

In this study, Rimel et al. set out to investigate the roles of CDK7 in transcription. Using SILAC-based phosphoproteomics with transcriptomics and biochemical assays, the authors identified high-confidence CDK7 substrates, a surprisingly widespread requirement for CDK7 activity in splicing, and unexpected aspects of CDK7 kinase regulation that involve its association with TFIIH.
Keywords: CDK12, CDK13, CDK7, CDK9, SF3B1, SILAC-MS, TFIIH, kinase inhibitor, splicing, transcription
Abstract
CDK7 associates with the 10-subunit TFIIH complex and regulates transcription by phosphorylating the C-terminal domain (CTD) of RNA polymerase II (RNAPII). Few additional CDK7 substrates are known. Here, using the covalent inhibitor SY-351 and quantitative phosphoproteomics, we identified CDK7 kinase substrates in human cells. Among hundreds of high-confidence targets, the vast majority are unique to CDK7 (i.e., distinct from other transcription-associated kinases), with a subset that suggest novel cellular functions. Transcription-associated factors were predominant CDK7 substrates, including SF3B1, U2AF2, and other splicing components. Accordingly, widespread and diverse splicing defects, such as alternative exon inclusion and intron retention, were characterized in CDK7-inhibited cells. Combined with biochemical assays, we establish that CDK7 directly activates other transcription-associated kinases CDK9, CDK12, and CDK13, invoking a “master regulator” role in transcription. We further demonstrate that TFIIH restricts CDK7 kinase function to the RNAPII CTD, whereas other substrates (e.g., SPT5 and SF3B1) are phosphorylated by the three-subunit CDK-activating kinase (CAK; CCNH, MAT1, and CDK7). These results suggest new models for CDK7 function in transcription and implicate CAK dissociation from TFIIH as essential for kinase activation. This straightforward regulatory strategy ensures CDK7 activation is spatially and temporally linked to transcription, and may apply toward other transcription-associated kinases.
TFIIH is essential for RNA polymerase II (RNAPII) transcription and is a component of the preinitiation complex (PIC), which assembles at transcription start sites of all RNAPII-regulated genes. TFIIH contains the ATPase/translocase XPB that appears to be essential for transcription because it “melts” the promoter to allow single-stranded DNA to enter the RNAPII active site (Tirode et al. 1999). Another catalytic subunit in TFIIH, the CDK7 kinase (Kin28 in yeast), also appears to be broadly required for proper regulation of RNAPII transcription. During early stages of transcription initiation, CDK7 phosphorylates the RNAPII CTD and this initiates a cascade of events that correlate with RNAPII promoter escape and transcription elongation (Corden 2013; Eick and Geyer 2013). CDK7 can also phosphorylate CDK9, which, together with CCNT1, comprises the P-TEFb complex. P-TEFb represents another transcription-associated kinase that is capable of phosphorylating the RNAPII CTD, and CDK7 phosphorylation of CDK9 enhances its kinase activity (Larochelle et al. 2012). Phosphorylation of the RNAPII CTD appears to be essential for proper processing of RNA transcripts (Kanin et al. 2007; Glover-Cutter et al. 2009; Hong et al. 2009), due in part to recruitment of factors involved in 5′ capping and splicing (Cho et al. 1997; McCracken et al. 1997; David et al. 2011; Ebmeier et al. 2017).
The biological roles for CDK7 remain incompletely understood, in part because identification of its kinase substrates has been limited. CDK7 can also function apart from TFIIH, as the three-subunit CDK activating kinase (CAK: CDK7, CCNH, and MAT1). In the cytoplasm, the CAK phosphorylates and activates other CDKs (e.g., CDK1 and CDK2) to regulate the cell cycle (Fisher 2005). It is not known whether the CAK may also function in the nucleus, but current models assume that nuclear CDK7 regulates transcription through its association with TFIIH.
Viruses are known to target TFIIH to hijack the RNAPII transcription response during infection (Qadri et al. 1996; Cujec et al. 1997; Le May et al. 2004), and TFIIH mutations (i.e., those that are not lethal) are linked to congenital and somatic diseases such as xeroderma pigmentosum, Cockayne syndrome, and various types of cancer (Schaeffer et al. 1993; Manuguerra et al. 2006). Numerous compounds have been developed that target the kinase activity of CDK7 (Kelso et al. 2014; Kwiatkowski et al. 2014; Olson et al. 2019), including several that advanced to clinical trials (Hu et al. 2019). Here, we used SY-351, a potent and selective covalent CDK7 inhibitor. By combining SILAC-based phosphoproteomics with transcriptomics and biochemical assays, we were able to identify high-confidence CDK7 substrates, a surprisingly widespread requirement for CDK7 activity in splicing and unexpected aspects of CDK7 kinase regulation that involve its association with TFIIH.
Results
SY-351: a potent, highly selective covalent CDK7 inhibitor
Potent covalent inhibition of CDK7 by SY-351 (Fig. 1A) has been recently described (Hu et al. 2019). Although in vivo properties, including high clearance, limited development of this compound, SY-351 is useful as a molecular probe to understand CDK7 inhibition in cells. Selectivity was evaluated in a panel of 252 kinases using KiNativ profiling (Supplemental Table S1). CDK7 was the top hit with >90% inhibition, and was the only kinase inhibited more than 50% with 0.2 µM SY-351. To minimize off-target effects, we used SY-351 at a concentration of 50 nM (0.05 µM) throughout this study (see below). At a higher concentration of 1 µM SY-351, only six other kinases were inhibited >50%, including CDK12 and CDK13 (Fig. 1B).
Figure 1.
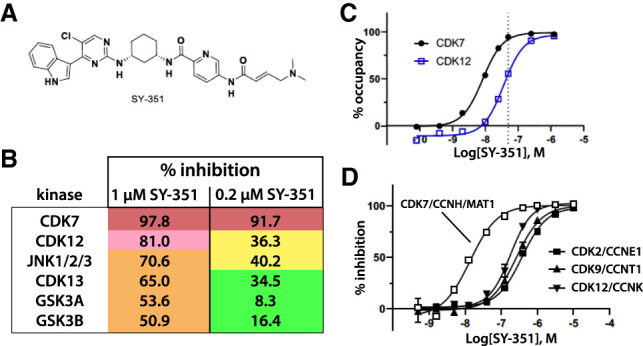
SY-351 is a potent and highly selective covalent inhibitor of human CDK7. (A) SY-351 structure. (B) Kinome selectivity in A549 cell lysate, with 0.2 µM and 1 µM SY-351. The top six hits shown are kinases inhibited > 50% by 1 µM SY-351. Note that SY-351 was used at 0.05 µM (50 nM) throughout this study. (C) CDK7 and CDK12 target occupancy in HL-60 cells after 1-h treatment. The SY-351 EC50 is 8.3 nM for CDK7 and 36 nM for CDK12. The SY-351 EC90 is 39 nM for CDK7 and 172 nM for CDK12. The dashed line indicates 50 nM SY-351, the dose used throughout this study. (D) SY-351 inhibition of active kinases CDK7/CCNH/MAT1, CDK2/CCNE1, CDK9/CCNT1, CDK12/CCNK at Km ATP for each enzyme. The best fit IC50 values are 23 nM, 321 nM, 226 nM, and 367 nM, respectively.
Similar to other compounds in this class (Kwiatkowski et al. 2014; Hu et al. 2019), the acrylamide moiety of SY-351 covalently reacts with cysteine residue 312 of CDK7 and exhibits time-dependent inhibition of CDK7 with a KI of 62.5 nM and kinact of 11.3 h−1 (Supplemental Fig. S1A). Due to the ability of SY-351 to covalently interact with cysteine residues, we sought to identify other proteins that might react in cells. We used activity-based protein profiling (ABPP) with a panel of alkyne functionalized analogues of SY-351 (Supplemental Fig. S2) in HL60 cells, as described (Cravatt et al. 2008; Lanning et al. 2014). Forty-six proteins were identified as competitive hits, including expected kinase targets CDK7 and the related CDK12 and CDK13 kinases, which have cysteine residues that align with CDK7: C1039 in CDK12 and C1017 in CDK13 (Supplemental Table S2).
The ability of SY-351 to covalently react with CDK12 and CDK13 must be considered to ensure that cellular effects in this study were due primarily to CDK7 inhibition. A 1-h treatment of 50 nM SY-351 reached the EC90 of CDK7 target engagement (39 nM), while minimizing CDK12 target engagement in HL60 cells (Fig. 1C). Moreover, as shown previously (Hu et al. 2019), SY-351 selectively inhibited catalytically active CAK complex (CDK7 in complex with CCNH and MAT1) over other cyclin-dependent kinases CDK2:CCNE1, CDK9:CCNT1, and CDK12:CCNK (Fig. 1D; Supplemental Fig. S1B). Based on these results, we concluded that inhibition of CDK7 predominates at 50 nM SY-351 and 1-h treatment. This condition was used for stable isotope labeling of amino acids in cell culture (SILAC)-based phosphoproteomics experiments in HL60 cells.
Identification of high-confidence CDK7 targets using SILAC phosphoproteomics
We performed a double-label SILAC phosphoproteomics experiment in which we metabolically labeled HL60 cells with “light”-labeled (Lys0 and Arg0) or “heavy”-labeled (Lys8 and Arg10) amino acids, which were treated with vehicle (DMSO) or 50 nM SY-351, followed by phosphopeptide enrichment and mass spectrometry analysis. The experimental design included two biological replicates of SY-351-treated heavy cells compared with DMSO treated light cells, a “label-flip” biological replicate of SY-351 light cells compared with DMSO treated heavy cells, and a “null” condition for which both heavy and light were treated with DMSO (Fig. 2A). The label-flip and null conditions control for systematic errors in phosphopeptide Heavy:Light SILAC ratios that are independent of the SY-351 treatment effect, which we observed after metabolic labeling of HL-60 cells using the SILAC protocol (Supplemental Fig. S3A). We used a linear statistical model to capture these systematic errors (see Materials and Methods), which improved our statistical power to detect phosphorylation sites that changed in abundance with SY-351 treatment (Supplemental Fig. S3B). We also confirmed that metabolic labeling did not transfer to proline through any salvage pathways (Supplemental Fig. S3C).
Figure 2.

SILAC phosphoproteomic overview and identification of phosphorylation sites significantly changed by SY-351 treatment. (A) Experimental design and workflow using SILAC double-labeled HL-60 cells (Light:Heavy) and phosphoproteomics. Labeled cell populations were treated with vehicle (DMSO) or 50 nM SY-351 as indicated, with two biological replicates of SY-351-treated heavy cells (vs. DMSO light cells), a “label-flip” biological replicate of SY-351 light cells versus DMSO heavy cells, and a “null” condition for which both heavy and light were treated with DMSO. Tryptic peptides were enriched using titanium dioxide beads and analyzed by high-resolution mass spectrometry to identify phosphopeptides and quantify their light:heavy SILAC ratios. (B) Volcano plot showing the statistical significance and magnitude of change with SY-351 treatment for phosphorylation site SILAC ratios. The adjusted P-value range for phosphorylation site ratios is indicated in color. (C) Venn diagram showing phosphorylation sites shared or mutually exclusive for transcription-associated kinases CDK7, CDK8, CDK9, and CDK12/CDK13. For each kinase, the top 400 identified phosphorylated proteins, based upon the largest negative log fold change values and P < 0.05, were used for comparisons, and the number of overlapping proteins was calculated for all combinations of samples. CDK7 targets identified here were compared with proteins identified as substrates for CDK8 in HCT116 (Poss et al. 2016), CDK9 in Raji B lymphocytes (Decker et al. 2019), and CDK12/13 in IMR-32 and Kelly cells (Krajewska et al. 2019).
Overall, we detected 621 phosphosites that decreased and 732 sites that increased (FDR < 0.05) (Fig. 2B) upon SY-351 treatment, with quantification of nearly 40,000 total phosphosites and 16,616 phosphosites quantified in all four replicates. Representative MS data are shown in Supplemental Figure S4A. Although decreased or increased phosphosite abundance could simply reflect changes in protein levels, this was not observed within the time frame of the experiment (Supplemental Fig. S4B). The large number of increasing sites was not unexpected and likely reflects compensatory functions of kinases and phosphatases (see the Discussion).
Comparison of CDK7 phosphosites with those from other transcription-associated kinases CDK8/CDK19 (Poss et al. 2016), CDK9 (Sansó et al. 2016; Decker et al. 2019), and CDK12/CDK13 (Krajewska et al. 2019) revealed few shared substrates (Fig. 2C; Supplemental Fig. S4C; Supplemental Table S3), suggesting nonredundant roles for these kinases. Although these MS-based analyses used different human cell types, the cellular targets for CDK7, CDK8/19, CDK9, or CDK12/13 are expected to be shared across cell types. A STRING analysis of the decreased sites (q value < 0.05) displays high-confidence CDK7 targets within known interaction networks (Fig. 3). Using a more stringent cutoff (log2 [SY-351/DMSO] < –0.5 and q-value < 0.01), 120 unique phosphorylation sites were identified that decreased with SY-351 treatment, corresponding to 72 phosphoproteins (Table 1). The CDK7 substrates identified in Figure 3 and Table 1 included many splicing and RNA processing factors (Supplemental Fig. S4D); most notable was SF3B1, with decreased phosphorylation at 18 high-confidence sites in SY-351-treated cells.
