

Abstract
CD8 T cell differentiation is a tightly regulated process generating effector and memory T cells over the course of acute infections. In cancer and chronic infection, this differentiation program is derailed, and antigen-specific CD8 T cells differentiate to a hyporesponsive state generally referred to as T cell exhaustion. Here, we review recent findings on heterogeneity of tumor-specific T cells and exhausted T cells during chronic infections, discussing distinct differentiation state dynamics, fate choices, and functional states. Delineating the regulatory mechanisms defining distinct T cell states and determining the requirements for therapeutic reprogramming of these states will provide needed insights for the design of effective immunotherapies for the treatment of cancer and chronic infections.
Introduction and terminology
Several terms are currently in use to describe hyporesponsive CD8 T cells, including tolerance, anergy, exhaustion, and dysfunction. Tolerance describes the central or peripheral inactivation of self-reactive T cells and serves to prevent autoimmunity [1]. Anergy is generally used to describe the hyporesponsive state of T cells stimulated in the absence of co-stimulatory signals [1]. In the context of chronic infections, hyporesponsive T cells are generally referred to as ‘exhausted,’ while T cells in the context of tumors have been described as ‘dysfunctional’ and/or exhausted. These different hyporesponsive states have shared and unique features. Here we will highlight (i) new insights into differentiation state dynamics and population heterogeneity of hyporesponsive T cells in chronic infections and cancer, (ii) how these states are determined by spatiotemporal factors, (iii) the underlying transcriptional and epigenetic regulation, and finally (iv) how these different states determine responses to immunotherapeutic interventions.
Phenotypic and functional traits of exhausted T cells in chronic viral infection
During chronic viral infections, virus-specific CD8 T cells enter a state of ‘exhaustion’—a state of functional hyporesponsiveness driven by chronic antigen stimulation [2]. Exhausted T cells lack full effector function, coinciding with the expression of numerous inhibitory receptors including PD1, LAG3, TIGIT, CD38, CD39, CD160, 2B4, TIM3, and CTLA4 [3]. These exhaustion-associated phenotypic and functional traits have distinct underlying transcriptional and epigenetic programs [2,4–9]. Virus-specific T cells initially acquire effector function during the early phase of the infection, but in the presence of persistent viral antigen and inflammation/infection, T cells become progressively exhausted, losing effector functions in a hierarchical manner (loss of proliferative capacity and IL-2 production first, followed by loss of TNFα, and ultimately loss of IFNγ production) [10]. Nevertheless, exhausted T cells are not completely unresponsive and retain some effector function, thereby allowing the host to control the pathogen without detrimental immunopathology. This is evidenced by the fact that depletion of exhausted T cells can cause fatal infection [11,12] while conversely, reinvigoration of exhausted T cells during chronic viral infection can result in fatal immunopathology [13,14]. Thus, T cell exhaustion is a state of ‘effective’ hyporesponsiveness, rather than a fully dysfunctional or non-responsive state, maintaining the host–pathogen stalemate [15].
Phenotypic and functional traits of hyporesponsive, tumor-reactive T cells in cancers
The study of established mouse and human tumors has demonstrated that tumor-infiltrating CD8 T cells (TIL) exhibit hallmark exhaustion features of T cells in chronic infection: TIL are impaired in the production of effector cytokines and/or cytotoxic molecules, express high levels of inhibitory receptors, and display alterations in TCR signaling pathways and transcription factor programming (including NFAT, TOX, TCF1, IRF4, BLIMP1) [16–23].
In spite of these overlapping phenotypic and functional traits, T cell differentiation during tumorigenesis is distinct from T cell differentiation in chronic infection: tumor-specific/neo-antigen-specific T cells generally do not differentiate through an early effector phase as seen with virus-specific T cells during a chronic infection; in developing tumors, tumor antigens are not presented ‘acutely’ in an inflammatory, stimulatory context. Instead, naїve tumor-reactive T cells are inadequately primed and/or activated in the draining lymph nodes or tumors, and enter an ‘anergy’-like hyporesponsive state, which progresses into an exhaustion-like state due to progressive tumor growth and persistence of tumor antigen [18,20,24–26].
