

Adamantinomatous craniopharyngiomas (ACPs) are tumours of the sellar region and although histologically benign, they frequently invade the hypothalamus, visual tracts and local vascular structures. This aggressive behaviour can result in profound chronic morbidity, poor quality of life and increased mortality in long‐term follow‐up [1].
ACPs are histologically complex tumours with variable cystic, calcified and solid components often surrounded by a florid glial and inflammatory reactive tissue. Within tumour epithelia, a peripheral layer of palisading epithelium (PE) encloses more loosely packed stellate cells (i.e. stellate reticulum (SR)) and epithelial whorls [2]. Since the first identification of activating CTNNB1 mutations in ACP [3], several cohorts have recapitulated this finding and mutations have been identified anywhere between 39 and 100% of the tumours. Nuclear accumulation of β‐catenin and immunohistochemical evidence of activation of the WNT pathway (e.g. Axin2 expression) is limited to only a small proportion of cells, which in many cases correlate with epithelial whorls (from now on referred to as clusters) [1, 2, 4].
Several studies have investigated the distribution of CTNNB1 mutations within the different tumour cell compartments. Using Sanger sequencing mutations were identified in both epithelial and ‘mesenchymal’ components [3]. However, Kato et al. did not identify mutations outside of the tumour epithelia [5]. More recently, using laser capture microdissection (LCM) and single‐strand conformational polymorphism analysis, CTNNB1 mutations were identified in all the eight cases [6]. Surprisingly, two cases harboured more than one CTNNB1 mutations and in four cases, different CTNNB1 mutations were found in β‐catenin‐accumulating and non‐accumulating tumour cells [6]. Subsequent studies have failed to identify ACP tumours harbouring more than one CTNNB1 mutation and only one report has described ACP with coexistence of CTNNB1 and BRAF‐V600E mutations [7]. Genetic tracing experiments in genetically engineered mouse models of ACP have revealed a non‐cell autonomous mechanism of pathogenesis, whereby not all tumour cells contain CTNNB1 mutations [1]. Together, these findings have brought into question whether CTNNB1 mutations are present in all ACP tumour cells or only in those accumulating nuclear β‐catenin.
To further investigate the occurrence and cell distribution of CTNNB1 and BRAF mutations in ACPs, we assessed the mutational status of the CTNNB1 exon 3 and the BRAF V600E locus, as well as other commonly mutated brain tumour genes including H3F3A, HIST1H3B, IDH1 and IDH2 in 22 tumours [8]. In addition to Sanger sequencing of exon 3 of CTNNB1, we used a highly sensitive next generation targeted amplicon sequencing panel (TAm‐Seq) [8]. Compared with conventional targeted sequencing TAm‐Seq replicate amplification of the regions of interest followed by barcoding of separate replicates and deep sequencing, enabling the identification of mutations at variant allele frequencies well below those reliably detected by Sanger sequencing (limited to ~20%) [9]. In three cases, in which cryopreserved tumour tissue was available, we performed LCM using the Zeiss PALM MicroBeam system. We analysed by TAm‐Seq specific tumour cell compartments including the clusters (C), PE and SR as well as local reactive glial tissue as previously described [10](Appendix S1). To confirm the sequencing results, we used immunofluorescence staining using antibodies against specific CTNNB1 mutations (Appendix S1).
Sanger sequencing analysis was successful in 19 of 22 cases of ACP, but confirmed the presence of a CTNNB1 mutation only in 12 of 19 tumours (Table 1). In contrast, TAm‐Seq identified CTNNB1 mutations in all cases (22 of 22), with mutation allele frequencies varying from 3 to 47% (Table 1). The higher detection rate reflects a greater sensitivity of the TAm‐Seq method relative to Sanger sequencing in the identification of mutations with lower allele frequency.
Table 1.
