

Abstract
Epigenetic regulation of gene expression in cancer cells has been extensively studied in recent decades, resulting in the FDA approval of multiple epigenetic agents for treating different cancer types. Recent studies have revealed novel roles of epigenetic dysregulation in altering the phenotypes of immune cells and tumor-associated stromal cells, including fibroblasts and endothelial cells. As a result, epigenetic dysregulation of these cells reshapes the tumor microenvironment (TME), changing it from an antitumor environment to an immunosuppressive environment. Here, we review recent studies demonstrating how specific epigenetic mechanisms drive aspects of stromal and immune cell differentiation with implications for the development of solid tumor therapeutics, focusing on the pancreatic ductal adenocarcinoma (PDA) TME as a representative of solid tumors. Due to their unique ability to reprogram the TME into a more immunopermissive environment, epigenetic agents have great potential for sensitizing cancer immunotherapy to augment the antitumor response, as an immunopermissive TME is a prerequisite for the success of cancer immunotherapy but is often not developed with solid tumors. The idea of combining epigenetic agents with cancer immunotherapy has been tested both in preclinical settings and in multiple clinical trials. In this review, we highlight the basic biological mechanisms underlying the synergy between epigenetic therapy and immunotherapy and discuss current efforts to translate this knowledge into clinical benefits for patients.
Keywords: Epigenetics, Immuno-modulation, Tumor microenvironment
Subject terms: Cancer microenvironment, Translational immunology
Introduction
Pancreatic ductal adenocarcinoma (PDA) is the third leading cause of cancer-related death in the United States, with a high mortality rate and an overall 5-year survival of ~9%.1,2 Despite recent advances in the management of PDA, for the majority of PDA patients, surgery remains the only chance of cure; however, only 10–20% of the total patient population are eligible for surgery.3 Immunotherapy for many tumor types is a research hotspot; however, it has yet to be effective in treating solid tumors, including PDA, primarily due to the immunosuppressive nature of the PDA tumor microenvironment (TME), with prominent dense stromal components.4,5 It has been shown that stromal and immune cells within the PDA TME undergo cell differentiation to acquire a more immunosuppressive phenotype that supports tumor growth.6 Different mechanisms are related to cellular programming in the TME, with increasing interest in epigenetic regulation. In this review, we highlight the major types of epigenetic modifications of different stromal components in the solid tumor TME with a focus on PDA and address the important roles that these modifications play in driving an immunosuppressive TME. In addition, we discuss the implications of epigenetic drugs used for sensitizing TME to cancer immunotherapy with a summary of the current states of the preclinical and clinical development of combination therapy based on epigenetic agents and cancer immunotherapy.
The tumor microenvironment of PDA
The PDA TME is an aggregate of tumor cells and nontumor stromal cells embedded in extracellular matrix (ECM) proteins.7 Different cell types constitute the PDA tumor stroma, including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), lymphocytes, and endothelial cells (Fig. 1).8 Together, they establish an overall immunosuppressive TME during tumorigenesis.
Fig. 1.
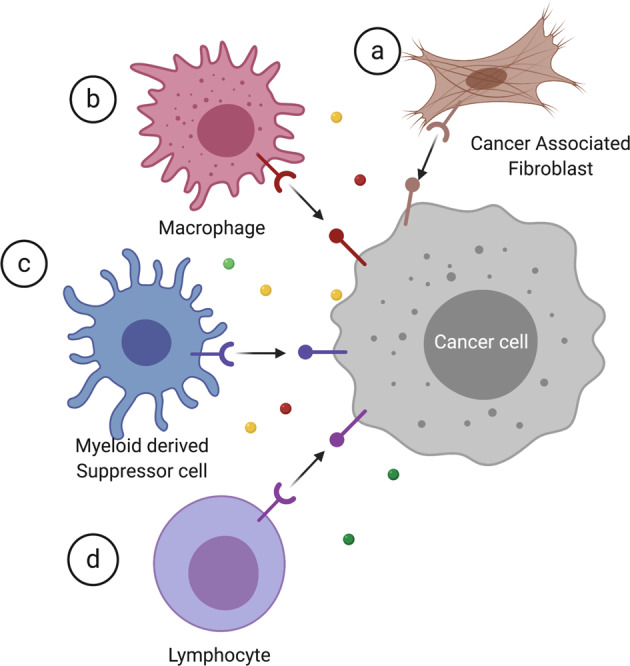
Stromal components within the tumor microenvironment. Major cell types include cancer-associated fibroblasts, macrophages, myeloid-derived suppressor cells, and lymphocytes
Cancer-associated fibroblasts
CAFs are major types of stromal cells that may account for as much as 90% of the whole PDA tumor mass.5 CAFs originate from activated pancreatic stellate cells and resident fibroblasts. They are activated in response to various stimuli from both tumor cells through direct contact and cytokines, including growth factors such as transforming growth factor-β (TGF-β) that are secreted within the TME.9,10 Upon activation, CAFs acquire tumor-promoting phenotypes that support tumor growth through various mechanisms. First, CAFs can secrete ECM proteins such as MMP2 to provide signals for supporting structures and tumor cells growth and migration.11,12 Second, CAFs can also secrete a variety of growth factors, cytokines, and chemokines such as CCL2 and CXCL12 to communicate with the surrounding tumor cells and other stromal components, leading to tumor growth, angiogenesis, cell stemness, and immunosuppression.13–16 Third, CAFs can promote tumor invasion by establishing direct contacts with tumor cells through the hedgehog signaling pathway.17 CAFs exhibit substantial heterogeneity with multiple proposed origins, including reprogrammed resident fibroblasts converted from adipocytes, endothelial or epithelial cells and differentiated from bone marrow-derived mesenchymal or hematopoietic stem cells.18 Activated CAFs are categorized into three types: myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs). MyCAFs and iCAFs are the most common types of CAFs in PDA, while apCAFs are among the subtypes newly discovered from single-cell RNA-sequencing.16,19 MyCAFs are located adjacent to tumor cells and express α-SMA, while iCAFs are located more distant from tumor cells within dense stroma, and they secrete inflammatory cytokines such as interleukin (IL)-6 and IL-11.20 ApCAFs express MHC class II molecules and CD74 and low levels of classical costimulatory molecules such as CD80, CD86, and CD40.19 MyCAFs are thought to directly interact with PDA tumor cells and are speculated to be induced by TGFβ signaling, while iCAFs may indirectly interact with tumor cells through paracrine IL1a signaling.16 ApCAFs are speculated to have an immune-modulatory capacity, as they can activate CD4+ T cells in an antigen-specific fashion.19 These three subtypes of CAFs have distinct transcriptomic profiles; however, gene expression analyses of CAFs in human breast and ovarian cancer revealed that somatic mutations are extremely infrequent in CAFs.16,19,21 Thus, it is more likely that the different CAF phenotypes are regulated at the epigenetic level, which we highlight in the next section.
Tumor-associated macrophages
Macrophages belong to the myeloid cell lineage. Tissue macrophages have two cell origins, either differentiate from circulating monocytes from the bloodstream or are seeded during the early development of different organs, where they resident as macrophages.22 Macrophages show great plasticity and are highly responsive to environmental cues, including pathogens, foreign antigens, cytokines, and chemokines. Activated macrophages can be categorized into M1 (classical activated) and M2 (alternative activated) phenotypes.23 Classical M1 activation occurs in response to bacterial infections such as lipopolysaccharide (LPS) and immune stimuli such as interferon γ (IFNγ). M1 macrophages can also mediate the innate immune response against intracellular parasites and tumors, which result in tissue disruption and local inflammation by secreting molecules such as tumor necrosis factor α (TNF-α), reactive nitrogen, and oxygen species. In addition, M1 macrophages promote T-helper-1 (Th1) responses.24 In contrast, alternative M2 macrophages are responsive to different stimuli, such as cytokines IL-4, IL-13, IL-10, and glucocorticoid hormones. M2 macrophages play key roles in mitigating inflammatory responses and wound healing. They also generally present with an immunosuppressive phenotype, as indicated through the release of immunosuppressive cytokines such as IL-10 that promote a Th2 immune response.25,26 TAMs were previously thought to adopt primarily the M2-like phenotype,27 through induction by tumor cells to promote cancer growth and metastasis and block T-cell function and proliferation.28 However, recent evidence suggests that TAMs undergo the M1–M2 transition during tumor progression. M1 macrophages are mainly abundant in chronic inflammatory sites, where tumors are initiated.29,30 During cancer progression, macrophages switch to an M2-like phenotype as the tumor begins to invade, vascularize, and develop.31,32 These findings were validated using human cancer patient samples in studies of M1 and M2 macrophage using CD68 and CD204 as respective M1 and M2 macrophage markers. More M2-like macrophages were found in patients with PDA than in patients with chronic pancreatitis. High numbers of M2 macrophages correlated with worse prognoses for the PDA patients.33 This study highlighted the plasticity of macrophages during PDA development. Various mechanisms are thought to regulate the reprogramming of M1-like TAMs to become M2-like TAMs; here, we discuss how epigenetic changes in macrophages regulate their phenotypes and functions.
Myeloid-derived suppressor cells
MDSCs constitute a heterogeneous population of immature myeloid cells (IMCs) that, with TAMs, CAFs, and T regulatory (Treg) cells, drive the immunosuppressive features of the TME.34 MDSCs are derived from IMCs in the bone marrow during the normal process of myelopoiesis.35 IMCs migrate into the circulatory system and to various peripheral organs, where they differentiate into granulocyte/macrophage progenitor (GMP) cells. In a cancer setting, rather than differentiating into monocytic/dendritic progenitor cells or myeloblasts, these GMPs are affected by the excess production of tumor-induced factors, such as IL-1β, within the TME and differentiate into MDSCs.36 MDSCs are characterized into three major types based on their phenotypic and morphological features or cell surface markers: Polymorphonuclear (PMN-MDSCs), which are similar to neutrophils; granulocytic MDSCs, which are similar to granulocytes; and mononuclear or monocytic MDSCs (M-MDSCs), which are similar to monocytes.37 In many cancer types, PMN-MDSCs comprise 80% of the total MDSCs.35 The major function of MDSCs in cancer is the establishment of the immunosuppressive TME through various mechanisms, including depletion of amino acids that are important for T-cell function, decreasing the trafficking of CD8+ effector T cells, increasing Treg cell responses, supporting tumor invasion, and angiogenesis.38,39 MDSCs also suppress T lymphocyte function directly or indirectly through the secretion of a set of immunosuppressive molecules, such as prostaglandin E2, TGF-β, and IL-10.40
PDA tumor cells can directly produce granulocyte macrophage colony-stimulating factor (GM-CSF) to promote MDSC accumulation. The hypoxic environment of the TME also plays a key role in driving immunosuppressive MDSC differentiation by elevating the level of secreted hypoxia-inducible factor 1 (HIF-1).41,42 Thus, understanding the mechanisms by which MDSCs are generated and function is important for the development therapeutics targeting MDSCs. Recent studies have suggested that epigenetic modifications of MDSCs play key roles in shaping MDSC formation and function. In this review, we highlight some recent studies that address the epigenetic regulation of MDSCs.