Identifying the precise differentiation state dynamics and functional states of tumor-infiltrating T cells has been difficult due to the many cell-intrinsic and extrinsic factors affecting T cell differentiation and dysfunction in tumors, including (i) antigen-specificity, (ii) TCR affinity, (iii) tumor antigen density, (iv) time present within tumor and/or exposure to tumor antigen, (v) tolerance mechanisms operating during the early, non-inflammatory phase of tumorigenesis, or (vi) microenvironmental immunosuppressive factors present within established tumors (hypoxia, nutrient deprivation etc.). Thus, TIL represent a highly heterogeneous T cell population with a wide range of T cell specificities, activation and functional/dysfunctional states with distinct requirements for therapeutic reprogramming. The complete responses seen in some cancer patients treated with checkpoint blockade antibodies have reinvigorated the field of cancer immunotherapy; however, significant clinical responses are only observed in a subset of patients and cancer types, and it is currently unknown why only certain cancers and/or patients respond to checkpoint immunotherapy. To address these clinical challenges and design predictably effective cancer treatments, current efforts are aimed at elucidating the underlying programs that define exhaustion states of TIL and their amenability to immunotherapeutic reprogramming. Recent technological advances including single-cell analysis, TCR-seq, as well as epigenomics and transcriptomics analyses are now beginning to yield startling new insights into the heterogeneity of antigen specificity, T cell repertoires, and T cell differentiation and functional states in tumors, and how these heterogeneities might define clinical responses to immunotherapy (see below) [27–38].
Heterogeneity of the exhausted T cell population during chronic infections
Exhausted T cells during chronic infections represent a heterogeneous T cell population. Paley et al. first demonstrated that virus-specific exhausted T cells consist of at least two subpopulations: a TBEThiPD1int progenitor CD8 T cell subset which proliferates and gives rise to an EOMEShiPD1hi terminally differentiated progeny [39]. EOMEShiPD1hi T cells do not replicate and display high expression of inhibitory receptors. The ultimate depletion of the progenitor pool and accumulation of terminally differentiated EOMEShiPD1hi T cells is thought to result in the loss of immune control of the infection. In support of this notion, livers of patients with chronic HCV infection show depletion of TBEThi precursors and accumulation of the terminally differentiated exhausted progeny, in contrast to patients with controlled infections [39].
Several other subsequent studies examined T cell heterogeneity during chronic infections and identified TCF1 as a critical transcription factor. Virus-specific TCF1+ (PD1+CXCR5+TIM3−) CD8 T cells have a memory/stem cell-like phenotype, self-renew and give rise to terminally differentiated TCF1low/neg (PD1+CXCR5−TIM3+) T cells [40–45] (Figure 1). Interestingly, these two populations are found in spatially distinct compartments: while the TCF1+ memory/stem cell-like progenitor population is mainly found in secondary lymphoid tissues, specifically in T cell zones (white pulp), the terminally differentiated, exhausted TCF1low/neg T cell population is predominantly found in peripheral tissues andthered pulp of spleens—the major sites and reservoirs of infected cells and/or antigen [41]. Thus, formation and maintenance of the TCF1+ progenitor population appear to be restricted to sites of low antigen/pathogen burden and virus replication.
Figure 1. Heterogeneity of exhausted CD8 T cells during chronic viral infections.
