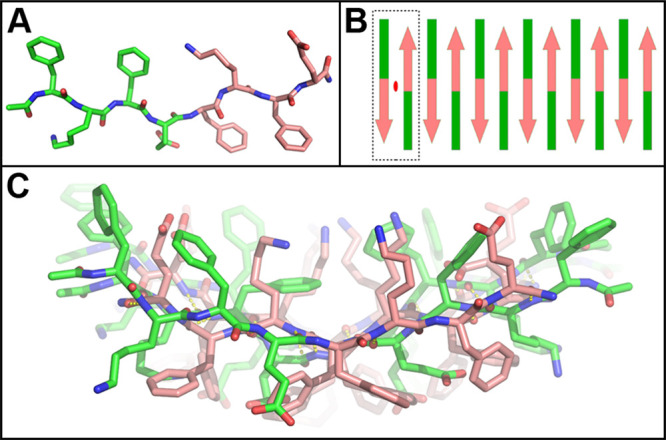
(A) MD modeling revealed that the energy-minimized ld fibril structure consists of peptides with an ∼160° rotation about the “l–d” (E4-f5) plane. (B) Peptides in the proposed structure assemble to form a fibril with a repeating unit (dashed box) that possesses internal twofold symmetry (red ellipse) between two antiparallel units. (C) The energy-minimized proto-filament model is a monolayer whose nonidentical faces include a Phe-stacked ridge (lower, middle) and a Lys-filled groove (upper, middle).
The proposed ld model maintains the typical antiparallel H-bond network. The model’s amino acid backbone is unchanged while the d-residues’ R-groups were relocated by a rotation of 120° about the peptide plane (Figure 5A). This positions the d-Phe phenyl rings below the β-sheet and introduces a kink at the Glu4-(d-Phe)5 l–d interface. This also introduces an internal strain countering the natural twist of the β-sheet and flattening it out, resulting in a much longer pitch than the homochiral KFE8 fibrils. The alternating hydrophobic and hydroscopic side chains no longer laminate to form a bilayer, and the heterochiral filament is a monolayer (Figure S8). Thus, our SAXS data and modeling both confirm the likelihood of a monolayer in agreement with theory regarding supramolecular helical tapes, suggesting that the ld supramolecular structures are likely tapes and not ribbons.
The imperceptibly small 0.13° twist per β-strand calculated from our geometric model is likely not H-bond induced, as in the homochiral peptides, but rather due to interactions between their terminal-capping acetyl and amide groups. The basic unit of the filament is an ld dimer, which has D2 symmetry perpendicular to the sheet and filament axes. The nonidentical upper and lower surfaces each have four Phe, two Glu, and two Lys stacks interspersed (Figures S8–S10). The lower surface has a central Phe-stacked ridge, while the upper surface has a Lys-filled groove. However, neither surface is predominantly hydrophobic, preventing the heterochiral filament from forming the laminated bilayers seen in homochiral KFE8 assemblies (Figure S10). It is possible that the wide sheets seen in TEM are the result of filament edge interactions (Figure S11).
The generality of this effect was examined using ll and ld analogs of RFE8 (FRFEFRFE) and RFD8 (FRFDFRFD). ll and ld analogs of KFERFD8 (FKFEFRFD) were also synthesized to determine whether enantiomeric blocks are explicitly required. In the homochiral variants, secondary structure (CD, Figure S12) and fibril morphology (TEM, Figure S13) appeared to be unaffected by the substitution of Lys with Arg (RFE8). In contrast, additionally substituting Glu with Asp (RFD8) greatly impacted hierarchical assembly, as evidenced by a large population of lower aspect ratio fibrils and loss of the characteristic CD signature of homochiral KFE8. ll KFERFD8 formed fibrils of intermediate length and produced a similarly shaped, lower-magnitude CD spectrum relative to KFE8, indicating that the presence of the FRFD block partially impedes assembly but that the homochiral peptides with nonenantiomeric blocks remain capable of fibrillizing. All ld variants formed tapes resembling those of heterochiral KFE8. Varying charged residue identities (ld RFE8 and RFD8) resulted in flat tapes without regularly spaced folds, suggesting that the observed helicity of KFE8 is not merely a surface adsorption artifact. Despite these morphological similarities, CD data indicated divergent secondary structure between the ld variants and complementary spectroscopic analyses may provide additional insight into the molecular-level effects of these substitutions.