T lymphocytes
T lymphocytes are major immune cells in the adaptive immune system that are classified into two major categories: MHC class-II-restricted CD4-expressing T cells (CD4), which can differentiate into different TH cell types, and MHC class-I-restricted CD8-expressing T cells (CD8), which can differentiate into cytotoxic effector T lymphocytes (CTLs) that kill virus-infected cells and tumor cells.43 T-cell activation can be divided into two phases, the acute phase and the memory phase. During acute infections, naive T cells are rapidly activated and differentiated into CTLs upon antigen stimulation. These differentiation and programming processes involve epigenetic regulation.44 After antigen clearance, most CTLs die, but a small differentiated fraction become memory T cells that can provide long-term protection upon a second exposure to the same antigen. Memory T-cell differentiation is also partially regulated by epigenetic changes.45 In the tumor setting, T-cell functions are dysregulated. Specifically, T cells undergo T-cell exhaustion due to repetitive stimulation by tumor antigens and various immunosuppressive factors within the TME.46 The process of T-cell exhaustion is also controlled by epigenetic regulation. T-cell presence is crucial in the TME. Enrichment of T lymphocytes within the TME is a prerequisite for the success of cancer immunotherapy.47 CD8+ T cells and FoxP3+ Treg cells are two major T-cell populations in the TME that play opposite roles. The major function of effector CD8+ T cells is the direct killing of tumor cells, and a small fraction of effector CD8+ T cells later become memory T cells to provide long-term protection. In contrast, FoxP3+ regulatory T cells have immunosuppressive roles that repress effector T-cell function.48 Interestingly, the localization and phenotypes of T lymphocyte population present great heterogeneity among human PDA patients. Studies have confirmed the presence of CD8+ T cells and Treg cells in human PDA patients are preferentially enriched in tertiary lymphoid structures; however, the majority of CD8+ T cells are not considered functional because of the lack of gene expression upon T-cell receptor signaling.49 The role of Treg cells in PDA is controversial. One study showed that depletion of Treg cells in a mouse model of PDA led to tumor progression rather than the reversal of immunosuppression.50 In this review, we discuss the epigenetic mechanisms that regulate effector and memory T-cell differentiation and T-cell exhaustion.
In conclusion, stromal and immune cells are differentiated and reprogrammed within the TME to adopt an immunosuppressive phenotype that promotes cancer growth and metastasis during tumorigenesis. As a result, targeting the mechanisms regulating stromal and immune cell differentiation presents great promise for cancer therapeutic development. In this review, we highlight how epigenetic regulation plays key roles in driving stromal and immune cell differentiation in the TME.
Introduction to basic epigenetic mechanisms
All cell types within a multicellular organism share homogenous genes but differentially expression them through epigenetic regulation, which gives rise to structurally and functionally heterogeneous cell types.51 Epigenetic regulation occurs during early development, such as genomic imprinting, is retained through mitosis and is heritable.52 Epigenetic regulation also occurs in a nonheritable fashion during plastic cell programming and differentiation, such as CAF activation and differentiation induced by tumor cells.53 Epigenetic regulation alters gene expression (activation and suppression) through the modification of DNA and histones and repositioning of the chromatin structure without changing the DNA itself.54 In this review, we cover the basic epigenetic regulatory mechanisms, including DNA methylation, histone modification, and chromatin remodeling.
DNA methylation and demethylation
DNA methylation, the most commonly studied epigenetic mechanism, is a modification in which a methyl group is added to a cysteine residue through covalent bonds. This process is controlled by the DNA methyltransferase (DNMT) family, which includes DNMT1, DNMT3A, DNMT3B, and DNMT demethylases, which are in the ten-eleven translocase (TET) group of enzymes.55 DNA methylation usually occurs at CpG sites where a cytosine residue is followed by a guanine residue.56 These CpG sites are often found in the promoter or other regulatory regions of genes.57 Hypermethylation of CpG sites at promoter or enhancer regions often leads to gene suppression, as DNA methylation causes changes in chromatin structure; that is, it transitions from the euchromatin form (loose or open, transcription-permissive) to the heterochromatin (condensed or closed, silent) form, which blocks the access of transcription factors to gene promoters.58,59 The methylation of CpG sites in gene bodies is found to activate gene expression in cancer cells.60 The DNMT family consists of DNMT1, DNMT3A, DNMT3B, and DNMT3L, where DNMT1 is critical for methylation maintenance, while DNMT3A and DNMT3B are crucial for de novo methylation.61,62 DNMT3L does not have enzymatic activity.63 DNMT1 specifically recognizes hemimethylated DNA single strands following DNA replication, while DNMT3A and DNMT3B regulate DNA methylation on both strands independent of DNA replication.64,65 Although DNMT1, DNMT3A, and DNMT3B control DNA methylation in different scenarios, they also cooperate in certain circumstances. Studies have shown that DNMT1 and DNMT3B work together to silence genes by coregulating DNA methylation patterns in human colorectal cancer cells.66,67 DNA demethylation is regulated by a TET group of enzymes. TETs oxidize 5-methyl-cytosine to form cytosine through a 5-hydroxymethyl-cytosine (5-hmC) intermediate. TETs play important roles in several solid tumors, in which loss of function promotes aberrant DNA methylation.68 Loss of 5-hmC regulated by TETs has been reported as an epigenetic hallmark of melanoma.69
Histone modification
Histone modification includes different types of posttranslational modifications (PTMs) at histone protein N-terminal tails, including acetylation, methylation, phosphorylation and, less commonly, ubiquitylation, SUMOylation, deamination, and lactylation.70 Here, we discuss the most commonly studied types of PTM, histone acetylation, histone methylation, and histone phosphorylation. These processes are regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), lysine-specific histone methyltransferases (HKMT), and lysine-specific demethylases (LSD), protein kinases and phosphatases.71 The result of histone modification is regulation of gene expression through the alteration of the chromatin structure to facilitate or impede the binding of transcriptional machinery that is involved in gene transcription.72 Histone acetylation is a dynamic and reversible process in which acetyl groups are added to positively charged histone lysine residues by HATs, while in the reverse reaction, acetyl groups are removed by HDACs such as HDAC6 through deacetylation.73 Histone acetylation leads to the “opening” of chromatin and is thus critical for gene activation.74 Histone (de)methylation is the (removal)/addition of methyl groups, most commonly found on lysine residues, by HKMTs such as enhancer of zeste homolog 1, 2 (EZH1, EZH2) and LSDs such as LSD1.71,75 This process can either activate or silence gene transcription depending on the histone, amino acid and residue methylated, degree of modification (mono-, di-, or trimethylation), attraction of additional function-specific protein cofactors to the site, and the existence of other methyl or acetyl groups in close proximity.76 For example, H3K9 methylation is a transcription repression mark, while H3K4 methylation is a transcription activation mark.77 Histone (de)phosphorylation is similar to histone acetylation, which modifies serine, threonine and tyrosine residues, requiring protein kinases and phosphatases to attach or remove phosphate groups.71,78 The functions of histone (de)phosphorylation not only regulate gene transcription but also other biological processes, such as DNA repair and cell cycle progression.79
Chromatin remodeling
Chromatin remodeling is a process in which chromatin-remodeling complexes (remodelers) together with other chromatin factors control chromatin organization through complete or partial nucleosome repositioning.80,81 This process is regulated by four major families of remodelers, namely, the SWI/SNF family, ISWI family, CHD family and INO80 family, and high-mobility group (HMG) proteins.82,83 The chromatin-remodeling process alters gene accessibility to transcription machinery to regulate gene expression and other processes, including DNA replication and DNA repair.82 Three major types of chromatin-remodeling processes are described. First, chromatin remodeling can be initiated via nucleosome sliding, which is the movement of a core histone octamer, which remains intact.84 The second type is nucleosome ejection, which causes a nucleosome to segregate away from the chromatin chain. The third type is histone eviction, which is the removal of histone H2A/H2B dimers from the DNA-associated nucleosome.85 These processes are regulated by a number of ATP-dependent chromatin remodelers with high binding affinity for modified core histone tails, through which they regulate both gene transcription activation and repression.86 SWI/SNF family remodelers have been found to be frequently altered in PDA; specifically, genomic sequencing showed that ARID1A, ARID1B, PBRM1, SMARCA2, and SMARCA4 are mutated in PDA.87 Mutations of individual subunits within the SWI/SNF complex are less frequent but together are found in approximately one-third of all PDA patients.87–89 As a result, SWI/SNF is considered a central tumor suppressive complex in PDA.
In conclusion, different epigenetic mechanisms work closely together to regulate gene expression by mediating the dynamic states of heterochromatin and euchromatin in different biological processes.
Epigenetic regulation of stromal and immune cells within the tumor microenvironment
DNA methylation in cancer-associated fibroblasts
As discussed above, CAFs show distinct transcriptomic profiling, yet rarely express somatic mutations.16,19,21 Thus, distinct gene expression signatures in CAFs are likely caused by the regulation at the epigenetic level. Multiple studies have revealed altered DNA methylation status of genes in CAFs isolated from prostate and breast cancer tissues.90,91 Here, we discuss how DNA methylation regulates CAF differentiation and function, focusing on four studies performed by different groups and some published and unpublished work by our own group. The first study discussed was performed by our group, who showed that CAFs adopt unique DNA methylation and expression patterns upon interaction with PDA tumor cells.53 In this study, a combined array analysis of DNA methylation and gene expression in human mesenchymal stem cells (MSCs), which are among the originators of CAFs, revealed that ~1585 genes were both methylated and downregulated, including the Suppressor of cytokine signaling 1 (SOCS1) gene, through direct contact with PDA tumor cells in cocultures. We also showed that SOCS1 inhibition by the tumor-induced methylation of CAFs activated activator of transcription 3 (STAT3) signaling, which induced IGF-1 expression and supported PDA cell growth in both an in vitro setting and in the tumor xenografts in mice. This process was prevented by the DNMT1 inhibitor (DNMTi) 5-aza-2′-deoxycytidine (decitabine). A recent study performed by Ohlund et al. also showed that myCAFs had the potential to be induced by TGFβ signaling upon direct contact with PDA tumor cells, while iCAFs may have been induced by paracrine IL1a signaling through indirect interaction with tumor cells. They also showed that myCAFs adopted a cancer-promoting phenotype, while iCAFs adopted an immunosuppressive phenotype.16 Inspired by this study, we sought to explain the distinct transcriptional profiles of myCAFs and iCAFs by studying epigenetic changes of genes related to myCAF and iCAF differentiation using data generated from a combined array analysis of human MSCs. Surprisingly, we found that human MSCs induced the DNA methylation of the IL1A and IL1B genes in coculture with PDA tumor cells, with which they directly interacted, a finding consistent with previously described direct interactions between myCAFs and tumor cells that regulated myCAF differentiation.16 From these findings, we hypothesized that methylation and downregulation of IL1A and IL1B induced by tumor cells, potentially through TGFβ signaling, locks CAFs into the myCAF phenotype and prevents the transformation of myCAFs into iCAFs, which in turn directly supports cancer growth. These findings also present a novel strategy to target CAFs; that is, targeting both IL1 signaling and TGFβ signaling may prevent the differentiation of both iCAFs and myCAFs.
