

Abstract
The APC tumour suppressor is the most commonly mutated gene in colorectal cancer. Loss of Apc in intestinal stem cells (ISCs) drives adenoma formation in mice via increased Wnt signalling 1, but reduced Wnt-ligand secretion surprisingly increases the ability of Apc-mutant ISCs to colonise a crypt (fixation) 2. Here, we investigate how Apc-mutant cells gain a clonal advantage over wild-type counterparts to achieve fixation. We find that Apc-mutant cells are enriched for transcripts encoding several secreted Wnt antagonists, with Notum being the most highly expressed. Conditioned medium from Apc-mutant cells suppresses the growth of wild-type organoids in a Notum-dependent manner. Furthermore, Notum-secreting Apc-mutant clones actively inhibit the proliferation of surrounding wild-type crypt cells and drive their differentiation, thereby outcompeting them from the niche. Importantly, genetic or pharmacological inhibition of Notum abrogates the ability of Apc-mutant cells to expand and form intestinal adenomas. Taken together, we identify Notum as a key mediator during the early stages of mutation fixation, which can be targeted to restore wild-type cell competitiveness, thus, offering novel preventative strategies for high-risk patients.
Loss-of-function mutations in the APC tumour suppressor are considered a key early event in colorectal cancer (CRC) initiation 5. For a mutation to be maintained within a crypt, it needs to become “fixed”, with mutant cells outcompeting wild-type intestinal stem cells (ISCs) from the crypt 6,7. Previous studies revealed Apc loss confers a clonal advantage to ISCs 7,8, increasing their probability of fixation/winning within the crypt and driving adenoma formation. Even though APC-deficient clones have an increased probability of “winning”, they can still be stochastically eliminated from the ISC pool i.e. lose. This suggests uncovering the molecular mechanisms whereby APC-deficient cells outcompete wild-type cells could lead to novel chemo-preventative approaches.
APC negatively regulates Wnt signalling by directing the phosphorylation and degradation of β-catenin 9. Since Apc-mutant tumours exhibit constitutive Wnt-pathway activation, we first sought to identify genes differentially upregulated in Apc-mutant cells relative to normal intestinal epithelium. Transcriptomic analysis of tumours that develop in VillinCreER;Apcfl/+ (hereafter VilCreER;Apcfl/+) mice following the sporadic loss of the remaining wild-type copy of Apc 10, akin to human CRC 11, show an upregulation of Wnt-target genes compared to normal intestine (Extended Data Fig. 1a). The most highly upregulated gene was Notum (Fig. 1a), which encodes a secreted Wnt deacylase that disrupts Wnt-ligand binding to Frizzled receptors 12 and as we previously showed, impairs self-renewal and tissue regeneration of wild-type ISCs during ageing 13. Therefore, to characterise the expression of Notum specifically in wild-type and Apc-mutant ISCs, we utilised the Lgr5-EGFP-IRES-CreERT2 mouse model, which drives the expression of GFP from the Lgr5 locus, allowing visualisation and lineage tracing of ISCs 14. In support of the tumour transcriptomic analysis (Fig. 1a), FACS isolated Apc-mutant Lgr5-GFP+ (hereafter Lgr5CreER;Apcfl/fl) cells show significant enrichment for Notum expression compared to wild-type Lgr5-GFP+ (Lgr5CreER;Apc+/+) cells already seven days post tamoxifen-induction (Fig. 1b), which we confirm in other Wnt-driven tumour models compared to normal intestinal epithelium (Fig. 1c, Extended Data Fig. 1b). Importantly, Notum expression mirrored nuclear β-catenin staining in serial tumour sections (Fig. 1d) and in both emerging and established Apc-mutant clones (Extended Data Fig. 1c, d), which positions Notum as a faithful surrogate marker of Apc-mutant clones 2. We previously showed that Apc-mutant cells are refractory to Wnt-ligand inhibitors in vivo 2, but that age-induced expression of Notum can inhibit Wnt signalling in wild-type ISCs 13. Moreover, Apc-mutant cells act as super-competitors in the developing fly, eliminating neighbouring cells in a Notum-dependent manner 15. Jointly, these data suggest emerging Apc-mutant clones may deploy paracrine effectors, such as Notum, to actively inhibit essential Wnt signalling in neighbouring wild-type ISCs.
Figure 1. Apc-mutant cells impair the growth of wild-type ISCs via Notum.

