

Abstract
Immunotherapies are revolutionizing the clinical management of a wide range of cancers. However, intrinsic or acquired unresponsiveness to immunotherapies does occur due to the dynamic cancer immunoediting which ultimately leads to immune escape. The evolutionarily conserved histone modifier enhancer of zeste 2 (EZH2) is aberrantly overexpressed in a number of human cancers. Accumulating studies indicate that EZH2 is a main driver of cancer cells’ immunoediting and mediate immune escape through downregulating immune recognition and activation, upregulating immune checkpoints and creating an immunosuppressive tumor microenvironment. In this review, we overviewed the roles of EZH2 in cancer immunoediting, the preclinical and clinical studies of current pharmacologic EZH2 inhibitors and the prospects for EZH2 inhibitor and immunotherapy combination for cancer treatment.
Keywords: : cancer immunotherapy, epigenetics, EZH2, immune editing
The emerging role of immune therapies in cancer
Drugs that enhance the patient's immune response against malignant cells are generically called ‘cancer immunotherapies’. These treatments are rapidly emerging as an effective therapeutic option for several solid tumors, most notably advanced melanoma, non-small-cell lung cancer, renal cell carcinoma and bladder cancer [1]. In these and other malignancies, immunotherapies display remarkable response rates and significantly improve patient survival. However, other common solid tumors (breast, prostate, colon) seem to be less susceptible to cancer immunotherapies. Even within the more ‘immune-susceptible’ cancers, oncologists have observed substantial inter-patient differences, with some exceptional responders and some intrinsically refractory patients. Finally, in some cases immunotherapies induce short-lived responses, which are followed by a rapid tumor progression [2,3]. All these clinical observations suggest that cancer cells and their microenvironment trigger molecular mechanisms that reduce or completely abrogate the efficacy of immunotherapies. Of note, the immune-resistant mechanisms in cancer immunotherapy overlap with the immunoediting process used by cancers to evade immunosurveillance, such as reduced antigenicity and immunogenicity, failure to recruit immune effector cells and decreased antitumor effector function caused by various immune checkpoints and suppressive tumor microenvironment [2–5].
In addition to DNA mutations, epigenetic alterations have been recently identified as driver of immunoediting and resistance to cancer immunotherapies. Epigenetic modifications are defined as heritable phenotypic changes that are not due to changes in DNA primary structure [6]. Unlike genetic mutations, epigenetic alterations are reversible and are targetable by clinically approved drugs. Hence, these alterations offer a unique possibility to modulate the gene expression profile of the tumor, in order to make it more susceptible to immunotherapies and to prevent the emergence of resistant clones. One particular epigenetic effector enhancer of zeste 2 (EZH2) is more and more recognized as a main driver of cancer cells’ immunoediting and mediates immune escape through downregulating immune recognition and activation, upregulating immune checkpoints and creating an immunosuppressive tumor microenvironment [7–10]. In this review, we will focus on the mechanisms of immunotherapy and immunotherapy resistance, the role of EZH2 in immunoediting and the potential of using EZH2 inhibitor to overcome immunotherapy resistance.
The interaction between cancer & the immune system
Cancer remains one of the leading causes of death in the world. According to the immunosurveillance theory, cancer initiation and development are constantly monitored and restrained by the immune system [11,12]. Cytotoxic effector cells play key roles in mediating cancer surveillance and elimination. In particular, CD8+ cytotoxic T lymphocytes (CTLs) are considered the most prominent tumoricidal effector cells. Upon priming and activation by antigen-presenting cells, CTLs can perform potent and tumor-specific killing in a MHC class I-restricted manner [13]. Besides, innate cytotoxic effector cells, such as natural killer (NK) cells, γδ T cells and tissue residing macrophages, are able to display rapid cytolytic activity in response to transformed cells [14–16]. Apart from the aforementioned cytotoxic effector cells, CD4+ T helper 1 (Th1) cells have a crucial role in anti-tumor immunity: Th1 cells provide critical assistance in the priming, activation and cytotoxicity of CTLs [17]; Th1 cytokines can promote the tumoricidal activity of macrophages and NK cells [18].
Theoretically, cancer development and outgrowth can be inhibited by cytotoxic innate and adaptive immune cells. However, despite the existence of a normal host immune system, many cancers manage to persist and progress. During this process, cancer cells evolve different mechanisms to avoid tumoricidal attack by negatively shaping anti-tumor immune responses [19,20].
Immune selection, immune editing & immune escape
Although cancer originates from a single transformed cell, it eventually develops into a heterogeneous population with various phenotypes due to genetic instability and epigenetic plasticity. When effective immunosurveillance posts a selection pressure on this heterogeneous population, some cancer clones are killed whereas other clones that are unsusceptible or resistant to antitumor immune responses are selected and maintained. Therefore, cancer development is considered as a Darwinian evolutionary process where the immune response is able to drive the cancer evolution toward an immune-resistant phenotype [21–24]. Particularly, it has been proposed that surviving epithelial cancers may acquire immune-suppressive preparties via a transdifferentiation process of ‘epithelial immune cell-like transition’, which enables the cancer cells to ‘communicate’ with immune cells [25]. Hence, the host immune system can both constrain and promote cancer development through the process of ‘cancer immunoediting’ [5,26].
Cancer immunoediting includes three sequential phases (‘three Es’, Figure 1), elimination, equilibrium and escape [4]. During these three phases, cancer gradually acquires active immune suppressive functions through reducing antigenicity (loss of neoantigens and/or poor antigen presentation), decreasing immunogenicity (failure to recruit and/or activate effector cells) and creating an immunosuppressive tumor microenvironment (TME) [27]. At the elimination phase, cancer cells can be effectively removed by immune effector cells. When certain variants survive but remain quiescent/dormant, cancer progresses into the equilibrium phase. At the equilibrium phase, cancer variants manage to avoid elimination mostly through reducing antigenicity [28,29]. Cancer then gradually shifts to the escape phase when more complicated immune evading phenotypes are acquired. First, cancer can further reduce neoantigen expression and acquire deficiencies in antigen presentation by loss of MHC I (human leukocyte antigen A, B and C, HLA-A, -B and -C) expression or dysregulation of antigen processing machinery [30,31]. By doing so, cancer cells can avoid the recognition and cytotoxic effect of T cells. Besides, some cancer can upregulate nonclassical HLA molecules (such as HLA-E and HLA-G), which are usually expressed at immune-privileged sites, to limit NK reactivity upon interaction with inhibitory receptors [31–33]. Second, cancer can upregulate co-inhibitory receptors/ligands to limit anti-cancer activity. Among them, programmed cell death protein 1 (PD-1), PD-1 ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) are potent immune checkpoints involving in the negative regulation of T-cell reactivity [34,35]. Besides, a growing list of other co-inhibitory receptors, including T-cell immunoreceptor with Ig and ITIM domains (TIGIT), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) and Lymphocyte-activation gene 3 (LAG-3), also play important roles in undermining T- and NK-cell responses [36–38]. Third, cancer is able to create an immunosuppressive TME flooded with immunosuppressive factors and immunosuppressive cells. The immunosuppressive factors include cytokines such as IL-4, IL-10, IL-13, IL-23 and TGF-β [39], as well as metabolites such as indoleamine 2,3-dioxygenase-1 (IDO) [40] and lactic acid [41,42]. The immunosuppressive cells, including Tregs, myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs, M2 macrophages), are capable of repressing effector activity and are either induced in situ or newly recruited and settled to the suppressive TME [43]. Gradually, the immune system becomes incompetent to suppress cancer growth. Cancer successfully escapes and progresses into clinical overt cancer.
Figure 1. . Schematic representation of the ‘3E’ theory.
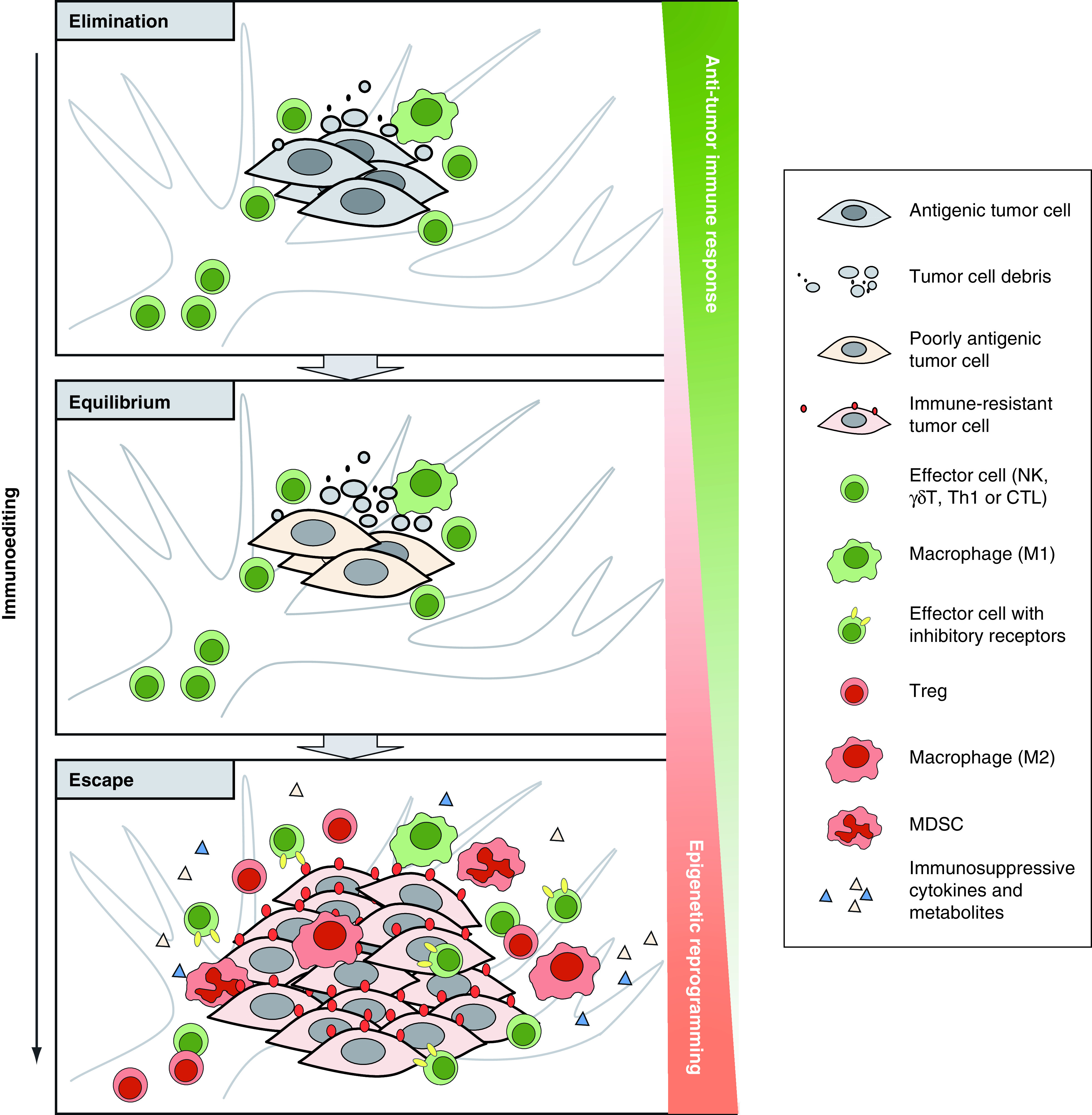
Cancer immunoediting includes three sequential phases: elimination, equilibrium and escape. At the elimination phase, antigenic cancer cells can be effectively removed by immune effector cells. At the equilibrium phase, some cancer cells which have a nonimmunogenic phenotype manage to evade the immune elimination and survive. As such, a balance is established between cancer and the immune system. At the escape phase, cancer evades the immune response through more complicated mechanisms, such as downregulating antigenicity, upregulating immune inhibitory molecules and generating an immunosuppressive TME flooded with suppressive cells and factors. Cancer manages to grow in an uncontrolled manner. In the whole process of immunoediting, the accumulation of epigenetic alterations leads to profound changes in tumor cells and tumor microenvironment that contribute to decreased antitumor immunity and tumor immune escape.