Epigenetic regulation of CAFs can be induced not only by tumor cells through direct contact but also indirectly through factors that are secreted. The second study we discuss was performed by Albrengues et al. They found that normal fibroblasts can be reprogrammed to adopt a pro-invasive phenotype by leukemia inducible factor (LIF), a proinflammatory cytokine secreted by tumor cells.10 LIF induced methylation through DNMT3B of the promoter region in the protein phosphatase regulator Src homology 2 domain-containing protein tyrosine phosphatase 1 (Shp-1). The repression of Shp-1 resulted in the constitutive activation of Janus-activated kinase 1/signal transducer (JAK1)/STAT3 signaling, which drove the reprogramming of normal fibroblasts into pro-invasive CAFs. This reprogramming process was prevented by the DNMTi decitabine, which restored Shp-1 expression and inhibited JAK1/STAT3 signaling.
In addition to hypermethylation, hypomethylation of CAFs was also identified by multiple groups. The third study we describe was based on an Affymetrix exon array analysis performed by Yu et al., who studied DNA methylation alterations in human PDA CAFs by comparing genes that were upregulated by DNMTi decitabine with those of pancreatic control fibroblasts using cultured cells isolated from PDA patients and nonneoplastic pancreas tissues.92 One gene was found to be overexpressed in the CAFs: a disintegrin and metalloprotease 12 (ADAM12). ADAM12 is a regulator of cell–cell and cell–matrix interactions that is overexpressed in CAFs and has been implicated in the support of tumor progression.93 This group later used bisulfate sequencing to confirm that overexpression of ADAM12 was regulated through hypomethylation at the gene promoter region. This study showed that aberrant hypomethylation is a mechanism through which gene activation reprograms PDA CAFs to support tumor growth. The fourth study described is an epigenomic analysis of patient-derived and de novo generated PDA CAFs performed by Bhagat et al., who demonstrated that a widespread loss of DNA methylation was associated with the overexpression of various inflammatory genes, including interleukins and chemokines, such as IL1A, CCL5, and CCL26, and cellular receptors, such as CXCR4 and ICAM3, which are all involved in critical cell signaling pathways in tumor progression.94 Interestingly, metabolite tracing used in this study revealed that the lactate produced by PDA tumor cells led to increased MSC production of alpha-ketoglutarate (aKG), an important cofactor of the DNA demethylase TET. Conversely, aKG mediated the activation of TET enzymes, which led to decreased DNA methylation during the de novo differentiation of MSCs to CAFs. This study presents a novel mechanism that links tumor metabolism to the epigenetic regulation of CAFs, where tumor cells induce the hypomethylation of genes in PDA CAFs by regulating the TET function via secreted metabolites.
In conclusion, both DNA hypermethylation and hypomethylation of different genes contribute to unique DNA methylation and gene expression signatures during CAF differentiation, resulting in reprogramming of CAFs to induce their acquisition of tumor-promoting phenotype.
Histone modification in cancer-associated fibroblasts
Histone modification is a commonly studied type of epigenetic regulation. To the best of our knowledge, no study has testing the role of histone modification in PDA CAFs, with the limited few conducted on breast cancer. Here, we discuss two studies that demonstrating that CAF differentiation is regulated by histone modifications. The first study was performed by Tyan et al., who demonstrated that histone demethylation was a result of HAT EZH2 repression associated with increased expression of an ECM-modifying proteoglycanase, ADAMTS, in CAFs.95 Tyan et al. first identified an increased level of ADAMTS1, both at the mRNA and protein level, in fibroblasts derived from normal human breast tissue cells cocultured with breast cancer cells and identified that this overexpression of ADAMTS1 was linked to a decrease in the repression marker H3K27me3 at the promoter but no change in DNA methylation. In addition, they found that the level of the histone methyltransferase critical for the H3K27me3 mark, EZH2, was decreased at this promoter, suggesting a role for EZH2 in the repression of the ADAMTS1 gene in normal fibroblasts that was inhibited in CAFs because of the effect of the breast cancer cells. In another study on breast cancer, Li et al. showed that, of breast cancer tissue, histone deacetylase 6 (HDAC6) was frequently upregulated in the CAFs, which promoted an immunosuppressive TME.96 They also showed that the upregulation of HDAC6 regulated the activation of STAT3, which initiated the expression of prostaglandin E2/cyclooxygenase-2 (COX2), resulting in the immunosuppressive action of the CAFs. To validate this study, they also pharmacologically inhibited HDAC6 through inhibitors and genetic knockdown of HDAC6 in CAFs and showed that the inhibition of HDAC6 suppressed the tumor recruitment of MDSCs and regulatory T cells and increased CD8+ and CD4+ T-cell activation, which together delayed tumor growth in vivo. Taken together, these findings demonstrate the role of histone modification in reprogramming CAFs, which acquire a tumor-promoting phenotype through the regulation of gene expression.
Chromatin remodeling in cancer-associated fibroblasts
Chromatin-remodeling complexes have not been extensively studied thus far. One study on prostate cancer by Zong et al. showed that the overexpression of Hmga2 (HMG AT-hook 2), which is a member of the HMG family, in prostate stromal cells was sufficient to induce dramatic neoplasia lesion formation of adjacent naive epithelial cells within the TME.97 They later discovered that this process was mediated by the overexpression of Wnt ligands upon paracrine signaling that activated Wnt/β-catenin signaling in the epithelial cells. This study revealed the role of chromatin remodeling in the acquisition of CAF phenotypes that drive tumorigenesis.
In summary, multiple epigenetic mechanisms, including DNA methylation, histone modification, and chromatin remodeling, together shape and reprogram the phenotypes of CAFs during tumorigenesis, which present opportunities for developing cancer therapeutics targeting these mechanisms (Fig. 2).
Fig. 2.
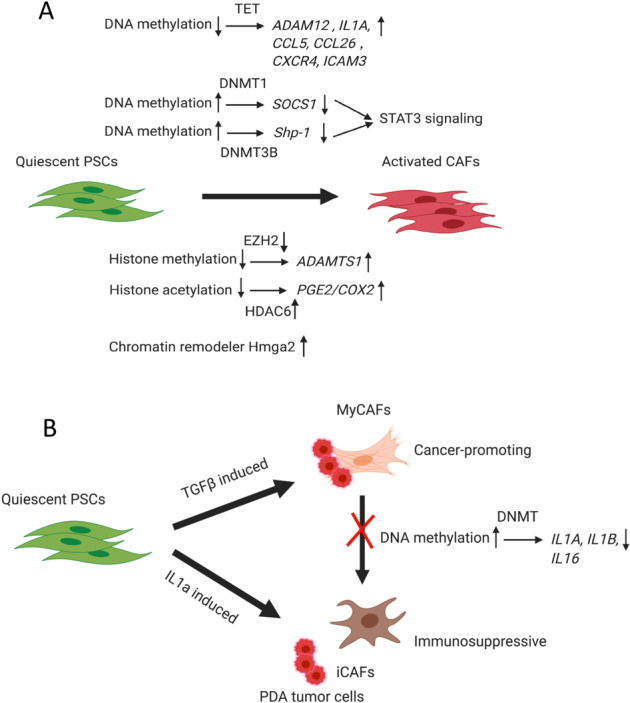
Epigenetic regulation in cancer-associated fibroblasts. a DNA methylation of SOCS1 regulated by DNMT1 and Shp-1 regulated by DNMT3B leads to gene repression, which activates STAT3 signaling during CAF differentiation and activation. DNA hypomethylation by TET, which leads to the upregulation of ADAM12, IL1A, CCL5, CCL26, CXCR4, and ICAM3 and triggers CAF differentiation. Decreased histone methylation regulated by EZH2 results in the upregulation of ADAMTS1 during CAF activation. Increased histone acetylation regulated by HDAC6 controls the upregulation of PGE2/COX2 during CAF activation. The chromatin remodeler Hmga2 is also induced to facilitate CAF activation. b Quiescent PSCs can be differentiated into either myCAFs or iCAFs, while myCAFs and iCAFs can also be reprogrammed interchangeably. MyCAFs can be induced by TGFβ signaling through direct contact with PDA tumor cells, while iCAFs can be induced by paracrine IL1a signaling through indirect interaction with tumor cells. MyCAFs have a cancer-promoting phenotype, while iCAFs have an immunosuppressive phenotype. DNA methylation of the IL1A and IL1B genes and their subsequent downregulation were observed in human MSCs cocultured with PDA tumor cells because of direct interaction, which potentially locked CAFs into the myCAF phenotype and prevented the transformation of myCAFs into iCAFs, supporting tumor growth
DNA methylation in tumor-associated macrophages
In the previous section, we discussed how macrophages show pronounced plasticity during cell differentiation and TAM reprogramming. Here, we explore DNA methylation changes in macrophages, focusing on four studies performed by different groups and some unpublished work performed by our group. The first study was performed by Schuyler et al., who identified unique DNA methylation patterns linked to myeloid lineages during development. They also observed an increased global methylation level during macrophage differentiation and activation by analyzing 112 whole-genome bisulfite-sequencing data sets created by the BLUEPRINT Epigenome Project.98 As unique DNA methylation signatures were observed during macrophage differentiation, the roles of DNMTs in macrophage differentiation were also tested. In another study, Yang et al. showed that DNMT3B was involved in M2 differentiation and phenotypic control, while the knockdown of DNMT3B in RAW264.7 macrophages and mouse bone marrow-derived macrophages (BMDMs) induced M2 polarization and prevented M1 marker expression.99 In contrast, DNMT1 was shown to positively regulate the M1 phenotype by silencing SOCS1 in LPS-stimulated RAW264.7 cells.100
DNA demethylation in macrophages has also been described. The fourth study was performed by Sun et al., who discovered a novel mechanism by which TNF-α overexpression in M1 macrophages can be regulated by DNA demethylation through the DNA demethylase TET1.101
DNA methylation in TAMs has not been widely studied. Preliminary research performed by our group revealed a novel mechanism by which PDA tumor cells induced DNA methylation changes in metabolic genes, such as NAD(P)H dehydrogenase (quinone-1) (NQO-1) and aldehyde dehydrogenase family 1 member a3 (ALDH1a3), in macrophages, which led to the metabolic reprogramming of macrophages, ultimately leading to the functional reprogramming of macrophages to promote the M1–M2 transition. This process was prevented by using the DNMTi decitabine to prevent tumor the cell migration induced by TAMs in vitro and to prevent tumor metastasis in vivo.
Histone modification in tumor-associated macrophages
Histone modification is the most common type of epigenetic regulation studied in macrophages. Histone methylation and demethylation have been found to regulate M1 and M2 differentiation, with studies showing different enzymes regulate these activities. The histone methyltransferase PRMT1 has been shown to positively regulate M2 macrophages while negatively regulating M1 macrophages through the induction of PPARγ in the M2 macrophages.102 Other HMTs, such as SMYD3, were found to be positive regulators of M2 polarization.103 JMJD3, an H3K27 demethylase, was also categorized as an essential regulator of M2, as indicated through its induction of Irf4, Arg1, and CD206.104 Here, we highlight two studies. The first study, performed by Yu et al., was novel in that it linked metabolism with histone methylation in macrophages. In this study, the authors revealed the synergistic effect of glucose with amino acid metabolism to fuel the production of S-adenosylmethionine, the methyl group donor of histone methylation, to support histone H3K36 trimethylation for inducing the IL-1β production that drives inflammatory macrophages.105 Histone methylation of macrophages was not well studied in PDA but has recently been studied in a breast cancer model. The second study was performed by Tan et al., who discovered that histone methylation played key roles in driving macrophage programming towards the acquisition of an antitumor M1-like phenotype, which is useful for treating cancer.106 They first showed that classical (M1) macrophages had reduced expression of histone demethylase LSD1, nuclear REST corepressor 1, and the zinc finger protein SNAIL in a murine triple-negative breast cancer model. Inspired by this observation, they then tested the effect of the LSD1 inhibitor phenelzine on the programming of the macrophage phenotype and demonstrated that phenelzine treatment increased the transcription and expression of M1-like signatures both in vitro and in vivo. This study suggested a role for modulating histone methylation to manipulate macrophage programming towards an antitumor M1-like phenotype for treating cancer.
