

Abstract
While the transition metal copper (Cu) is an essential nutrient that is conventionally viewed as a static cofactor within enzyme active sites, a nontraditional role for Cu as a modulator of kinase signaling is emerging. We discovered that Cu is required for the activity of the autophagic kinases ULK1/2 through a direct Cu-ULK1/2 interaction. Genetic loss of the Cu transporter Ctr1 or mutations in ULK1 that disrupt Cu-binding reduced ULK1/2-dependent signaling and autophagosome complex formation. Elevated intracellular Cu levels are associated with starvation induced autophagy and sufficient to enhance ULK1 kinase activity and in turn autophagic flux. The growth and survival of lung tumors driven by KRASG12D is diminished in the absence of Ctr1, depends on ULK1 Cu-binding, and is associated with reduced autophagy levels and signaling. These findings suggest a molecular basis for exploiting Cu-chelation therapy to forestall autophagy signaling to limit proliferation and survival in cancer.
Introduction
By default, the dynamic and adaptive nature of signaling networks allows them to respond and, in some cases, sense extracellular and intracellular inputs1. While growth factors, nutrients, and metabolites are well-appreciated regulators of cell proliferation, the contribution of transition metals to cellular processes that support proliferation and contribute to malignant transformation are understudied. The transition metal copper (Cu) is essential for a diverse array of biological processes from cellular proliferation, neuropeptide processing, free radical detoxification, and pigmentation2. The importance of intact Cu homeostatic mechanisms for cell growth control is underscored by the stunted growth and failure to thrive associated with Cu deficiency in Menkes disease patients3 and the increased prevalence of cancer in patients and animal models with hereditary Cu overload in Wilson disease4–6. However, the dysregulated growth phenotypes associated with alterations in Cu availability cannot be fully explained by the limited number of Cu-dependent enzymes that traditionally harness the redox potential of Cu as a structural or catalytic cofactor.
Several recent studies have illuminated functions for Cu as a non-structural intracellular mediator of signaling by directly connecting Cu to signaling pathway components that modulate cell proliferation or lipid metabolism7–9. We previously showed that in response to growth factors, Cu contributes to the amplitude of canonical MAPK signaling through an interaction between Cu and the kinases MEK1 and MEK27. Examination of this signaling mechanism in the context of cancer revealed that BRAFV600E-driven tumors depend on MEK1/2 association with Cu, and moreover, are sensitive to Cu-chelating drugs8,10,11. In Wilson disease, Cu influences 3’,5’-cyclic AMP (cAMP)-mediated lipolysis downstream of β-adrenergic receptor signaling by directly inhibiting the phosphodiesterase PDE3B9, serving as one molecular mechanism to explain the connection between aberrant Cu accumulation and altered lipid metabolism5. These findings bolster a paradigm linking Cu levels to signaling pathway components that are essential for normal physiology, but that become dysregulated in multiple disease states including cancer. Thus, uncovering additional kinases that are regulated by Cu levels is needed to further define Cu-dependent cellular processes that may contribute to the efficacy of Cu chelators for cancer therapy12.
Results
Cu binds to ULK1 and ULK2 and is required for kinase activity.
To explore other Cu-dependent kinases, we first performed an alignment of the Cu-binding sequence in MEK18 against kinase domains and identified the autophagy regulatory kinases ULK1 and ULK2 as sharing significant sequence similarity at the amino acids (H188, M230, and H239) required for Cu-binding in MEK1 (Fig. 1a). Both ULK1 and ULK2 bind to a Cu-charged resin, but not a metal free resin control, Fe-, or Zn-charged resin (Fig. 1b,c). To investigate the impact of Cu-binding to ULK1 and ULK2 function, we first tested whether Cu is directly required for ULK1 and ULK2 kinase activity. ULK1 and ULK2 in vitro kinase activity was enhanced in response to increasing concentrations of CuCl2 (Fig. 1d,e and Extended Data Fig. 1a,b) and inhibited when treated with increasing concentrations of the Cu chelator tetrathiomolybdate (TTM) (Fig. 1f,g and Extended Data Fig. 1c,d). The reduced ULK1 and ULK2 kinase activity in presence of TTM was similar to inhibition achieved with the ULK1/2 inhibitor MRT6892113 (Fig. 1f,g and Extended Data Fig. 1c,d).
Figure 1. Cu binds to ULK1 and ULK2 and is required for kinase activity.
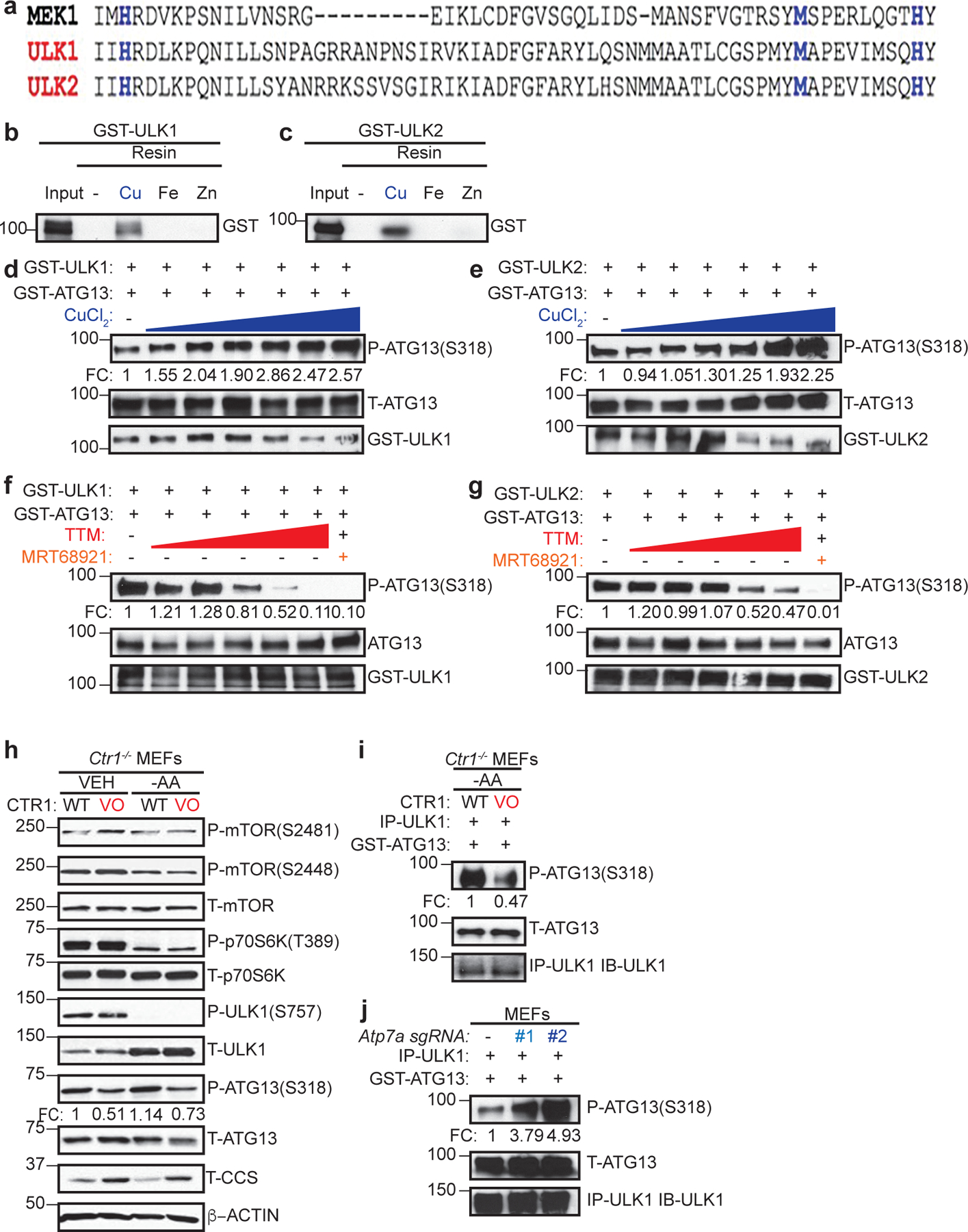
a, The amino-acid sequence of human MEK1 aligned to human ULK1 and human ULK2. Black letters, amino acids; blue letters, the four amino acids mutated in Cu-binding mutant (CBM) of MEK1 to decrease Cu binding and those conserved between ULK1 and ULK2. b,c, Immunoblot detection of recombinant GST-ULK1 or GST-ULK2 bound to a resin charged with or without Cu, Fe, or Zn compared to input. n=3 biologically independent experiments. d,e,f,g, Immunoblot detection of recombinant phosphorylated (P)-ATG13, total (T)-ATG13, and GST-ULK1 or GST-ULK2 from ULK1 or ULK2 in vitro kinase assays treated with or without increasing concentrations of CuCl2 from 0 to 10 μM (d,e), with or without increasing concentrations of TTM from 0 to 50 μM, or 10 μM MRT68921 (f,g). Quantification: ΔP-ATG13/T-ATG13 normalized to GST-ULK1 and GST-ATG13 alone. d,e,g, n=3 biologically independent experiments. g, n=4 biologically independent experiments. h, Immunoblot detection of P- and/or T- mTOR, p70S6K, ULK1, ATG13, CCS, or β-ACTIN from Ctr1−/− MEFs stably expressing CTR1WT (WT) or empty vector (VO) treated with vehicle (VEH) or amino acid deprivation (-AA). Quantification: ΔP-ATG13/T-ATG13 normalized to WT, VEH control. n=3 biologically independent experiments. i,j, Immunoblot detection of recombinant P-ATG13, T-ATG13, or immunoprecipitated (IP)-ULK1 from immunocomplex ULK1 kinase assays from Ctr1−/− MEFs stably expressing WT or VO treated with VEH or -AA or MEFs stably expressing sgRNA against Rosa (−) or Atp7a (#1 or #2). Quantification: ΔP-ATG13/T-ATG13 normalized to WT, VEH control or Rosa (−) control. n=3 biologically independent experiments.
Reducing or increasing intracellular Cu levels modulates ULK1/2 kinase activity.
ULK1/2 are well-established downstream targets of the protein kinase mammalian target of rapamycin (mTOR)14–17. When associated with rapamycin-sensitive mTOR complex 1 (mTORC1)18, mTOR controls cellular energetics by driving anabolic processes, such as lipid, nucleotide, and protein synthesis19, and suppressing catabolic processes14–17, like autophagy. mTOR kinase activity is blocked in response to amino acid deprivation20–22, low cellular ATP levels23,24, or rapamycin treatment25,26. This in turn relieves the inhibitory phosphorylation of ULK1 and ULK2 and initiates the formation of an autophagosome14–17, which degrades and recycles proteins, lipids, and organelles to restore nutrient balance in the form of building blocks for metabolic pathways. To investigate the relationship between Cu and mTORC1-ULK1/2 signaling in mammalian cells, Cu-replete and Cu-deficient cells were generated by expressing the high affinity Cu transporter CTR1 (WT) or an empty vector (VO) in immortalized Ctr1−/− mouse embryonic fibroblasts (MEFs). Intracellular Cu levels in CTR1WT expressing Ctr1-null MEFs are comparable to Ctr1+/+ MEFs as confirmed by reduced CCS protein stability27, which is degraded in a Cu-dependent fashion, increased Cu levels via inductively coupled plasma mass spectrometry (ICP-MS), and elevated fluorescence from the CF4 probe28, which detects labile Cu (Fig. 1d and Extended Data Fig. 1e–h). Ctr1 loss reduced ULK1-mediated phosphorylation of ATG13 under basal conditions and the elevated phosphorylation of ATG13 that occurs upon amino acid deprivation14–17 was only observed in the Cu-replete conditions (Fig. 1h and Extended Data Fig. 1i). In contrast, PI3K/AKT signaling-mediated phosphorylation29 of mTORS2448 and mTOR phosphorylation30–32 of ULK1S757, p70S6KT389, and mTORS2481 remained unchanged upon reduced Cu levels (Fig. 1h). These data suggest that Cu is required for ULK1 activity but not its upstream regulation by mTORC1 or mTOR itself. In agreement, ULK1 kinase activity is elevated upon amino acid withdrawal in Cu-replete cells but is blunted in the Cu-deficient cells (Fig. 1i and Extended Data Fig. 1j). Additionally, MEFs lacking the Cu exporter Atp7a, which have elevated intracellular Cu levels, also had increased ULK1 kinase activity (Fig. 1j and Extended Data Fig. 1k). These findings that reduced Cu levels decrease ULK1/2-dependent signaling downstream of mTORC1 inhibition (Fig. 1h–j) suggest that autophagy was a yet to be identified Cu-dependent process.
Cu is both necessary and sufficient for autophagic flux in an ULK1/2-dependent fashion.
The ULK1-ATG13 complex senses low-energy and nutrient depleted cellular states and initiates autophagosome formation14–17. To determine whether changes in levels of Cu affect ULK1/2-dependent autophagy, Cu-replete or Cu-deficient MEFs were exposed to amino acid deprivation20–22 or rapamycin25,26, which inhibit mTOR. Specifically, autophagic flux was measured by three techniques, including i) Western blot analysis of the processing of LC3 from its cleaved form (LC3-I) to phosphatidylethanolamine-conjugated form (LC3-II)33, ii) flow cytometry measurement of the ratio of autolysosomes (mCherry-LC3B) to autophagosomes (EGFP-LC3B)34, and iii) immunofluorescence detection of the abundance of autophagosomes (mCherry-EGFP-LC3 positive puncta) and autolysosomes (mCherry-LC3 positive puncta)35. These studies revealed that loss of Ctr1 significantly reduced LC3-I processing (Fig. 2a and Extended Data Fig. 2a) and prevented autophagosome to autolysosome conversion upon autophagy induction (Fig. 2b–d). Further, under basal conditions the number of mCherry-EGFP-LC3 positive puncta was significantly reduced, indicating a potential block in autophagosome formation and in turn recruitment of LC3B (Fig. 2c,d). In contrast, CRISPR/Cas9-mediated knockdown of endogenous Atp7a (Extended Data Fig. 2b) caused elevated Cu levels that were sufficient to enhance LC3-II levels (Fig. 2e and Extended Data Fig. 2c) and autophagic flux (Fig. 2f–h). Collectively, these data suggest that the levels of Cu could serve as a rheostat for ULK1/2-dependent autophagy by regulating ULK1/2 kinase activity in an mTOR-dependent fashion.
Figure 2. Genetic ablation or enhancement in intracellular Cu levels modulates autophagic flux.