a, Volcano plot showing log2 fold change (x-axis) and –log2-transformed p-value (y-axis) of genes differentially expressed between Apc-mutant (VilCreER;Apcfl/+) tumour tissue and wild-type small intestine. Significantly altered genes shown in red, negative Wnt regulators highlighted in green using Wald test (two-tailed). (n=3 WT mice, n=5 VilCreER;Apcfl/+mice). b, Notum expressed in Lgr5-GFP+ cells, isolated from wild-type (Lgr5CreER;Apc+/+; +/+) and Apc-mutant intestines (Lgr5CreER;Apcfl/fl; fl/fl), 5–7 days following tamoxifen-induction. n=3 +/+ mice, n=5 fl/fl mice. c, Notum-ISH in wild-type (3–5 month-old Lgr5CreER;Apc+/+) small intestinal epithelium. Scale bar, 100 μm. d, Serial sections of intestinal tumour epithelium from Lgr5CreER;Apcfl/fl (3 month-old) mice stained for Notum (ISH) and β-catenin (IHC). Scale bar, 200 μm. e, Top, schematic illustrating experimental pipeline for wild-type (WT) organoid treatments. Bottom, representative images of WT small intestinal organoids, grown for 5 days in conditioned medium (CM) collected from wild-type (WT) or Apc-mutant (Apc-/-) organoids, derived from 3 mice of each genotype. WT organoids, treated with CM ± NOTUMi, were derived from n=6 mice. Treatments were repeated twice. Red arrows indicate organoid atrophy/death. Scale bar, 100 μm. f, Quantification of the number of organoids, formed over multiple passages (P1, P2, and P3), during culture in WT or Apc-/- CM supplemented with NOTUMi. n=6 mice per condition. g, qPCR for Wnt-target genes (Notum, Axin2), ISC markers (Lgr5, Ascl2), and cell-lineage markers (Lyz1, Krt20) expressed in organoids described in e. h, Quantification of WT organoids, formed over multiple passages (P1, P2, and P3), during culture in Apc-/-;Notum-/- CM supplemented with recombinant NOTUM. n=3 mice per condition. Mann–Whitney one-tailed U-test. Data are mean ± s.e.m. In b, f, g, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
To functionally test whether Apc-mutant cells can suppress their wild-type neighbours via secreted factors, we grew wild-type small intestinal organoids in conditioned medium (CM) collected from organoids derived from VilCreER;Apc+/+ (wild-type; hereafter WT CM) and VilCreER;Apcfl/fl (Apc-mutant; hereafter Apc-/- CM) mice (Fig. 1e). The growth and viability of wild-type organoids cultured in Apc-/- CM was profoundly inhibited (Fig. 1e and Extended Data Fig. 1e, f). Functionally, this corresponded to strikingly fewer organoids over time (Fig. 1f), suggesting that Apc-/- CM inhibited stemness. In support, these organoids exhibited decreased expression of Wnt-target genes and ISC markers and, conversely, increased expression of differentiation markers (Fig. 1g), reminiscent of the decrease in stemness caused by age-associated Notum induction 13. Complementary to our data, the accompanying manuscript by van Neerven et al. describes the rapid atrophy of wild-type organoids when co-cultured with Apc-mutant cells. Together, these findings underscore the importance of paracrine interactions between neighbouring competing cells and suggest factors, secreted by Apc-mutant cells, can impair the stemness of wild-type ISCs by suppressing their capacity for growth and promoting their differentiation.
We next assayed the growth kinetics of wild-type organoids, cultured in WT or Apc-/- CM, supplemented with a selective Notum inhibitor (10nM LP-922056, hereafter NOTUMi) 16,17. Interestingly, the addition of NOTUMi rescued the growth-arrest phenotypes observed in wild-type organoids treated with Apc-/- CM (Fig. 1e). Remarkably, by P3, the number of organoids cultured in Apc-/- CM containing NOTUMi had increased to levels comparable to those observed in WT CM (Fig. 1f). Importantly, upon NOTUMi supplementation, the expression of Wnt-target genes and markers of ISCs and differentiation in Apc-/- CM-treated organoids also reached a level comparable with WT CM (Fig. 1g). However, Lgr5 expression was notably higher after NOTUMi treatment in both Apc-/- CM and WT CM, demonstrating NOTUMi’s potency and suggesting even modest Notum expression impacts ISCs. To confirm the robustness of Notum-mediated ISC suppression, we interbred Lgr5CreER;Apcfl/fl mice with mice carrying a conditional Notum allele 18 (hereafter Lgr5CreER;Apcfl/fl;Notumfl/fl mice) and collected the conditioned medium from tamoxifen-induced Lgr5CreER;Apcfl/fl;Notumfl/fl organoids (Apc-/-;Notum-/- CM) (Extended Data Fig. 1g). Importantly, wild-type organoids treated with Apc-/-;Notum-/- CM displayed comparable growth kinetics to vehicle control, which was reversed/blocked in cells treated with Apc-/-;Notum-/- CM supplemented with recombinant NOTUM (Fig. 1h and Extended Data Fig. 1g).
Besides Notum, several other Wnt inhibitors were also upregulated by Apc-mutant cells compared to wild-type (Fig. 1a and Extended Data Fig. 2a, b). To test whether these genes are also implicated in the competitive advantage of Apc-mutant cells, wild-type organoids were treated with recombinant WIF1 and/or DKK3 and assayed for changes in morphology and growth (Extended Data Fig. 2c). Interestingly, compared to NOTUM-treated cells, which potently suppressed organoid expansion, WIF1 and DKK3 failed to perturb organoid growth when added individually or in combination to wild-type cells, despite being upregulated in Lgr5CreER;Apcfl/fl;Notumfl/fl organoids (Extended Data Fig 2c, d). In line with its effects on organoid growth, NOTUM supressed Wnt signalling more robustly than WIF1 and DKK3 (Extended Data Fig. 2e). Of note, recombinant NOTUM, WIF1, DKK3 or NOTUMi had no effect on the growth of Apc-mutant organoids (Extended Data Fig. 2f, g), indicating NOTUM preferentially suppresses the growth of wild-type rather than Apc-mutant cells. Taken together, these findings establish NOTUM as the key paracrine negative regulator of Wnt signalling produced by Apc-mutant cells to inhibit wild-type ISC function.