Histone acetylation and deacetylation are also prevalent in macrophages. HDAC9 and HDAC11 are negative regulators of M2 polarization.107,108 HDAC SIRT2 and HDAC4 act as positive M2 phenotype regulators by inducing Gata3, Arg1, and Cd11c expression in IL4-stimulated mouse BMDMs.109,110 Here, we discuss one study that demonstrated a novel role for SIRT1/2 in regulation coupled with DNA methylation via DNMT3B during macrophage differentiation.111 SIRT1/2 were significantly upregulated during macrophage differentiation. The authors then discovered that SIRT1/2 mediated gains of methylation accompanied by decreases in activating histone marks at inflammatory loci in genes, such as ADORA2A, RUNX3, and JAK, which resulted in general gene repression. Inhibition of SIRT1/2 upon macrophage activation stimulated by LPS challenge abrogated the DNA methylation gains mediated by SIRT1/2, resulting in the upregulation of these inflammation-related genes. Together, this study confirms the role of SIRT1/2 in restricting the premature activation of proinflammatory genes through the mediation of methylation gains concomitant with activating histone mark reduction at inflammatory gene loci. HDACs are also found to link metabolism with epigenetics. Here, we discuss one study that identified HDAC7 as a key molecular link between Toll-like receptor (TLR)-inducible aerobic glycolysis and macrophage inflammatory responses in murine macrophages.112 The overexpression of HDAC7 in transgenic mice can amplify LPS-inducible lactate secretion and promote a glycolysis-associated inflammatory signature. Through proteomic screening, the group also identified the glycolytic enzyme pyruvate kinase M isoform 2 (Pkm2) as a partner of HDAC7 in murine macrophages. The Hdac7–Pkm2 complex serves as an immunometabolism signaling hub, whereby Pkm2 deacetylation initiates its proinflammatory functions. Disrupting this complex was found to suppress inflammatory responses both in vitro and in vivo. Histone modification was also studied in TAMs. One group showed that tumor cells can induce the deacetylation of histones at promoter regions in the gene of a master regulator of MHC-II expression, the class II transactivator, in TAMs, which resulted in the downregulation of MHC class-II-dependent antigen presentation genes.113
A novel type of histone modification, histone lactylation, was recently discovered in macrophages, which also links metabolism with epigenetics. Zhang et al. showed that lactate-derived lactylation of histone lysine residues served as an epigenetic modification that directly activated gene transcription.114 Interestingly, they used M1 macrophages exposed to bacteria in a model system and found that histone lactylation showed temporal dynamics and that increased histone lactylation induced the expression of homeostatic genes involved in wound healing, including Arg1, in the late phase of M1 macrophage polarization. The role of histone lactylation in regulating M1 homeostasis suggests the possibility of targeting histone lactylation for preventing the M1–M2 macrophage transition in the tumors.
In summary, we discuss the roles of DNA methylation and histone modifications in macrophages that reprogram their phenotype to one that supports tumors and promotes cancer progression (Fig. 3). Drugs that can block or reverse these processes hold great potential as novel cancer therapies.
Fig. 3.
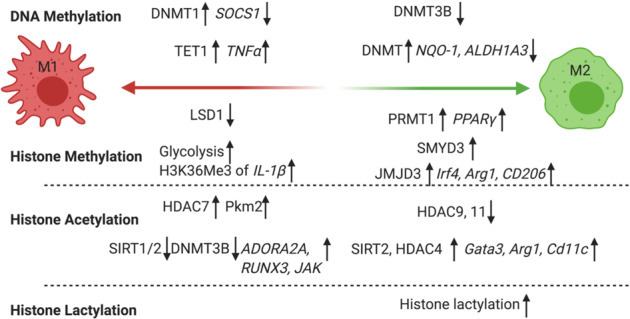
Epigenetic regulation in macrophages. DNMT3B is a negative regulator of M2. Tumor cells induced DNA methylation of NQO-1 and ALDH1A3, which promotes the M1–M2 transition. HMT PRMT1 is a positive regulator of M2 macrophages through induction of PPARγ. HMT SMYD3 is a positive regulator of M2 polarization. JMJD3, an H3K27 demethylase, is a positive regulator of M2 through its induction of Irf4, Arg1, and CD206. HDAC9 and HDAC11 are negative regulators of M2 polarization. HDAC SIRT2 and HDAC4 act as positive M2 regulators by inducing Gata3, Arg1, and Cd11c expression. Histone lactylation links metabolism with epigenetics in regulating M1 homeostasis and preventing M1–M2 macrophage transition in tumors. DNMT1 is a positive regulator of the M1 phenotype because it silences SOCS1. TET1 regulates M1 differentiation by promoting the overexpression of TNF-α. A reduced level of LSD1 is observed in M1 macrophages and is thought to maintain the M1 inflammatory gene expression signature. Glycolysis causes the accumulation of S-adenosylmethionine (SAM), which leads to increased histone H3K36 trimethylation for IL-1β production, which drives inflammatory macrophage differentiation. SIRT1/2 with DNMT3B prevent M1 activation through the repression of inflammatory genes
DNA methylation in myeloid-derived suppressor cells
DNA methylation of MDSCs is frequently studied in multiple cancer types. Here, we introduce findings from two studies. In one study, Sasidharan et al. studied how DNA methylation regulated gene expression changes in different subtypes of MDSCs in colorectal cancer patients. They first discovered that both immature MDSCs and PMN-MDSCs levels were increased in tumor tissues compared with normal control tissues.115 Interestingly, 17 genes associated with DNA methylation were downregulated in the PMN-MDSCs, while 50 DNA-mediated transcriptional silencing genes were upregulated in the tumor-infiltrating immature MDSCs. These results suggest a potential role of DNA methylation in regulating MDSC differentiation. In another study with ovarian cancer patients, Rodriguez et al. showed that MDSCs and DCs had different DNA methylation patterns.116 Inspired by this observation, they then discovered that prostaglandin E2, a proinflammatory mediator, induced the upregulation of DNMT3A in the MDSCs. As a result of DNMT3A upregulation, a number of genes, including S1PR4, RUNX1, IL1RN, and CCR2, were found to be hypermethylated specifically in the MDSCs compared with the levels in the corresponding peripheral monocytes. In the same study, the authors discovered that DNA was demethylated during MDSC generation, suggesting that a function-specific DNA methylation pattern could be established during MDSC generation.
Histone methylation in myeloid-derived suppressor cells
Histone methylation is less commonly studied in MDSCs. One paper by Redd et al. demonstrated that the histone methyltransferase SETD1B acted as a novel regulator of iNOS expression in MDSCs in a murine colorectal cancer model.117 In this paper, the authors first showed that tumor-induced MDSCs exhibited increased SETD1B expression. Later, they identified the target genes of SETD1B through chromatin immunoprecipitation and revealed that H3K4me3 was enriched at the nos2 promoter in the tumor-induced MDSCs, which upregulated iNOS expression. They also demonstrated that iNOS expression in MDSCs was abrogated after SETD1B was inhibited or silenced with inhibitors or RNA interference, respectively. This study presents a novel mechanism showing that iNOS expression is regulated through epigenetic regulation via SETD1B.
Histone acetylation in myeloid-derived suppressor cells
Among the different histone modifications, histone acetylation and deacetylation are the most commonly studied in MDSCs. Here, we discuss findings from four studies. First, in the same MDSC transcriptomic profiling study performed by Sasidharan et al., genes associated with HDAC activation and HAT-related genes were found to be upregulated in the tumor-infiltrating immature MDSCs in colorectal cancer patients.115 In contrast, the genes related to HDAC binding were downregulated in the PMN-MDSCs. To discern the role of HDAC in shaping MDSC function, the authors used HDAC inhibitors to treat cells isolated from human tumor tissue and found that these HDAC inhibitors significantly reduced the expression of ARG1, CCR2, and ITGAL, which are all genes related to immunosuppression and myeloid cell recruitment. Based on these findings, they concluded that HDAC activation played important roles in regulating the functions of immature MDSCs during the facilitation of immunosuppression and myeloid cell recruitment. In another study, Sahakian et al. revealed a specific role for HDAC11 as a key regulator of IL-10 gene expression in MDSCs.118 They first discovered that MDSCs isolated from EL4 thymoma tumor-bearing mice expressed high levels of HDAC11, while IMCs in the tumor-bearing mice expressed low levels of HDAC11, implying that the transition of IMCs to MDSCs requires a decrease in the expression of HDAC11. Using tumor-bearing HDAC11-knockout mice, they discovered that, without HDAC11 expression, the phenotype of the MDSCs was more suppressive than that of the wild-type (WT) during tumor-bearing control, and the HDAC11-KO tumor-bearing mice also exhibited faster tumor growth than the WT control mice. Together, this study suggests the role of HDAC11 as a negative regulator of MDSC expansion/function. The third study we present here, performed by Youn et al., revealed HDAC2 as another key regulator of MDSC activity.119 In this paper, the authors first showed that the majority of PMN-MDSCs were differentiated from M-MDSCs using multiple cancer models, including EL-4 thymoma, Lewis lung carcinoma and 4T1 mammary carcinoma. This differentiation process was regulated by HDAC2 through transcriptional silencing of the retinoblastoma (Rb) gene upon histone deacetylation at its promoter region. Then they demonstrated that Rb1 was important for the immunosuppressive function of MDSCs, showing that the MDSCs isolated from Rb1-deficient mice failed to inhibit the T-cell response. This study presents a novel mechanism by which HDAC2 regulates MDSC differentiation by modulating Rb1 expression. The last study we discuss here linked histone acetylation with chromatin remodeling for controlling MDSC function.120 Nagata et al. revealed that the bromodomain (BRD) of the CREB (cyclic-AMP response element binding protein)-binding protein (CBP) and E1A-binding protein of 300 kDa (EP300) (CBP/EP300) acted as a master regulator of H3K27 acetylation in MDSCs, modifying promoters, and enhancers of tumorigenic target genes in a CT26 colorectal cancer model. CBP/EP300-BRD has intrinsic HAT activity that controls H3K27 acetylation. Using a CBP/EP300-BRD inhibitor (CBP/EP300-BRDi) in vivo altered the intratumoral MDSCs and reprogrammed the tumor-associated MDSCs such that they transitioned from exhibiting a suppressive phenotype to an inflammatory phenotype through downregulation of STAT pathway-related genes, such as Ly6C2, Ccr2, Mmp9, and NOS2, and the inhibition of Arg1 and iNOS. As a result, CBP/EP300-BRD inhibition slowed tumor growth in immunocompetent tumor-bearing mice and in MDSC-dependent xenograft models. These studies suggest a mechanism in which histone modifications regulating MDSCs can potentially be targeted to reverse the immunosuppressive phenotype of MDSCs for cancer treatment.