The emerging role of immunotherapies: restoring immunosurveillance & counteracting immune escape mechanisms
Nowadays, escape from immunosurveillance is increasingly recognized as a hallmark event in cancer initiation and progression. Immunotherapies aiming at restoring immunosurveillance and counteracting immune escape mechanisms are revolutionizing the clinical management of a wide range of cancers [20,27].
The most straightforward way to restore cancer immunity is by increasing the number of effector cells. Anti-cancer effector cells can either be activated and expanded in vivo through cancer vaccine immunization or be expanded and manipulated in vitro and then transferred to cancer patients as seen in adoptive cell therapy and engineered chimeric antigen receptor (CAR)-T cell therapy [44,45]. Besides, antibody-based targeting therapeutics are able to recruit effector cells to the tumor site [46]. For cancer types that exclude efficient immune cell infiltration (immunologically cold), a series of targeted therapies can be used to reprogram the TME and make it more permissive to effector cells. These therapies include: Toll-like receptor (TLR) agonists [47,48], VEGF neutralizing antibodies [49], signal transducer and activator of transcription 3 (STAT3) inhibitors [50,51], Wnt/β-catenin pathway inhibitors [52] and stimulator of interferon genes agonists [2,53,54]. Some of these therapies are currently approved for the treatment of specific malignancies. For example, the anti-VEGF antibody bevacizumab is employed for the treatment of metastatic renal, colorectal and ovarian cancer and for recurrent gliomas [55]. Other targeted therapies are being tested in Phase I/II clinical trials [56].
Another effective strategy that enhances the antitumor immune response is to counteract immune inhibitory mechanisms. Co-inhibitory receptors (particularly immune checkpoint receptors) upregulated on effector T and NK cells can dampen anti-cancer responses. Accordingly, immune checkpoint blockade (ICB) therapies targeting PD-1 and CTLA-4 have demonstrated clinical efficacy in a variety of solid tumors and are now the standard treatment for advanced melanoma and non-small cell lung carcinoma (NSCLC) [57,58]. In addition, antibodies that block other co-inhibitory receptors such as TIGIT, TIM-3 and LAG-3, either alone or in combination with ICB, have also shown satisfactory efficacies in mouse models and are now being tested in Phase I/2/3 clinical trials for the treatment of melanoma, renal cell carcinoma, NSCLC and other solid tumors [2,43,59–61].
Apart from co-inhibitory receptors, the anticancer activity of effector cells is also strongly inhibited by the immunosuppressive TME. Various antagonists that block the action of immunosuppressive cytokines (e.g., IL-23 [62,63] and TGF-β [64,65]) and metabolites (e.g., IDO [40]) have been shown to effectively prevent the induction and enrichment of the suppressive cells in TME in preclinical studies. Among them TGF-β and IDO antagonists, either alone or in combination with ICB, chemotherapy or radiotherapy, are being tested in Phase I/II trials for the treatment of patients with solid tumors [40,66]. In addition, the depletion or reprogramming of TME suppressive cells (e.g., Tregs [67,68], myeloid-derived suppressor cells [69,70] and M2-TAMs [71,72]) have been reported to enhance anticancer immune responses in both preclinical and clinical settings.
Of interest, cancer prefers aerobic glycolysis over the more energy-efficient oxidative phosphorylation pathway, a property known as the ‘Warburg effect’ [73,74]. This altered metabolism results in the accumulation of lactic acid and consequent acidification of TME. Notably, the acidic TME has been linked to multiple mechanisms of immune suppression and become an attractive target for cancer immunotherapy [41,75,76]. Blocking lactic acid transportation through monocarboxylate transporter 1 (MCT 1) and/or 4 (MCT 4) have been shown to inhibit tumor growth in preclinical studies [75,77,78]. Moreover, MCT1 inhibitor AZD3965 is currently being tested in a Phase I trial for the treatment of patients with small-cell lung carcinoma [79].
Considering the complexity and diversity of the immune context of the TME, the response rate to certain immunotherapies may differ substantially between cancer patients. For this reason, Binnewies et al. have stratified tumor immune microenvironment into three classes according to the composition of the immune infiltrate and the character of the inflammatory response [43]. Similarly, O'Donnell et al. have proposed a four-type TME stratification model, which also takes tumor mutational burden into consideration, along with tumor inflammatory gene signature [2]. Although both models are valuable in predicting clinical responsiveness and guiding the proper selection of immunotherapeutic strategies, given the fact that altered metabolism (particularly elevated lactic acid production) in TME can cause general immune suppression and dampen the clinical efficacy of cancer immunotherapies [41,80], it is important to also consider the metabolic signature of TME in this respect.
Epigenetic immune editing: an early mechanism of therapy resistance
Intrinsic & acquired resistance to immunotherapy
As discussed above, owing to the complex immunoediting and immune escape mechanisms, some cancers can be effectively controlled by immunotherapies, whereas others are intrinsically unresponsive or resistant to it. Significantly, even for those cancers that respond well to immunotherapy, the responses may be transient due to potent acquired resistance mechanisms [81–84]. For example, intrinsic ICB resistance is most likely due to low tumor mutational burden and/or poor immune cell infiltration which leads to minimal immune recovery with both CTLA-4 and PD-1/PD-L1 blockade [85–87]. Whereas acquired ICB resistance has been linked to more complex mechanisms of adaptation, like loss of neoantigen, poor antigen presentation, failure to recruit and activate T cells and upregulation of alternative immune checkpoints [88–92].
Of note, immunoediting used by cancer to evade immunosurveillance also occurs in cancer undergoing immunotherapy. While effective immunotherapy can suppress cancer progression, it may also lead to immunoselection and outgrowth of resistant cell clones. Thus, to fully eradicate a clinically overt cancer, it is necessary to effectively disrupt the evolutionary processes leading to cancer cell adaptation and therapeutic resistance.
The role of epigenetics in immunoediting
Intratumoral heterogeneity is a necessary precondition for cancer progression and for immunoselection. Emerging evidence suggests that epigenetic modifications are key mediators of cancer heterogeneity [93]. It has been suggested that epigenetic modifications often precede genetic alterations and that these reversible alterations confer lineage plasticity to cancer cells [94]. Epigenetic mechanisms include DNA methylation and histones post translational modifications. Histones are basic proteins that compact the DNA into structural units known as nucleosomes. Each nucleosome is composed of an octamer containing two copies of each core histone (H2A, H2B, H3 and H4). Approximately 147 bp of DNA are wrapped around each nucleosome. Each core histone contains a long N-terminal tail, which can be modified by a number of post translational modifications (acetylation, methylation, phosphorylation) [95]. While DNA methylation of promoters is associated with gene silencing, histones post translational modifications can enhance or suppress local gene expression, depending on the histone residue affected and on the nature of the chemical modification. For example, histone acetylation favors a transcriptionally active chromatin structure and is therefore associated with active gene expression. These modifications are able to affect the genomic structure and regulate gene expression without affecting the DNA sequence [96].
Aberrant epigenetic modifications are widely described as essential players in cancer immunoediting and immune escape [97–99]. For example, it has been shown that the loss of HLA and the depletion of expressed neoantigens are due to increased DNA methylation and reduced histone acetylation [23,29]. In addition, DNA hypomethylation and increased histone acetylation status of the CTLA-4, PD-1 and PD-L1 promoter regions have been shown to be associated with the upregulation of these genes in the TME [100–102]. Moreover, growing evidence suggests that cancer can shape the immune landscape of TME through changing the epigenetic modification portfolio of relevant genes. Thereby, a suppressive cytokine environment can be established and suppressive cellular components can be induced and recruited [90,103–107].
Unlike genetic alterations, epigenetic alterations can be reversed pharmacologically [108]. Notably, epigenetic modifiers, like inhibitors of DNA methyltransferase or histone deacetylase was shown to be able to synergize with immunotherapy for the eradication of mouse tumor models [109–111]. Moreover, epigenetic therapy in combination with ICB therapy was proven to produce longer periods of disease-free survival and increased overall survival in NSCLC patients [112]. Therefore, epigenetic therapies have the potential to reprogram the immunosurveillance- and immunotherapy-driven cancer evolution. When combined, epigenetic therapy may potentiate current immunotherapy to maximize benefits and overcome acquired therapy resistance.