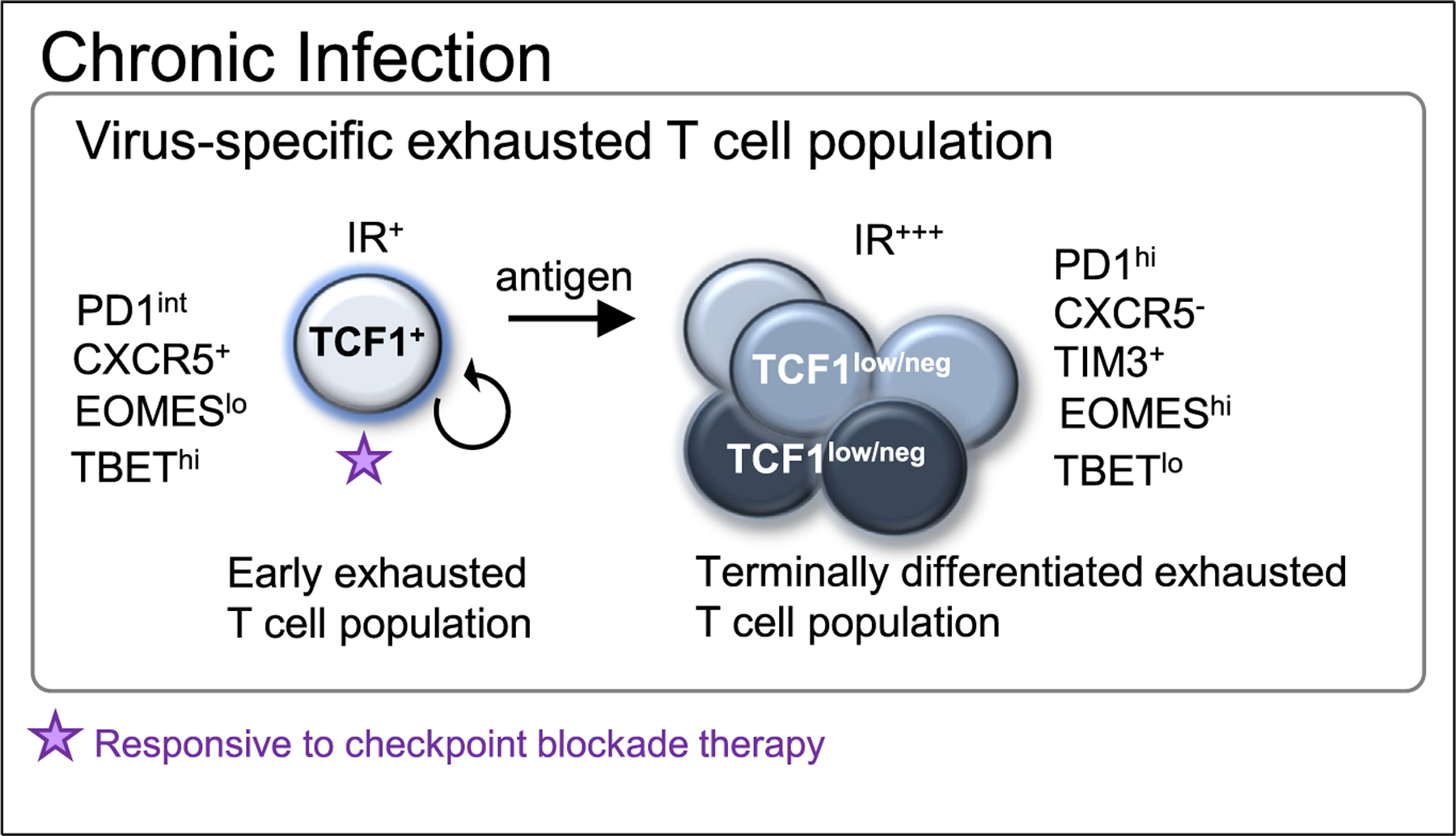
Virus-specific exhausted CD8 T cells consist of an exhausted TCF1+ PD1int T cell population with memory/stem cell-like characteristics which gives rise to a terminally differentiated, exhausted TCF1low/neg PD1hiT cell population. PD1/PDL checkpoint blockade reinvigorates the TCF1+ PD1int progenitor population but not the terminally differentiated TCF1 low/neg T cell population.