In conclusion, DNA methylation and histone modification, including histone methylation and acetylation, on a wide range of functionally related genes in MDSCs contribute significantly to MDSC differentiation during tumorigenesis, and these mechanisms may be targeted for cancer therapy development (Fig. 4).
Fig. 4.
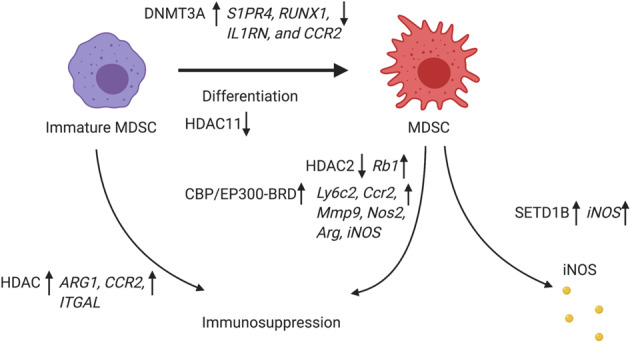
Epigenetic regulation in myeloid-derived suppressor cells. DNMT3A is upregulated in MDSCs and methylates genes, including S1PR4, RUNX1, IL1RN, and CCR2, to create unique DNA methylation patterns during MDSC differentiation. HDAC11 is a negative regulator of MDSC expansion/function. HMT SET1B controls MDSC function through upregulation of iNOS. HDAC activation facilitates immunosuppression and myeloid cell recruitment of immature MDSCs through the activation of ARG1, CCR2, and ITGAL. HDAC2 regulates MDSC differentiation and immunosuppressive function by modulating Rb1 expression. CBP/EP300-BRD, with its intrinsic histone acetyltransferase activity, reprograms tumor-associated MDSCs from expressing a suppressive to expressing an inflammatory phenotype through downregulation of STAT pathway-related genes such as Ly6C2, Ccr2, Mmp9, and NOS2 and the inhibition of Arg1 and iNOS
DNA methylation in T lymphocytes
Various epigenetic modifications have been discovered during effector T-cell differentiation in cancer. Here, we will introduce three studies highlighting DNA methylation in T lymphocytes. One study, performed by Peng et al., linked effector T-cell differentiation with both DNA methylation and histone methylation in human ovarian cancer.121 The authors discovered that EZH2-mediated histone H3 lysine 27 trimethylation (H3K27me3) and DNA methyltransferase 1 (DNMT1)-mediated DNA methylation repressed the expression of chemokines CXCL9 and CXCL10 in Th1 cells and subsequently prevented effector T-cell trafficking to the TME. These findings were validated by using EZH2 and DNMT inhibitors to upregulate the expression of CXCL9 and CXCL10, increase effector T-cell tumor infiltration, and slow tumor progression in vivo. In human ovarian patients, the researchers found that the expression of EZH2 and DNMT1 was negatively associated with the number of tumor-infiltrating CD8(+) T cells and patient outcomes. This study proves that epigenetic reprogramming can reshape T-cell function in cancer and may be targeted for developing cancer therapy. The idea was confirmed by a second, clinical study, in which Li et al. demonstrated that low-dose DNMTi decitabine was an effective treatment option for a subgroup of solid tumor patients, specifically by promoting an antitumor T-cell response.122 They showed that low-dose decitabine was able to enhance the activation and proliferation of human IFNγ+ T cells and promote Th1 polarization and function of cytotoxic T cells both in vivo and in vitro due to the upregulation of IFNγ-stimulated genes IFI27, IFI44, IFIT3, IFITM1, PSMB9, and IRF1. As a result, patients achieved clinical benefits from decitabine and showed inhibited cancer progression. This study provides evidence that epigenetic modulators can be used as antitumor therapies with the rationale of targeting T-cell activation and function in the TME. Another study in colorectal cancer also revealed unique DNA methylation features in tumor-reactive CD8+ T cells.123 In a genome-wide DNA methylation analysis, Yang et al. compared tumor-reactive and tumor-infiltrating bystander CD8+ lymphocytes (TILs) with naive and effector memory CD8+ T-cell subtypes, serving as controls, from colorectal cancer patients to study their transcriptome and methylome characteristics. Whole-genome methylation profiling identified a distinct methylome pattern for the tumor-reactive CD8+ T cells, with tumor-reactive markers CD39 and CD103 being specifically demethylated. Signature genes for cytotoxic T cells, including PRF1, IFNG, GZMB, CCL3, CCL4, NKG7, and CST7, were found to be highly methylated in the naive subtype and then became demethylated during the naive cell differentiation into TEM cells, indicating that the hypomethylation programming was established following the differentiation of the tumor-reactive CD8+ T cells from naive T cells. Finally, for the exhausted T-cell signature genes, two inhibitory receptors, PDCD1 and CTLA4, were found to be specifically demethylated within the induction of tumor-reactive CD8+ T cells. Another inhibitory receptor, LAG3, was initially methylated in naive cells and was demethylated in the later stages of the T-cell subtype transition. Together, these studies build a strong argument for using DNA methylation modulators to mediate T-cell function and prevent T-cell exhaustion for treating cancer.
Histone methylation in T lymphocytes
Histone modifications are also involved in the regulation of T-cell differentiation. We discuss one study highlighting the role of histone methylation in T-cell differentiation. In a single-cell RNA-sequencing analysis, Kakaradov et al. revealed a previously unknown role of EZH2 in T-cell differentiation.124 In this paper, they first discovered that EZH2 was highly expressed in terminal effector cells compared with the level expressed in T cells, which progressed through the first round of cell division during activation. From an immunoprecipitation sequencing analysis of EZH2-related genes, the authors first identified H3K27me3 peaks, and then mapped them onto ~6000 genes detected with single-cell RNA-sequencing of naive, terminal effector, and total memory CD8+ T cells. The effector cells exhibited significant gains in H3K27me3 coverage that correlated with gene repression, whereas the promoter regions in memory cells exhibited considerable losses in H3K27me3 coverage, suggesting that epigenetic silencing was crucial for the differentiation of the terminal effector cells. These findings suggest a mechanism by which terminal effector cell differentiation is regulated through the epigenetic silencing of transcripts, predominately by HMT EZH2, which was associated with memory lymphocytes.
Histone acetylation in T lymphocytes
Histone acetylation can regulate T-cell differentiation synergistically with metabolic changes and cytokines produced inside T cells. Here, we discuss two studies that highlight histone acetylation in T cells. One study showed that lactate dehydrogenase A (LDHA) enhanced the histone acetylation and transcriptional activation of IFNγ through the induction of acetyl-coenzyme A during aerobic glycolysis in activated T cells.125 Deletion of LDHA in T cells protected mice from immunopathological conditions triggered by excessive IFNγ expression. These findings revealed an epigenetic mechanism that explains why aerobic glycolysis augments effector T-cell differentiation and responses. In another study, a histone deacetylase inhibitor (HDACi) was shown to reprogram differentiated human CD8+ T cells into central memory-like T cells in synergy with IL21.126 The transition from CD8+ effector T cells to memory T cells was initiated by the increased H3 acetylation at the CD28 promoter region and resulted in increased chromatin accessibility. Increased chromatin accessibility allowed the binding of pSTAT3 to the CD28 region activated by IL21 and the subsequent upregulation of surface CD28 and CD62L, which are markers of central memory T cells. This study also showed that these reprogrammed cells exhibited enhanced proliferation in response to both IL2 and IL15 treatment and a stable memory-associated transcriptional signature with increased Lef1 and Tcf7. These findings suggest a novel mechanism by which epigenetic modifications, together with cytokines, can drive memory T-cell formation from effector T cells, a transition that can be potentially used in the generation of highly persistent T-cell populations for tumor treatments, particularly adoptive-transferred T-cell therapy.
Chromatin remodeling in T lymphocytes
The chromatin-remodeling mechanism is studied with respect to regulating T-cell exhaustion. One study discovered a chromatin-remodeling mechanism that regulated PD1 expression through a chromatin-organizing special AT-rich sequence-binding protein-1 (Satb1).127 Satb1 restrained PD-1 expression upon T-cell activation by recruiting a nucleosome-remodeling deacetylase complex to Pdcd1 regulatory regions. In Stab1-deficient mice, Satb1-deficient T cells exhibited a 40-fold increase in PD-1 expression. Tumor-infiltrating T lymphocytes also showed reduced Satb1 expression in human ovarian cancer patients, which correlated with reduced effector T-cell activity. This study suggested a role for the chromatin remodeler Satb1 in preventing premature T-cell exhaustion by regulating PD1 expression upon T-cell activation. In another study, Philip et al. showed that tumor-specific CD8 T (TST) cells in solid tumors presented chromatin dynamics with different states during tumorigenesis.128 They identified two discrete chromatin states of TSTs: state 1, a plastic dysfunctional state from which T cells can be rescued, and state 2, a fixed dysfunctional state in which the cells are resistant to reprogramming. State 1 dysfunction was reversible, but further chromatin remodeling between days 7 and 14, the cells entered state 2 and were fixed. These states were identified with surface markers, CD101 and CD38, which are associated with discrete dysfunctional chromatin states, and higher expression of these two markers indicated the reduced reprogrammability of high PD1-expressing T cells within heterogeneous TIL populations. This study suggested a novel mechanism to explain why patients with high PD1 levels present different responses to checkpoint blockade therapies, as some of the patients harbored TST with state 1 and some with state 2, as regulated by the chromatin-remodeling process. It also suggested opportunities for to increasing the reprogrammability of TSTs by modulating the chromatin-remodeling process.
In conclusion, epigenetic mechanisms play important roles in facilitating T-cell differentiation with functional phenotypes, the memory phenotype and even the exhaustive phenotype (Fig. 5). Epigenetic modulations hold the potential for manipulating these processes to facilitate the acquisition of the effector phenotype by T cells to induce the killing of tumor cells.
Fig. 5.
