

EST12/Rv1579c activates a RACK1–NLRP3–GSDMD pyroptosis–IL-1β immune pathway to increase mycobacterial clearance.
Abstract
Pyroptosis, an inflammatory form of programmed cell death, has been implicated in eliminating pathogenic infections. However, macrophage pyroptosis–related proteins from Mycobacterium tuberculosis (M.tb) have largely gone unexplored. Here, we identified a cell pyroptosis–inducing protein, Rv1579c, named EST12, secreted from the M.tb H37Rv region of difference 3. EST12 binds to the receptor for activated C kinase 1 (RACK1) in macrophages, and the EST12-RACK1 complex recruits the deubiquitinase UCHL5 to promote the K48-linked deubiquitination of NLRP3, subsequently leading to an NLRP3 inflammasome caspase-1/11–pyroptosis gasdermin D–interleukin-1β immune process. Analysis of the crystal structure of EST12 reveals that the amino acid Y80 acts as a critical binding site for RACK1. An EST12-deficient strain (H37RvΔEST12) displayed higher susceptibility to M.tb infection in vitro and in vivo. These results provide the first proof that RACK1 acts as an endogenous host sensor for pathogens and that EST12-RACK1–induced pyroptosis plays a pivotal role in M.tb-induced immunity.
INTRODUCTION
Tuberculosis (TB), which is caused by Mycobacterium tuberculosis (M.tb), is the leading cause of death by an infectious disease worldwide. The World Health Organization reported that approximately 10.0 million new TB cases occurred in 2018 worldwide and that approximately one-quarter of the world’s population is infected with M.tb (1). A vaccine based on an attenuated live strain of Mycobacterium bovis, termed bacille Calmette-Guérin (BCG), is the only available TB vaccine and has limited protective efficacy in adults against TB. Because of the current lack of an effective vaccine, extensive drug-resistant M.tb, and side effects of toxic chemotherapeutic agents, further research into understanding the interaction between host and mycobacterium in the immune response is crucial.
Macrophages are the primary cellular niche for M.tb during infection. M.tb induces both a macrophage protective immune response and immune escape through multiple strategies during acute and chronic or latent infection (2). Gasdermin D (GSDMD), which mediates a regulated lytic cell death mode termed pyroptosis, is identified as a substrate for murine caspase-1 and caspase-11 and human caspase-1, caspase-4, and caspase-5 (3–6). Pyroptosis is a type of proinflammatory programmed cell death mediated by caspase-1–cleaved GSDMD (N-terminal fragment), which binds to the membrane to form membrane pores and promote the release of proinflammatory mediators, notably IL-1β. GSDMD and gasdermin E (GSDME) are two effectors of pyroptosis downstream from caspase activation, and GSDME is specifically cleaved by caspase-3 to induce pyroptosis in both human and murine cells (3, 7). Recent studies have reported that pyroptosis is an important component of the innate immune response and plays an important role in eliminating pathogenic infections and endogenous risk signals (4). However, the susceptibility of mice lacking GSDMD or other gasdermin proteins to the infectious agents, especially M.tb, has not been investigated. The mechanism underlying the immune sensing mechanism of M.tb leading to pyroptosis of macrophages remains unknown.
Comparative genomic analysis has identified more than 100 coding sequences missing from BCG but present in the virulent strains of M.tb complex–specific genomic regions of deletion (RDs), designated RD1 to RD16 (8, 9). Recent evidence indicates that RDs may encode potential new functional antigens important for TB immunity and pathogenesis. For example, supplementation with RD antigens encoded by RD1, RD2, or RD13 enhances the protective efficacy of BCG in tuberculosis mice, and CFP10 (culture filtered protein 10) and ESAT-6 (6-kDa early secreted antigen target) are the most widely used biomarkers in the clinic for diagnosing TB (10–14). However, the functions of most RD-encoded proteins are largely unexplored to date. Therefore, it is important to study the functions of RD-coding proteins to increase TB immunity.
Here, we screened and identified macrophage pyroptosis–related proteins from M.tb H37Rv RD-encoded proteins. We found an RD3-encoded secreted protein Rv1579c that had a substantial pyroptotic effect on macrophages but limited or no effect on T cells. Rv1579c (molecular weight: 12 kDa, named EST12) and its critical amino acid Y80 directly interact with receptor for activated C kinase 1 (RACK1) and trigger macrophage pyroptosis. An EST12-deficient strain (H37RvΔEST12) displayed higher susceptibility to M.tb infection in vitro and in vivo. Our data demonstrate that the EST12-RACK1 interaction activates an NLRP3 (NACHT, LRR AND PYD domains-containing protein 3)–caspase-1/11–GSDMD–interleukin-1β (IL-1β) immune process and plays a pivotal role in M.tb-induced immunity. Our study highlights the role played by M.tb-EST12 in activating the host immune response to increase mycobacterial clearance.
RESULTS
Rv1579c/EST12 from M.tb H37Rv RD3 induced GSDMD-mediated pyroptosis of macrophages
Forty RD-encoded recombinant proteins were purified (Rv1579c, named EST12, 12 kDa, as an example shown in fig. S1A) and used to treat murine peritoneal macrophages by lactate dehydrogenase (LDH) release assay. We found an RD3-encoded protein, Rv1579c/EST12, as a secreted protein of H37Rv (fig. S1B), which had the most significant cytotoxic effect (>75%) on murine peritoneal macrophages at less than 3.5 μM among other RD-encoding proteins, such as Rv1577 and Rv3875 (ESAT6) (Fig. 1, A and B, and fig. S1C). The EST12 protein caused the death of murine peritoneal macrophages (Fig. 1, A and B), human monocytic THP-1–derived macrophages (Fig. 1C), and mouse bone marrow–derived macrophages (BMDMs) (Fig. 1D) in a dose- and time-dependent manner. Our data showed that EST12 had strong cytotoxicity effects on macrophages and BMDCs, as well as a minor cytotoxicity effect on neutrophils, but had limited or no cytotoxicity effects on CD4+ T and CD8+ T cells (fig. S1D), suggesting that EST12 has no direct effect on T cells. The addition of polymyxin B, a neutralizer of lipopolysaccharide (LPS) (15), had no effect on cell death induced by different concentrations of EST12 (fig. S1E), suggesting that the EST12 protein causes cell pyroptosis unrelated to LPS.
Fig. 1. Identification of a macrophage pyroptosis–inducing protein EST12.
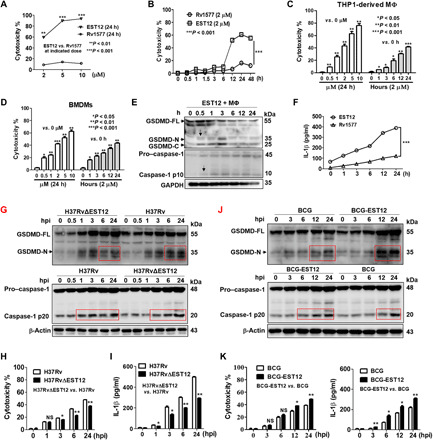
(A to D) Mouse peritoneal macrophages (A and B), PMA-differentiated THP1 cells (C), and BMDMs (D), treated with EST12 (A to D) and Rv1577 (A and B) as indicated, were analyzed by LDH release assay. Unpaired t test versus 0 hours or 0 μM. (E and F) Peritoneal macrophages treated with 2 μM EST12 or Rv1577 were detected for the GSDMD activation by WB (E) and IL-1β secretion by ELISA (F). Two-way ANOVA with Bonferroni’s multiple comparison test versus Rv1577 was used for comparison. (G to I) BMDMs infected with H37Rv or H37RvΔEST12 were detected for caspase-1/GSDMD activation by WB (G), cytotoxicity by LDH release assay (H), and IL-1β secretion by ELISA (I). Two-way ANOVA with Bonferroni’s multiple comparison test versus H37Rv was used. (J and K) BMDMs infected with BCG and BCG-EST12 were detected for caspase-1/GSDMD activation by WB (J) and cytotoxicity by LDH and IL-1β secretion by ELISA (K). Two-way ANOVA with Bonferroni’s multiple comparison test versus BCG was used. The data are expressed as the mean ± SEM (n = 3); NS, not significant (P > 0.05), *P < 0.05, **P < 0.01, or ***P < 0.001. The anti–caspase-1 p10 (E and F) and p20 (G to J) pAbs (polyclonal antibodies) were used.
The gene product of the open reading frame of EST12 in M.tb H37Rv is annotated as a conserved hypothetical protein in the database (https://mycobrowser.epfl.ch/), and its function has not been reported. EST12 was only detected in genomic DNA from the M.tb strains H37Rv and H37Ra but not in genomic DNA from BCG, Mycolicibacterium smegmatis (M. smeg), Mycobacterium intracellulare, Mycobacterium marinum, or Mycobacterium avium (fig. S1F).
To directly determine whether GSDMD and GSDME are involved in EST12-induced cell death, murine peritoneal macrophages were stimulated with 2 μΜ EST12 for different time periods, and cell lysates were probed for active cleavage products of GSDMD and GSDME. We observed that full-length GSDMD (55 kDa) was clearly released as the cleaved GSDMD–N-terminal (35 kDa) and –C-terminal (25 kDa) domains after treatment with 2 μΜ EST12 for 0.5 hours in mouse peritoneal macrophages (Fig. 1E), as well as an increase in activation of the cleavage of the p20 fragment of caspase-1 after a 0.5-hour treatment (Fig. 1E) and increased secretion of the proinflammatory cytokine IL-1β of macrophages by EST12 (Fig. 1F), suggesting that EST12 induced inflammatory pyroptosis of the macrophages. However, caspase-3 and GSDME were not activated for cleavage by EST12 stimulation (fig. S1G), suggesting that caspase-3 and GSDME are not involved in EST12-induced pyroptosis in mouse macrophages. We also investigated whether EST12-induced human macrophage pyroptosis is mediated by GSDMD. Human THP-1 monocyte-derived macrophages were further treated with EST12, and we found that caspase-1 and GSDMD were cleaved by EST12 (fig. S1H); however, the cleavage of caspase-4/5 and caspase-3/GSDME was not observed (fig. S1I). Thus, the pyroptosis induced by EST12 is mediated by caspase-1/GSDMD activation in both mouse and human macrophages but not by caspase-3 and GSDME.
An EST12-deficient H37Rv strain (H37RvΔEST12) was successfully constructed by the homologous recombination method (fig. S1J) and evaluated by Western blot (WB) (fig. S1K), and the growth of H37RvΔEST12 showed no difference compared with wild-type (WT) H37Rv (fig. S1L). Then, we detected the activation of caspase-1 and GSDMD of macrophages infected with H37Rv and H37RvΔEST12 (Fig. 1G); the results showed that there was no significant change between the H37Rv∆EST12 group and the H37Rv group within 1 hour post infection (hpi). However, H37Rv induced more cleaved caspase-1 p20 and GSDMD–N-terminal fragments in BMDMs after 6 hours of stimulation compared with H37Rv∆EST12 (Fig. 1G). Similar results showed that lower levels of LDH release and inflammatory cytokine IL-1β were observed in the BMDMs of the H37RvΔEST12 group than in those of the H37Rv group (Fig. 1, H and I), suggesting that H37RvΔEST12 caused less inflammatory pyroptosis compared with the H37Rv group. BCG-EST12 consistently induced more cleaved caspase-1 p20 and GSDMD–N-terminal fragment in BMDMs (Fig. 1J) and much higher cytotoxicity and IL-1β secretion of BMDMs compared with WT BCG (Fig. 1K). Moreover, BCG infection induced a lower level of IL-1β compared with M.tb (fig. S1M). The above data suggest that recombinant BCG-EST12 or H37Rv could induce significantly higher levels of IL-1β and cytotoxicity compared with BCG. Similarly, we also found that M. smeg–EST12 induced much higher cytotoxicity and IL-1β secretion of BMDMs compared with WT M. smeg (fig. S1N). All the above data strongly demonstrate that EST12 triggers GSDMD-mediated pyroptosis and inflammatory cytokine IL-1β secretion in macrophages.