Figure 3.

Protein–protein interaction network of phosphoproteins with significantly decreasing phosphorylation sites upon SY-351 treatment. Proteins with decreasing phosphorylation sites (log2FC < 0 and FDR < 0.05) were visualized using the STRING database web application, and clustered using the MCL algorithm, with clusters indicated by node color.
Table 1.
High-confidence CDK7 targets

Finally, to visualize significantly changing phosphorylation sites with known kinase-substrate relationships in the PhosphoSitePlus database (Hornbeck et al. 2015), we mapped significant sites (q-value < 0.05) onto a kinase-substrate network generated in Cytoscape. A subnetwork was selected from the larger parent network to emphasize only kinases and substrates within a few degrees of separation from CDK7, CDK9, and POLR2A (Supplemental Fig. S5). CDK9 was included here because others have shown that CDK7 can activate CDK9 through phosphorylation of the CDK9 T-loop (Larochelle et al. 2012). Whereas phosphorylation at the CDK9 T-loop site (Thr186) was detected in our SILAC-MS data, it did not significantly change in SY-351-treated HL60 cells (Supplemental Table S4). As expected, the kinase-substrate subnetwork showed known CDK7 phosphorylation sites on CDK1 (Thr161) and within the RNAPII CTD (Ser1878). Notably, known T-loop sites in CDK12 (Thr893) and CDK13 (Thr871) were also identified, suggesting that CDK7 might directly activate these kinases (see below).
Inhibition of CDK7 causes diverse and widespread splicing changes
To probe potential CDK7-dependent effects on RNA processing, we completed spike-in normalized (Supplemental Fig. S6A) RNA-seq analyses in HL60 cells treated with SY-351 (50 nM for 5 h) along with DMSO controls. Biological replicates (CTRL vs. SY-351) (Supplemental Fig. S6B) were sequenced to high depth (>120 million mapped reads/replicate) to facilitate splicing analysis, and the 5-h treatment time was determined empirically by assessing total RNA and cell viability across a series of time points (Supplemental Fig. S6C,D). At 5 h, total cellular RNA levels remained relatively unchanged versus t = 0 controls; moreover, a 5-h time point enabled potential splicing changes to manifest in steady-state mRNA. Significant changes in mRNA abundance (P < 0.05; fold change > 2) were identified by DEseq2 in SY-351-treated samples, with an excess of down-regulated transcripts over up-regulated transcripts (Fig. 4A). Gene set enrichment analysis (GSEA) (Fig. 4B) showed decreases in proliferative hallmarks with SY-351 treatment (e.g., E2F targets, MYC targets, G2/M checkpoint), as expected.
Figure 4.

CDK7 inhibition with SY-351 causes widespread defects in splicing. (A) Differentially expressed mRNAs following SY351 treatment (DESeq2). Replicate RNA-seq data sets were generated from rRNA-depleted total RNA from HL60 cells treated with DMSO (CTRL) or SY-351 (50 nM in DMSO) for 5 h. (B) GSEA analysis. (C) SY-351 favors exon inclusion over exon skipping, splicing over retention of introns, and alters 5′ and 3′ splice site selection equally, without preference for upstream or downstream sites. Alternative splicing events significantly affected by SY-351 were identified in replicate RNA-seq data sets using MAJIQ (Vaquero-Garcia et al. 2016). (D) IGV genome browser Sashimi plot of a segment of the LTV1 gene. Normalized read numbers for DMSO control and SY-351 treated samples are shown on the Y-axis. Splice junction read numbers for sense strand transcripts are shown in blue and red. Note reduced exon inclusion (green arrow) and retention of both flanking introns (black arrows) in SY-351. (E) Immunofluorescence microscopy of endogenous SF3B1 (green) and Hoechst (blue) in DMSO and SY-351 conditions. (F) Quantitation of SF3B1 puncta per nucleus in DMSO and SY-351 conditions, P = 0.001 (n = 3 biological replicates).
SY-351 is highly selective for CDK7, although some inhibition of CDK12 and CDK13 was observed at higher concentrations in kinome-wide profiling (Fig. 1B). Whereas our experimental design minimized potential confounding effects from CDK12/13 inhibition (Fig. 1C,D), we nevertheless probed the RNA-seq data for evidence that CDK12 inhibition was occurring. Others have shown that CDK12 inhibition results in premature, intronic polyadenylation (iPA) (Dubbury et al. 2018; Krajewska et al. 2019; Fan et al. 2020), which causes reduced exon read counts toward gene 3′ ends. A DEXSeq analysis (Anders et al. 2012) showed no evidence for iPA genome-wide in cells treated with SY-351 (Supplemental Fig. S6E), which further validated our experimental strategy and confirmed the selectivity of SY-351 for CDK7.
To investigate possible effects of SY-351 on alternative splicing, we used MAJIQ (Vaquero-Garcia et al. 2016), which detects altered exon skipping and intron retention events as well as more complex splicing changes. We identified 11,348 changes in exon inclusion upon SY-351 treatment versus DMSO controls (ΔPSI [percent spliced in] ≥ 0.2, P < 0.05) (Fig. 4C), and SY-351 treatment increased inclusion/reduced skipping of alternative exons more frequently than it decreased their inclusion/increased skipping (6738 vs. 4610) (Fig. 4C). The 5′ and 3′ splice sites at alternative exons whose inclusion was affected by SY-351 did not significantly differ in strength (Yeo et al. 2004) from those that were unaffected (Supplemental Fig. S7A).
Notably, both reduced and increased exon skipping were frequently associated with abnormal retention of introns flanking the alternative exon; this phenomenon was the major source of abnormal transcripts detected in SY-351-treated cells. Reduced exon inclusion with retention of flanking introns is illustrated in Figure 4D (see also Supplemental Fig. S7B). Increased exon inclusion with (1) retention of a downstream flanking intron (Supplemental Fig. S7C,D), (2) an upstream flanking intron (Supplemental Fig. S7E), or (3) both downstream and upstream introns (Supplemental Fig. S7F) was also observed. We detected significant effects of SY-351 on use of annotated alternative 5′ and 3′ splice sites in approximately equal numbers (753 5′ss vs. 790 3′ss), with similar numbers of either upstream of or downstream from alternative splice sites (Fig. 4C). In addition, SY-351 significantly altered the splicing of over 1500 annotated retained introns with a strong bias toward increased (Supplemental Fig. S8A–D) versus decreased (Supplemental Fig. S8E) splicing of these introns (1151 vs. 522 cases) (Fig. 4C).
In summary, SY-351 inhibited splicing of introns flanking many alternative exons, but did not generally inhibit splicing. In fact, SY-351 actually increased splicing of many retained introns (Supplemental Fig. S8A–D).
Splicing changes can be linked in part to SF3B1
SF3B1 is a component of the U2 small nuclear ribonucleoprotein (snRNP) and facilitates hybridization of the U2 snRNA with the pre-mRNA branch point sequence within the spliceosome (Maji et al. 2019). Because 18 different SF3B1 sites were identified by SILAC phosphoproteomics, we asked whether defects in SF3B1 function would be evident in cells treated with SY-351. As shown in Table 1, all 18 high-confidence phosphorylation sites reside between SF3B1 residues 207–434. Previous studies (Eilbracht and Schmidt-Zachmann 2001) linked this region to SF3B1 nuclear localization (residues 196–216) and SF3B1 association with nuclear speckles (TP-rich domain; residues 208–440). Therefore, we tested whether SF3B1 nuclear localization or speckle association would be affected by SY-351 treatment. As shown in Supplemental Figure S9A,B, nuclear versus cytoplasmic localization of SF3B1 did not appear to be altered in SY-351-treated cells, at least under our assay conditions (4 h SY-351, 50 nM). In contrast, immunofluorescence (IF) experiments showed changes in SF3B1 association with nuclear speckles in SY-351-treated cells versus untreated controls (Fig. 4E,F), although total nuclear IF signal remained largely unchanged (Supplemental Fig. S9C). Moreover, the SY-351 effects were distinct from THZ-531, an inhibitor of CDK12/13 (Supplemental Fig. S9D,E). Although we cannot decouple the changes in SF3B1 speckle localization from transcriptional changes triggered by SY-351, the IF data further implicate the CDK7 kinase as a regulator of SF3B1 function.
We also compared splicing changes induced by SY-351 with those induced by the SF3B1 inhibitor Pladienolide B (PladB) (Kotake et al. 2007). Using MAJIQ2 (Vaquero-Garcia et al. 2016), we identified 1152 local splicing variations (LSVs) in common between SY-351 and PladB. These events met the following criteria: (1) P-value < 0.05, (2) absolute ΔPSI ≥ 0.2, (3) used in >10% of reads, and (4) the coordinates of the 5′ and 3′ splice sites were within 10 bases in both cell lines (HL60 for SY-351 and K562 for PladB). Among these 1152 events, the ΔPSI value was altered by both inhibitors in the same direction in 765 cases (P-value = 2.3307 × 10−79, hypergeometric test). Examples of inhibitor effects on splicing of individual introns are shown in Supplemental Figure S9F. From this comparative analysis, we conclude that a subset of the splicing defects observed in SY-351-treated cells result from changes in SF3B1 function.
CDK7 kinase activity is regulated by TFIIH
We performed in vitro kinase assays with the 10-subunit human TFIIH complex (Fig. 5A) to test whether high-confidence CDK7 substrates were, in fact, direct targets. Substrates tested were DSIF, NELF, SF3B1, U2AF2, TFIIF, MYC, and the RNAPII CTD (Supplemental Fig. S10), which were identified as CDK7 kinase targets from the phosphoproteomics data (Fig. 3). Contrary to expectations, TFIIH was unable to efficiently phosphorylate these substrates in vitro, with the exception of the RNAPII CTD (Fig. 5B). Because CDK7 can activate other CDKs (Rimel and Taatjes 2018), these data suggested that DSIF, NELF, SF3B1, U2AF2, TFIIF, and MYC may be indirect targets of CDK7. It was also plausible, however, that CDK7 might be modifying these substrates in the context of the three-subunit CAK module (CDK7, CCNH, and MNAT1) (Fig. 5C), which can exist as a stable complex apart from TFIIH (Rimel and Taatjes 2018). We tested whether the same set of substrates would be differentially modified by the three-subunit CAK module versus TFIIH (Fig. 5B,D). The data revealed that CDK7 efficiently phosphorylated DSIF, TFIIF, U2AF2, and SF3B1 within the three-subunit kinase module, in stark contrast to the 10-subunit TFIIH complex. In addition, the comparison of TFIIH versus CAK showed that CDK7 was slightly more active toward the RNAPII CTD within TFIIH versus the CAK (Fig. 5D). TFIIH also phosphorylated the RNAPII CTD within promoter-assembled, transcriptionally active preinitiation complexes (PICs), as expected (Supplemental Fig. S11A). NELF and MYC were not efficiently phosphorylated by CDK7 within the CAK or TFIIH (Supplemental Fig. S11B,C), suggesting these are not directly targeted by CDK7.
Figure 5.

CDK7 kinase activity is negatively regulated by TFIIH. (A) SYPRO-stained gel of human TFIIH. (B) TFIIH activates CDK7 kinase toward the CTD, but reduces or prevents CDK7 phosphorylation of DSIF, SF3B1, U2AF2, and TFIIF. Within the CAK, CDK7 is activated toward these substrates. (C) SYPRO-stained gel of human CAK complex (MAT1 is GST-tagged). (D) Quantitation of CDK7 kinase results across biological replicates (n = 3) with autorad signal normalized to CAK phosphorylated DSIF; P-values as shown.
We conducted Western blotting experiments with antibodies specific to the Ser2, Ser5, or Ser7 phosphorylated CTD to assess whether selectivity for these sites might differ between the CAK and TFIIH. Consistent with the bulk 32P-ATP kinase assays, the data revealed increased modification of each site by TFIIH (vs. CAK); however, the Ser2, Ser5, and Ser7 phosphorylated CTD species migrated differently in each case (CAK vs. TFIIH), suggesting that a different number or different pattern of sites within the 52-repeat sequence were being modified (Supplemental Fig. S11D,E).
The data shown in Figure 5 established distinct substrate specificities for CDK7 in the free kinase module versus TFIIH. To test this further, we compared the three-subunit CAK module with the 10-subunit TFIIH complex in positional scanning peptide array experiments, which were completed with 32P-ATP (Begley et al. 2015). Although peptide arrays are limited in their ability to match the structure of native, physiological substrates, the assay provided an independent means to compare CDK7 substrate preferences in the context of the CAK versus TFIIH. Consistent with the in vitro kinase assays with native substrates (Fig. 5), the peptide array data showed differential phosphorylation site preferences (Supplemental Fig. S12A), with TFIIH showing greater selectivity compared with the three-subunit CAK.