CTNNB1 exon 3 sequencing analysis of DNA from 22 cases of ACP
| Case No: | Age at diagnosis (years)* |
Tumour content (% nuclear area) |
DNA conc (copies/ul) |
CTNNB1 mutation by Tam‐Seq |
CTNNB1 mutation by Sanger Seq |
Average mutation allele frequency |
|---|---|---|---|---|---|---|
| 1 | 8 | 20 | 1100 | T41I | T41I | 17% |
| 2 | 14 | 70 | 2315 | T41I | T41I | 28% |
| 3 | 6 | 70 | 390 | S37F | No mutation | 15% |
| 4 | 14 | 60 | 1155 | S45F | Failed** | 35% |
| 5 | 8 | 10 | 525 | S33F | No mutation | 3% |
| 6 | 12 | 50 | 680 | S37A | No mutation | 16% |
| 7 | 46 | 70 | 770 | T41I | No mutation | 21% |
| 8 | 71 | 80 | 8854 | T41I | T41I | 35% |
| 9 | adult | 80 | 395 | S33C | S33C | 39% |
| 10 | adult | 80 | 2273 | T41I | T41I | 29% |
| 11 | 60 | 70 | 143 | T41I | T41I | 15% |
| 12 | 83 | 45 | 2087 | S33C | S33C | 19% |
| 13 | 22 | 80 | 439 | T41I | T41I | 26% |
| 14 | 87 | 90 | 1573 | D32N | D32N | 31% |
| 15 | 61 | 50 | 195 | S33C | S33C | 30% |
| 16 | 65 | 40 | 237 | T41A | T41A | 25% |
| 17 | 53 | 40 | 520 | S37A | S37A | 17% |
| 18 | adult | 90 | 470 | I35S | Failed** | 43% |
| 19 | adult | 60 | 260 | S33F | No mutation | 25% |
| 20 | adult | 25 | 1256 | S37F | No mutation | 3% |
| 21 | adult | 20 | 1460 | S33F | No mutation | 18% |
| 22 | adult | 70 | 4505 | S37C | Failed** | 47% |
Age not available in all patients.
Sanger sequencing reaction failed to give readable trace.
All CTNNB1 mutations have previously been described in ACP, that is D32, S33, I35, S37 and T41 substitutions, which are expected to prevent phosphorylation and therefore disrupt the degradation of β‐catenin [1, 4]. No tumours were found to carry more than one CTNNB1 pathogenic mutation and no mutations in the hotpots of BRAF, H3F3A, HIST1H3B, IDH1 or IDH2 were identified in ACP.
The CTNNB1 mutation allele frequency in ACP correlated with the percentage of tumour epithelia as assessed by nuclear content (r = 0.70, P = 0.0004; Appendix S1, Table S1; Figure S1). Indeed, the CTNNB1 mutation allele frequencies observed across the tumour samples were consistent with the presence of heterozygous CTNNB1 mutations throughout all tumour epithelial cells. For instance, in case 9, the variant allele frequency of the S33C mutation was 39%, consistent with 78% of the cells harbouring a heterozygous mutation and closely correlating with the histological assessment of tumour epithelia of around 80% (Table 1).
The previous results were confirmed by TAm‐Seq analysis of specific tumour cell compartments isolated by LCM (Appendix S1; Figure S2). CTNNB1 mutation frequencies were 47–55%, which are consistent with heterozygosity within all tumour compartments (clusters, PE, SR) but nearly zero within the surrounding glial tissue (max 0.65%) and germline DNA (available in two of the three cases) (Table S1).
Finally, we used immunofluorescence staining with specific antibodies against the S33F, S37F and T41I substitutions in β‐catenin on cases of ACP (cases: S33F, n = 2; S37F, n = 4; T41I, n = 6; Figure 1). These studies confirmed the expression mutant β‐catenin protein across the tumour epithelium with nuclear accumulation limited to the cell clusters (Figure 1). Negative control immunofluorescence staining on ACP tumours separately known to harbour other CTNNB1 mutations confirmed the specificity of the antibodies used (Figure 1) [10].
Figure 1.
