

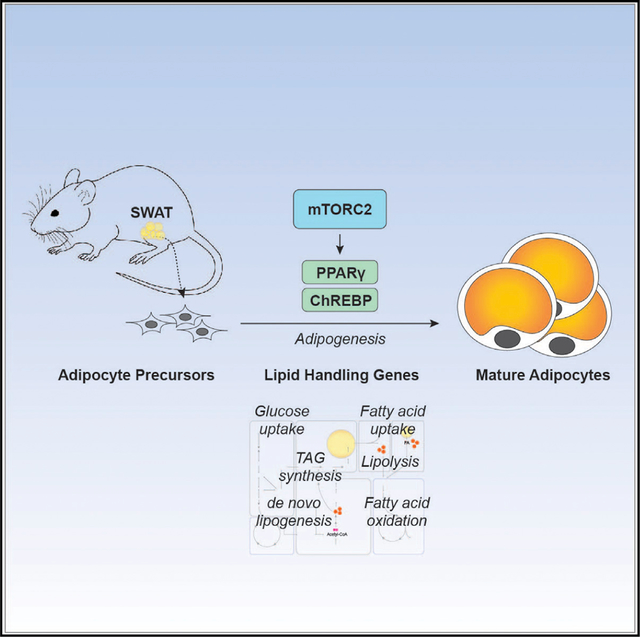
SUMMARY
Overweight and obesity are associated with type 2 diabetes, non-alcoholic fatty liver disease, cardiovascular disease and cancer, but all fat is not equal, as storing excess lipid in subcutaneous white adipose tissue (SWAT) is more metabolically favorable than in visceral fat. Here, we uncover a critical role for mTORC2 in setting SWAT lipid handling capacity. We find that subcutaneous white preadipocytes differentiating without the essential mTORC2 subunit Rictor upregulate mature adipocyte markers but develop a striking lipid storage defect resulting in smaller adipocytes, reduced tissue size, lipid re-distribution to visceral and brown fat, and sex-distinct effects on systemic metabolic fitness. Mechanistically, mTORC2 promotes transcriptional upregulation of select lipid metabolism genes controlled by PPARγ and ChREBP, including genes that control lipid uptake, synthesis, and degradation pathways as well as Akt2, which encodes a major mTORC2 substrate and insulin effector. Further exploring this pathway may uncover new strategies to improve insulin sensitivity.
Graphical Abstract
In Brief
Storing excess lipid in subcutaneous fat is more metabolically favorable than in visceral fat. Hsiao et al. show that mTORC2 is required during subcutaneous fat development to establish lipid handling capacity. Without mTORC2, developing subcutaneous fat is reduced and lipid is redistributed to other depots, causing gender-specific consequences.
INTRODUCTION
White adipose tissue (WAT) stores energy and secretes endocrine factors that control metabolism (Guilherme et al., 2019; Lee et al., 2017; Lefterova et al., 2014; Scherer, 2019). WAT expands in response to over-nutrition so that the excess calories can be safely stored as triacylglycerol (TAG), preventing toxic lipid accumulation in non-adipose tissues (Almandoz et al., 2013; Snel et al., 2012; Unger et al., 2010). However, in over-weight and obese individuals, white adipocytes become dysregulated and contribute, through mechanisms incompletely understood, to obesity-related comorbidities including type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), cardiovascular diseases (CVDs), and cancer (Van Gaal et al., 2006). Thiazolidinediones (TZDs) are oral insulin sensitizing drugs used to treat T2D. They act by stimulating PPARγ, the master transcriptional regulator of adipogenesis, to enhance insulin sensitivity and promote glucose use and lipid synthesis and storage (Hauner, 2002). Although TZDs are commonly prescribed, serious side effects have limited their efficacy (Cariou et al., 2012). Thus, a better understanding of PPARγ regulation may lead to improved therapies.
Importantly, not all WAT depots play equal roles in metabolism. For example, the health risks of metabolic syndrome and cardiovascular events for overweight patients with excessive visceral WAT (VWAT) are higher than for individuals with excess subcutaneous WAT (SWAT) (Ferrara et al., 2019; Lessard and Tchernof, 2012; McLaughlin et al., 2011). An individual’s body fat set point and ability to grow adipose tissue during development and upon over-nutrition are also variable in the population and between sexes (Fitzgerald et al., 2018; Tchoukalova et al., 2010; Tramunt et al., 2020). Such complexities suggest that anti-obesity therapies will likely have greater success when personalized. Thus, understanding depot and sex differences in adipose tissue biology is also clinically relevant.
In mature white adipocytes, the mechanistic target of rapamycin complex 2 (mTORC2) regulates glucose uptake and de novo lipogenesis (DNL) in vivo in part through regulating the carbohydrate response element binding protein (ChREBP) transcription factor (Guo et al., 2019; Guri et al., 2017; Jung et al., 2019; Tang et al., 2016). In humans, a positive correlation between DNL in SWAT and systemic insulin sensitivity has been shown (Eissing et al., 2013; Roberts et al., 2009; Smith and Kahn, 2016). Consistently, conditionally deleting Rictor in mice with Adiponectin-Cre, which targets all mature adipocytes, causes insulin resistance (Tang et al., 2016; Yu et al., 2019). The AKT kinases (AKT1, AKT2, and AKT3) are phosphorylated by mTORC2 in their C-terminal hydrophobic motif (HM) sites (Ser473, Ser474, and Ser472, respectively) (Hresko and Mueckler, 2005; Sarbassov et al., 2005). However, global downstream AKT signaling appears minimally affected in vivo in Adiponectin-Cre;Rictor-knockout (KO) mice (RictorAdipoq-Cre) despite the lack of AKT HM phosphorylation (Tang et al., 2016). AKT2 is the major AKT isoform in adipocytes. Mutating AKT2-S474 to alanine in vitro in 3T3L1 adipocytes also revealed that HM phosphorylation is dispensable for insulin-stimulated glucose uptake and mTORC1 activity (Beg et al., 2017), while another study using AKT2-S474A mutant 3T3L1 adipocytes showed that HM phosphorylation is required for maximal AKT signaling to TSC2, PRAS40, FoxO1/3, and AS160 (Kearney et al., 2019). Possible explanations for the observed differences between models, which are not necessarily exclusive, are that AKT signaling compensation occurs with prolonged Rictor loss in vivo, but not equally across all AKT substrates or functions; that individual AKT substrates inherently differ in their mTORC2 dependency; and that AKT-independent mechanisms contribute to WAT dysfunction. Moreover, mutating AKT2 S474 is not identical to deleting Rictor, as mTORC2 regulates other AKT phosphorylation sites and AGC family kinases (Facchinetti et al., 2008; Hiraoka et al., 2011; Ikenoue et al., 2008), and thus variables in experimental strategy also likely contribute to some of these differences. Nevertheless, previous studies focus largely on mTORC2’s role in mature white adipocytes and whether Rictor/mTORC2 is required for WAT development is not known.
Here, we investigate the role of mTORC2 in subcutaneous fat development using both in vitro and in vivo models. In both primary and immortalized cells, we find that Rictor/mTORC2 is not required to induce PPARγ during differentiation but that it is required for the expression of specific PPARγ target genes that encode regulators of lipid uptake and storage. mTORC2 may not stimulate these PPARγ genes through ChREBP but rather in coordination with ChREBP to promote maximum lipid storage capacity. To show physiological relevance, we also deleted Rictor in vivo in precursor cells that give rise to SWAT, but not to VWAT or brown adipose tissue (BAT). Consistent with our in vitro findings, this impaired the expression of select PPARγ target genes that encode regulators of lipid handling, in addition to attenuating expression of ChREBP/SREBP1c target genes that control de novo lipid synthesis. This resulted in reduced subcutaneous white adipocyte size, reduced overall SWAT mass, and re-distribution of lipids to the visceral and brown fat depots. Interestingly, this caused insulin resistance in males. However, females were able to maintain normal insulin sensitivity despite Rictor loss causing a similar but milder effect on SWAT mass and lipid re-distribution. Overall, these data suggest a model in which mTORC2 acts upstream of the adipogenic transcriptional machinery during SWAT development to program lipid handling capacity. As the ability to store lipid in SWAT is correlated with improved metabolic health, these findings may have important implications for developing T2D treatments.
RESULTS
mTORC2 Promotes Lipid Filling during Subcutaneous White Adipogenesis In Vitro
To investigate the role of mTORC2 in SWAT development, we first generated a primary subcutaneous white adipocyte differentiation model by isolating stromal vascular fraction (SVF) cells, which contain preadipocytes, from the inguinal WAT depots of UBC-CreERT2;RictorloxP/loxP mice and briefly treating them with 4-hydroxy tamoxifen (4-OHT) to induce Rictor deletion (Figure 1A). Following 4-OHT washout, primary Rictor-inducible KO SVF cells (Rictor-iKOprimary preadipocytes hereafter) and their isogenic vehicle-treated controls were differentiated following a standard protocol (Zebisch et al., 2012). Staining of differentiated primary adipocytes with oil red O (Figure 1B) or by LipidTOX and Perilipin 1 (PLIN1) immunofluorescence (Figure 1C) indicates decreased lipid droplet accumulation in the Rictor-iKOprimary cells. Quantification of the oil red O-stained lipid droplets after isopropanol extraction indicates ~20% less neutral lipid in the Rictor-iKOprimary cells (Figure S1A). The total cell number (Figure S1B) and percentage of PLIN1-positive cells (Figure S1C) is unchanged by Rictor loss, suggesting a defect in intracellular lipid accumulation. We also induced CreERT2 activity in otherwise wild-type SVF cells (i.e., having no floxed Rictor alleles) to confirm that neither brief tamoxifen exposure nor temporal recombinase activity alone in the undifferentiated cells affects oil red O staining, RICTOR level, or AKT phosphorylation upon differentiation (Figures 1A, S1D, and S1E). These data suggest that mTORC2 positively regulates subcutaneous white adipocyte lipid accumulation.
Figure 1. mTORC2 Promotes Lipid Filling during Subcutaneous White Adipogenesis In Vitro.
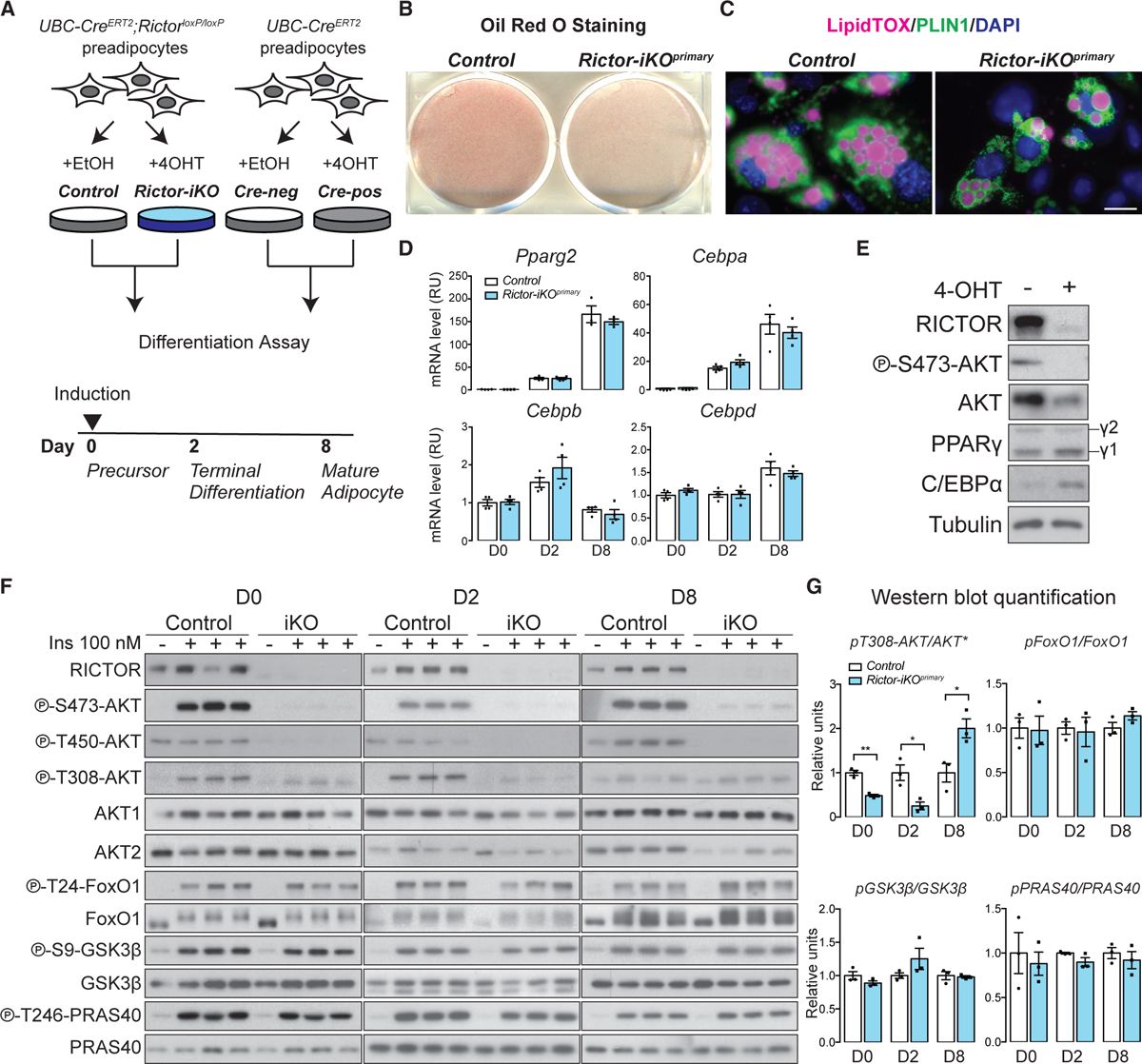
(A) Model of in vitro experimental strategy. 4-OHT, 4-hydroxytamoxifen; Cre-neg, Cre-negative cells; Cre-pos, Cre-positive cells.
(B) Oil red O (ORO) staining of differentiated (day 8) isogenic control and Rictor-iKO primary (Rictor-iKOprimary) cells.
(C) LipidTOX and Perilipin 1 (PLIN1) immunofluorescence staining of differentiated (day 8) control and Rictor-iKOprimary cells. Scale bar, 100 μm.
(D) Relative mRNA expression by RT-PCR of differentiation marker genes at the indicated differentiation days (n = 4; data represent mean ± SEM).
(E) Western blot of lysates from differentiated (day 8) cells. γ1, PPARγ1 isoform, γ2, PPARγ2.
(F) Western blot of the indicated total and phospho-proteins in lysates with or without 100 nM insulin (ins) stimulation at days 0, 2, and 8 of differentiation.
(G) Quantification of the indicated total and phosphorylated protein levels. Total AKT (asterisk) reflects AKT1 and AKT2 levels (n = 3; data represent mean ± SEM; *p < 0.05 and **p < 0.01).