EZH2 functions in immune editing
Emerging evidence indicates that several of the aforementioned mechanisms of epigenetic editing are mediated by the evolutionarily conserved histone modifier EZH2. The EZH2 locus encodes the catalytic subunit of the polycomb repressive complex 2 (PRC2), a multimeric complex that mediates gene silencing via histone H3 lysine 27 trimethylation (H3K27me3) [113]. Other PRC2 components include SUZ12 (suppressor of zeste 12) and EED (embryonic ectoderm development), which stabilize the complex and promote the dissemination of the H3K27me3 to nearby nucleosomes (Figure 2). Gene duplication in higher vertebrates has generated the EZH1 locus, which encodes another methyl-transferase with partially redundant functions [114]. Protein sequence alinement revealed that EZH2 and EZH1 are highly conserved in vertebrates and that these proteins are the only enzymes capable of catalysing the H3K27me3 modification. It is still not clear to which extent EZH1 and EZH2 functions differ in physiological conditions. Notably, the gene silencing activity of PRC2 can be directed to specific loci by interactions with long noncoding RNAs (lncRNAs), such as HOTAIR and NEAT1 [115]. In addition to the PRC2-dependent gene silencing, several non-canonical functions of EZH2 have been recently identified. These include gene silencing via H3K9me3 [116,117], the activation of the transcription factors STAT3 [118] and NFKB [119]. Hence, EZH2 functions include both gene repression and activation and seem to be highly dependent on the local interaction with other macromolecules.
Figure 2. . Canonical (left) and non-canonical (right) EZH2 mechanisms of action.
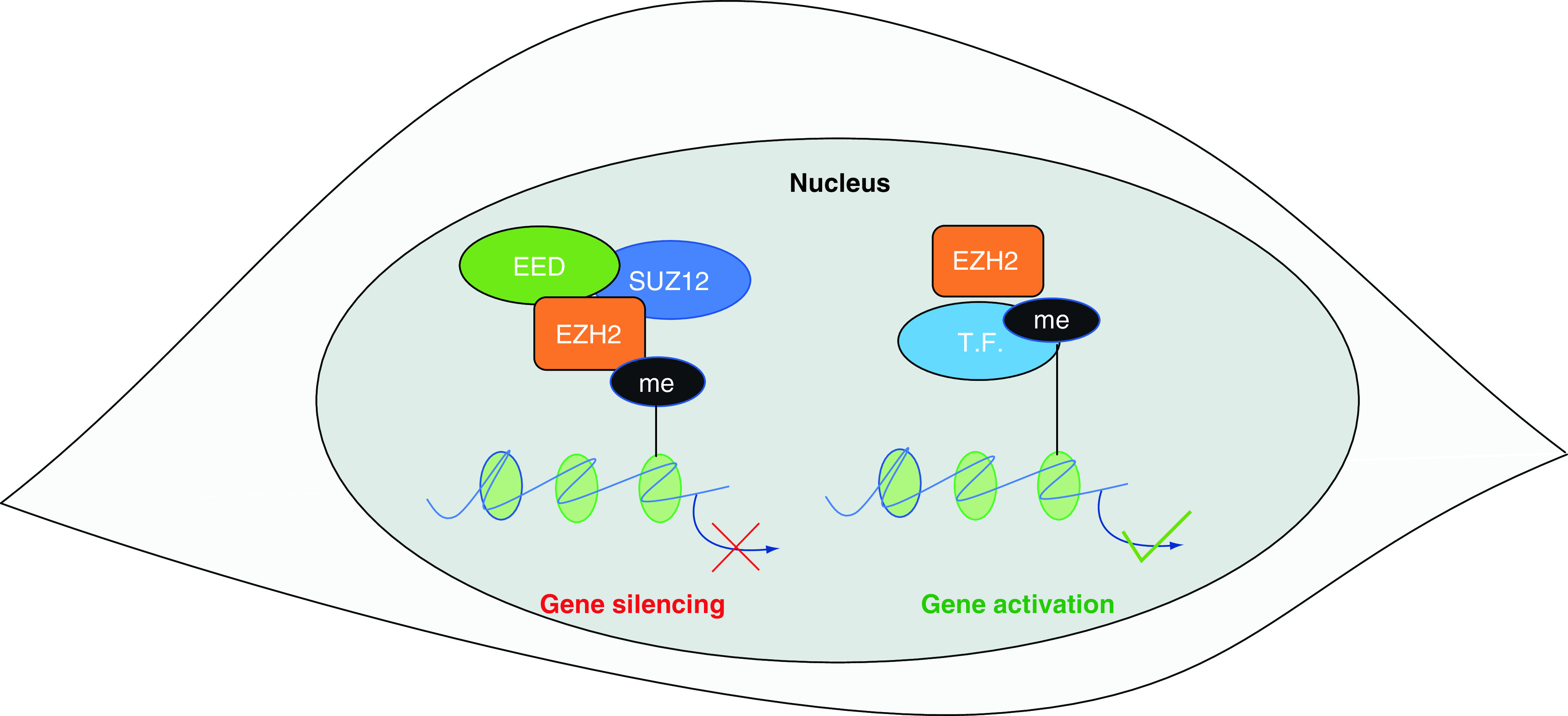
The canonical mechanism of action of EZH2 involves gene silencing via H3K27 tri-methylation. In this case, EZH2 acts as the catalytic subunit of polycomb repressive complex 2 (which includes SUZ12 and EED). Emerging evidence indicates that EZH2 can also act via PRC2-independent mechanisms. These noncanonical mechanisms of action include the methylation and activation of some transcription factors.
PRC2 proteins regulate the self-renewal of embryonic and hematopoietic stem cells [120] and the correct development of the immune systems [121]. PRC2 target genes are highly dependent on the cellular context; for example, in embryonic stem cells PRC2 silences the expression of cell cycle inhibitors and lineage-specific genes [120]; in lymphoid cells PRC2 controls the timely expression of cytokines and other factors that regulate the correct maturation and activation of B and T cells [121].
The importance of PRC2 proteins in embryo development is confirmed by the observation that homozygous mutations in any of the PRC2 components cause early embryonic lethality in mice [122]. In humans, congenital heterozygous mutations of EZH2 cause a wide spectrum of pathologic phenotypes, ranging from tall stature without intellectual disability to typical Weaver syndrome (tall stature, facial dysmorphia and intellectual disability) [123].
Beyond its physiological functions, EZH2 has been shown to regulate the activity of cancer cells. EZH2 overexpression has been identified in a wide variety of solid malignancies, including breast, prostate and lung cancers [113]. Notably, EZH2 over-expression is almost invariably associated with worse prognosis in different cancers [124,125]. Mechanistically, EZH2 controls the coordinated inactivation of several tumor suppressor genes, thereby promoting cancer growth, metastasis and drug resistance [126,127]. Moreover, EZH2 expression in the TME can indirectly promote cancer progression. For example, EZH2 has been shown to stimulate the production of pro-angiogenic factors by tumor associated fibroblasts [128]. Considering the pro-oncogenic roles of EZH2 in both cancer cells and tumor-associated cells, it is not surprising that a variety of EZH2 inhibitors are being tested in cancer clinical trials (as discussed in the next section). It is interesting to note that, even though EZH1 has partially redundant physiological functions, the oncogenic role of this gene is much less investigated. However, some reports suggest that EZH1 cooperates with EZH2 to drive a malignant phenotype [129]. As will be discussed in the next section, most of the currently available PRC2 inhibitors preferentially target EZH2. Hence, EZH1 overexpression could be a mechanism of intrinsic or acquired resistance to these drugs. It will be therefore important to test dual EZH1/EZH2 inhibitors for cancer treatment [130].
The recent success of cancer immune therapies has attracted attention on a less investigated feature of PRC2, in other words, its ability to regulate the relationship between cancer and the immune system. To understand the complex roles of PRC2 in these contexts, we must bear in mind that the functions of most epigenetic effectors are cell specific. For this reason, we will discuss the immune-editing functions of PRC2 in the context of the adaptive and innate immune response.
EZH2 suppresses the activation of the adaptive immune response
A growing body of evidence indicates that, in addition to its oncogenic functions, intratumoral EZH2 expression inhibits the adaptive immune response (Figure 3A). One of the main mechanisms of cancer immune evasion is the suppression of the MHC-I antigen processing pathway (MHC-APP). A recent genome-wide functional screening revealed that PRC2 genes are essential for promoting the coordinated silencing of MHC-APP genes in both leukemia and solid cancer cells [7]. The study showed that increased H3K27me3 at MHC-APP loci had a double effect: it reduced the basal expression of these genes; it inhibited the activation of these genes, which is normally induced by IFN-γ. These mechanisms of epigenetic repression are essential for evading the immune surveillance of effector T cells. Upon analysis of a wide range of cancer cell lines, the authors of this study found that neuroendocrine cancers show the lowest expression of MHC-APP proteins and the highest expression of PRC2 genes. Interestingly neuroendocrine trans-differentiation is emerging as a common mechanism of resistance to targeted therapies in prostate and lung cancers [131]. We have shown that PRC2 genes drive neuroendocrine trans-differentiation in prostate cancer [132]. Hence it is conceivable that the ability to evade the immune response is one of the microevolutionary pressures driving the PRC2-dependent neuroendocrine trans-differentiation.
Figure 3. . EZH2 regulates several aspects of the adaptive and innate antitumor immune response.

(A) Intratumoral EZH2 expression suppresses the adaptive antitumor immune response. EZH2 expression in cancer cells inhibits T-cell activation by suppressing the MHC-I antigen presentation pathway and upregulating PD-L1 expression. Besides cancer cells, EZH2 expression in Treg cells is crucial for maintaining their regulatory phenotype and function, which also results in reduced T-cell activation. (B) Intratumoral EZH2 expression suppresses the innate antitumor immune response. EZH2 expression in cancer cells silences the expression of NKG2D ligands, thereby blocking the activation of NK cells and favors the secretion of CCL5, which recruits immunosuppressive M2-type macrophages to TME. Besides cancer cells, EZH2 expression in NK cells directly inhibits their maturation, activation and antitumor activity.
NK: Natural killer.
Another mechanism by which cancer cells inhibit the T cell-mediated immune response is the upregulation of the immune checkpoint protein PD-L1. In lung cancer cells, EZH2 promotes the expression of PD-L1 by activating the hypoxia-inducible factor 1α [133]. A clinical study on resected lung adenocarcinomas confirmed the positive correlation between EZH2 and PD-L1 expression [8]. However, an opposite correlation between these two genes has been identified in hepatocellular carcinoma. In this case, EZH2 directly induces H3K27me3 at the promoter of PD-L1, thereby triggering gene silencing [134]. These results highlight the cell-specificity of PRC2 proteins, which will need to be considered in future translational studies.