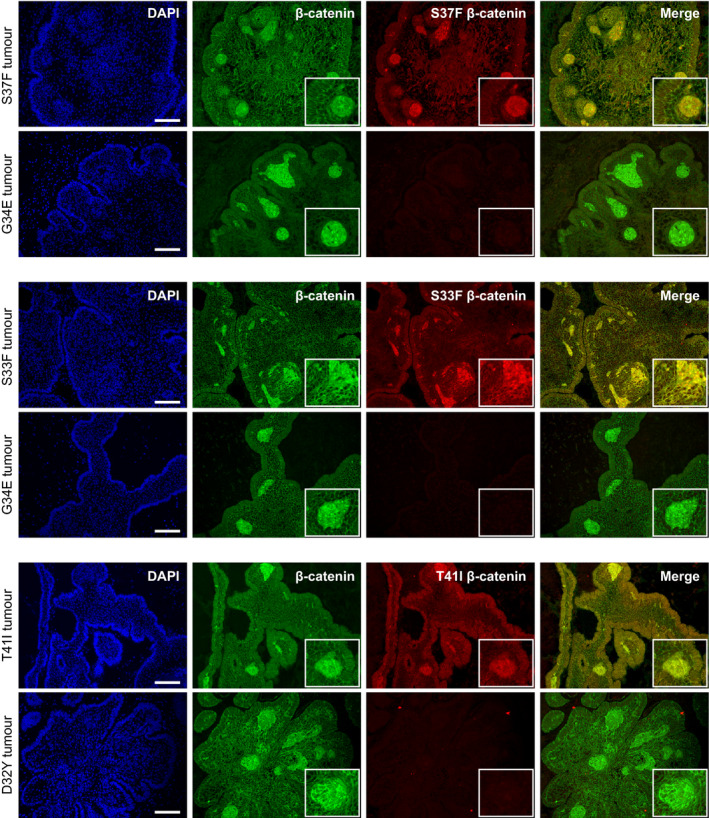
Mutant β‐catenin is expressed throughout the tumour epithelia. Double immunofluorescence stainings on FFPE histological section of ACP tumours harbouring different mutations (shown on the left) using specific antibodies against either S37F, S33F or T41I mutant β‐catenin (red) as well as mutation non‐specific β‐catenin antibody (shown within the panels). Boxes highlight nuclear accumulation within clusters. Scale bar = 100 µm.
In conclusion, we confirm a high prevalence of CTNNB1 mutations in ACP. The high sensitivity of TAm‐Seq has helped identify CTNNB1 mutations in all ACP samples analysed, including those with very low allele frequencies. This suggests that failure to identify CTNNB1 mutations in a low proportion of ACP tumours in previous studies using Sanger sequencing, single‐strand conformation polymorphism analysis, exome sequencing and targeted next generation sequencing may reflect methodological limitations.
We reveal that all cellular components of the tumour epithelium harbour the same CTNNB1 mutation and found no evidence of second CTNNB1 mutations, even at low allele frequencies. Additionally, we show the presence of CTNNB1 mutations throughout the tumour epithelia, which is consistent with the early stages of tumourigenesis in the embryonic mouse model of ACP. In this model, oncogenic Ctnnb1 mutation is present throughout the entire tumoural pituitary, but nuclear accumulation of β‐catenin is limited to only a small proportion of cells mostly forming clusters, which are molecularly analogous to the human clusters [11, 12]. The mechanism behind why β‐catenin is accumulated only in single cells or clusters despite the presence of CTNNB1 mutations in other epithelial tumour cells (i.e. palisading epithelium and stellate reticulum) is not known.
Ethics
Experiments were performed under NHS Research Ethics Committee approval (14/LO/2265) or approval from individual biobanks. Where required, informed consent was obtained from all individual participants included in the study.
Funding
Funding for this research was provided by Cancer Research UK, the Children’s Cancer and Leukaemia Group, Children with Cancer UK (15/190), MRC (MR/M125/1), the Brain Tumour Charity (SIGNAL and EVEREST), Great Ormond Street Hospital Children’s Charity and the National Institute of Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. J.R.A. is supported by a Cancer Research UK Clinical Trials Fellow. JMGM is part of the Translational OMICS Strategic Focus Group at Tecnologico de Monterrey. J.P.M.‐B. is a Great Ormond Street Hospital for Children’s Charity Principal Investigator.
Conflict of Interest
The Editors of Neuropathology and Applied Neurobiology are committed to peer‐review integrity and upholding the highest standards of review. As such, this article was peer‐reviewed by independent, anonymous expert referees, and the authors (including TSJ and JPMB) had no role in either the editorial decision or the handling of the paper.
Supporting information
Figure S1 . A. Correlation between mutation allele frequency and histologically assessed tumour content (%nuclei). Arrows indicate cases 9 and 17. B. Examples of ACP FFPE histological sections stained with haematoxylin and eosin.