We observed no difference in Pparg2, Cebpa, Cebpb, and Cebpd mRNA expression during differentiation in Rictor-iKOprimary cells (Figure 1D), and consistently, PPARγ2 protein expresses normally (Figure 1E). We did observe that the PPARγ2 cofactor C/EBPα expresses at higher than normal protein level following differentiation in Rictor-iKOprimary cells despite having an unchanged mRNA expression profile (Figures 1D and 1E). Primary cells lacking Rictor have decreased AKT HM phosphorylation (S473 on AKT1, S474 on AKT2) and AKT turn motif phosphorylation (T450 on AKT1, T451 on AKT2) throughout differentiation, confirming Rictor ablation (Figures 1E and 1F). AKT1-T308/AKT2-T309 phosphorylation decreases by ~50% in Rictor-iKOprimary cells at day 0 (D0) and D2 of differentiation but increases at D8 relative to controls (Figures 1F and 1G). This is consistent with previous in vivo observations in adipocytes that mTORC2 facilitates but is not essential for AKT-T308 phosphorylation (Hung et al., 2014; Jung et al., 2019; Tang et al., 2016). Interestingly, isoform-specific AKT1 and AKT2 analysis shows a decrease in AKT2 mRNA induction during differentiation, resulting in reduced AKT2 protein at D8 (Figures 1F and S1F). Transcriptional regulation of Akt2 was not observed in Rictor-deleted mature adipocytes (Tang et al., 2016). Nevertheless, the decrease in AKT2 level does not prevent insulin (100 nM) from stimulating phosphorylation of AKT substrates such as FoxO1 (T24), GSK3β (S9), or PRAS40 (T246) in the Rictor-deficient cells (Figures 1F and 1G). Collectively, these data suggest that lipid accumulation during adipogenesis, but not differentiation, requires mTORC2.
We also immortalized UBC-CreERT2;RictorloxP/loxP SVF cells (hereafter Rictor-iKOimmortal preadipocytes) to test whether immortalization alters differentiation dynamics and mTORC2 dependency. Like their primary cell counterparts, Rictor-iKOimmortal preadipocytes have a lipid filling defect, exhibiting a 60% decrease in oil red O staining after differentiation compared with controls (Figure S1G). Similar to the Rictor-iKOprimary cells, the D8 Rictor-iKOimmortal cells show reduced RICTOR and AKT-S473/4 phosphorylation, slightly higher p-AKT-T308/9, decreased AKT2 protein, and increased C/EBPα protein (Figure S1H). Notably, AKT1 mRNA and protein levels also increase in the D8 Rictor-iKOimmortal cells, which is not detected in the Rictor-iKOprimary cells, and this appears at least partly due to increased Akt1 transcription (Figures S1H and S1I). There is also a slight difference in PPARγ2 induction in the immortalized cells, which transcriptionally induces normally at D2, as in primary cells, but fails to maximally amplify thereafter (Figures S1J and S1K). Nevertheless, Rictor-iKOimmortal preadipocytes exhibit many of the same features as Rictor-iKOprimary preadipocytes, with the observed differences likely resulting from the immortalization procedure.
mTORC2 Promotes Expression of Lipid Handling Genes
To begin exploring how mTORC2 regulates lipid accumulation during differentiation, we generated RNA sequencing (RNA-seq) transcriptomes from the control and Rictor-iKOprimary SVF cells in the precursor stage (D0), the terminal differentiation stage (D2, when PPARγ2 is induced), and the mature adipocyte stage (D8) (Figure 1A). By first making pairwise comparisons between the control and KO cells at each differentiation day examined, we found that most of the differential gene expression occurs between D2 and D8 (Figure 2A). For example, we identified 141 genes significantly downregulated in Rictor-iKOprimary cells at D8 compared with only 52 and 38 genes at D0 and D2, respectively and 37, 76, and 78 upregulated genes at D0, D2, and D8 respectively (Figure 2A; Table S5). We classified the downregulated genes as requiring Rictor for normal induction (Rictor-required genes) and the upregulated genes as being suppressed by Rictor (Rictor-suppressed genes) (Figure 2A). Gene Ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for D0 and D2 Rictor-required genes (those downregulated) reveals genes thought to function in cell adhesion (n = 8) and extracellular matrix receptor interaction pathways (n = 6) (Table S1). In contrast, the D8 Rictor-required genes are enriched for metabolic processes especially lipid metabolism (n = 22) (Table S1). Among the Rictor-suppressed genes is an over-representation of inflammation pathway genes (Table S1). Notably, genes that suppress adipogenesis, such as Pref1 and Pdgfra, are not increased by Rictor loss, consistent with Rictor-iKOprimary cells’ having a defect in metabolic gene expression but not in differentiation. Thus, mTORC2 is a positive regulator of lipid metabolic gene expression during adipocyte differentiation.
Figure 2. mTORC2 Promotes Expression of Lipid-Handling Genes.
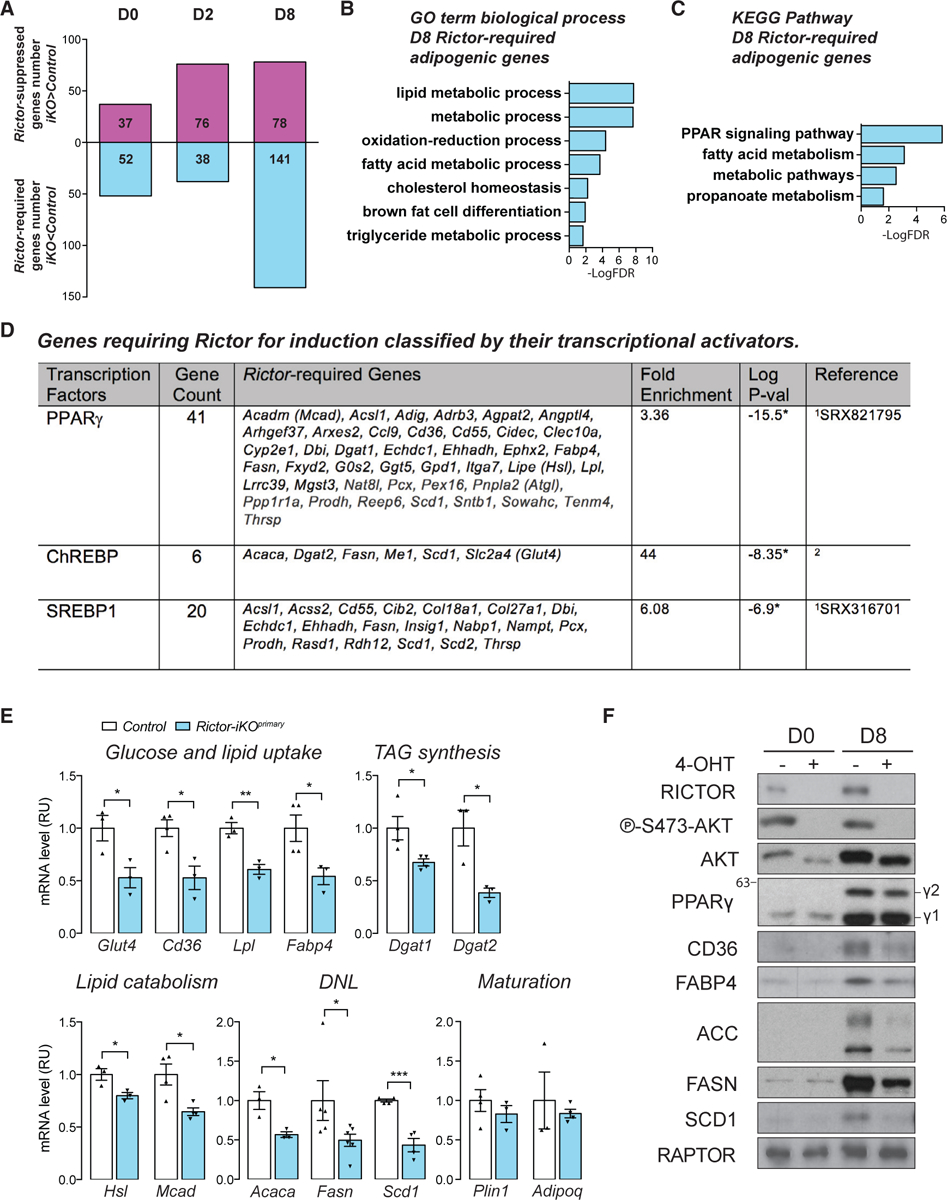
(A) Upregulated (Rictor-suppressed) and downregulated (Rictor-required) genes at the indicated differentiation days (fold change > 1.4, adjusted p value < 0.05).
(B) Gene Ontology (GO) term enrichment analysis of day 8 Rictor-required adipogenic genes analyzed by the Database for Annotation, Visualization and Integrated Discovery (DAVID).
(C) KEGG pathway enrichment analysis of D8 Rictor-required adipogenic genes analyzed by DAVID.
(D) Genes requiring Rictor for induction classified by their published transcriptional activators. 1Compared with ChIP-Atlas database (Oki et al., 2018); 2compared with the ChREBP targets listed in Iizuka (2017). *p < 0.05. Full gene names are listed in Table S5 and Figure S2A legend.
(E) Relative mRNA expression by RT-PCR of the indicated genes in day 8 control and Rictor-iKOprimary cells (n ≥ 3; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001).
(F) Western blot of the indicated total proteins in lysates from undifferentiated (D0) and differentiated (D8) control and Rictor-iKOprimary cells. γ1, PPARγ1 isoform; γ2, PPARγ2. Numbers at the left are protein sizes in kilodaltons.
We also compared the D8 and D0 control transcriptomes to identify adipogenic genes, which we defined as genes upregulated >1.4 fold at a false discovery rate (FDR) < 0.05 in D8 versus D0. This identified 825 adipogenic genes in the control primary cells (Table S5). Among the D8 Rictor-required genes, 77 of them (54.6%) are also adipogenic genes on the basis of this analysis (Table S5). GO analysis identified lipid metabolism genes as being highly overrepresented among the D8 Rictor-required adipogenic genes (Figure 2B). KEGG analysis further identified the PPAR signaling pathway as the top scoring pathway among the D8 Rictor-required genes (KEGG results on the basis of 10 of 77 genes) (Figure 2C). Notably, Rictor-required genes in the PPAR signaling pathway encode regulators of both anabolic and catabolic lipid metabolism such as fatty acid uptake, fatty acid oxidation, DNL, and TAG synthesis (Figure S2A). By comparing the D8 Rictor-required genes with a published database of PPARγ targets, we identified several additional Rictor-required genes (41 of 141) as likely PPARγ targets (Figure 2D). In addition, 6 and 20 of Rictor-required genes are also classified as ChREBP and/or SREBP1 targets, respectively, on the basis of published data (Oki et al., 2018; Ortega-Prieto and Postic, 2019) (Figure 2D). The complete gene lists for each category in this section are available in Table S5.
For several of the Rictor-required genes identified by RNA-seq, we developed RT-PCR assays to confirm their D8 expression differences in primary cells (Figure 2E). We focused on genes that regulate lipid metabolism, including previously reported PPARγ target genes (Scd1, Dgat1, Glut4, Cd36, Lpl, Fabp4, Hsl, and Mcad), DNL genes (Acaca, Fasn, and Scd1), and as controls, Rictor-independent adipogenic genes (Plin1 and Adipoq). As predicted from the RNA-seq data, genes encoding regulators of several lipid anabolic pathways, such as fatty acid and TAG synthesis (Acaca, Fasn, Scd1, Dgat1, and Dgat2) and glucose and fatty acid uptake (Glut4, Cd36, Lpl, and Fabp4) are decreased in D8 Rictor-iKOprimary cells (Figure 2E). We also confirmed attenuation of genes that encode regulators of lipid catabolic processes such as lipolysis (Hsl) and beta-oxidation (Mcad) in Rictor-iKOprimary cells (Figure 2E). We further confirmed decreased CD36, FABP4, ACC, FASN, and SCD1 protein expression by western blot (Figure 2F). In contrast, the PPARγ targets Plin1 and Adipoq are unaffected by Rictor loss (Figure 2E), which is also consistent with normal PLIN1-positive staining in the D8 adipocytes (Figures 1C and S1C). Importantly, the control CreERT2 cells (Figure 1A) show no defects in Acaca, Fasn, Scd1, Cd36, Lpl, Fabp4, or Glut4 expression (Figure S2B). We confirmed that decreased expression of the PPARγ targets Cd36 and Fabp4 requires Rictor deletion prior to differentiation, as deleting Rictor after differentiation did not attenuate their expression despite ablating AKT-S473 phosphorylation (Figures S2C and S2D). In contrast, DNL genes (Acaca and Fasn) require Rictor both during differentiation and in mature adipocytes (Figure S2D) consistent with our previous in vivo findings (Tang et al., 2016). Thus, without Rictor, differentiating SWAT preadipocytes cannot establish their normal lipid metabolic gene expression program, which includes regulators of lipid synthesis, uptake, breakdown, and oxidation pathways.
To confirm that the observed gene expression differences reflect metabolic changes, we performed functional assays. Using 14C-glucose, we show that lipid synthesis increases 65-fold from D0 to D8 in control cells and confirmed that D8 Rictor-iKOprimary cells have a 92% reduction in de novo lipid synthesis, with the D0 cells also showing a slight decrease (Figure S2E). Consistent with reduced Glut4 expression, we also measured 25% and 35% decreases in insulin-stimulated 3H-2-DG glucose uptake at D0 and D8, respectively (Figure S2F). However, non-insulin-stimulated glycolysis and glycolytic capacity measured on a Seahorse Extracellular Flux Analyzer exhibited higher and normal capacity, respectively, on the basis of extracellular acidification rate (Figure S2G), which is consistent with RNA-seq data showing normal glycolysis gene expression in Rictor-iKOprimary cells (Table S5; Figure S2A). Using BODIPY as a probe to measure lipid uptake, we also measured 25% and 35% decreases in lipid uptake after 10 and 30 min of labeling, respectively, in Rictor-iKOprimary cells (Figure S2H). These data are consistent with a model in which mTORC2 sets the general lipid handling capacity of SWAT during adipogenesis.
Specific PPARγ Targets Require Rictor for Full Induction
To further explore the connection between mTORC2 and PPARγ, we expressed a PPARγ activity reporter construct that contains three PPRE elements in the luciferase promoter (Kim et al., 1998) in control and Rictor-iKOimmortal cells and quantified reporter gene activity at D2 of differentiation. As expected, reporter activity increases 2-fold when the activity at D2 is compared with that in undifferentiated cells (D0) (Figure S3A). Reporter activity decreases by 53% when Rictor is deleted (Figure S3A), and while supplementing the PPARγ agonist rosiglitazone enhances reporter activity 1.8-fold in control cells, it has no effect in the Rictor-iKOimmortal cells (Figure S3A). Similarly, over-expressing recombinant HA-PPARγ2 enhances reporter activity 2-fold over baseline in D2 control cells, and this is reduced by 58% in the Rictor-iKOimmortal cells despite recombinant HA-PPARγ2 expressing at similar levels in the control and KO cells (Figures S3A and S3B). Moreover, overexpressing HA-PPARγ2 does not rescue lipid droplet accumulation in Rictor-deficient cells upon differentiation (Figure S3C). It also fails to restore expression of Cd36, Lpl, Fabp4, Acaca, Fasn, or Scd1 (Figure S3D) or ATP citrate lyase (ACLY), ACC, or FASN protein expression (Figure S3E), although lipid content and Cd36 expression do show minor increases relative to Rictor-deficient cells expressing the empty vector control. We also tested whether supplementing rosiglitazone during the full differentiation assay would improve the Rictor-KO phenotype. Rosiglitazone does increase oil red O staining and target gene expression in both control and Rictor-iKOimmortal cells, but lipid accumulation and gene expression remain significantly attenuated in Rictor-iKOimmortal cells relative to control (Figures S3F–S3H). Overall, these data are consistent with Rictor loss impairing PPARγ activity.