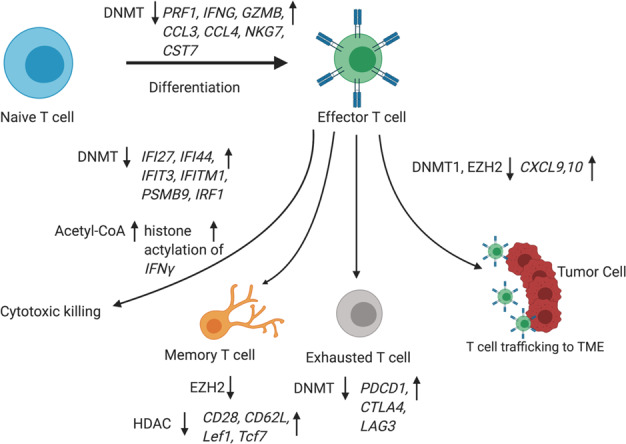
Epigenetic regulation of T lymphocytes. DNMT downregulation promotes Th1 polarization and activation of cytotoxic T cells because of the upregulation of IFNγ-stimulated genes, such as IFI27, IFI44, IFIT3, IFITM1, PSMB9, and IRF1. EZH2-mediated histone H3 lysine 27 trimethylation (H3K27me3) and DNA methyltransferase 1 (DNMT1)-mediated DNA methylation repress the expression of chemokines CXCL9 and CXCL10 and subsequently prevent effector T-cell trafficking to the TME. DNMT downregulation leads to the upregulation of signature genes in cytotoxic T cells, including PRF1, IFNG, GZMB, CCL3, CCL4, NKG7, and CST7, during naive T-cell to effector T-cell differentiation. Three inhibitory receptors, PDCD1, CTLA4, and LAG3, were found to be specifically demethylated within tumor-reactive CD8+ T cells and in late stages of T-cell subtype activation. HMT EZH2 negatively regulates the differentiation of memory T cells from effector T cells. Induction of acetyl-coenzyme A during aerobic glycolysis enhances the histone acetylation and transcriptional activation of Ifng in activated T cells. Histone deacetylase inhibition was shown to reprogram differentiated human CD8+ T cells into central memory-like T cells synergistically with IL21 and the upregulation of central memory T-cell surface CD28 and CD62L, showing a stable memory-associated transcriptional signature with increased Lef1 and Tcf7
Rationale for combining epigenetic therapies with cancer immunotherapy
Cancer Immunotherapy, specifcially, immune checkpoint inhibitors (ICIs) with anti-PD1 inhibitors has revolutionized cancer therapy in recent years and has been shown to be effective in many cancer types, including melanoma and lung cancer.129 However, the majority of cancer patients do not respond to anti-PD1 therapy, particularly patients with solid tumors.130 The main reason is that most solid tumors, such as PDA, are “cold” tumors with a low tumor-mutation burden and a lack of effector CD8+ T cells in the TME.131 As a result, the inhibition of PD1 is not effective in treating these solid tumors. Recent studies have focused on testing combinations of immunotherapies with different therapies, including chemotherapy, radiation therapy, and targeted therapy, with the rationale of converting “cold” tumors to “hot” tumors to increase effector CD8+ T-cell infiltration into the TME.132 Epigenetic therapy presents a unique opportunity to reshape the TME from being immunosuppressive to being an antitumor through the regulation of different stromal and immune cells via the various mechanisms discussed in the previous sections. In this review, we discuss the mechanisms of epigenetic immune modulation in the TME with an emphasis on its significance in priming and sensitizing the host immune system to ICI.
Before discussing how epigenetic agents may prime the TME for immunotherapy sensitization, we need to explain how ICI works. Chronic interactions between tumor cells and subsets of immune cells with PD-L1/CD80 or CD86 expression induce the expression of PD-1/CTLA-4 on cytotoxic CD8+ T cells, which renders cytotoxic CD8+ tumor-infiltrating lymphocytes ineffective at killing tumor cells.133–135 Thus, the rationale for immune checkpoint inhibition is to use antibodies targeting PD-1, PD-L1, or CTLA-4 to reverse this inhibitory action and maintain the tumor killing function of cytotoxic CD8+ tumor-infiltrating lymphocytes.136,137 Based on the mechanisms of action of ICI, people have demonstrated that a clinical response to ICI is dependent upon the immune status of the tumor, which is identified by meeting three criteria: first, antigen-specific CD8+ lymphocytes must be the TME;138 second, an immune-permissive state must be facilitated by resident stromal and immune cell populations139,140; and third, MHC class-I-mediated antigen presentation must be functionally competent on tumor cells.141 Surprisingly, epigenetic modulators can potentially address all three criteria to sensitize the TME to ICI. Now, using representative preclinical studies in different cancer types, we discuss the key synergistic mechanisms of epigenetic modulators with ICI.
Modulation of immune composition within the TME
Multiple preclinical studies have demonstrated that epigenetic agents can modulate the immune composition of the TME by decreasing the abundance of TAMs and MDSCs and increasing the numbers of CD8+ effector T cells and memory T cells. Here, we introduce two preclinical studies that utilized epigenetic agents to modulate TME components. In the first study, Christmas et al. tested the combination of an HDAC inhibitor, entinostat (ENT), with anti-PD-1, anti-CTLA-4, or both in Panc02 metastatic pancreatic cancer and HER2/neu transgenic breast cancer mouse models.142 They demonstrated that this combination of ENT with ICI led to significantly improved tumor-free survival in both cancer mouse models. Flow cytometry and functional analysis revealed that the antitumor effect of this combination therapy was due to a transition of M-MDSCs to G-MDCs with decreased suppression because of reduced STAT3 signaling in the TME and an increased number of activated granzyme-B-producing CD8+ T effector cells in both tumor cell types. The gene expression profiling of both MDSCs and TILs revealed significant changes in immune-related pathways. In another study using an ovarian cancer model, Stone et al. showed that DNMTi (5-azacitidine, AZA) and HDACi improved the efficacy of ICI by reducing the immune suppression of TME through type I IFN signaling.143 The action of AZA was dependent on the initiation of the type I IFN response for the antitumor response in vivo, as the use of an antibody targeting IFN alpha and beta receptor subunit 1 abrogated the antitumorigenic actions of AZA. Through type I IFN signaling, AZA increased the number of CD45+ immune cells and the percentages of active CD8+ T and natural killer (NK) cells and reduced the percentages of macrophages and MDSCs in the TME. The addition of an HDACi to AZA further increased T-cell and NK cell activation and reduced the extent of TAM presence. In the end, the researchers showed that a triple combination of DNMTi/HDACi plus the ICI PD-1 conferred the best antitumor effect and longest overall survival. These findings suggest that epigenetic agents can reshape the immune composition to make TME more permissive to ICI.
Facilitated differentiation of CD8+ TILs towards the acquisition of effector/memory phenotypes
As previously discussed, DNA methylation and histone acetylation have been associated with the regulation of T-cell differentiation (Fig. 5). Inspired by these findings, multiple studies have tested the roles of epigenetic modulators in the reprogramming functions of T cells combined with ICI. In one study, Topper et al. tested the effects of combining DNMTi (AZA) and HDACi (ITF-2357) in treating lung cancer.144 They first showed that the combination of Aza and ITF-2357 significantly induced the expression of IFNα/β pathway-related genes, including those associated with antigen presentation, such as IRF7, STAT1, IFNB1, and OASL. The activation of the IFNα/β pathway by Aza partially relied on increasing double-stranded RNA (dsRNA) species, including endogenous retrovirus (ERV) transcripts. In addition, by looking at the TIL transcriptome of 3698 differentially expressed TIL genes, they discovered the downregulation of T-cell exhaustion-associated genes and the induction of activation- and memory-associated genes of TILs. These result suggested that an expansion of memory and/or effector T cells can be induced by epigenetic agents within the TME, and this effect can be combined with those of ICI to augment the antitumor response.
Reduced immune exhaustion
Unique DNA methylation patterns were previously discovered in exhausted T cells (Fig. 5).123 Similarly, a study performed by Ghoneim et al. revealed exhaustion-associated DNA methylation patterns in tumor-infiltrating CD8 T cells expressing a high level of PD-1 in a prostate cancer Tramp-C2 murine model and during chronic viral infection.46 These de novo DNA methylation patterns were progressively acquired in antigen-specific CD8 T cells at the effector and exhaustion stages upon PD-1 blockade treatment and identified by whole-genome bisulfite sequencing. In addition, the researchers determined the consequences of blocking de novo DNA methylation with DNMTi (decitabine) combined with ICI in vivo. Sequential decitabine and PD-L1 blockade treatments were able to induce the proliferation of both polyclonal and antigen-specific tumor-infiltrating CD8 T cells, which resulted in controlled tumor growth. These findings highlight the role of DNA methylation in programming T-cell exhaustion and can be targeted to prevent T-cell exhaustion and, through synergism with ICI treatments, to augment the antitumor response.
Augmentation of innate immune signaling for facilitating T-cell infiltration
DNA demethylating agents can abrogate the repression of genes related to immunomodulatory signaling.145 One study performed by Chiappinelli et al. revealed that DNMTi (AZA) upregulated the expression of ERV genes, which are normally hypermethylated in ovarian cancer cells.146 ERV expression activated the viral defense pathway in these cancer cells. As a result, DNMTi triggered the cytosolic sensing of dsRNA through dsRNA sensors TLR3 and MAVS, triggering a type I interferon response and apoptosis. In addition, they discovered that a high viral defense expression signature in tumors was significantly associated with a durable clinical response in melanoma patients treated with ICIs. DNMTi treatment also sensitized the TME to anti-CTLA4 therapy in a preclinical melanoma model. Another study also found similar results in colorectal cancers, while DNMTi (AZA) induced a viral defense response through the induction of dsRNAs derived partially from ERV.147 Together, these studies prove the concept that epigenetic agents can induce innate immune signaling to facilitate T-cell infiltration in cancer.
Reprogramming the phenotypes of TAMs and MDSCs drives their transition from being immunosuppressive to being permissive
Epigenetic modifications can modify myeloid cell differentiation as previously described (Figs. 3 and 4). Here, we introduce one preclinical study with clinical implications and some unpublished work performed by our own group. In a preclinical study, Zhihao et al. revealed the function of DNMTi (AZA) and HDACi (entinostat) in treating pulmonary metastases.148 Using mouse models of pulmonary metastases, they discovered that DNMTi and HDACi disrupted the premetastatic niche by inhibiting the trafficking of MDSCs through the downregulation of CCR2 and CXCR2 and by promoting the MDSC acquisition of a more interstitial macrophage-like phenotype, with the gene expression signatures EGR1, EGR2, MAFB, MAF, and PPARγ. This preclinical work was also translated into a clinical investigation in which they showed that low-dose adjuvant epigenesis modulation therapy inhibited both the formation and growth of lung metastases through its selective effects on MDSC in lung, breast, and esophageal cancers after surgical removal of the primary tumor. These findings present a mechanism by which epigenetic modulators can reprogram MDSC phenotypes to become more immunopermissive. Research performed by our own group also showed that unique DNA methylation patterns with specificity on metabolic genes can be induced in TAMs by PDA tumor cells. This tumor-induced DNA methylation mechanism reshaped the metabolic states of the macrophages, leading o functional changes to promote the M1–M2 transition. The use of DNMTi decitabine only partially reversed this M1–M2 transition of macrophages in PDA. Thus, epigenetic modulators have potential to prevent the immunosuppressive reprogramming of stromal and immune cells within the TME to create a TME permissive to ICI.
Improvements to immune recognition
Tumor cells escape immune surveillance by inhibiting the expression of tumor-specific antigens and MHC I molecules, which are essential for T-cell activation.149,150 Epigenetic modulators induce the upregulation of cancer testis antigen (CTA) and MHC I molecules in multiple cancer types. Here, we introduce two preclinical studies performed by different groups and one preclinical study performed by our own group. Work from Siebenkas et al. showed that DNMTi (decitabine) increased the expression of both antigen processing and presentation and CTAs such as B2M, CALR, CD58, PSMB8, and PSMB9 at both the mRNA and protein levels in colon and ovarian cancer cell lines.151 Inspired by this research, a study performed in our lab first confirmed that decitabine upregulated a broader range of CTAs in colorectal cancer.152 In addition, we found that decitabine was able to sensitize tumor cells to the whole-cell cancer vaccine GVAX through upregulation of CTAs that prolonged the survival of mice serving as metastatic colorectal cancer models. We also proved that this vaccine sensitization improved the antitumor response of the combination therapy through an augmented T-cell response to specific antigens, which had been upregulated by decitabine. Taken together, these findings prove that epigenetic therapy may sensitize the TME to immunotherapy through the upregulation of CTAs to improve immune recognition. In another study, Ritter et al. showed that Merkel cell carcinoma tumor cells had reduced HLA class-I surface expression because of impaired expression of key components of the antigen-processing machinery (APM), which included LMP2, LMP7, TAP1, and TAP2.150 They later discovered that silencing of the HLA class-I APM was due to histone deacetylation. The inhibition of HDACs with inhibitors not only induced the acetylation of histones in the promoter regions of APM-related genes but also allowed the re-expression of APM components both in vitro and in a xenograft mouse model. These findings present a mechanism showing how epigenetic therapy improves immune recognition of tumor cells by inducing APM-regulated protein expression. Together, DNMTi and HDACi may potentially boost the antitumor response of ICIs through upregulation of CTA and APM expression in tumor cells to augment immune recognition by T cells.