EST12 induces macrophage inflammatory pyroptosis through interaction with RACK1
Next, to search for potential cellular EST12-associated proteins in macrophages, we performed pull-down assays with macrophage membrane proteins or cytoplasmic proteins and EST12-His-Ni-agarose beads, followed by mass spectrometry (MS) analysis (fig. S2A and table S1). We identified the RACK1 protein, which had the highest protein score according to liquid chromatography–tandem MS (LC-MS/MS) analysis, as the most likely host binding partner for EST12 (table S1). The binding of EST12 to RACK1 was further confirmed by immunoblotting, in which both macrophage cytoplasmic and membrane proteins, especially major cellular cytoplasmic proteins, were blotted with anti-RACK1 after the addition of and pulldown by the Ni-agarose-His-EST12 protein (Fig. 2A). We also transfected EST12–green fluorescent protein (GFP) and RACK1-Red expression plasmids into macrophage RAW264.7, and confocal fluorescence microscopy showed that EST12 and RACK1 colocalized in the cytoplasm, producing a yellow color (Fig. 2B). Furthermore, reverse transcription–quantitative real-time polymerase chain reaction (RT-qPCR) (Fig. 2C, top) and WB (Fig. 2C, bottom) analysis revealed that EST12-stimulated macrophage RACK1 expression, which peaked at 6 hours and then decreased (Fig. 2C), suggesting that EST12 promotes RACK1 expression at early stage.
Fig. 2. EST12 induces macrophage pyroptosis through interaction with RACK1.
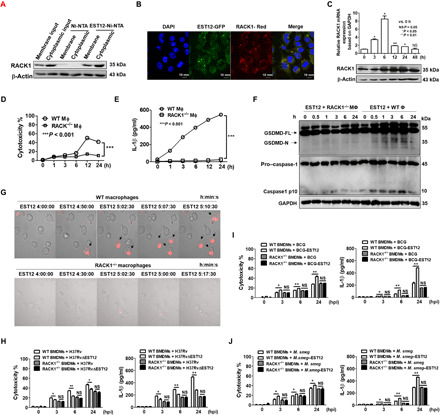
(A) An EST12-His pull-down assay was performed with RAW264.7 cells and analyzed by WB. (B) RAW264.7 cells transfected with pEGFP-C1-EST12 and pAsRED2-C1-RACK1, respectively, were analyzed by confocal microscopy. (C) RAW264.7 cells stimulated with 2 μM EST12 as indicated were analyzed by WB (top) and RT-qPCR (bottom). Unpaired t test, versus 0 hours was used for analysis. (D to F) Both WT and RACK1−/− peritoneal macrophages were treated with 2 μM EST12; supernatants were analyzed by LDH and ELISA (D and E), while cell lysates were analyzed for GSDMD and caspase-1 by WB (F). Two-way ANOVA with Bonferroni’s multiple comparison test versus WT MΦ. (G) Both WT and RACK1−/− peritoneal macrophages treated with 2 μΜ EST12 and PI staining were imaged using a Zeiss LSM 880. (H to J) Both WT and RACK1−/− BMDMs infected with H37Rv or H37RvΔEST12 (H), BCG or BCG-EST12 (I), and M. smeg or M. smeg–EST12 (J), respectively, were evaluated by LDH and ELISA. Unpaired t test, H37RvΔEST12 versus H37Rv (H), BCG-EST12 versus BCG (I), M. smeg–EST12 versus M. smeg (J). The data are expressed as the mean ± SEM, n = 3; NS (P > 0.05), *P < 0.05, **P < 0.01, or ***P < 0.001.
To study the role of the RACK1-EST12 interaction in macrophages, we generated a macrophage-specific conditional knockout mouse model of RACK1 (named the RACK1ΔMΦ mouse) (fig. S2B). WB was used to analyze the expression and translation of RACK1, respectively, in different cells from RACK1ΔMϕ mice, and the results showed that RACK1 was successfully and specifically knocked out in macrophages but neither in B cells nor in T cells (fig. S2C). In addition, the hair color and body movement of RACK1ΔMΦ mice were observed to be similar to those of WT mice.
Peritoneal macrophages collected from WT mice and RACK1ΔMΦ mice were treated with 2 μM EST12 for different amounts of time (from 0 to 24 hours). EST12-induced cytotoxicity (Fig. 2D) and IL-1β secretion (Fig. 2E) were significantly decreased in RACK1−/− peritoneal macrophages relative to WT macrophages, and the cleavage of GSDMD N fragment (Fig. 2F, top) and caspase-1 p10 (Fig. 2F, bottom) induced by EST12 were strongly suppressed in RACK1−/− macrophages compared with WT macrophages. Real-time confocal microscopy analysis showed that EST12 triggered peritoneal macrophages to display a typical morphology of cell pyroptosis: plasma membrane disruption, multiple bubble-like protrusions, and the appearance of a “fried egg” within the cell’s nucleus located above the plane of the cell body (Fig. 2G, top, and movie S1). Furthermore, real-time confocal microscopy observation of RACK1−/− macrophages treated with EST12 indicated very little propidium iodide (PI) uptake during the observation period (Fig. 2G, bottom, and movie S2). EST12 induced phosphorylation of inhibitor of nuclear factor κB alpha (IκBα), nuclear factor κB (NF-κB) p65 subunit, and c-Jun N-terminal kinase, leading to up-regulation of total c-Jun expression and activation of c-Jun by phosphorylation (fig. S2D). EST12 also significantly increased Lys63 (K63)–linked ubiquitination of NF-κB essential modulator [NEMO, also known as IκB kinase–γ (IKK-γ)], which is involved in IKK complex recruitment for NF-κB activation (fig. S2E). The secretion of IL-1β induced by EST12 was decreased markedly in the presence of PDTC (ammonium pyrrolidine dithiocarbamate) (NF-κB inhibitor) and curcumin [AP-1 (activating protein-1) inhibitor] (fig. S2F). In contrast, the ability of EST12 to activate the NF-κB and AP-1 pathways was markedly reduced in RACK1−/− peritoneal macrophages by WB analysis (fig. S2D).
Furthermore, the pyroptosis and IL-1β secretion of WT BMDMs by H37RvΔEST12 infection showed no differences compared with H37Rv infection in RACK1−/− BMDMs (Fig. 2H). No difference in cytotoxicity or IL-1β secretion was observed between BCG-EST12 and WT BCG in RACK1−/− BMDMs (Fig. 2I). Similar results were observed between M. smeg–EST12 and WT M. smeg in RACK1−/− BMDMs (Fig. 2J). The above data strongly suggest that EST12 causes macrophage inflammatory pyroptosis via RACK1.
We further determined that cytotoxicity was markedly decreased after the addition of cytochalasin D, an endocytosis inhibitor (fig. S2G) (16), suggesting that the cytotoxic effect of EST12 on murine peritoneal macrophages was largely relative to endocytosis. Macrophages treated with EST12 for 1 to 6 hours of stimulation were further analyzed by confocal fluorescence microscopy analysis using phycoerythrin (PE)–labeled anti-EST12 (red color) and 4′,6-diamidino-2-phenylindole (DAPI) nuclear dye (blue color) staining, and the results showed that EST12 was internalized and migrated from the extracellular space to the intracellular space of the macrophages (fig. S2H). We also observed that EST12-GFP was colocalized with EEA1 (early endosome antigen marker), LAMP1 (lysosomal-associated membrane protein 1, a lysosomal marker), and large multifunctional peptidase 2 (LMP2) (large mulifunctional peptidase 2), but not with calnexin (an endoplasmic reticulum membrane marker) (fig. S2I). These results suggest that endocytosed EST12 was located in the phagosome and then released from the phagosome into the cytosol, where it encountered cytoplasmic RACK1.
The EST12 C terminus and Y80 are the critical sites for RACK1 binding
To determine the precise binding site of the EST12 protein for RACK1, we successfully solved the crystal structure of EST12, which clearly showed the presence of three α helices and two β strands. The resolution of the crystal structure of EST12 is 1.9 Å (Fig. 3A). The crystal diffraction data and refinement statistics of the EST12 protein are shown in table S2 and the Protein Data Bank (PDB) x-ray structure validation report (data file S1). We analyzed the interaction sites of EST12 and RACK1 through the prediction of the complex structure by the Rosetta server (17–19). Cartoon representations of EST12 (magenta) contain three α helices and two β strands. The structure of RACK1 (cyan) was presented as previously described (PDB code: 4AOW). The potential interface of EST12 is indicated with green (E55), blue (F76), and cyan (Y80) spheres, with the posterior two sites being located on the third α helix (C-terminal fragment, last 24 amino acids on the third helix) of the EST12 protein, which is flexible (Fig. 3B). Hence, the interaction of EST12 with RACK1 may depend on the third α helix of EST12 (Fig. 3B).
Fig. 3. EST12 Y80 is a critical binding site for RACK1.
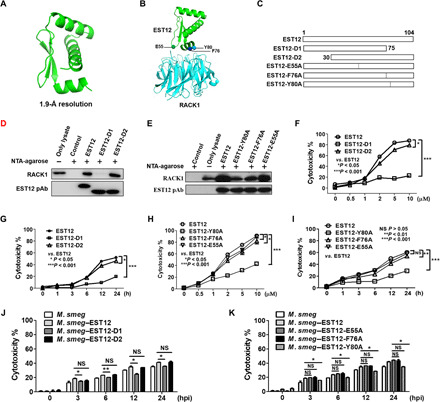
(A) The crystal structure of EST12 was solved at 1.9-Å resolution. (B) The predicted structure of the EST12-RACK1 complex using the Rosetta server. (C) Schematic representation of truncated and mutant recombinant proteins: EST12 C-terminal deletions (EST12-D1), N-terminal deletions (EST12-D2), and the three mutants of EST12 (EST12-E55A, EST12-F76A, and EST12-Y80A). (D and E) Recombinant His-fusion proteins (EST12-D1 and EST12-D2) (D) and (EST12-E55A, EST12-F76A, and EST12-Y80A) (E) coupled with Ni-agarose beads for pulldown were incubated with RAW264.7 cells and analyzed by WB using anti-RACK1, anti-His, or anti-EST12. (F to I) Peritoneal macrophages treated with EST12/EST12-D1/EST12-D2 (F and G) or EST12/EST12-E55A/EST12-F76A/EST12-Y80A (H and I) at different doses (F and H) and for different periods of time (G and I), as indicated, were analyzed for LDH assay. Two-way ANOVA with Bonferroni’s multiple comparison test versus EST12 was used. (J and K) BMDMs infected with M. smeg and M. smeg–EST12/EST12-D1/EST12-D2 (J) or M. smeg–EST12/EST12-E55A/EST12-F76A/EST12-Y80A (K) were evaluated by LDH assay. Unpaired t test versus M. smeg–EST12 was used for analysis. The data are expressed as the mean ± SEM of n = 3 cultures. NS (P > 0.05), *P < 0.05, **P < 0.01, and ***P < 0.001.
To further analyze the interaction between EST12 and RACK1, we introduced the E55A, F76A, and Y80A mutations into EST12, producing the D1 and D2 EST12 deletion proteins (Fig. 3C). We performed pull-down assays with RAW264.7 cell lysates using WT/mutated/deleted-EST12-His fusion proteins conjugated to Ni-agarose beads, followed by immunoblotting analysis. Compared with WT EST12 and EST12-D2 proteins, the C-terminal deficient EST12 (EST12-D1) barely interacted with RACK1 (Fig. 3D); three mutants (Y80A, F76A, and E55A), especially Y80A and F76A, showed a significant decrease in binding to RACK1, compared with EST12-RACK1 interaction (Fig. 3E). The cytotoxic effects of EST12-D1 were also significantly decreased compared with EST12 in a dose- and time-dependent manner (Fig. 3, F and G). Compared with EST12, the cytotoxic effects induced by EST12-E55A/F76A/Y80A were reduced in a dose- and time-dependent manner, with EST12-Y80A in particular decreased the most (Fig. 3, H and I). These results strongly suggest that both theY80 and F76 sites of EST12 are obviously involved in the binding between EST12 and RACK1, and the Y80 is the most critical site for EST12-induced cytotoxicity. Consistently, the pyroptosis induced by the M. smeg–EST12-D1 and M. smeg–EST12-Y80A infections was reduced compared with M. smeg–EST12 (Fig. 3, J and K), suggesting that the EST12 C terminus and Y80 are the critical functional sites for EST12. Together, our results indicate that Y80 at the EST12 C terminus is a critical binding site for RACK1 that leads to pyroptosis.