To investigate whether our in vitro observations translate to intra-cryptal competition in vivo, we administered low-level tamoxifen induction (0.15 mg) to clonally delete Apc and Notum in only occasional Lgr5+ cells per crypt. As the Apc locus does not recombine at the same frequency with the widely used Rosa26 reporter 2, we used nuclear β-catenin staining as a surrogate to measure Apc-mutant clonal outgrowth in the presence or absence of Notum (Fig. 2a) and measured the relative ratio of fully-to-partially fixed crypts 2 and the total number of mutant (β-catenin+) clones emerging over time (Fig. 2b, c). In Lgr5CreER;Apcfl/fl;Notum+/+ mice, Apc-mutant clones became rapidly fixed so that, by 14 days post induction, over 50% of lesions were fixed within a crypt (Fig. 2b) whereas only 10–15% of double mutant Lgr5CreER;Apcfl/fl;Notumfl/fl clones were fixed at this time-point (Fig. 2b), consistent with the observed reduction in the total number of double mutant clones over time (Fig. 2c). Next, we evaluated the size distribution of Apc-mutant clones at the crypt base over time, as detected by nuclear β-catenin+/lysozyme+ staining (Fig. 2d). Compared to Lgr5CreER;Apcfl/fl;Notum+/+ mice, which showed fully clonal crypts from 21 days post-tamoxifen, clones from Lgr5CreER;Apcfl/fl;Notumfl/fl mice failed to expand at the same rate, indicative of impaired clone fixation following Notum deletion (Fig. 2e). Collectively, these data show that in the early phases of clonal competition, Notum-mediated Wnt inhibition is essential for Apc-mutant cells to win over neighbouring wild-type cells and fix the crypt with mutant progeny.
Figure 2. Notum is required for Apc-mutant fixation in vivo.

a, Representative β-catenin IHC reveals fully and partially Apc-mutant fixed crypts in Lgr5CreER;Apcfl/fl (Notum+/+) and Lgr5CreER;Apcfl/fl;Notumfl/fl (Notumfl/fl) mice. n=4 mice per genotype were stained. Scale bar, 200 μm. b, Ratio of fully-to-partially fixed crypts in Notum+/+ and Notumfl/fl mice induced with 0.15 mg tamoxifen and sampled at 10, 14, and 21 days post induction (d.p.i). n=4 mice/genotype per timepoint. c, Total number of nuclear β-catenin+ lesions in Notum+/+ and Notumfl/fl mice over 10, 14, and 21 days post induction. n=4 mice/genotype per timepoint. d, Immunofluorescence staining for β-catenin (red) and lysozyme (green) of representative whole-mount Apc-mutant clones (red) from Notum+/+ and Notumfl/fl mice 21 days post induction. Nuclei were labelled with DAPI (blue). n=4 mice per genotype were stained. Scale bar, 200 μm. e, Left, heat maps depict the relative frequency of mutant clones of the indicated size (columns) at various timepoints (rows) for both Notum+/+ and Notumfl/fl mice. Right, graph displays the average clone size of Notum+/+ and Notumfl/fl mice over time post tamoxifen-induction. n=4 mice/genotype per timepoint. Data are mean ± s.e.m. In b, c, e, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
We next asked whether co-deletion of Apc and Notum in Lgr5+ ISCs could affect long-term clonal expansion and tumour formation. Unlike our observations at earlier time points, we observed no difference in the survival of Lgr5CreER;Apcfl/fl;Notumfl/fl mice or their overall tumour burden compared to controls (Extended Data Fig. 3a–c). However, most large adenomas that grew out had escaped Notum deletion and expressed robust levels of Notum (Extended Data Fig. 3d), suggesting that retaining Notum function confers a survival/clonal advantage during adenoma development and, conversely, that Notum loss-of-function bestows a clonal disadvantage upon Apc-mutant cells. To further validate our findings, we generated a novel Notum conditional knockout allele (designated Notumc to distinguish it from Notumfl) by inserting two loxP sites flanking exon 8 (Extended Data Fig. 4a, b) and crossed the offspring to Lgr5CreER;Apcfl/fl mice (Lgr5CreER;Apcfl/fl;Notumc/c) (Extended Data Fig. 4c). Using this novel mouse model, we observed robust Notum recombination in Apc-mutant cells, which again translated to a reduction in fully-clonal Apc-mutant crypts and an overall decrease in small- but not large- intestinal Apc-mutant (β-catenin+) clones following Notum deletion (hereafter NotumcKO) (Extended Data Fig. 4c-h). With previous results (Fig. 2), this underscores a key requirement for Notum in Apc-mutant clonal fixation. However, Notum did not change the frequency or proliferation of Apc-mutant Lgr5-GFP+ cells, indicating the reduced multiplicity of emergent mutant clones was indeed due to changes in early ISC clonal competition (WT vs Apc-mutant) and not tumour intrinsic mechanisms (Extended Data Fig. 4i). Of note, Notum deletion did not grossly alter intestinal homeostasis in non-Apc mutant mice (Extended Data Fig. 5a-c), but did render cultured Lgr5-GFP+ ISCs cells more clonogenic in vitro (Extended Data Fig. 5d, e), consistent with its in vivo function as a lowly expressed mTOR-responsive Wnt regulator 13.