Current clinical trials: testing combination strategies using epigenetic modulators and PD-1/PD-L1 blockades
The basic research studies and preclinical research data we have presented thus far support the idea that epigenetic agents affect both tumor stroma and tumor cells to prime the TME for adopting an immunopermissive environment. This induction of an immunopermissive TME is crucial to the effectiveness of ICI therapy in cancer patients, particularly for patients with solid tumors. Recently, a number of clinical trials have tested the efficacy of PD-1/PD-L1 antibodies combined with epigenetic agents based on DNMTi and HDACi treatment of patients with multiple cancer types. Representative studies specifically focusing on solid tumors are summarized in Table 1.
Table 1.
Current clinical trials combining checkpoint inhibitors and epigenetic drugs in different solid tumor types
| ClinicalTrials.gov identifier | Recruiment status | Phase | Cancer type | Epigenetic drug/s | Epigenetic target | Immune checkpoint inhibitor/s |
|---|---|---|---|---|---|---|
| NCT02437136 | Active, not recruiting | 1a/b | NSCLC, melanoma and mismatch repair-proficient CRC | Entinostat | HDAC1,2,3 | Pembrolizumab |
| NCT02638090 | Recruiting | 1/2 | Stage IV NSCLC | Vorinostat | HDAC1,2,3,7,11 | Pembrolizumab |
| NCT02512172 | Active, not recruiting | 1 | MSS CRC | Romidepsin, oral azacitidine | HDAC1,2; DNMT1 | Pembrolizumab |
| NCT03264404 | Recruiting | 2 | PDA | Azacitidine | DNMT1 | Pembrolizumab |
| NCT03854474 | Recruiting | 1/2 | Advanced urothelial carcinoma, locally advanced urothelial carcinoma, metastatic bladder urothelial carcinoma | Tazemetostat | EZH2 | Pembrolizumab |
| NCT03590054 | Recruiting | 1 | Stage III cutaneous melanoma, stage IV cutaneous melanoma, locally advanced melanoma, locally advanced solid neoplasm | Abexinostat | Pan-HDAC inhibitor | Pembrolizumab |
| NCT03250273 | Recruiting | 2 | PDA and cholangiocarcinoma | Entinostat | HDAC1,2,3 | Nivolumab |
| NCT01928576 | Recruiting | 2 | NSCLC | Azacitidine with/without entinostat | DNMT1; HDAC1,2,3 | Nivolumab |
| NCT02635061 | Active, not recruiting | 1 | NSCLC | ACY-421 | HDAC6 | Nivolumab |
| NCT02032810 | Active, not recruiting | 1 | Unresectbale III/IV melanoma | Panobinostat | HDAC | Ipilimumab |
| NCT02708680 | Active, not recruiting | 1b | TNBC | Entinostat | HDAC1,2,3 | Atezolizumab |
| NCT02805660 | Active, not recruiting | 1/2 | Advanced solid tumors and NSCLC | Mecotinostat | HDAC1,2,3,11 | Durvalumab |
| NCT03308396 | Recruiting | 1/2 | Advanced kidney cancer, renal cell cancer | Guadecitabine | DNMT1 | Durvalumab |
| NCT02915523 | Active, not recruiting | 1b/2 | Advanced epithelial OC | Entinostat | HDAC1,2,3 | Avelumab |
| NCT03812796 | Recruiting | 2 | GI cancer | Domatinostat | HDAC1,2,3 | Avelumab |
| NCT02961101 | Recruiting | 1/2 | Non-Hodgkins lymphoma, Hodgkins lymphoma, GI cancer, HPC, BC, OC, lung cancer, renal cell cancer, PDA, bile duct cancer | Decitabine | DNMT1, DNMT3A, DNMT3B | Anti-PD-1 antibody |
All information on current clinical trials was obtained from ClinicalTrials.gov
NSCLC non-small cell lung cancer, BC breast cancer, OC ovarian cancer, MSS microsatellite stable, CRC colorectal cancer, TNBC triple-negative breast cancer, GI gastrointestinal, PDA pancreatic ductal adenocarcinoma
Concluding remarks
Recent studies have revealed the novel roles of epigenetic agents in the regulation of cancer stromal and immune cell differentiation and reprogramming, including CAFs, myeloid cells, and T cells. As a result, epigenetic therapy has emerged as a promising immunotherapy partner because it can sensitize solid tumors based on the rationale of creating a less immunosuppressive TME that is favorable for effector CD8+ T-cell function. Epigenetic reprogramming of the TME involves modulation of the TME with an increased number of effector and memory T cells and a decreased number of TAMs and MDSCs; prevention of T-cell exhaustion; reprogramming of TAMs and MDSCs to acquire less immunosuppressive phenotypes; augmentation of innate immune signaling through the activation of the viral defense pathway; and improvements of the immune system for recognition of tumor cells through upregulation of CTAs and MHC I molecules. Many preclinical studies testing different combinations of DNMTis and HDACis with ICI have demonstrated improved efficacy compared with treatments with ICI alone and prolonged survival in multiple murine cancer models. Encouraged by these preclinical findings, a growing number of clinical trials for multiple cancer types have been conducted with the focus on testing the established epigenetic agents, DNMT inhibitors and HDAC inhibitors (alone or in combination) together with ICI. These combination therapies are also being tested with ICI in refractory patients after immune checkpoint inhibition based on the rationale of reversing immunotherapy resistance. Future directions will focus on the development of next-generation novel epigenetic agents in addition to DNMTi and HDACi, such as EZH2, LSD1, G9a, and BET inhibitors.153,154 Combination therapy with epigenetic agents and other types of immunotherapies, such as cancer vaccines and adoptive T-cell therapies, may also present promising opportunities.
Acknowledgements
This work was supported in part by NIH grant T32 CA126607 (L.Z.); NIH grant R01 CA169702 (L.Z.); NIH grant R01 CA197296 (L.Z.); the Viragh Foundation and the Skip Viragh Pancreatic Cancer Center at Johns Hopkins (L.Z.); the Sol Goldman Pancreatic Cancer Research Center (L.Z.); NCI SPORE P50 CA062924 (L.Z.); and Cancer Center Support Grant P30 CA006973.
Competing interests
L.Z. receives grant support from Bristol-Myers Squibb, Merck, iTeos, Amgen, NovaRock, InxMed, and Halozyme. L.Z. is a paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Akrevia, DataRevive, and Mingruzhiyao. L.Z. holds shares of Alphamab and Mingruzhiyao.
References
- 1.Rahib L, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Ducreux M, et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015;26(Suppl 5):v56–v68. doi: 10.1093/annonc/mdv295. [DOI] [PubMed] [Google Scholar]
- 4.Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Medicine. 2016;95:e5541. doi: 10.1097/MD.0000000000005541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karagiannis GS, et al. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012;10:1403–1418. doi: 10.1158/1541-7786.MCR-12-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo S, Deng C-X. Effect of stromal cells in tumor microenvironment on metastasis initiation. Int J. Biol. Sci. 2018;14:2083–2093. doi: 10.7150/ijbs.25720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks DL, Olson RL, Fernandez-Zapico ME. Epigenetic control of the tumor microenvironment. Epigenomics. 2016;8:1671–1687. doi: 10.2217/epi-2016-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murakami T, et al. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019;3:130–137. doi: 10.1002/ags3.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Q, et al. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics. 2018;8:5072–5087. doi: 10.7150/thno.26546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albrengues J, et al. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014;7:1664–1678. doi: 10.1016/j.celrep.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 11.Erdogan B, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017;216:3799–3816. doi: 10.1083/jcb.201704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bates AL, et al. Stromal matrix metalloproteinase 2 regulates collagen expression and promotes the outgrowth of experimental metastases. J. Pathol. 2015;235:773–783. doi: 10.1002/path.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao W, et al. Galectin-3 mediates tumor cell-stroma interactions by activating pancreatic stellate cells to produce cytokines via integrin signaling. Gastroenterology. 2018;154:1524–1537.e1526. doi: 10.1053/j.gastro.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 15.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 16.Ohlund D, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choe C, et al. Tumor-stromal interactions with direct cell contacts enhance motility of non-small cell lung cancer cells through the hedgehog signaling pathway. Anticancer Res. 2013;33:3715–3723. [PubMed] [Google Scholar]
- 18.Shiga K, et al. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers. 2015;7:2443–2458. doi: 10.3390/cancers7040902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elyada E, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Öhlund D, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu W, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat. Genet. 2008;40:650–655. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 24.Mielgo A, Schmid MC. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013;46:131–138. doi: 10.5483/BMBRep.2013.46.3.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karp CL, Murray PJ. Non-canonical alternatives: what a macrophage is 4. J. Exp. Med. 2012;209:427–431. doi: 10.1084/jem.20120295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015;125:3365–3376. doi: 10.1172/JCI80006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greten FR, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 31.Qian B, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE. 2009;4:e6562–e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshikawa K, et al. Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012;103:2012–2020. doi: 10.1111/j.1349-7006.2012.02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat. Rev. Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu S, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thyagarajan A, et al. Myeloid-derived suppressor cells and pancreatic cancer: implications in novel therapeutic approaches. Cancers. 2019;11:1627. doi: 10.3390/cancers11111627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anani W. & Shurin M. R. Targeting myeloid-derived suppressor cells in cancer. In: Kalinski P., ed. Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy. 105–128 (Springer International Publishing, Cham, 2017). [DOI] [PubMed]
- 39.Tartour E, et al. Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev. 2011;30:83–95. doi: 10.1007/s10555-011-9281-4. [DOI] [PubMed] [Google Scholar]
- 40.Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol. Cell Biol. 2013;91:493–502. doi: 10.1038/icb.2013.29. [DOI] [PubMed] [Google Scholar]
- 41.Liu G, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. 2014;74:727. doi: 10.1158/0008-5472.CAN-13-2584. [DOI] [PubMed] [Google Scholar]
- 42.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidl C, Delacher M, Huehn J, Feuerer M. Epigenetic mechanisms regulating T-cell responses. J. Allergy Clin. Immunol. 2018;142:728–743. doi: 10.1016/j.jaci.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 44.Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T cell dysfunction in cancer immunity and immunotherapy. Front. Immunol. 2019;10:1719–1719. doi: 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tough DF, Rioja I, Modis LK, Prinjha RK. Epigenetic regulation of T cell memory: recalling therapeutic implications. Trends Immunol. 2020;41:29–45. doi: 10.1016/j.it.2019.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Ghoneim HE, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170:142–157.e119. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang M, Yin B, Wang HY, Wang R-F. Current advances in T-cell-based cancer immunotherapy. Immunotherapy. 2014;6:1265–1278. doi: 10.2217/imt.14.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct. Biotechnol. J. 2018;17:1–13. doi: 10.1016/j.csbj.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 2017;5:978–991. doi: 10.1158/2326-6066.CIR-16-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 2020;10:422. doi: 10.1158/2159-8290.CD-19-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 52.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 53.Xiao Q, et al. Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 2016;76:5395–5404. doi: 10.1158/0008-5472.CAN-15-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waddington CH. Embryology, epigenetics and biogenetics. Nature. 1956;177:1241–1241. [Google Scholar]
- 55.Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2:607–617. doi: 10.1177/1947601910393957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mathieu O, Picard G, Tourmente S. Methylation of a euchromatin-heterochromatin transition region in Arabidopsis thaliana chromosome 5 left arm. Chromosome Res. 2002;10:455–466. doi: 10.1023/a:1020936229771. [DOI] [PubMed] [Google Scholar]
- 59.Huff JT, Zilberman D. Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell. 2014;156:1286–1297. doi: 10.1016/j.cell.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang X, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 62.Egger G, et al. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc. Natl Acad. Sci. USA. 2006;103:14080–14085. doi: 10.1073/pnas.0604602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kareta MS, Botello ZM, Ennis JJ, Chou C, Chedin F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006;281:25893–25902. doi: 10.1074/jbc.M603140200. [DOI] [PubMed] [Google Scholar]