The results summarized in Figure 5 suggested that CDK7 phosphorylates substrates other than the RNAPII CTD as part of the CAK, not TFIIH. Many high-confidence CDK7 substrates (Table 1; Supplemental Table S3) represent nuclear factors associated with elongating RNAPII. Because TFIIH assembles at transcription start sites as part of the RNAPII preinitiation complex (Rimel and Taatjes 2018), it is plausible that CDK7 might dissociate from TFIIH to access such substrates. To test this idea, we performed size exclusion chromatography on HCT116 nuclear or cytoplasmic fractions. As shown in Supplemental Figure S12B, CDK7 was detected at molecular weights consistent with the CAK complex in cytoplasmic fractions, as expected. In contrast, CDK7 migrated only at molecular weights consistent with the 10-subunit TFIIH complex (∼500 kDa) in nuclear extracts (Supplemental Fig. S12B). These results suggest that the CAK remains associated with TFIIH in cell nuclei; however, we cannot rule out that a small percentage of CDK7 (below detection limit) completely dissociates as the CAK complex and remains in the nucleus.
CDK7 directly activates transcription-associated kinases CDK9, CDK12, and CDK13
CDK7 has been shown to activate another transcription-associated kinase, CDK9, via phosphorylation of its T-loop residue T186 (Larochelle et al. 2012). The SILAC phosphoproteomics data with the CDK7 inhibitor SY-351 implicated CCNK, CDK12, and CDK13 as direct targets of CDK7 (Table 1); in fact, high-confidence sites in CDK12 and CDK13 included T-loop residues (T893 and T871, respectively). This suggested that, as with CDK9 (Larochelle et al. 2012), CDK7 might activate CDK12 and CDK13 as well. CDK12 and CDK13 are essential for normal cotranscriptional phosphorylation of the RNAPII CTD (primarily in gene bodies, at Ser2) and therefore help regulate splicing and RNAPII termination (Greenleaf 2019; Chou et al. 2020).
Because CDK12:CCNK and CDK13:CCNK are highly active in vitro and efficiently autophosphorylate, we could not reliably assess whether CDK12, CDK13, or CCNK were direct targets of CDK7 in kinase assays. However, we were able to test whether CDK7 was capable of activating CDK12 and/or CDK13 using an experimental strategy outlined in Figure 6A. CDK12:CCNK or CDK13:CCNK were prephosphorylated by the CAK and subsequently tested versus mock-treated complexes (identical incubation but without added CAK). As shown in Figure 6B,C, CDK7-dependent prephosphorylation of CDK12:CCNK or CDK13:CCNK activated these kinases toward a common substrate, the RNAPII CTD. Importantly, the CAK was removed prior to these kinase assays to ensure no contaminating CDK7 was present, which would otherwise confound the analysis (Supplemental Fig. S13A). In parallel, we tested P-TEFb (CDK9, CCNT1) complexes and verified CDK7-dependent activation of CDK9 (Fig. 6B,C), in agreement with previous studies (Larochelle et al. 2012).
Figure 6.
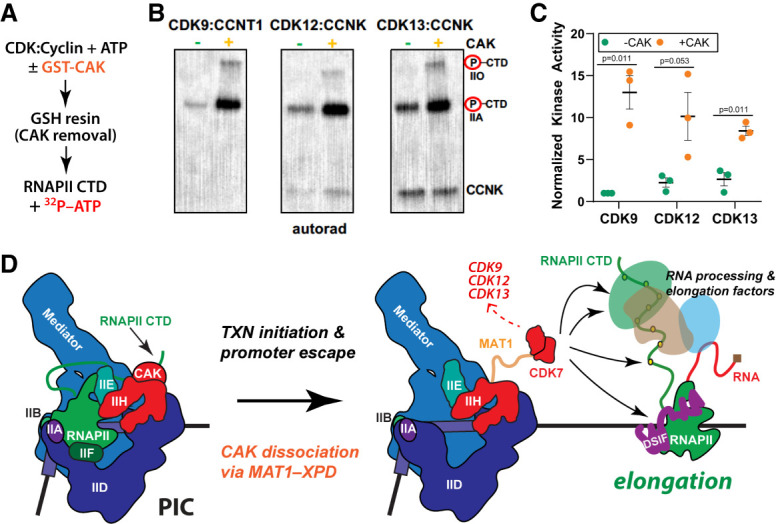
CDK7 is a master regulator of transcription-associated kinases; model. (A) Experimental overview; CDK:cyclin complexes were incubated with the CAK, followed by CAK removal. CDK:cyclin complexes were then tested for activity against the RNAPII CTD. (B) Representative kinase data in which purified CDK9:CCNT1, CDK12:CCNK, or CDK13:CCNK were tested against a common substrate, the RNAPII CTD. Each kinase complex was prephosphorylated by CDK7 (vs. controls) as shown. (C) Quantitation of kinase results across biological replicates (n = 3) with autorad signal normalized to CDK9 CAK pretreatment; P-values as shown. (D) Working model for TFIIH/CDK7 function in RNAPII transcription. Within the PIC, the CAK is assembled with TFIIH and CDK7 is activated toward the RNAPII CTD. The CDK7 phosphorylated CTD can be released from Mediator (Søgaard and Svejstrup 2007) and helps recruit chromatin modifiers, capping enzymes, and other RNA-processing factors (Ebmeier et al. 2017). Following promoter escape, the CAK may dissociate from TFIIH while maintaining interaction with XPD in the TFIIH core, via the flexible CAK subunit MAT1 (Greber et al. 2019). This “CAK release” allows access to cotranscriptional substrates (e.g., splicing and elongation factors) and also activates CDK7 toward these substrates. In this way, CDK7 activity is spatially and temporally regulated by TFIIH. Moreover, CDK7-dependent activation of CDK9, CDK12, and CDK13 would promote coordinated regulation of promoter-proximal pausing, transcription elongation, and mRNA biogenesis.
We next tested whether the 10-subunit TFIIH complex would similarly enable CDK7-dependent kinase activation, following the protocol outlined in Supplemental Figure S13B. As shown in Supplemental Figure S13C–E, TFIIH was unable to activate the CDK9 or CDK12 kinases, in contrast to the CAK. These results are consistent with data summarized in Figure 5 and further implicate CAK structural reorganization or dissociation from TFIIH as essential for CDK7 kinase activation. To further probe these results, we tested whether a distinct kinase, ERK, could activate CDK9, CDK12, or CDK13 in a manner similar to the CAK. As shown in Supplemental Figure S13, F–H, ERK was unable to activate CDK9, CDK12, or CDK13 in the same series of assays, demonstrating a selective role for the CAK.
Combined with the phosphoproteomics results that identified T-loop sites in CDK12 and CDK13 as CDK7 substrates, the results summarized in Figure 6 revealed that CDK7 directly activates the CDK12 and CDK13 kinases, as well as CDK9. Taken together, these results implicate CDK7 as a master regulator of transcription-associated kinases, analogous to its role as the master regulator of cell cycle kinases (Larochelle et al. 2007).
Discussion
The regulatory roles of CDK7 in RNAPII transcription have remained elusive and enigmatic. Various methods to inhibit CDK7 activity have been implemented over the years, in both yeast and mammalian systems. Confounding issues have included cytotoxicity (Kwiatkowski et al. 2014), incomplete CDK7 inhibition (e.g., with analog-sensitive alleles, which must compete with cellular ATP) (Kanin et al. 2007; Hong et al. 2009), chemical probes that inhibit other transcription-associated kinases (Kwiatkowski et al. 2014), and masking of transcriptional inhibitory effects through global mRNA stabilization (Rodríguez-Molina et al. 2016). Additionally, because of differing transcription regulatory mechanisms, data from yeast (e.g., Kin28 in S. cerevisiae) may have limited relevance to human CDK7. Despite these limitations, a basic role for CDK7 in transcription initiation, elongation, and pre-mRNA capping has emerged. Our results, which involved a combination of biochemistry, chemical biology, transcriptomics, and the first large-scale identification of CDK7 kinase substrates, build and expand upon these themes.
CDK7 as a master regulator of transcriptional kinases
Although CDK7 is a well-known activator of cell cycle CDKs (Larochelle et al. 2007; Schachter et al. 2013), our biochemical and phosphoproteomics data suggest CDK7 is also a master regulator of transcription-associated kinases, with the potential to activate CDK9, CDK12, and CDK13. Whereas the Mediator kinases CDK8 and CDK19 represent other transcription-associated kinases, these lack evidence of activation via T-loop phosphorylation; in fact, the T-loops of CDK8 and CDK19 have T-to-D substitutions, which mimic a phosphorylated state. The Fisher laboratory (Larochelle et al. 2012) previously implicated CDK7 in the activation of CDK9, the P-TEFb kinase, through phosphorylation of the CDK9 activation loop. We confirmed this observation for CDK9 and also identified high-confidence CDK7 sites in the activation loops of CDK12 and CDK13, which suggested that CDK7 activates these kinases. This hypothesis was verified with in vitro kinase assays using purified CDK12:CCNK or CDK13:CCNK (Fig. 6B,C). Importantly, this CDK7-dependent activation of transcription-associated kinases appeared to be blocked by TFIIH; only within the CAK was CDK7 capable of activating CDK9, CDK12, or CDK13. These results were consistent with kinase assays with other substrates that showed negative regulation of CDK7 function by TFIIH (Fig. 5). A key exception was the RNAPII CTD, which was efficiently phosphorylated by CDK7 as part of the CAK or TFIIH. CDK7 activation of the RNAPII Ser2 CTD kinases CDK12 and CDK13 is consistent with reduced Ser2 phosphorylation observed toward gene 3′ ends in CDK7-inhibited cells (Ebmeier et al. 2017).
The identification of CDK7 as a master regulator of other transcriptional kinases suggests a larger and more central role for CDK7 in controlling transcription and RNA processing. The ability to activate CDK9, CDK12, and CDK13 also reveals a new mechanistic basis for CDK7 amplification in certain cancers; CDK7 inhibitors may therefore have increased effects on gene expression, which could be both beneficial and counterproductive in a clinical setting. With respect to the data in this study, the large-scale splicing defects we observed could represent the combined effect of blocking CDK7 activity and reducing kinase activity of CDK9, CDK12, and CDK13, each of which is linked to splicing regulation (Chou et al. 2020). Furthermore, it is likely that a subset of high-confidence substrates identified in the phosphoprotemics data represent indirect targets of CDK7 that result from CDK7-dependent reduction in CDK9, CDK12, and/or CDK13 activity. However, it appears that our experimental strategy minimized indirect effects (see below). The ability of CDK7 to activate CDK9, CDK12, and CDK13 will make it challenging to completely decouple CDK7 kinase-specific functions in mammalian cells.
CDK7 inhibition: distinct targets and cellular compensation
Although phosphoproteomics experiments revealed a large number of decreased phosphosites in SY-351-treated cells (50 nM, 1 h), a significant number of phosphosites also increased (Fig. 2B). This likely reflects a combination of phosphatase activity changes and mobilization of other cellular kinases upon CDK7 inhibition. A common theme among CDK knockout studies has been functional compensation by other kinases (Malumbres et al. 2004; Santamaría et al. 2007). This phenomenon may also explain an intriguing finding reported for the covalent CDK7 inhibitor YKL-5-124, which was evaluated primarily in HAP-1 cells (Olson et al. 2019). The data revealed that CDK7 inhibition had no major effect on global levels of RNAPII CTD phosphorylation. Although these data contradict current models of CDK7 function, it is noteworthy that analysis of RNAPII CTD phosphorylation occurred 6 h after treatment, which would allow time for other kinases to compensate. Many kinases have been shown to be capable of CTD phosphorylation, including ERK, DYRK1A, PLK3, CDK9, CDK12, and CDK13.
Kinome profiling data indicated SY-351 is among the most potent and selective CDK7 inhibitors (Supplemental Table S1); however, low-level inhibition persists for several other kinases, including CDK12. Compared with other transcription-associated kinases CDK8, CDK9, and CDK12/CDK13, CDK7 phosphorylates a distinct set of proteins (Fig. 2C). Because CDK7 itself activates CDK9, CDK12, and CDK13, the distinct set of substrates identified for CDK7 indicated that our experimental strategy (50 nM SY-351, 1 h) minimized secondary or off-target effects. Moreover, analysis of exon usage across long genes (Supplemental Fig. S6E) lacked any hallmarks of CDK12 inhibition (Dubbury et al. 2018; Krajewska et al. 2019) in cells treated with SY-351. Nevertheless, we cannot rule out the possibility that some identified phosphorylation sites result from reduced activity of CDK9, CDK12, or CDK13, due to loss of CDK7-dependent activation.
The most well-represented classes of proteins whose phosphorylation levels decreased upon CDK7 inhibition included cell cycle and transcription factors and regulators of mRNA biogenesis. These targets are consistent with known biological roles for CDK7 but reveal potential mechanisms by which CDK7 controls these basic cellular processes. We also identified factors important for cell motility, vesicle trafficking, stress granule formation, and endocytosis, suggesting novel cellular roles for CDK7 that warrant further investigation.