Next, we used chromatin immunoprecipitation (ChIP) assay to examine endogenous PPARγ target gene promoters in primary cells for both PPARγ binding to PPRE elements and for histone H3K9 acetylation, which is associated with PPARγ target gene activity (Lefterova et al., 2008; Salma et al., 2004; Steger et al., 2010; Wang et al., 2019). We examined the PPRE regions in Rictor-dependent (Cd36 and Fabp4) and a Rictor-independent (Pkm2) PPARγ target (Figures S4A and S4B; Table S4). At differentiation D2, PPARγ-PPRE binding is unchanged at the promoters of Cd36, Fabp4, and Pkm2 when Rictor is absent; however, at D8, PPARγ binding at the Cd36 and Fabp4 PPREs decreases by 40% and 33%, respectively, in the absence of Rictor (Figure 3A). In contrast, but consistent with our gene expression analysis, PPARγ binding to the Pkm2 promoter (Panasyuk et al., 2012) is unaffected by Rictor loss (Figure 3A). Deleting Rictor also decreased H3K9ac by 40% and 44%, respectively, in the PPREs of Cd36 and Fabp4 at D2, preceding measurable loss of PPARγ binding (Figure 3B). H3K9ac further decreases at both promoters by 73% and 80%, respectively, at D8 while remaining unaffected in the Pkm2 PPRE throughout differentiation (Figure 3B). Similar results were obtained using the Rictor-iKOimmortal system; for example, PPARγ binding to the Fabp4-PPRE decreases in Rictor-iKOimmortal cells, although the defect occurs 2 days earlier in the immortalized cells than in the primary cells (Figure S4C). This is consistent with the immortalized cells but not the primary cells, showing greater dependency on Rictor for PPARγ amplification (Figures S1J and S1K). Total histone H3 levels and global H3K9 acetylation appear unaffected by Rictor loss (Figure S4D). These data are consistent with specific PPARγ targets requiring Rictor for full induction during differentiation.
Figure 3. Specific PPARγ Targets Require Rictor for Full Induction.
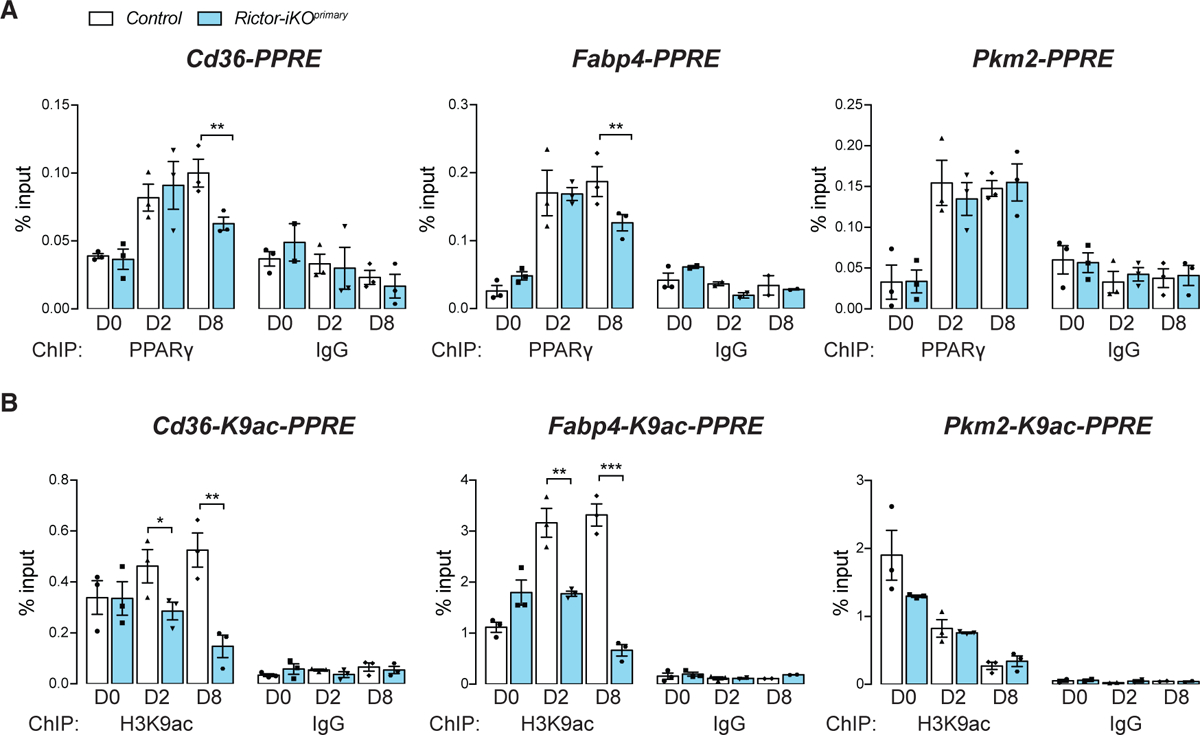
(A) PPARγ/PPAR-responsive element (PPRE) interaction identified by chromatin immunoprecipitation (ChIP) at Cd36, Fabp4, and Pkm2 promoters in control and Rictor-iKOprimary cells (n = 3 for PPARγ ChIP, n = 3 for IgG ChIP; data represent mean ± SEM; **p < 0.01). ChIP with IgG were used as negative controls.
(B) H3K9 acetylation (H3K9ac) by ChIP analysis at Cd36, Fabp4, and Pkm2 promoters in control and Rictor-iKOprimary cells (n = 3 for PPARγ ChIP, n = 3 for IgG ChIP; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001). ChIP with IgG were used as negative controls.
Neither ChREBPβ nor SREBP1n Overexpression Is Sufficient to Rescue PPARγ Target Genes
In mature adipocytes, mTORC2 positively regulates expression of the transcription factor ChREBPβ and its target genes in the DNL pathway (e.g., Acly, Acc, Fasn) (Tang et al., 2016). ChREBPβ has also been shown to regulate PPARγ expression and activity in 3T3L1 cells during adipogenesis (Witte et al., 2015). Therefore, we asked whether overexpressing recombinant ChREBPα or ChREBPβ in Rictor-iKOimmortal cells could rescue lipid accumulation and PPARγ gene expression. In the control cells, overexpressing recombinant ChREBPβ increases lipid amount by 20% determined by oil red O staining (Figure 4A); however, lipid accumulation is unaffected in Rictor-iKO cells overexpressing ChREBPα or ChREBPβ (Figure 4A). Notably, expressing ChREBPβ partially restored the mRNA and protein expression of ACLY, ACC, and FASN (Figures 4B and 4C), but it had no impact on the PPARγ target genes Cd36, Lpl, Fabp4, Dgat1, and Dgat2 (Figure 4C). Expressing ChREBPα had no effect on PPARγ target genes and minimal effects on DNL gene expression (Figures 4A–4C). The SREBP1 lipogenic transcription factor shares targets with ChREBP, and its activity is positively linked to mTORC2 in the liver (Hagiwara et al., 2012; Yuan et al., 2012). Interestingly, however, we observe an increase in the level of nuclear SREBP1 (SREBP1n) in both Rictor-iKOprimary and Rictor-iKOimmortal cells (Figures 4D and 4F), suggesting increased SREBP1 processing. Consistent with this, the gene encoding the SREBP-processing inhibitor INSIG1 is a Rictor-required gene (Figure 2D; Table S5). Moreover, overexpressing the transcriptionally active SREBP1n cleavage product in Rictor-iKOimmortal cells had little effect on PPARγ target gene expression and failed to restore lipid droplet formation (Figures 4E–4G). Thus, neither ChREBPα/β nor SREBP1n overexpression is sufficient to restore defects in lipid metabolic gene expression when cells differentiate in the absence of Rictor.
Figure 4. Overexpressing ChREBPβ or SREBP1n Does Not Rescue Rictor Loss.
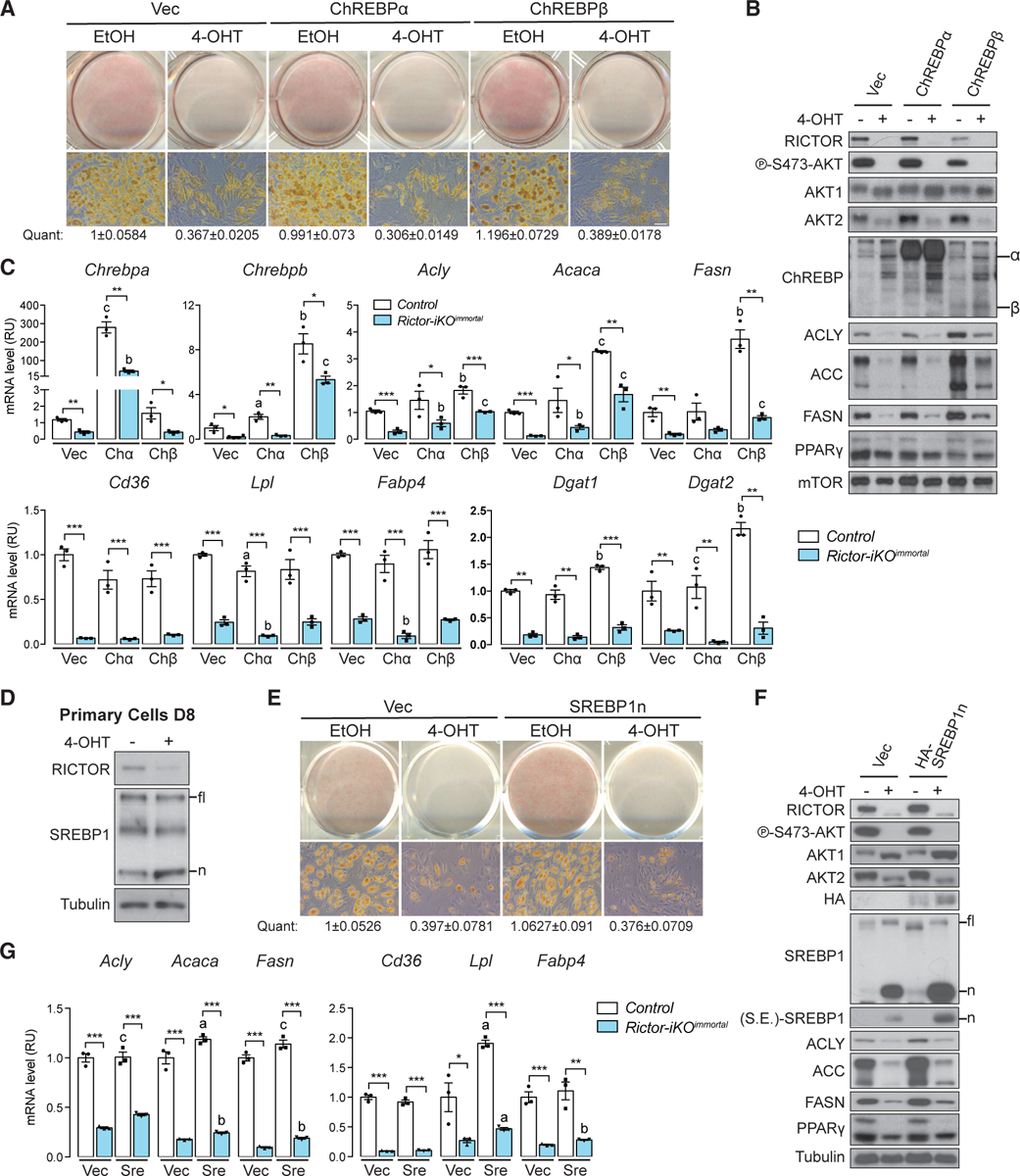
(A) Oil red O (ORO) staining of differentiated control (EtOH) and Rictor-iKOimmortal (4-OHT) cells expressing empty vector (Vec), ChREBPα, or ChREBPβ. The number below represents quantification (quant) of oil red O after isopropanol extraction (scale bar, 50 μm; data represent mean ± SEM).
(B) Western blot of lysates corresponding to (A).
(C) Relative mRNA expression by RT-PCR of the indicated genes corresponding to (A) (n = 3; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001). a–c denote comparison of overexpressing cells to vector-containing cells: a, p < 0.05; b, p < 0.01; c, p < 0.001.
(D) Western blot of lysates from differentiated (D8) control and Rictor-iKOprimary cells. fl, full-length SREBP1; n, processed nuclear SREBP1 product.
(E) Oil red O (ORO) staining of differentiated control and Rictor-iKOimmortal cells expressing empty vector (Vec) or SREBP1n. The number below represents quantification (quant) of oil red O after isopropanol extraction (data represent mean ± SEM).
(F) Western blot of lysates from differentiated control and Rictor-iKOimmortal cells corresponding to (E). S.E., shorter exposure.
(G) Relative mRNA expression by RT-PCR of indicated genes in control and Rictor-iKOimmortal cells corresponding to (E) (n = 3; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001). a–c denote comparison of overexpressing cells to vector-containing cells: a, p < 0.05; b, p < 0.01; c, p < 0.001.
In contrast to our findings in this study, previous work using a brown preadipocyte differentiation model showed that Rictor is required for PPARγ2 induction (Calejman et al., 2020; Hung et al., 2014), suggesting that brown and white preadipocyte differentiation may have different mTORC2 requirements in vitro. Consistent with this notion, we recently showed that overexpressing ACLY or ACLY-S455D partially and completely rescues Rictor loss in the brown preadipocyte model (Calejman et al., 2020); however, in Rictor-deficient subcutaneous white preadipocytes, stably overexpressing recombinant ACLY, ACLY-S455D, or ACLY-S455E does not rescue lipid accumulation (Figure S4E), Pparg2, Cd36, or Fabp4 gene expression (Figure S4F), or ACC protein levels during differentiation (Figure S4G). In fact, overexpressing ACLY enhances the suppressive effect of Rictor loss on gene expression in the white preadipocyte model (Figure S4F). This is consistent with these models of brown and subcutaneous white adipogenesis having different mTORC2 requirements.
AKT1-S473D Restores PPARγ Target Gene Expression
Next, we asked whether rescuing AKT HM phosphorylation could restore PPARγ target gene expression by generating Rictor-iKOimmortal cells expressing recombinant HA-AKT1-S473D or HA-AKT2-S474D phospho-mimetics or their HA-AKT1 and HA-AKT2 wild-type and HA-AKT1-S473A phospho-deficient controls. Only HA-AKT1-S473D restores lipid accumulation in differentiating Rictor-iKOimmortal cells (Figures 5A and 5B). Western blot confirms expression of each recombinant AKT construct (Figure 5C). Overexpressing HA-AKT1-S473D also increases Chrebpb, Acaca, Pparg2, Fabp4, and Cd36 expression as well as ACC protein expression in Rictor-iKOimmortal cells (Figures 5C and 5D). HA-S474D-AKT2 and to a lesser extent HA-AKT2 wild-type also increases Chrebpb expression consistent with a recent study linking AKT2 and ChREBP-dependent DNL in brown fat (Figure 5D) (Sanchez-Gurmaches et al., 2019) and suggesting that AKT1 and AKT2 may cooperate or compensate for each other in ChREBP regulation. Thus, restoring AKT HM phosphorylation is sufficient to rescue Rictor-dependent lipid metabolic gene expression.