EST12-RACK1 triggers macrophage pyroptosis and IL-1β secretion via NLRP3 and GSDMD
Inflammasome activation is a key defense mechanism against bacterial infection that induces innate immune responses, such as caspase-1 activation and inflammatory cell death (20). To determine the downstream molecules that are activated after EST12 stimulation, RAW264.7 cells were treated with the EST12 protein for 3 hours and then analyzed by mRNA microarrays (fig. S3A). Data from the mRNA microarrays showed that NLRP3, IL-1β, IL-6, and tumor necrosis factor–α (TNF-α) expression were up-regulated. Among the eight mouse PYRIN-domain proteins (21, 22) that were tested (NLRP1b, NLRP2, NLRP3, NLRP5, NLRP6, NLRP9, NLRP12, and PYRIN) by treatment with EST12, only NLRP3 mRNA expression was significantly altered in macrophages, while mRNA expression of NLRP1b, NLRP2, NLRP3, NLRP6, NLRP12, and Pyrin was unchanged, and mRNA expression of NLRP6 and NLRP9 was undetectable (Fig. 4A). Immunoblotting analysis confirmed that protein expression of NLRP3 was the most up-regulated and reached a peak 6 hours after EST12 stimulation (Fig. 4B). Confocal microscopy analysis showed that the expression of NLRP3 was the highest at 6 hours after EST12 stimulation (Fig. 4C). NLRP3 expression was markedly impaired in both RACK1−/− (Fig. 4D) after EST12 treatment. These results demonstrate that EST12 causes NLRP3 expression dependent on RACK1.
Fig. 4. EST12 induces NLRP3 inflammasome activation via RACK1.
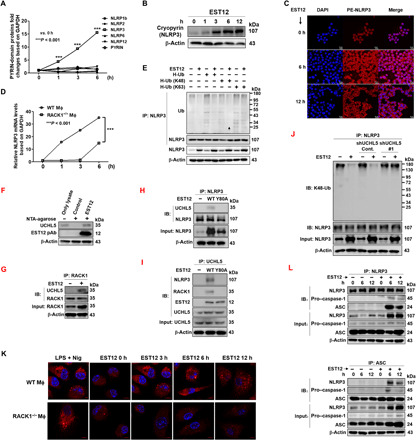
(A) Eight PYRIN-domain–containing protein mRNA expression levels in peritoneal macrophages pretreated with EST12 were analyzed by RT-qPCR. Unpaired t test. (B and C) RAW264.7 cells stimulated with EST12 were detected by WB (B) and confocal microscopy (C). (D) Both WT and RACK1−/− peritoneal macrophages were treated with EST12, and NLRP3 mRNA expression was analyzed by RT-qPCR. Two-way ANOVA with Bonferroni’s multiple comparison test was used. (E) RAW264.7 cells transfected with the indicated plasmids and then stimulated with EST12 were subjected to IP with anti-NLRP3 and WB analysis. (F) His-EST12 pull-down assay with the lysates of BMDMs was analyzed by WB. (G to J) BMDMs treated with EST12 were immunoprecipitated with anti-RACK1 (G), anti-NLRP3 (H and J), and anti-UCHL5 (I) and analyzed by immunoblotting (IB). (K) Both WT and RACK1−/− peritoneal macrophages treated with EST12 were analyzed by confocal microscopy with anti-ASC (red) and nuclear staining with DAPI (blue). (L) Peritoneal macrophages pretreated with or without EST12 were subjected to IP using anti-NLRP3 and anti-ASC and IB analysis. The data are expressed as the mean ± SEM of n = 3 cultures. ***P < 0.001.
Recent findings have shown that priming rapidly stimulates mouse NLRP3 inflammasome activation by inducing the activation of NF-κB and the deubiquitination of NLRP3 (23). In the above, we determined the activation of NF-κB p65 after stimulation with EST12 (fig. S2D). Here, we transfected RAW264.7 cells with the plasmids encoding WT ubiquitin (H-Ub), the ubiquitin mutant H-Ub (K48), and H-Ub (K63), and found that NLRP3 ubiquitination was decreased in the presence of EST12 (Fig. 4E) and that EST12 promoted NLRP3 deubiquitination in the presence of H-Ub (K48), suggesting that EST12 mediates K48-linked deubiquitination of NLRP3 (Fig. 4E). We observed similar results by confocal microscopy analysis that NLRP3 protein expression was markedly increased while K48-linked ubiquitination was reduced in BMDMs after EST12 stimulation for 6 to 12 hours compared with those in untreated BMDMs (fig. S3B). Moreover, EST12-Y80A lost its effect on K48-linked deubiquitination of NLRP3 (fig. S3C), indicating that the increase of NLRP3 deubiquitination after EST12 stimulation was dependent on EST12-Y80.
To investigate EST12-related deubiquitinase, we analyzed the results from MS [EST12-6×His-Ni-NTA (nitrilotriacetic acid) agarose pulldown of EST12-treated RAW 264.7 cell lysates and, subsequently, of MS analysis] (fig. S2A); we found a deubiquitinase-UCHL5 (ubiquitin carboxyl-terminal hydrolase isozyme L5), which belongs to the ubiquitin C-terminal hydrolases (UCHs) family, as one of the candidates for interacting with EST12. We further confirmed the interaction between the deubiquitinase-UCHL5 and EST12 by EST12-6×His-Ni-NTA pulldown of EST12-treated RAW 264.7 cell lysates and, subsequently, WB analysis with anti-UCHL5 (Fig. 4F). Furthermore, through co-immunoprecipitation (IP) analysis, we observed that both NLRP3 and RACK1 directly interacted with UCHL5 after EST12 stimulation (Fig. 4, G to I), and EST12-Y80A stimulation could not promote the interaction of RACK1 with UCHL5 and NLRP3 (Fig. 4, H and I). However, UCHL5 did not interact with NLRP3 or with EST12 in RACK1-deficient macrophages upon EST12 stimulation (fig. S3D), suggesting that RACK1 could serve as a scaffold protein or a bridge between EST12 and UCHL5-NLRP3. Knockdown of UCHL5 by its specific short hairpin RNA (shRNA) markedly decreased the K48 deubiquitination of NLRP3 after EST12 stimulation in RAW264.7 cells for 6 hours (Fig. 4J and fig. S3E), suggesting that UCHL5 mediates K48 deubiquitination of NLRP3 after EST12 stimulation. RACK1 protein expression was up-regulated at 24 hours and then decreased after EST12 stimulation (Fig. 2C), and a co-IP assay showed that EST12 stimulation for 24 hours induced RACK1 ubiquitinated at K48; however, knockdown of UCHL5 did not affect K48-linked ubiquitination of RACK1 with EST12 stimulation (fig. S3F). The above data strongly suggested that EST12 stimulates NLRP3 activation by UCHL5 mediation of the K48-linked deubiquitination of NLRP3.
Inflammasomes are multiprotein complexes that are often composed of intracellular NOD (nucleotide-binding oligomerization domain)–like receptors (NLRs), which are apoptosis-associated speck-like proteins that contain a caspase recruitment domain (ASC) and caspase-1 precursor (24). A hallmark of inflammasome activation is the ASC speck, a micrometer-sized structure formed by the inflammasome adaptor protein ASC (24, 25). We performed immunofluorescence assays to investigate ASC speck formation in peritoneal macrophages after treatment with 2 μΜ EST12 and with LPS (1 μg/ml) for 4 hours, followed by 20 μΜ nigericin treatment for 45 min (LPS + Nig) as a positive control of NLRP3 inflammasome activation. We observed ASC speck formation in the presence of EST12 but not in the EST12-treated RACK1−/− peritoneal macrophages (Fig. 4K) or in the truncated EST12-D1–treated group (fig. S3G). Through IP and WB experiments, we identified that NLRP3 could precipitate both ASC and pro–caspase-1 (Fig. 4L, top). ASC could also precipitate NLRP3 and pro–caspase-1, as shown in Fig. 4L (bottom). These data strongly suggest that these three components (NLRP3, ASC, and pro–caspase-1) can be assembled as an NLRP3 inflammasome after EST12 treatment.
It has been reported that activation of the NLRP3 inflammasome in response to diverse stimuli requires Ca2+ influx and mitochondrial reactive oxygen species (mtROS) accumulation (26). Similarly, we found that the levels of mtROS and intracellular Ca2+ in WT BMDMs were increased and significantly higher compared with those in RACK1−/− BMDMs after EST12 treatment (fig. S3, H and I). Together, our results show that the EST12-RACK1 complex induces the activation of NLRP3 inflammasomes.
The cytotoxic effect of peritoneal macrophages (Fig. 5, A and B) and BMDMs (fig. S4, A and B) from GSDMD−/− mice or NLRP3−/− mice after EST12 treatment was markedly decreased compared with those from WT mice, regardless of different times or dosages. The secreted IL-1β was undetectable in the supernatants of both peritoneal macrophages (Fig. 5C) and BMDMs (fig. S5C) from GSDMD−/− or NLRP3−/− mice after stimulation with the EST12 protein for different periods of time. Moreover, we investigated the effects of EST12 on caspase-1 and GSDMD cleavage in NLRP3−/− peritoneal macrophages and found that both showed no activation effect (Fig. 5D). GSDMD was not cleaved by EST12-Y80A treatment for different periods of time in WT macrophages (Fig. 5E), which suggests that EST12-Y80 triggers macrophage GSDMD-mediated pyroptosis and IL-1β secretion via NLRP3.
Fig. 5. EST12 triggers macrophage pyroptosis and IL-1β secretion via NLRP3 and GSDMD.
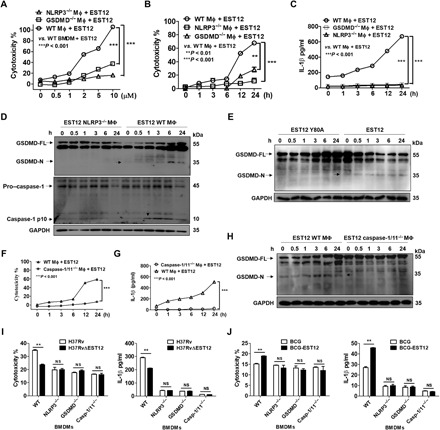
(A to C) WT, NLRP3−/−, and GSDMD−/− peritoneal macrophages treated with EST12 at the indicated doses (A) or for the indicated time periods (B and C) were analyzed for cytotoxicity by LDH assay (A and B) and IL-1β secretion by ELISA (C). Two-way ANOVA with Bonferroni’s multiple comparison test. (D) WT and NLRP3−/− peritoneal macrophages treated with EST12 were detected for GSDMD and caspase-1 activation by WB. (E) Peritoneal macrophages treated with EST12 and EST12-Y80A were detected for GSDMD activation by WB. (F to H) WT and caspase-1/11−/− peritoneal macrophages treated with EST12 were detected for LDH assay (F), secreted IL-1β by ELISA (G), and GSDMD activation by WB (H). Two-way ANOVA with Bonferroni’s multiple comparison test was used. (I and J) WT, NLRP3−/−, GSDMD−/−, and caspase-1/11−/− BMDMs infected with H37Rv and H37RvΔEST12 (I) or BCG and BCG-EST12 for 6 hours (J) were analyzed by LDH assay and ELISA. Unpaired t test versus H37Rv (I) or BCG (J). The data are expressed as the mean ± SEM of n = 3. NS (P > 0.05), **P < 0.01, and ***P < 0.001.
Peritoneal macrophages were pretreated with Z-VAD-FMK (a pancaspase inhibitor), belnacasan (VX-765) (a caspase-1 inhibitor), Z-DEVD-FMK (a caspase-3 inhibitor), or KCl (an NLRP3 inflammasome inhibitor). Compared with the control groups (the macrophages-only and dimethyl sulfoxide–treated macrophages groups), all of the inhibitors blocked EST12-induced cytotoxicity except for Z-DEVD-FMK (which inhibited caspase-3–mediated apoptosis) (fig. S4D). These data verify that EST12 triggers caspase-1–mediated pyroptosis and NLRP3 inflammasome activation but not caspase-3–mediated apoptosis.