CDK7 governs alternative splicing
Unexpectedly, SY-351 caused profound and widespread changes in alternative mRNA splicing, and we hypothesize that reduced phosphorylation of the universal splicing factor SF3B1 contributes to these defects (Kfir et al. 2015). In support, we observed that SF3B1 association with nuclear speckles was altered in cells treated with SY-351 versus controls (Fig. 4E,F). Furthermore, a comparative analysis of splicing defects in SY-351 versus PladB-treated cells showed some commonalities (Supplemental Fig. S9F); however, the variety and scope of splicing defects was greater with SY-351, suggesting that additional factors contribute to the splicing changes caused by CDK7 inhibition.
SF3B1 is implicated in alternative splicing, as revealed through the impact of oncogenic SF3B1 mutations on the transcriptome in leukemias and other cancer types (Darman et al. 2015; Obeng et al. 2016; Seiler et al. 2018). Although SF3B1 (and U2AF2) functions in 3′ splice site specification, our results showed that SY-351 alters use of both alternative 3′ and 5′ splice sites equally (Fig. 4C). SY-351 preferentially favored inclusion versus skipping of alternative exons, with frequent inhibition of splicing of introns flanking alternative exons (Fig. 4C,D). SY-351 also frequently enhanced splicing of retained introns (Fig. 4C). These diverse effects suggest complex mechanisms by which CDK7 regulates splicing, likely through direct phosphorylation of SF3B1 and other splicing factors, as well as the RNAPII CTD. The surprisingly large effect of this “transcription-associated” kinase on mRNA processing probably reflects strict mechanistic coupling between transcription and splicing (Bentley 2014; Herzel et al. 2017).
Mechanistic model for regulation of CDK7 function during transcription
A simple way to ensure that CDK7 phosphorylates its substrates at the appropriate time (e.g., transcription initiation vs. elongation) and place (e.g., genomic loci being actively transcribed) is to restrict its activity at gene promoters. In our efforts to verify high-confidence CDK7 substrates, we noted stark differences between the three-subunit CAK and the 10-subunit TFIIH complex (Fig. 5). As part of TFIIH, CDK7 was unable to efficiently phosphorylate any substrate tested, except for the RNAPII CTD. In contrast, CDK7 efficiently phosphorylated transcription elongation and splicing factors SPT5 (DSIF subunit), SF3B1, U2AF2, and the RNAPII CTD as part of the CAK complex. The CAK, but not TFIIH, was also capable of directly activating CDK9, CDK12, and CDK13 in vitro, presumably through T-loop phosphorylation of each kinase. These results suggest that TFIIH, which associates with the PIC at transcription start sites, represses CDK7 function toward substrates except the RNAPII CTD; this repression then appears to be relieved upon CDK7 dissociation from TFIIH (Fig. 6D). In this way, CDK7 activity could be responsive to distinct transcriptional stages, such that RNAPII CTD phosphorylation will occur within the PIC, whereas substrates relevant to elongation and RNA processing will be phosphorylated only after RNAPII initiation and promoter escape.
CDK7 dissociation could result from complete separation from TFIIH as the CAK; however, size exclusion chromatography showed no evidence of the free CAK in the nucleus (Supplemental Fig. S12B). Alternately, a structural reorganization could occur in which the CAK remains flexibly tethered to core TFIIH. The CAK associates with core TFIIH through its MAT1 subunit (Luo et al. 2015), which interacts with both XPB and XPD (Greber et al. 2017, 2019). Although it is not known how CAK dissociation from TFIIH is controlled, evidence for CAK dissociation during transcription was recently reported by the Egly group (Compe et al. 2019), and structural data from the Nogales laboratory (Greber et al. 2019) suggest that the MAT1–XPB interaction is disrupted upon TFIIH binding to promoter DNA. Loss of the MAT1-XPB interaction could release the CAK from core TFIIH, while retaining a long, flexible tether to XPD. This would enable CDK7 to freely sample a large volume around the promoter (and a diverse array of kinase substrates) without requiring its complete dissociation from TFIIH (Fig. 6D). Based on cryoEM structural data (Greber et al. 2019), we modeled how the CAK may dissociate from TFIIH while retaining the MAT1–XPD interaction (Supplemental Movie S1). Further structural and functional studies will be required to test this model.
Similar to CDK7, other transcription-associated kinases assemble within different protein complexes. For example, CDK9 is a component of several biochemically distinct complexes (Luo et al. 2012), and the CDK8 module reversibly associates with the Mediator complex (Knuesel et al. 2009a). CDK12 and CDK13 also appear to assemble into compositionally distinct complexes that may be cell type-specific (Bartkowiak and Greenleaf 2015; Liang et al. 2015; Huttlin et al. 2017). Our results with CDK7 suggest that other transcription-associated kinases may be regulated by similar means; that is, substrate preference may be dependent upon CDK-associated factors. Although further research is needed to rigorously test this hypothesis, it is noteworthy that CDK9 activity increases within the Super Elongation Complex (Luo et al. 2012) and CDK8 modifies chromatin templates (histone H3) only upon association with Mediator (Knuesel et al. 2009b).
CDK7 as a therapeutic target
Given its central role in cell cycle regulation and transcription, CDK7 has broad biomedical relevance. An important step in understanding the biological roles for any kinase is to define its substrates; this effort led us toward unexpected insights about CDK7 function and its regulation that may advance development of next-generation molecular therapeutics. For instance, given the distinct substrate preferences, it may be possible to develop CDK7 inhibitors that are selective for TFIIH versus the CAK.
Recent data (Olson et al. 2019), including this study, suggest that other kinases can compensate for CDK7 inhibition. Here, we identified CDK7 as a master regulator of transcription-associated kinases, which mirrors its role as an activator of cell cycle kinases (Larochelle et al. 2007; Schachter et al. 2013). This new understanding suggests that CDK7 inhibitors may more effectively circumvent compensatory mechanisms by other transcription-associated kinases, which may yield therapeutic advantages.
Materials and methods
Activity assay: KI/kinact and selectivity
SY-351 biochemical potency and selectivity was determined as in Hu et al. (2019). Briefly, covalent potency was determined by measuring the KI and kinact with CDK7/CCNH/MAT1 complex. Selectivity over a panel of CDK enzymes were determined at both 2mM ATP and Km ATP concentration as determined for each CDK complex. The Km ATP concentrations were as follows for each enzyme: 50 µM with CDK7/CCNH/MAT1, 100 µM with CDK2/CCNE1, 30 µM with CDK9/CCNT1, and 30 µM with CDK12/CCNK.
Kinase selectivity
Broad kinase selectivity was determined by KiNativ in situ profiling (ActivX Biosciences, Inc.), as described (Patricelli et al. 2011). SY-351 was screened at both 1.0 µM and 0.2 µM concentrations in A549 cell lysate to determine percentage inhibition against 252 kinases. Compound was incubated for 15 min at room temperature in cell lysate, followed by probe addition and a 10-min incubation, also at room temperature.
Synthesis of chemical compounds
For synthesis of chemical compounds, see the Supplemental Material.
Cell treatment for ABPP experiments
Three separate experiments, giving 27 paired samples, were run and analyzed. The first consisted of three paired samples, one for each of the probes at 1 µM, with (light medium) or without (heavy medium) SY-351 preincubation. The second and third each consisted of duplicate samples for each of the three probes, at both 100 nM and 1 µM, with or without 10 µM SY-351 preincubation. For each sample, a total of 10 million cells in 20 mL of medium, in a T25 flask were used. Twenty microliters of DMSO or 10 mM SY-351 inhibitor (1000× 0.1% DMSO) was added directly to heavy or light cells, respectively. After 1 h of incubation, 20 µL of the appropriate probe solution was added (1000× the final concentration, 0.2% DMSO final) for another hour. Cells were washed twice with 10 mL of cold Dulbecco's PBS (D-PBS), lacking calcium and magnesium (Wisent) and then lysed in 200 µL freshly prepared ice cold D-PBS plus 1% IGEPAL CA-630 (Sigma; referred to below as NP40) plus Complete protease inhibitors without EDTA (Roche). After 30 min on ice with occasional mixing using a P-1000 pipette tip, supernatants were recovered following spinning at 1000×g for 5 min. Total protein concentration was determined using BCA assay (Thermo) with BSA standards, and samples were frozen at −80°C. The procedure typically yielded 1 mg of protein at ∼5 µg/µL.
Lysates were treated following the reaction protocol described by Lanning et al. (2014). In brief, 0.5 mg (∼100 µL each) of corresponding light and heavy lysates were mixed with room temperature D-PBS in a 1.5-mL microtube to give 500 µL of suspension at 2 mg/mL total protein. Click reactions were initiated immediately by adding biotin-azide (Thermo) to a final concentration of 200 µM, followed by TCEP to 1 mM final (added and vortexed). TBTA dissolved in 4:1 DMSO/t-butanol (30 µL per sample) and CuSO4 (10 µL per sample) were premixed and added to reach 100 µM TBTA/1 mM CuSO4. Reactions were allowed to proceed in a Thermomixer for 1 h at 25°C in the dark with gentle vortexing after 30 min (some visible protein precipitation was typically observed). Excess reagents were then removed by adding 2 mL of methanol, 1.5 mL of water, and 0.5 mL of chloroform (HPLC grade) and mixing vigorously by vortexing in 15-mL tubes. The biphasic solution was then centrifuged at 4000 rpm for 20 min at 4°C. The protein precipitant is found at the phase interface as a solid disk. Both the bottom organic and upper aqueous layers were carefully removed as completely as possible. A second wash was performed by adding 600 μL of methanol, 600 μL of water, and 150 μL of chloroform, vortexing, then transferring to a 1.5-mL tube. Samples were spun at 20,000g for 10 min at 4°C to repellet and then air-dried for ∼15 min. Proteins were resuspended in 500 μL of 6 M urea (Sigma) plus 25 mM NH4HCO3 (Sigma) freshly prepared. Cysteines were reduced and alkylated by first DTT to 10 mM for 30 min at room temperature in the dark, followed by adding iodoacetamide to a concentration of 25 mM, with a further 30-min incubation at room temperature in the dark. The mixture was transferred to a 15-mL tube containing 6 mL of PBS (pH 7.4; Wisent) plus 0.25% sodium deoxycholate (Sigma) freshly prepared and 100 µL of a 50% slurry of Streptavidin agarose beads (Thermo) previously washed and equilibrated in the same buffer. After allowing biotinylated proteins to bind for 2 h in the dark with agitation, streptavidin beads were washed once with 10 mL of freshly prepared binding buffer and twice with 10 mL of PBS alone to remove detergent. Finally, the beads are resuspended in 200 μL of 25 mM ammonium bicarbonate/2 M urea and transferred to a microfuge tube for trypsin digestion.
Next, 2 μg of sequencing grade trypsin (Promega) was added to each sample and digestion was allowed to proceed overnight in a Thermomixer set at 1000 rpm and 30°C. The digest supernatant was then collected, and the resin was washed twice using 50 μL of PBS. The combined supernatants for each sample were then acidified with 15 μL of 10% trifluoroacetic acid, followed by addition of 15 µL of acetonitrile (final concentration 5%). Protein digests were desalted using Pierce C-18 spin columns following the manufacturer's instructions. The final eluate was dried in a SpeedVac, and the dried samples were stored at −20°C for LC-MS/MS analysis.
LC-MS/MS analysis (ABPP)
For the first two experiments, liquid chromatography-tandem MS (LC-MS/MS) analyses were performed on a Thermo EASY nLC II LC system coupled to a Thermo LTQ Orbitrap Velos mass spectrometer equipped with a nanospray ion source. Tryptic peptides were resuspended in 25 μL of solubilization solution containing 97% water, 2% acetonitrile (ACN) and 1% of formic acid (FA). A volume of 2 μL of each sample containing ∼200 ng tryptic peptides was injected onto a 10-cm × 75-μm column packed with Phenomenex Jupiter C18 stationary phase (3-μm particle diameter and 100 Å pore size). Peptides were eluted using a 120-min gradient at a flow rate of 400 nL/min with mobile phase A (96.9% water, 3% ACN, 0.1% FA) and B (97% ACN, 2.9% water, 0.1% FA). The gradient started at 2% B, with linear increases to 8% B at 14 min, to 12% B at 41 min, to 24% B at 100 min, to 32% B at 108 min, and to 87% B at 111 min, followed by isocratic with 87% B for 3 min, and finally with 2% B for 6 min to re-equilibrate the column. A full MS spectrum (m/z 400–1400) was acquired in the Orbitrap at a resolution of 60,000, then the 10 most abundant multiple charged ions were selected in data-dependent acquisition mode for MS/MS sequencing in linear trap with the option of dynamic exclusion. Peptide fragmentation was performed using collision induced dissociation at a normalized collision energy of 35% with activation time of 10 msec. Data were acquired using the Xcalibur software. For the first experiment, a 70-min gradient was used, but the procedures were otherwise identical. Analyses for the third experiment were performed on a Q-Exactive Orbitrap mass spectrometer coupled with a Thermo EASY nLC 1000 with the similar settings as described above.
Prior to injection of project samples onto either Orbitrap, instrument performance was evaluated by analyzing a standard BSA tryptic digest (New England Biolabs); experimental samples was injected only if >30 unique peptides identified. To monitor instrument performance, the BSA digest was injected every 12 h and at the end of the run.