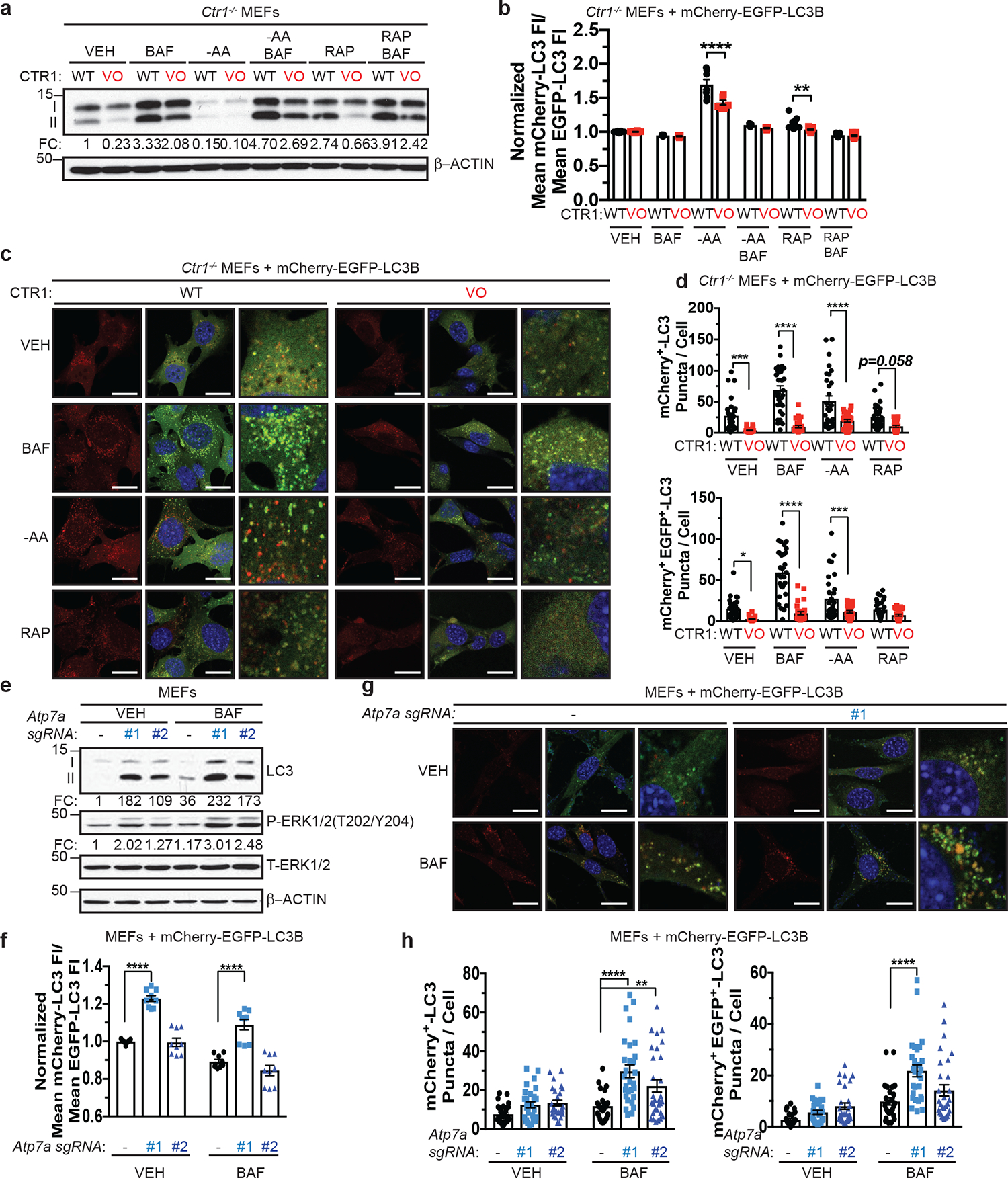
a, Immunoblot detection of proteins from treated MEFs. Quantification: ΔLC3-II/β-ACTIN normalized to WT, VEH control. n=3 biologically independent experiments. b, Scatter dot plot with bar at mean mCherry-LC3 fluorescent intensity (FI)/mean EGFP-LC3 FI ± s.e.m. analyzed by flow cytometry from treated MEFs. N represents number[AU please clarify what the number is here.] of biologically independent samples, VEH n=16; BAF n=7; -AA n=7; -AA+BAF n=7; RAP, WT n=16, VO n=15; RAP+BAF n=13. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. **, P=0.0035; ****, P<0.0001. c, Representative fluorescence images of EGFP, mCherry, or the merge from treated MEFs. Scale bar, 20 μm. d, Scatter dot plot with bar at mean mCherry+-LC3 or mCherry+ EGFP+-LC3 puncta per cell ± s.e.m. from treated MEFs. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. n represents number of [AU please clarify the number which n represents] fields of view. Top, VEH n=30; BAF, WT n=30, VO n=29; -AA, WT n=30, VO n=28; RAP n=30; ***, P=0.0006; ****; P<0.001; Bottom, VEH, WT n=29, VO n=30; BAF, WT n=30, VO n=29; -AA, WT n=30, VO n=29; RAP, WT n=32, VO n=31; *, P=0.0147, ***; P=0.0009; ****, P<0.0001. e, Immunoblot detection of proteins from treated MEFs. Quantification: ΔLC3-II/T- β-ACTIN and ΔP-ERK1/2/T-ERK1/2 normalized to Rosa (−), VEH control. n=4 biologically independent experiments. f, Scatter dot plot with bar at mean mCherry-LC3 fluorescent intensity (FI)/mean EGFP-LC3 FI ± s.e.m. analyzed by flow cytometry from treated MEFs. n=9 biologically independent samples. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. ****, P<0.0001. g, Representative fluorescence images of EGFP, mCherry, or the merge from treated MEFs. Scale bar, 20 μm. h, Scatter dot plot with bar at mean mCherry+-LC3 or mCherry+ EGFP+-LC3 puncta per cell ± s.e.m. from treated MEFs. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. n represents number of [AU please clarify this number] fields of view. Left, VEH, Rosa n=30, #1 n=29, #2 n=26, BAF, Rosa n=24, #1 n=29, #2 n=30; **, P=0.00911; ****, P<0.0001; Right, VEH, Rosa n=30, #1 n=27, #2 n=30; BAF, Rosa n=26, #1 n=29, #2 n=30; **, P=0.0042; ****, P<0.0001.
Since altering Cu levels has been shown to affect MAPK signaling7,8, we interrogated the contribution of the MAPK pathway to Cu-mediated regulation of autophagy via genetic and pharmacologic approaches. First, we directly tested whether disrupting MEK1 Cu-binding was sufficient to reduce autophagy in the presence (Ctr1flox/flox) or absence (Ctr1−/−) of Cu transport. While expression of Cu-binding mutant (CBM) of MEK18 reduced phosphorylation of ERK1/2 (Extended Data Fig. 2d), LC3-II levels were unchanged and Cu-dependent autophagic flux was still observed in Ctr1−/− MEFs (Extended Data Fig. 2e,f). Second, treatment with the MEK1/2 inhibitor, trametinib, which abolished phosphorylation of ERK1/2, did not phenocopy the reduced autophagic flux in the absence of Ctr1 (Extended Data Fig. 2g,h). Consistent with these findings, expression of a gain-of-function (GOF) ERK2 mutant8, which bypasses the ability of Cu to influence MEK1/2 activity in Ctr1−/− MEFs (Extended Data Fig. 2i), did not increase LC3-II levels in cells devoid of significant Cu transport (Extended Data Fig. 2j,k). Finally, while increasing Cu levels via Atp7a knockdown is sufficient to increase phosphorylation of ERK1/2 (Fig. 2e), loss of Atp7a in MEFs lacking the key autophagy regulator Atg5 (Extended Data Fig. 2b), which is part of the ubiquitin-like conjugation system that lipidates LC333, failed to re-establish autophagy (Extended Data Fig. 2l). Collectively, these data indicate that Cu-dependent autophagy is mechanistically independent of Cu-regulated MEK1/2 activity and upstream of ATG5, which is required for autophagosome nucleation.
Elevated intracellular Cu levels are associated with starvation induced autophagy and are required for autophagosome complex formation.
Since our findings suggest that Cu is required for ULK1/2-dependent signaling and autophagic flux in response to mTOR inhibition, we next hypothesized that Cu levels may fluctuate in an acute manner after autophagy induction. Interestingly, a statistically significant elevation in labile intracellular Cu levels, as measured by increased fluorescence of the Cu probe CF428 and not its control (Ctrl-CF4)28, was detected in a time-dependent fashion post amino acid withdrawal (Fig. 3a,b, Extended Data Fig. 3a,b, and Supplementary Video 1–4). Thus, the requirement for Cu for efficient autophagic flux is driven by Cu fluctuations that support ULK1/2 kinase activity. While ULK1/2 activation is typically a prerequisite for LC3 processing by the ubiquitin-like conjugation system on the forming autophagosome14, several upstream ULK1/2-dependent molecular events are essential to initiate autophagosome formation36,37 (Extended Data Fig. 3c). Specifically, active ULK1 translocates to the ER with its constitutive binding partners ATG13 and FIP200 to drive phagophore formation by phosphorylating core components of the class III PI3K VPS34 complex, which includes BECLIN-1, PIK3R4 (p150), and VPS34 itself14–17,38–40. The ULK1-mediated phosphorylation of BECLIN-1 or ATG14 within VPS34 complex 1 stimulates VPS34 activity, which converts phosphatidylinositol to PI3P40,41. The generation of PI3P serves as a docking site for FYVE domain containing proteins, like DFCP1 and WIPI1, that recruit the ubiquitin like conjugation complex ATG5-ATG12-ATG16 to lipidate LC3B36,42. To dissect the contribution of Cu to the molecular events directly downstream of ULK1-dependent autophagosome formation, we visualized ULK1 complex translocation to puncta, (EGFP-ATG13 or EGFP-FIP200), VPS34 complex PI3P generation on puncta (EGFP-DFCP1), and PI3P effector translocation to puncta (EGFP-WIPI1) in response to amino acid deprivation in the presence (Ctr1flox/flox) or absence (Ctr1−/−) of Cu transport. Cu deficiency significantly decreased both ATG13, FIP200, WIPI1, and DFCP1 puncta formation in response to amino acid deprivation, indicating that Cu transport contributes to the early steps of phagophore nucleation (Fig. 3c–j). Defective recruitment of these initiating complexes and PI3P production, limited the number of EGFP-LC3 positive autophagosomes measured by flow cytometry43 (Fig. 3k and Extended Data Fig. 3d), an effect that was rescued by loss of the Cu exporter Atp7A (Fig. 3l). A selective requirement for Cu in the regulation of autophagosome formation was observed, as the Cu chelator TTM reduced EGFP-LC3 positive autophagosome number, while the iron (Fe) chelator desferoxamine (DFO) had no effect (Extended Data Fig. 3e,f). These findings suggest that ULK1/2-driven autophagy initiation requires Cu transport.
Figure 3. Cu is both necessary and sufficient for autophagosome formation.
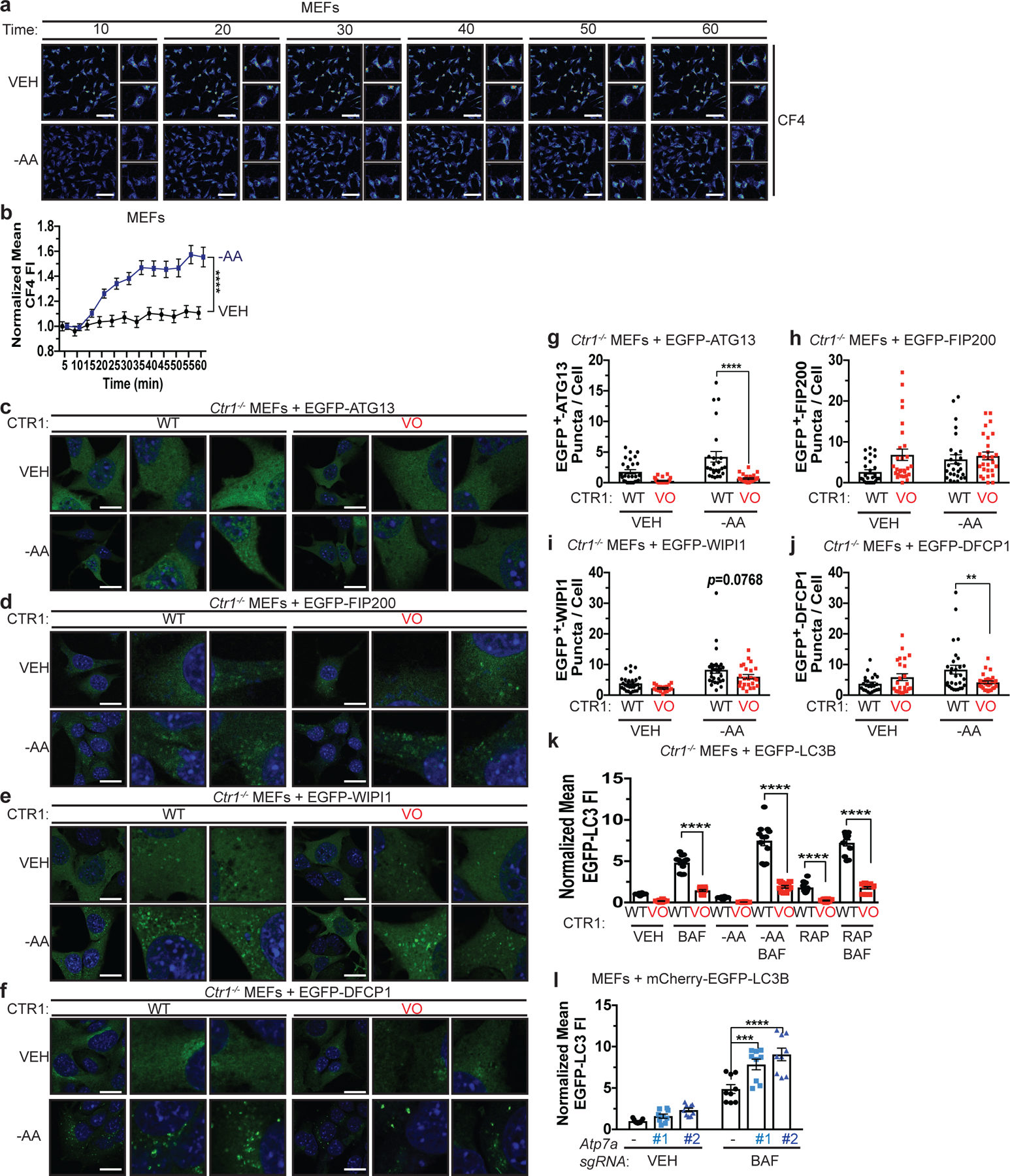
a, Representative live cell imaging of the Cu probe CF4 every ten minutes for 60 minutes from MEFs treated with vehicle (VEH) or amino acid deprivation (-AA). Scale bar, 100 μm. b, Mean CF4 fluorescence intensity (FI) ± s.e.m. versus time (minutes, min) from MEFs treated with VEH or -AA normalized to t=0, five minutes. n=30 individual cells. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. ****, P<0.0001. c,d,e,f, Representative fluorescence images of EGFP-ATG13 (c), EGFP-FIP200 (d), EGFP-WIP1 (e), or EGFP-DFCP1 (f) from Ctr1−/− MEFs stably expressing CTR1WT (WT) or empty vector (VO) and EGFP-ATG13, EGFP-FIP200, EGFP-WIP1, or EGFP-DFCP1 treated with VEH or -AA. Scale bar, 20 μm. g,h,i,j, Scatter dot plot with bar at mean EGFP+ puncta per cell ± s.e.m. from Ctr1−/− MEFs stably expressing WT or VO and EGFP-ATG13 (g), EGFP-FIP200 (h), EGFP-WIP1 (i), or EGFP-DFCP1 (j) treated with VEH or -AA. N represents number of[AU please clarify this number] fields of view. EGFP-ATG13, VEH, WT n=27, VO n=26; -AA, WT n=26, VO n=27. EGFP-FIP200, VEH n=28; -AA, WT n=27, VO n=28. EGFP-WIP1, VEH, WT n=29, VO n=18; -AA, WT n=29, VO n=24. EGFP-DFCP1, VEH n=24, -AA n=29. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. **, P=0.0057; ****, P<0.0001. k, Scatter dot plot with bar at mean EGFP-LC3 FI ± s.e.m. analyzed by flow cytometry from Ctr1−/− MEFs stably expressing WT or VO and EGFP-LC3B treated with VEH, -AA, or RAP with or without BAF normalized to WT, VEH control. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. n=12 biologically independent samples. ****, P<0.0001. l, Scatter dot plot with bar at mean EGFP-LC3 FI ± s.e.m. analyzed by flow cytometry from MEFs stably expressing sgRNA against Rosa (−) or Atp7a (#1 or #2) and EGFP-LC3B treated with VEH or BAF normalized to Rosa (−), VEH control. n=9 biologically independent samples. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. ***, P=0.0002; ****, P<0.0001.