In addition, the cytotoxicity (Fig. 5F) and IL-1β secretion (Fig. 5G) were obviously reduced after EST12 treatment in the caspase-1/11−/− peritoneal macrophages and the caspase-1/11−/− BMDMs (fig. S4, E and F) relative to the WT BMDM group, and no cleavage of GSDMD was observed in caspase-1/11−/− macrophages (Fig. 5H), suggesting that EST12 triggers GSDMD-mediated pyroptosis and IL-1β secretion of macrophages via caspase-1. Furthermore, no differences in the cytotoxicity or IL-1β secretion were observed between H37Rv and H37RvΔEST12 in NLRP3−/−, GSDMD−/−, and caspase-1/11−/− BMDMs, except in WT BMDMs (Fig. 5I). Similar results were observed between BCG-EST12 and BCG (Fig. 5J) and between M. smeg–EST12 and M. smeg (fig. S4G) in NLRP3−/−, GSDMD−/−, and caspase-1/11−/− BMDMs. All the above results strongly demonstrate that EST12-Y80 triggers macrophage pyroptosis and IL-1β secretion via RACK1, NLRP3, GSDMD, and caspase-1.
EST12 activates the host immune response to increase mycobacterial clearance
The pyroptosis of infected macrophages is essential for activation of the host’s immune system during a bacterial infection. Thus, we used H37Rv infection mouse models to evaluate the role of M.tb EST12. Both M.tb H37Rv and H37RvΔEST12 strains were used to infect C57BL/6 mice via intranasal administration. A total of 5 × 104 CFUs (colony-forming units) of M.tb/mouse (intranasal infection) (27, 28) was used in our acute infection mouse model. After infection for 30 days, significantly higher lung bacterial CFUs and lower serum IL-1β levels were observed in the H37RvΔEST12 group relative to those of the H37Rv group by bacterial colony counting and acid-fast staining (Fig. 6, A and C) as well as by enzyme-linked immunosorbent assay (ELISA) (Fig. 6B). Histopathological analysis of alveolar tissue from the H37RvΔEST12 infection group showed increased lymphocyte infiltration and smaller intact alveolar spaces compared with the H37Rv group (Fig. 6D).
Fig. 6. EST12 activates the host immune response to increase mycobacterial clearance.
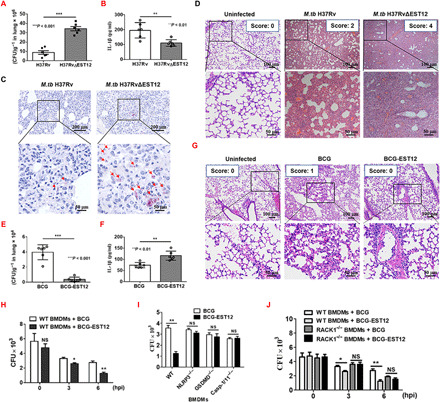
(A to D) WT mice (n = 6 per group) were infected with H37Rv and H37RvΔEST12 intranasally. On day 30 after infection, the mice were euthanized and evaluated for lung CFUs (A) and serum IL-1β by ELISA (B). Unpaired t test. (C) Lung tissue sections from WT mice infected with H37Rv and H37RvΔEST12 were analyzed with acid-fast stain. The arrows indicate acid-fast staining positive bacteria. (D) Lung tissue sections in the H37Rv and H37RvΔEST12 groups stained with hematoxylin and eosin (H&E) were evaluated by light microscopy. (E and F) WT mice (n = 6 per group) were challenged with BCG and BCG-EST12 intranasally. Lung CFUs were enumerated (E) and serum IL-1β was measured by ELISA (F). Unpaired t test. (G) Lung tissue sections in the BCG or BCG-EST12 groups stained with H&E were evaluated by light microscopy. (H to J) WT (H) and RACK1−/− (J) BMDMs infected with BCG and BCG-EST12 for the indicated time (H and J) or WT, NLRP3−/−, GSDMD−/−, and caspase-1/11−/− BMDMs (I) infected for 6 hours were enumerated for CFUs. The data are expressed as the mean ± SEM. NS (P < 0.05), *P < 0.05, **P < 0.01, and ***P < 0.001.
Consistently, BCG-EST12 or M. smeg–EST12 caused lower bacterial loads in the lung or spleen tissues and higher levels of serum IL-1β in WT mice compared with BCG (Fig. 6, E and F) or M. smeg after infection (fig. S5, A to C). Histopathological analysis of alveolar tissue from the BCG infection group showed more serious injuries compared with the BCG-EST12 group according to the histology score (Fig. 6G).
Moreover, we counted CFUs in WT BMDMs after infection with different bacteria, as described in Materials and Methods. We observed that BCG-EST12 and M. smeg–EST12 infections led to fewer CFUs at 3 and 6 hpi in WT BMDMs compared with the BCG (Fig. 6H) and M. smeg groups (fig. S5D). No differences in CFUs in RACK1−/−, NLRP3−/−, GSDMD−/−, and caspase-1/11−/− BMDMs after infection between BCG-EST12 and BCG (Fig. 6, I and J) and M. smeg–EST12 and M. smeg were found (fig. S5, E and F). Together, the above results strongly suggest that EST12 increases resistance to M.tb infection in both macrophages and mice, and it plays a pivotal role in M.tb-induced immunity via RACK1-, NLRP3-, GSDMD-, and caspase-1/11–mediated inflammatory pyroptosis (fig. S6).
DISCUSSION
The interaction between bacteria and host has led to the evolution of diverse mechanisms for bacteria to overcome attack by host immune cells and for host cells to sense and initiate anti-M.tb responses, including the capacity to induce host cell death programs, such as macrophage pyroptosis. Pyroptosis is critical for host defenses against infection and danger signals, and excessive pyroptosis causes immunological diseases and septic shock. Thus, pyroptosis might be a double-edged sword that is critical for both host defense and immunological diseases.
Emerging evidence indicates that cellular GSDMD-induced pyroptosis contributes to immune defense against various bacterial and viral infections, including the intracellular bacteria Salmonella, Bacillus, Listeria, Shigella, and Legionella (29–32). However, susceptibility profile of mice lacking GSDMD or other gasdermin proteins to infectious agents, especially M.tb, has not been investigated, and the mechanism by which M.tb, especially M.tb RD-encoded proteins, manipulates host cell pyroptosis remains elusive.
Here, we performed a screen analysis of 40 M.tb RD-encoded recombinant proteins that initiated macrophage pyroptosis. We found that the Rv1579c (EST12) protein from the H37Rv RD3 region induced pyroptosis of macrophages through an interaction with cytoplasmic RACK1 protein. EST12 binds to RACK1 and activates NF-κB/AP1 (fig. S2D) in macrophages, which subsequently leads to an NLRP3 inflammasome–caspase-1–pyroptosis GSDMD–IL-1β immune pathway (fig. S6). Moreover, EST12 activated both human and mouse caspase-1 and GSDMD to induce the pyroptosis of macrophages but did not activate either human or mouse caspase-3/GSDME or human caspase-4/5. EST12 increased resistance to M.tb infection in both macrophages and mice and played a pivotal role in M.tb-induced immunity (fig. S6). Our present study elucidates the macrophage pyroptosis induced by EST12 and its mechanism in anti-TB innate immunity at both cellular and animal levels. There might also be the contribution of the other mechanism by EST12 stimulation such as increased Ca2+ influx and mtROS accumulation (fig. S3, I and J). Our findings expand the understanding of host cell pyroptosis/death and interactions between the host cell and mycobacterial effectors.
RACK1 has been reported to serve as a scaffold protein for many kinases and receptors and plays a pivotal role in a wide range of biological responses, including signal transduction, tumor cell invasion, angiogenesis, and immune responses (33–35). However, the role of RACK1 during pathogenic mycobacterial infection has remained largely unknown. Our study indicates that RACK1 is involved in M.tb-mediated pyroptosis and immunity. Our findings show that the recognition and binding of M.tb EST12 to RACK1 contributes to the pyroptosis of macrophages. The EST12 of M.tb H37Rv promotes resistance to M.tb infection and IL-1β secretion in mice dependent on RACK1. Our data delineate the mechanism underlying the M.tb EST12-RACK1 interaction directing NLRP3 inflammasome–mediated pyroptosis.
There was only a very weak GSDMD-N cleavage at 24 hours in WT macrophages after EST12 treatment as measured by WB analysis (Fig. 2F), and we suspect that this might be due to the degradation of EST12 after 24 hours. Nevertheless, EST12 still induced RACK1-dependent cytotoxicity at 24 hours (Fig. 2D), although there was less GSDMD cleaved at 24 hours. The reason might be that the gasdermin pores, formed by the previously cleaved GSDMD N terminus, might still lead to elicit cell death (36).
Similarly, the RACK1 signaling scaffold protein interacts with several other bacterial proteins, including Helicobacter pylori VacA cytotoxin and Yersinia pseudotuberculosis (37, 38). Inhibition of RACK1 does not affect Drosophila or human cell viability and proliferation, but it is an essential determinant of hepatitis C virus translation and infection (39). RACK1 also promotes the progression and invasion of several cancers (40). RACK1 has been identified as a virus-interacting host factor in promoting their replication. During infectious bursal disease virus infection, RACK1 forms a complex with VDAC2 (voltage-dependent anion channel 2) and VP5 (viral protein 5) to inhibit apoptosis and enhance viral replication (41); RACK1 and the CCT (chaperonin TRiC) and OST (oligosaccharyltransferase) complexes are required for dengue virus replication and play distinct roles in the viral life cycle (42). More detailed study of molecular mechanisms and structural analysis of RACK1 would be important for providing a target for TB vaccine and drug development.
Several inflammasome activators have been shown to induce pyroptosis. For example, NLRC4 indirectly senses bacterial flagellin and type III secretion systems and responds by assembling an inflammasome complex that promotes caspase-1 activation and pyroptosis (43). Different PRRs (pattern recognition receptors) are involved in the recognition of M.tb and can induce the activation of different inflammasomes in macrophages, mostly NLRP3 and AIM2 inflammasomes (44–47). However, few studies have shown that M.tb can induce downstream caspase-1–GSDMD activation of inflammasomes in vivo and in vitro. Our studies showed that M.tb EST12 induces macrophage pyroptosis by binding to RACK1 and activating NLRP3-ASC complex formation and the NLRP3–caspase-1–GADMD–IL-1β pathway. M.tb EST12-induced pyroptosis promoted resistance to M.tb infection and IL-1β secretion in mice. Previous studies have shown that IL-1β directly promotes antimicrobial immunity in murine and human macrophages (48, 49). We demonstrated that EST12 increased resistance to M.tb infection in macrophages and mice by activating a RACK1–NLRP3–gasdermin D pyroptosis–IL-1β immune pathway. Our data delineate the mechanism underlying M.tb EST12-RACK1 interaction–mediated NLRP3-GSDMD pyroptosis and the IL-1β secretion process.
NEMO is a key regulator in NF-κB–mediated signaling. K63-linked pUb (polyubiquitination) chain formation has been reported to be important for recruiting NEMO to activate signaling transduction (50). The EST12-RACK1 interaction induces NEMO K63 ubiquitination (fig. S2E) and NLRP3 K48 deubiquitination (Fig. 4, E and J, and fig. S3, C and D). The mRNA and protein expressions of NLRP3, which is located downstream of NEMO, were the most up-regulated and reached a peak at 6 hours after EST12 stimulation, suggesting that EST12 might play a role during early and acute infection. Deubiquitination of NLRP3 has been suggested to contribute to inflammasome activation (51). Ubiquitination plays an important role in modulating the stability and functions of various proteins. In our study, the EST12-RACK1 complex interacted with the deubiquitinase UCHL5 to mediate K48-linked deubiquitination of NLRP3. UCHL5 is associated with the 26S proteasome, where it serves to remove distal ubiquitin moieties from polyubiquitylated proteins. UCHL5’s proteasome-associated activity was shown to liberate proteins from destruction (52). A previous study reported that the role of UCHL5 is to remove K48-linked ubiquitin to prevent proteasomal degradation of ubiquitinated substrates (53). Consistently, the EST12-Y80 site mutant lost its induction effect on the NLRP3 deubiquitination by UCHL5 (fig. S3D). Collectively, RACK1 as a scaffold protein interacted with EST12-Y80, and it further recruited the deubiquitinase UCHL5 to promote the K48-linked deubiquitination of NLRP3.