MS data were processed using Thermo Proteome Discoverer software (v2.1, SP1) with the SEQUEST search engine. Peptide sequence data were searched against the UniProt Human proteome database. The enzyme for database search was chosen as trypsin (full) and maximum missed cleavage sites was set at 2. Mass tolerances of the precursor ion and fragment ion were set at 10 ppm and 0.7 Da, respectively, for Thermo LTQ Orbitrap Velos mass spectrometer. Mass tolerances of the precursor ion and fragment ion were set at 10 ppm and 0.02 Da, respectively, for Thermo Q-Exactive mass spectrometer.
Dynamic modifications on Methionine (oxidation, +15.994915 Da) and Cysteine (carbamidomethyl, +57.021464 Da) were allowed. Only peptides and proteins with high confidence (false discovery rate <1%) were reported. In addition to the false discovery rate, proteins were considered to be identified if they had at least one unique peptide, and they were considered quantified if they had at least one quantified SILAC pair. The quantification method SILAC 2plex (Arg10 and Lys8) was chosen for SILAC experiments and the light peptide was selected as the control channel. Also, peptides detected as singletons, for which only the heavy or light isotopically labeled peptide was detected and sequenced, but which passed all other filtering parameters, were given an arbitrary ratio of 100 if only heavy isotopically labeled peptide was detected and an arbitrary ratio of 0.01 if only light peptide was detected.
In order to evaluate the significance of reported ratios, control experiments were performed using a 1:1 mixture of heavy and light cells treated with 1 µM of each probe, which should theoretically give a heavy:light ration of 1.0 for all proteins. This helped to establish that a cut-off ratio of three is highly significant, since only a single protein gave a ratio greater than three (data not shown).
To combine data sets obtained on the two different instruments for final SY-351 competed protein ranking, an Excel macro was created using the VBA editor. The primary ranking criterion was the number of independent samples for which the protein was found with a heavy:light ratio ≥ 3. In addition, proteins had a higher rank if they were identified in at least one incubation with 100 nM probe. A further criterion was the average number of unique peptides (total detected unique peptides divided by the number of data sets for which at least one unique peptide was identified).
CDK7 and CDK12 target engagement in HL60 cells
The SY-351 target occupancy in HL-60 cells was determined as described (Hu et al. 2019). Briefly, cells were treated with DMSO or SY-351 for 1 h before protein samples were harvested in MPER mammalian protein extraction reagent (78501, ThermoFisher) with 1× Halt protease and phosphatase inhibitor cocktail (100×;Life Technologies 78440) and 1× Benzonase nuclease (1000×; Sigma-Aldrich E1014-25KU). Unoccupied CDK7 or unoccupied CDK12 in the lysates was precipitated by incubating with biotinylated SY-314 (Hu et al. 2019) overnight at 4°C. MSD bare plate (Meso Scale Diagnostics L15XA-3) and MSD streptavidin plates (Meso Scale Diagnostics L15SA-5) were used to capture input and free CDK7 or CDK12, respectively, following the vendor's protocol.
To determine percent occupancy in tumor lysate samples, the following equation was used: %CDKocc = (1-free-CDK/free-CDK of vehicle treated sample) × 100%. This calculation assumes the occupancy in vehicle-treated tumors to be 0%. The target occupancy data derived from experiments in which both CDK7 and CDK12 occupancy were measured side by side.
Cell viability determination with SY-351 treatment
Samples of HL-60 cells were taken at 3, 4, 5, and 6 h after treatment with either 50 nM SY-351 or DMSO. To determine viability, four biological replicates each were sampled for all treatment conditions (DMSO or SY-351 at each time of treatment), and cell counting technical replication was performed in duplicate for every sample. All samples were counted on a Bio-Rad TC20 cell counter according to the manufacturer's instructions. Unpaired t-tests were conducted for SY-351 and vehicle treatment each time of treatment using GraphPad Prism 6.
SILAC labeling of HL-60 cells
Stable isotope labeling of HL-60 cellular protein was conducted as described (Xiong and Wang 2010). Cells were cultured as two distinct populations in Iscove's modified Dulbecco medium (IMDM) lacking arginine and lysine (Thermo Scientific 88367) supplemented with either (1) 84 mg/L Arg0 (Sigma-Aldrich A6969) and 146 mg/L Lys0 (Sigma-Aldrich L9037) for the “light” population, or (2) molar equivalents of Arg10 (CIL CNLM-539-H) and Lys8 (CIL CNLM-291-H) for the “heavy” population. All populations of cells were maintained in 10% dialyzed FBS (Thermo Fisher 26400044) and 1× antibiotic–antimycotic (Thermo Fisher). Cells were passaged five times at a ratio of 1:4 and isotopic amino acid incorporation and Arg → Pro conversion were evaluated using LC-MS/MS prior to performing experiments.
Proteomics and phosphoproteomics sample preparation
Sample preparation was performed essentially as described (Poss et al. 2016) with select modifications. HL-60 cells used for experiments were passaged a total number of seven times in SILAC media in T175 suspension flasks. For each replicate, ∼20–25 mg of total protein (4× T175 flasks per population, at a cell density of ∼750,000 cells/mL and a final volume of 28 mL per flask) was harvested after treatment with either 50 nM SY-351 or 0.1% DMSO for 1 h. In one replicate, the treatment regimen was reversed, and light cells were SY-351-treated, while heavy cells were DMSO-treated. In the null experiment, both heavy and light populations were DMSO-treated. Cells were harvested in 50-mL conical tubes on ice and spun at 1100 rpm for 5 min at 4°C, and the supernatant was discarded. Cells were then washed with cold PBS and spun again, the supernatant was removed, and pellets were flash-frozen in liquid N2 and stored at −80°C until sample preparation.
For MS sample preparation, frozen cell pellets were thawed in 3 mL of 95°C ST buffer (4% [w/v] SDS, 100 mM Tris at pH 8.5) and heated for 10 min at 95°C. Samples were then probe sonicated three times each in cycles of 20 sec on and 20 sec off. Total protein concentration was determined using the BCA assay kit (Pierce), and lysates were subsequently mixed 1:1 based on total protein. Disulfide bonds were reduced simultaneously with alkylation using final concentrations of 10 mM TCEP and 40 mM chloroacetamide, respectively, with 25-min incubation time at room temperature . Mixed samples were then diluted 10-fold to 12-fold in buffer UA (8 M urea, 100 mM Tris at pH 8.5) and split between two 50-mL conical ultrafiltration devices (10-kD MWCO, Millipore), with 10–12 mg of total protein loaded onto each filter. Samples were transferred into UB (1.5 M urea, 100 mM Tris at pH 8.5) by spinning samples down at 4000g at room temperature to ∼1 mL in the filter, and then filling the filter three times in UB. Lysyl endopeptide C (LysC) (Wako) was added at ∼1:100 (wt:wt protein, total of 100 µg per sample) in UB and incubated rocking for 4 h at 35°C, followed by 125 µg of trypsin (Pierce) incubated on an orbital shaker overnight at 35°C. Samples were retrieved from the filter and quantified in UB using a nanodrop; recovered total peptide ranged from 7 to 9 mg. Tryptic peptides were desalted, samples of peptide inputs were taken for whole-proteome analysis, and phosphopeptides were enriched as described (Poss et al. 2016).
For high pH reversed-phase C18 phosphopeptide prefractionation, phosphopeptide-enriched fractions were separated on an in-house fabricated reversed-phase C18 column (Microchrom 1.8 µm, 130A rpC18, nanolcms solutions) using the buffer 10 mM ammonium formate (pH 10.0) and a Waters M-class UPLC equipped with a UV detector. Samples were loaded directly on column in 2% (v/v) acetonitrile and resolved with a 25-min gradient to 50% (v/v) acetonitrile. Twelve fractions were concatenated seven times over the gradient elution using a SunCollect fraction collector (SunChrom), and then dried using a speedvac. Samples for both phosphoproteomics and proteomics were fractionated in this manner, resulting in 12 concatenated samples.
For nanoUPLC mass spectrometry data acquisition and analysis, sample fractions were suspended in 7 µL of 3% (v/v) acetonitrile with 0.1% (v/v) trifluoroacetic acid, and ∼1 µg of enriched and fractionated phosphopeptides were directly injected onto a column, rpC18 1.7 µm, 130 Å, 75-µm × 250-mm M-class BEH column (Waters), using a Waters M-class UPLC. For phosphopeptides, 2 µL of 7 µL total was run in two technical replicates. For proteomics experiments, 1 µL of 7-µL total was run once for each of the 12 fractions per sample. Peptides were eluted at 300 nL/min with a gradient from 3% to 20% acetonitrile over 100 min into an Orbitrap Fusion tribrid mass spectrometer (Thermo Scientific). Precursor mass spectra (MS1) were acquired at a resolution of 120,000 from 380 to 1500 m/z with an AGC target of 2E5 and a maximum injection time of 50 msec. Dynamic exclusion was set for 20 sec with a mass tolerance of ±10 ppm. Precursor peptide ion isolation for MS2 fragment scans was 1.6 Da using the quadrupole, and the most intense ions were sequenced using top speed with a 3-sec cycle time. All MS2 sequencing was performed using higher-energy collision dissociation (HCD) at 35% collision energy and all fragment ions were scanned in the linear ion trap. An AGC target of 1E4 and a 35-msec maximum injection time was used for all linear ion trap scans.
All raw MS files for quantitative phosphoproteomics and proteomics were searched using the MaxQuant (v1.6.3.4) software package. Samples were searched individually against the Uniprot human proteome database (downloaded on August 30, 2017) using the default MaxQuant parameters, except multiplicity was set to 2 (heavy/light) with Arg10 and Lys8 selected, LysC/P was selected as an additional enzyme, “requantify” was unchecked, and Phospho (STY) was selected as a variable modification in both runs. For initial phosphosite sample processing, the Phospho (STY) table was processed with Perseus using the following workflow: Reverse and contaminant reads were removed, the site table was expanded to accommodate differentially phosphorylated peptides, and rows without quantification were removed after site table expansion. Valid value filter for phosphoproteomics were set to four, meaning a quantified ratio had to be present in all three biological replicates, as well as the null experiment. For protein quantification, the protein group table was processed similarly except that there was no expansion of the site table. Phosphoproteomics and proteomics samples were then further processed in R as described.
Statistical analysis of SILAC phosphoproteome data
All R code to reproduce the phosphoproteomic analysis and generate figures is in the Supplemental Material. The SILAC phosphorylation site-level SILAC ratios from the MaxQuant Phospho(STY).txt file were annotated with primary sequence level annotations using the Perseus software application (Tyanova et al. 2016), providing information on kinase substrate relationships and functions from the PhosphoSitePlus database (Hornbeck et al. 2015). The experimental design comprised four SILAC comparisons (samples): a null condition in which both heavy and light cell populations were treated with DMSO (“null”), two biological replicates (“rep1” and ”rep2”) in which the heavy population was treated with SY-351, and a “label flip” condition (“rep3LF”). The no-treatment “null” condition and the label-flip “rep3LF” condition control for systematic effects superimposed on the phosphopeptide SILAC ratios that are not related to the SY-351 treatment effect. The SILAC labeling experimental design for each sample (analyzed by multidimensional LC/MS/MS) is summarized as follows, in which “L” designates light treatment and “H” designates heavy treatment: Null = DMSO (L) and DMSO (H); rep1 = DMSO (L) and SY-351 (H); rep2 = DMSO (L) and SY-351 (H); rep3LF = SY-351 (L) and DMSO (H).
The experimental design allows for improved statistical power to detect SY-351-specific effects. To identify SILAC ratios for phosphorylation sites that exhibit statistically significant change due to SY-351 treatment, we used a linear modeling approach based on the empirical Bayes moderated t-test approach in the R package limma (Ritchie et al. 2015). Assuming ygi is the observed log2(heavy/light) SILAC ratio for phosphorylation site g in sample i, the phosphorylation site-wise linear models satisfy E(yg) = Xβg, where E(yg) is the column vector of expected log2 ratios for the four samples, X is the 4 × 2 design matrix, and β0 is an unknown coefficient vector that parameterizes the average log2(heavy/light) SILAC ratios in each experimental condition. More explicitly,
The design matrix, Χ (the first matrix on the right side of the equation above), allows for modeling the SY-351-specific effect and isotope effect simultaneously, where the first and second columns of X correspond to the βg0 and βg1 coefficients, respectively. The rows of X correspond to each sample. The coefficient βg0 captures the “isotope” effect for each phosphorylation site g, and represents systematic bias in the SILAC ratios not due to drug treatment (and present in all samples), for example, because of isotope-labeled cell population-specific effects. The coefficient βg1 captures the SY-351 treatment effect, which is the average log2(SY-351/DMSO) ratio for each phosphorylation site g.
We found that incorporating the “isotope” coefficient βg0 into the model provided a better fit to the residual error in the data relative to a simpler model (data not shown) with only one coefficient for the SY-351 effect βi1 (“SY351” in design matrix). Model comparison with the Bayesian information criterion (BIC) showed that the two coefficient model identified 12,279 site ratios with a higher BIC score compared with 4335 site ratios with a higher BIC score. We applied a filter that removes phosphorylation sites with SILAC ratios in the “null” no-treatment condition to remove sites that have large systematic shifts in SILAC ratios not due to the drug treatment, out of concern that these sites are more likely to be unreliably estimated. This had the side effect of increasing the number of phosphorylation sites deemed significant at a q-value < 0.05, due to reduced number of hypothesis tests.