Genetic ablation of Ctr1 decreases autophagy and proliferation to reduce tumorigenesis and sensitizes cancer cells to starvation in a mouse model of KrasG12D-driven lung cancer.
Increased levels of basal autophagy in some cancers represents a vulnerability to autophagy inhibition44. We previously demonstrated that genetic ablation of Ctr1 reduced BRAFV600E-driven lung tumor development, leading to a survival advantage8. While the reliance of BRAFV600E-driven tumors on both autophagy45 and Cu transport8 could be exploited therapeutically, activating mutations in KRAS are the most common driver event in this disease46. Importantly, oncogenic KrasG12D-driven lung adenocarcinomas also depend on efficient autophagy for tumor maintenance47. Deletion of the critical autophagy component Atg7 converts KrasG12D-driven adenomas and adenocarcinomas to oncocytomas, a benign tumor type characterized by an accumulation of dysfunctional mitochondria47. To investigate the relationship between Cu, oncogenic KRAS-driven lung cancer, and autophagy, we investigated whether loss of Ctr1 disrupts autophagy in an autochthonously arising KRASG12D-driven lung adenocarcinoma model. We transduced cohorts of LSL-KrasG12D/+;Trp53flox/flox;Rosa26::LSL-Luc (KPLuc) mice via intratracheal administration of lentivirus expressing Cre recombinase, Cas9, and sgRNA targeting either β-GAL or Ctr1. Tumor growth was monitored over time with luminescence imaging. Strikingly, we found that genetic targeting of Cu transport blunts the development of KRASG12D-driven lung tumors (Fig. 4a,b,d,g and Extended Data Fig. 4a–c). Tumor volume at 16 weeks was significantly decreased in mice with CRISPR-mediated deletion of Ctr1 (Fig. 4a,b,g). Ctr1 loss was confirmed by increased CCS levels in the tumor tissue (Fig. 4c,e,h) and correlated with reduced in vivo tumor cell proliferation (Ki67-positive staining, Fig. 4f,i). Molecularly, the established Ctr1 deficient KPLuc tumors had decreased LC3-positive staining, increased p62-positive staining, and reduced autophagosome number, indicative of diminished autophagy (Fig. 5a–f). Thus, the loss of Ctr1 mitigates KRASG12D-mediated autophagy that is necessary for lung tumor growth.
Figure 4. Genetic ablation of Ctr1 decreases proliferation to reduce tumorigenesis in a mouse model of KrasG12D-driven lung cancer.
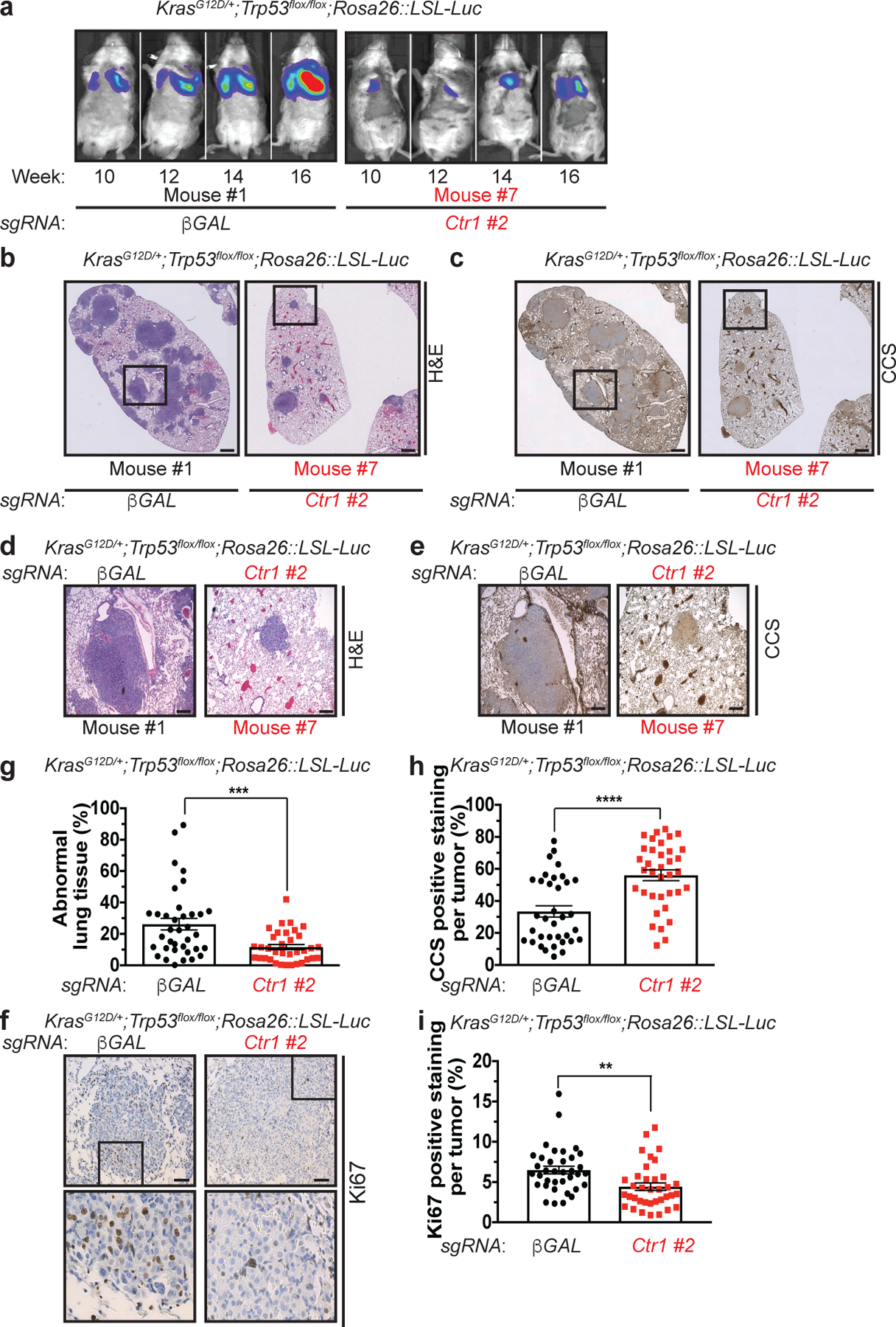
a, Normalized representative images of in vivo luminescence of KrasG12D/+;Trp53flox/flox;Rosa26::LSL-Luc (KPLuc) mice introduced with either sgRNA against β-GAL or Ctr1 at indicated time points. n=6 biologically independent animals. b,c,d,e,f, Representative images of H&E stained (b,−5x stitched images, d-5x); immunohistochemical detection of CCS (c-5x stitched images, e-5x), or Ki67 (f-40x) of lungs from KPLuc mice expressing sgRNA against β-GAL or Ctr1 (5x stitched scale bar: 250 μm; 5x scale bar: 250 μm; 40x scale bar: 50 μm). g,h,i, Scatter dot plot with bar at mean ± s.e.m. % abnormal lung tissue (g, β-GAL, n=36 images; Ctr1, n=35 images), % CCS positive staining per tumor (h, β-GAL, n=35 images; Ctr1, n=36 images), or % Ki67 positive staining per tumor (i, n=36 images) from KPLuc mice expressing sgRNA against β-GAL or Ctr1. Results were compared using an unpaired, one-tailed Student’s t-test. **, P=0.0025; ***, P=0.0006; ****, P<0.0001.
Figure 5. Genetic ablation of Ctr1 decreases autophagy to reduce tumorigenesis and sensitizes cancer cells to starvation in a mouse model of KrasG12D-driven lung cancer.
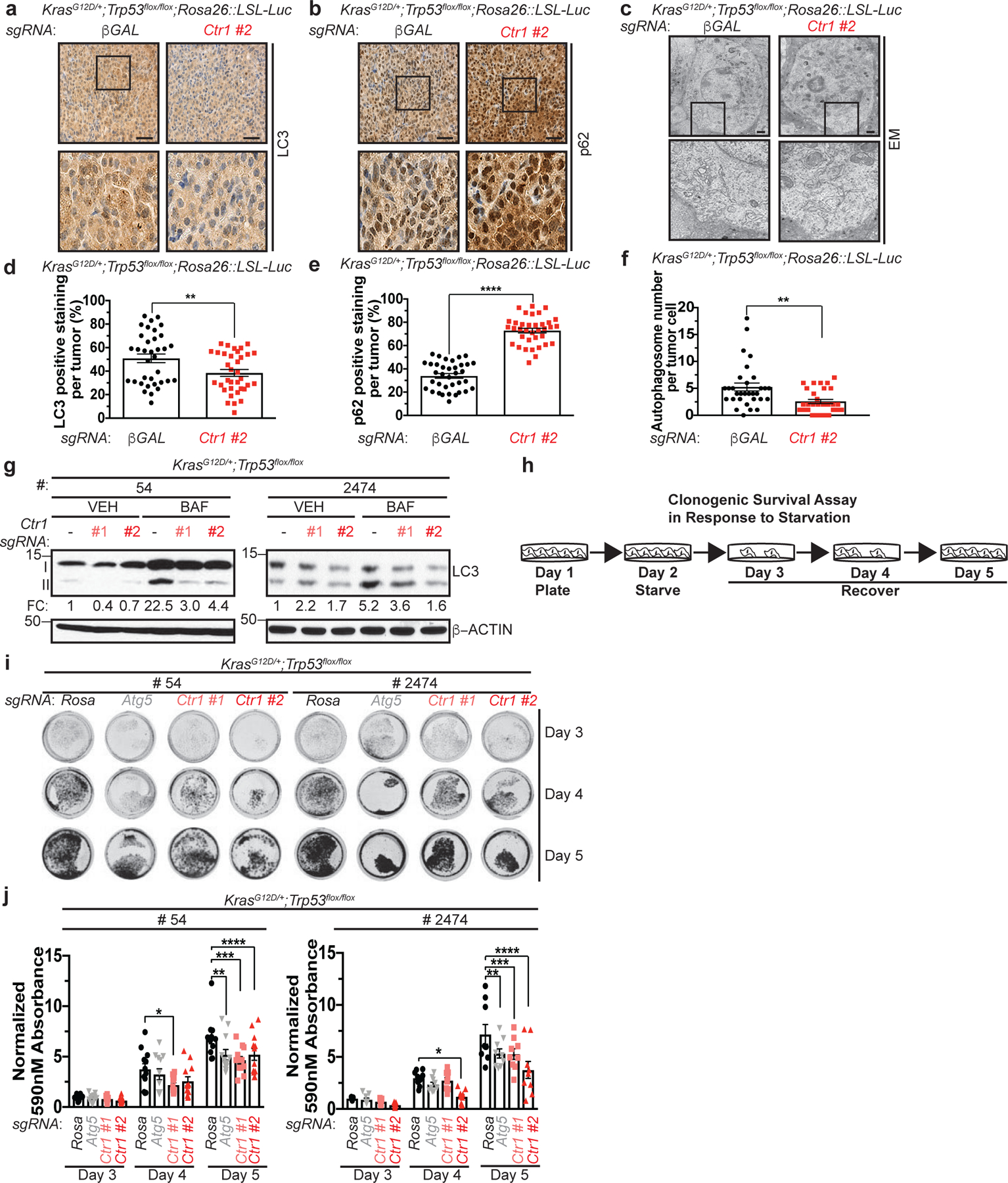
a,b,c, Representative images of immunohistochemical detection of LC3 (a-40x) or p62 (b-40x); or electron microscopy (EM) (c) of lungs from KPLuc mice expressing sgRNA against β-GAL or Ctr1 (40x scale bar: 50 μm; EM scale bar: 1 μm). d,e,f, Scatter dot plot of mean ± s.e.m. % LC3 positive staining per tumor (d, n=35 images), % p62 positive staining per tumor (e, n=35 images), or autophagosome number per tumor cell (f, β-GAL, n=31 images; Ctr1, n=30 images) from KPLuc mice expressing sgRNA against β-GAL or Ctr1. Results were compared using an unpaired, one-tailed Student’s t-test. d, **, P=0.0098; e, ****, P<0.0001; f, **, P=0.0024. g, Immunoblot detection of LC3-I, LC3-II, or β-ACTIN from KP lung adenocarcinoma cell lines #54 (KP #54) and #2474 (KP #2474) stably expressing sgRNA against Rosa (−) or Ctr1 (#1 or #2) treated with vehicle (VEH) or bafilomycin (BAF). Quantification: ΔLC3-II/β-ACTIN normalized to Rosa (−), VEH control. n=3 biologically independent experiments. h, Schematic of clonogenic survival assay in response to starvation. KP #54 and KP #2474 cells stably expressing sgRNA against Rosa, Atg5, or Ctr1 following one day of starvation (EBSS) were recovered for three days in normal medium (DMEM). i, Representative crystal violet images of KP #54 and KP #2474 cells stably expressing sgRNA against Rosa, Atg5, or Ctr1 (#1 or #2) from days 3, 4, and 5 of recovery. j, Scatter dot plot with bar at mean absorbance of extracted crystal violet at 590nM ± s.e.m. of KP #54 and KP #2474 cells stably expressing sgRNA against Rosa, Atg5, or Ctr1 (#1 or #2) from days 3, 4, and 5 of recovery normalized to Rosa, day 3 control. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. KP #54, n represents number of biologically independent samples; Day 3 n=12, Day 4 n=12, Day 5, Rosa n=11, Atg5 n=12, #1 n=12, #2 n=12. *, P=0.0344; **, P=0.0209; ***, P=0.0010; ****, P=0.0229. KP #2474, n=9 biologically independent samples; *, P=0.0228; **, P=0.0191; ***, P=0.0148; ****, P<0.0001.