In addition, EZH2 controls the adaptive response via regulation of Treg activity. As discussed, Tregs are a subtype of T cells that induce immunotolerance toward cancer cells. In particular, Treg cells produce a range of cytokines that suppress the activation and recruitment of both cytotoxic (CD8+) and helper (CD4+) T effector T cells. It has been shown that EZH2 is essential for the maintenance of the Treg phenotype. EZH2 inhibition in Treg cells leads to the recruitment of CD4+ and CD8+ effector T cells, which elicit a potent antitumor response [9]. Hence, EZH2 expression in both cancer cells and tumor-infiltrating T cells seem to elicit an immune-tolerant phenotype and to promote cancer progression, thus positioning EZH2 inhibitors as attractive potential adjuvants for immunotherapy.
EZH2 controls the phenotype of TAMs & NKs
Intratumoral EZH2 expression has also been shown to control the innate immunity (Figure 3B). NKs are lymphocytes that recognize and kill cancer cells in an antigen-independent manner [135]. NKs express activating NKG2D receptors on their surface. When an NKG2D receptor binds its ligand, which can be expressed in cancer cells, NKs initiate a cytotoxic immune response. Cancer cells expressing EZH2 can suppress the expression of NKG2D ligands, thereby escaping the NK cytotoxic response [136]. Notably, EZH2 can also be expressed by NK cells. In this case, EZH2 directly inhibits NK differentiation and activation [137], hence EZH2 inhibition could have a double positive impact in eliciting a NK-mediated immune response against cancers.
Another important component of the TME is represented by TAMs. Two main types of TAMs have been described in the TME: M1-TAMs are characterized by the expression of inflammatory cytokines and by cytotoxic capabilities; M2-TAMs express anti-inflammatory mediators and tend to reduce the local immune response, thereby favoring cancer progression. Hence, cancer cells tend to recruit M2-TAMs in the TME. EZH2 has been implicated also in this phenomenon. For example, EZH2 expression in lung cancer cells increases the secretion of the chemokine CCL5, which triggers the recruitment of TAMs [10]. This process promotes invasion and metastasis. In gliomas, EZH2 expression shifts macrophages and microglia from M1 to M2 phenotype, thereby reducing the anticancer immune response [138].
In addition to these mechanisms of direct immune response inhibition, EZH2 has been also implicated in cancer metabolic reprogramming. For example, in glioblastomas EZH2 favors the production of lactic acid via the Warburg effect [139]. As discussed in the previous paragraphs, the creation of an acidic TME is a generalized mechanism of immune suppression. Taken together, these results indicate that intratumoral EZH2 expression is a potent inhibitor of the immune response.
Overview of pharmacologic EZH2 inhibitors
Pharmacologic inhibition of EZH2 has become an active field of research following numerous studies that have clearly highlighted the pathophysiological implications of PRC2 activation in numerous human malignancies. The first EZH2 inhibitor with demonstrated antitumor activity was 3-deazaneplanocin A (DZNeP), a drug initially developed to target the Ebola virus [140]. Mechanistically, DZNeP inhibits the enzyme S-Adenosyl-L-homocysteine thereby reducing global H3K27me3 levels [141]. Initial studies have shown that DZNeP induces significant apoptotic cell death in cancer cells but not in normal cells [142] while effectively targeting subpopulations of cells with stem-like properties [143]. However, DZNeP carries little clinical value as a drug due to its hydrophilic nature and rapid renal excretion [144]. Thus, an increasing number of more potent and cell-permeable compounds targeting EZH2 have been developed and evaluated in clinical trials.
Tazemetostat
To date, Tazemetostat (Tazverik™) is the most extensively studied EZH2 inhibitor in the clinical setting. Notably, it has received accelerated approval in January 2020 in the USA as frontline therapy for selected cases of epithelioid sarcoma [145]. It was derived from EPZ005687 following optimization of pharmacokinetic properties [146]. Preclinical data indicate that Tazemetostat has a 35-fold higher affinity for EZH2 over EZH1 and with Ki value of <2.5 nM (data from Epizyme). Preclinical studies in cell lines have shown IC50 in the nanomolar range for both wild type (WT) and mutant EZH2 [147] with marked tumor reduction in xenograft models and concurrent H3K27me3 depletion [148].
In total, 22 clinical trials involving Tazemetostat have been initiated including two Phase III trials that are currently recruiting patients. In a Phase I study, Tazemetostat demonstrated a favorable safety profile (NCT01897571). Adverse events were generally of grade 1 and 2 severity and consisted mostly of asthenia (33%), anemia (14%), anorexia (6%), muscle spasms (14%), nausea (20%) and vomiting (9%) [149]. No deaths were recorded and only one grade 4 thrombocytopenia occurred. This study established the dose of 800 mg oral administration, twice a day (PO BID) for completion of Phase II clinical trials involving Tazemetostat. The anticancer activity of Tazemetostat was evaluated in this study, which showed durable objective responses in 38% of patients with B-cell non-Hodgkin lymphoma, including some complete responses. However, only 5% of patients with solid tumors exhibited a durable objective response [149].
In a Phase II trial, Tazemetostat induced responses in 15% of patients with 67% of the responses lasting more than 6 months [150]. This was a multicenter, double-blind, placebo-controlled, randomized Phase III study leading to the recent accelerated approval of Tazemetostat in epithelioid sarcoma, thus positioning Tazemetostat as the first molecularly targeted therapy for this aggressive type of cancer (NCT04204941).
In another Phase Ib clinical trial, Tazemetostat has been investigated in combination with the chemotherapy regimen R-CHOP in patients with aggressive diffuse large B-Cell lymphoma (DLBCL) [151]. The study consisted of 17 patients with ages ranging from 60 to 80 years old. Only two dose-limiting toxicities were recorded. In total, 47% of patients experienced grade 3–4 hematologic adverse events. Other commonly observed side effect included constipation (24%), nausea (12%), as well as hypokalemia (12%). This study also established 800 mg bid as the recommended Phase II dose [151].
To date, we are still awaiting publicly available results from eight clinical trials involving Tazemetostat. Notably, there is a completed Phase II trial investigating the use of Tazemetostat in advanced mesothelioma with BAP1 loss of function (NCT02860286). In addition, another Phase Ib/III is double-blind, randomized, of Tazemetostat or placebo in combination with Lenalidomide plus Rituximab is recruiting patients diagnosed with recurrent follicular lymphoma (NCT04224493).
GSK2816126
GSK2816126 is another EZH2 inhibitor that has made its way into clinical testing. It was initially developed in for the treatment of lymphoma with EZH2-activating mutations. In preclinical studies, GSK2816126 displayed high selectivity for EZH2 (over 100-fold over EZH1) with Ki ranging in the low nanomolar range for both wild type and mutant EZH2 and restored the expression of pathologically silenced PRC2 target genes [152]. Furthermore, GSK2816126 induced cancer cell death in vitro at nanomolar concentrations and displayed significant anti-tumor activity in xenograft models of EZH2-mutant DLBCL [152].
To date, one clinical trial has investigated GSK2816126. It consisted of an open-label, dose escalation study aimed at assessing the pharmacodynamics and pharmacodynamics and clinical activity of GSK2816126 in a total 41 patients with either advanced non-Hodgkin’s lymphoma (20) or refractory solid tumors (21) [153]. It was of a two-part study to determine the recommended Phase II dose (RP2D) for GSK2816126 given biweekly via intravenous (IV) infusion in a 3-weeks-on/1-week-regimen. The most restricting factor was liver transaminitis, which limited the maximum-tolerated dose to 2400 mg. All patients had at least one adverse effect with the most common being nausea and vomiting in about half of patient. Moreover, 32% of patients experienced a significant complication of grade III or less including pyrexia and fever. With regards to anticancer activity, only one patient with lymphoma had a partial radiologic response while 34% of patients achieved stable disease and 51% with progressive disease [153].
CPI-1205
CPI-1205 is an orally bioavailable, indole-based, small component EZH2 inhibitor that selectively binds to the EZH2 catalytic pocket [154]. CPI-1205 displays a biochemical IC50 = 0.002 μM a cellular EC50 = 0.032 μM) for mutant EZH2 [154]. Moreover, CPI-1205 inhibited tumor growth in vitro and in xenograft models of EZH2 mutant DLBCL [154].
These studies paved the way for three clinical trials. The first trial started in 2015 and is currently finished with results pending. This study was a Phase I sequential dose escalation and expansion study of CPI-1205 in patients with progressive B-cell lymphomas (NCT02395601). Two other clinical trials involving CPI-1205 are currently ongoing for which results are currently unavailable. One is ProStar, a Phase Ib/II trial investigating oral administration of CPI-1205 combined with either enzalutamide or abiraterone/prednisone in metastatic CRPC (NCT03480646). The second is a Phase I/II trial assessing CPI-1205 + ipilimumab in patients advanced solid tumors (NCT03525795).
Emerging EZH2 inhibitors
In addition to the compounds discussed above, three more compounds are in clinical trials currently recruiting patients. CPI-0209 is a second generation EZH2 inhibitor which demonstrated preclinical evidence of anticancer activity in bladder cancer models (data from Constellation Pharmaceuticals). It will be evaluated taken orally once daily alone or in combination with Irinotecan in a sequential dose escalation and expansion study in patients with advanced solid tumors (NCT04104776). Secondly, PF-06821497 is a potent and orally available EZH2 antagonist currently in Phase I testing for advanced malignancies (NCT03460977). Another orally available EZH2 inhibitor, SHR2554, is also in a Phase I clinical trial currently recruiting patients with recurrent neoplasms but no preclinical data could be found in the public domain (NCT03603951).
Considering the recent interest in epigenetic therapies, a number of groups actively continue to develop novel EZH2 inhibitors. Notably, there are ongoing efforts to optimize pre-existing compounds through rational structure-activity and drug docking methods to improve pharmacokinetic and pharmacodynamic properties [155]. In addition, another emerging therapeutic strategy is to design small molecules that disrupt the interaction between EZH2 and EED, which leads to PRC2 inactivation [156,157]. Thus, the rapid development and success of EZH2-targeted therapies is likely to continue in the future and will serve as the standard for other epigenetic drug targets.