Importantly, TCF1+ (and/or TBEThiPD1int) exhausted progenitor T cells, but not terminally differentiated exhausted TCF1low/neg (and/or EOMEShiPD1hi) T cells, proliferate in response to PD1/PDL1 checkpoint blockade revealing that the memory/stem cell-like progenitor population is the prime target of immunotherapeutic interventions during chronic infections [40,41] (Figure 1).
Heterogeneity of tumor-infiltrating lymphocytes (TIL)
The identification of a memory/stem cell-like progenitor T cell population in chronic infections amenable to reprogramming by checkpoint blockade antibodies has sparked the search for similar progenitor T cell populations in tumors. Previous studies demonstrated that the induction of WNT/β-catenin signaling arrests T cell differentiation and drives the generation of self-renewing, memory stem cell-like T cells [46]. More recent studies characterizing TIL populations from human tumors indeed demonstrated the presence of TCF1+ TIL with stem cell-like characteristics and cytotoxic potential [29,43,47–49], and the frequency of TCF1+ CD8 T cells in tumor tissue correlated with responses to immunotherapy. However, it is unclear whether these TCF1+ T cells are truly tumor-reactive and/or represent a truly stable, self-renewing memory/stem cell-like progenitor T cell population as seen in chronic infections. TCF1+ TIL may include non-tumor reactive, bystander, cytotoxic T cells abundant in human tumor infiltrates and phenotypically distinct, lacking hallmarks of chronic antigen stimulation [18,21,30,35] (Figure 2). Interestingly, a recent study combining single-cell RNA-seq and TCR-seq, and assessing tumor reactivity of TIL from melanoma patients, revealed that TCF1+ TIL include bystander non-tumor reactive cytotoxic T cell populations, while T cell clones with tumor-reactivity had dysfunctional features including high expression of PD1, LAG3, CD39, and TOX, and low expression of TCF1 [28].
Figure 2. Heterogeneity of tumor-infiltrating lymphocyte (TIL) populations.
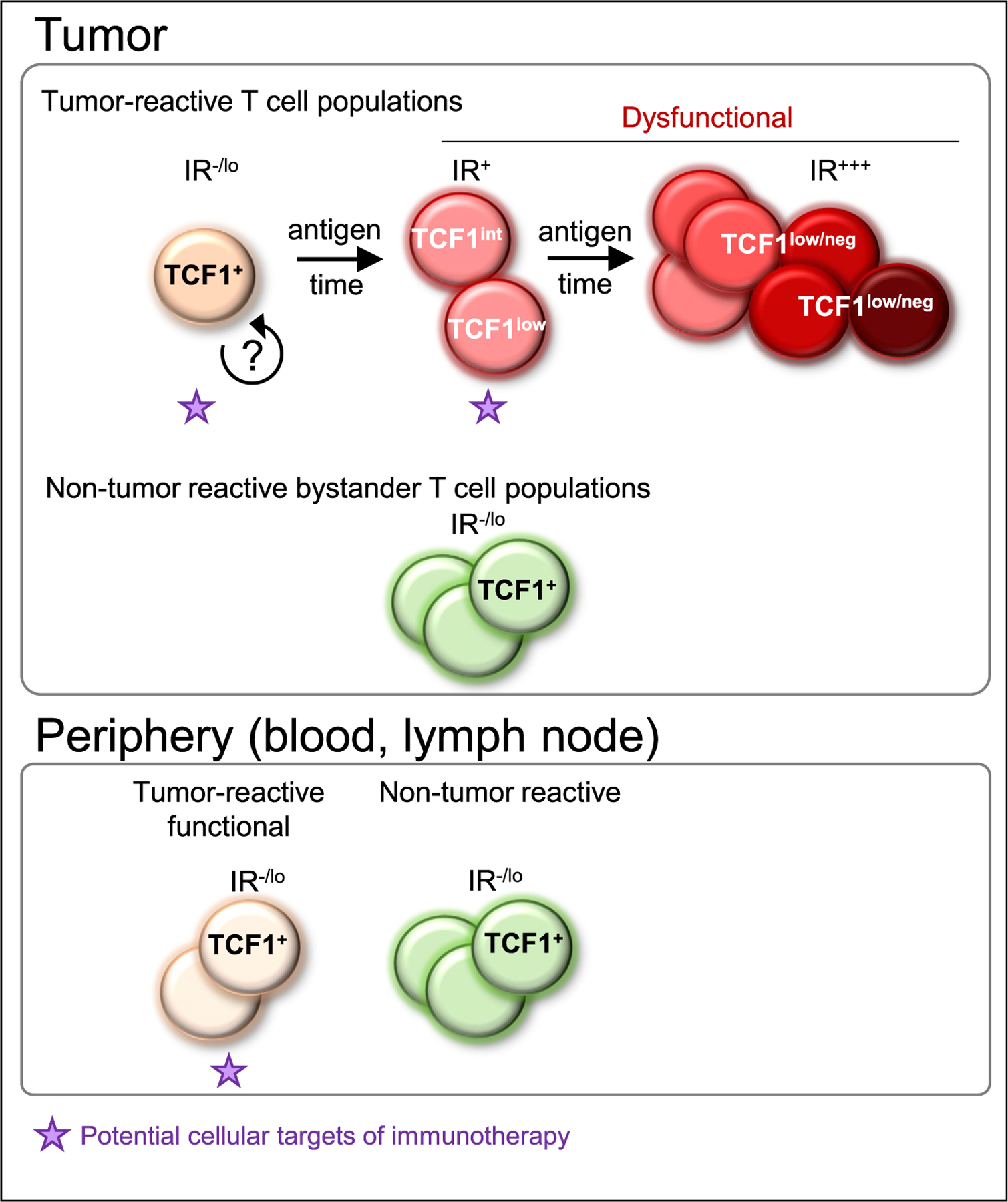