Figure 5. AKT1-S473D Is Sufficient to Rescue Lipid Accumulation Defect in Rictor-KO Cells.
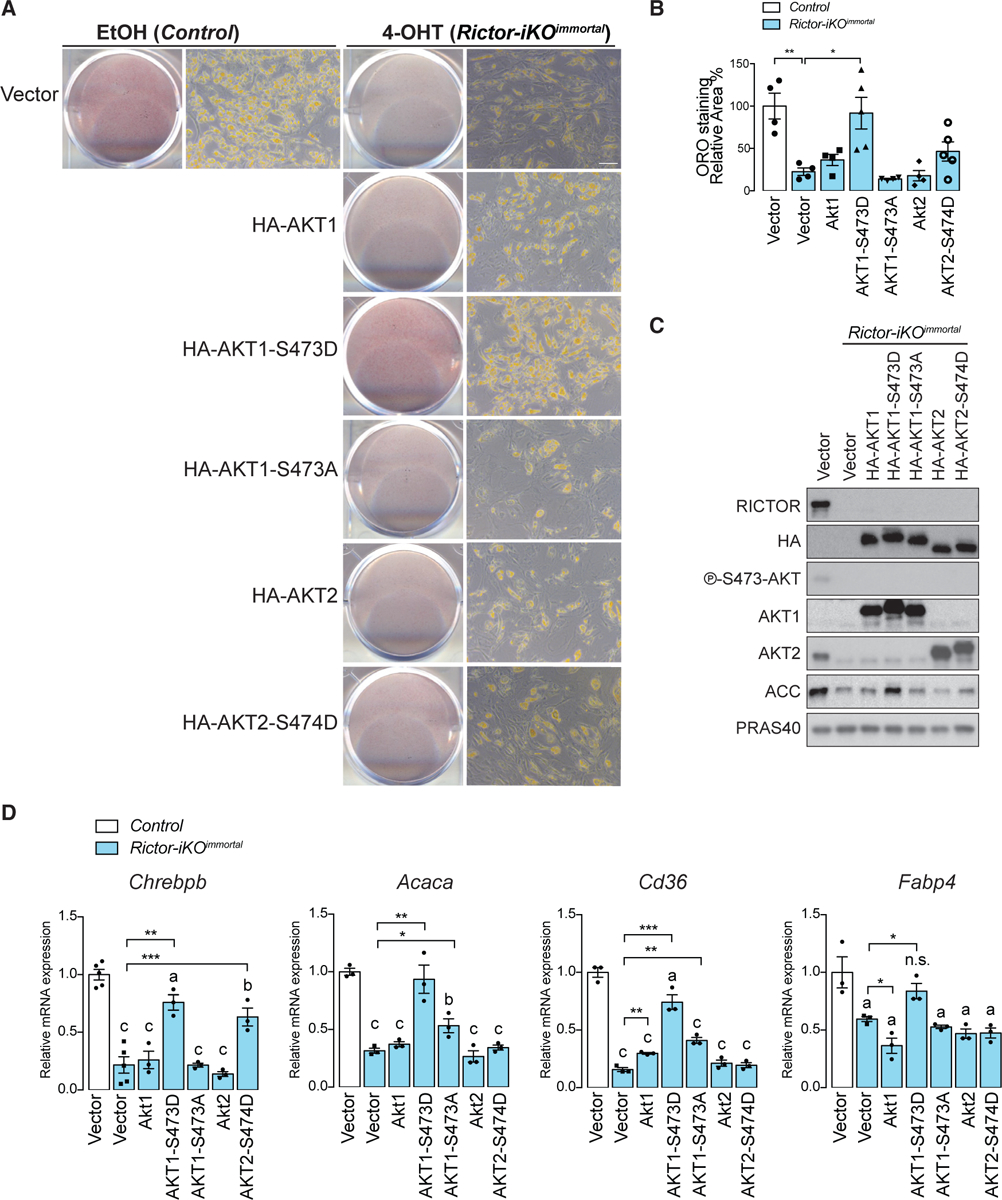
(A) Oil red O (ORO) staining of differentiated control (EtOH) and Rictor-iKOimmortal (4-OHT) cells expressing empty vector, HA-AKT1, HA-AKT1-S473D, HA-AKT1-S473A, HA-AKT2, or HA-AKT2-S474D.
(B) Quantification of oil red O from (A) after isopropanol extraction (scale bar, 50 μm; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001).
(C) Western blot of lysates from differentiated control (EtOH) and Rictor-iKOimmortal (4-OHT) cells expressing empty vector, HA-tagged AKT1, AKT1-S473D, AKT1-S473A, AKT2, or AKT2-S474D.
(D) Relative mRNA expression by RT-PCR of the indicated genes corresponding to (A) (n = 3; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001). a–c denote comparison of overexpressing cells to vector-containing cells: a, p < 0.05; b, p < 0.01; c, p < 0.001.
SWAT Development Requires mTORC2 In Vivo
To examine the physiological relevance of these findings, we generated Prx1-Cre;Rictorfl/fl mice (RictorPrx1-Cre) in which Rictor is deleted in vivo in a precursor cell population that gives rise to posterior SWAT but not to VWAT or BAT (Krueger et al., 2014; Sanchez-Gurmaches et al., 2015). RictorPrx1-Cre mice weigh significantly less than controls starting at 6 and 12 weeks of age for females and males, respectively (Figure 6A). Food intake is equivalent between groups (Figure S5A). In both sexes, the SWAT weighs significantly less in RictorPrx1-Cre mice (65% less in males and 57% less in females) (Figures 6B and 6C). H&E staining and imaging of the whole SWAT depot show reduced adipocyte size in RictorPrx1-Cre mice (Figures 6D and 6E). Reciprocally, VWAT and BAT masses in the male RictorPrx1-Cre mice increase by 60% and 35%, respectively, as a result of adipocyte hypertrophy (Figures 6B–6E). On the other hand, VWAT mass increases by only 40% in the female RictorPrx1-Cre mice (Figures 6B and 6C), because of milder cell hypertrophy (Figures 6D and 6E), but there is no significant difference in female BAT mass (Figures 6B and 6C). We determined that reduced tissue mass is mainly a result of smaller cell size by calculating total depot cellularity (Parlee et al., 2014), which reveals a linear relationship between tissue weight and average adipocyte volume (r2 = 0.97 in male SAT and r2 = 0.80 in female SAT) suggesting no significant difference in cellularity (Figure S5B). Moreover, adipocyte precursor cell (APC) number is unchanged between the SWAT of RictorPrx1-Cre mice and controls (Figure S5C) consistent with the SWAT partial-lipodystrophy phenotype originating from a lipid accumulation defect during adipogenesis. Western blotting confirms that RictorPrx1-Cre mice lack RICTOR and p-AKT-S473 in posterior SWAT, but not in VWAT or BAT (Figure 6F). Neither male nor female RictorPrx1-Cre mice have enlarged livers (Figure S5D) or evidence of hepatic steatosis on the basis of direct TAG measurement (Figure S5E). Analysis of neonates indicates that SWAT lipodystrophy occurs as early as postpartum D7 (P7), at which point the SWAT weighs 40% less and contains smaller adipocytes (Figures S5F–S5H). This is in stark contrast to deleting Rictor in mature adipocytes (e.g., with Adiponectin-Cre), which does not affect adipose tissue mass or adipocyte size through 20 weeks of age on standard chow (Tang et al., 2016). These data are consistent with Rictor’s also being required for SWAT development in vivo and further reveals a sex difference in how adipose tissue lipids are redistributed following Rictor loss.
Figure 6. Subcutaneous White Adipose Tissue Growth Requires mTORC2 In Vivo.
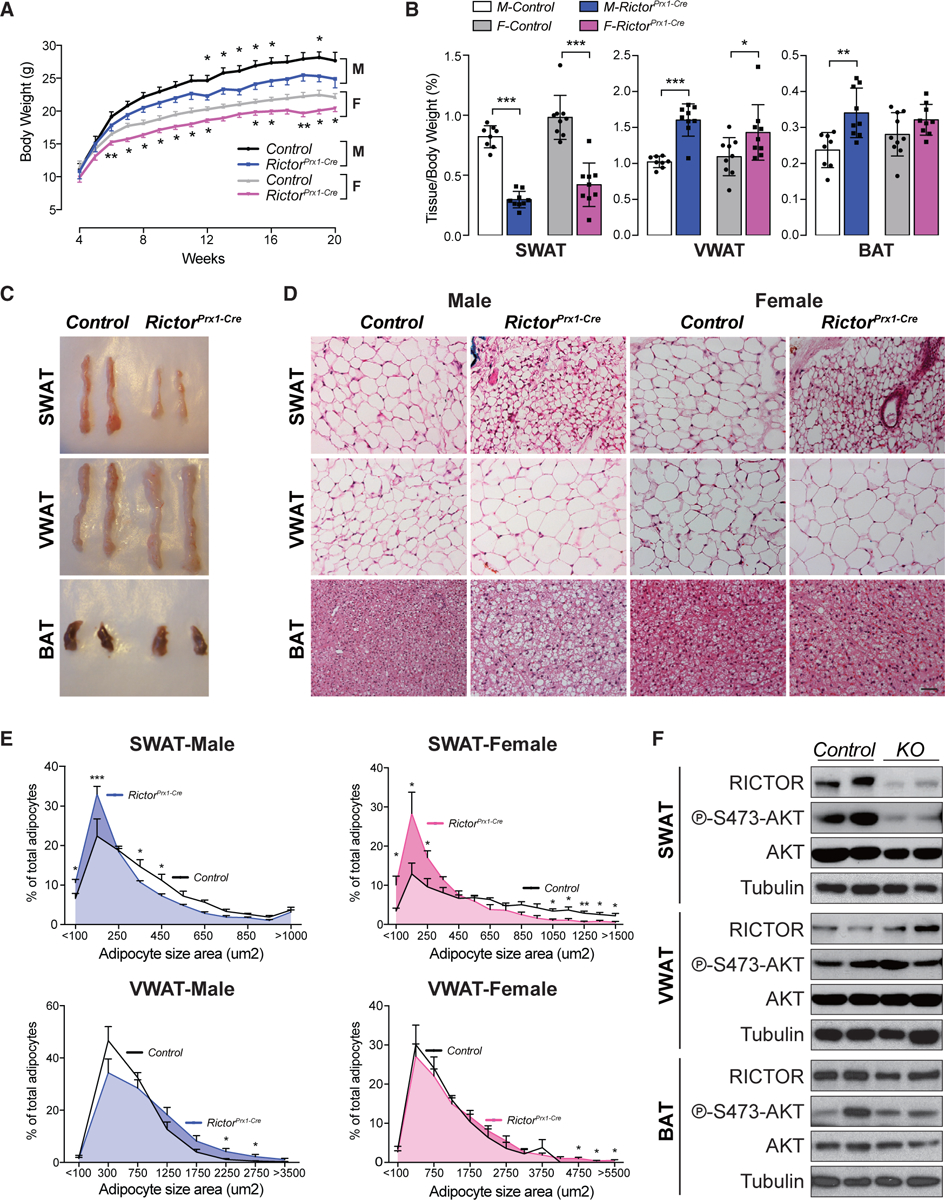
(A) Growth curves of male (M) and female (F) control and RictorPrx1-Cre mice (n = 10–14; data represent mean ± SEM; t test; *p < 0.05).
(B) Tissue weight relative to body weight of subcutaneous white adipose tissue (SWAT), visceral white adipose tissue (VWAT), and brown adipose tissue (BAT) (n = 8–10; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001).
(C) Representative images of the indicated fat depots from 8-week-old male control and RictorPrx1-Cre mice.
(D) H&E stains corresponding to the tissues in (C) for both male and female mice (scale bar, 100 μm).
(E) Individual adipocyte cell size distribution in each indicated depot (n = 4 mice; >1,000 and 500–1,000 individual adipocytes measured from SWAT and VWAT of each mouse, respectively; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001).
(F) Representative western blot of lysates from SWAT, VWAT, and BAT of 8-week-old male control and RictorPrx1-Cre mice.
Prx1-Cre-expressing precursors also give rise to some bone marrow adipocytes as well as osteoblasts and chondrocytes (Krueger et al., 2014; Logan et al., 2002). Consequently, computed tomographic (CT) scanning shows that the femur and tibia of male RictorPrx1-Cre mice are 10% and 5% shorter, respectively, correlating with thinner cortical and trabecular bone, which is more prominent in males (Figure S5I; Table S2) (Chen et al., 2015; Liu et al., 2016; Sun et al., 2016). Reduced bone length may explain why the quadriceps also weighs slightly less in RictorPrx1-Cre mice despite the muscle’s having normal morphology and RICTOR expression (Figures S5J–S5L). RictorPrx1-Cre male mice also have less bone marrow volume (MV) in the proximal region and a trending decrease in marrow adipose tissue (MAT) especially in females, as shown by osmium staining combined with CT scanning (Table S3) (Scheller et al., 2015). Gene expression analysis confirms reduced Rictor mRNA expression but normal Pparg2 expression in the marrow adipocytes of both male and female RictorPrx1-Cre mice (Figure S5M). These data are consistent with previous studies showing that Prx1-Cre also targets bone marrow mesenchyme (Krueger et al., 2014) and MAT (Sun et al., 2016).
Male Rictorprx1-Cre Mice Become Insulin Resistant
We next asked if SWAT dysfunction due to Rictor loss causes insulin resistance. Interestingly, 8-week-old male RictorPrx1-Cre mice develop insulin intolerance, as indicated by a 30% increase in glucose AUC relative to controls (Figure S6A). This correlates with a trending increase in serum insulin level in males (Figure S6E). This is not observed in females (Figure S6B), and neither sex shows defects in glucose tolerance (Figures S6C and S6D). Adiponectin, leptin, and non-esterified fatty acids (NEFAs) are unchanged in RictorPrx1-Cre male mice fed ad libitum (Figure S6E). The propensity for male RictorPrx1-Cre to develop insulin resistance is consistent with their greater accumulation of visceral fat.
Rictor Is Required In Vivo during SWAT Development for Lipid Metabolic Gene Expression
Similar to what we observed in vitro (i.e., in the D8 primary cells that were differentiated in the absence of Rictor), the SWAT of RictorPrx1-Cre mice expresses PPARγ and C/EBPα in vivo as well as insulin receptor beta (IRβ; which is elevated over control) (Figure 7A). Also, similar to the in vitro models, the SWAT from Rictorprx1-Cre mice has reduced AKT2 mRNA and protein expression (Figures 7A and S7A). In vivo, reduced AKT2 expression correlates with reduced p-AKT-T308, which is consistent with AKT2’s being the major AKT isoform in mature adipocytes. Interestingly, despite reduced p-AKT-T308, p-AS160-T642, p-GSK3β-S9, and p-FoxO1-T24 remain intact; however, p-PRAS40-T246 is reduced (Figure 7A). PRAS40 phosphorylation relieves its negative effect on mTORC1, and consistently, p-S6K1-T389 (a direct mTORC1 substrate) is also reduced (Figure 7A). We did not observe this effect in vitro (Figures 1F and S7B) or in vivo when Rictor is deleted in mature adipocytes with Adiponectin-Cre (Tang et al., 2016), suggesting the mTORC1-S6K1 signaling defect is a secondary effect caused by reduced AKT2 induction during differentiation.