This “isotope” effect captured by the coefficient βg0, and the SY-351-specific effect captured in the coefficient βg1, is shown in scatter plots of the log2 ratios in Supplemental Figure S3B as on-diagonal gray points and red points, respectively, superimposed on the log2 SILAC ratios for each sample. The effect of the isotope label-flip comparison for differentially expressed phosphosites can be seen as the negative correlation of red points when compared with the nonswapped conditions. The isotope bias effect can be seen as a nonzero correlation in the comparisons with the null condition (first column of scatter plots), and the superposition of these correlated sites in the flipped sample (rep3LF) comparisons.
STRING network analysis
Proteins with decreasing phosphorylation sites (log2FC < 0 and FDR < 0.05) were visualized using the STRING database web application, filtered using a minimum required interaction score of 0.9. The thickness of network edges indicates the confidence score for the strength of interaction based on active interaction sources set to “Experiments,” “Databases,” and “Neighborhood.” Disconnected nodes were hidden, and the network was clustered using the MCL algorithm with the inflation parameter set to default of 3.
GO analysis of phosphosites
Analysis of differentially phosphorylated sites upon SY-351 treatment (qval < 0.05) was conducted with Metascape (Zhou et al. 2019) with decreased sites and increased sites submitted separately. Unique gene symbols were submitted through the web tool, and H. sapiens was selected for both “input as species” and “analysis as species.” From the analysis report page, the bar graph summary was exported as an image, and included GO terms, as well as pathways that were enriched in the top 20 results.
Assessment of shared CDK sites (CDK7 vs. CDK8, CDK9, and CDK12/13)
Overlapping SILAC MS sites were determined by considering all sites with the largest negative log fold change values and P < 0.05. The top 400 of these sites were used to compare shared protein targets, and the number of overlapping proteins was calculated for all combinations of samples. CDK7 sites identified here were compared with targets identified for CDK8 (Poss et al. 2016), CDK9 (Sansó et al. 2016; Decker et al. 2019), and CDK12/13 (Krajewska et al. 2019).
RT-qPCR
RNA was harvested from HL-60 cells using TRIzol reagent according to the manufacturer's instructions, with subsequent RNA cleanup using the RNeasy kit (Qiagen) according to the instructions. For cDNA synthesis, 1 µg of RNA was used as input for the qScript cDNA Synthesis Kit (QuantaBio) according to the manufacturer's instructions. qPCR was performed on a BioRad C1000 series instrument with 384 well plates and 10 µL reactions. For data analysis, primer standard curves were used, efficiencies were calculated, and quantifications were calculated using the method described (Pfaffl 2001). Primer sequences were as follows: 18S rRNA (Fwd: 5′-GCCGCTAGAGGTGAAATTCTTG-3′ and Rev: 5′-CTTTCGCTCTGGTCCGTCTT-3′), GAPDH (Fwd: 5′-CGTGGAAGGACTCATGACCA-3′ and Rev: 5′-CAGTCTTCTGGGTGGCAGTGA-3′), and MYC (Fwd: 5′-AGTGGAAAACCAGCAGCCTC-3′ and Rev: 5′-TTCTCCTCCTCGTCGCAGTA-3′).
RNA-seq
Approximately 15 million early-passage HL-60 cells in 15 mL of media were treated in T-25 suspension flasks with either 0.1% DMSO (0.1% final vol:vol) or 50 nM SY-351 for 5 h. Cells were harvested on ice by moving to 15-mL conical tubes and centrifugation at 1100 rpm for 2 min at 4°C. RNA was extracted using the RNeasy kit (Qiagen), with additions of both DTT and SUPERase-In (ThermoFisher), according to all manufacturer's instructions. DNaseI digestion was performed using the on-column protocol. RNA was eluted in 50 µL of water, and all samples were analyzed on a nanodrop prior to further processing.
Prior to sequencing, 1 µL of 1:10 diluted ERCC ExFold RNA SPIKE-IN (Thermo Fisher) mixes were added to 5 µg of total RNA per sample. ExFold Mix 2 was spiked into DMSO-treated samples, and ExFold Mix 1 was spiked into SY-351-treated samples. All samples were analyzed on a BioAnalyzer and found to have RIN values >8.7. To conduct deep mRNA-seq and detect splicing alterations potentially due to CDK7 phosphorylation of splicing factors, poly(A) selection was performed with the Universal Plus mRNA-seq library preparation kit (NuGEN) with 500 ng of RNA. Sequencing was performed on the NovaSEQ 6000, using a paired-end 150-bp cycle (2 × 150).
Reads were mapped using the Star mapper v2.5.2a, and well-correlated biological replicate data sets were selected for further analysis each with >140 million mapped reads. Differential splicing was analyzed using MAJIQ v1.1.3a (Vaquero-Garcia et al. 2016) using default settings. Changes in alternative splicing have ΔPSI ≥ 0.20 with P-value < 0.05. The strengths of 5′ and 3′ splice sites at exons with differential inclusion/skipping were calculated using the maximum entropy method (Yeo et al. 2004). For differentially used alternative 5′ and 3′ splice sites identified by MAJIQ, we determined whether SY-351 caused a shift in favor of the upstream or downstream site.
Processing of sequencing data
The initial processing of all sequencing data was performed using the RNAseq-Flow Pipeline, a data processing pipeline written in the Groovy programming language. The code for this pipeline can be found at https://github.com/Dowell-Lab/RNAseq-Flow, with analysis for this experiment performed at commit 3fe1b7. Data were mapped to the hg38 reference genome.
Per-exon differential expression analysis was performed using DEXSeq (Anders et al. 2012) with counts generated using featureCounts (Liao et al. 2014) and the Ensembl hg38 reference genome. Box plots were generated after normalizing the log2 fold change data using the pseudo-log10 transformation [asinh(x/2)/log(10)]. Box plots were generated using ggplot in R after filtering out genes with <10 exons and binning exons into their nearest decile based on the total number of exons for each gene.
Isoform resolution
For the remainder of the analysis, only the maximally expressed isoform of each gene was considered. The maximally expressed isoform was determined by calculating the RPKM normalized expression over each isoform and selecting the one with the maximum RPKM expression.
Metagene analysis
Each gene in the isoform-resolved reference sequence was divided into a fixed number of bins, and the utility featurecounts was used to determine the total counts in those regions. The mean count and standard deviation of that mean were calculated, and all bins were then plotted along with that standard deviation.
Principal component analysis
Principal component analysis was performed using the standard “prcomp” function provided by the “sva” package for the R programming language. Batch effects from the different days replicates were generated on were corrected using the “removeBatchEffect” function provided by the “limma” package from the R programming language.
Differential expression analysis
Differential expression analysis was performed using the “DESeq2” package for the R programming language. Counts were generated using the utility featurecounts across resolved isoforms in the RefSeq hg38 gene annotation.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed with the Broad Institute's GSEA software on the GenePattern server using the preranked module. Log2 fold change values were used as the rank metric for all genes and compared against the Hallmark gene set database for enrichment.
Splicing analysis from pladB-treated cells
RNA-seq data (Kotake et al. 2007) from PladB-treated K562 cells (4 h; GEO accession GSE148768) were compared with the RNA-seq data from SY-351 HL60 cells described here. Splicing changes were detected using MAJIQ2 and were filtered for a P-value < 0.05 with an absolute DPSI ≥ 0.2. The splicing events selected were used in >10% of reads, and the coordinates of the 5′ and 3′ splice sites in both cell lines agreed within 10 bases. The number of local splicing variations (LSVs) affected by SY-351 was determined to be 13,984, whereas the number of LSVs for PladB was 9830.
Purification of DSIF, NELF, SF3B1, U2AF2, Pol II CTD, TFIIF, MYC, and STAT1
DSIF
The human DSIF complex (SPT4 and SPT5) was expressed in Rosetta2(DE3)pLysS cells (Novagen 71403). The expression plasmid, which was a gift from Dr. Rob Fisher, was customized by adding a C-terminal Strep tag II to Spt5. Cells were lysed in B-PER cell lysis reagent (Thermo Scientific 78266). Following sonication and benzonase treatment, the clarified lysate was subjected to tandem affinity purification, using the N-terminal HIS tag on Spt4 and the Strep tag II on Spt5. Briefly, Ni affinity purification was performed using Ni-NTA agarose (Qiagen 30210). Eluted protein was purified again over a second Ni-NTA agarose resin. This was followed by Strep-Tactin XT affinity purification according to the manufacturer's protocol (IBA 2-4998-000).
NELF
The four-subunit NELF complex (NELF-A, NELF-B, NELF-C, and NELF-E) was expressed in Rosetta2(DE3)pLysS (Novagen 71403) cells. The expression plasmid was a gift from Dr. Bryan Gibson and Dr. Lee Kraus. The lysate from bacterial expression was treated with benzonase, clarified, and run over a Ni-NTA agarose (Invitrogen R90101) column.
SF3B1
SF3B1 (SF3B155) was expressed in HEK293-T cells. The expression plasmid was purchased from Addgene (pCDNA3.1-FLAG-SF3B1-WT 82576). Cells were transfected and harvested after 24 h. Whole-cell extract was generated and SF3B1 immunoprecipitated with α-FLAG M2-agarose beads (Sigma A2220-1ML).
U2AF2
U2AF2 (U2AF65) was expressed in BL21 cells. The expression plasmid was a gift from Dr. Ravinder Singh (University of Colorado at Boulder). Whole-cell extract was generated from bacterial expression and GST-U2AF2 was purified over a glutathione resin and eluted in buffer containing 30 mM glutathione.
Pol II CTD
The Pol II GST-CTD was expressed in BL21 cells and purified as described (Ebmeier et al. 2017). Briefly, cells were induced and whole-cell extract was generated from bacterial expression and GST-CTD immunoprecipitated with GSH beads. Eluted material was then run over an S200 sizing column to ensure purification of only full-length GST-CTD.
TFIIF
TFIIF was purified as described (Knuesel et al. 2009a).
MYC
A c-Myc cDNA expression vector was purchased from Openbiosystems.com and cloned into pET17 with a 6-Histidine tag for expression in BL21 E. coli. After induction with IPTG, cells were harvested and lysed using lysozyme and sonication. Cleared lysates were loaded onto a Q fastflow column and the flow-through, containing c-Myc (determined by Western blots), was then loaded onto a Mono S column, in which most of c-Myc flowed through. The flowthrough fraction was then supplemented with NaCl (to 250 mM) and imidazole (to 10 mM), and urea was added to 6 M. A Ni+2 affinity resin was then equilibriated with binding buffer (10 mM imidazole, 250 mM NaCl, 6 M urea), followed by binding with mixing for 1 h at 4°C. The resin was then washed with high-salt wash buffer (1M NaCl, 20 mM imidazole, 6 M urea, 0.1% NP40) and then a low-salt wash buffer (0.2M NaCl, 20 mM imidazole, 0.02% NP40). Bound protein was eluted first with 150 mM imidazole in Tris buffer (0.2M NaCl, 0.02% NP40) and then with 500 mM imidazole (0.2M NaCl, 0.02% NP40 at pH 2). The elutes were dialyzed against 100 mM KCl HEMG (20 mM HEPES at pH 7.9, 0.2 mM EDTA, 2 mM MgCl2, 10% glycerol) and MYC was detected with SDS-PAGE and Western blots.
STAT1
GST-Stat1 was expressed in BL21 E. coli cells. After induction with IPTG, Stat1 was isolated with a glutathione (GSH) affinity column and eluted with 30 GSH, as described (Pelish et al. 2015).
Purification of CDK complexes, ERK, and CDC25.
CDK8 module
The four-subunit CDK8 module (CDK8, CCNC, MED12, and MED13) was expressed and purified as described (Knuesel et al. 2009a).
P-TEFb (CDK9 and CCNT1)
P-TEFb was purchased from Fisher Scientific (PV4131).
CDK12:CCNK and CDK13:CCNK
CDK12:CCNK complexes and CDK13:CCNK complexes were purified as described (Bösken et al. 2014; Greifenberg et al. 2016).
ERK
ERK purification was completed as described (Pegram et al. 2019).
CDC25
CDC25 was expressed in E. coli BL21 cells. The expression plasmid was purchased from Addgene (cat# 10969). Whole-cell extract was generated and GST-Cdc25 was precipitated with a glutathione (GSH) conjugated resin, washed extensively, and eluted with 30 mM GSH.
Purification of CAK and TFIIH
The CAK complex was purchased from Millipore (CDK7/CCNH/MAT1; CAK complex; 250 µg of 14-476M). The TFIIH purification was completed as described (Fant et al. 2020).