Our in vitro findings suggest Notum inhibits wild-type ISCs by decreasing stemness and promoting their differentiation. Therefore, we next sought to determine whether the same processes underpinned Apc-mutant fixation in vivo. To test this, we quantified the frequency of EdU+ wild-type cells within Apc-mutant crypts, with and without Notum (NotumWT vs NotumcKO respectively) following high-dose tamoxifen (3 mg) (Fig. 3a). Consistent with our earlier results (Fig. 1), wild-type cells residing within NotumWT-mutant crypts (Lgr5CreER;Apcfl/fl;Notum+/+) had reduced proliferation compared to wild-type crypts positioned remotely from a mutant clone (Fig. 3a). In striking contrast, wild-type cells cohabiting NotumcKO-mutant crypts (Lgr5CreER;Apcfl/fl;Notumc/c) showed similar proliferation to distant wild-type crypts. We next measured the relative nuclear expression of Wnt-regulated Sox9 19,20 in ISCs (adjoining Paneth cells) and transit-amplifying (TA) progenitor cells (not adjoining Paneth cells) in Apc-mutant crypts and the surrounding wild-type epithelium (Fig. 3b). Importantly, Sox9 expression in wild-type ISCs was most dramatically reduced in crypts containing Apc-mutant clones and closer to levels detected in TA cells, suggestive of a partial differentiation and loss of stemness (Fig. 3b). However, Sox9 expression in wild-type ISCs, either sharing the crypt with a NotumcKO-mutant clone or located in neighbouring crypts, was unchanged. These data suggest Notum promotes Apc-mutant clone expansion by driving the differentiation of wild-type ISCs consistent with its function in the ageing intestine13 and in vitro (Fig. 1). Indeed, the number of Muc2+ secretory crypt cells was increased in NotumWT-mutant compared to NotumcKO-mutant animals (Fig. 3c). To examine whether Apc-mutant cells eliminate wild-type cells via apoptosis, as in Drosophila 15, we quantified the numbers of apoptotic cells within Apc-mutant clones and the neighbouring wild-type epithelium and found similar levels, independent of Notum status (Extended Data Fig. 6a). Moreover, VillinCreER;Baxfl/fl;Bak-/- intestinal organoids (lacking two key pro-apoptotic regulators, BAX and BAK, thereby compromising apoptosis) experienced robust organoid atrophy following Notum-proficient Apc-/- CM treatment, which was rescued by the addition of NOTUMi (Fig. 3d), suggesting Notum eliminates wild-type ISCs via differentiation rather than by inducing apoptosis.
Figure 3. Notum inhibits cell proliferation and drives differentiation of wild-type ISCs.

a, Representative images (left) and quantification (right) of wild-type crypt proliferation, marked by EdU incorporation (red) in Lgr5CreER;Apcfl/fl;Notum+/+ (NotumWT) and Lgr5CreER;Apcfl/fl;Notumc/c (NotumcKO) mice at 14 d.p.i. Distance refers to the location of analysed wild-type crypts relative to the closest β-catenin+ clones (white, dashed yellow line). “0” refers to wild-type cells within the Apc-mutant crypt. Proliferation is represented in relation to distant crypts, >3 three crypts away from Apc-mutant clones. n=5 per genotype. Scale bar, 50 μm. b, Representative confocal images showing nuclear SOX9 expression (mean fluorescence intensity, MFI) in ISCs (green arrows) and transit-amplifying (TA) cells (blue arrows) neighbouring Apc-mutant clones (yellow border). Nuclei labelled with DAPI (white). Analysis of ISC nuclear SOX9 intensity is shown in relation to TA cells, with the same distance parameters described in panel a. n=5 mice/genotype. Scale bar, 20 μm. c, Analysis of Muc2+ (green) cells in animals described in panel a. Scoring of positively labelled cells in clonal crypts was performed as described in panel a. n=5 NotumWT and n=4 NotumcKO mice. Scale bar, 50 μm. d, Left, quantification of tamoxifen-induced VilCreER;Baxfl/fl;Bak-/- organoids (Baxfl/fl;Bak-/-) following treatment with Apc-/- CM ± NOTUMi. Right, representative images of Baxfl/fl;Bak-/- organoids 5 days following corresponding treatments. Organoid lines were derived from n=3 mice. Mann–Whitney one-tailed U-test. Scale bar, 100 μm. e, Representative β-catenin immunofluorescence (left) and quantification of wild-type crypt cross-sectional area (right) adjacent to or remote from Apc-mutant clones in whole-mount small intestine from Lgr5CreER;Apcfl/fl;Notum+/+ (Notum+/+) and Lgr5CreER;Apcfl/fl;Notumfl/fl (Notumfl/fl) mice at 14 d.p.i. (0.15 mg tamoxifen). Nuclei labelled with DAPI (white). n=4 Notum+/+ and n=3 Notumfl/fl mice. Scale bar, 50 μm. Data are mean ± s.e.m. In a, b, c, e, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
Interestingly, Notum’s effects extend beyond its production source (Apc-mutant crypts), i.e Notum can influence the proliferation and differentiation of wild-type ISCs in crypts immediately adjacent to Apc-mutant clones (Fig. 3a, c). This was mirrored in the diminished size of wild-type crypts, immediately adjacent to Apc-mutant clones (as measured by lateral cross-sectional area), compared to those at least two- crypt- diameters away (Remote) (Fig. 3e). However, wild-type crypts adjacent to Apc/Notum-mutant crypts were of comparable size to their remote counterparts (Fig. 3e), demonstrating Notum can indeed function over inter-cryptal distances. Collectively, these data confirm and demonstrate Notum promotes Apc-mutant clone expansion and fixation by directly suppressing Wnt signalling and the proliferation of neighbouring wild-type ISCs, while promoting their differentiation (Fig. 3a-c).