Figure 7. Rictor Regulates Expression of Lipid-Handling Genes during SWAT Growth.
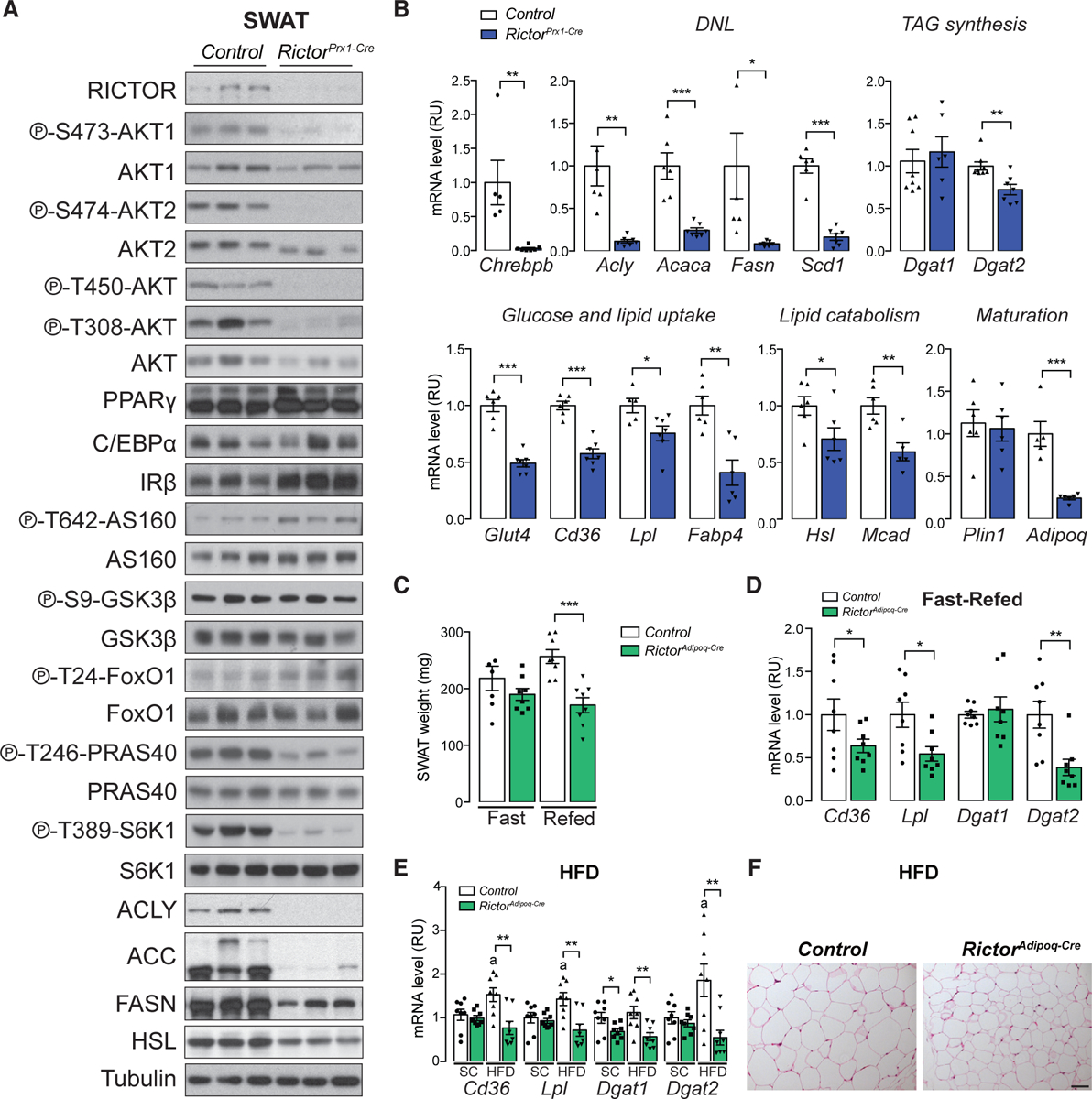
(A) Western blot of lysates from the subcutaneous white adipose tissue (SWAT) of 8-week-old control and RictorPrx1-Cre mice.
(B) Relative mRNA expression by RT-PCR of indicated genes from SWAT of 8-week-old control and RictorPrx1-Cre mice (n = 6–8; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001).
(C) Tissue weight of SWAT of 8-week-old control and RictorAdipoq-Cre mice refed for 6 h following 24 h fasting (n = 8; data represent mean ± SEM; ***p < 0.001).
(D) Relative mRNA expression by RT-PCR of indicated genes from SWAT of 8-week-old control and RictorAdipoq-Cre mice refed for 6 h following 24 h fasting (n = 8; data represent mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001).
(E) Relative mRNA expression by RT-PCR of indicated genes from SWAT of control and RictorAdipoq-Cre mice after 12 week HFD or standard chow (SC) feeding (n = 7 or 8; data represent mean ± SEM; *p < 0.05 and **p < 0.01; a, p < 0.05 when HFD samples were compared with SC samples).
(F) H&E stains of SWAT from control and RictorAdipoq-Cre mice after 12 week HFD or chow (SC) feeding (scale bar, 100 μm).
We next asked whether SWAT lipid-handling genes require Rictor during development for their expression in vivo. This is indeed the case for all of the Rictor-required anabolic and catabolic lipid metabolism genes as well as their products that we examined and that were previously identified by primary cell RNA-seq (Figures 7A and 7B). Adiponectin, however, is reduced in expression in vivo but not in the primary cell model (Figure 7B). This is consistent with previous observations showing that adiponectin levels may be sensitive to prolonged adipocyte Rictor loss in vivo (Cybulski et al., 2009; Tang et al., 2016). Notably, SWAT primary SVF preadipocytes isolated from Rictorprx1-Cre mice and differentiated in vitro also show reduced lipid accumulation (Figure S7C), decreased lipogenic and TAG synthesis gene and/or protein expression (Figures S7D and S7E), and decreased AKT2 expression (Figures S7D and S7E) after differentiation. Moreover, Adiponectin mRNA expression is unaffected in primary RictorPrx1-Cre preadipocytes that are differentiated (Figure S7E), consistent with downregulation of Adiponectin mRNA occurring secondary to Rictor loss. Consistent with previous data, Rictor-deficient SWAT also has defective insulin-stimulated glucose uptake and increased basal lipolysis (Figures S7F and S7G). These data confirm the physiological relevance of our in vitro findings and the role of mTORC2 in establishing the lipid handling capacity of SWAT.
In our previous study of RictorAdipoq-Cre mice, in which Rictor was deleted in mature adipocytes rather than in precursors as in this study, the expression of the PPARγ target genes Cd36, Lpl, and Fabp4 were unchanged between the control and Rictor-deficient SWAT depots when mice were eating standard chow ad libitum (Tang et al., 2016). Reasoning that the difference could be explained by SWAT development’s placing a greater demand on mTORC2-regulated PPARγ activity, we wondered whether challenging RictorAdipoq-Cre mice to store more lipid in SWAT would reveal the PPARγ gene expression defects. To this end, we re-examined PPARγ gene expression in RictorAdipoq-Cre mice that were refed following a fast or given a high-fat diet (HFD). Indeed, challenging RictorAdipoq-Cre mice with a prolonged fast followed by 6 h of refeeding resulted in 36%, 50%, and 60% reductions in Cd36, Lpl, and Dgat2 expression, respectively, corresponding to a 33% reduction in tissue mass relative to controls (Figures 7C and 7D). Similarly, placing RictorAdipoq-Cre on HFD for 12 weeks failed to increase Cd36, Lpl, and Fabp4 expression in the Rictor-KO fat (Figure 7E), consistent with reduced adipose tissue lipid accretion and overall smaller adipocytes (Figure 7F) (Tang et al., 2016). We conclude that mTORC2 is required to maximally stimulate expression of PPARγ-dependent lipid storage genes in subcutaneous white adipocytes when they are challenged with physiological states that draw high demand on lipid storage pathways, such as tissue development, refeeding after a fast, and chronic obesogenic diets.
DISCUSSION
In this study, we investigate the role of mTORC2 in SWAT development. Our findings support a model in which differentiating subcutaneous white adipocytes require Rictor/mTORC2 to establish maximum lipid handling capacity during development. This is mediated in part by positively regulating expression of specific PPARγ genes that encode regulators of lipid storage. One possibility is that mTORC2 regulates PPARγ’s ability to identify and/or remain associated with specific targets through a factor that cooperates with PPARγ (such as a protein or metabolite), by acting downstream (e.g., by regulating the chromatin state), or through direct regulation, although possibly arguing against the latter is the observation that only select PPARγ targets are mTORC2 dependent. Regardless, rescue experts suggest mTORC2 acts on these PPARγ genes at least in part through AKT signaling. Moreover, mTORC2 is required for maximum PPARγ target gene expression in mature subcutaneous white adipocytes, particularly during dietary challenges that promote rapid lipid storage and adipose tissue growth, such as consuming an HFD. TZDs, which are drugs used to treat T2D, work by binding and stimulating PPARγ, but they have negative side effects, including potential heart failure (Cariou et al., 2012). Because mTORC2 appears to promote only a subset of PPARγ activities, our study may help identify alternative mechanisms to stimulate safe lipid storage and insulin sensitivity.
In contrast to our present findings in white preadipocyte differentiation, Rictor is required for PPARγ mRNA induction in a brown preadipocyte differentiation model, and therefore, Rictor-deficient brown preadipocytes are completely incapable of differentiating and synthesizing lipid droplets when subjected to standard in vitro differentiation assays. In brown preadipocytes, this defect is linked to a deficiency in ACLY phosphorylation (S455) and acetyl-coA production. This phenotype is partially rescued by over-expressing recombinant ACLY and completely rescued by overexpressing the phospho-mimetic construct ACLY-S455D (Calejman et al., 2020). A recent study using a non-adipocyte model (primary bovine mammary epithelial cells) also showed blocked PPARγ2 expression when Rictor was knocked down by short hairpin RNA (shRNA) (Guo et al., 2019). Consistent with the brown and white preadipocyte differentiation models showing different requirements for mTORC2 (i.e., Rictor is not required for PPARγ2 mRNA induction here), overexpressing ACLY-S455D did not rescue gene expression and lipid accumulation defects in the Rictor-deficient subcutaneous white preadipocytes in this study. One explanation for why these brown and white adipocyte differentiation models differ is that they are different cell types derived from the natural precursor cell population in their respective depots (i.e., interscapular BAT and inguinal WAT). Alternatively, the brown preadipocytes are immortalized using the SV40-Large T antigen protocol (Fasshauer et al., 2000), which could change the metabolic requirements for PPARγ induction. Determining how mTORC2 signaling regulates metabolism and gene expression across different cell types is an important ongoing area of research.
How might mTORC2 regulate PPARγ activity? One possibility is that a PPARγ post-translational modification(s), such as phosphorylation, acetylation, SUMOylation, or O-GlcNAcylation, could be sensitive to mTORC2 signaling (Brunmeir and Xu 2018; Floyd and Stephens, 2004; Jennewein et al., 2008; Ji et al., 2012; Ohshima et al., 2004; Pascual et al., 2005). For example, phosphorylation of PPARγ increases or decreases its activity depending on the sites and the upstream regulators (Choi et al., 2014, 2010; Compe et al., 2005; Helenius et al., 2009; Hu et al., 1996; Iankova et al., 2006; Adams et al., 1997). Acetylation of PPARγ, on the other hand, has been shown to positively regulate lipid synthesis (Tian et al., 2014). Another possibility is that mTORC2 regulates the ability of PPARγ to bind certain co-factors (Miard and Fajas, 2005), which could include histone acetyltransferases (HATs) or histone deacetylases (HDACs) (Miard and Fajas, 2005). mTORC2 could also regulate PPARγ’s ability to bind certain targets by affecting the chromatin landscape or chromatin remodeling factors, evidenced by decreased H3K9ac in Rictor-KO PPREs shown here (Lefterova et al., 2010; Zhang et al., 2012). Combining global gene expression analysis with chromatin modification profiling, especially in vivo, will be important to differentiate among these possibilities (Roh et al., 2017). The detailed mechanism of how mTORC2 regulates PPARγ activity is still under investigation.
Another interesting finding in our study is that male and female mice respond differently to Prx1-Cre-mediated Rictor deletion with respect to fat re-distribution and insulin sensitivity. For example, only male RictorPrx1-Cre mice develop insulin resistance. Interestingly, although RictorAdipoq-Cre mice do not exhibit lipodystrophy, they are also insulin resistant, and this is more pronounced in male mice (Tang et al., 2016; Yu et al., 2019). Thus, adipose mTORC2 may play a greater role in controlling systemic insulin sensitivity in males than in females, and the SWAT may be particularly important to this phenomenon. The mechanism controlling this is currently unknown. However, these observations add to the growing appreciation for sex differences in adipose tissue metabolic regulation. For example, female Adiponectin-Cre;Aclyfloxed mice were recently reported to have a more severe metabolic phenotype than their male KO counterparts (Fernandez et al., 2019). Understanding sex differences in adipose tissue metabolism is an exciting ongoing research area.
One limitation of our study is that Prx1-Cre targets other cell lineages in addition to SWAT, including some bone lineages and marrow adipocytes. Importantly for this study, however, Prx1-Cre does not target precursors of brown fat or visceral white fat (Krueger et al., 2014; Sanchez-Gurmaches et al., 2015). Our use of three different in vitro differentiation models that exhibit overlapping phenotypes with one another and the in vivo model, including primary SVF preadipocytes derived from the RictorPrx1-Cre mice, greatly strengthens the ability to distinguish the mTORC2 functions that are specific to SWAT development. Nevertheless, effects caused by Rictor loss in non-adipose tissue cells cannot be ruled out in the in vivo model. Unfortunately, there is also no reciprocal Cre driver that is as robust at targeting only VWAT precursors. Therefore, we cannot make conclusions about how mTORC2 might function in VWAT development. Although our AKT rescue experiments support a model in which Rictor is acting through mTORC2 to promote lipid gene expression, we cannot rule on the possibility that Rictor might also have mTOR-independent functions in adipose tissues (Gao et al., 2010; Hagan et al., 2008). This aspect of Rictor biology remains poorly understood.
Overall, our study reveals previously unknown mTORC2 functions in regulating SWAT growth, adipose tissue gene expression, and sex-dependent metabolic homeostasis. These conclusions may have important implications for understanding and treating T2D and other obesity-related metabolic diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David A. Guertin (david.guertin@umassmed.edu).
Materials Availability
The plasmids and mice in this study were generated from the materials available in Addgene and Jackson Labs, respectively. Please contact the Lead Contact for further information.