As supramolecular peptide assemblies have been used as scaffolds for in vitro cell culture and in vivo tissue regeneration, we sought to compare the rheological properties of the homochiral and block heterochiral analogs. The steady flow curves for all peptide gels show decreasing viscosity with increasing shear rate, demonstrating shear-thinning behavior (Figure 6A). This can be quantitatively compared by fitting the viscosity vs shear rate curves to a power law model (Table S3).25 Higher flow consistency indices for the ll and dd gels confirm their stability and strong cross-linking relative to ld and dl. All gels exhibited excellent shear thinning properties, with power law indices of −0.15 and −0.13 (negative values likely indicate sample slip) for ll and dd, respectively, and 0.03 for ld and dl.26 The block heterochiral peptide gels possessed approximately 3-fold lower storage moduli at all shear rates (Figure 6A, inset). In contrast, all gels maintained their stability and showed consistent mechanical recovery after being subjected to multiple low- or high-strain cycles (Figure 6B). This is a desirable property for hydrogels intended for biomedical applications, as viscosity reduces under the shear stress of injection before recovering in situ.27
Figure 6.
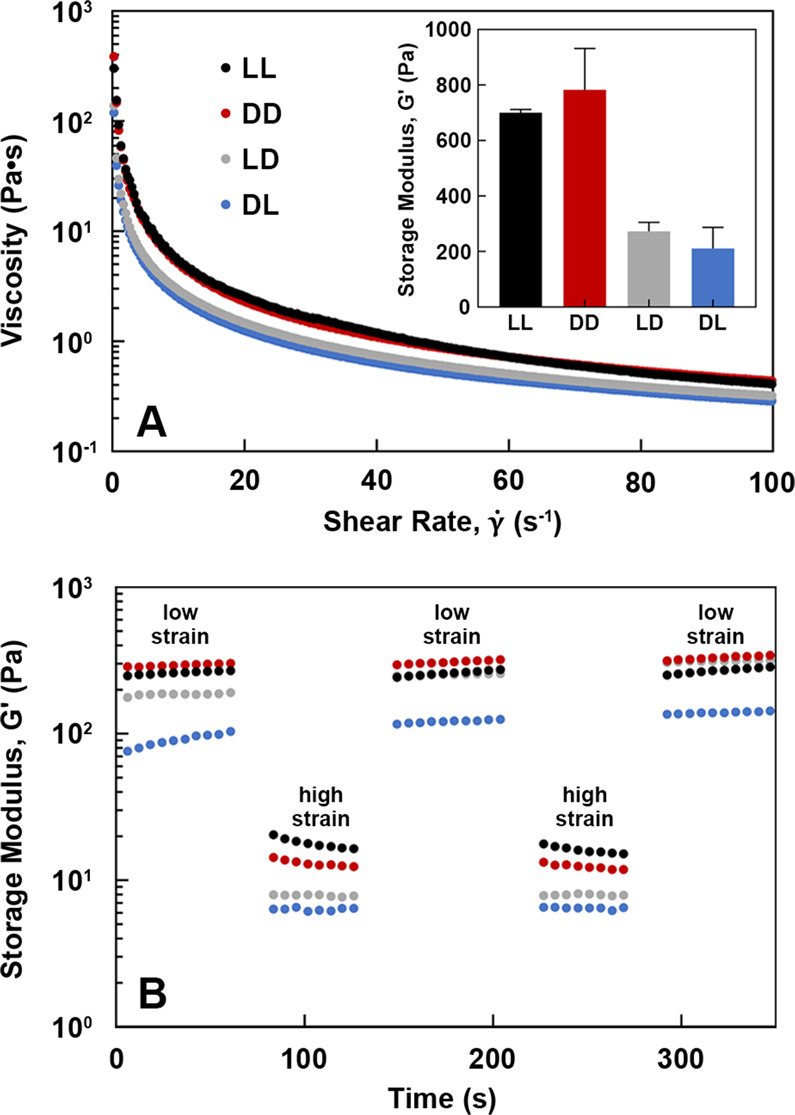
Rheological studies of peptide gels (10 mM in water). (A) Shear thinning behavior and storage moduli (inset). (B) Mechanical recovery after cyclic strain conditions.
In this communication, we present block heterochirality as a complementary strategy for the design of self-assembling peptides with alternating polar and nonpolar residues. While the use of chirality in peptide-based biomaterials has centered primarily around controlling in vivo degradation, there is an emerging interest in its utility to alter assembly mechanics and modulate bulk material properties. The Nilsson and Schneider groups have used enantiomeric mixtures of the peptides KFE8 and MAX1 to demonstrate the role of stereocomplexation (i.e., energetic favorability of interactions between enantiomers) in rippled β-sheet formation and enhanced hydrogel rigidity.13,14 This work indicates that incorporating both l- and d-repeat motifs into a single peptide facilitates unique intermolecular interactions that direct the assembly of alternate fibril morphologies. Based on the model we have proposed, rotation at the l–d interface allows for an H-bond network to be maintained and for both hydrophobic (Phe) and charged (Lys, Glu) groups to be presented on each face of a β-sheet monolayer. One can envision extending this design pattern to amphipathic peptides of various lengths and amino acid compositions to develop materials with unique morphological, physicochemical, and biological properties.