Here, our data clearly demonstrated that EST12 caused macrophage pyroptosis by releasing the cleaved GSDMD–N-terminal domain. Furthermore, the Y80 amino acid of EST12 acts as a critical functional binding site with RACK1. Recent reports indicate a toxin-like behavior of M.tb MPT63 that results in membrane pore formation by toxic oligomers and macrophage cell death (54). The M.tb effector proline-proline-glutamate (PPE)60 regulates host cell pyroptosis and cytokine production through NF-κB signaling (55). Consistently, we also observed the cleaved GSDMD–N-terminal fragment by H37RvΔEST12 stimulation (Fig. 1G), and the differences in cytotoxicity and IL-1β secretion induced by H37Rv and H37RvΔEST12 (Fig. 1, H and I) accounted for ~15% of the effect, which indicates that EST12 may not be the only contributing factor for pyroptosis: There may be other components of M.tb H37Rv∆EST12 or H37Rv (e.g., MPT63 and PPE60) that could also induce the pyroptosis of macrophages in addition to EST12. Moreover, we found that BCG induced less pro–caspase-1 and GSDMD cleavage, secreted lower IL-1β, and exhibited less cytotoxicity with recombinant BCG-EST12 (Figs. 1, I and K, and 5J). BCG infection alone was able to induce the pyroptosis of macrophages, suggesting that some effectors of BCG may also cause IL-1β secretion and pyroptosis in macrophages (Fig. 1, I and K). Other reports have also shown that BCG could lead to an increased secretion of IL-1β and the expression of caspase-1 (56). We compared IL-1β levels between BCG and H37Rv and found that BCG was a much lower IL-1β secretion and pyroptosis inducer compared with M.tb H37Rv.
EST12 is represented as a secreted protein of M.tb H37Rv (fig. S1B). Amino acid sequence analysis has revealed that the EST12 protein contains the ESX-1 (ESAT-6 secretion systems 1)/type VII secretion (T7S) locus with the Y80xxxD/E motif. There are up to five mycobacterial T7S systems, named ESX-1 to ESX-5, of which the T7S substrates or substrate complexes are defined by the general secretion motif, YxxxD/E (57). However, according to the resolved crystal structure, the EST12 is characterized to have three α helices and two β strands, followed by the YxxxD/E motif, indicating that its structure might differ from the ESX secretion system proteins, which have a typical helix-turn-helix structure. Future investigations will be needed to identify the kind of mycobacterial secretion system to which EST12 belongs.
Numerous mycobacterial secreted factors [such as SapM (secretory acid phosphatase), CpsA, and ESAT-6], which regulates M.tb infection to escape phagosomes, have been identified (58–60), but it remains unclear which pathways are involved in the escape of M.tb components from phagosomes. The M.tb-secreted protein EST12 was endocytosed by the macrophages (fig. S2, H and I), and the endocytosed EST12 was then colocalized with early endosome, lysosome, and proteasome (fig. S2I). These results suggest that endocytosed EST12 was then possibly located in the phagosome and then possibly released from the phagosome into the cytosol, where it encountered the cytoplasmic RACK1. We speculate that the effect of EST12 could potentially be similar to how the mycobacterial ESX-1 cosecretes proteins CFP-10/ESAT-6, which are also released from the phagosome (61). However, the pathway of phagosomal release in which the mechanism of EST12 is involved needs to be identified in the future.
M.tb is an extremely successful intracellular pathogen that can interfere (either by activation or by inhibition) with host cellular functions (such as phagocytosis, autophagy, apoptosis, and inflammasome activation) to help the pathogen evade or circumvent the host’s innate immunity for its chronic infection. For example, the binding of M.tb mannose-capped lipoarabinomannan (ManLAM) to the mannose receptor is involved in the inhibition of phagosome maturation (62), and the interaction of ManLAM and Toll-like receptor 2 induces IL-10 production by B cells that facilitate mycobacterial survival in vivo (63). Other studies have also suggested that M.tb could activate host immunity through its proteins. M.tb Rv1468c promotes xenophagic clearance of M.tb in macrophages and decreases bacterial loads in mice (64); MPT53 and its disulfide oxidoreductase activity is required for M.tb to induce the host inflammatory cytokines, and the deletion of MPT53 causes higher bacterial burdens and exacerbates tissue impairment in the lungs of infected mice (65); PPE38 (Rv2352c) is required for the secretion of M.tb PE (proline-glutamate)_PGRS (polymorphic GC-rich repetitive sequences) proteins, and a deletion in the ppe38 locus contributes to the increased in vivo growth of M.tb (66). On the basis of the increasing experimental evidence and clinical observations, it is clear that M.tb controls a host’s innate immunity by dictating a sophisticated program that involves multiple host signaling pathways and cellular functions (67). Here, our study revealed that M.tb EST12 caused the macrophage pyroptosis, induced IL-1β secretion, decreased bacterial loads in mice, and activated the host’s immunity (fig. S6). Other mechanisms might also be contributed by EST12 stimulation such as increased Ca2+ influx and mtROS accumulation. Thus, our study suggested that M.tb’s pathogenesis strategy is to reduce its virulence possibly for its chronic or persistent infection, and the EST12-induced pyroptosis might be one of M.tb’s sophisticated pathogenesis strategies.
We also observed that the differences in the bacterial colonies between the H37Rv group and H37RvΔEST12 were not large (Fig. 6A), and we speculate that some other components within the complex components from M.tb H37Rv, except EST12, might also affect the outcome of the infection and lead to smaller differences between M.tb H37Rv and M.tb H37RvΔEST12.
Our study identified that EST12 caused pyroptosis through interaction with RACK1 in macrophages. RACK1 acts as a major host factor in sensing M.tb EST12 and recruits the deubiquitinase UCHL5 to promote the deubiquitination of NLRP3, subsequently activating the macrophage NLRP3 inflammasome–caspase-1–GSDMD pyroptosis–IL-1β immune pathway. EST12-induced pyroptosis contributed to resistance to M.tb infection via RACK1, NLRP3, caspase-1, GSDMD, and IL-1β. This study adds a new insight into understanding M.tb-macrophage interactions and the innate immune response to M.tb. Our findings will help to understand the interaction between macrophage and the M.tb RD-encoding protein EST12 and highlight the pivotal role of the EST12-RACK1-induced pyroptosis in M.tb-induced immunity.
MATERIALS AND METHODS
Reagents are described in table S3.
Bacterial strains
M.tb H37Rv [strain American Type Culture Collection (ATCC) 27294] was maintained on Lowenstein-Jensen medium. M.tb H37RvΔEST12 was constructed by Shanghai Gene-optimal Science & Technology Co., Ltd. according to previous publications (68, 69). Briefly, Rv1579c (EST12) gene in H37Rv genome was replaced by screening gene cassette (SacB-Hygromycin B) by a homologous recombination method. M. bovis BCG (Pasteur strain ATCC 35734) was harvested while in log-phase growth. M. smegmatis (strain ATCC 19420), M. marinum (strain ATCC 927), M. intracellulare (strain ATCC 13950), M. avium (strain ATCC 25291), Escherichia coli DH5α (strain ATCC 25922), and E. coli BL-21 (stain ATCC BAA-1025) were propagated from laboratory stocks (School of Medicine, Wuhan University, Wuhan, China). E. coli DH5a and BL21 (DE3) were grown in flasks using LB medium. The mycobacterial strains were grown in Middlebrook 7H9 broth (7H9) supplemented with 10% oleic acid–albumin–dexrose–catalase (OADC) and 0.05% Tween 80 (Sigma-Aldrich) or on Middlebrook 7H10 agar (BD Biosciences) supplemented with 10% OADC. The shuttle vector pMV261 (provided by Jiaoyu Deng, Chinese Academy of Sciences) was used to overexpress M.tb Rv1579 (EST12) in BCG or M. smeg. BCG-EST12 and M. smeg–EST12 were plated on 7H10 containing kanamycin (20 μg/ml). The resulting colonies were grown on 7H9 plates containing kanamycin (20 μg/ml).
Animals
NLRP3−/− mice were purchased from the Jackson Laboratory (B6.129S6-Nlrp3tm1Bhk/J, stock no: 021302). GSDMD−/− and caspase-1/11−/− mice were provided in kind by Feng Shao (National Institute of Biological Sciences, Beijing). Mice carrying the floxed gnb2l1 allele (Gnb2l1F/F) (gnb2l1 encodes RACK1) were provided in kind by Jiyan Zhang (Department of Molecular Immunology, Institute of Basic Medical Sciences). Mice expressing Cre recombinase (LysMcre) were purchased from the Jackson Laboratory [B6.129P2-Lyz2tm1(cre)Ifo/J, stock no: 004781]. The Cre-loxP system was used to disrupt the 172–base pair region in exon 2 of the gnb2l1 gene (which encodes RACK1). The mouse mating was performed by crossing mice carrying the floxed gnb2l1 allele with mice expressing Cre recombinase (LysMcre) to generate a macrophage-specific RACK1 knockout mouse model (referred to as RACK1ΔMΦ). RACK1ΔMΦ mice were born at Mendelian frequency and proportional male/female ratios.
Cell culture
The murine macrophage cell line RAW264.7 was purchased from the China Center for Type Culture Collection and was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS). Murine peritoneal macrophages were harvested from peritoneal exudates and resuspended in RPMI 1640 with 10% (v/v) heat-inactivated FBS (Gibco). BMDMs were differentiated from isolated mouse bone marrow cells by incubation in 10% FBS plus macrophage colony-stimulating factor (M-CSF; 40 ng/ml) for 7 days. BMDCs were differentiated from isolated mouse bone marrow cells by being cultured for 7 days in complete RPMI 1640 (Gibco) containing granulocyte M-CSF (GM-CSF; 20 ng/ml; Peprotech) and IL-4 (20 ng/ml; Peprotech). Mouse B cells were isolated and purified from splenocytes using CD19+ magnetic activated cell sorting (Miltenyi Biotec), and mouse CD4+ T and CD8+ T cells were isolated and purified from splenocytes using an EasySepTM mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies). Murine neutrophils were harvested from mouse peritoneal exudates 3 hours after intraperitoneal injection of fluid thioglycollate medium (BD Biosciences) (70). Human THP-1 monocyte cell line was suspended in RPMI 1640 culture medium and distributed to 48-well plates at a density of 105 cells per well and differentiated into macrophages during 24 hours of culture with 20 ng/ml of phorbol 12-myristate 13-acetate (PMA) (37°C; 5% CO2). THP-1 monocyte cell line–derived macrophages were attached to the plastic surface of the plates and confirmed by light microscopy.
Construction, expression, and purification of recombinant RD proteins
Forty genes for RD1 to RD3 and RD11 to RD14 were amplified from the genome of M.tb iH37Rv (inactivated at 65°C for 2 hours). Bacterial expression plasmids were constructed by inserting the genes into pET28a, and Rv1579c (EST12) was also constructed in pMV261 and pEGFP-C1 plasmids. RACK1 was constructed in the pAsRED2-C1 plasmid. The pSilencer 1.0-U6 plasmid was used to construct shRNA of UCHL5 and the targeting sequences of sh-UCHL5 #1: 5′-GGAGTGGTGTCTCATGGAAAG-3′ and sh-UCHL5 #2: 5′-GGGTCTTCACCGAGCTCATTA-3′. All primers used for cloning are listed in table S4.
E. coli BL21 (DE3) cells were transformed with each expression plasmid pET-28a with 6×His-tag. Expression of the recombinant proteins was induced by the addition of isopropyl-β-d-thiogalactopyranoside for 12 hours at 25°C. The cell pellets were resuspended in phosphate-buffered saline (PBS) and disrupted by dynamic high-pressure homogenization, and the lysates were clarified by centrifugation (12,000 rpm for 15 min at 4°C). The supernatants were incubated with Ni-agarose pre-equilibrated with 10 mM imidazole solution [25 mM tris-HCl, 200 mM NaCl, and 10 mM imidazole (pH 7.4)]. The recombinant proteins were eluted with 20, 40, 80, and 200 mM imidazole buffer, with 25 mM tris-HCl and 200 mM NaCl (pH 7.4). The purified recombinant proteins were further purified using endotoxin-free purification polymyxin B columns and analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE).