Data and Code Availability
Unprocessed data from this manuscript have been deposited to Gene Expression Omnibus (GEO; GSE146470).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
We utilized the white preadipocytes residing in stromal vascular fraction (SVF) of Ubc-CreERT2;Rictorfloxed mice for in vitro studies. The SVFs are isolated by digesting the inguinal WAT in digestion buffer (123 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 5 mM glucose, 100 mM HEPES, 1% antibiotics and 4% BSA at pH 7.4 containing 1.5 mg/mL of collagenase A). The isolated cells were cultured directly as primary cells or immortalized by 3T3 immortalization protocol as previously described to generate cell lines (Tang et al., 2016). Cells were maintained in 25mM glucose (high-glucose), pyruvate and glutamine-containing DMEM in incubators at 37°C and 5% CO2. For adipocyte differentiation (Zebisch et al., 2012), cells were seeded at medium density and allowed to proliferate to confluence in the presence of high-glucose DMEM containing 10% FBS and 1% antibiotics (called complete medium). Two days after the cells reached confluency, cells were induced to differentiate by adding induction media (high-glucose DMEM containing 10% FBS, 1% antibiotics, 100 nM insulin, 2 μg/mL dexamethasone, 0.5mM 3-isobutyl-1-methylxanthine (IBMX) and 1 nM Rosiglitazone) for 2 days and the medium was replaced with complete medium containing 100 nM insulin for another two days and the cells were maintained in complete medium since then until day 8. Deletion of Rictor in preadipocytes was achieved by treating the cells with one dose of 4-hydroxytamoxifen (4-OHT, 1 μM) for two constitutive days before induction for early deletion as previous described (Hung et al., 2014; Tang et al., 2016). The cells were exposed to 4-OHT for only 2 days before differentiation and remained 4-OHT-free thereafter, which also minimizes the effects of 4-OHT on the cells. For Rictor deletion in differentiating adipocytes (Ad-Rictor-iKO), 4-OHT was supplemented in culture medium for two constitutive days from D2 to D4 after the differentiation was induced. Control cells received equivalent volume of ethanol (EtOH) as vehicle-treated controls. SVF isolated from Ubc-CreERT2 mice was treated either by EtOH or 4-OHT to address the effect of Cre. At different time points during differentiation, cells were collected for protein, mRNA or Oil-Red-O staining analysis. To analyze the signaling, cells were serum starved in high-glucose DMEM for 3 hours and stimulated with 100 nM insulin for 15 minutes.
Mice and Mice Housing
Rictor-floxed mice were described previously (Shiota et al., 2006) and were crossed with mice expressing either Prx1-Cre (JAK #005584), Adiponectin-Cre (JAK #028020) or Ubc-CreERT2 (JAX #007001) mice to generate conditional or inducible KO models. Floxed Cre-negative mice were used as controls. All the mice were in C57BL/6 background. The mice used for all studies were between 8–20 weeks old. Mice were housed in the Animal Medicine facilities of the UMMS in a clean room set at 22°C and 45% humidity under daily 12h light/dark cycles in ventilated racks with cages changed every two weeks, and fed a normal chow diet (Prolab® Isopro® RMH 3000) from Lab Diet ad libitum. For HFD challenge, diet was switched from normal chow to 60% HFD (D12492 Harlan Laboratories) when the mice were 8 weeks old. The mice were monitored for 12 weeks and the body weight was recorded weekly. Both male and female mice were utilized in this study. All animal experiments were approved by Institutional Animal Care and Use Committee of University (IACUC) of Massachusetts Medical school (UMMS). No animals were excluded from any experiments, unless they displayed obvious wounds from fighting as determined by our veterinarians.
METHOD DETAILS
Immunofluorescence and LipidTOX staining
Cells seeded on coverslips were fixed with iced-cold methanol at −20C for 15 min. Fixed cells were then blocked with PBS containing 3% BSA and 0.3% Triton for 30 min at room temperature, and incubated with primary anti-perilipin 1 (CST, 1:200 diluted in 1% BSA and 0.1% Triton) at 4°C overnight. After washed with PBS three times, the coverslips were stained with secondary antibodies (AlexaFluor-488-conjugated goat anti-rabbit IgG, Invitrogen, 1:400) mixed with HCS LipidTOX deep red neutral lipid stain (Invitrogen, H34477) at room temperature for an hour followed by DAPI staining and mounted on glass slides with Prolong Gold. Cells were examined with a laser-scanning microscope (Zeiss Axio imager). At least 6 images were obtained for each condition and the images were analyzed by ImageJ.
Oil Red O staining
The differentiated cells were washed three times with PBS and fixed with 10% buffered formalin at 4°C overnight. Cells were incubated in propylene glycol and then stained with a filtered Oil Red O solution (0.5% Oil Red O in propylene glycol) for 10 min at 37°C, washed with 85% propylene glycol and three times with distilled water, and visualized under a microscope (Zeiss). Oil Red O contents were then quantified by direct extraction of Oil Red O from stained cells using isopropanol and absorbance at 510 nM using a microplate reader (Tecan Safire2).
Western blot analysis and immunoprecipitation assays
Cells were harvested in cold PBS and lysed in protein lysis buffer (1% Triton X-100, 50 mM HEPES at pH 7.4, 150 mM NaCl, 5% glycerol, 2 mM EDTA, protease/phosphatase inhibitor cocktails). For immunoblot analysis of surgically dissected fat tissue depots, tissues were homogenized and lysed in RIPA buffer (150 mM NaCl, 50 mM HEPES at pH 7.4, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 0.5% Na-deoxycholate) containing protease and phosphatase inhibitor cocktails. Protein lysates were mixed with 5X SDS sample buffer and boiled, separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane filters and subjected to immunoblot analysis. Antibodies used in this study are listed in Key Resources Table. The PPARγ antibody was validated by transiently expressing recombinant PPARγ1 (with pSV Sport PPAR gamma) 1 or PPARγ2 (pSV Sport PPAR gamma 2) in HEK293T cells and matching the resulting recombinant protein bands in side-by-side western blots with endogenous PPARγ1 and PPARγ2, respectively, in primary and differentiated SVF cells.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mTOR | Cell Signaling Technology | Cat# 2972; RRID: AB_330978 |
| RICTOR | Cell Signaling Technology | Cat# 2140; RRID: AB_561245 |
| PPARγ (Western blot) | Cell Signaling Technology | Cat# 2443; RRID: AB_823598 |
| Perilipin1 (IF staining) | Cell Signaling Technology | Cat# 9349; RRID: AB_10829911 |
| C/EBP0α | Santa Cruz Biotechnology | Cat# sc-365318; RRID: AB_10846948 |
| α-Tubulin | Cell Signaling Technology | Cat# 2125; RRID: AB_2619646 |
| Phospho-S473-AKT | Cell Signaling Technology | Cat# 4058; RRID: AB_331168 |
| Phospho-T450-AKT | Cell Signaling Technology | Cat# 9267; RRID: AB_823676 |
| Phospho-T308-AKT | Cell Signaling Technology | Cat# 4056; RRID: AB_331163 |
| Phospho-S473-AKT1 | Cell Signaling Technology | Cat# 9018; RRID: AB_2629283 |
| Phospho-S474-AKT2 | Cell Signaling Technology | Cat# 8599; RRID: AB_2630347 |
| AKT1 | Cell Signaling Technology | Cat# 2938; RRID: AB_915788 |
| AKT2 | Cell Signaling Technology | Cat# 3063; RRID: AB_2225186 |
| AKT | Cell Signaling Technology | Cat# 9272; RRID: AB_329827 |
| Phospho-T24-FoxO1 | Cell Signaling Technology | Cat# 9464; RRID: AB_329842 |
| FoxO1 | Cell Signaling Technology | Cat# 2880; RRID: AB_2106495 |
| Phospho-S9-GSK3β | Cell Signaling Technology | Cat# 9322; RRID: AB_2115196 |
| GSK3bβ | Cell Signaling Technology | Cat# 12456; RRID: AB_2636978 |
| Phospho-T246-PRAS40 | Cell Signaling Technology | Cat# 2997; RRID: AB_2258110 |
| PRAS40 | Cell Signaling Technology | Cat# 2691; RRID: AB_2225033 |
| Phospho-T642-AS160 | Cell Signaling Technology | Cat# 8881; RRID: AB_2651042 |
| AS160 | EMD Millipore | Cat# 07–741; RRID: AB_492639 |
| Phospho-T389-S6K1 | Cell Signaling Technology | Cat# 9205; RRID: AB_330944 |
| S6K1 | Santa Cruz Biotechnology | Cat# sc8418 RRID: AB_628094 |
| Insulin Receptor(IR)β | Cell Signaling Technology | Cat# 3025; RRID: AB_2280448 |
| ACC | Cell Signaling Technology | Cat# 3676; RRID: AB_2219397 |
| ACLY | Cell Signaling Technology | Cat# 4332; RRID: AB_2223744 |
| FASN | Cell Signaling Technology | Cat# 3180; RRID: AB_2100796 |
| ChREBP | Novus | Cat# NB400–135; RRID: AB_10002435 |
| SREBP | EMD Millipore | Cat# 04–469, RRID:AB_612072 |
| HA | Cell Signaling Technology | Cat# 2367; RRID: AB_10691311 |
| Histone H3 | Cell Signaling Technology | Cat# 4499; RRID: AB_10544537 |
| Acetyl-Histone H3 (lys9) | Cell Signaling Technology | Cat# 9649; RRID: AB_823528 |
| PPARg | Santa Cruz Biotechnology | Cat# sc-7196; RRID: AB_654710 |
| CD36 | NOVUS | Cat#: NB400–144SS; RRID: AB_920879 |
| SCD1 | Abclonal | Cat# A16429 |
| FABP4 | Cell Signaling Technology | Cat# 2120; RRID: AB_2102466 |
| Goat anti-Rabbit IgG (H+L) CrossAdsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11011; RRID: AB_143157 |
| PE-Cy7-conjugated anti-CD31 | eBioscience, Thermo Fisher Scientific | Cat# 25-0311-82; RRID: AB_2716949 |
| PE-Cy7-conjugated anti-CD45 | eBioscience, Thermo Fisher Scientific | Cat# 25-0451-82; RRID: AB_2734986 |
| A700-conjugated anti-CD29 | BioLegend | Cat# 102218; RRID: AB_493711 |
| A647-conjugated anti-CD34 | BioLegend | Cat# 119314; RRID: AB_604089 |
| LybA/E-conjugated anti-Sca1 | eBioscience, Thermo Fisher Scientific | Cat# 45-0242-82; RRID: AB_1210701 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-hydroxy Tamoxifen (4-OHT) | Toronto research chemicals | H954729 |
| Rosiglitazone | Cayman Chemical | 71740 |
| Oil Red O | Sigma-Aldrich | O0625 |
| Human insulin, regular | Novo Nordisk | #183302 |
| Insulin | Sigma-Aldrich | I2643 |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma-Aldrich | I5879 |
| Dexamethasone | Sigma-Aldrich | D1756 |
| Osmium tetroxide 2% aqueous solution | Polysciences Inc | #23311 |
| HCS LipidTOX Deep Red Neutral Lipid Stain | Invitrogen | H34477 |
| Trichostatin A (TSA) | Sigma-Aldrich | T1952 |
| BODIPYFLC16 | Invitrogen | D3821 |
| Propidium iodide | Invitrogen | P3566 |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter assay system | Promega | E1910 |
| Free Glycerol Reagent | Sigma-Aldrich | F6428 |
| NEBNext® Ultra Directional RNA Library Prep Kit for Illumina® | New England Biolabs | E7760 |
| Seahorse XF Glycolysis stress test kit | Agilent Technologies | Cat# 103020-100 |
| Seahorse XF Palmitate Oxidation stress test kit and FAO substrate | Agilent Technologies | Cat# 103693-100 Cat# 102720-100 |
| Deposited Data | ||
| Raw and analyzed data | This study | GSE146470 |
| Experimental Models: Cell Lines | ||
| Immortalized subcutaneous white preadipocytes | (Tang et al., 2016) | N/A |
| Primary subcutaneous white preadipocytes | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Prx1-Cre | Jackson Labs | 005584 |
| Mouse: Adiponectin-Cre | Jackson Labs | 028020 |
| Mouse: Rictorfloxed | Jackson Labs | 020649 |
| Mouse: UBC-creERT2 | Jackson Labs | 007001 |
| Oligonucleotides | ||
| Mouse primers | IDT | See Table S4 |
| Recombinant DNA | ||
| pMSCV-Puro | (Akama-Garren et al., 2016) | RRID: Addgene_68469 |
| pMSCV-ChREBPα-Puro | (Witte etal., 2015) | N/A |
| pMSCV-ChREBPβ-Puro | (Witte etal., 2015) | N/A |
| pMSCV-HA-PPARγ2-Hygro | This study | N/A |
| pMSCV-Myc-ACLY-Puro | (Calejman et al., 2020) | N/A |
| pMSCV-Myc-ACLY-S455D-Puro | (Calejman et al., 2020) | N/A |
| pMSCV-Myc-ACLY-S455E-Puro | This study | N/A |
| pMSCV-Myc-ACLY-S455A-Puro | (Calejman et al., 2020) | N/A |
| PPRE X3-TK-luc | (Kim etal., 1998) | RRID: Addgene_1015 |
| pMSCV-HA-AKT1-Hygro | (Calejman et al., 2020) | N/A |
| pMSCV-HA-AKT1-S473D-Hygro | (Calejman et al., 2020) | N/A |
| pMSCV-HA-AKT1-S473A-Hygro | (Calejman et al., 2020) | N/A |
| pMSCV-HA-AKT2-Hygro | (Calejman et al., 2020) | N/A |
| pMSCV-HA-AKT2-S474D-Hygro | (Calejman et al., 2020) | N/A |
| pSV Sport PPAR gamma 1 | Addgene | RRID: Addgene_8886 |
| pSV Sport PPAR gamma 2 | Addgene | RRID: Addgene_8862 |
| Software and Algorithms | ||
| ImageJ (Fiji) | (Schindelin et al., 2012) | https://imagej.nih.gov/ij/ |
| Adiposoft Fiji plugin) | (Galarraga et al., 2012) | N/A |
| star_2.5.3a | (Dobin et al., 2013) | N/A |
| Ensembl annotation GRCm38.94 | (Zerbino et al., 2018) | N/A |
| featureCounts_1.5.2 | (Liao, Smyth, and Shi 2014) | N/A |
| DESeq2_1.20.0 | (Love, Huber, and Anders 2014) | N/A |
| Other | ||
| ChIP-Atlas | (Okietal. 2018) | N/A |
| DAVID Bioinformatics Resources 6.8 | (Huang, Sherman, and Lempicki 2009a; 2009b) | https://david.ncifcrf.gov/home.jsp |
| Cytation™ 5 Image reader | Biotek | N/A |
| LSRII A-5 laser flow cytometer | BD Biosciences | N/A |
| Seahorse XFe96 Analyzer | Agilent | N/A |
Gene expression analysis
Total RNA was isolated from cells or tissues using QIAzol (QIAGEN, #79306) and an RNeasy kit (QIAGEN). Equal amounts of RNA were retro-transcribed to cDNA using a High capacity cDNA reverse transcription kit (#4368813, Applied Biosystems). Quantitative RT-PCR (qRT-PCR) was performed in 10 μL reactions using a StepOnePlus real-time PCR machine from Applied Biosystems using SYBR Green PCR master mix (#4309156, Applied Biosystems, or 2XUlrtraSYBR from CWBio) according to manufacturer instructions. TATA-box binding protein (Tbp) gene expression was used as a normalization gene in all conventional RT-PCR experiments. Data analysis was performed on web-based software provided by the manufacturer. Primer sequences are shown in Key Resources Table and Table S4.