Interestingly, several recent studies reported that genetic disruption of oncogenic KRAS or MAPK pathway inhibition elicits protective autophagy via an ULK1-dependent signaling mechanism and indicate that dual targeting of the canonical MAPK pathway and autophagy may be necessary for anti-tumorigenic activity in KRAS-driven tumors48–50. In agreement with our findings that Cu is necessary for MEK1/28 and ULK1/2 kinase activity (Fig. 1d–g and Extended Data Fig. 1a–d), loss of Ctr1 in KRASG12D lung tumors diminished ERK1/2 activation (Extended Data Fig. 5a,b), while also blunting compensatory autophagy induction via ULK1/2 (Fig. 5a–f). These data suggest that the dual inhibition of these kinase signaling nodes contributes to the reduced in vivo tumor cell proliferation (Ki67-positive staining, Fig. 4f,i) associated with Ctr1 deficiency. However, these results failed to address the extent to which Cu-regulated MAPK signaling versus Cu-mediated autophagy contributes to lung tumor cell growth and survival during KRASG12D-driven tumorigenesis. To begin to interrogate whether the requirement for Cu to induce autophagy is necessary for oncogenic KRASG12D lung tumor cell phenotypes, we established lung adenocarcinoma cell lines isolated from LSL-KrasG12D/+;Trp53flox/flox (KP) mice in which Ctr1 was inactivated via CRISPR/Cas9 (Extended Data Fig. 5c). Targeted disruption of Ctr1 reduced LC3-II levels in two KP lung adenocarcinoma cell lines as compared to cells transduced with a non-targeting control sgRNA (Fig. 5g and Extended Data Fig. 5d). The dependency of KRAS-driven cancer cell lines on autophagy for growth and survival has been previously evaluated in vitro with clonogenic survival assays (Fig. 5h), in which cancer cells are plated in normal media, subsequently starved of amino acids for one day to induce autophagy, and then recovered in normal media for three days, and the ability of the cancer cells to survive is assessed51. In agreement with Guo et al.51, CRISPR/Cas9 knockout of Atg5 in KP lung adenocarcinoma cells significantly reduced the clonogenic survival of cells post starvation, an effect that was phenocopied by Ctr1 deletion (Fig. 5i,j). Importantly, expression of ERK2GOF was not sufficient to rescue the decrease in clonogenic survival in the absence Atg5 or Ctr1 (Extended Data Fig. 5e,f), which suggests that the survival of these lung cancer cells depends on autophagy in a MAPK-independent fashion. Nevertheless, our collective results demonstrate that the dependence of KRASG12D-driven tumors on MAPK signaling and autophagy events can be inhibited by altering Cu levels, which could be achieved with Cu chelators alone.
Binding of Cu to ULK1 is required for autophagy signaling and induction.
To mechanistically interrogate the contribution of Cu-binding to ULK1 signal transduction and autophagy initiation, we next mutated H136, M188, and H197 to alanine, which share conserved primary sequence with the Cu-binding mutant (CBM) of MEK18 (Fig. 1a). Compared to wild-type ULK1, the ULK1CBM had reduced in vitro ability to bind a Cu-charged resin and phosphorylate ATG13 (Fig. 6a,b). While the lack of ULKCBM in vitro kinase activity could suggest a Cu-binding independent finding, we previously demonstrated that introduction of mutations at these conserved residues in MEK5, which is highly similar to MEK1/2 and does not bind Cu, did not alter MEK5 kinase activity8. Along these lines, the ULK1CBM immunoprecipitated from Ulk1/2−/− MEFs deprived of amino acids failed to phosphorylate ATG13 as compared to the exogenous ULK1WT (Fig. 6c). While both ULK1WT and ULK1CBM were efficiently phosphorylated by AMPK at serine 555 and mTORC1 at serine 757 when stably expressed in Ulk1/2−/− MEFs (Extended Data Fig. 6a), the ULK1CBM exhibited a mobility shift that corresponded to reduced ULK1 autophosphorylation due to defective kinase activity (Extended Data Fig. 6b). Further, despite decreased kinase activity the ULK1CBM retained strong interactions with canonical binding partners ATG13, ATG101, and FIP200, suggesting that the three-dimensional structure of ULK1 is not destabilized upon introduction of mutations into ULK1CBM (Extended Data Fig. 6c). To evaluate the consequence of disrupting ULK1 Cu-binding on autophagic flux, signaling, and autophagosome formation, Ulk1 and Ulk2 knockout MEFs were generated via CRISPR/Cas9 and validated to have reduced autophagic flux in response to amino acid deprivation, but as previously reported still retain visible levels of LC3-II52 (Extended Data Fig. 6d–f). Only expression of ULK1WT partially rescued LC3-II levels, phosphorylation of ATG13, and autophagosome formation in the context of Ulk1/2 deficiency, indicating that Cu binding is required to restore autophagy downstream of ULK1 (Fig. 6d,e, Fig. 7a–f, and Extended Data Fig. 6g,h). Finally, consistent with Cu-dependent autophagy acting solely through ULK1/2, we found that loss of Ctr1 in MEFs deficient in Ulk1/2 does not further alter autophagic flux (Fig. 6f and Extended Data Fig. 6i), as measured by LC3-II levels. This further bolsters the conclusion that Cu levels mechanistically contribute to autophagy via ULK1/2 and not another Cu-dependent parallel pathway.
Figure 6. Binding of Cu to ULK1 is required for autophagy signaling and induction.
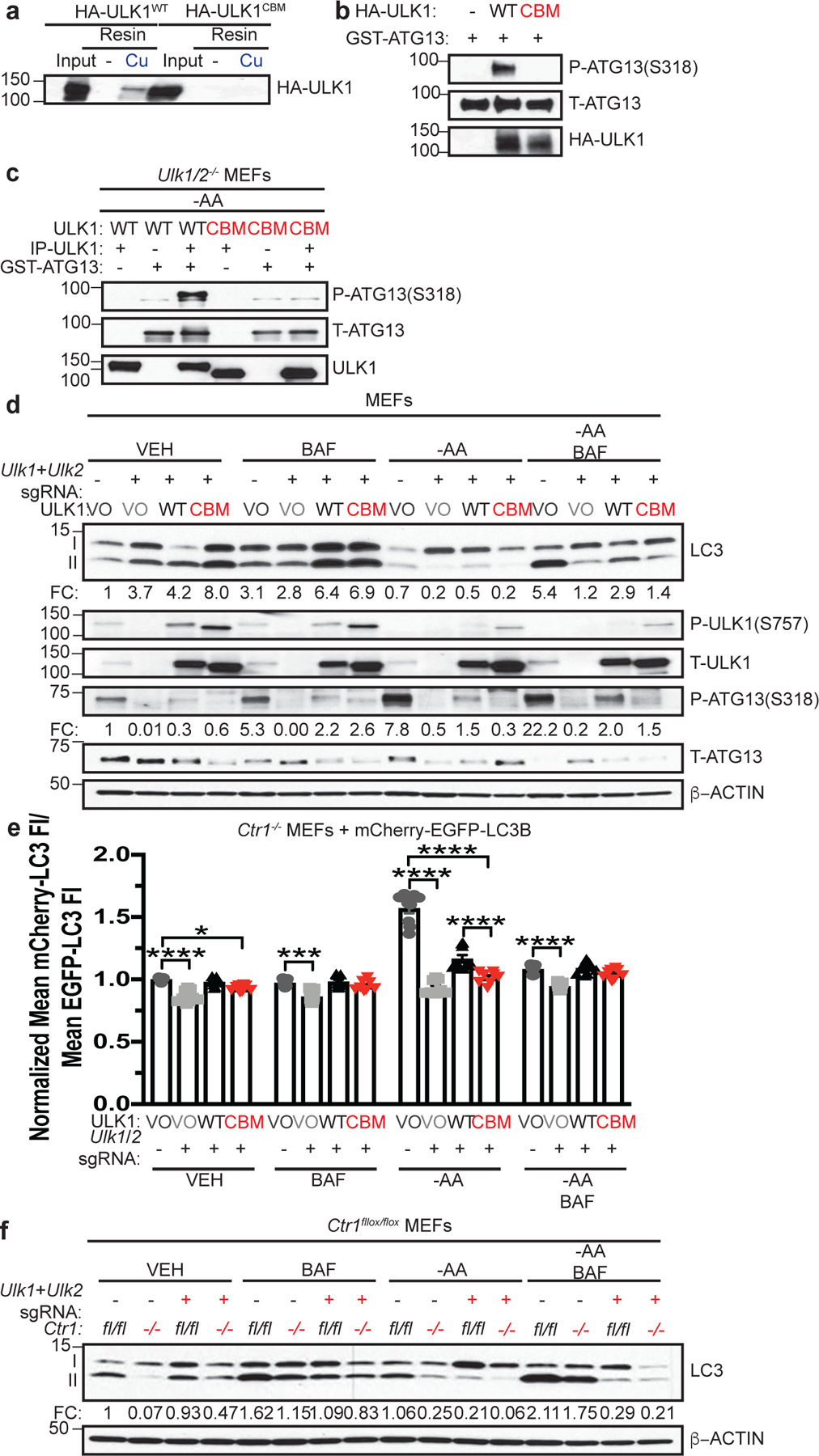
a, Immunoblot detection of recombinant HA-ULK1WT or Cu binding mutant HA-ULK1CBM bound to a resin charged with or without Cu compared to input. n=3 biologically independent experiments. b, Immunoblot detection of recombinant phosphorylated (P)-ATG13, total (T)-ATG13, and HA-ULK1WT or HA-ULK1CBM from ULK1 in vitro kinase assays. n=3 biologically independent experiments. c, Immunoblot detection of recombinant P-ATG13, T-ATG13, or immunoprecipitated (IP)-ULK1 from Ulk1/2−/− MEFs stably expressing HA-ULK1WT (WT) or HA-ULK1CBM (CBM) treated with amino acid deprivation (-AA). n=3 biologically independent experiments. d, Immunoblot detection of LC3-I, LC3-II, P-ULK1, T-ULK1, P-ATG13, T-ATG13, or β-ACTIN from MEFs stably expressing sgRNA against Rosa (−) reconstituted with empty vector (VO) or sgRNA against Ulk1 and Ulk2 reconstituted with empty vector (VO), HA-ULK1WT (WT), or HA-ULK1CBM (CBM) treated with vehicle (VEH) or -AA with or without bafilomycin (BAF). Quantification: ΔLC3-II/β-ACTIN and ΔP-ATG13/T-ATG13 normalized to Rosa (−), VO, VEH control. n=3 biologically independent experiments. e, Scatter dot plot with bar at mean mCherry-LC3 fluorescent intensity (FI)/mean EGFP-LC3 FI ± s.e.m. analyzed by flow cytometry from MEFs stably expressing sgRNA against Rosa (−) reconstituted with VO or sgRNA against Ulk1 and Ulk2 reconstituted with VO, WT, or CBM and mCherry-EGFP-LC3B treated with VEH or -AA with or without BAF normalized to Rosa (−), VO, VEH control. n= number of biologically independent samples; VO=9, VO=9, WT=9, CBM=8. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. *, P=0.0335; ***, P=0.0002; ****, P<0.0001. f, Immunoblot detection of LC3-I, LC3-II, or β-ACTIN from Ctr1flox/flox (fl/fl) or Ctr1−/− (−/−) MEFs stably expressing sgRNA against Rosa (−) or sgRNA against Ulk1 and Ulk2 (+) treated with VEH or -AA with or without BAF. Quantification: ΔLC3-II/β-ACTIN normalized to Ctr1flox/flox, Rosa (−), VEH control. n=4 biologically independent experiments.
Figure 7. Binding of Cu to ULK1 is required for autophagosome complex formation.