Preparation of rabbit polyclonal antibody
A New Zealand rabbit (2 to 3 kg) was purchased from the Wuhan University Center for Animal Experiment/A3-Lab (Wuhan, China). Emulsified recombinant EST12 protein [PBS (500 μg/ml)] was injected hypodermically at five or six different sites in the nuchal region of the rabbit every week for 4 weeks. One week after the final injection, blood was collected from the central artery. Serum was obtained and stored at −80°C. The titer evaluation was performed by ELISA in recombinant protein-coated microtiter plates, with serial dilutions of rabbit serum as the primary antibody and horseradish peroxidase (HRP)–conjugated goat anti-rabbit immunoglobulin G (IgG) as the secondary antibody. The maximum antibody dilution that fulfilled the criteria (OD450positive/OD450negative > 2.1) was considered as the antibody titer. Serum collected before immunization was used as a negative control.
LDH release assay
The LDH release was measured by a CytoTox 96 NonRadioactive Cytotoxicity Assay (Promega Corporation), according to the manufacturer’s protocol. Macrophages/CD4+ T cells/CD8+ T cells/neutrophils/BMDCs were plated in 96-well plates (1 × 105 cells/100 μl per well) and incubated with EST12 as the indicated conditions, and the supernatants were harvested to quantify the release of LDH from the cells. The percentage of specific killing was calculated as follows: specific killing% = (experimental release − spontaneous release)/(total release − spontaneous release) × 100%. The experiments were performed in triplicate.
ELISA for cytokine measurement
Peritoneal macrophages were stimulated with 2 μΜ EST12 from 0 to 24 hours or with 0 to 10 μM EST12 for 24 hours. Supernatants were harvested, and the levels of cytokines were evaluated using ELISA by measurement of OD450. Mouse serum TNF-α, IL-6, and IL-1β or conditioned medium was determined using the mouse Quantikine ELISA kit (Dakewe Biotechnology, Beijing, China).
Cell transfection
RAW264.7 cells were cultured in DMEM supplemented with 10% (v/v) heat-inactivated FBS. Transient transfection was performed with Neofect DNA transfection reagent (Neofect) for RAW264.7 cells following the manufacturer’s instructions. The transfection of peritoneal macrophages was performed with jetPEI-Macrophage (Polyplus).
Stimulation of macrophages
Macrophages were treated with different doses of EST12 or EST12 truncated and mutant for 24 hours or with 2 μΜ EST12 or EST12 truncated and mutant for different periods of time. To study the activation of the NLRP3 inflammasome, macrophages were primed for 4 hours with LPS (100 ng/ml) and stimulated for 30 to 45 min with 20 μΜ nigericin. In some experiments, the cells were pretreated with various stimuli including curcumin (20 μΜ), PDTC (25 μΜ), cytochalasin D (2.5 μΜ), Z-VAD-FMK (20 μΜ), Z-DEVD-FMK (20 μΜ), VX-765 (12.5 μΜ), KCl (50 mM) for 30 min, and polymyxin B (10 μg/ml) for 1 hour.
Culture and infection of macrophages
BMDMs were cultivated overnight and treated with H37Rv and H37RvΔEST12 [(multiplicity of infection) MOI = 10] or BCG and BCG-EST12 (MOI = 10) or M. smegmatis and EST12–M. smegmatis (MOI = 10). The supernatants were collected for LDH and inflammatory cytokine assay, and the cell lysates were used for WB assay.
WT, RACK1−/−, caspase-1−/−, NLRP3−/−, and GSDMD−/− BMDMs were harvested and infected with BCG strains or M. smegmatis strains at an MOI = 10. After 2 hours of incubation in DMEM, the medium was discarded, and the cells were washed three times with 1× PBS to exclude extracellular bacteria. Then, the cells were incubated again with fresh DMEM supplemented with gentamicin (10 μg/ml) to kill nonwashed extracellular bacteria. At different time points of postinfection, cell culture supernatants were used for IL-1β or LDH detection. The cells were washed three times with 1× PBS and harvested for CFU counting. Cells used for bacterial counting were lysed in 7H9 broth containing 0.05% SDS for 10 min. Three sets of serial 10-fold dilutions of the lysates from each time point were prepared in 0.05% Tween 80, and portions were plated on 7H10 agar plates.
WB analysis
For WB analysis, 2 μM EST12 and EST12-Y80A stimulated peritoneal macrophages from 0 to 24 hours were lysed in cell lysis buffer [radioimmunoprecipitation assay (RIPA)] containing 10 mM Hepes (pH 7.5), 50 mM NaCl, 1% Triton X-100, and 2 mM EDTA. LPS was used as a control at a final concentration of 10 μg/ml. The proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). To detect the secretion of EST12 in H37Rv, the supernatants (two batches) and whole bacterial lysates from 5 ml of H37Rv cultures were collected, concentrated, and analyzed using a rabbit anti-EST12 polyclonal antibody. To validate the macrophage-specific conditional RACK1 knockout mouse model, peritoneal macrophages from RACK1ΔMϕ mice and WT mice and B cells and CD4+ T cells from RACK1ΔMϕ mice were isolated and purified from splenocytes; RAW264.7 cells were used as controls, and all the above cells were lysed to detect RACK1 expression by WB. The blots were blocked with 5% nonfat dry milk or bovine serum albumin (BSA) in tris-buffered saline (TBS) for 1 hour at room temperature and subsequently incubated overnight at 4°C with primary antibodies in TBS–Tween 20 [0.5% (v/v)] (TBST). Following three washes of 10 min each with TBST, the blots were incubated with goat anti-mouse IgG or goat anti-rabbit IgG conjugated to HRP at a dilution of 1:5000 in blocking buffer for 1 hour at room temperature. After three washes with TBST, the blots were developed by Immobilon WesternBright ECL HRP substrate (K-12045-D10, Advansta).
Immunoprecipitation
Peritoneal macrophages were harvested at 0, 6, and 24 hours after treatment with or without recombinant protein (2 μM EST12) and then lysed in RIPA with protease inhibitor phenylmethylsulfonyl fluoride (PMSF). The cell lysates were then incubated with anti-ASC/NLRP3 monoclonal antibody (mAb) at 4°C for 8 hours. The ASC/NLRP3 complexes were captured using protein A + G magnetic beads and then washed three times with TBS [50 mM tris, 150 mM NaCl, 0.1 to 0.5% Triton X-100, or Tween 20 (pH 7.5)] to remove nonspecific binding protein. Elution buffer (0.1 to 0.2 M glycine and 0.1 to 0.5% Triton X-100) was used to elute the antigen. The eluted proteins were subjected to WB analysis with anti-ASC/NLRP3/caspase-1 mAbs.
To detect the polyubiquitination of NEMO and NLRP3, RAW264.7 cells were transfected with the indicated plasmids encoding WT ubiquitin (H-Ub), mutant ubiquitin H-Ub (K48), or H-Ub (K63). At 48 hours after transfection, the RAW264.7 cells were stimulated with EST12 for 0 or 6 hours. The supernatants of the cell lysates were subjected to immunoprecipitation with antibodies against NEMO and NLRP3 and then analyzed by immunoblotting.
To detect the role of UCHL5 in K48-linked deubiquitination of NLRP3, shRNAs-pSilence1.0-U6 of UCHL5 (sh-UCHL5) were transfected into RAW264.7 cells for 24 hours and then treated with EST12 for 0 or 6 hours. The cell lysates were subjected to immunoprecipitation with antibodies against NLRP3 and were then analyzed by immunoblotting.
To detect the role of UCHL5 in K48-linked ubiquitination of RACK1, shRNAs of UCHL5 were transfected into RAW264.7 cells for 24 hours and then treated with EST12 for 0 or 24 hours. The cell lysates were subjected to immunoprecipitation with antibodies against RACK1 and were then analyzed by immunoblotting.
To detect the interaction of RACK1 with UCHL5, BMDMs were stimulated with EST12 for 6 hours, and the cell lysates were subjected to immunoprecipitation with antibodies against RACK1. They were then analyzed by immunoblotting.
To detect the interaction of UCHL5 with RACK1 and NLRP3, BMDMs were stimulated with EST12 for 6 hours, and the cell lysates were subjected to immunoprecipitation with antibodies against UCHL5. They were then analyzed by immunoblotting.
EST12-Ni-NTA bead pull-down assay and MS analysis
The RAW264.7 cell lysates were incubated with 10 μg of the purified recombinant EST12 or EST12 mutant and deletion proteins with 6×His-tags at 4°C overnight. The reaction mixtures were then incubated with 20 μl of Ni-NTA agarose at 4°C for 4 hours. After intensive washing with washing buffer A [50 mM Hepes (pH 7.5), 300 mM NaCl, 20 mM imidazole, and 0.1 mM PMSF], the proteins that were bound to the agarose were eluted with elution buffer (elution buffer containing 250 mM imidazole). The eluted proteins were analyzed by SDS-PAGE or WB. For SDS-PAGE, the specific bands were excised and subjected to LC-MS (TripleTOF 5600+, SCIEX, USA).
Resolving the crystal structure of EST12
The purified EST12 protein was desalted and concentrated to approximately 35 mg/ml in 10 mM tris-HCl (pH 8.0) and 5 mM dithiothreitol for crystallization. All crystallization experiments were performed with the hanging-drop vapor-diffusion method. The protein solution (0.5 μl) and the reservoir solution (0.5 μl) were mixed and equilibrated with 30 μl of reservoir solution in each well at 277 K. Crystallization was performed in 6% (v/v) Tacsimate (pH 6.0), 0.1 M MES monohydrate (pH 6.0), and 25% (w/v) polyethylene glycol 4000 at 277 K in space group P21.
All crystals were flash-cooled in liquid nitrogen after a flash-soak in a cryoprotection solution consisting of the reservoir solution with an additional concentration of glycerol 25%. X-ray diffraction data were collected at the BL17U1 of Shanghai Synchrotron Radiation Facility (Shanghai, China) under liquid-nitrogen cryoconditions at 100 K. The data were processed using CCP4 (collaborative computational project number 4) program suite (71). The structures were determined by molecular replacement using a standard long helix, and the rest of the model was autobuilt using PHENIX (72).
Confocal microscopy and internalization assay
To test the cellular colocalization of EST12 and RACK1, RAW264.7 cells were transfected with a plasmid expressing GFP-EST12 and RED-RACK1, either alone or with both plasmids. After transfection for 48 hours, cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100 and blocked with 10% BSA. The cells were then stained with DAPI for 10 min at room temperature, followed by washing three times with 1× cold PBS (pH 7.4). The colocalization of EST12 and RACK1 was visualized with a Leica SP2 confocal system (Leica Microsystems, Wetzlar, Germany). To test the internalization of EST12, RAW264.7 cells were stimulated with purified EST12 protein for 1 and 6 hours, stained with anti-EST12 rabbit antibodies and PE-conjugated goat anti-rabbit IgG and DAPI and observed by the Leica SP2 confocal system (Leica Microsystems, Wetzlar, Germany). To test the NLRP3 expression after EST12 treatment of RAW264.7 cells, the RAW264.7 cells were treated with 2 μΜ EST12 for 0 to 12 hours and then incubated with anti-NLRP3 antibody and PE-labeled goat anti-rabbit secondary antibody. The cell nuclei DNA was stained with DAPI.
To test the K48-linked deubiquitination of NLRP3, BMDMs were treated with 2 μΜ EST12 for 0, 6, and 12 hours or primed for 4 hours with LPS (100 ng/ml) and stimulated for 30 to 45 min with 20 μΜ nigericin as a positive control, and then incubated with anti-NLRP3 and K48-linked ubiquitin antibody, followed by incubation with anti-mouse IgG (H + L), F(ab′)2 fragment (Alexa Fluor 594 conjugate) and anti-rabbit IgG (H + L), F(ab′)2 fragment (Alexa Fluor 488 conjugate). The expression and colocalization of NLRP3 and K48-linked ubiquitin were analyzed using a confocal laser scanning microscope (Zeiss LSM 880 with a 40×), and the integrated densities of NLRP3 and K48-linked ubiquitin were quantified with ImageJ software.