Combining EZH2 inhibition with immunotherapy: opportunities & challenges
As discussed in the previous paragraphs, EZH2 is a key regulator of cancer immunoediting. In addition, EZH2 inhibitors are being tested in a variety of clinical trials. Hence, it is worth considering the advantages of combining immunotherapies with clinically available EZH2 inhibitors. Given the pro-oncogenic role of EZH2 and its ability to suppress both the innate and adaptive immune response, these drug combinations could be highly synergistic. However, we should consider that EZH2-target genes are tissue- and cancer-specific and that the TME is a diverse and dynamic assembly of cells interacting with an evolving malignancy. For these reasons, the efficacy and safety of these combinations should be carefully tested in the context of a specific cancer type and tumor stage. One should also consider the potential side effects caused by the combination of two treatments that can affect several physiological systems.
Despite these caveats, emerging evidence indicates that the pharmacological inhibition of EZH2 is a promising strategy to enhance the efficacy of some clinically available immunotherapies, in particular ICB therapies. While ICB therapeutic strategies targeting the CTLA-4 and PD-1/PD-L1 axis have revolutionized the management of melanoma and many solid tumor malignancies, many patients do not respond or develop resistance to these interventions [158]. In vitro and in vivo experiments on melanoma models have demonstrated the role of EZH2 in immunotherapy resistance [159,160]. In a melanoma mouse model, anti-CTLA-4 therapy was shown to upregulate EZH2 expression in melanoma cells, which in turn led to their decreased antigen presentation and immunogenicity. EZH2 inhibitor could reverse these effects, synergize with anti-CTLA-4 and effectively suppress melanoma growth [159]. Besides cancer cells, anti-CTLA-4 therapy was shown to increase EZH2 expression in peripheral T cells from treated patients. Pharmacological inhibition of EZH2 in human T cells elicited phenotypic and functional alterations of the Tregs and enhanced cytotoxic activity of T effectors [160]. Accordingly, anti-CTLA-4 and EZH2 inhibitor combination therapy in the melanoma-bearing mice significantly reduced tumor growth and increased mouse survival compared with anti-CTLA-4 monotherapy [160]. In addition to melanoma, an ovarian cancer preclinical study showed that EZH2 and DNA methyltransferase inhibitors could increase effector T cell tumor infiltration through upregulating Th1-type chemokines CXCL9 and CXCL10 in TME, hence significantly augmented the therapeutic efficacy of anti-PD-L1 therapy as well as T cell transfusion therapy [90]. Taken together, all the above-mentioned immunotherapy resistance is associated with the coordinated silencing of genes involved in antigen processing, T cell reprograming and cytokine secretion, which can be reversed by EZH2 inhibitors. This observation suggests that EZH2 targeting drugs could prevent the resistance to a variety of immunotherapies. In accordance with these findings, other preclinical studies showed that the combination of EZH2 inhibitors and ICBs is highly synergistic in hepatocellular carcinoma [161] and head and neck cancers [162]. While most of these studies have tested clinically approved EZH2 inhibitors, one of them employed DZNeP [161], which is the first described EZH2-targeting drug [143]. Subsequent studies have shown that DZNeP is a global histone methyl-transferase inhibitor [141]. Moreover, while DNZeP anticancer activity is greater than that of more specific EZH2 inhibitors [163], this drug might have a suboptimal pharmacokinetic profile [164]. Hence, the effects of DZNeP cannot be entirely ascribed to EZH2 inhibition. This example underscores the importance of using clinically available drugs, whose mechanism of action is fully elucidated, in order to increase the translational significance of preclinical studies.
Based on these results, it would be interesting to test the efficacy of EZH2 inhibitors in combination with ICBs, particularly in those ICB nonresponsive or resistant cancers. A search on clinicaltrials.gov revealed that currently only two Phase I studies are registered to test this combination. Unfortunately, none of these studies is currently recruiting patients. It would be interesting to understand why this promising combination is not being investigated more widely. One potential hindrance is represented by the most common adverse events associated with the use of ICBs. These events are due to the generalised activation of the immune response and range from a relatively mild skin rash to the emergence of severe autoimmune diseases (e.g., type I diabetes and myasthenia gravis) [165]. It is worth noting that the most severe adverse events are relatively rare. However, considering that EZH2 inhibition has been shown to favor the generalized activation of the adaptive and innate immune system, it is conceivable that its combination with ICBs could increase the incidence of autoimmune diseases. This potential drawback should be carefully considered when designing Phase I clinical trials.
Combinations between EZH2 inhibitors and other immunotherapies have been less investigated. However, recent evidence suggests that EZH2 targeting could be an almost universal strategy to improve the efficacy of immunotherapies. For example, a study conducted on Ewing sarcoma models showed that EZH2 inhibitors significantly increased the expression of tumor-associated antigens, thereby improving the effect of CAR-T therapy [166]. CAR-T therapies are being tested in a wide variety of solid and hematological malignancies but are already a feasible option in diffuse large B-cell lymphomas [167]. Interestingly, EZH2 inhibitors are approved for the treatment of these lymphomas [168]. Hence, we believe that this combination should be investigated in this disease.
EZH2 inhibitors could also enhance the efficacy of targeted therapies that have also an immunomodulatory function. For example, it has been shown that EZH2 activates the VEGF-dependent signalling in several cancer types, thereby promoting angiogenesis and metastasis [169]. Similarly, EZH2 has been implicated in STAT3-dependent progression of melanoma [170], prostate [171] and bladder cancers [172]. Despite these promising results, no study has investigated the immunological consequences of combining EZH2 inhibitors with targeted therapies that inhibit these signalling molecules.
In conclusion, the current evidence strongly suggests that EZH2 inhibitors could improve the efficacy of ICBs in melanoma and head and neck cancer. The combination with other immunotherapies should be further investigated in preclinical studies, to assess the best therapeutic strategy and the potential adverse events. In the next section, we will try to discuss an approach to design these clinical studies within the context of personalized cancer therapy.
Future perspective: personalized epi-immunotherapy
In this review, we have discussed emerging evidence supporting the hypothesis that EZH2 is a main driver of cancer cells’ immune editing. Interestingly, EZH2 is expressed in cancer cells and in most immune cells that compose the TME. Hence, EZH2 inhibitors could simultaneously target different components of the malignancy. Despite the promising results of most preclinical studies, several questions need to be addressed before testing EZH2/immunotherapy combinations in advanced clinical trials. Considering the increasing number of immunotherapies available and the fact that EZH2 regulates a different set of genes in different tissues, preclinical trials should be carefully designed to test simultaneously a wide range of treatments, in order to identify the best combination for each cancer type (and for each disease stage). In addition, preclinical studies should employ animal models that mirror as closely as possible the human immune system. This would enable identification of combinations that are potentially associated with a higher risk of adverse autoimmune events.
One important aspect to consider when designing new EZH2 inhibitor is their toxicological profile. While the expression patterns of EZH2 have not been fully investigated in all normal human cells, it is generally well accepted that EZH2 activity remains at a lower level in non-neoplastic cells [173]. For example, a number of publications have demonstrated that EZH2 levels are highly increased in aggressive cancer cells compared with normal cells [174,175]. Furthermore, EZH2 activity appears to be more important in embryonic development where it plays a role in proliferation and differentiation [176], thereby making EZH2 less functionally relevant in mature, differentiated cells. Although generally well tolerated, current EZH2 inhibitors are associated with relevant adverse events, including liver toxicity and fever. These effects are probably due to the basal expression of EZH2 in normal tissues and to its relevant functions in the maintenance and specification of different stem cell lineages. Future EZH2-targeting drugs should be designed to specifically penetrate cancer cells and/or to be administered selectively in the neoplastic tissue.
When an optimal and safe combination is identified, the clinical trial design should consider the wide variety of molecular alterations observed in human malignancies. Next generation sequencing data from different cohorts have extensively demonstrated that most cancer types are characterized by an elevated degree of intratumoral and intertumoral heterogeneity [177]. Given the complex and dynamic relationship between the TME and the malignant cells, it will be important to develop personalized diagnostic tools that aim to: identify the patients more likely to benefit from the EZH2/immunotherapy combination; follow-up the therapeutic response. We believe that the best approach to obtain these goals is to design clinical trials aimed at identifying biomarkers that predict the response to both immunotherapy and EZH2-targeting drugs. The markers of response to immunotherapy will be different, depending on the specific therapeutic strategy. For example, it has been shown that the mutational burden of cancers is a good indicator of response to ICBs [178]. As expected, circulating biomarkers of angiogenesis have been identified as better predictors of response to therapies that inhibit VEGF-dependent pathways [179]. In any case, it will be important to identify minimally invasive diagnostic tools, such as those that allow the measurement of biomarkers from blood, urine and other biological fluids. This strategy will enable the clinicians to reassess the patient's response to the combination periodically and to modify the therapeutic regimen accordingly. Interestingly, the tumor mutational burden can be estimated by techniques that enable the sequencing of circulating tumor DNA [180]. At present, there is no validated method to measure intratumoral EZH2 activity noninvasively. However, we believe that a recently developed technique could be modified for this purpose.
Cell free (cf) circulating nucleosomes have been recognized as novel, noninvasive biomarkers in oncology. Increased levels of nucleosomes in circulation have been associated both with tumor burden and progression in multiple cancers [181,182]. Nucleosomes and their associated DNA can be released upon apoptotic cell death and associated chromatin fragmentation [183] as well as necrotic cell death and degradation of larger chromatin fragments [184]. The histone content of the nucleosome protects the associated DNA from circulating DNase activity resulting in a peak size of around 167 bp.
While detection of total cf nucleosomes has been possible for some time [185] recently developed innovative immunoassays have enabled detection and genome wide profiling of cf nucleosomes containing histone and DNA modifications as well as histone variants associated with tumor-specific epigenetic changes, not only at the tumor site, but also noninvasively in the circulation in an approach referred to as Nucleosomics™ [186,187]. Using this technology, cancer associated epigenetic changes of cf nucleosomes have been identified as putative diagnostic panels for pancreatic cancer [188] and colorectal cancer [189] as well as lung disease [190] using serum samples. Circulating H3K27me3 has been measured in serum using a first generation qualitative Nu.Q™ assay [188]. Our recent in vitro data [Unpublished data] developed with a prototype second generation quantitative Nu.Q H3K27me3 assay, shows the feasibility of monitoring cellular EZH2 activity in cell extracts and supernatants. Nu.Q research use assays consist of epitope specific antibodies coated in a 96-well plate format with an anti-nucleosome detection antibody. Nucleosomes are detected using a colorimetric readout. Analytically validated kits are now commercially available and a pipeline of additional kits is in development for research use and in clinical format. The kits include a recombinant nucleosome standard to allow quantitative assessment of specific nucleosome levels. This strongly supports the utility of using the Nu.Q H3K27me3 assay to quantify the effects of EZH2 activity in tumor derived cf circulating nucleosomes.