RNA-sequencing (RNA-seq) and Bioinformatics analysis
RNAs were extracted from primary culture cells as described. Extracted RNA (3ug) was processed for mRNA isolation using NEB-Next Poly(A) mRNA Magnetic Isolation Module (NEB, #E7490). Isolated mRNA was used to generate a cDNA library using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina according to the manufacturer’s instructions. For the multiplex purpose, the libraries were barcoded using commercially available primers for Illumina system (NEB). The quantity and quality were checked using Qubit and a fragment analyzer (a service provided by Molecular Biology Core Lab, MCBL at UMMS), respectively. The sequence was done by paired end read 100 bases using HiSeq 4000. For bioinformatics analysis,
Bioinformatics analysis
With star_2.5.3a (Dobin et al., 2013), paired-end reads were aligned to mouse genome mm10 (GRCm38.p6), which is annotated with Ensembl annotation GRCm38.94 (Zerbino et al., 2018). Aligned exon fragments with mapping quality higher than 20 were counted toward gene expression with featureCounts_1.5.2 (Liao et al., 2014). Differential expression (DE) analysis was performed with DESeq2_1.20.0 (Love et al., 2014). Within DE analysis, mouse was taken as a known batch variable. Also, ‘ashr’ was used to create log2 Fold Change (LFC) shrinkage for each comparison (Stephens, 2017). Significant DE genes (DEGs) were filtered with the criteria FDR < 0.05 and absolute log2 fold change (|LFC|) > 0.485 (fold change > 1.4). The analyzed data is listed in Table S5. Gene set enrichment analysis was performed using DAVID Bioinformatics Resources online (Huang et al., 2009a, 2009b) and ChIP-atlas for transcription factor interaction (Oki et al., 2018).
Chromatin immunoprecipitation (ChIP) analysis
Nuclei were isolated using nuclei preparation buffer supplemented with protease inhibitor and deacetylase inhibitor (Trichostatin A, TSA), and nuclei were pelleted by centrifugation. Cells were cross-linked with 1% paraformaldehyde for 10 minutes at room temperature and quenched for 10 minutes by adding 0.125 M glycine. After three washes with cold PBS, cells were lysed with lysis buffer (1% SDS, 20 mM EDTA pH8, 50 mM Tris-HCl pH8) supplemented with protease inhibitor and deacetylase inhibitor and placed on ice for 10 minutes. Fractionation of chromatin was done by Bioruptor (setting: 2 cycles, 15 minutes each with high intensity 30–30 s on-off interval). ChIP was done using the indicated primary antibodies and incubated overnight. Pull down of antibody-bound fragments was done by adding agarose protein G beads (Prometheus), followed by serial washes two times each with RIPA low salt buffer (0.1% SDS, 1% Triton x-100, 1 mM EDTA, 2 0mM Tris-HCl pH 8, 140 mM NaCl and 0.1% Na Deoxycholate), RIPA high salt buffer (0.1% SDS, 1% Triton x-100, 1 mM EDTA, 2 0mM Tris-HCl pH 8, 500 mM NaCl and 0.1% Na Deoxycholate), LiCl buffer (250 mM LiCl, 0.5% NP40, 0.5% Na Deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH8) and TE buffer (10mM Tris-HCl pH8 and 1 mM EDTA). DNA fragments were eluted by 100 mM NaHCO3 and 1% SDS, uncrosslinked, and treated with protease K and RNase. Fragments were eluted by phenol-chloroform extraction. Eluted fragments were analyzed by RT-PCR using the primers listed in Key Resources Table.
Luciferase Reporter Gene assay
The luciferase reporter containing PPRE (PPRE X3-TK-luc) was a gift from Bruce Spiegelman (Kim et al., 1998). The PPRE-firefly luc-containing plasmid is transiently co-transfected with RL-TK (containing Renilla-Luc as reference for transfection efficiency) into preadipocytes. Rictor deletion was deleted 24 hours after the transfection and differentiation was done as previously described. At day 2 of differentiation, cells were lysed and enzymic reactions were done using Dual Luciferase reporter Assay (DLR from Promega). The relative firefly luciferase/Renilla luciferase signals were determined by a microplate reader (Tecan Safire2).
Construction of overexpression by retroviral infection
To generate retroviruses, HEK293T cells were transfected with pMSCV-retroviral vectors subcloned with PPARγ2, ChREBPα, ChREBPβ, SREBP1n, ACLY with mutants, and AKT with mutants in combination with the retroviral packaging DNA (pCL-Ampho). Culture media was changed 12 hours after transfection and the virus-containing supernatant was collected 48 hours after transfection and passed through a 0.45 μm (PVDF) filter. Preadipocytes were transduced in medium containing 8 μg/mL of polybrene by centrifugation at 1700 RPM for 30 min. After 24 hours, cells were subjected to antibiotic selection and future analysis. The plasmids used in this study are listed in Key Resources Table.
Glucose uptake measurement
Cells were preincubated for 3 h in KRH medium without glucose (120 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 1.3 mM MgSO4, 1.3 mM KH2PO4 and HEPES 25mM with pH 7.4 plus 0.5% BSA + 2 mM pyruvate). For insulin stimulated group, cells were treated with 100 μM insulin for 15 minutes at the end of incubation. Deoxy-D-glucose 2-[1,2-3H(N)] mixed with unlabeled 2-DOG was then added, and incubation was continued for an additional 10 min. The medium was then removed and cells were washed three times with KRH medium without glucose and BSA to terminate the assay. Cells were then lysed in 1% triton, mixed with scintillation buffer, and the up-take of 3H glucose was quantified in counts per minute (cpm) using a scintillation counter. The cpm values were normalized to the protein concentration of each sample. For in vivo glucose uptake measurement, mice were fasted for 6 hours and were received an intraperitoneal injection of 10 mCi of Deoxy-D-glucose 2-[1,2-3H(N)] in a total volume of 150 mL. Two hours following the injection, mice were euthanized and tissue samples were collected, weighed and homogenized. Specific fractional uptakes of 3H-deoxyglucose were determined by scintillation counter.
De novo lipogenesis assay
Cells were incubated with 25 mM DMEM in which 0.01% of the total glucose concentration of the medium was comprised of D-[U-14C]-glucose for three days. Chloroform extraction was performed, and labeled lipids were measured using a scintillation counter. Each sample was normalized to total protein concentration.
BODIPY FL C16 uptake
BODIPY FL C16, a fluorescence analog of palmitic acid, were utilized for measuring lipid uptake as previously described (Dubikovskaya et al., 2014). Primary preadipocytes were cultured and induced into differentiation as previously described. At the end of differentiation, cultured medium was replaced by HBSS with 0.1% fatty acid free BSA containing 2 μM BODIPY FL C16 and the cells were incubated for 10 minutes or 30 minutes. The reaction was stopped by washing with cold PBS with 0.2% BSA. Cells were then trypsinized and diluted with FACS buffer (HBSS with 10% FBS, 10 mM EDTA, 50 μg/mL propidium iodide) and proceeded to FACS analysis (LSRII A-5 Laser).
Glycolytic stress test
Glycolytic ability was measured using a Seahorse XFe96 analyzer (Agilent Technologies). Mouse primary preadipocytes were seeded in a XFe96 cell culture microplate (Agilent Technologies) and differentiation was induced as previously described. At differentiation day 8, cells were washed and incubated with assay medium for 1 h at 37°C in a CO2-free incubator. Plates were then transferred to a Seahorse Bioscience XFe96 analyzer. Extracellular acidification rate (ECAR) were measured at baseline, followed by adding 10 mM glucose, oligomycin (1.5 μM), and 50 mM 2-deoxyglucose. indicates glycolytic capacity. The ECAR was normalized to cell number determined by Cytation™ 5.
Measurement of lipolysis in adipose tissue
For measurement of lipolysis, cultured adipocytes or adipose tissues from mice were harvested and incubated in DMEM with or without isoproterenol at 10 μM for 4 or 6 hours and the medium were collected to measure glycerol concentration using commercial kit (Free glycerol reagent, Sigma). The glycerol level was normalized with protein concentration of the tissue mass.
Histone extraction
Cells were washed 2 times with PBS and lysed with Triton extraction buffer (PBS containing 0.5% Triton X-100, protease inhibitor and 1 μM TSA) for 10 minutes at 4°C with gentle swirling. Lysates were pelleted and the pellets were resuspended in 0.2N HCl and incubated at 4°C overnight. Centrifugation was done in the next day and supernatants were collected for western blots.
Tissue harvest and histology
Adipose tissue depots and other organs/tissues were carefully dissected to avoid contamination from surrounding tissues. Organs/tissues were weighed by a microscale (XS105, Mettler Toledo). Samples for RNA or protein were frozen down immediately in liquid nitrogen and stored at 80°C for further analysis. For signaling, mice were fasted for 6 hours in the morning without changing other husbandry conditions. The same amount of normal chow diet was provided for an hour after fasting period and the indicated tissues were harvested and stored as described above. For histology, tissue pieces were fixed by 10% formalin. Embedding, sectioning and Hematoxylin & Eosin (H&E) staining were done by the UMass Medical School Morphology Core. Liver samples were embedded in O.C.T. compound (Tissue-Tek) before sectioning and Oil Red O staining. Images were taken by Zeiss Axio microscope. For cell size measurements, tissue slices were scanned by Zeiss Axio Scan.Z1 (N = 4 for wild-type and 4 for conditional KOs) and the adipocyte size was automatically measured by ImageJ with plug-in (Adipose). More than 1000 cells were analyzed for each depot. For estimating adipocyte number in a depot, average adipocyte volume was calculated from more than 800 cells which were imaged as described above. The adiposity was then determined by depot weight-to-average adipocyte volume ratio.
Glucose Tolerance Test / Insulin Tolerance Test
Animals subjected for glucose tolerance test (GTT) were fasted overnight, followed by intraperitoneal glucose (2 g/kg of body weight) injection. For insulin tolerance test animals were fasted for 5 hours in the morning without changing other husbandry conditions, followed by intraperitoneal insulin (0.75 U/kg of body weight, Novolin) injection. Blood glucose levels were measured by tail bleeding with a commercially available glucose meter (GE) at indicated time points.
Liver TAG measurement
The protocol is modified from a previously described method (Jouihan, 2012). For each sample, 100–300mg of liver was taken and lysed in ethanolic KOH (2 parts of ethanol and 1 part of 30% KOH) at 55 overnight. Digested lysates were diluted with 50% EtOH and centrifuge for 5 minutes. 200 uL of lysate were taken and mixed with 215 uL 1M MgCl2, then the mixture was left on ice for 10 minutes and followed by centrifugation for 5 minutes. Mixtures were mixed with Free Glycerol Reagent (Sigma) and incubated for 15 minutes at 37°C. The samples were measured by a microplate reader at 540 nm absorbance.
Serology
Blood was collected from animals by cardiocentesis. Serum was collected from the supernatant after centrifugation. The analysis of insulin, NEFA, leptin and adiponectin was performed by National Mouse Metabolic Phenotyping Center (MMPC) at UMMS.
Marrow fat quantification by osmium staining and CT
The protocol was modified from a previously described method (Scheller et al., 2014). Bones were fixed for 24–48 hours in 10% neutral-buffered formalin (VWR, Radnor, PA; cat. no. 16004–128), washed with water and decalcified in 14% EDTA, pH 7.4, for 14 days. After washing again with water, 600 μl Sorensen’s phosphate buffer (pH 7.4) was added to one bone (tibia) in a 1.5 mL microtube. Four per cent osmium tetroxide (200 μl) solution (Electron Microscopy Services, Hatfield, PA; cat.no. 19170) was added to each tube to make a 1% solution. Bones were stained in the fume hood 48 hours at room temperature. Osmium solution was carefully removed to a small liquid waste container that had been filled with corn oil to ~25% of the volume. Any used pipet tips were ‘rinsed’ of active osmium tetroxide by pipeting corn oil. All tips and tubes were discarded as osmium solid waste. Bones were washed, in the same tube, by incubating in 1 ml of Sorensen’s buffer for 3 h at room temperature. This was repeated twice and the last wash was left in the hood overnight. Stained bones were then moved to a fresh set of 1.5 ml microtubes containing 1 ml Sorensen’s buffer each. The used tubes were discarded as solid osmium waste. At this point, the bones and tubes were removed from the fume hood and used for CAT scan.
MicroCT
Specimens were embedded in 1% agarose and placed in a 19-mm diameter tube. The length of the bone was scanned using a μCT system (μCT100 Scanco Medical, Bassersdorf, Switzerland). Scan settings are as follows: voxel size 12 μm, medium resolution, 70 kVp, 114 μA, 0.5 mm AL filter and integration time 500 ms. Density measurements were calibrated to the manufacturer’s hydroxy-apatite phantom. Analysis was performed using the manufacturer’s evaluation software and a threshold of 400 for MAT.
APC quantification
APCs were isolated as previously described (Rodeheffer et al., 2008). In brief, stromal-vascular fraction (SVF) was prepared from SWAT by collagenase (1.5mg/mL) treatment and the pellets were resuspended in erythrocyte lyses buffer (0.15 M NH4Cl and 0.01 M KHCO3 in water). Cells were then pelleted and resuspended in staining media (Hanks’ balanced salt solution (HBSS) with 2% fetal bovine serum) and labeled with the following primary antibodies: PE-Cy7-conjugated anti-CD31, PE-Cy7-conjugated anti-CD45, A700-conjugated anti-CD29, A647-conjugated anti-CD34 and LybA/E-conjugated anti-Sca1 Antibodies. After staining, cells were filtered through a 35-μm cell-strainer capped tube (#352235, BD Falcon) to ensure single-cell suspension and stained with live/dead Blue (L34962, Invitrogen). Live single cells were gated according to the expression of surface markers (CD31−CD45−CD29+CD34+Sca1+) in a BD LSRII analyzer. Data were analyzed with FlowJo.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean + SEM, unless stated otherwise. Student’s t test, or non-parametric Mann-Whitney test, were used to determine statistical significance. Statistical analysis was done using GraphPad Prism. The number of mice used per experiment is stated in each figure legend.
Supplementary Material
Highlights.