Figure S2 . Representative images of laser capture microdissection (LCM): A. Areas selected for LCM are highlighted by colours: Clusters (green), stellate reticulum (black), palisading epithelium (blue), reactive glial tissue (yellow). B. Example showing the excision of a cluster. C. Example of multiple clusters pooled before DNA extraction. Scale bar =100μm.
Table S1 . Laser capture microdissection identifies CTNNB1 mutations in all ACP tumour compartments.
Appendix S1 . Supplementary Methods
Acknowledgements
Tissue samples were obtained from Plymouth Hospitals NHS trust as part of the UK Brain Archive Information Network (BRAIN UK), which is funded by the Medical Research Council and brains trust. We thank the CCLG Tissue Bank for access to samples, and contributing CCLG Centres, including members of the ECMC Paediatric network. The CCLG Tissue Bank is funded by Cancer Research UK and CCLG. Tissue samples were provided by the Imperial College Healthcare NHS Trust Tissue Bank. Other investigators may have received samples from these same tissues. The research supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. This work is supported by the NIHR GOSH BRC. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health
References
- 1. Muller HL, Merchant TE, Puget S, Martinez‐Barbera JP. New outlook on the diagnosis, treatment and follow‐up of childhood‐onset craniopharyngioma. Nat Rev Endocrinol 2017; 13: 299–312 [DOI] [PubMed] [Google Scholar]
- 2. Martinez‐Barbera JP, Buslei R. Adamantinomatous craniopharyngioma: pathology, molecular genetics and mouse models. J Pediatr Endocrinol Metab 2015; 28: 7–17 [DOI] [PubMed] [Google Scholar]
- 3. Sekine S, Sato S, Takata T, Fukuda Y, Ishida T, Kishino M, et al Beta‐catenin mutations are frequent in calcifying odontogenic cysts, but rare in ameloblastomas. Am J Pathol 2003; 163: 1707–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larkin SJ, Ansorge O. Pathology and pathogenesis of craniopharyngiomas. Pituitary 2013; 16: 9–17 [DOI] [PubMed] [Google Scholar]
- 5. Kato K, Nakatani Y, Kanno H, Inayama Y, Ijiri R, Nagahara N, et al Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol 2004; 203: 814–21 [DOI] [PubMed] [Google Scholar]
- 6. Holsken A, Kreutzer J, Hofmann BM, Hans V, Oppel F, Buchfelder M, et al Target gene activation of the Wnt signaling pathway in nuclear beta‐catenin accumulating cells of adamantinomatous craniopharyngiomas. Brain Pathol 2009; 19: 357–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larkin SJ, Preda V, Karavitaki N, Grossman A, Ansorge O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1‐mutated adamantinomatous craniopharyngioma. Acta Neuropathol 2014; 127: 927–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stone TJ, Keeley A, Virasami A, Harkness W, Tisdall M, Izquierdo Delgado E, et al Comprehensive molecular characterisation of epilepsy‐associated glioneuronal tumours. Acta Neuropathol 2018; 135: 115–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012; 4: 136ra68 [DOI] [PubMed] [Google Scholar]
- 10. Apps JR, Carreno G, Gonzalez‐Meljem JM, Haston S, Guiho R, Cooper JE et al Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol 2018; 135: 757–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gonzalez‐Meljem JM, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, et al Stem cell senescence drives age‐attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun 2017; 8: 1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaston‐Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, et al Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci USA 2011; 108: 11482–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 . A. Correlation between mutation allele frequency and histologically assessed tumour content (%nuclei). Arrows indicate cases 9 and 17. B. Examples of ACP FFPE histological sections stained with haematoxylin and eosin.
Figure S2 . Representative images of laser capture microdissection (LCM): A. Areas selected for LCM are highlighted by colours: Clusters (green), stellate reticulum (black), palisading epithelium (blue), reactive glial tissue (yellow). B. Example showing the excision of a cluster. C. Example of multiple clusters pooled before DNA extraction. Scale bar =100μm.
Table S1 . Laser capture microdissection identifies CTNNB1 mutations in all ACP tumour compartments.
Appendix S1 . Supplementary Methods