Acknowledgments
This work was supported by Washington University in St. Louis Department of Biomedical Engineering and by grants from the National Institute of Allergy and Infectious Diseases (R01 AI130278-01A1) and the National Institute of General Medical Sciences (8 P41 GM103422). We acknowledge Sealy Center for Structural Biology and Molecular Biophysics at the University of Texas Medical Branch at Galveston and the Washington University Center for Cellular Imaging for providing research resources. Thanks to Wonmuk Hwang (Texas A&M University) for providing the LL KFE8MD model.8
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b09755.
Experimental procedures; representative pitch measurements; LD and DL tape width distributions; additional TEM images; second derivative FT-IR spectra; filament bend and twist calculations; SAXS characteristics; WAXS d-spacing; visual representations, APBS electrostatic surfaces, and possible edge interactions of the energy-minimized LD KFE8 model; CD spectroscopic data and TEM images for LL and LD KFE8, RFE8, RFD8, and KFERFD8; shear thinning power law fit parameters; mass and purity characterization; HPLC chromatograms; MALDI-TOF-MS spectra (PDF)
Author Contributions
∇ T.M.C. and C.L.O. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Hamley I. W. Self-assembly of amphiphilic peptides. Soft Matter 2011, 7 (9), 4122. 10.1039/c0sm01218a. [DOI] [Google Scholar]
- Bowerman C. J.; Nilsson B. L. Self-assembly of amphipathic β-sheet peptides: Insights and applications. Biopolymers 2012, 98 (3), 169–184. 10.1002/bip.22058. [DOI] [PubMed] [Google Scholar]
- Lee N. R.; Bowerman C. J.; Nilsson B. L. Effects of varied sequence pattern on the self-assembly of amphipathic peptides. Biomacromolecules 2013, 14 (9), 3267–3277. 10.1021/bm400876s. [DOI] [PubMed] [Google Scholar]
- Marini D. M.; Hwang W.; Lauffenburger D. A.; Zhang S.; Kamm R. D. Left-handed helical ribbon intermediates in the self-assembly of a β-sheet peptide. Nano Lett. 2002, 2 (4), 295–299. 10.1021/nl015697g. [DOI] [Google Scholar]
- Lee N. R.; Bowerman C. J.; Nilsson B. L. Sequence length determinants for self-assembly of amphipathic β-sheet peptides. Biopolymers 2013, 100 (6), 738–750. 10.1002/bip.22248. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Deng L.; Wang Y.; Lu J. R.; Xu H. Different nanostructures caused by competition of intra- and inter-beta-sheet interactions in hierarchical self-assembly of short peptides. J. Colloid Interface Sci. 2016, 464, 219–228. 10.1016/j.jcis.2015.11.030. [DOI] [PubMed] [Google Scholar]
- Melchionna M.; Styan K. E.; Marchesan S. The unexpected advantages of using d-amino acids for peptide self- assembly into nanostructured hydrogels for medicine. Curr. Top. Med. Chem. 2016, 16 (18), 2009–2018. 10.2174/1568026616999160212120302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z.; Zhang S. Designer nanomaterials using chiral self-assembling peptide systems and their emerging benefit for society. Chem. Soc. Rev. 2012, 41 (13), 4736. 10.1039/c2cs15360b. [DOI] [PubMed] [Google Scholar]
- Appavu R.; Chesson C. B.; Koyfman A. Y.; Snook J. D.; Kohlhapp F. J.; Zloza A.; Rudra J. S. Enhancing the magnitude of antibody responses through biomaterial stereochemistry. ACS Biomater. Sci. Eng. 2015, 1 (7), 601–609. 10.1021/acsbiomaterials.5b00139. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Qu Y.; Waddington L. J.; Easton C. D.; Glattauer V.; Lithgow T. J.; McLean K. M.; Forsythe J. S.; Hartley P. G. Self-assembly of ciprofloxacin and a tripeptide into an antimicrobial nanostructured hydrogel. Biomaterials 2013, 34 (14), 3678–3687. 10.1016/j.biomaterials.2013.01.096. [DOI] [PubMed] [Google Scholar]
- Vargiu A. V.; Iglesias D.; Styan K. E.; Waddington L. J.; Easton C. D.; Marchesan S. Design of a hydrophobic tripeptide that self-assembles into amphiphilic superstructures forming a hydrogel biomaterial. Chem. Commun. 2016, 52 (35), 5912–5915. 10.1039/C5CC10531E. [DOI] [PubMed] [Google Scholar]