In vitro kinase assays
The endpoint kinase assays comparing TFIIH-CDK7 and CAK-CDK7 activity were performed with 50 nM kinase (TFIIH or CAK) and 10 nM substrate of interest with 100 µM ATP and [32P-γ]ATP for 2 h at 37°C. These concentrations were chosen based upon prior titration experiments. Titration experiments also enabled TFIIH versus CAK comparisons at identical activity (i.e., same total RNAPII CTD phosphorylation levels). Although this required different concentrations (TFIIH vs. CAK), the results remained consistent with the assays with fixed kinase (50 nM) and substrate (10 nM) concentrations. Kinase reactions were quenched with 2× Laemmli buffer, boiled for 5 min, and run on a 4%–20% gradient protein gel (BioRad 4%–20% Mini-Protean TGX gel, 15-well, 15 µL of 456-1096). Gels were dried and exposed to evaluate autorad signal. ImageJ software was used to measure autorad signal and Prism8 used to plot the data normalized to CAK-DSIF signal.
In vitro PIC kinase assays were performed with a reconstituted transcription system, assembled on the native HSPA1B promoter as described (Fant et al. 2020). Briefly, 10 µL of PIC mix (TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH, RNAPII, and Mediator at ∼5–100 nM each; TFIIH estimated 5 nM) was mixed with 10 µL HSPA1B promoter template (5 nM in 10 μL, plus 400 nM HSF1) and incubated for 15 min at 30°C to allow for PIC assembly. Kinase reactions were started by adding C/G/UTP (to 450 μM), ATP (22.5 μM), and [32P-γ]ATP. The reactions were quenched after a 30-min incubation at 30°C by addition of 4× Laemmli buffer and boiled for 5 min. Gels were run and imaged as described above.
Endpoint kinase assays were performed to compare TFIIH-CDK7 and CAK-CDK7 phosphosite preference on the RNAPII CTD. Assays were performed with 50 nM kinase, 10 nM RNAPII CTD, and 100 µM ATP for 2 h at 37°C. Reactions were quenched with 2× Laemmli buffer, boiled for 5 min, run on a 4%–20% gradient protein gel, and transferred to a nitrocellulose membrane. The membrane was then blocked in 5% BSA, cut, and incubated 1:5000 for PSer2 and PSer7, 1:10,000 for PSer5, and 1:2000 for CDK7 (antibody information below). Quantitation and analysis were performed with ImageJ and Prism8, with normalization to CDK7 signal.
CDK activation assays were performed by first incubating 50 nM GST-Cdc25 (phosphatase) separately with 50 nM CDK9:CCNT, 400 nM CDK12:CCNK, and 200 nM CDK13:CCNK to dephosphorylate their T-loops (Bösken et al. 2014; Greifenberg et al. 2016). The concentration of each kinase was determined by first performing a titration against the RNAPII CTD and normalizing for activity. GST-Cdc25 was depleted with GSH resin and rigorously tested by Western blot to confirm its complete removal. Dephosphorylated CDK complexes were then incubated with GST-CAK and 100 µM ATP for 1 h at 37°C. The GST-CAK complex was then depleted with GSH beads and the flow-through was then rigorously tested by Western to confirm its complete removal. Kinase complexes were then incubated with RNAPII GST-CTD and [32P-γ]ATP for 2 h at 37°C. Reactions were quenched with 2× Laemmli buffer, boiled for 5 min, and run on a 4%–20% gradient protein gel (BioRad 4%–20% Mini-Protean TGX Gel, 15 well, 15 µL of 456-1096). Gels were dried and exposed to evaluate autorad signal. ImageJ software was used to measure autorad signal and Prism8 used to plot the data normalized to CDK9:CCNT1 RNAPII CTD phosphorylation signal without CAK activation.
For TFIIH kinase activation assays, purified TFIIH was cross-linked to Protein A agarose beads. Experiments were completed as described above, with immobilized TFIIH instead of GST-CAK. As TFIIH was already immobilized on beads, the depletion/removal step was omitted. ImageJ and Prism8 were used to quantify the data normalized to TFIIH-only conditions.
Activated ERK2 (pTpY) was also tested in the CDK activation assays. Experiments were completed as described above, substituting 50 nM ERK for the CAK. The final endpoint kinase assay, however, was preincubated with the ERK-specific inhibitor SCH772984 prior to the addition of the RNAPII CTD and [32P]ATP. Control experiments confirmed that SCH772984 did not inhibit CDK9, CDK12, or CDK13 under the conditions of these assays. ImageJ and Prism8 were used to quantify the data normalized to CDK13 conditions.
HCT116 cell fractionation and size exclusion chromatography
HCT116 cells were grown in McCoy's 5A medium supplemented with 10% FBS and 1% Pen-Strep. Cells were harvested and nuclei isolated from the cytoplasmic fraction, as described (Fant et al. 2020). Nuclei were then sonicated in a Bioruptor in RIPA buffer and the nuclear pellet separated through centrifugation. The nuclear pellet was then prepared by resuspension in RIPA with the addition of benzonase, RNase A, and DNase I to release bound proteins. Concentrated fractions were then passed over an S6 Increase sizing column and immunoblots done probing for core subunit XPB (p89) and CAK subunit CDK7. Relative complex size was determined by comparing elution volume of known protein standards to the elution volume of the proteins of interest in the cellular fractions. Similar fractionation experiments were attempted with HL60 cell extracts, but these were inconclusive due to limited protein amounts.
SF3B1 cellular localization
HCT116 cells were treated with 50 nM SY-351 or DMSO for 4 h. Cytoplasmic and nuclear fractions were then isolated, and total protein concentration was determined by BCA. Samples were normalized for 5 µg of total protein, loaded on a 4%–20% acrylamide gradient protein gel, and transferred to a nitrocellulose membrane. The membrane was then blocked in 5% milk, cut, and incubated 1:4000 for SF3B1 or ACTB, 1:1000 for CDK7, and 1:12000 for Histone H3. Quantitation and analysis was performed with ImageJ and Prism8 with +SY-351 signal normalized to DMSO conditions.
Western blot antibodies
The following Pol II CTD phospho-specific antibodies were used to compare TFIIH-associated and CAK phosphosite preference: anti-RNA polymerase II subunit B1 (phospho-CTD Ser-7; Millipore Sigma, clone 4E12 04-1570), anti-RNA polymerase II subunit B1 (phospho-CTD Ser-5; Millipore Sigma clone 3E8 04-1572), and anti-RNA polymerase II subunit B1 (phospho-CTD Ser-2; Millipore Sigma clone 3E10 04-1571). Signals were normalized to the CDK7 antibody (sc-856 lot#C1914), as described (Ebmeier et al. 2017). For SF3B1 quantitative Westerns: SF3B1 (CST D7L5T, 14434), Actin B (sc-47778, AB_2714189), and Histone H3 from A. Shilatifard, as described (Ebmeier et al. 2017).
Immunofluorescence (IF)
HCT116 cells were grown in laboratory-made imaging dishes (35-mm cell culture dishes with a hole in the center, with a standard #1.5 coverslip affixed to the dish using the Sylgard 184 silicone elastomer kit (Dow Corning 3097366-1004)). For compound treatments, SY-351 (50 nM) or THZ-531 (200 nM) were added (or DMSO control) 4 h prior to analysis. Cells were fixed and stained using standard protocols. Briefly, cells were fixed using 4% paraformaldehyde, permeabilized using 0.2% Triton-X 100, and blocked with 3% BSA. Primary SF3B1 antibody [EPR11987(B), Abcam ab170854] staining was done overnight at 4°C at a 1:100 dilution in 3% BSA. Secondary antibody staining was done using goat anti-rabbit AlexaFluor 647 (Thermo Fisher Scientific A21244, lot 1156625) at a 1:5000 dilution in 3% BSA for 2 h at room temperature. Hoechst 33258 (Millipore Sigma 861405, lot 02026ME) was added at a final concentration of 10 μg/mL to counterstain nuclei during the final 10 min of secondary antibody incubation. Dishes were either promptly imaged or stored in PBS overnight at 4°C until imaging.
IF image acquisition
Images were acquired on a Nikon TI-2 microscope equipped with a Hamamatsu Orca Fusion sCMOS camera (6.5-μm pixels) using a 100× 1.49 NA oil immersion objective. Cells were imaged using the wide-field modality with illumination provided by an Excelitas X-Cite 120-LED light engine and individual excitation and emission filter sets specific for DAPI and Cy5. To ensure imaging coverage through the entire depth of the nucleus, we acquired Z-stacks at 200-nm intervals spanning a total range of 10 μm. Images were acquired using 5% lamp power at 50-msec exposure times for both the DAPI and Cy5 channels, with 4× averaging to reduce background camera noise.
IF image analysis
All image analysis was done using Matlab R2020a (Mathworks). Briefly, Z-stacks were collapsed into maximum intensity projections and the resulting images were subjected to background subtraction using a rolling ball algorithm (radius = 500 px). Nuclei were segmented using global thresholding and watershedding to separate overlapping objects. The nuclear mask was dilated to generate a cytosolic ring for calculating cytosolic SF3B1 intensity. For thresholding and scoring puncta in individual nuclei, we first cropped the puncta image using the bounding box that defined individual nuclei. This enhanced the robustness of the adaptive thresholding to detect SF3B1 puncta and minimized false positives from diffuse signal. All raw image files and analysis scripts are available upon request.
Statistical analysis
Statistical analysis of the in vitro autorad kinase data, Western kinase data, and quantitative Westerns was performed using the Prism8 paired multiple t-tests Holm-Sidak method, α = 0.05, with p-adjusted values reported. Statistical analysis of the overall relative fluorescence (RFU) among the IF conditions was performed using Prism8 standard ANOVA with Tukey's multiple comparisons test. Analysis of the ratio of fluorescence between the cytoplasm and nucleus was performed with the Prism8 Brown-Forsythe and Welch ANOVA and Dunnett's T3 multiple comparisons test. The puncta-per-nucleus statistical analysis was run with the Kruskal–Wall nonparametric test and uncorrected Dunn's test.
TFIIH animation
The animation shows the cryo-EM structure of human TFIIH core complex (PDB 6NMI) (Greber et al. 2019). CDK7 and Cyclin H of the CAK module was used from the cryo-EM structure of human holo-PIC in the closed state (PDB 6O9L) (Yan et al. 2019). Volume maps of these structures were obtained from the molecular visualization application, UCSF Chimera (Pettersen et al. 2004), and imported into the 3D graphics application, Autodesk Maya for animation (https://www.autodesk.com/products/maya). The hydrophobic region of MAT1 corresponding to 100 amino acids, which interacts with the CDK7–Cyclin H dimer, was modeled in Autodesk Maya. This dynamic model was rendered into a series of images and imported into the postproduction application, Adobe After Effects (https://www.adobe.com/products/aftereffects) for compositing and creation of the final video.
Data availability
RNA-seq data have been uploaded on GEO GSE151699. Proteomics data have been uploaded on the MassIVE repository with accession MSV000085575.
Competing interest statement
D.J.T. is a member of the SAB at Dewpoint Therapeutics. The laboratory of D.J.T. receives some support from Syros Pharmaceuticals. L.C.C. is a founder and member of the SAB of Agios Pharmaceuticals and of Petra Pharmaceuticals. These companies are developing novel therapies for cancer. L.C.C.’s laboratory also receives some financial support from Petra Pharmaceuticals. J.L.J. reports consultant activities for Petra Pharmaceuticals. T.M.Y. is a stockholder and on the board of directors of Destroke, Inc., an early-stage start-up developing mobile technology for automated clinical stroke detection. M.J.B., K.B.H., S.H., G.M., and J.J.M. have or had an equity position in Syros Pharmaceuticals, Inc.
Supplementary Material
Acknowledgments
We thank R. Tjian for ERCC3 antibodies, W.L. Kraus for NELF expression plasmids, R. Fisher for SPT4 and SPT5 expression plasmid, and the UC-Boulder BioFrontiers Computing Core (National Institutes of Health [NIH] OD12300). Funding support was provided by the NIH (GM118051 to D.L.B., GM110064 to D.J.T., and T32GM065103 to L.J.D.), the National Cancer Institute (R21CA205912 to W.M.O. and D.J.T., and F31CA250432 to J.K.R.), the German Research Foundation (DFG; DE 3069/1-1 to T.M.D., and GE 976/9-2 to M.G.), and the National Science Foundation (MCB1818147 to D.J.T., ABI1759949 to R.D.D., and MCB1903300 to J.I.).
Author contributions: J.K.R., Z.C.P., W.M.O., and D.J.T. designed the study. M.J.B., K.B.H., S.H., G.M., J.J.M., P.W,W., M.B., L.T., P.D., C.C., and J.K.R. characterized SY-351. J.K.R. and T.-M.D. performed biochemical assays. Z.C.P., C.C.E., and W.M.O. performed mass spectrometry and data analysis. P.W.W., M.B., L.T., T.-M.D., H.B., I.H.K., and M.G. were responsible for key reagents. Z.C.P., B.E., and Z.L.M. performed RNA-seq and data analysis. L.J.D. performed IF analysis. J.L.J. and T.M.Y. performed peptide arrays. S.N. and J.I. produced the animation. L.C.C., R.D.D., D.L.B., W.M.O., and D.J.T. acquired funding and supervised the study. J.K.R., D.L.B., and D.J.T. wrote the manuscript with input from all authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.341545.120.
Freely available online through the Genes & Development Open Access option.