Rictor is deleted specifically in developing subcutaneous but not visceral or brown fat
Developing subcutaneous fat requires mTORC2 to establish maximum lipid storing capacity
mTORC2 regulates expression of diverse lipid metabolism genes during adipogenesis
Inhibiting mTORC2 during SWAT development has sex-specific consequences
ACKNOWLEDGMENTS
We thank members of the Guertin lab for valuable discussions. We thank Dr. Tony Imbalzano for reading the manuscript and Dr. Yung Hwang, Dr. Norm Kennedy, and Dr. Hyun Cheol Roh for technical assistance. D.A.G. is supported by NIH grants R01DK094004 and R01CA196986 and a Leukemia and Lymphoma Society Career Development Award, and D.A.G. and K.E.W. are supported by NIH grant 1R01DK116005. S.M.J. is supported by a postdoctoral fellowship from the American Diabetes Association (1-18-PDF-128). J.S.-G. was supported by a postdoctoral fellowship from the American Heart Association (15POST25550079). C.M.C. was supported by a postdoctoral fellowship from the American Diabetes Association (1-16-PMF-008). A.K.L. is supported by a post-doctoral fellowship from the American Cancer Society (PF-19-103-01-TBE).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108223.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams M, Reginato MJ, Shao D, Lazar MA, and Chatterjee VK (1997). Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem 272, 5128–5132. [DOI] [PubMed] [Google Scholar]
- Akama-Garren EH, Joshi NS, Tammela T, Chang GP, Wagner BL, Lee D-Y, Rideout WM 3rd, Papagiannakopoulos T, Xue W, and Jacks T (2016). A Modular Assembly Platform for Rapid Generation of DNA Constructs. Sci. Rep 6, 16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almandoz JP, Singh E, Howell LA, Grothe K, Vlazny DT, Smailovic A, Irving BA, Nelson RH, and Miles JM (2013). Spillover of fatty acids during dietary fat storage in type 2 diabetes: relationship to body fat depots and effects of weight loss. Diabetes 62, 1897–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg M, Abdullah N, Thowfeik FS, Altorki NK, and McGraw TE (2017). Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. eLife 6, e26896 10.7554/eLife.26896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R, and Xu F (2018). Functional regulation of PPARs through post-translational modifications. Int. J. Mol. Sci 19, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calejman CM, Trefely S, Entwisle SW, Luciano A, Jung SM, Hsiao W, Torres A, Hung CM, Li H, Snyder NW, et al. (2020). Author correction: mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat. Commun 11, 4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariou B, Charbonnel B, and Staels B (2012). Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol. Metab 23, 205–215. [DOI] [PubMed] [Google Scholar]
- Chen J, Holguin N, Shi Y, Silva MJ, and Long F (2015). mTORC2 signaling promotes skeletal growth and bone formation in mice. J. Bone Miner. Res 30, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M, et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Choi S-S, Kim ES, Jedrychowski MP, Yang YR, Jang H-J, Suh P-G, Banks AS, Gygi SP, and Spiegelman BM (2014). Thrap3 docks on phosphoserine 273 of PPARγ and controls diabetic gene programming. Genes Dev. 28, 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compe E, Drané P, Laurent C, Diderich K, Braun C, Hoeijmakers JHJ, and Egly J-M (2005). Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Mol. Cell. Biol 25, 6065–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski N, Polak P, Auwerx J, Rüegg MA, and Hall MN (2009). mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc. Natl. Acad. Sci. U S A 106, 9902–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubikovskaya E, Chudnovskiy R, Karateev G, Park HM, and Stahl A (2014). Measurement of long-chain fatty acid uptake into adipocytes. Methods Enzymol. 538, 107–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissing L, Scherer T, Tödter K, Knippschild U, Greve JW, Buurman WA, Pinnschmidt HO, Rensen SS, Wolf AM, Bartelt A, et al. (2013). De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun 4, 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al. (2008). The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 27, 1932–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer M, Klein J, Ueki K, Kriauciunas KM, Benito M, White MF, and Kahn CR (2000). Essential role of insulin receptor substrate-2 in insulin stimulation of Glut4 translocation and glucose uptake in brown adipocytes. J. Biol. Chem 275, 25494–25501. [DOI] [PubMed] [Google Scholar]
- Fernandez S, Viola JM, Torres A, Wallace M, Trefely S, Zhao S, Affronti HC, Gengatharan JM, Guertin DA, Snyder NW, et al. (2019). Adipocyte ACLY facilitates dietary carbohydrate handling to maintain metabolic homeostasis in females. Cell Rep. 27, 2772–2784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara D, Montecucco F, Dallegri F, and Carbone F (2019). Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell. Physiol 234, 21630–21641. [DOI] [PubMed] [Google Scholar]
- Fitzgerald SJ, Janorkar AV, Barnes A, and Maranon RO (2018). A new approach to study the sex differences in adipose tissue. J. Biomed. Sci 25, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd ZE, and Stephens JM (2004). Control of peroxisome proliferator-activated receptor gamma2 stability and activity by SUMOylation. Obes. Res 12, 921–928. [DOI] [PubMed] [Google Scholar]
- Galarraga M, Campión J, Muñoz-Barrutia A, Boqué N, Moreno H, Martínez JA, Milagro F, and Ortiz-de-Solórzano C (2012). Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J. Lipid Res 53, 2791–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wan L, Inuzuka H, Berg AH, Tseng A, Zhai B, Shaik S, Bennett E, Tron AE, Gasser JA, et al. (2010). Rictor forms a complex with Cullin-1 to promote SGK1 ubiquitination and destruction. Mol. Cell 39, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Henriques F, Bedard AH, and Czech MP (2019). Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat. Rev. Endocrinol 15, 207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Zhao K, Feng X, Yan D, Yao R, Chen Y, Bao L, and Wang Z (2019). mTORC2 regulates lipogenic gene expression through PPARγ to control lipid synthesis in bovine mammary epithelial cells. BioMed Res. Int 2019, 5196028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, Jenoe P, Heim MH, Riezman I, Riezman H, and Hall MN (2017). mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32, 807–823.e12. [DOI] [PubMed] [Google Scholar]
- Hagan GN, Lin Y, Magnuson MA, Avruch J, and Czech MP (2008). A Rictor-Myo1c complex participates in dynamic cortical actin events in 3T3-L1 adipocytes. Mol. Cell. Biol 28, 4215–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Rüegg MA, and Hall MN (2012). Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 15, 725–738. [DOI] [PubMed] [Google Scholar]
- Hauner H (2002). The mode of action of thiazolidinediones. Diabetes Metab. Res. Rev 18 (Suppl 2), S10–S15. [DOI] [PubMed] [Google Scholar]
- Helenius K, Yang Y, Alasaari J, and Mäkelä TP (2009). Mat1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipocyte differentiation. Mol. Cell. Biol 29, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka D, Okumura E, and Kishimoto T (2011). Turn motif phosphorylation negatively regulates activation loop phosphorylation in Akt. Oncogene 30, 4487–4497. [DOI] [PubMed] [Google Scholar]
- Hresko RC, and Mueckler M (2005). mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem 280, 40406–40416. [DOI] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, and Spiegelman BM (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274, 2100–2103. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009a). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009b). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C-M, Calejman CM, Sanchez-Gurmaches J, Li H, Clish CB, Hettmer S, Wagers AJ, and Guertin DA (2014). Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep. 8, 256–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iankova I, Petersen RK, Annicotte J-S, Chavey C, Hansen JB, Kratchmarova I, Sarruf D, Benkirane M, Kristiansen K, and Fajas L (2006). Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol 20, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K (2017). The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta Mol. Basis Dis 1863, 474–485. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Inoki K, Yang Q, Zhou X, and Guan K-L (2008). Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 27, 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein C, Kuhn A-M, Schmidt MV, Meilladec-Jullig V, von Knethen A, Gonzalez FJ, and Brüne B (2008). Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol 181, 5646–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S, Park SY, Roth J, Kim HS, and Cho JW (2012). O-GlcNAc modification of PPARγ reduces its transcriptional activity. Biochem. Biophys. Res. Commun 417, 1158–1163. [DOI] [PubMed] [Google Scholar]
- Jouihan H (2012). Measurement of liver triglyceride content. Bio Protoc. 2 (13). [Google Scholar]
- Jung SM, Hung C-M, Hildebrand SR, Sanchez-Gurmaches J, Martinez-Pastor B, Gengatharan JM, Wallace M, Mukhopadhyay D, Martinez Calejman C, Luciano AK, et al. (2019). Non-canonical mTORC2 signaling regulates brown adipocyte lipid catabolism through SIRT6-FoxO1. Mol. Cell 75, 807–822.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney AL, Cooke KC, Norris DM, Zadoorian A, Krycer JR, Fazakerley DJ, Burchfield JG, and James DE (2019). Serine 474 phosphorylation is essential for maximal Akt2 kinase activity in adipocytes. J. Biol. Chem 294, 16729–16739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Wright HM, Wright M, and Spiegelman BM (1998). ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. U S A 95, 4333–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger KC, Costa MJ, Du H, and Feldman BJ (2014). Characterization of Cre recombinase activity for in vivo targeting of adipocyte precursor cells. Stem Cell Reports 3, 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PL, Jung SM, and Guertin DA (2017). The complex roles of mechanistic target of rapamycin in adipocytes and beyond. Trends Endocrinol. Metab 28, 319–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert CJ Jr., Liu XS, and Lazar MA (2008). PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22, 2941–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, Manduchi E, Grant GR, and Lazar MA (2010). Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol. Cell. Biol 30, 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterova MI, Haakonsson AK, Lazar MA, and Mandrup S (2014). PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab 25, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, and Tchernof A (2012). Depot- and obesity-related differences in adipogenesis. Clin. Lipidol 7, 587–596. [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu D-M, Zhao L, Liu T-T, Jiao P-L, Zhao D-D, Shih M-S, Tao B, Sun L-H, Zhao H-Y, and Liu J-M (2016). Rictor/mTORC2 loss in osteoblasts impairs bone mass and strength. Bone 90, 50–58. [DOI] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, and Tabin CJ (2002). Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Lamendola C, Liu A, and Abbasi F (2011). Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J. Clin. Endocrinol. Metab 96, E1756–E1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miard S, and Fajas L (2005). Atypical transcriptional regulators and cofactors of PPARgamma. Int. J. Obes 29 (Suppl 1), S10–S12. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Koga H, and Shimotohno K (2004). Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. J. Biol. Chem 279, 29551–29557. [DOI] [PubMed] [Google Scholar]
- Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, Kawaji H, Nakaki R, Sese J, and Meno C (2018). ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 19, e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Prieto P, and Postic C (2019). Carbohydrate sensing through the transcription factor ChREBP. Front. Genet 10, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasyuk G, Espeillac C, Chauvin C, Pradelli LA, Horie Y, Suzuki A, Annicotte J-S, Fajas L, Foretz M, Verdeguer F, et al. (2012). PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun 3, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlee SD, Lentz SI, Mori H, and MacDougald OA (2014). Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol. 537, 93–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, and Glass CK (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R, Hodson L, Dennis AL, Neville MJ, Humphreys SM, Harnden KE, Micklem KJ, and Frayn KN (2009). Markers of de novo lipogenesis in adipose tissue: associations with small adipocytes and insulin sensitivity in humans. Diabetologia 52, 882–890. [DOI] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, and Friedman JM (2008). Identification of white adipocyte progenitor cells in vivo. Cell 135, 240–249. [DOI] [PubMed] [Google Scholar]
- Roh HC, Tsai LT-Y, Lyubetskaya A, Tenen D, Kumari M, and Rosen ED (2017). Simultaneous transcriptional and epigenomic profiling from specific cell types within heterogeneous tissues in vivo. Cell Rep. 18, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salma N, Xiao H, Mueller E, and Imbalzano AN (2004). Temporal recruitment of transcription factors and SWI/SNF chromatin-remodeling enzymes during adipogenic induction of the peroxisome proliferator-activated receptor gamma nuclear hormone receptor. Mol. Cell. Biol 24, 4651–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Hsiao W-Y, and Guertin DA (2015). Highly selective in vivo labeling of subcutaneous white adipocyte precursors with Prx1-Cre. Stem Cell Reports 4, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Martinez Calejman C, Jung SM, Li H, and Guertin DA (2019). Brown fat organogenesis and maintenance requires AKT1 and AKT2. Mol. Metab 23, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Scheller EL, Troiano N, VanHoutan JN, Bouxsein MA, Fretz JA, Xi Y, Nelson T, Katz G, Berry R, Church CD, et al. (2014). Use of osmium tetroxide staining with microcomputerized tomography to visualize and quantify bone marrow adipose tissue in vivo. Methods Enzymol. 537, 123–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, Wu B, Ding S-Y, Bredella MA, Fazeli PK, et al. (2015). Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat. Commun 6, 7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer PE (2019). The many secret lives of adipocytes: implications for diabetes. Diabetologia 62, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota C, Woo J-T, Lindner J, Shelton KD, and Magnuson MA (2006). Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 11, 583–589. [DOI] [PubMed] [Google Scholar]
- Smith U, and Kahn BB (2016). Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med 280, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel M, Jonker JT, Schoones J, Lamb H, de Roos A, Pijl H, Smit JWA, Meinders AE, and Jazet IM (2012). Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int. J. Endocrinol 2012, 983814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Grant GR, Schupp M, Tomaru T, Lefterova MI, Schug J, Manduchi E, Stoeckert CJ Jr., and Lazar MA (2010). Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 24, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M (2017). False discovery rates: a new deal. Biostatistics 18, 275–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Shi Y, Lee W-C, Lee S-Y, and Long F (2016). Rictor is required for optimal bone accrual in response to anti-sclerostin therapy in the mouse. Bone 85, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Wallace M, Sanchez-Gurmaches J, Hsiao W-Y, Li H, Lee PL, Vernia S, Metallo CM, and Guertin DA (2016). Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat. Commun 7, 11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchoukalova YD, Koutsari C, Votruba SB, Tchkonia T, Giorgadze N, Thomou T, Kirkland JL, and Jensen MD (2010). Sex- and depot-dependent differences in adipogenesis in normal-weight humans. Obesity (Silver Spring) 18, 1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Wang C, Hagen FK, Gormley M, Addya S, Soccio R, Casimiro MC, Zhou J, Powell MJ, Xu P, et al. (2014). Acetylation-defective mutant of Pparγ is associated with decreased lipid synthesis in breast cancer cells. Oncotarget 5, 7303–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal J-F, Montagner A, and Gourdy P (2020). Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 63, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Clark GO, Scherer PE, and Orci L (2010). Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 1801, 209–214. [DOI] [PubMed] [Google Scholar]
- Van Gaal LF, Mertens IL, and De Block CE (2006). Mechanisms linking obesity with cardiovascular disease. Nature 444, 875–880. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Z, Wang Q, Liang H, and Liu D (2019). Trichostatin A and vorinostat promote adipogenic differentiation through H3K9 acetylation and dimethylation. Res. Vet. Sci 126, 207–212. [DOI] [PubMed] [Google Scholar]
- Witte N, Muenzner M, Rietscher J, Knauer M, Heidenreich S, Nuotio-Antar AM, Graef FA, Fedders R, Tolkachov A, Goehring I, and Schupp M (2015). The glucose sensor ChREBP links de novo lipogenesis to PPARγ activity and adipocyte differentiation. Endocrinology 156, 4008–4019. [DOI] [PubMed] [Google Scholar]
- Yu D, Tomasiewicz JL, Yang SE, Miller BR, Wakai MH, Sherman DS, Cummings NE, Baar EL, Brinkman JA, Syed FA, and Lamming DW (2019). Calorie-restriction-induced insulin sensitivity is mediated by adipose mTORC2 and not required for lifespan extension. Cell Rep. 29, 236–248.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Pino E, Wu L, Kacergis M, and Soukas AA (2012). Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J. Biol. Chem 287, 29579–29588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebisch K, Voigt V, Wabitsch M, and Brandsch M (2012). Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem 425, 88–90. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. (2018). Ensembl 2018. Nucleic Acids Res. 46 (D1), D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ramlee MK, Brunmeir R, Villanueva CJ, Halperin D, and Xu F (2012). Dynamic and distinct histone modifications modulate the expression of key adipogenesis regulatory genes. Cell Cycle 11, 4310–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed data from this manuscript have been deposited to Gene Expression Omnibus (GEO; GSE146470).