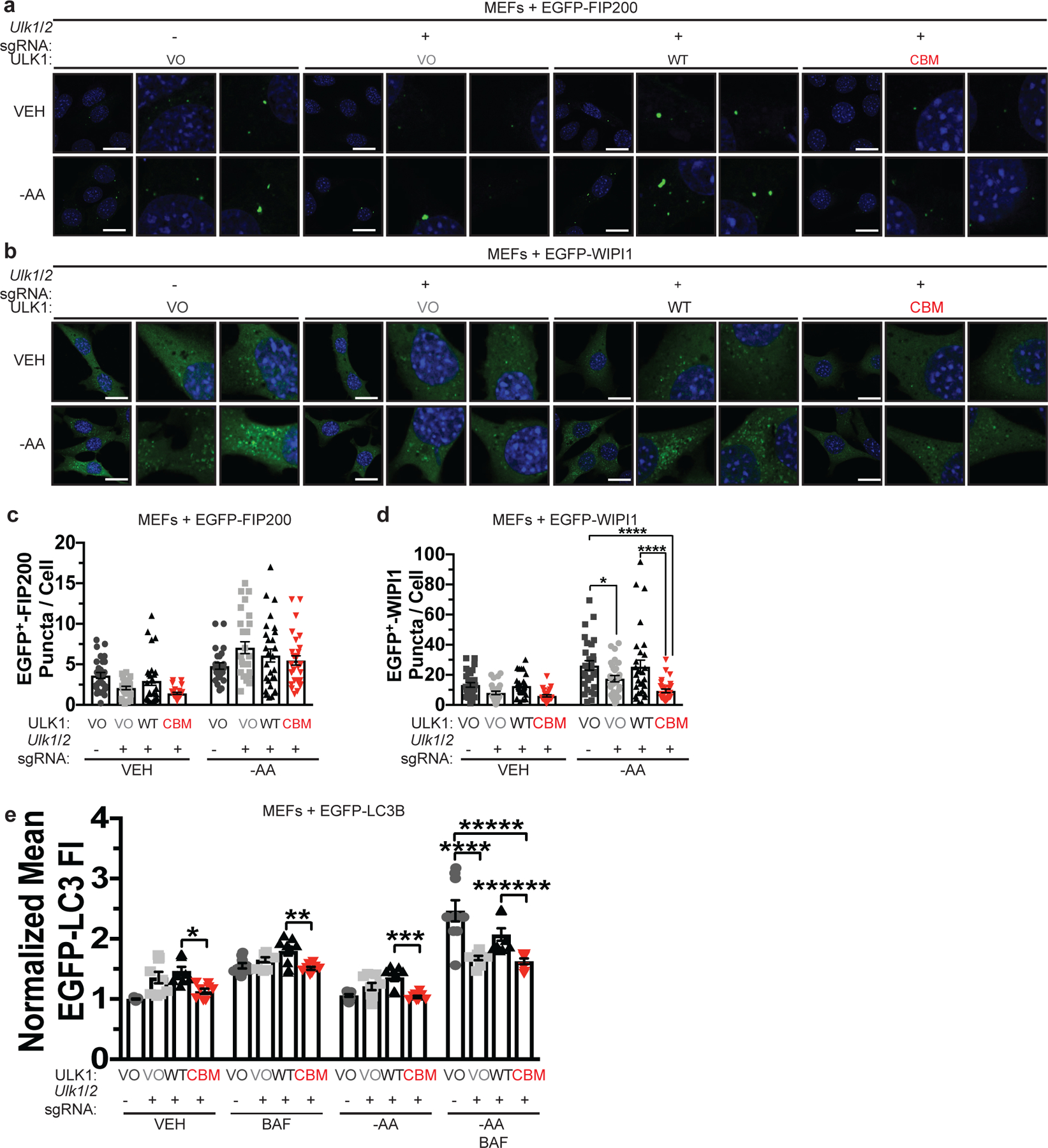
a,b, Representative fluorescence images of EGFP-FIP200 (a) or EGFP-WIP1 (b) from MEFs stably expressing sgRNA against Rosa (−) reconstituted with VO or sgRNA against Ulk1 and Ulk2 reconstituted with VO, WT, or CBM and EGFP-FIP200 or EGFP-WIP1 treated with VEH or -AA. Scale bar, 20 μm. c,d, Scatter dot plot with bar at mean mean EGFP+ puncta per cell ± s.e.m. from MEFs stably expressing sgRNA against Rosa (−) reconstituted with VO or sgRNA against Ulk1 and Ulk2 reconstituted with VO, WT, or CBM and EGFP-FIP200 (c) or EGFP-WIP1 (d) treated with VEH or -AA. n represents number offields of view. EGFP-FIP200, VEH, VO=30, VO=27, WT=28, CBM=27; -AA, VO=26, VO=28, WT=28, CBM=29; EGFP-WIP1, VEH, VO=30, VO=28, WT=30, CBM=28; -AA, VO=29, VO=30, WT=29, CBM=30. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. *, P=0.0394; ****, P<0.0001. e, Scatter dot plot with bar at mean EGFP-LC3 FI ± s.e.m. analyzed by flow cytometry from MEFs stably expressing sgRNA against Rosa (−) reconstituted with VO or sgRNA against Ulk1 and Ulk2 reconstituted with VO, WT, or CBM and EGFP-LC3B treated with VEH or -AA with or without BAF normalized to Rosa (−), VO, VEH control. N represents number of biologically independent samples. VO=9, VO=9, WT=9, CBM=8. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. *, P=0.0083; **, P=0.0321; ***, P=0.0125; ****, P<0.0001; *****, P<0.0001; ******, P=0.0002.
Binding of Cu to ULK1 is required for KRASG12D-driven tumor growth and cancer cell survival in response to starvation.
To translate our finding that Cu-binding to ULK1 is required for autophagy to KRAS-driven tumorigenesis, we next tested and found that blocking ULK1 Cu-binding was associated with reduced tumor growth kinetics and endpoint tumor weight of KRASG12D-transformed Ulk1−/− immortalized MEFs stably expressing the ULK1CBM compared to ULK1WT (Extended Data Fig. 7a–c). To investigate whether these results could be extrapolated to the setting of endogenous oncogenic KRAS tumorigenesis, Ulk1 and Ulk2 were knocked out by CRISPR/Cas9 in mouse KRAS mutant lung adenocarcinoma cell lines (Extended Data Fig. 7d). As expected, loss of endogenous Ulk1/2 decreased the levels of LC3-II and phosphorylation of ATG13 (Fig. 8a,b and Extended Data Fig. 7e,f). Targeted disruption of Ulk1/2 reduced in vivo tumor growth to a similar extent to loss of Atg5 (Fig. 8c–e). In agreement with Cu being required for ULK1 kinase activity, only ULK1WT, but not ULK1CBM could partially restore autophagic flux, signaling, and efficient subcutaneous tumor growth (Fig. 8a–e). Given that Cu mechanistically contributes to autophagy via ULK1/2, the clonogenic survival of KP adenocarcinoma cells was dependent on ULK1/2 but was not further exacerbated by deletion of Ctr1 (Fig. 8f,g and Extended Data Fig. 7g). Together, these data support a mechanism to regulate ULK1 activity via Cu binding to promote nutrient deprived or oncogene-driven autophagy that is required for tumorigenesis.
Figure 8. Binding of Cu to ULK1 is required for KRASG12D-driven tumor growth and cancer cell survival in response to starvation.
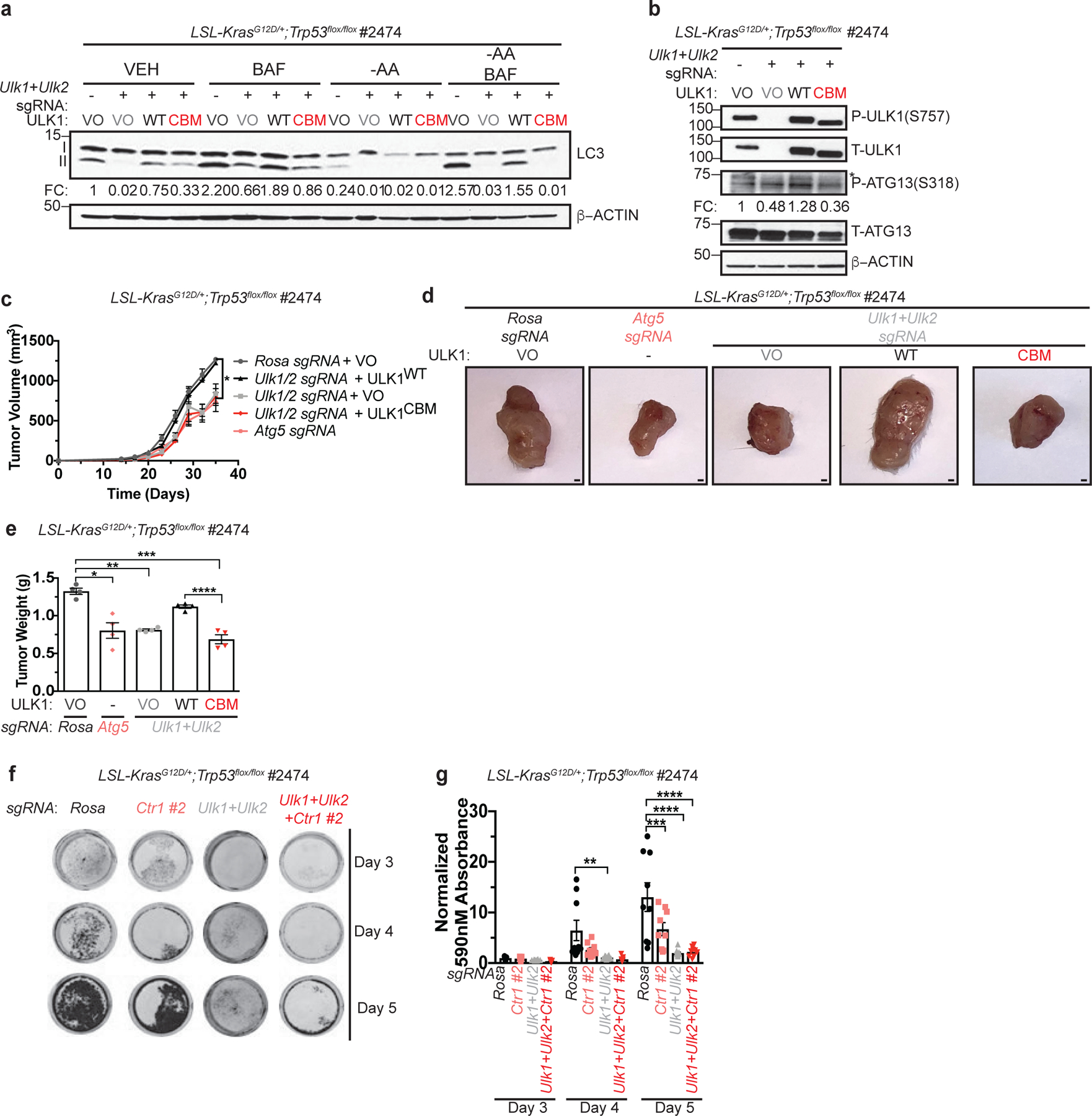
a, Immunoblot detection of proteins from treated KP cells. Quantification: ΔLC3-II/β-ACTIN normalized to Rosa (−), VO, VEH control. n=3 biologically independent experiments. b, Immunoblot detection of proteins from treated KP cells. Quantification: ΔP-ATG13/T-ATG13 normalized to Rosa (−), VO, VEH control. n=3 biologically independent experiments. c, Mean tumor volume (mm3) ± s.e.m. versus time (days) in mice injected with KP cells. n=4 biologically independent animals. Results were compared using a paired, two-tailed Student’s t-test. Rosa vs. Atg5, *, P=0.0228; Rosa vs. Ulk1/2, *, P=0.0176; Ulk1/2 vs. Ulk1/2 + ULK1WT, *, P=0.0199; Rosa vs. Ulk1/2 + ULK1CBM, *, P=0.0116; Ulk1/2 + ULK1WT vs. Ulk1/2 + ULK1CBM, *, P=0.0157. d, Representative dissected tumors from mice injected with KP cells. Scale bar, 100 μm. e, Scatter dot plot with bar at mean tumor weight (g) ± s.e.m. of KP tumors at endpoint. n=4 biologically independent animals. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. *, P=0.0001; **, P=0.0001; ***, P<0.0001; ****, P=0.0007. f, Representative crystal violet images of KP cells from days 3, 4, and 5 of recovery. g, Scatter dot plot with bar at mean absorbance of extracted crystal violet at 590nM ± s.e.m. of KP cells from days 3, 4, and 5 of recovery normalized to Rosa, day 3 control. N represents number of biologically independent samples. Rosa=9, Ctr1 #2 n=9, Ulk1/2 n=12, Ctr1 #2 + Ulk1/2 n=9. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. **, P=0.0010; ***, P=0.0002; ****, P<0.0001.
Discussion
Despite evidence that Cu homeostasis is essential for life2, relatively little is known about cellular pathways that respond directly to Cu levels. We show here that Cu directly modulates the activity of the autophagic kinases ULK1 and ULK2 to function as an endogenous rheostat to control autophagy during nutrient deprivation. These findings highlight a molecular basis for a Cu-dependent cellular process to support energy homeostasis. One enticing hypothesis is that sensing Cu abundance through dynamic signaling networks may help cells adapt to environmental changes that would require differential cellular metabolism. By extension, limiting Cu availability or decreasing the binding of Cu to ULK1 may be an attractive therapeutic opportunity to reduce the growth of oncogene-driven lung adenocarcinomas that in part rely on autophagy for tumor maintenance and cell survival. Interestingly, models of KRAS-driven lung cancer have differing levels of autophagy dependence based on which tumor suppressor is dysregulated, suggesting that the utility of reducing Cu levels to dually target autophagy and MAPK signaling could be exploited more broadly. Further, while targeting protein kinase catalytic activity is a proven approach in the landscape of cancer therapeutics, orthogonal approaches that exploit the Cu-dependency of kinases that are critical for maintaining cellular processes essential for tumor growth, such as autophagy, represents an untapped therapeutic opportunity.
Extended Data
Extended Data 1. Cu is both necessary and sufficient for ULK1 and ULK2 kinase activity.