We believe that the Nu.Q assay approach, in combination with a pharmacogenetic assay for immunotherapy would enable clinicians to select the best EZH2-immunotherapy combination and to follow-up the response to both drugs (Figure 4). Given the broad range of immuno-activating functions displayed by EZH2 inhibitors, we think several of these combinations will improve the survival of patients with different cancers.
Figure 4. . A strategy for personalized epi-immunotherapy.
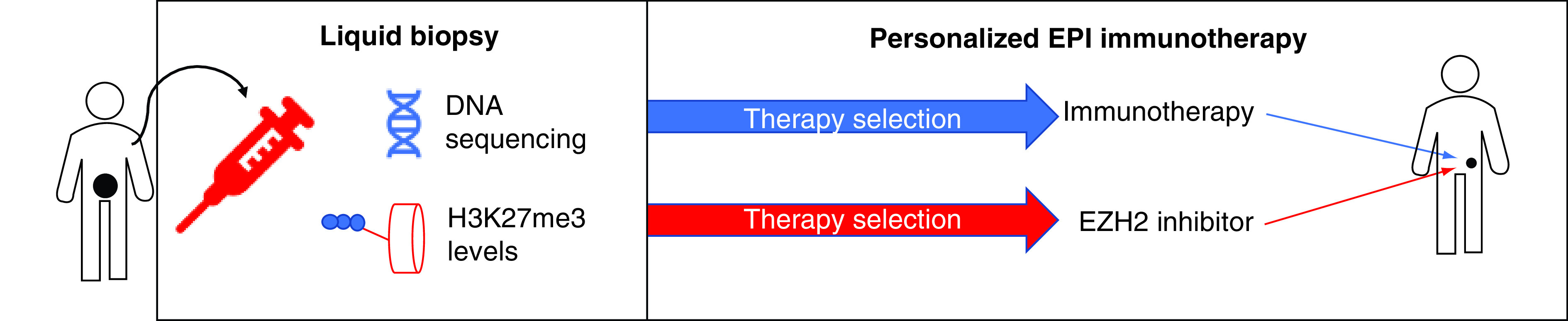
Using a liquid biopsy approach (e.g., from a blood sample) relevant information about cancer-specific mutations and epigenetic alterations (H3K27me3) levels can be obtained. The first information will enable clinicians to select the optimal immunotherapy, while the latter is useful to identify cancers with higher EZH2 activity and therefore more sensitive to EZH2 inhibitors. The same approach can be employed for dynamic monitoring of cancer progression during the therapy.
Executive summary.
Immunotherapies are emerging as an effective treatment for incurable malignancies. However, some patients display acquired or intrinsic resistance to these treatments.
In addition to genetic mutations, epigenetic alterations are emerging as key mechanisms of resistance to cancer immunotherapies. Notably, epigenetic modifications are reversible and can be targeted by clinically approved drugs.
EZH2 is an epigenetic mediator that induces epigenetic gene silencing via histones post translational modifications. EZH2 can also methylate other proteins (e.g., transcription factors) that in turn control gene expression.
EZH2 is overexpressed in many solid tumors and associated with worse prognosis. Higher EZH2 expression in cancer cells and in the tumor microenvironment suppresses the adaptive and innate immune response.
EZH2 inhibition is a promising strategy to improve the efficacy of cancer immunotherapies and to prevent the occurrence of resistance.
Three EZH2 inhibitors have entered clinical trials for the treatment of human malignancies. These inhibitors are generally well tolerated and have shown some anticancer activity.
A recently developed technology enables the measurement of EZH2 activity in patients’ blood samples. We envisage that this technology could be coupled with the use of EZH2 inhibitors and with DNA-based liquid biopsies to develop personalized epi-immunotherapies.
Footnotes
Financial & competing interests disclosure
The authors would like to thank the following sources of financial support: Higher Education Innovation Fund (HEIF-UK; to F Crea); Canadian Foundation for Translational Immunology (to N Kang, YZ Wang); CIHR operating grant (141635, 144159 and 153081 to YZ Wang), TFRI program project (1062 to YZ Wang), MITACS (IT14958 to YZ Wang) and National Cancer Institute (a pilot project of P50CA097186 to YZ Wang). M Eccleston is an employee of OncoLytika Ltd which provides consultancy services to Belgian Volition. Both M Eccleston and OncoLytika Ltd. hold stock in VolitionRx Plc. M Eccleston is an inventor on issued and pending patents assigned to Belgium Volition. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Vaddepally RK, Kharel P, Pandey R. et al. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN Guidelines with the level of evidence. Cancers (Basel) 12(3), 738–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16(3), 151–167 (2019). [DOI] [PubMed] [Google Scholar]; •• Focuses on the mechanisms of resistance to immunotherapy.
- 3.Sharma P, Hu-Lieskovan S, Wargo JA. et al. Primary, adaptive and acquired resistance to cancer immunotherapy. Cell 168(4), 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]; •• Focuses on the three phases (3Es) of cancer immunoediting.
- 5.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331(6024), 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Deans C, Maggert KA. What do you mean, “epigenetic”. Genetics 199(4), 887–896 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burr ML, Sparbier CE, Chan KL. et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36(4), 385–401.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toyokawa G, Takada K, Tagawa T. et al. A positive correlation between the EZH2 and PD-L1 expression in resected lung adenocarcinomas. Ann. Thorac. Surg. 107(2), 393–400 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Quiros J, Mahuron K. et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 23(11), 3262–3274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia L, Zhu X, Zhang L, Xu Y, Chen G, Luo J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burnet FM. The concept of immunological surveillance. Prog. Exp. Tumor Res. 13, 1–27 (1970). [DOI] [PubMed] [Google Scholar]
- 12.Thomas L. On immunosurveillance in human cancer. Yale J. Biol. Med. 55(3–4), 329–333 (1982). [PMC free article] [PubMed] [Google Scholar]
- 13.Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat. Med. 14(6), 623–628 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Malmberg KJ, Carlsten M, Björklund A. et al. Natural killer cell-mediated immunosurveillance of human cancer. Semin. Immunol. 31, 20–29 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Iannello A, Thompson TW, Ardolino M. et al. Immunosurveillance and immunotherapy of tumors by innate immune cells. Curr. Opin. Immunol. 38, 52–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayday AC. γδ T cell update: adaptate orchestrators of immune surveillance. J. Immunol. 203(2), 311–320 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Melssen M, Slingluff CL. Vaccines targeting helper T cells for cancer immunotherapy. Curr. Opin. Immunol. 47, 85–92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 10(5), 588–594 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Pawelec G. Tumour escape: antitumour effectors too much of a good thing. Cancer Immunol. Immunother. 53(3), 262–274 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vinay DS, Ryan EP, Pawelec G. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 35(Suppl.), S185–S198 (2015). [DOI] [PubMed] [Google Scholar]
- 21.McGranahan N, Swanton C. Cancer evolution constrained by the immune microenvironment. Cell 170(5), 825–827 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Haq R. Trapping cancers as they adapt to survive. Cancer Discov. 7(11), 1216–1217 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Rosenthal R, Cadieux EL, Salgado R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567(7749), 479–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell 164(6), 1233–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SY, Gout PW, Collins CC. et al. Epithelial immune cell-like transition (EIT): a proposed transdifferentiation process underlying immune-suppressive activity of epithelial cancers. Differentiation 83(5), 293–298 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Dunn GP, Bruce AT, Ikeda H. et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3(11), 991–998 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21(4), 687–692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talukdar S, Bhoopathi P, Emdad L. et al. Dormancy and cancer stem cells: an enigma for cancer therapeutic targeting. Adv. Cancer Res. 141, 43–84 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Garrido F, Aptsiauri N, Doorduijn EM. et al. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han LY, Fletcher MS, Urbauer DL. et al. HLA class I antigen processing machinery component expression and intratumoral T-Cell infiltrate as independent prognostic markers in ovarian carcinoma. Clin. Cancer Res. 14(11), 3372–3379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bukur J, Jasinski S, Seliger B. The role of classical and non-classical HLA class I antigens in human tumors. Semin. Cancer Biol. 22(4), 350–358 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Adrián Cabestré F, Moreau P, Riteau B. et al. HLA-G expression in human melanoma cells: protection from NK cytolysis. J. Reprod. Immunol. 43(2), 183–193 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Seliger B, Jasinski-Bergner S, Quandt D. et al. HLA-E expression and its clinical relevance in human renal cell carcinoma. Oncotarget 7(41), 67360–67372 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12(4), 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlößer HA, Theurich S, Shimabukuro-Vornhagen A. et al. Overcoming tumor-mediated immunosuppression. Immunotherapy 6(9), 973–988 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3 and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44(5), 989–1004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marin-Acevedo JA, Dholaria B, Soyano AE. et al. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J. Hematol. Oncol. 11(1), 39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnell A, Bod L, Madi A. et al. The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res. 30, 285–299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burkholder B, Huang RY, Burgess R. et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 1845(2), 182–201 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Prendergast GC, Malachowski WP, DuHadaway JB. et al. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 77(24), 6795–6811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi SY, Collins CC, Gout PW. et al. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite. J. Pathol. 230(4), 350–355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrot A, da Fonseca LM, Salustiano EJ. et al. Metabolic symbiosis and immunomodulation: how tumor cell-derived lactate may disturb innate and adaptive immune responses. Front Oncol 8, 81 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Binnewies M, Roberts EW, Kersten K. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24(5), 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Focuses on the complexity and diversity of the immune context of the tumor microenvironment and its influence on response to therapy.