To mitigate the strong negative selection against Notum-deficient clones during adenoma formation observed using the Lgr5CreER mouse, we crossed Notumfl/fl mice to VillinCreER;ApcMin/+ mice (hereafter VilCreER;ApcMin/+), which develop tumours following spontaneous loss-of-heterozygosity of the remaining Apc allele 11. Given Notum is minimally expressed in the intestine of young mice 13 (Fig. 1c) and that ApcMin/+ mice develop tumours from 60 days, we reasoned that deleting Notum prior to loss of second copy of Apc and consequent tumour formation would remove the strong selection against Notum-deficient cells. To do this, we administered tamoxifen (2 mg) at 6 and 8 weeks of age to ensure robust deletion of Notum throughout the epithelium. In contrast to VilCreER;ApcMin/+;Notum+/+ controls, VilCreER;ApcMin/+;Notumfl/fl mice had a striking extension in survival (Fig. 4a) and reduced intestinal tumour burden when harvested prior to the onset of clinical symptoms (85 days) (Fig. 4b, c). Interestingly, we observed a mixture of Notum+ and Notum- adenomas in VilCreER;ApcMin/+;Notumfl/fl mice (Fig. 4d and Extended Data Fig. 7a), which is consistent with the variable recombination efficiency of VilCreER in ApcMin/+ tumour epithelia (Extended Data Fig. 7b) and a strong selection against Notum-deficient cells during early tumour formation. We also noted increased Wif1, but not Dkk3, expression in areas of Notum- tumour epithelium, suggesting selective pressure to upregulate other Wnt inhibitors in Notum’s absence (Extended Data Fig. 7c). This is consistent with earlier observations (Extended Data Fig. 3) that establish Notum as the primary mediator driving Apc-mutant clone fixation.
Figure 4. Inhibition of Notum limits Apc-mutant fixation and intestinal tumour progression.

a, Survival plot for VilCreER;ApcMin/+;Notum+/+, VilCreER;ApcMin/+;Notumfl/+ and VilCreER;ApcMin/+;Notumfl/fl mice aged until clinical endpoint following induction with 2 mg tamoxifen at 6 and 8 weeks of age. n=30 Notum+/+, n=13 Notumfl/+, n=9 Notumfl/fl mice, where 5 still alive at the time of submission. Of note, animal sampled at 101 days was tumour-free (censored). P calculated using log-rank test. b, c, Total tumour number (b) and burden (c) per mouse of genotypes shown. Mice were sampled at 85 days of age. n=11 Notum+/+, n=8 Notumfl/fl mice. d, Notum-ISH on small intestinal tissue from mice described in b. Scale bar, 10 mm. e, Representative β-catenin immunofluorescence (red) of Lgr5CreER;Apcfl/fl mice induced with 0.15 mg tamoxifen followed by twice daily treatment with vehicle or NOTUMi (30 mg/kg) for 21 days post induction. Nuclei were labelled with DAPI (white). Scale bar, 50 μm. f, Ratio of fully-to-partially fixed crypts in mice described in e. n=4 per treatment group. g, Quantification and classification of β-catenin+ clonal lesions from mice described in e. n=4 per treatment group. h, Representative images of NOTUM-ISH and β-catenin IHC on serial sections from human colonic adenoma and surrounding normal mucosal tissue. Note, NOTUM/β-catenin double-positive cells only observed in the adenoma tissue. Staining was performed on >10 patient samples. Scale bar, 20 μm. i, Relative percentages of NOTUM-intensity within positive regions of adenoma and neighbouring mucosal tissue from human FAP-patient samples. j, Schematic depicting the proposed model of Notum-mediated Wnt inhibition of wild-type ISCs (green) by Apc-mutant cells (brown). Curved arrows indicate activation and blunt-ended arrows inhibition. Arrow thickness resembles strength of activity. Data are mean ± s.e.m. In b, c, f, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
In the accompanying manuscript, van Neerven and colleagues investigate the role of the Wnt agonist LiCl in reducing the competitive fitness of Apc-mutant cells. Here, we undertook an alternative approach to assess whether pharmacological intervention of Notum expression could yield therapeutic efficacy in our intestinal tumour models. To this end, Lgr5CreER;Apcfl/fl mice received daily NOTUMi treatment (30 mg/kg) following low-dose (0.15 mg) tamoxifen-induction and were sampled 21 days post tamoxifen-induction. Encouragingly, NOTUMi treatment significantly reduced Apc-mutant fixation and the overall number of β-catenin+ lesions compared to vehicle treated mice (Fig. 4e-g and Extended Data Fig. 8a, b), recapitulating our genetic models, thus positioning Notum as a novel therapeutic target for Apc-driven intestinal tumours.