ASC speck imaging
Peritoneal macrophages of WT and RACK1ΔMφ mice were treated with 2 mM EST12 or EST12-D1 for the indicated time. Peritoneal macrophages were primed with LPS (100 ng/ml) for 4 hours followed by 20 μΜ nigericin treatment for 45 min as a positive control. Cells were fixed in 4% paraformaldehyde at room temperature for 15 min. After washing three times with PBS, the cells were permeabilized with PBS containing 0.1% Triton X-100 for 5 min, washed, and, lastly, blocked with PBS containing 5% BSA for 1 hour. The cells were then incubated with the anti-ASC mAb overnight at 4°C, followed by incubation with anti-mouse IgG (H + L), F(ab′)2 fragment (Alexa Fluor 594 conjugate) for 1 hour. After washing three times, the cells were incubated with DAPI solution for 5 min. The cells were then analyzed using a confocal laser scanning microscope [Zeiss LSM 880 with a 63×/1.49 NA (numerical aperture) oil objective ].
For time-lapse microscopy, peritoneal macrophages of WT and RACK1ΔMφ mice were plated onto a 35-mm glass bottom dish (Nest) and primed and treated with 2 μΜ EST12 for 4 hours. PI (5 ng/ml) was added to the medium for monitoring cell membrane integrity. Imaging was carried out using Zeiss LSM 880 with a 63×/1.49 NA oil objective.
Flow cytometry analysis
For detection of mtROS, WT and RACK1−/− BMDMs were treated with 2 μM EST12 for the indicated time, and 25 μΜ H2O2 treatment was used as a positive control. BMDMs were harvested with PBS and then stained with 5 μM MitoSOX Red (mtROS superoxide) at 37°C for 30 min in the dark. Samples were immediately analyzed on a BD Accuri C6 cytometer (BD Biosciences), and the data were presented as the percentages of the mtROS median fluorescence intensities (MFIs) and processed by CFlowPlus software.
For the detection of intracellular Ca2+, WT and RACK1−/− BMDMs were treated with 2 μM EST12 for the indicated time. In addition, LPS (100 ng/ml) was primed for 4 hours, followed by nigericin treatment for 45 min as a positive control. BMDMs were harvested with PBS and then stained with 5 μM Fluo-4 AM at 37°C for 30 min in the dark, and then the cells were washed with PBS. Samples were analyzed by FCM (flow cytometry), and the data were presented as the percentages of intracellular Ca2+ MFIs and processed by CFlowPlus software.
Reverse transcription–quantitative real-time polymerase chain reaction
To detect the mRNA expression of RACK1 and eight PYRIN-domain–containing protein mRNA expression levels, the adherent murine peritoneal macrophages (1.5 × 106 in 1.5 ml of culture medium) were treated with 2 μM EST12 protein for different periods. Total RNA was extracted from the cells using TRIzol reagent (Life Technologies Corporation, USA). The ReverTra Ace-α-First-Strand cDNA Synthesis Kit (Toyobo Biologics Inc., Japan) was used according to the manufacturer’s instructions to synthesize the first-strand complementary DNA (cDNA) from the mRNA in the total RNA sample. The resulting cDNA was stored at −20°C until use in RT-qPCR experiments.
To detect NLRP3 mRNA expression, RAW264.7 macrophage cells were transfected with shRNA-RACK1 for 24 hours and then treated with 2 μM EST12 protein for different time periods. RACK1−/− peritoneal macrophages were also stimulated with 2 μM EST12 protein for different time periods to detect the mRNA expression of NLRP3. Total RNA was extracted and used for RT-qPCR experiments.
The RT-qPCR reactions were run on an ABI StepOnePlus (Applied Biosystems) using the standard cycling conditions. Target gene expression levels were normalized based on glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences of individual genes were as follows: RACK1, 5′-TGACCGAGCAGATGACCC-3′ (forward) and 5′-GTGGTGCCCGTTGTGAGA-3′ (reverse); NLRP3, 5′-GATGGGTTTGCTGGGATA-3′ (forward) and 5′-AGCTGCGTGTAGCGACTG-3′ (reverse); NLRP1b, 5′-AGGACAGCAGGGACATAA-3′ (forward) and 5′-CATTCAACAAGCCACAAA-3′ (reverse); NLRP2, 5′-CAGATGATGCCTACGAGA-3′ (forward) and 5′-TTCTGAGCAGCAGTAGAAC-3′ (reverse); NLRP5, 5′-TGACAGAGGCAGATAAAGA-3′ (forward) and 5′-CCCAGCCAAGAACAATAC-3′ (reverse); NLRP6, 5′-ACCCAGAATGAGACCAGTT-3′ (forward) and 5′-CCAGTGTAGCCATAAGCAG-3′ (reverse); NLRP9b, 5′-CCCAGTGGCTCCAGTTAG-3′ (forward) and 5′-TCGGCTGTATTTCGTCTTT-3′ (reverse); NLRP12, 5′-TTGAGAGCCAATCAGCGT-3′ (forward) and 5′-CTGGAGCGTTCCCACTCT-3′ (reverse); PYRIN, 5′-AAGGGCCACTCTAAGATC-3′ (forward) and 5′-GGACAGAACCACCAACTC-3′ (reverse); GAPDH, 5′-TGTTTCCTCGTCCCGTAG-3′ (forward) and 5′-CAATCTCCACTTTGCCACT-3′ (reverse). Relative RNA levels were calculated by the comparative cycle threshold (CT) method (2−ΔΔCT method), where CT indicates the amplification cycle number at which the fluorescence generated within a reaction rises above a defined threshold fluorescence, and ΔΔCT = experimental sample (Cttarget gene − CtGAPDH) − control sample (Cttarget gene − CtGAPDH).
Mouse models
Each C57BL/6 mouse (male, ages between 8 and 10 weeks) was infected with H37Rv, H37RvΔEST12, BCG, and BCG-EST12 (5 × 104 CFUs of M.tb/50 μl diluted with PBS) by intranasal administration on day 0. On day 30, the mice were euthanized. The bacterial loads in the lungs of infected mice were assessed as previously described (73). The serum cytokine production was assessed by ELISA. The sections of lung tissues and spleen tissues were fixed in 4% paraformaldehyde, and pathological sections were examined with hematoxylin and eosin (H&E) stain. The Ziehl-Neelsen acid fast staining was performed to analyze the bacterial load in the lung sections. An overall histology score was assigned to the lungs of mice based on the extent of granulomatous inflammation as follows: 0 = no lesion, 1 = minimal lesion (1 to 10% area of tissue in the section involved), 2 = mild lesion (11 to 30% area involved), 3 = moderate lesion (30 to 50% area involved), 4 = marked lesion (50 to 80% area involved), and 5 = severe lesion (>80% area involved).
In the M. smegmatis challenge model, WT C57BL/6 mice were intravenously infected with WT M. smeg and M. smeg–EST12 with 108 CFUs per mouse. The bacterial loads in the lungs and spleens of infected mice were assessed, and the serum cytokine production was assessed by ELISA.
All mice were maintained in microisolator cages, fed with standard laboratory diet and water, and housed in the animal colony at the animal center of Wuhan University. All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication no. 86-23, revised 1985). All animal protocols were approved by the Institutional Animal Care and Use Committee of the Institute of Model Animals of Wuhan University (nos. S01317101C, S01317012S, and 2018022).
Statistical analysis
Data are presented as the mean ± SEM. All data met the assumptions of the tests. Student’s unpaired t test was used to compare the means of the two groups. One-way analysis of variance (ANOVA) with Bonferroni posttests was used to compare the means between multiple groups. GraphPad Prism software (version 6.0) was used to determine statistical significance. P values less than 0.05 were considered statistically significant. The exact value of n, representing the number of experimental replications or numbers of mice used in the experiments, is indicated in the figure legends.
Supplementary Material
Acknowledgments
We acknowledge F. Shao (National Institute of Biological Sciences, Beijing, China) for providing GSDMD−/− and caspase-1/11−/− mice and rabbit polyclonal antibody anti-GSDMD. We acknowledge J. Zhang (Department of Molecular Immunology, Institute of Basic Medical Sciences, Beijing, China) for providing (Gnb2l1)fl/fl mice. We acknowledge H. Shu and Y. Wang (Wuhan University, Wuhan, China) for providing plasmids encoding NEMO, WT ubiquitin (H-Ub), ubiquitin mutant H-Ub (K48), and H-Ub (K63). Funding: This work was supported by grants from the National Grand Program on Key Infectious Disease of China (2017ZX10201301-006 and 2012ZX10003002-015), the National Key R&D Program of China (2018YFA0507603), the National Natural Science Foundation of China (91740120 and 21572173), the National Outstanding Youth Foundation of China (81025008), the Major Projects of Technological Innovation of Hubei Province (2016ACA150), the Natural Science Foundation Project of Hubei Province (2016CFA062), the Medical Science Advancement Program (Basic Medical Sciences) of Wuhan University (TFJC 2018002), and the Fundamental Research Funds for the Central Universities. Author contributions: X.-L.Z. designed and supervised the research. Z.Q., J.W., and G.L. conducted the experiments. L.Y., J.Z., Y.J., and Z.L. performed key crystal structure of EST12 experiments. Z.Q., Y.Z., Y.X., and X.-L.Z. wrote the manuscript, and X.-L.Z. revised the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eaba4733/DC1
REFERENCES AND NOTES
- 1.World Health Organization (WHO), Global Tuberculosis Report 2019 (WHO, 2019); https://who.int/tb/publications/global_report/en/#userconsent#.