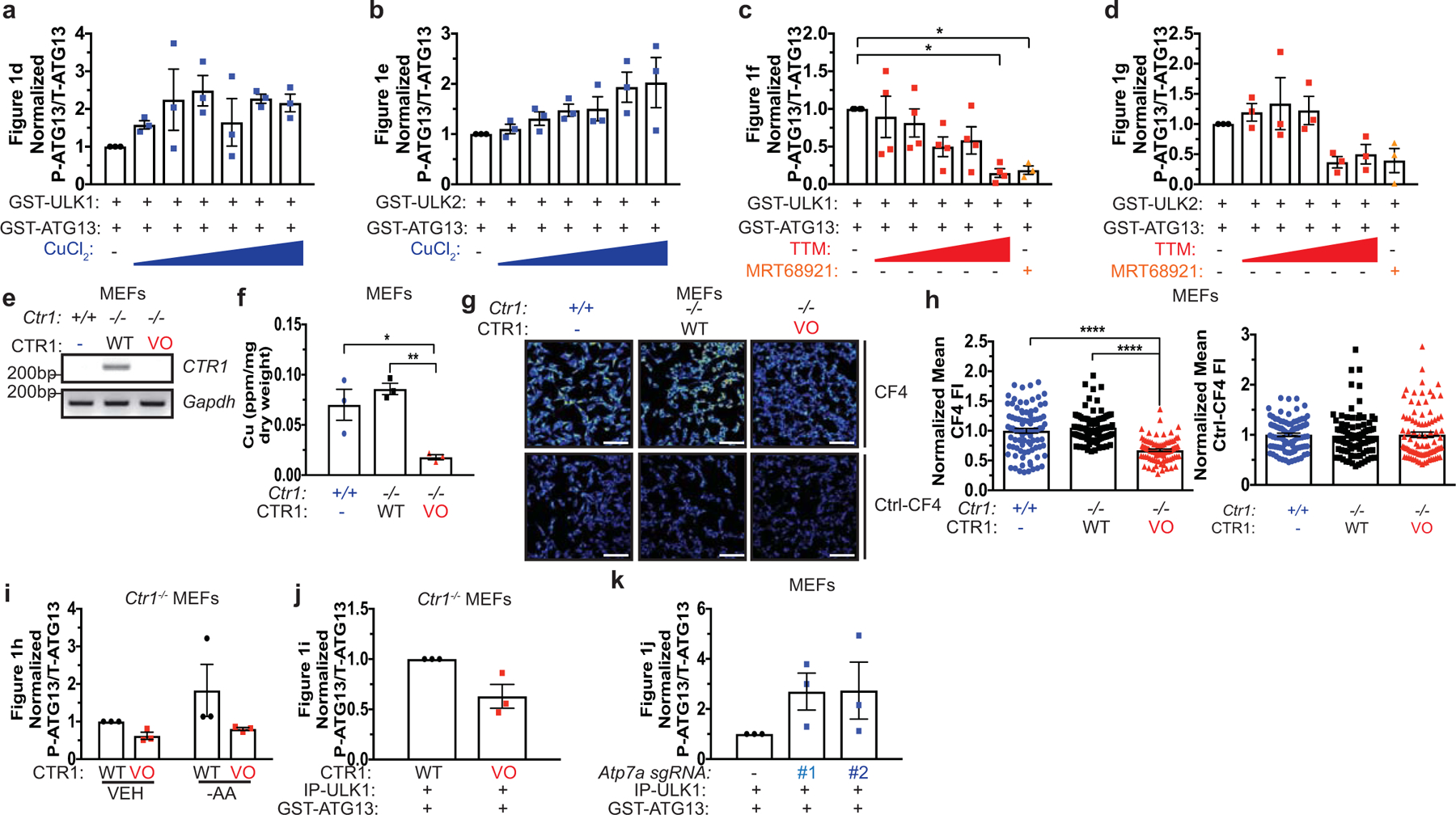
a,b,c,d, Scatter dot plot with bar at mean normalized ΔPhosphorylated (P)-ATG13/Total (T)-ATG13 from Figure 1d,e,g. n=3 biologically independent experiments, Figure 1d,e,g, or n=4 biologically independent experiments, Figure 1f. Results were compared using a one-way ANOVA followed by a Tukey’s multi-comparisons test. 0 vs. 50 μM TTM, *, P=0.0151; 0 vs. 10 μM MRT68921, *, P=0.0395. e, RT-PCR detection of indicated mRNAs from Ctr1+/+ MEFs or Ctr1−/− MEFs stably expressing CTR1WT (WT) or empty vector (VO). n=1 biologically independent sample. f, Scatter dot plot of inductively coupled plasma mass spectrometry (ICP-MS) detection with bar at mean Cu (parts per million, ppm) from Ctr1+/+ MEFs (+/+) or Ctr1−/− MEFs stably expressing WT or VO per sample weight ± s.e.m. n=3 biologically independent samples. Results were compared using a one-way ANOVA followed by a Tukey’s multi-comparisons test. *, P=0.05 0.0202; **, P=0.0059. g, Representative live cell imaging of the Cu probe CF4 or control Cu probe Ctrl-CF4 from Ctr1+/+ MEFs (+/+) or Ctr1−/− MEFs stably expressing WT or VO. Scale bar, 100 μm. h, Scatter dot plot with bar at mean CF4 or Ctrl-CF4 fluorescence intensity (FI) ± s.e.m. from Ctr1+/+ MEFs (+/+) or Ctr1−/− MEFs stably expressing WT or VO. n=90 individual cells. Results were compared using a one-way ANOVA followed by a Tukey’s multi-comparisons test. ****, P<0.0001. i,j,k, Scatter dot plot with bar at mean normalized ΔP-ATG13/T-ATG13 from Figure 1h,i,j. n=3 biologically independent experiments. Results were compared using a one-way ANOVA followed by a Tukey’s multi-comparisons test, an unpaired, two-tailed Student’s t-test, or two-way ANOVA followed by a Sidak’s multi-comparisons test. ns.
Extended Data 2. Copper is both necessary and sufficient for autophagy induction and signaling in a MAPK signaling independent fashion, upstream of ATG5.
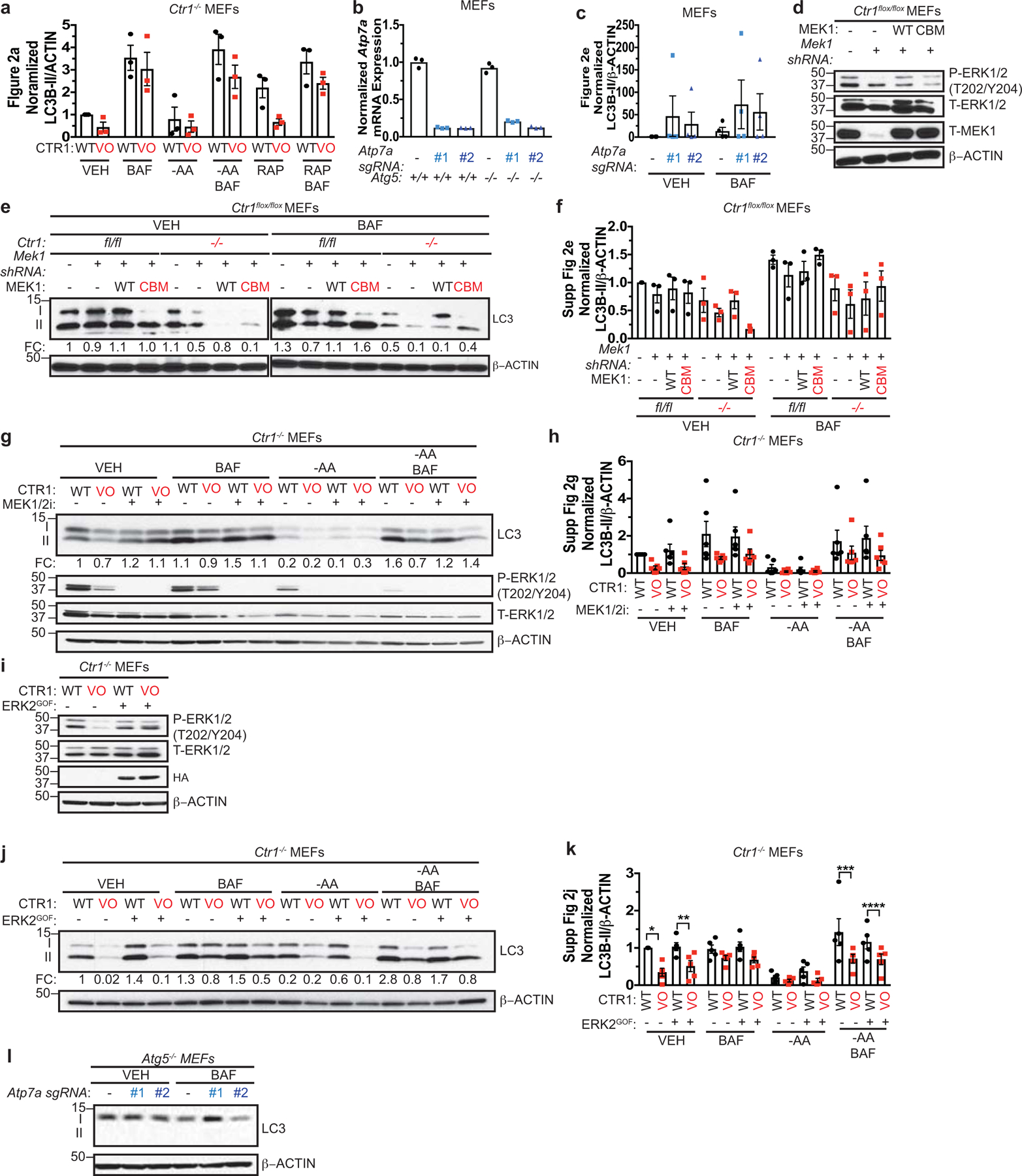
a, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN from Figure 2a. n=3 biologically independent experiments. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. ns. b, Scatter dot plot with bar at mean normalized expression of Atp7a mRNA from MEFs n=1 biologically independent experiment performed in technical triplicate. c, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN from Figure 2e. n=4 biologically independent experiments. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. ns. d, Immunoblot detection of proteins from MEFs. e, Immunoblot detection of proteins from treated MEFs. Quantification: ΔLC3-II/β-ACTIN normalized to fl/fl, empty vector (−), VEH control. f, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN from Extended Data 2e. n=3 biologically independent experiments. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. ns. g, Immunoblot detection of proteins from treated MEFs. Quantification: ΔLC3-II/β-ACTIN normalized to WT, VEH control. h, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN from Extended Data 2g. n=6 biologically independent experiments. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. ns. i, Immunoblot detection of proteins from MEFs. j, Immunoblot detection of proteins from treated MEFs. Quantification: ΔLC3-II/β-ACTIN normalized to WT, VEH control. k, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN from Extended Data 2j. n=5 biologically independent experiments. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. *, P=0.0071; **, P=0.0434; ***, P=0.0026; ****, P=0.0021. l, Immunoblot detection of proteins from treated MEFs. Western blot images are representative of at least three biological replicates.
Extended Data 3. Cu but not Fe is required for autophagosome formation and is associated with fluctuations in the Cu labile pool.
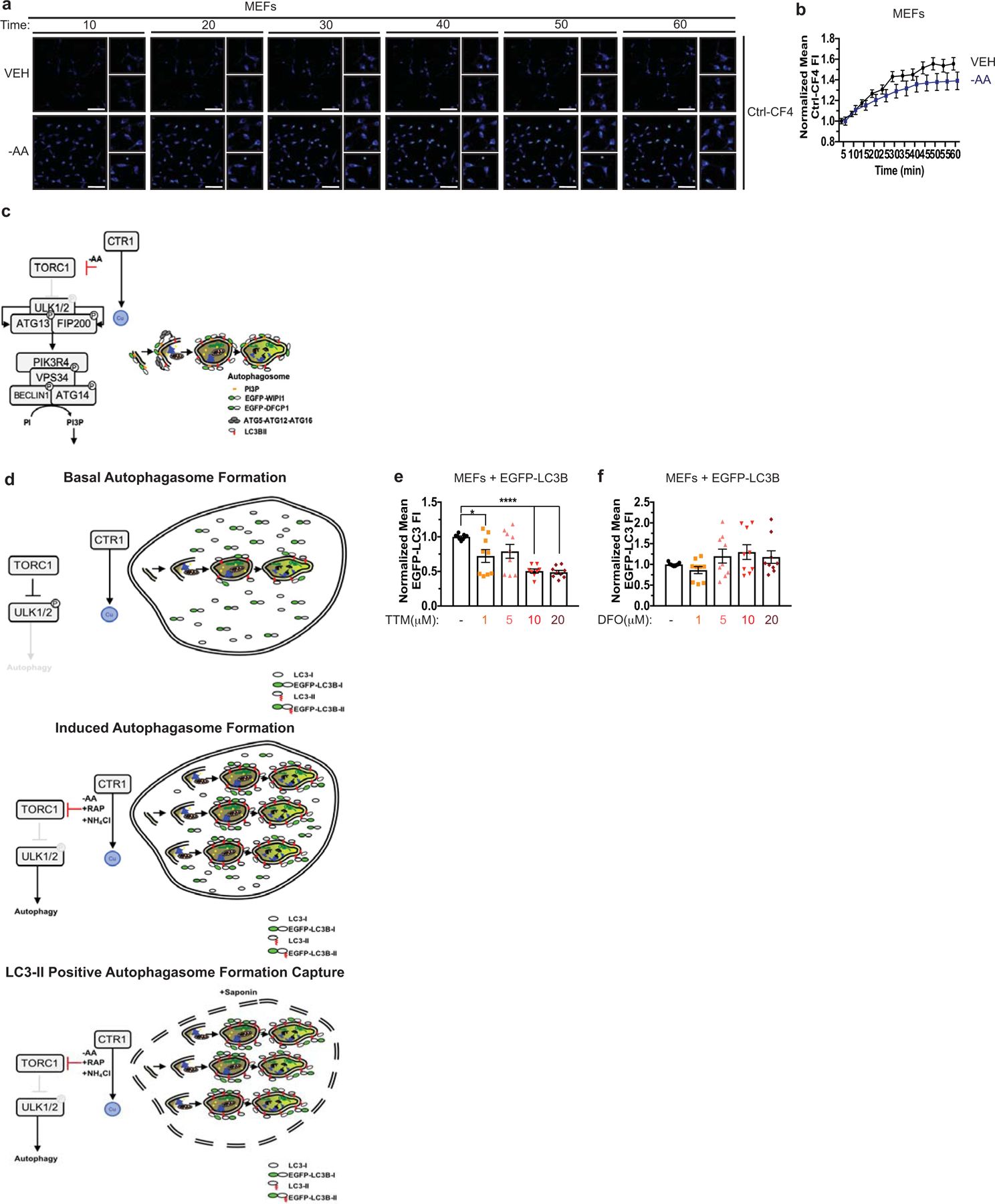
a, Representative live cell imaging of the Cu probe Ctrl-CF4 every ten minutes for 60 minutes from MEFs treated with vehicle (VEH) or amino acid deprivation (-AA). Scale bar, 100 μm. b, Mean Ctrl-CF4 fluorescence intensity (FI) ± s.e.m. versus time (minutes, min) from MEFs treated with VEH or -AA normalized to t=0, five minutes. n=30 individual cells. Results were compared using a two-way ANOVA followed by a Sidak’s multi-comparisons test. c, Schematic of immunofluorescence-based approach to access autophagosome formation. d, Schematic of flow cytometry-based approach to access autophagosome number. e,f, Scatter dot plot with bar at mean GFP-LC3 fluorescent intensity ± s.e.m. analyzed by flow cytometry from MEFs stably expressing EGFP-LC3B treated VEH or increasing concentrations of Cu chelator TTM (e) or Fe chelator DFO (f). n=9 biologically independent samples. Results were compared using a one-way ANOVA followed by a Dunnett’s multi-comparisons test. *, P=0.0148; ****, P<0.0001.