- 64.Hervouet E, Peixoto P, Delage-Mourroux R, Boyer-Guittaut M, Cartron P-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics. 2018;10:17. doi: 10.1186/s13148-018-0450-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jair KW, et al. De novo CpG island methylation in human cancer cells. Cancer Res. 2006;66:682–692. doi: 10.1158/0008-5472.CAN-05-1980. [DOI] [PubMed] [Google Scholar]
- 66.Liang G, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rhee I, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 68.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lian Christine G, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–1146. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klymenko Y, Nephew KP. Epigenetic crosstalk between the tumor microenvironment and ovarian cancer cells: a therapeutic road less traveled. Cancers. 2018;10:295. doi: 10.3390/cancers10090295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol. Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 73.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 74.Hassan AH, et al. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- 75.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 76.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jambhekar A, Dhall A, Shi Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019;20:625–641. doi: 10.1038/s41580-019-0151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rossetto D, Avvakumov N, Côté J. Histone phosphorylation. Epigenetics. 2012;7:1098–1108. doi: 10.4161/epi.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Banerjee T, Chakravarti D. A peek into the complex realm of histone phosphorylation. Mol. Cell. Biol. 2011;31:4858. doi: 10.1128/MCB.05631-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Becker PB, Hörz W. ATP-dependent nucleosome remodeling. Annu. Rev. Biochem. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- 81.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 82.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 83.Ozturk N, Singh I, Mehta A, Braun T, Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev. Biol. 2014;2:5–5. doi: 10.3389/fcell.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hamiche A, Sandaltzopoulos R, Gdula DA, Wu C. ATP-dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell. 1999;97:833–842. doi: 10.1016/s0092-8674(00)80796-5. [DOI] [PubMed] [Google Scholar]
- 85.Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017;18:407–422. doi: 10.1038/nrm.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lorch Y, Maier-Davis B, Kornberg RD. Mechanism of chromatin remodeling. Proc. Natl Acad. Sci. 2010;107:3458. doi: 10.1073/pnas.1000398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bailey P, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 88.Zinzalla G. A new way forward in cancer drug discovery: inhibiting the SWI/SNF chromatin remodelling complex. Chembiochem. 2016;17:677–682. doi: 10.1002/cbic.201500565. [DOI] [PubMed] [Google Scholar]
- 89.Shain AH, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl Acad. Sci. USA. 2012;109:E252–E259. doi: 10.1073/pnas.1114817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hanson JA, et al. Gene promoter methylation in prostate tumor-associated stromal cells. J. Natl Cancer Inst. 2006;98:255–261. doi: 10.1093/jnci/djj051. [DOI] [PubMed] [Google Scholar]
- 91.Hu M, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005;37:899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 92.Yu J, et al. Unlike pancreatic cancer cells pancreatic cancer associated fibroblasts display minimal gene induction after 5-aza-2′-deoxycytidine. PLoS ONE. 2012;7:e43456–e43456. doi: 10.1371/journal.pone.0043456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iba K, Albrechtsen R, Gilpin BJ, Loechel F, Wewer UM. Cysteine-rich domain of human ADAM 12 (meltrin alpha) supports tumor cell adhesion. Am. J. Pathol. 1999;154:1489–1501. doi: 10.1016/s0002-9440(10)65403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bhagat TD, et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife. 2019;8:e50663. doi: 10.7554/eLife.50663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tyan SW, et al. Breast cancer cells induce stromal fibroblasts to secrete ADAMTS1 for cancer invasion through an epigenetic change. PLoS ONE. 2012;7:e35128. doi: 10.1371/journal.pone.0035128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li A, Chen P, Leng Y, Kang J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene. 2018;37:5952–5966. doi: 10.1038/s41388-018-0379-9. [DOI] [PubMed] [Google Scholar]
- 97.Zong Y, et al. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signaling. Proc. Natl Acad. Sci. 2012;109:E3395. doi: 10.1073/pnas.1217982109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schuyler RP, et al. Distinct trends of DNA methylation patterning in the innate and adaptive immune systems. Cell Rep. 2016;17:2101–2111. doi: 10.1016/j.celrep.2016.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang X, et al. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014;28:565–574. doi: 10.1210/me.2013-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cheng C, et al. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol. Lett. 2014;225:488–497. doi: 10.1016/j.toxlet.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 101.Sun F, et al. TET1 is an important transcriptional activator of TNFalpha expression in macrophages. PLoS ONE. 2019;14:e0218551. doi: 10.1371/journal.pone.0218551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tikhanovich I, et al. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor gamma-dependent macrophage differentiation. J. Biol. Chem. 2017;292:6882–6894. doi: 10.1074/jbc.M117.778761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kittan NA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE. 2013;8:e78045. doi: 10.1371/journal.pone.0078045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Satoh T, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 105.Yu W, et al. One-carbon metabolism supports S-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol. Cell. 2019;75:1147–1160.e1145. doi: 10.1016/j.molcel.2019.06.039. [DOI] [PubMed] [Google Scholar]
- 106.Tan AHY, et al. Lysine-specific histone demethylase 1A regulates macrophage polarization and checkpoint molecules in the tumor microenvironment of triple-negative breast cancer. Front Immunol. 2019;10:1351. doi: 10.3389/fimmu.2019.01351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cao Q, et al. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb. Vasc. Biol. 2014;34:1871–1879. doi: 10.1161/ATVBAHA.114.303393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Villagra A, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang Q, et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol. Cell Biol. 2015;35:63–75. doi: 10.1128/MCB.00805-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoshizaki T, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010;298:E419–E428. doi: 10.1152/ajpendo.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li T, et al. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2020;48:665–681. doi: 10.1093/nar/gkz1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Das Gupta K, et al. Class IIa histone deacetylases drive toll-like receptor-inducible glycolysis and macrophage inflammatory responses via pyruvate kinase M2. Cell Rep. 2020;30:2712–2728.e2718. doi: 10.1016/j.celrep.2020.02.007. [DOI] [PubMed] [Google Scholar]
- 113.Chang YC, et al. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood. 2008;111:5054–5063. doi: 10.1182/blood-2007-12-130609. [DOI] [PubMed] [Google Scholar]
- 114.Zhang D, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sasidharan Nair V, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin. Epigenetics. 2020;12:13. doi: 10.1186/s13148-020-0808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rodríguez-Ubreva J, et al. Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Rep. 2017;21:154–167. doi: 10.1016/j.celrep.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 117.Redd PS, et al. SETD1B activates iNOS expression in myeloid-derived suppressor cells. Cancer Res. 2017;77:2834. doi: 10.1158/0008-5472.CAN-16-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sahakian E, et al. Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015;63:579–585. doi: 10.1016/j.molimm.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Youn J-I, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013;14:211–220. doi: 10.1038/ni.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Almeida Nagata DE, et al. Regulation of tumor-associated myeloid cell activity by CBP/EP300 bromodomain modulation of H3K27 acetylation. Cell Rep. 2019;27:269–281.e264. doi: 10.1016/j.celrep.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 121.Peng D, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li X, et al. Increased IFNgamma(+) T cells are responsible for the clinical responses of low-dose DNA-demethylating agent decitabine antitumor therapy. Clin. Cancer Res. 2017;23:6031–6043. doi: 10.1158/1078-0432.CCR-17-1201. [DOI] [PubMed] [Google Scholar]
- 123.Yang R, et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019;21:2. doi: 10.1186/s13059-019-1921-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kakaradov B, et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol. 2017;18:422–432. doi: 10.1038/ni.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Peng M, et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484. doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang, J. et al. Histone deacetylase inhibitors and IL21 cooperate to reprogram human effector CD8+ T cells to memory T cells. Cancer Immunol. Res. 2020:canimm.0619.2019. [DOI] [PMC free article] [PubMed]
- 127.Stephen TL, et al. SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity. 2017;46:51–64. doi: 10.1016/j.immuni.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Philip M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 130.Zhao X, Subramanian S. Intrinsic resistance of solid tumors to immune checkpoint blockade therapy. Cancer Res. 2017;77:817–822. doi: 10.1158/0008-5472.CAN-16-2379. [DOI] [PubMed] [Google Scholar]
- 131.Bonaventura P, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front. Immunol. 2019;10:168–168. doi: 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yan Y, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front. Immunol. 2018;9:1739–1739. doi: 10.3389/fimmu.2018.01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev. Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv. Immunol. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 136.Iwai Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lanitis E., Dangaj D., Irving M. & Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 28(suppl_12), xii18–xii32 (2017). [DOI] [PubMed]
- 139.Peranzoni E, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl Acad. Sci. USA. 2018;115:E4041–e4050. doi: 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Woo EY, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- 141.de Charette M, Marabelle A, Houot R. Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? Eur. J. Cancer. 2016;68:134–147. doi: 10.1016/j.ejca.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 142.Christmas, B. J. et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol. Res. 2018:canimm.0070.2018. [DOI] [PMC free article] [PubMed]
- 143.Stone ML, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl Acad. Sci. USA. 2017;114:E10981–e10990. doi: 10.1073/pnas.1712514114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Topper MJ, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171:1284–1300.e1221. doi: 10.1016/j.cell.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li H, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chiappinelli KB, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roulois D, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lu Z, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579:284–290. doi: 10.1038/s41586-020-2054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McKee MD, Roszkowski JJ, Nishimura MI. T cell avidity and tumor recognition: implications and therapeutic strategies. J. Transl. Med. 2005;3:35. doi: 10.1186/1479-5876-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ritter C, et al. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017;7:2290. doi: 10.1038/s41598-017-02608-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Siebenkas C, et al. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE. 2017;12:e0179501. doi: 10.1371/journal.pone.0179501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim VM, et al. Neoantigen-based EpiGVAX vaccine initiates antitumor immunity in colorectal cancer. JCI Insight. 2020;5:e136368. doi: 10.1172/jci.insight.136368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020;17:75–90. doi: 10.1038/s41571-019-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sheahan, A. V. et al. Targeting EZH2 increases therapeutic efficacy of check-point blockade in models of prostate cancer. https://www.biorxiv.org/content/10.1101/730135v1 (2019).