We recently described human CRC stratification into ligand -dependent (RSPO-fusion/RNF43-mutant) and –independent (APC-mutant) subtypes, based on the expression of Wnt-target genes, which can inform Wnt-targeted therapy suitability 21. As such, we confirmed strong NOTUM expression in polyps and carcinoma samples from two independent patient cohorts, including patients with familial adenomatous polyposis (FAP), but not in known RSPO-fusion mutant tumour epithelium or normal mucosa 21 (Fig. 4h, i and Extended Data Fig. 8c, d). To model ligand-dependent Wnt transformation and assess potential Notum deregulation, we deleted the E3 ubiquitin ligases Rnf43 and Znrf3, which function as negative feedback regulators of Wnt signalling 22,23, from the intestinal epithelium. Importantly, in contrast to Apc loss, we did not observe expression of Notum (or Wif1 and Dkk3; not shown) in VilCreER;Rnf43fl/fl;Znrf3fl/fl mice 14 days following tamoxifen-induction, despite almost all epithelial cells expressing nuclear β-catenin (Extended Data Fig. 8e). More importantly, the lack of functional Notum in Rnf43/Znrf3 mutant epithelia translated to no alterations in wildtype organoid growth following treatment with conditioned medium, harvested from tamoxifen-induced VilCreER;Rnf43fl/fl;Znrf3fl/fl organoids (R/Z-/- CM), and was only perturbed following addition of recombinant NOTUM (Extended Data Fig. 8f). These data confirm the mutational routes taken to activate Wnt signalling in CRC (APC vs RSPO or RNF43) deploy distinct transcriptional outputs that impose different molecular mechanisms to achieve mutant-clone fixation in the intestine.
Cell competition was first identified in the developing fly, where cells that hyper-proliferate due to elevated Myc levels, or express Notum due to high Wg, act as super-competitors that induce apoptosis of their neighbours 15 24. Here, we discover a variant of these cell competition processes, where differentiation, rather than cell death, determines the dominance and retention of clones during early adenoma development. Apc-mutant cells act as super-competitors and secrete Notum to attenuate the Wnt-rich stem cell niche, thereby outcompeting wild-type ISCs from the intestinal crypt by reducing their self-renewal. Importantly, NOTUM expression is significantly enriched in Wnt ligand-independent human intestinal tumours, in contrast to ligand-dependent subgroups where NOTUM is silenced by methylation 21. Together, this supports a model whereby paracrine Notum signalling is deployed to suppress the stemness of neighbouring wild-type ISCs, while not impacting Wnt ligand-independent Apc-mutant cell growth (Fig. 4j). Finally, given that multiple NOTUM inhibitors are currently under development, our robust pre-clinical data argue NOTUM inhibition could provide an efficacious strategy for preventing clonal fixation and mutant expansion underpinning adenoma development in patients predisposed to CRC. 17.
Methods
Mouse studies
All animal experiments were performed in accordance with UK Home Office regulations (under project licence 70/8646) and adherence to the ARRIVE guidelines and were subject to review by the Animal Welfare and Ethical Review Board of the University of Glasgow and the Finnish National Animal Experimentation Board. All mice were maintained on a mixed C57BL/6 background. Mice were housed in conventional cages in an animal room at constant temperature (19–23 °C) and humidity (55%±10%) under a 12-h light–dark cycle and were allowed access to standard diet and water ad libitum. Mice of both genders, aged 2-6 months, were induced with a single intraperitoneal (i.p) injection of 0.15, 2 or 3 mg tamoxifen (Sigma-Aldrich, #T5648) as indicated. For Lgr5CreER;Notumc/c studies, mice were fed tamoxifen food (Harlan, #TD55125) for 10 days and analysed 4 or 8 months after. The transgenes/alleles used for this study were as follows: VilCreER 25, Lgr5-EGFP-IRES-CreER (designated Lgr5CreER) 14, Apc580S 26, ApcMin 11, Apc1322T 27, Notumfl 18, Ctnnb1E×3 28, Rnf43fl 23, Znrf3fl 23, Rosa26tdTomato29 and Baxfl/Bakko 30. For tumour growth studies, mice were aged until they showed clinical signs of intestinal disease (anaemia, hunching, and/or weight loss). For Notum inhibition studies mice were dosed with LP-922056/NOTUMi (30 mg/kg, twice daily oral gavage) or equivalent volume of vehicle (distilled water+0.5% Tween-20).