Extended Data 4. Genetic ablation of Ctr1 reduces KrasG12D-driven lung tumorigenesis.
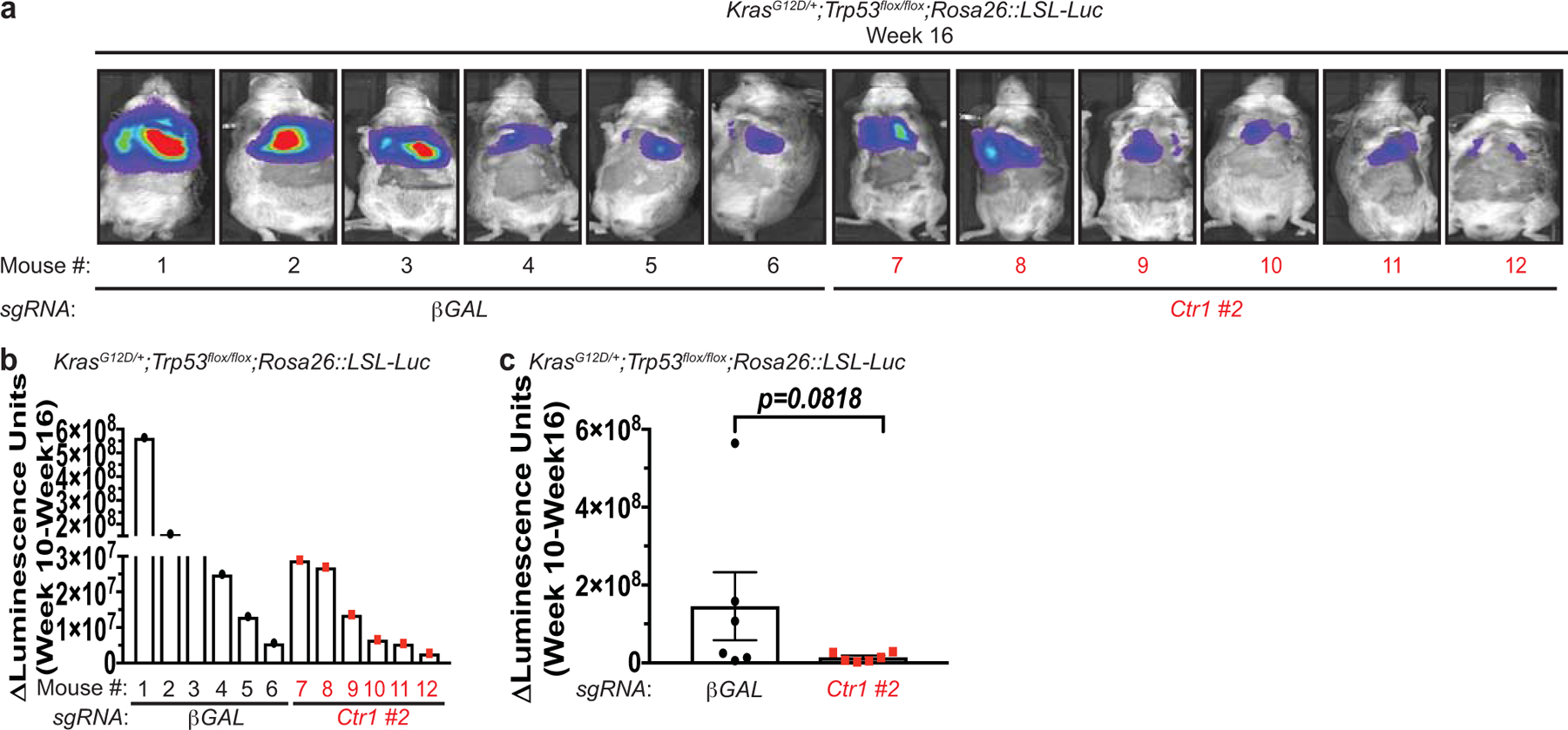
a, Normalized representative images of in vivo luminescence of KrasG12D/+;Trp53flox/flox;Rosa26::LSL-Luc (KPLuc) mice introduced with either sgRNA against β-GAL or Ctr1 at week 16 endpoint. b,c, Scatter dot plot with bar at mean ΔLuminescence units from week 10 to week 16 from KPLuc mice introduced with either sgRNA against β-GAL or Ctr1. n=6 biologically independent animals. Results were compared using an unpaired, one-tailed Student’s t-test. P=0.0818.
Extended Data 5. Genetic ablation of Ctr1 decreases MAPK signaling to reduce KrasG12D-driven lung tumorigenesis, while survival in response starvation is independent of MAPK signaling.
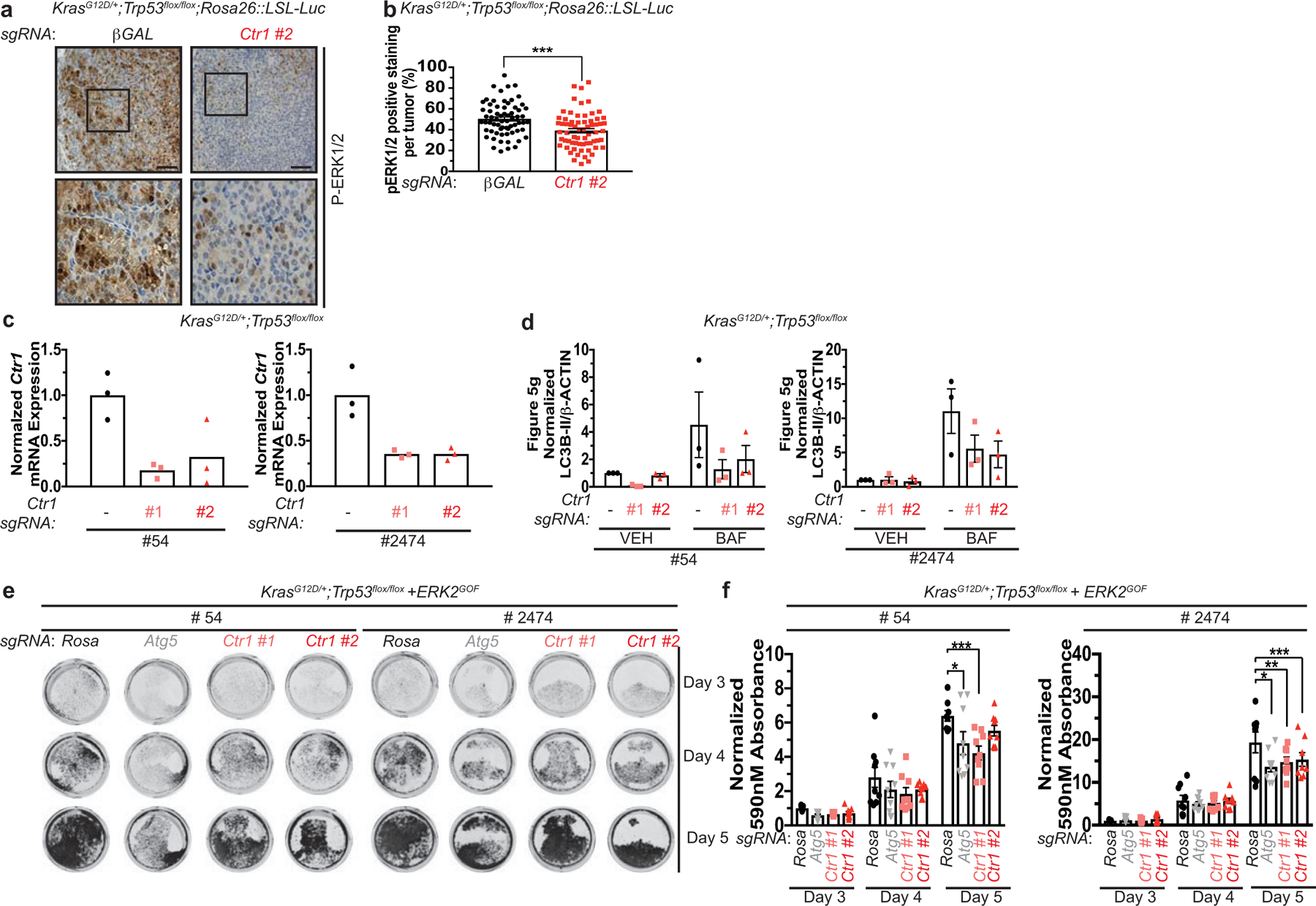
a, Representative 40x images of immunohistochemical detection of phosphorylated (P)-ERK1/2 of lungs from KPLuc mice expressing sgRNA against β-GAL or Ctr1. (40x scale bar: 50 μm). b, Scatter dot plot with bar at mean ± s.e.m. % P-ERK1/2 positive staining per tumor (β-GAL and Ctr1) from KPLuc mice expressing sgRNA against β-GAL or Ctr1. n=66 images. Results were compared using an unpaired, two-tailed Student’s t-test. ***, P=0.0002. c, Scatter dot plot with bar at mean normalized quantitative PCR (qPCR) expression of Ctr1 mRNA from KP lung adenocarcinoma cell lines #54 (KP #54) and #2474 (KP #2474) stably expressing sgRNA against Rosa (−) or Ctr1 (#1 or #2). n=1 performed in technical triplicate. d, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN from Figure 5g. n=3 independent experiments. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. ns. e, Representative crystal violet images of KP #54 and KP #2474 cells stably expressing sgRNA against Rosa, Atg5, or Ctr1 (#1 or #2) and ERK2GOF from days 3, 4, and 5 of recovery. f, Scatter dot plot with bar at mean absorbance of extracted crystal violet at 590nM ± s.e.m. of KP #54 and KP #2474 cells stably expressing sgRNA against Rosa, Atg5, or Ctr1 (#1 or #2) and gain-of-function (GOF) HA-ERK2 (ERK2GOF) from days 3, 4, and 5 of recovery normalized to Rosa, day 3 control. KP #54, n=# biologically independent samples; Day 3=9, Day 4=9, Day 5, Rosa=8, Atg5=9, #1=9, #2=9. *, P=0.0140; ***, P=0.0003. KP #2474, n=# biologically independent samples; Day 3=9, Day 4=9, Day 5, Rosa=9, Atg5=9, #1=8, #2=8. *, P=0.0011; **, P=0.0158; ***, P=0.0462.
Extended Data 6. Binding of Cu to ULK1 is required for kinase activity but not substrate association or phosphorylation.
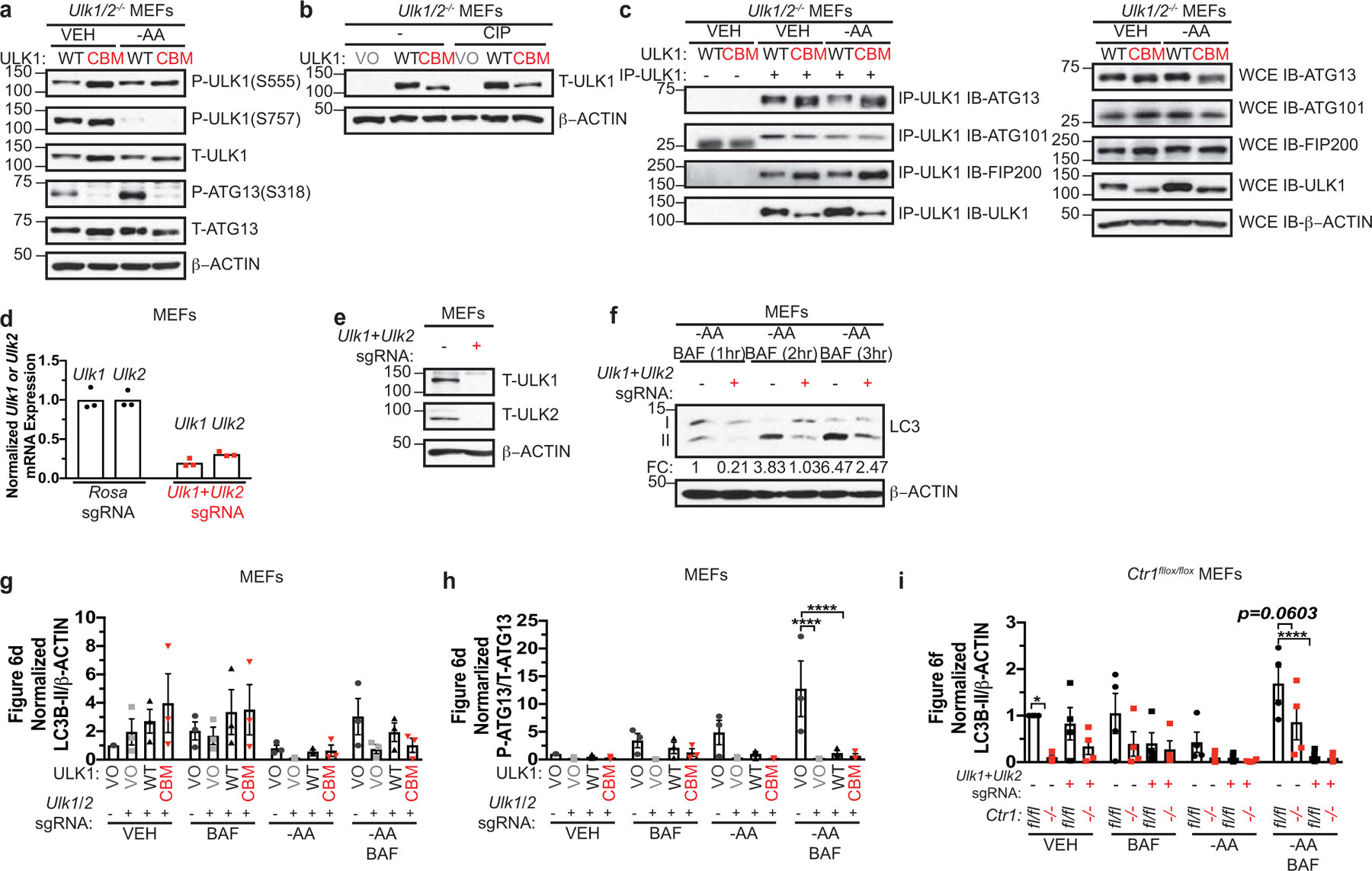
a, Immunoblot detection of phosphorylated (P) ATG13, total (T)- ATG13, P-ULK1 (S555), P-ULK1 (S757), T-ULK1, or β-ACTIN from Ulk1/2−/− MEFs stably expressing HA-ULK1WT (WT) or HA-ULK1CBM (CBM) treated with vehicle (VEH) or amino acid deprivation (-AA). b, Immunoblot detection of T-ULK1 or β-ACTIN from cell lysates treated with or without calf alkaline phosphatase (CIP) from Ulk1/2−/− MEFs stably expressing empty vector (VO), HA-ULK1WT (WT), or HA-ULK1CBM (CBM). c, Immunoblot detection of T-ATG13, T-ATG101, T-FIP200, T-ULK1, or β-ACTIN from immunoprecipitated (IP)-ULK1 or whole cell extracts (WCE) from Ulk1/2−/− MEFs stably expressing HA-ULK1WT (WT) or HA-ULK1CBM (CBM) treated with VEH or -AA. d, Scatter dot plot with bar at mean normalized quantitative PCR (qPCR) expression of Ulk1 or Ulk2 mRNA from MEFs stably expressing sgRNA against Rosa or Ulk1 and Ulk2. n=1 performed in technical triplicate. e, Immunoblot detection of T-ULK1, T-ULK2, or β-ACTIN from MEFs stably expressing sgRNA against Rosa (−) or Ulk1 and Ulk2 (+). f, Immunoblot detection of LC3-I, LC3-II, or β-ACTIN from MEFs stably expressing sgRNA against Rosa (−) or Ulk1 and Ulk2 (+) treated with -AA and bafilomycin (BAF) for 1 hour (hr), 2 hr, and 3hr. g,h,i, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN or ΔP-ATG13/T-ATG13 from Figure 6d,f. n=3 independent experiments. Results were compared using a two-way ANOVA followed by a Tukey’s multi-comparisons test. g, ns; h, ****, P<0.0001. i, *, P=0.0365; ****, P<0.0001. Western blot images are representative of at least three biological replicates.