References
- Anders S, Reyes A, Huber W. 2012. Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017. 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B, Greenleaf AL. 2015. Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J Biol Chem 290: 1786–1795. 10.1074/jbc.M114.612226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley MJ, Yun CH, Gewinner CA, Asara JM, Johnson JL, Coyle AJ, Eck MJ, Apostolou I, Cantley LC. 2015. EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nat Struct Mol Biol 22: 983–990. 10.1038/nsmb.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. 2014. Coupling mRNA processing with transcription in time and space. Nature reviews Genetics 15: 163–175. 10.1038/nrg3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, Geyer M. 2014. The structure and substrate specificity of human Cdk12/Cyclin K. Nat Commun 5: 3505 10.1038/ncomms4505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Takagi T, Moore CR, Buratowski S. 1997. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 11: 3319–3326. 10.1101/gad.11.24.3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Quigley DA, Robinson TM, Feng FY, Ashworth A. 2020. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov 10: 351–370. 10.1158/2159-8290.CD-19-0528 [DOI] [PubMed] [Google Scholar]
- Compe E, Genes CM, Braun C, Coin F, Egly JM. 2019. TFIIE orchestrates the recruitment of the TFIIH kinase module at promoter before release during transcription. Nat Commun 10: 2084 10.1038/s41467-019-10131-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL. 2013. RNA polymerase II C-terminal domain: tethering transcription to transcript and template. Chem Rev 113: 8423–8455. 10.1021/cr400158h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Wright AT, Kozarich JW. 2008. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem 77: 383–414. 10.1146/annurev.biochem.75.101304.124125 [DOI] [PubMed] [Google Scholar]
- Cujec TP, Okamoto H, Fujinaga K, Meyer J, Chamberlin H, Morgan DO, Peterlin BM. 1997. The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genes Dev 11: 2645–2657. 10.1101/gad.11.20.2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. 10.1016/j.celrep.2015.09.053 [DOI] [PubMed] [Google Scholar]
- David CJ, Boyne AR, Millhouse SR, Manley JL. 2011. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65–Prp19 complex. Genes Dev 25: 972–983. 10.1101/gad.2038011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker TM, Forné I, Straub T, Elsaman H, Ma G, Shah N, Imhof A, Eick D. 2019. Analog-sensitive cell line identifies cellular substrates of CDK9. Oncotarget 10: 6934–6943. 10.18632/oncotarget.27334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbury SJ, Boutz PL, Sharp PA. 2018. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 564: 141–145. 10.1038/s41586-018-0758-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebmeier CC, Erickson B, Allen BL, Allen MA, Kim H, Fong N, Jacobsen JR, Liang K, Shilatifard A, Dowell RD, et al. 2017. Human TFIIH kinase CDK7 regulates transcription-associated chromatin modifications. Cell Rep 20: 1173–1186. 10.1016/j.celrep.2017.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick D, Geyer M. 2013. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 113: 8456–8490. 10.1021/cr400071f [DOI] [PubMed] [Google Scholar]
- Eilbracht J, Schmidt-Zachmann MS. 2001. Identification of a sequence element directing a protein to nuclear speckles. Proc Natl Acad Sci 98: 3849–3854. 10.1073/pnas.071042298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Devlin JR, Hogg SJ, Doyle MA, Harrison PF, Todorovski I, Cluse LA, Knight DA, Sandow JJ, Gregory G, et al. 2020. CDK13 cooperates with CDK12 to control global RNA polymerase II processivity. Sci Adv 6: eaaz5041 10.1126/sciadv.aaz5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fant CB, Levandowski CB, Gupta K, Maas ZL, Moir J, Rubin JD, Sawyer A, Esbin MN, Rimel JK, Luyties O, et al. 2020. TFIID enables RNA polymerase II promoter-proximal pausing. Mol Cell 78: 785–793.e8. 10.1016/j.molcel.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP. 2005. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 118: 5171–5180. 10.1242/jcs.02718 [DOI] [PubMed] [Google Scholar]
- Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. 2009. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol 29: 5455–5464. 10.1128/MCB.00637-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Nguyen THD, Fang J, Afonine PV, Adams PD, Nogales E. 2017. The cryo-electron microscopy structure of human transcription factor IIH. Nature 549: 414–417. 10.1038/nature23903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Toso DB, Fang J, Nogales E. 2019. The complete structure of the human TFIIH core complex. Elife 8: e44771 10.7554/eLife.44771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenleaf AL. 2019. Human CDK12 and CDK13, multi-tasking CTD kinases for the new millenium. Transcription 10: 91–110. 10.1080/21541264.2018.1535211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greifenberg AK, Hönig D, Pilarova K, Düster R, Bartholomeeusen K, Bösken CA, Anand K, Blazek D, Geyer M. 2016. Structural and functional analysis of the Cdk13/cyclin K complex. Cell Rep 14: 320–331. 10.1016/j.celrep.2015.12.025 [DOI] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, Neugebauer KM. 2017. Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol 18: 637–650. 10.1038/nrm.2017.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SW, Hong SM, Yoo JW, Lee YC, Kim S, Lis JT, Lee DK. 2009. Phosphorylation of the RNA polymerase II C-terminal domain by TFIIH kinase is not essential for transcription of Saccharomyces cerevisiae genome. Proc Natl Acad Sci 106: 14276–14280. 10.1073/pnas.0903642106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. 2015. Phosphositeplus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520. 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Marineau JJ, Rajagopal N, Hamman KB, Choi YJ, Schmidt DR, Ke N, Johannessen L, Bradley MJ, Orlando DA, et al. 2019. Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res 79: 3479–3491. 10.1158/0008-5472.CAN-19-0119 [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. 2017. Architecture of the human interactome defines protein communities and disease networks. Nature 545: 505–509. 10.1038/nature22366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanin EI, Kipp RT, Kung C, Slattery M, Viale A, Hahn S, Shokat KM, Ansari AZ. 2007. Chemical inhibition of the TFIIH-associated kinase cdk7/kin28 does not impair global mRNA synthesis. Proc Natl Acad Sci 104: 5812–5817. 10.1073/pnas.0611505104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TW, Baumgart K, Eickhoff J, Albert T, Antrecht C, Lemcke S, Klebl B, Meisterernst M. 2014. Cyclin-dependent kinase 7 controls mRNA synthesis by affecting stability of preinitiation complexes, leading to altered gene expression, cell cycle progression, and survival of tumor cells. Mol Cell Biol 34: 3675–3688. 10.1128/MCB.00595-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfir N, Lev-Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, Ast G. 2015. SF3B1 association with chromatin determines splicing outcomes. Cell Rep 11: 618–629. 10.1016/j.celrep.2015.03.048 [DOI] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. 2009a. The human CDK8 subcomplex is a molecular switch that controls mediator co-activator function. Genes Dev 23: 439–451. 10.1101/gad.1767009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ. 2009b. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol Cell Biol 29: 650–661. 10.1128/MCB.00993-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. 2007. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 3: 570–575. 10.1038/nchembio.2007.16 [DOI] [PubMed] [Google Scholar]
- Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, Sharma B, Day DS, Kwiatkowski N, Pomaville M, et al. 2019. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat Commun 10: 1757 10.1038/s41467-019-09703-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. 2014. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511: 616–620. 10.1038/nature13393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson TO, Joslyn C, Kath JC, Niessen S, et al. 2014. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol 10: 760–767. 10.1038/nchembio.1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP. 2007. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell 25: 839–850. 10.1016/j.molcel.2007.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Amat R, Glover-Cutter K, Sansó M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. 2012. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 19: 1108–1115. 10.1038/nsmb.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May N, Dubaele S, Proietti De Santis L, Billecocq A, Bouloy M, Egly JM. 2004. TFIIH transcription factor, a target for the rift valley hemorrhagic fever virus. Cell 116: 541–550. 10.1016/S0092-8674(04)00132-1 [DOI] [PubMed] [Google Scholar]
- Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, Shilatifard A. 2015. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol 35: 928–938. 10.1128/MCB.01426-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. 2012. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol 32: 2608–2617. 10.1128/MCB.00182-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Cimermancic P, Viswanath S, Ebmeier CC, Kim B, Dehecq M, Raman V, Greenberg CH, Pellarin R, Sali A, et al. 2015. Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol Cell 59: 794–806. 10.1016/j.molcel.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji D, Grossfield A, Kielkopf CL. 2019. Structures of SF3b1 reveal a dynamic Achilles heel of spliceosome assembly: implications for cancer-associated abnormalities and drug discovery. Biochim Biophys Acta Gene Regul Mech 1862: 194440 10.1016/j.bbagrm.2019.194440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Sotillo R, Santamarı´a D, Galán J, Cerezo A, Ortega S, Dubus P, Barbacid M. 2004. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118: 493–504. 10.1016/j.cell.2004.08.002 [DOI] [PubMed] [Google Scholar]
- Manuguerra M, Saletta F, Karagas MR, Berwick M, Veglia F, Vineis P, Matullo G. 2006. XRCC3 and XPD/ERCC2 single nucleotide polymorphisms and the risk of cancer: a HuGE review. Am J Epidemiol 164: 297–302. 10.1093/aje/kwj189 [DOI] [PubMed] [Google Scholar]
- McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Amgen EST Program, Shuman S, et al. 1997. 5'-capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev 11: 3306–3318. 10.1101/gad.11.24.3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. 2016. Physiologic expression of Sf3b1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 30: 404–417. 10.1016/j.ccell.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson CM, Liang Y, Leggett A, Park WD, Li L, Mills CE, Elsarrag SZ, Ficarro SB, Zhang T, Düster R, et al. 2019. Development of a selective CDK7 covalent inhibitor reveals predominant cell-cycle phenotype. Cell Chem Biol 26: 792–803.e10. 10.1016/j.chembiol.2019.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. 2011. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol 18: 699–710. 10.1016/j.chembiol.2011.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram LM, Liddle JC, Xiao Y, Hoh M, Rudolph J, Iverson DB, Vigers GP, Smith D, Zhang H, Wang W, et al. 2019. Activation loop dynamics are controlled by conformation-selective inhibitors of ERK2. Proc Natl Acad Sci 116: 15463–15468. 10.1073/pnas.1906824116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, Caruso BT, Arefolov A, Fadeyi O, Christie AL, et al. 2015. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 526: 273–276. 10.1038/nature14904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Odell AT, Tangpeerachaikul A, Lee T, Pelish HE, Shair MD, Dowell RD, Old WM, Taatjes DJ. 2016. Identification of mediator kinase substrates in human cells using cortistatin A and quantitative phosphoproteomics. Cell Rep 15: 436–450. 10.1016/j.celrep.2016.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadri I, Conaway JW, Conaway RC, Schaack J, Siddiqui A. 1996. Hepatitis B virus transactivator protein, HBx, associates with the components of TFIIH and stimulates the DNA helicase activity of TFIIH. Proc Natl Acad Sci 93: 10578–10583. 10.1073/pnas.93.20.10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimel JK, Taatjes DJ. 2018. The essential and multi-functional TFIIH complex. Protein Sci 27: 1018–1037. 10.1002/pro.3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Molina JB, Tseng SC, Simonett SP, Taunton J, Ansari AZ. 2016. Engineered covalent inactivation of TFIIH-kinase reveals an elongation checkpoint and results in widespread mRNA stabilization. Mol Cell 63: 433–444. 10.1016/j.molcel.2016.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansó M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, Ali A, Ghosh A, Larochelle S, Rana TM, et al. 2016. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev 30: 117–131. 10.1101/gad.269589.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M, Barbacid M. 2007. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448: 811–815. 10.1038/nature06046 [DOI] [PubMed] [Google Scholar]
- Schachter MM, Merrick KA, Larochelle S, Hirschi A, Zhang C, Shokat KM, Rubin SM, Fisher RP. 2013. A Cdk7–Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell 50: 250–260. 10.1016/j.molcel.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, Chambon P, Egly JM. 1993. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science 260: 58–63. 10.1126/science.8465201 [DOI] [PubMed] [Google Scholar]
- Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG, The Cancer Genome Atlas Research Network, Buonamici S, Yu L, et al. 2018. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep 23: 282–296.e4. 10.1016/j.celrep.2018.01.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søgaard TMM, Svejstrup JQ. 2007. Hyperphosphorylation of the C-terminal repeat domain of RNA polymerase II facilitates dissociation of its complex with mediator. J Biol Chem 282: 14113–14120. 10.1074/jbc.M701345200 [DOI] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. 1999. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell 3: 87–95. 10.1016/S1097-2765(00)80177-X [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. 2016. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740. 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, Barash Y. 2016. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5: e11752 10.7554/eLife.11752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Wang Y. 2010. Quantitative proteomic analysis reveals the perturbation of multiple cellular pathways in HL-60 cells induced by arsenite treatment. J Proteome Res 9: 1129–1137. 10.1021/pr9011359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Dodd T, He Y, Tainer JA, Tsutakawa SE, Ivanov I. 2019. Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat Struct Mol Biol 26: 397–406. 10.1038/s41594-019-0220-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Hoon S, Venkatesh B, Burge CB. 2004. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci 101: 15700–15705. 10.1073/pnas.0404901101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. 2019. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10: 1523 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been uploaded on GEO GSE151699. Proteomics data have been uploaded on the MassIVE repository with accession MSV000085575.