- Swanekamp R. J.; Welch J. J.; Nilsson B. L. Proteolytic stability of amphipathic peptide hydrogels composed of self-assembled pleated β-sheet or coassembled rippled β-sheet fibrils. Chem. Commun. 2014, 50 (70), 10133–10136. 10.1039/C4CC04644G. [DOI] [PubMed] [Google Scholar]
- Swanekamp R. J.; DiMaio J. T. M.; Bowerman C. J.; Nilsson B. L. Coassembly of enantiomeric amphipathic peptides into amyloid-inspired rippled β-sheet fibrils. J. Am. Chem. Soc. 2012, 134 (12), 5556–5559. 10.1021/ja301642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy K. J.; Giano M. C.; Jin A.; Pochan D. J.; Schneider J. P. Enhanced mechanical rigidity of hydrogels formed from enantiomeric peptide assemblies. J. Am. Chem. Soc. 2011, 133 (38), 14975–14977. 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraban M. B.; Feng Y.; Hammouda B.; Hyland L. L.; Yu Y. B. Chirality-mediated mechanical and structural properties of oligopeptide hydrogels. Chem. Mater. 2012, 24 (12), 2299–2310. 10.1021/cm300422q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesan S.; Easton C. D.; Styan K. E.; Waddington L. J.; Kushkaki F.; Goodall L.; McLean K. M.; Forsythe J. S.; Hartley P. G. Chirality effects at each amino acid position on tripeptide self-assembly into hydrogel biomaterials. Nanoscale 2014, 6 (10), 5172–5180. 10.1039/C3NR06752A. [DOI] [PubMed] [Google Scholar]
- Garcia A. M.; Iglesias D.; Parisi E.; Styan K. E.; Waddington L. J.; Deganutti C.; De Zorzi R.; Grassi M.; Melchionna M.; Vargiu A. V.; Marchesan S. Chirality effects on peptide self-assembly unraveled from molecules to materials. Chem. 2018, 4 (8), 1862–1876. 10.1016/j.chempr.2018.05.016. [DOI] [Google Scholar]
- Schneider J. P.; Pochan D. J.; Ozbas B.; Rajagopal K.; Pakstis L.; Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124 (50), 15030–15037. 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Styan K. E.; Easton C. D.; Waddington L.; Vargiu A. V. Higher and lower supramolecular orders for the design of self-assembled heterochiral tripeptide hydrogel biomaterials. J. Mater. Chem. B 2015, 3 (41), 8123–8132. 10.1039/C5TB00858A. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Holmes T.; Lockshin C.; Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (8), 3334. 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggeli A.; Nyrkova I. A.; Bell M.; Harding R.; Carrick L.; McLeish T. C. B.; Semenov A. N.; Boden N. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (21), 11857–11862. 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesan S.; Waddington L.; Easton C. D.; Winkler D.; Goodall L.; Forsythe J.; Hartley P. G. Unzipping the role of chirality in nanoscale self-assembly of tripeptide hydrogels. Nanoscale 2012, 4, 6752–6760. 10.1039/c2nr32006a. [DOI] [PubMed] [Google Scholar]
- Toyama B. H.; Weissman J. S. Amyloid structure: Conformational diversity and consequences. Annu. Rev. Biochem. 2011, 80, 557–585. 10.1146/annurev-biochem-090908-120656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelletto V.; Ryumin P.; Cramer R.; Hamley I. W.; Taylor M.; Allsop D.; Reza M.; Ruokolainen J.; Arnold T.; Hermida-Merino D.; Garcia C. I.; Leal M. C.; Castaño E. Self-assembly and anti-amyloid cytotoxicity activity of amyloid beta peptide derivatives. Sci. Rep. 2017, 7, 43637. 10.1038/srep43637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan G.; Verma A.; Das A.; Ojha K. Rheological studies and optimization of herschel-bulkley flow parameters of viscous karaya polymer suspensions using ga and pso algorithms. Rheol. Acta 2018, 57 (3), 267–285. 10.1007/s00397-017-1060-x. [DOI] [Google Scholar]
- Fraiha M.; Biagi J. D.; Ferraz A. C. d. O. Rheological behavior of corn and soy mix as feed ingredients. Cienc. Tecnol. Aliment. 2011, 31 (1), 129–134. 10.1590/S0101-20612011000100018. [DOI] [Google Scholar]
- Guvendiren M.; Lu H. D.; Burdick J. A. Shear-thinning hydrogels for biomedical applications. Soft Matter 2012, 8 (2), 260–272. 10.1039/C1SM06513K. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.