Extended Data 7. Binding of Cu to ULK1 is required for tumorigenesis by oncogenic KRASG12D.
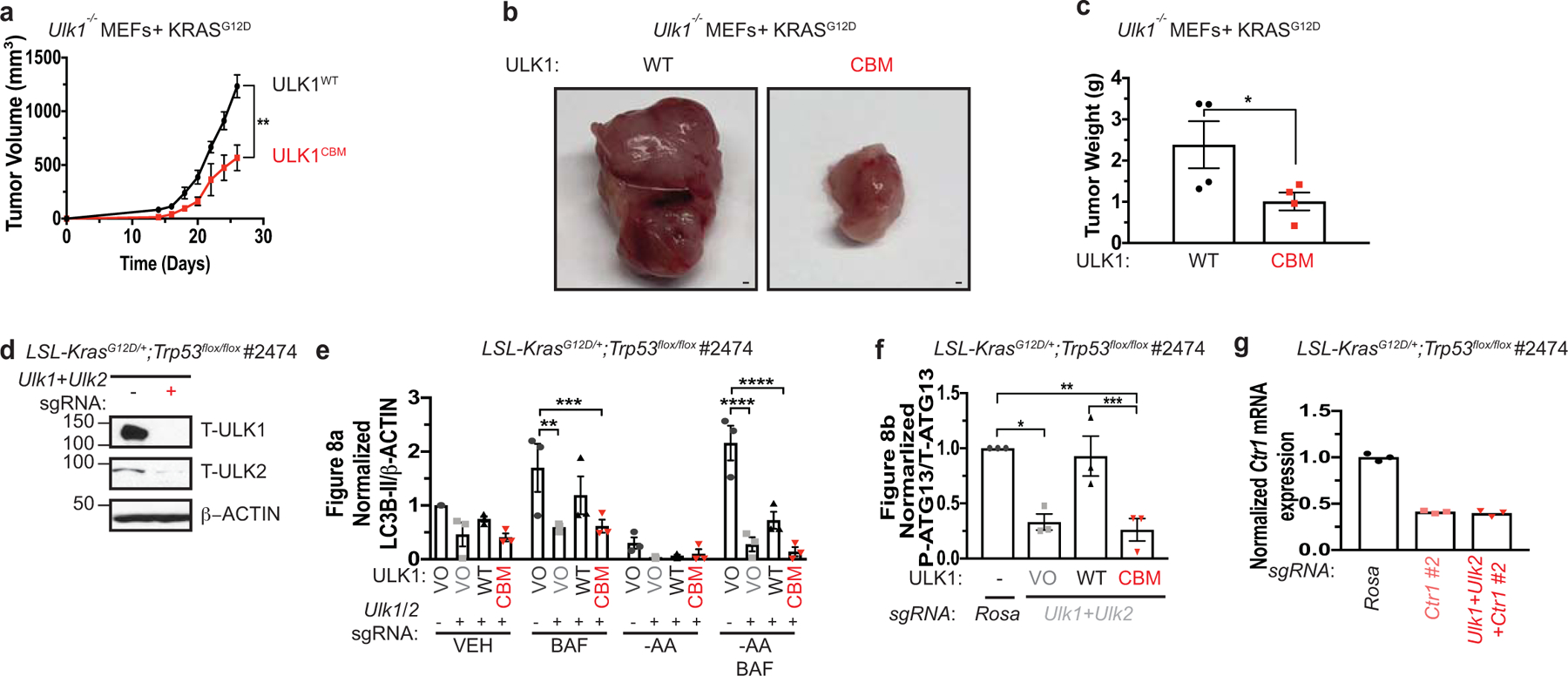
a, Mean tumor volume (mm3) ± s.e.m. versus time (days) in mice injected with Ulk1−/− MEFs stably expressing either HA-ULK1WT or HA-ULK1CBM and transformed with KRASG12D. n=4 biologically independent animals. Results were compared using a paired, one-tailed Student’s t-test. **, P=0.0095. b, Representative dissected tumors from mice injected with Ulk1−/− MEFs stably expressing either HA-ULK1WT (WT) or HA-ULK1CBM (CBM) and transformed with KRASG12D. Scale bar, 100 μm. c, Scatter dot plot with bar at mean tumor weight (g) ± s.e.m. of tumors at endpoint from Ulk1−/− MEFs stably expressing either WT or CBM and transformed with KRASG12D. n=4 biologically independent samples. Results were compared using an unpaired, one-tailed Student’s t-test. *, P=0.0325. n=4. d, Immunoblot detection of T-ULK1, T-ULK2, or β-ACTIN from KrasG12D/+;Trp53flox/flox (KP) lung adenocarcinoma cell line #2474 (KP #2474) stably expressing sgRNA against Rosa (−) or Ulk1 and Ulk2 (+). e,f, Scatter dot plot with bar at mean normalized ΔLC3-II/β-ACTIN or ΔP-ATG13/T-ATG13 from Figure 8a,b. n=3 independent experiments. Results were compared using a one-way ANOVA or a two-way ANOVA followed by a Tukey’s multi-comparisons test. e, **, P=0.0013; ***, P=0.0016; ****, P<0.0001; f, *, P=0.0113; **, P=0.0063; ***, P=0.0113. g, Scatter dot plot with bar at mean normalized quantitative PCR (qPCR) expression of Ctr1 mRNA from KP #2474 cells stably expressing sgRNA against Rosa, Ctr1 #2, or Ulk1, Ulk2, and Ctr1 #2. n=1 performed in technical triplicate.
Supplementary Material
Supplementary Files Legends
Supplementary Video 3. Representative live cell imaging of the Cu probe Ctrl-CF4 every ten minutes for 60 minutes from MEFs treated with vehicle (VEH) from 3 independent fields of view. Reproduced in 2 independent experiments.
Supplementary Video 1. Representative live cell imaging of the Cu probe CF4 every ten minutes for 60 minutes from MEFs treated with vehicle (VEH) from 3 independent fields of view. Reproduced in 2 independent experiments.
Supplementary Video 2. Representative live cell imaging of the Cu probe CF4 every ten minutes for 60 minutes from MEFs treated with amino acid deprivation (-AA) from 3 independent fields of view. Reproduced in 2 independent experiments.
Supplementary Video 4. Representative live cell imaging of the Cu probe Ctrl-CF4 every ten minutes for 60 minutes from MEFs treated with amino acid deprivation (-AA) from 3 independent fields of view. Reproduced in 2 independent experiments.
Acknowledgements
We thank D.J. Thiele (Duke University), C.J. Chang (University of California Berkeley), R.K. Amaravadi (University of Pennsylvania), and S.A. Tooze (The Francis Crick Institute) for reagents and R.K. Amaravadi, I.A. Asangani, L. Busino, C.V. Dang, J.M. Davis, T.P. Gade, K.E. Hamilton, B. Keith, M.E. Murphy, R. Natesan, A.M. O’Reilly, S.W. Ryeom, M.C. Simon, B.Z. Stanger, J. Tobias, N.A. Tripp, A.T. Weerartna, K.E. Wellen, and E.S. Witze for technical support, discussions, and/or review of the manuscript. This work was supported by American Cancer Society Postdoctoral Fellowship 131203 PF 17 147 01 CCG (J.M.P), NIH grants GM124749 (D.C.B.), CA193603 (D.M.F), CA222503 (D.M.F), ES019851 (M.C), CA243294 (T.T), Pew Scholars Program in Biomedical Science Award #50359 (D.C.B.), and V Foundation Scholar Award 3C59 8ABS 3424 3BDA (D.C.B).
Footnotes
Competing interests. D.C.B holds ownership in Merlon Inc. D.C.B. is an inventor on the patent application 20150017261 entitled “Methods of treating and preventing cancer by disrupting the binding of copper in the MAP kinase pathway”. No potential conflicts of interest were disclosed by the other authors.
Publisher’s note. Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kolch W, Halasz M, Granovskaya M & Kholodenko BN The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 15, 515–527 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Festa RA & Thiele DJ Copper: an essential metal in biology. Curr. Biol 21, R877–83 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chelly J et al. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet 3, 14–19 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Mercer JFB et al. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet 3, 20–25 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Huster D et al. Consequences of copper accumulation in the livers of the Atp7b−/−(Wilson disease gene) knockout mice. Am. J. Pathol 168, 423–434 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfeiffenberger J et al. Hepatobiliary malignancies in Wilson disease. Liver Int. 35, 1615–1622 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Turski ML et al. A novel role for copper in Ras/MAPK signaling. Mol. Cell. Biol 32, 1284–1295 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brady DC et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 496–496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krishnamoorthy L et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol 12, 586–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brady DC, Crowe MS, Greenberg DN & Counter CM Copper chelation inhibits BRAFV600E-driven melanomagenesis and counters resistance to BRAFV600E and MEK1/2 inhibitors. Cancer Res. 77, 6240–6252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu MM, Casio M, Range DE, Sosa JA & Counter CM Copper chelation as targeted therapy in a mouse model of oncogenic BRAF-driven papillary thyroid cancer. Clin. Cancer Res 24, 4271–4281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodman VL, Brewer GJ & Merajver SD Control of copper status for cancer therapy. Curr. Cancer Drug Targets 5, 543–549 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Petherick KJ et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem 290, 11376–11383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan EYW, Kir S & Tooze SA siRNA Screening of the Kinome Identifies ULK1 as a Multidomain Modulator of Autophagy. J Biol Chem 282, 25464–25474 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Ganley IG et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284, 12297–12305 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosokawa N et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung CH et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20, 1992–2003 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D-H et al. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell 110, 163–175 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Thoreen CC et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hara K et al. Amino Acid Sufficiency and mTOR Regulate p70 S6 Kinase and eIF-4E BP1 through a Common Effector Mechanism. J Biol Chem 273, 14484–14494 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Kim E, Goraksha-Hicks P, Li L, Neufeld TP & Guan K-L Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10, 935–945 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sancak Y et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoki K, Zhu T & Guan K-L TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 115, 577–590 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Gwinn DM et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30, 214–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P & Snyder SH RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78, 35–43 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Brown EJ et al. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 369, 756–758 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Brady GF et al. Regulation of the copper chaperone CCS by XIAP-mediated ubiquitination. Mol Cell Biol 30, 1923–1936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao T et al. Copper regulates rest-activity cycles through the locus coeruleus-norepinephrine system. Nat. Chem. Biol 14, 655–663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiang GG & Abraham RT Phosphorylation of Mammalian Target of Rapamycin (mTOR) at Ser-2448 Is Mediated by p70S6 Kinase. J Biol Chem 280, 25485–25490 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Brown EJ et al. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature 377, 441–446 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Soliman GA et al. mTOR Ser-2481 Autophosphorylation Monitors mTORC-specific Catalytic Activity and Clarifies Rapamycin Mechanism of Action. J Biol Chem 285, 7866–7879 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Kundu M, Viollet B & Guan K-L AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichimura Y et al. A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Gump JM et al. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol 16, 47–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pankiv S et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem 282, 24131–24145 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Itakura E & Mizushima N Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mercer TJ, Gubas A & Tooze SA A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem 293, 5386–5395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karanasios E et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci 126, 5224–5238 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Egan DF et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 59, 285–297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russell RC et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15, 741–750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park YS et al. AKT-induced PKM2 phosphorylation signals for IGF-1-stimulated cancer cell growth. Oncotarget 7, 48155–48167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dooley HC et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 55, 238–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eng KE, Panas MD, Hedestam GBK & McInerney GM A novel quantitative flow cytometry-based assay for autophagy. Autophagy 6, 634–641 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Amaravadi RK, Kimmelman AC & Debnath J Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 9, 1167–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strohecker AM et al. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BrafV600E–Driven Lung Tumors. Cancer Discov. 3, 1272–1285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo JY et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27, 1447–1461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bryant KL et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25, 628–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinsey CG et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25, 620–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee C-S et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. U. S. A 116, 4508–4517 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo JY et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McAlpine F, Williamson LE, Tooze SA & Chan EYW Regulation of nutrient-sensitive autophagy by uncoordinated 51-like kinases 1 and 2. Autophagy 9, 361–373 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Files Legends
Supplementary Video 3. Representative live cell imaging of the Cu probe Ctrl-CF4 every ten minutes for 60 minutes from MEFs treated with vehicle (VEH) from 3 independent fields of view. Reproduced in 2 independent experiments.
Supplementary Video 1. Representative live cell imaging of the Cu probe CF4 every ten minutes for 60 minutes from MEFs treated with vehicle (VEH) from 3 independent fields of view. Reproduced in 2 independent experiments.
Supplementary Video 2. Representative live cell imaging of the Cu probe CF4 every ten minutes for 60 minutes from MEFs treated with amino acid deprivation (-AA) from 3 independent fields of view. Reproduced in 2 independent experiments.
Supplementary Video 4. Representative live cell imaging of the Cu probe Ctrl-CF4 every ten minutes for 60 minutes from MEFs treated with amino acid deprivation (-AA) from 3 independent fields of view. Reproduced in 2 independent experiments.