- 44.Guo C, Manjili MH, Subjeck JR. et al. Therapeutic cancer vaccines: past, present and future. Adv. Cancer Res. 119, 421–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 257(1), 56–71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat. Rev. Cancer 12(4), 278–287 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Gallotta M, Assi H, Degagné É. et al. Inhaled TLR9 agonist renders lung tumors permissive to PD-1 blockade by promoting optimal CD4. Cancer Res. 78(17), 4943–4956 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Kauffman EC, Liu H, Schwartz MJ. et al. Toll-like receptor 7 agonist therapy with imidazoquinoline enhances cancer cell death and increases lymphocytic infiltration and proinflammatory cytokine production in established tumors of a renal cell carcinoma mouse model. J. Oncol. 2012, 103298 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu X, Li J, Connolly EM. et al. Combined anti-VEGF and Anti-CTLA-4 therapy elicits humoral immunity to galectin-1 which is associated with favorable clinical outcomes. Cancer Immunol. Res. 5(6), 446–454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kujawski M, Zhang C, Herrmann A. et al. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 70(23), 9599–9610 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu C, Talukder A, Savage NM. et al. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 6(3), e1291106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luke JJ, Bao R, Sweis RF. et al. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res. 25(10), 3074–3083 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang H, Lee WS, Kong SJ. et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest. 130, 4350–4364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell 31(3), 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonifazi M, Rossi M, Moja L. et al. Bevacizumab in clinical practice: prescribing appropriateness relative to national indications and safety. Oncologist 17(1), 117–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anwar MA, Shah M, Kim J. et al. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 39(3), 1053–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers and combinations. Sci. Transl. Med. 8(328), 328rv4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy and limitations. Front. Oncol. 8, 86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 200(2), 108–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedlaender A, Addeo A, Banna G. New emerging targets in cancer immunotherapy: the role of TIM3. ESMO Open 4(Suppl. 3), e000497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long L, Zhang X, Chen F. et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer 9(5–6), 176–189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheng S, Zhang J, Ai J. et al. Aberrant expression of IL-23/IL-23R in patients with breast cancer and its clinical significance. Mol. Med. Rep. 17(3), 4639–4644 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Baird AM, Leonard J, Naicker KM. et al. IL-23 is pro-proliferative, epigenetically regulated and modulated by chemotherapy in non-small-cell lung cancer. Lung Cancer 79(1), 83–90 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Soares KC, Rucki AA, Kim V. et al. TGF-β blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget 6(40), 43005–43015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Terabe M, Robertson FC, Clark K. et al. Blockade of only TGF-β 1 and 2 is sufficient to enhance the efficacy of vaccine and PD-1 checkpoint blockade immunotherapy. Oncoimmunology 6(5), e1308616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Gramont A, Faivre S, Raymond E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 6(1), e1257453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morse MA, Hobeika AC, Osada T. et al. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood 112(3), 610–618 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target. Cancer Sci 110(7), 2080–2089 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miret JJ, Kirschmeier P, Koyama S. et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J. Immunother. Cancer 7(1), 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fleming V, Hu X, Weber R. et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 9, 398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol. Cancer 18(1), 94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front. Oncol. 8, 49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.WARBURG O. On respiratory impairment in cancer cells. Science 124(3215), 269–270 (1956). [PubMed] [Google Scholar]
- 74.Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 92(3), 329–333 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Marchiq I, Pouysségur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. (Berl) 94(2), 155–171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kouidhi S, Ben Ayed F, Elgaaied Benammar A. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front. Immunol. 9, 353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi SYC, Ettinger SL, Lin D. et al. Targeting MCT4 to reduce lactic acid secretion and glycolysis for treatment of neuroendocrine prostate cancer. Cancer Med. 7(7), 3385–3392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Todenhöfer T, Seiler R, Stewart C. et al. Selective inhibition of the lactate transporter MCT4 reduces growth of invasive bladder cancer. Mol. Cancer Ther. 17(12), 2746–2755 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Polański R, Hodgkinson CL, Fusi A. et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 20(4), 926–937 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H. et al. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 7, 52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol. 20(9), 1100–1109 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Hodi FS, O'Day SJ, McDermott DF. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363(8), 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wolchok JD, Chiarion-Sileni V, Gonzalez R. et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377(14), 1345–1356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luke JJ, Flaherty KT, Ribas A. et al. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 14(8), 463–482 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Rizvi NA, Hellmann MD, Snyder A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small-cell lung cancer. Science 348(6230), 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Snyder A, Makarov V, Merghoub T. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371(23), 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hugo W, Zaretsky JM, Sun L. et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165(1), 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anagnostou V, Smith KN, Forde PM. et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small-cell lung cancer. Cancer Discov. 7(3), 264–276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gettinger S, Choi J, Hastings K. et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 7(12), 1420–1435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng D, Kryczek I, Nagarsheth N. et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527(7577), 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Suggests that epigenetic silencing of Th1-type chemokines is a novel immune-evasion mechanism of tumors. Selective epigenetic reprogramming alters the T-cell landscape in cancer and may enhance the clinical efficacy of cancer therapy.
- 91.Cristescu R, Mogg R, Ayers M. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362(6411), eaar3593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koyama S, Akbay EA, Li YY. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 10501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chapuy B, Stewart C, Dunford AJ. et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood 134(26), 2369–2382 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction. Nat. Rev. Cancer 6(2), 107–116 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Morales V, Richard-Foy H. Role of histone N-terminal tails and their acetylation in nucleosome dynamics. Mol. Cell Biol. 20(19), 7230–7237 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Portela A, Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 28(10), 1057–1068 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin. Genet. 81(4), 303–311 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qamra A, Xing M, Padmanabhan N. et al. Epigenomic promoter alterations amplify gene isoform and immunogenic diversity in gastric adenocarcinoma. Cancer Discov. 7(6), 630–651 (2017). [DOI] [PubMed] [Google Scholar]
- 99.Wierzbinska JA, Toth R, Ishaque N. et al. Methylome-based cell-of-origin modeling (Methyl-COOM) identifies aberrant expression of immune regulatory molecules in CLL. Genome Med. 12(1), 29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berglund A, Mills M, Putney RM. et al. Methylation of immune synapse genes modulates tumor immunogenicity. J. Clin. Invest. 130(2), 974–980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wylie B, Chee J, Forbes CA. et al. Acquired resistance during adoptive cell therapy by transcriptional silencing of immunogenic antigens. Oncoimmunology 8, 1609874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu J, He D, Cheng L. et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene 39(19), 3939–3951 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Floess S, Freyer J, Siewert C. et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5(2), e38 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lu Z, Zou J, Li S. et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 579(7798), 284–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mitra S, Lauss M, Cabrita R. et al. Analysis of DNA methylation patterns in the tumor immune microenvironment of metastatic melanoma. Mol. Oncol. 14(5), 933–950 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Topper MJ, Vaz M, Marrone KA. et al. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 17(2), 75–90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chiappinelli KB, Zahnow CA, Ahuja N. et al. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 76(7), 1683–1689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kelly AD, Issa JJ. The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr. Opin. Genet. Dev. 42, 68–77 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Símová J, Polláková V, Indrová M. et al. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. Br. J. Cancer 105(10), 1533–1541 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Christmas BJ, Rafie CI, Hopkins AC. et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol. Res. 6(12), 1561–1577 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park J, Thomas S, Munster PN. Epigenetic modulation with histone deacetylase inhibitors in combination with immunotherapy. Epigenomics 7(4), 641–652 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Juergens RA, Wrangle J, Vendetti FP. et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small-cell lung cancer. Cancer Discov. 1(7), 598–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Crea F, Clermont PL, Mai A. et al. Histone modifications, stem cells and prostate cancer. Curr. Pharm. Des. 20(11), 1687–1697 (2014). [DOI] [PubMed] [Google Scholar]; •• Discusses the crucial role of histone modification in prostate cancer initiation and progression and the potential of epigenetic therapy in prostate cancer management.