Generation of Notum conditional allele
The Notum conditional loss-of-function allele (Notumc) was generated using genOway customised mouse model services. Briefly, a homology fragment for the C57BL/6 mouse Notum gene locus was isolated and a targeting vector containing loxP sites flanking Notum exon 8 and an FRT-flanked neomycin selection cassette was generated. The NheI linearised targeting vector was electroporated into C57BL/6 mouse embryonic stem (ES) cells followed by G418 selection. Correctly targeted ES cell clones were identified by PCR and Southern blotting (Extended Data Fig. 4b). Validated ES cell clones were injected into blastocysts to generate chimeras. Viable chimeras were crossed with C57BL/6 Flp deleter mice to remove the neomycin cassette and generate mice with the desired conditional Notum allele (Extended Data Fig. 4a). Upon Cre-mediated deletion, Notum exon 8 is lost causing a frameshift that introduces a premature stop codon in exon 9 and destroying the catalytic triad required for Notum pectin acetylesterase activity.
Patient material
Formalin-fixed paraffin embedded intestinal polyps and carcinoma tissue were collected from anonymised patients who had undergone curative surgery and completed adjuvant therapy for stage III CRC. All patients gave informed consent for the storage and analysis of their samples (Victor Trial study; PMID: 20837956, REC reference: 17/NW/0252). Familial adenomatous polyposis (FAP) patient material was collected from a study approved by the Ethics Committee of the Helsinki University Central Hospital (DNRO 239/13/03/02/2011) and the samples were anonymized prior to use for the study.
Organoid culture
Mouse small intestinal crypts were isolated from wild-type (VilCreER;Apc+/+), Lgr5CreER;Notumc/c, VilCreER;Baxfl/fl;Bak-/-, VilCreER;Rnf43fl/fl;Znrf3fl/fl, Lgr5CreER;Apcfl/fl;Notumfl/fl and VilCreER;Apcfl/fl mice as previously described 31. Isolated crypts were resuspended in Matrigel (BD Bioscience), plated in 6-well plates, and overlaid with ENR growth medium comprising (Advanced DMEM/F12 (#12634010) supplemented with penicillinstreptomycin (#15070063), 10 mM HEPES (#15630056), 2 mM glutamine (#25030081), N2 (#17502048), B27 (# 17504044) (all from Gibco, Life Technologies), 100 ng/ml Noggin (#250-38), 500 ng/ml R-Spondin (#315-32), and 50 ng/ml EGF (#315-09) (all from PeproTech). Conditioned medium (CM), derived from wild-type (WT CM), VilCreER;Apcfl/fl (Apc-/- CM), Lgr5CreER;Apcfl/fl;Notumfl/fl (Apc-/-;Notum-/- CM), VilCreER;Baxfl/fl;BakKO (Bax.Bak-/- CM) and VilCreER;Rnf43fl/fl;Znrf3fl/fl (R/Z-/- CM) organoids, was collected over the course of 5 days, centrifuged at 1500 rpm for 5 min, and supplemented with the growth factors EGF, Noggin, and R-Spondin before use. Notum inhibition was achieved in vitro by adding 10 nM NOTUMi (Lexicon Pharmaceuticals) 17 to the CM, and was replenished every other day. Recombinant NOTUM (1 μg/ml; R&D Systems, #9150-NO-050), WIF1 (1 μg/ml; R&D Systems, #135-CW-050) and DKK3 (1 μg/ml; R&D Systems, #1118-DK-050) was mixed with standard growth medium and replenished every other day. For organoid growth rate experiments, ~100 wild-type organoids were resuspended in Matrigel and plated into 96-well plates. Following treatment, organoids were isolated, passaged and replated into 96-well plates. Organoids were quantified 3-5 days following passage. For clonogenicity experiments, 1,000 isolated Lgr5hi cells were plated into 60% Matrigel and overlaid with ENR medium containing 10 μM Chir99021 (GSK3 inhibitor; Tocris, #4423) and 10 μM Y27632 (ROCK inhibitor; Tocris, #1245) and changed after 2 days. Medium was replaced to regular ENR after 5 days of culture and colony numbers were quantified on day 7 after plating.
Organoid viability
Organoid viability was measured as previously described 32. Briefly, organoids were mechanically disrupted and resuspended in TrypLE™ Express (Thermo Fisher Scientific, #12604013) with 100–200 U DNAse (Sigma, #4716728001) for 1 h at 37 °C. Cells were passed through a 40-μm strainer, counted, and seeded in 100 μl Matrigel/PBS (1:1 mixture) in a 96-well plate and overlaid with standard growth medium or CM plus treatments 48 h after seeding (described above). Viability was quantified using CellTiter-Blue® (Promega, #G8080) at indicated time points in combination with morphological grading/scoring of organoids with clear central lumen. The number of crypts/organoids was scored from a total of 30-50 organoids from 3 representative images per condition in n=2 independent experiments.
Single-cell sorting and analysis
Single cells from small intestinal crypts were isolated and analysed by flow cytometry as described in 13.