- 2.Dallenga T., Repnik U., Corleis B., Eich J., Reimer R., Griffiths G. W., Schaible U. E., M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 22, 519–530.e3 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F., Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 4.He W. T., Wan H., Hu L., Chen P., Wang X., Huang Z., Yang Z. H., Zhong C. Q., Han J., Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25, 1285–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma D., Kanneganti T. D., The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 213, 617–629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kayagaki N., Stowe I. B., Lee B. L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q. T., Liu P. S., Lill J. R., Li H., Wu J., Kummerfeld S., Zhang J., Lee W. P., Snipas S. J., Salvesen G. S., Morris L. X., Fitzgerald L., Zhang Y., Bertram E. M., Goodnow C. C., Dixit V. M., Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Wang Y., Gao W., Shi X., Ding J., Liu W., He H., Wang K., Shao F., Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Mahairas G. G., Sabo P. J., Hickey M. J., Singh D. C., Stover C. K., Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 178, 1274–1282 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behr M. A., Wilson M. A., Gill W. P., Salamon H., Schoolnik G. K., Rane S., Small P. M., Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284, 1520–1523 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Kalra M., Grover A., Mehta N., Singh J., Kaur J., Sable S. B., Behera D., Sharma P., Verma I., Khuller G. K., Supplementation with RD antigens enhances the protective efficacy of BCG in tuberculous mice. Clin. Immunol. 125, 173–183 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Pai M., Zwerling A., Menzies D., Systematic review: T-cell–based assays for the diagnosis of latent tuberculosis infection: An update. Ann. Intern. Med. 149, 177–184 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pai M., Denkinger C. M., Kik S. V., Rangaka M. X., Zwerling A., Oxlade O., Metcalfe J. Z., Cattamanchi A., Dowdy D. W., Dheda K., Banaei N., Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin. Microbiol. Rev. 27, 3–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo W., Qu Z. L., Xie Y., Xiang J., Zhang X. L., Identification of a novel immunodominant antigen Rv2645 from RD13 with potential as a cell-mediated immunity-based TB diagnostic agent. J. Infect. 71, 534–543 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Luo W., Qu Z., Zhang L., Xie Y., Luo F., Tan Y., Pan Q., Zhang X. L., Recombinant BCG::Rv2645 elicits enhanced protective immunity compared to BCG in vivo with induced ISGylation-related genes and Th1 and Th17 responses. Vaccine 36, 2998–3009 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Cavaillon J. M., Haeffner-Cavaillon N., Polymyxin-B inhibition of LPS-induced interleukin-1 secretion by human monocytes is dependent upon the LPS origin. Mol. Immunol. 23, 965–969 (1986). [DOI] [PubMed] [Google Scholar]
- 16.Vanaja S. K., Russo A. J., Behl B., Banerjee I., Yankova M., Deshmukh S. D., Rathinam V. A. K., Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165, 1106–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyskov S., Gray J. J., The RosettaDock server for local protein–protein docking. Nucleic Acids Res. 36, W233–W238 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyskov S., Chou F. C., Conchúir S. Ó., Der B. S., Drew K., Kuroda D., Xu J., Weitzner B. D., Renfrew P. D., Sripakdeevong P., Borgo B., Havranek J. J., Kuhlman B., Kortemme T., Bonneau R., Gray J. J., Das R., Serverification of molecular modeling applications: The Rosetta Online Server that Includes Everyone (ROSIE). PLOS ONE 8, e63906 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruiz Carrillo D., Chandrasekaran R., Nilsson M., Cornvik T., Liew C. W., Tan S. M., Lescar J., Structure of human Rack1 protein at a resolution of 2.45 Å. Acta Cryst. 68, 867–872 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo H., Callaway J. B., Ting J. P., Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seshadri S., Duncan M. D., Hart J. M., Gavrilin M. A., Wewers M. D., Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1β processing and release. J. Immunol. 179, 1274–1281 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Yu J.-W., Wu J., Zhang Z., Datta P., Ibrahimi I., Taniguchi S., Sagara J., Fernandes-Alnemri T., Alnemri E. S., Cryopyrin and pyrin activate caspase-1, but not NF-κB, via ASC oligomerization. Cell Death Differ. 13, 236–249 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Py B. F., Kim M. S., Vakifahmetoglu-Norberg H., Yuan J., Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Heneka M. T., McManus R. M., Latz E., Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Mariathasan S., Weiss D. S., Newton K., McBride J., O’Rourke K., Roose-Girma M., Lee W. P., Weinrauch Y., Monack D. M., Dixit V. M., Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Elliott E. I., Sutterwala F. S., Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 265, 35–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J., Thorson L., Strokes R. W., Santosuosso M., Huygen K., Zganiacz A., Hitt M., Zhou X., Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 173, 6357–6365 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Pepponi I., Stylianou E., van Dolleweerd C., Diogo G. R., Paul M. J., Drake P. M. W., Ma J. K.-C., Reljic R., Immune-complex mimics as a molecular platform for adjuvant-free vaccine delivery. PLOS ONE 8, e60855 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Man S. M., Karki R., Kanneganti T. D., Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miao E. A., Leaf I. A., Treuting P. M., Mao D. P., Dors M., Sarkar A., Warren S. E., Wewers M. D., Aderem A., Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karki R., Lee E., Place D., Samir P., Mavuluri J., Sharma B. R., Balakrishnan A., Malireddi R. K. S., Geiger R., Zhu Q., Neale G., Kanneganti T. D., IRF8 regulates transcription of Naips for NLRC4 inflammasome activation. Cell 173, 920–933.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J., Fernandes-Alnemri T., Alnemri E. S., Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams D. R., Ron D., Kiely P. A., RACK1, a multifaceted scaffolding protein: Structure and function. Cell Commun. Signal 9, 22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duff D., Long A., Roles for RACK1 in cancer cell migration and invasion. Cell. Signal. 35, 250–255 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Zhang X., Jain R., Li G., Roles of Rack1 proteins in fungal pathogenesis. Biomed. Res. Int. 2016, 4130376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Broz P., Pelegrín P., Shao F., The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Hennig E. E., Butruk E., Ostrowski J., RACK1 protein interacts with helicobacter pylori VacA cytotoxin: The yeast two-hybrid approach. Biochem. Biophys. Res. Commun. 289, 103–110 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Thorslund S. E., Edgren T., Pettersson J., Nordfelth R., Sellin M. E., Ivanova E., Francis M. S., Isaksson E. L., Wolf-Watz H., Fällman M., The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLOS ONE 6, e16784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majzoub K., Hafirassou M. L., Meignin C., Goto A., Marzi S., Fedorova A., Verdier Y., Vinh J., Hoffmann J. A., Martin F., Baumert T. F., Schuster C., Imler J. L., RACK1 controls IRES-mediated translation of viruses. Cell 159, 1086–1095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J. J., Xie D., RACK1, a versatile hub in cancer. Oncogene 34, 1890–1898 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Lin W. C., Zhang Z., Xu Z., Wang B., Li X., Cao H., Wang Y., Zhang S., The association of receptor of activated protein kinase C 1 (RACK1) with infectious bursal disease virus viral protein VP5 and voltage-dependent anion channel 2 (VDAC2) inhibits apoptosis and enhances viral replication. J. Biol. Chem. 290, 8500–8510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hafirassou M. L., Meertens L., Umana-Diaz C., Labeau A., Dejarnac O., Bonnet-Madin L., Kummerer B. M., Delaugerre C., Roingeard P., Vidalain P.-O., Amara A., A global interactome map of the dengue virus NS1 identifies virus restriction and dependency host factors. Cell Rep. 21, 3900–3913 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Qu Y., Misaghi S., Izrael-Tomasevic A., Newton K., Gilmour L. L., Lamkanfi M., Louie S., Kayagaki N., Liu J., Kömüves L., Cupp J. E., Arnott D., Monack D., Dixit V. M., Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 (2012). [DOI] [PubMed] [Google Scholar]
- 44.van de Veerdonk F. L., Netea M. G., Dinarello C. A., Joosten L. A., Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 32, 110–116 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Wassermann R., Gulen M. F., Sala C., Perin S. G., Lou Y., Rybniker J., Schmid-Burgk J. L., Schmidt T., Hornung V., Cole S. T., Ablasser A., Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17, 799–810 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Wawrocki S., Druszczynska M., Inflammasomes in Mycobacterium tuberculosis-driven immunity. Can. J. Infect. Dis. Med. Microbiol. 2017, 2309478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Briken V., Ahlbrand S. E., Shah S., Mycobacterium tuberculosis and the host cell inflammasome: A complex relationship. Front. Cell. Infect. Microbiol. 3, 62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jayaraman P., Sada-Ovalle I., Nishimura T., Anderson A. C., Kuchroo V. K., Remold H. G., Behar S. M., IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J. Immunol. 190, 4196–4204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verway M., Bouttier M., Wang T. T., Carrier M., Calderon M., An B. S., Devemy E., McIntosh F., Divangahi M., Behr M. A., White J. H., Vitamin D induces interleukin-1β expression: Paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLOS Pathog. 9, e1003407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y., Li Y., Yang X., Wang J., Wang R., Qian X., Zhang W., Xiao W., Uev1A-Ubc13 catalyzes K63-linked ubiquitination of RHBDF2 to promote TACE maturation. Cell. Signal. 42, 155–164 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Ren G., Zhang X., Xiao Y., Zhang W., Wang Y., Ma W., Wang X., Song P., Lai L., Chen H., Zhan Y., Zhang J., Yu M., Ge C., Li C., Yin R., Yang X., ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 38, e100376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lam Y. A., Xu W., DeMartino G. N., Cohen R. E., Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737–740 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Lee Y.-T. C., Chang C.-Y., Chen S.-Y., Pan Y.-R., Ho M.-R., Hsu S.-T. D., Entropic stabilization of a deubiquitinase provides conformational plasticity and slow unfolding kinetics beneficial for functioning on the proteasome. Sci. Rep. 7, 45174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sannigrahi A., Nandi I., Chall S., Jawed J. J., Halder A., Majumdar S., Karmakar S., Chattopadhyay K., Conformational switch driven membrane pore formation by Mycobacterium secretory protein MPT63 induces macrophage cell death. ACS Chem. Biol. 14, 1601–1610 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Gong Z., Kuang Z., Li H., Li C., Ali M. K., Huang F., Li P., Li Q., Huang X., Ren S., Li J., Xie J., Regulation of host cell pyroptosis and cytokines production by Mycobacterium tuberculosis effector PPE60 requires LUBAC mediated NF-κB signaling. Cell. Immunol. 335, 41–50 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Antas P. R. Z., Ponte C. G. G., Almeida M. R., Albuquerque L. H. P., Sousa-Vasconcelos P. S., Pedro T., Gomes N. L. S., Moreira O. C., Silva F. C., Castello-Branco L. R. R., Pinho R. T., The in vitro Mycobacterium bovis BCG moreau infection of human monocytes that induces caspase-1 expression, release and dependent cell death is mostly reliant upon cell integrity. J. Inflamm. 16, 18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phan T. H., Ummels R., Bitter W., Houben E. N., Identification of a substrate domain that determines system specificity in mycobacterial type VII secretion systems. Sci. Rep. 7, 42704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Puri R. V., Reddy P. V., Tyagi A. K., Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues. PLOS ONE 8, e70514 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koster S., Upadhyay S., Chandra P., Papavinasasundaram K., Yang G. Z., Hassan A., Grigsby S. J., Mittal E., Park H. S., Jones V., Hsu F. F., Jackson M., Sassetti C. M., Philips J. A., Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. U.S.A. 114, E8711–E8720 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Romagnoli A., Etna M. P., Giacomini E., Pardini M., Remoli M. E., Corazzari M., Falasca L., Goletti D., Gafa V., Simeone R., Delogu G., Piacentini M., Brosch R., Fimia G. M., Coccia E. M., ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8, 1357–1370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu J. J., Laine O., Masciocchi M., Manoranjan J., Smith J., Du S. J., Edwards N., Zhu X., Fenselau C., Gao L. Y., A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol. Microbiol. 66, 787–800 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Kang P. B., Azad A. K., Torrelles J. B., Kaufman T. M., Beharka A., Tibesar E., DesJardin L. E., Schlesinger L. S., The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202, 987–999 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan C. H., Qu Z. L., Tang X. L., Liu Q., Luo W., Huang C., Pan Q., Zhang X. L., Mycobacterium tuberculosis mannose-capped lipoarabinomannan induces IL-10-producing B cells and hinders CD4+ Th1 immunity. iScience 11, 13–30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chai Q. Y., Wang X. D., Qiang L. H., Zhang Y., Ge P. P., Lu Z., Zhong Y. Z., Li B. X., Wang J., Zhang L. Q., Zhou D. W., Li W., Dong W. Z., Pang Y., Gao G. F., Liu C. H., A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat. Commun. 10, 1973 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L., Liu Z., Wang J., Liu H., Wu J., Tang T., Li H., Yang H., Qin L., Ma D., Chen J., Liu F., Wang P., Zheng R., Song P., Zhou Y., Cui Z., Wu X., Huang X., Liang H., Zhang S., Cao J., Wu C., Chen Y., Su D., Chen X., Zeng G., Ge B., Oxidization of TGFβ-activated kinase by MPT53 is required for immunity to Mycobacterium tuberculosis. Nat. Microbiol. 4, 1378–1388 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Ates L. S., Dippenaar A., Ummels R., Piersma S. R., van der Woude A. D., van der Kuij K., Le Chevalier F., Mata-Espinosa D., Barrios-Payán J., Marquina-Castillo B., Guapillo C., Jiménez C. R., Pain A., Houben E. N. G., Warren R. M., Brosch R., Hernández-Pando R., Bitter W., Mutations in ppe38 block PE_PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat. Microbiol. 3, 181–188 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Liu C. H., Liu H., Ge B., Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 14, 963–975 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jain P., Hsu T., Arai M., Biermann K., Thaler D. S., Nguyen A., González P. A., Tufariello J. M., Kriakov J., Chen B., Larsen M. H., Jacobs W. R. Jr., Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. MBio 5, e01245–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bardarov S., Bardarov S., Pavelka M. S., Sambandamurthy V., Larsen M., Tufariello J., Chan J., Hatfull G., Jacobs W. R., Specialized transduction: An efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Ma Y., Chen H. D., Wang Y., Wang Q., Li Y., Zhao Y., Zhang X. L., Interleukin 24 as a novel potential cytokine immunotherapy for the treatment of Mycobacterium tuberculosis infection. Microbes Infect. 13, 1099–1110 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun X., Pan Q., Yuan C., Wang Q., Tang X. L., Ding K., Zhou X., Zhang X. L., A single ssDNA aptamer binding to mannose-capped lipoarabinomannan of Bacillus Calmette-Guérin enhances immunoprotective effect against tuberculosis. J. Am. Chem. Soc. 138, 11680–11689 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/43/eaba4733/DC1