- 114.Völkel P, Bary A, Raby L. et al. Ezh1 arises from Ezh2 gene duplication but its function is not required for zebrafish development. Sci. Rep. 9(1), 4319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Q, Liu L, Zhang S. et al. Long noncoding RNA NEAT1 suppresses hepatocyte proliferation in fulminant hepatic failure through increased recruitment of EZH2 to the LATS2 promoter region and promotion of H3K27me3 methylation. Exp. Mol. Med. 52(3), 461–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Curry E, Green I, Chapman-Rothe N. et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenetics 7, 84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coward WR, Feghali-Bostwick CA, Jenkins G. et al. A central role for G9a and EZH2 in the epigenetic silencing of cyclooxygenase-2 in idiopathic pulmonary fibrosis. FASEB J. 28(7), 3183–3196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim E, Kim M, Woo DH. et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23(6), 839–852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lawrence CL, Baldwin AS. Non-canonical EZH2 transcriptionally activates RelB in triple negative breast cancer. PLoS ONE 11(10), e0165005 (2016).27764181 [Google Scholar]
- 120.Jones A, Wang H. Polycomb repressive complex 2 in embryonic stem cells: an overview. Protein Cell 1(12), 1056–1062 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nutt SL, Keenan C, Chopin M. et al. EZH2 function in immune cell development. Biol. Chem. 401(8), 933–943 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Deevy O, Bracken AP. PRC2 functions in development and congenital disorders. Development 146(19), dev181354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cyrus S, Burkardt D, Weaver DD. et al. PRC2-complex related dysfunction in overgrowth syndromes: a review of EZH2, EED and SUZ12 and their syndromic phenotypes. Am. J. Med. Genet. C Semin. Med. Genet. 181(4), 519–531 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Matsubara T, Toyokawa G, Takada K. et al. The association and prognostic impact of enhancer of zeste homologue 2 expression and epithelial-mesenchymal transition in resected lung adenocarcinoma. PLoS ONE 14(5), e0215103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu X, Scott H, Carlsson SV. et al. Increased EZH2 expression in prostate cancer is associated with metastatic recurrence following external beam radiotherapy. Prostate 79(10), 1079–1089 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stazi G, Taglieri L, Nicolai A. et al. Dissecting the role of novel EZH2 inhibitors in primary glioblastoma cell cultures: effects on proliferation, epithelial-mesenchymal transition, migration and on the pro-inflammatory phenotype. Clin. Epigenetics 11(1), 173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 127.Avan A, Crea F, Paolicchi E. et al. Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-deazaneplanocin A with gemcitabine in pancreatic cancer cells. Mol. Cancer Ther. 11(8), 1735–1746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang B, Huang M, Li Q. Cancer-associated fibroblasts promote angiogenesis of hepatocellular carcinoma by VEGF-mediated EZH2/VASH1 pathway. Technol. Cancer Res. Treat 18, (2019). 10.1177/1533033819879905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zeng Z, Yang Y, Wu H. MicroRNA-765 alleviates the malignant progression of breast cancer via interacting with EZH1. Am. J. Transl. Res. 11(7), 4500–4507 (2019). [PMC free article] [PubMed] [Google Scholar]
- 130.Nakagawa M, Kitabayashi I. Oncogenic roles of enhancer of zeste homolog 1/2 in hematological malignancies. Cancer Sci. 109(8), 2342–2348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Quintanal-Villalonga Á, Chan JM, Yu HA. et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 17(6), 360–371 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Clermont PL, Lin D, Crea F. et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenetics 7, 40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao Y, Wang XX, Wu W. et al. EZH2 regulates PD-L1 expression via HIF-1α in non-small-cell lung cancer cells. Biochem. Biophys. Res. Commun. 517(2), 201–209 (2019). [DOI] [PubMed] [Google Scholar]
- 134.Xiao G, Jin LL, Liu CQ. et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J. Immunother. Cancer 7(1), 300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu J, Lanier LL. Natural killer cells and cancer. Adv. Cancer Res. 90, 127–156 (2003). [DOI] [PubMed] [Google Scholar]
- 136.Bugide S, Green MR, Wajapeyee N. Inhibition of enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl Acad. Sci. USA 115(15), E3509–E3518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yin J, Leavenworth JW, Li Y. et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc. Natl Acad. Sci. USA 112(52), 15988–15993 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yin Y, Qiu S, Li X. et al. EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. J. Neuroinflammation 14(1), 220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pang B, Zheng XR, Tian JX. et al. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 7(29), 45134–45143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bray M, Driscoll J, Huggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antiviral Res. 45(2), 135–147 (2000). [DOI] [PubMed] [Google Scholar]
- 141.Miranda TB, Cortez CC, Yoo CB. et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 8(6), 1579–1588 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tan J, Yang X, Zhuang L. et al. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 21(9), 1050–1063 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Crea F, Hurt EM, Mathews LA. et al. Pharmacologic disruption of polycomb repressive complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 10, 40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hung SW, Mody H, Marrache S. et al. Pharmacological reversal of histone methylation presensitizes pancreatic cancer cells to nucleoside drugs: in vitro optimization and novel nanoparticle delivery studies. PLoS ONE 8, e71196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hoy SM. Tazemetostat: first approval. Drugs 80(5), 513–521 (2020). [DOI] [PubMed] [Google Scholar]
- 146.Knutson SK, Warholic NM, Wigle TJ. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl Acad. Sci. USA 110(19), 7922–7927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Knutson SK, Warholic NM, Wigle TJ. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl Acad Sci. USA 110(19), 7922–7927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Knutson SK, Kawano S, Minoshima Y. et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13(4), 842–854 (2014). [DOI] [PubMed] [Google Scholar]
- 149.Italiano A, Soria JC, Toulmonde M. et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, Phase I study. Lancet Oncol. 19(5), 649–659 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Leslie M. First EZH2 inhibitor approved-for rare sarcoma. Cancer Discov. 10(3), 333–334 (2020). [DOI] [PubMed] [Google Scholar]
- 151.Sarkozy C, Morschhauser F, Dubois S. et al. A LYSA Phase Ib study of tazemetostat (EPZ-6438) plus R-CHOP in patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL) with poor prognosis features. Clin. Cancer Res. 26(13), 3145–3153 (2020). [DOI] [PubMed] [Google Scholar]
- 152.McCabe MT, Ott HM, Ganji G. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492(7427), 108–112 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Yap TA, Winter JN, Giulino-Roth L. et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin. Cancer Res. 25(24), 7331–7339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vaswani RG, Gehling VS, Dakin LA. et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for Phase I clinical trials for B-Cell lymphomas. J. Med. Chem. 59(21), 9928–9941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lu B, Shen X, Zhang L. et al. Discovery of EBI-2511: a highly potent and orally active EZH2 inhibitor for the treatment of non-hodgkin's lymphoma. ACS Med. Chem. Lett. 9(2), 98–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang Y, Zhang J, Yu Z. et al. Discovery of first-in-class, potent and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J. Med. Chem. 60(6), 2215–2226 (2017). [DOI] [PubMed] [Google Scholar]
- 157.He Y, Selvaraju S, Curtin ML. et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 13(4), 389–395 (2017). [DOI] [PubMed] [Google Scholar]
- 158.Marabondo S, Kaufman HL. High-dose interleukin-2 (IL-2) for the treatment of melanoma: safety considerations and future directions. Expert Opin. Drug Saf. 16(12), 1347–1357 (2017). [DOI] [PubMed] [Google Scholar]
- 159.Zingg D, Arenas-Ramirez N, Sahin D. et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 20(4), 854–867 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Goswami S, Apostolou I, Zhang J. et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest. 128(9), 3813–3818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrates that modulating EZH2 expression in T cells can improve antitumor responses elicited by anti-CTLA-4 therapy, which provides a strong rationale for a combination trial of EZH2 inhibitor plus anti-CTLA-4 in patients with primary or adaptive resistance to anti-CTLA-4 therapy.
- 161.Hong YK, Li Y, Pandit H. et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell. Immunol. 336, 66–74 (2019). [DOI] [PubMed] [Google Scholar]
- 162.Zhou L, Mudianto T, Ma X. et al. Targeting EZH2 enhances antigen presentation, antitumor immunity and circumvents anti-PD-1 resistance in head and neck cancer. Clin. Cancer Res. 26(1), 290–300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Momparler RL, Côté S, Marquez VE. et al. Comparison of the antineoplastic action of 3-deazaneplanocin-A and inhibitors that target the catalytic site of EZH2 histone methyltransferase. Cancer Rep. Rev. 3, 1–4 (2020). [Google Scholar]
- 164.Sun F, Li J, Yu Q. et al. Loading 3-deazaneplanocin A into pegylated unilamellar liposomes by forming transient phenylboronic acid-drug complex and its pharmacokinetic features in Sprague-Dawley rats. Eur. J. Pharm. Biopharm 80(2), 323–331 (2012). [DOI] [PubMed] [Google Scholar]
- 165.Chamoto K, Hatae R, Honjo T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 25(5), 790–800 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kailayangiri S, Altvater B, Lesch S. et al. EZH2 inhibition in ewing sarcoma upregulates G. Mol. Ther. 27(5), 933–946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 21(3), e168–e178 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 131(19), 2125–2137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Crea F, Fornaro L, Bocci G. et al. EZH2 inhibition: targeting the crossroad of tumor invasion and angiogenesis. Cancer Metastasis Rev. 31(3–4), 753–761 (2012). [DOI] [PubMed] [Google Scholar]
- 170.Xu Z, Sun Y, Guo Y. et al. NF-YA promotes invasion and angiogenesis by upregulating EZH2-STAT3 signaling in human melanoma cells. Oncol. Rep. 35(6), 3630–3638 (2016). [DOI] [PubMed] [Google Scholar]
- 171.Luo J, Wang K, Yeh S. et al. LncRNA-p21 alters the antiandrogen enzalutamide-induced prostate cancer neuroendocrine differentiation via modulating the EZH2/STAT3 signaling. Nat Commun 10(1), 2571 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chen Z, Du Y, Liu X. et al. EZH2 inhibition suppresses bladder cancer cell growth and metastasis via the JAK2/STAT3 signaling pathway. Oncol. Lett. 18(1), 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat. Med. 22(2), 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yamamoto Y, Ning G, Howitt BE. et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J. Pathol. 238(4), 519–530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xu B, Abourbih S, Sircar K. et al. Enhancer of zeste homolog 2 expression is associated with metastasis and adverse clinical outcome in clear cell renal cell carcinoma: a comparative study and review of the literature. Arch. Pathol. Lab. Med. 137(10), 1326–1336 (2013). [DOI] [PubMed] [Google Scholar]
- 176.O'Carroll D, Erhardt S, Pagani M. et al. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 21(13), 4330–4336 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wei L, Wang J, Lampert E. et al. Intratumoral and intertumoral genomic heterogeneity of multifocal localized prostate cancer impacts molecular classifications and genomic prognosticators. Eur. Urol. 71(2), 183–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chan TA, Yarchoan M, Jaffee E. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30(1), 44–56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fornaro L, Vivaldi C, Orlandi P. et al. Longitudinal evaluation of angiogenesis-related circulating biomarkers during second-line treatment with paclitaxel and ramucirumab in advanced gastroesophageal cancer. Ann. Oncol. 30(Suppl. 4), iv88 (2019). [Google Scholar]
- 180.Forschner A, Battke F, Hadaschik D. et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma - results of a prospective biomarker study. J. Immunother. Cancer 7(1), 180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Holdenrieder S, Stieber P. Apoptotic markers in cancer. Clin. Biochem. 37(7), 605–617 (2004). [DOI] [PubMed] [Google Scholar]
- 182.Holdenrieder S, Nagel D, Schalhorn A. et al. Clinical relevance of circulating nucleosomes in cancer. Ann. NY Acad. Sci. 1137, 180–189 (2008). [DOI] [PubMed] [Google Scholar]
- 183.Gabler C, Blank N, Hieronymus T. et al. Extranuclear detection of histones and nucleosomes in activated human lymphoblasts as an early event in apoptosis. Ann. Rheum. Dis. 63(9), 1135–1144 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thierry AR, El Messaoudi S, Gahan PB. et al. Origins, structures and functions of circulating DNA in oncology. Cancer Metastasis Rev. 35(3), 347–376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Holdenrieder S, Stieber P, Bodenmüller H. et al. Nucleosomes in serum as a marker for cell death. Clin. Chem. Lab. Med. 39(7), 596–605 (2001). [DOI] [PubMed] [Google Scholar]
- 186.Holdenrieder S, Dharuman Y, Standop J. et al. Novel serum nucleosomics biomarkers for the detection of colorectal cancer. Anticancer Res. 34(5), 2357–2362 (2014). [PubMed] [Google Scholar]
- 187.McAnena P, Brown JA, Kerin MJ. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers (Basel) 9(1), 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bauden M, Pamart D, Ansari D. et al. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin. Epigenetics 7, 106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rahier JF, Druez A, Faugeras L. et al. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin. Epigenetics 9, 53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Guiot J, Struman I, Chavez V. et al. Altered epigenetic features in circulating nucleosomes in idiopathic pulmonary fibrosis. Clin. Epigenetics 9, 84 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]