TIL are heterogeneous and include tumor-reactive and non-tumor reactive T cells. Non-tumor reactive, bystander T cells appear functional and cytotoxic, express high levels of TCF1 and no or low levels of inhibitory receptors (IR). Tumor-induced T cell dysfunction is progressive and various dysfunctional states exist depending on spatiotemporal factors including antigen burden and duration of tumor antigen exposure. Tumor-specific T cells are initially TCF1+ but with time lose TCF1 expression, become TCF1 low/neg, and upregulate numerous inhibitory receptors (IR+++). It is currently not known if TCF1+ tumor-specific T cells represent a stable, self-renewing population. We hypothesize that, in human tumors, as seen in autochthonous tumor mouse models, reprogrammable and non-reprogrammable dysfunctional T cells may be present. Tumor-reactive T cells are also found in the periphery (e.g. blood and lymph nodes) and typically do not have the ‘exhausted’ phenotype, and these T cells maybe the population most amenable to immunotherapy.
Following differentiation of a naїve tumor-specific T cell population over the course of tumorigenesis in an autochthonous tumor mouse model demonstrated that tumor-specific TIL within malignant lesions are initially TCF1hi/int, but over the course of tumorigenesis and with continued tumor antigen encounter, gradually lose TCF1 expression, become TCF1low/int and ultimately TCF1neg. This gradual loss of TCF1 coincided with the progressive upregulation of canonical inhibitory receptors and the inability to undergo functional rescue [18,20]. Thus, tumor-specific T cell dysfunction is progressive, ultimately resulting in a severe state of dysfunction/exhaustion which is highly resistant to therapeutic reprogramming. In human tumors, T cell tumor-infiltration and exposure time to tumor antigens (as well as specificity, affinity etc.; see above) is variable; thus TIL are in distinct differentiation, activation and dysfunctional states. Understanding whether and which dysfunctional T cell state(s) within tumors can be reinvigorated through immunotherapeutic interventions, or whether clinical responses require the recruitment of functional (potentially TCF1+) tumor-reactive T cells from ‘outside’ (e.g. draining lymph nodes, blood etc.) is being intensely investigated and these findings will be critically important to understand and design effective immunotherapeutic strategies (Figure 2).
Epigenetic programs defining dysfunctional and exhausted T cell states and therapeutic reprogrammability
Distinct functional CD8 T cell states, such as the naїve, effector and memory states, are associated with specific epigenetic programs that regulate transcription and define functional and phenotypic properties [50]. Recent technological advances including ATAC-seq [51] allowed the identification of chromatin states of exhausted T cells in chronic infections and tumor-specific T cells in mouse and human cancers [6–8,20,21,29]. These analyses reveal that both exhausted/dysfunctional T cells in chronic infections and tumors harbor chromatin accessibility patterns distinct from those of naїve, effector or memory T cells, with thousands of uniquely differentially accessible peaks, including in loci of genes encoding critical exhaustion-associated transcription factors and inhibitory receptors.
Studies investigating chromatin state dynamics of tumor-specific T cells over the course of tumorigenesis demonstrated that naїve tumor-specific T cells enter an epigenetically encoded program of dysfunction after encountering tumor antigen in early malignant lesions. This epigenetic landscape (chromatin state 1) was markedly distinct from that of early effector T cells during an acute infection and from early ‘exhausted’ T cells during a chronic infection [20,21]. Thus, naїve tumor-specific T cells encountering tumor antigen in early malignant lesions in a non-inflammatory context follow a distinct differentiation pathway from naїve T cells encountering antigen during an infection. With continued tumor antigen encounter and tumor progression, PD1hi tumor-specific T cells undergo further chromatin remodeling, entering another distinct chromatin state (state 2). These two chromatin accessibility patterns correlated temporally with the T cells’ amenability to therapeutic reprogramming [20]. Thus tumor-specific T cells initially differentiate through a plastic dysfunctional state that is functionally rescuable but ultimately enter a severe and fixed state of dysfunction that appears to be resistant to reprogramming. Interestingly, plastic and fixed dysfunctional T cells expressed similar levels of PD1 and LAG3 but could be distinguished by expression of other surface membrane proteins, including CD38, CD39, 2B4, and CD101, which were shown to be predictive biomarkers for therapeutic reprogrammability. Thus, while some inhibitory receptors such as PD1 more broadly might define TIL with tumor-reactivity, other inhibitory receptors can be utilized to determine functional states and reprogrammability of PD1hi TIL within mouse and human tumors [18,20,29,52]. In chronic viral infection, interestingly, it was shown that despite functional reinvigoration of exhausted virus-specific T cells by PD1 checkpoint blockade, chromatin states only changed minimally and ultimately drove T cells to reenter their previous functional and transcriptional exhausted state [6].
Together, these findings demonstrate that exhaustion of virus-specific T cells during chronic infection and tumor-specific T cells in tumors and their therapeutic reprogrammability are epigenetically encoded and suggest that effective immunotherapeutic strategies might require targeting the epigenome of T cells [50,53].
Concluding remarks
Recent technological advances have provided important insights into the heterogeneity and programming of hyporesponsive T cell populations in chronic infection and cancer. It has become increasingly clear that distinct T cell subsets with distinct transcriptional and epigenetic programs and functional states harbor distinct requirements for therapeutic reprogramming. Future studies are needed to identify (i) the precise spatiotemporal factors that determine these distinct functional states, and (ii) which T cell differentiation states and population subsets represent the critical cellular target(s) for immunotherapy, especially in the context of cancer.
Acknowledgements
This work was supported by National Institutes of Health grant DP2 CA225212, Josie Robertson Young Investigator Award, MSK Cancer Center Core Grant P30 CA008748 (to A.S) and V Foundation for Cancer Research, and Serodino Family Adventure Allee Fund, (to M.P.).
Footnotes
Conflict of interest statement
Nothing delcared.
References
- 1.Schietinger A, Greenberg PD: Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014, 35:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wherry EJ et al. : Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27:670–684. [DOI] [PubMed] [Google Scholar]
- 3.Blackburn SD et al. : Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009, 10:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shin H et al. : A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009, 31:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez GJ et al. : The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015, 42:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pauken KE et al. : Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354:1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sen DR et al. : The epigenetic landscape of T cell exhaustion. Science 2016, 354:1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott-Browne JP et al. : Dynamic changes in chromatin accessibility occur in CD8(+) T cells responding to viral infection. Immunity 2016, 45:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Man K et al. : Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity 2017, 47:1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- 10.Wherry EJ et al. : Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 2003, 77:4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin X et al. : Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med 1999, 189:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmitz JE et al. : Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 1999, 283:857–860. [DOI] [PubMed] [Google Scholar]
- 13.Frebel H et al. : Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med 2012, 209:2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zinselmeyer BH et al. : PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med 2013, 210:757–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cornberg M et al. : Clonal exhaustion as a mechanism to protect against severe immunopathology and death from an overwhelming CD8 T cell response. Front Immunol 2013, 4:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baitsch L et al. : Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest 2011, 121:2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giordano M et al. : Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J 2015, 34:2042–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schietinger A et al. : Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 2016, 45:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waugh KA et al. : Molecular profile of tumor-specific CD8+ T cell hypofunction in a transplantable murine cancer model. J Immunol 2016, 197:1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Philip M et al. : Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mognol GP et al. : Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A 2017, 114:E2776–E2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J et al. : NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567:530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X et al. : Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567:525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willimsky G, Blankenstein T: Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 2005, 437:141–146. [DOI] [PubMed] [Google Scholar]
- 25.Spranger S et al. : Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523:231–235. [DOI] [PubMed] [Google Scholar]
- 26.Speiser DE et al. : Regulatory circuits of T cell function in cancer. Nat Rev Immunol 2016, 16:599–611. [DOI] [PubMed] [Google Scholar]
- 27.Azizi E et al. : Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174:1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H et al. : Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2019, 176:775–789.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sade-Feldman M et al. : Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018, 175:998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simoni Y et al. : Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557:575–579. [DOI] [PubMed] [Google Scholar]
- 31.Tirosh I et al. : Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng C et al. : Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017, 169:1342–1356.e16. [DOI] [PubMed] [Google Scholar]
- 33.Chevrier S et al. : An immune atlas of clear cell renal cell carcinoma. Cell 2017, 169:736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavin Y et al. : Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017, 169:750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheper W et al. : Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med 2019, 25:89–94. [DOI] [PubMed] [Google Scholar]
- 36.Guo X et al. : Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018, 24:978–985. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L et al. : Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564:268–272. [DOI] [PubMed] [Google Scholar]
- 38.Savas P et al. : Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 2018, 24:986–993. [DOI] [PubMed] [Google Scholar]
- 39.Paley MA et al. : Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338:1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He R et al. : Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature 2016, 537:412–428. [DOI] [PubMed] [Google Scholar]
- 41.Im SJ et al. : Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Utzschneider DT et al. : T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 2016, 45:415–427. [DOI] [PubMed] [Google Scholar]
- 43.Wu T et al. : The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 2016, 1. [DOI] [PMC free article] [PubMed]
- 44.Wieland D et al. : TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat Commun 2017, 8:15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leong YA et al. : CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol 2016, 17:1187–1196. [DOI] [PubMed] [Google Scholar]
- 46.Gattinoni L et al. : Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 2009, 15:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brummelman J et al. : High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med 2018, 215:2520–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siddiqui I et al. : Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019, 50:195–211.e10. [DOI] [PubMed] [Google Scholar]
- 49.Kurtulus S et al. : Checkpoint blockade immunotherapy induces dynamic changes in PD-1(−)CD8(+) tumor-infiltrating T cells. Immunity 2019, 50:181–194.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henning AN et al. : Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 2018, 18:340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buenrostro JD et al. : Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013, 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thommen DS et al. : A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 2018, 24:994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghoneim HE et al. : De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 2017, 170:142–157.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]