Clonal analysis and adenoma scoring
Small intestinal tissue was isolated, flushed with water, cut longitudinally and fixed in 10% neutral buffered formalin overnight at 4°C (β-catenin clonal scoring) or room temperature (en face scoring). For en face processing, small (~ 2 x 2 cm) pieces of tissue were paraffin embedded en face and cut into 5 μm sections. En face sections were stained and imaged using a Zeiss 710 confocal or an Opera Phenix high-content imaging platform (Perkin Elmer). For β-catenin IHC clonal analysis, the following criteria were used measure the ratio of fully fixed to partially fixed crypts and the relative percentage of clonal crypts; full - both walls of the crypt nuclear β-catenin+ and clones in contact with a Paneth cell; partial - one wall of the crypt nuclear β-catenin+ and clones in contact with a Paneth cell; non-base - Nuclear β-catenin+ clone/s within crypt, but not in contact with a Paneth cell; undefined: Nuclear β-catenin+ clone/s not directly observable within main epithelial chain. The distribution and size of mutant clones and intestinal crypts was analysed and calculated using Harmony and Image J software respectively. Intestinal lesions were scored using the following criteria; microadenoma – small cluster of dysplastic cells contained within single crypt; lesion – 2 or more fully dysplastic crypts contained within a villus structure; small adenoma – collection of 2-5 connected fully dysplastic crypts that extend into the villus; large adenoma – fully dysplastic glands occupying >5 gland diameters and extend above villus.
RNA isolation and qPCR
RNA isolation was performed using the RNeasy mini kit (Qiagen, #74104) or TRIzol reagent (Thermo Fisher Scientific) for the sorted cells and cDNA was synthesised using high capacity cDNA reverse transcription kit, (#4368813) or for sorted cells RevertAid First Strand cDNA synthesis kit (#K1622) both (Thermo Fisher Scientific). SYBR Green (Thermo Fisher Scientific, #F410XL) qPCR reactions were performed with the BioRad system under standard conditions. Relative fold change in gene expression was calculated using the 2-ΔΔCt method.
All ΔΔCt values were normalised to housekeeping gene Gapdh. Primers used for qPCR: Gapdh forward, 5’-GAAGGCCGGGGCCCACTTGA-3’; Gapdh reverse, 5’- CTGGGTGGCAGTGATGGCATGG-3’; Axin2 forward, 5’-GCGACGCACTGACCGACGAT-3’; Axin2 reverse, 5’-GCAGGCGGTGGGTTCTCGGA-3’; Lgr5 forward, 5’- GACAATGCTCTCACAGAC-3’; Lgr5 reverse, 5’-GGAGTGGATTCTATTATTATGG-3’; Notum forward, 5’-CTGCGTGGTACACTCAAGGA-3’; Notum reverse, 5’- CCGTCCAATAGCTCCGTATG-3’; Ascl2 forward, 5’-CTACTCGTCGGAGGAAAG-3’; Ascl2 reverse, 5’-ACTAGACAGCATGGGTAAG-3’; Lyzl forward, 5’- GAGACCGAAGCACCGACTATG-3’; Lyzl reverse, 5’-CGGTTTTGACATTGTGTTCGC-3’; Krt2O forward, 5’-AGTTTTCACCGAAGTCTGAGTTC-3’; Krt2O reverse, 5’- GTAGCTCATTACGGCTTTGGAG-3’. Wif1 forward, 5’-TCTGGAGCATCCTACCTTGC-3’; Wif1 reverse, 5’-ATGAGCACTCTAGCCTGATGG-3’. Dkk3 forward, 5’- CTCGGGGGTATTTTGCTGTGT-3’; Dkk3 reverse, 5’-TCCTCCTGAGGGTAGTTGAGA-3’.
RNA sequencing and analysis
Whole tissue from the small intestine was used for RNA purification. RNA integrity was analysed with a NanoChip (Agilent RNA 6000 Nanokit #5067-1511). A total of 2 μg of RNA was purified via poly(A) selection. The libraries were run on an Illumina NextSeq 500 sequencing system using the High-Output kit (75 cycles). Analysis of RNAseq data was performed as previously described 33.
RNA in situ hybridisation
In situ hybridisation for Notum (#472548), Wif1 (#412368), Dkk3 (#400938), Lgr5 (#312178), and human NOTUM (#430311) mRNA (all from Advanced Cell Diagnostics) was performed using RNAscope 2.5 LS Reagent Kit–BROWN (Advanced Cell Diagnostics) on a BOND RX autostainer (Leica) according to the manufacturer’s instructions. BaseScope (Advanced Cell Diagnostics) ApcEx14 (Advanced Cell Diagnostics, #701641) was used to identify cells exhibiting Cre-mediated deletion of the wild-type Apc allele according to the manufacturer’s instructions. Positive control probes (Mm-Ppib, Ba-Ppib; Advanced Cell Diagnostics, #313918) were included in each run to ensure RNA integrity and staining specificity.
Immunohistochemistry and immunofluorescence
Intestines were flushed with water, cut open longitudinally, pinned out onto silicone plates and fixed in 10% neutral buffered formalin overnight at 4°C. Fixed tissue was rolled from proximal to distal end into swiss-rolls and processed for paraffin embedding. Tissue blocks were cut into 5 μm sections and stained with haematoxylin and eosin (H&E). Immunohistochemistry (IHC) and immunofluorescence (IF) were performed on formalin-fixed intestinal sections according to standard staining protocols. Primary antibodies used for IHC and IF were against: β-catenin (1:100; BD Biosciences, #610154), Lysozyme (1:500; Dako, #A0099), RFP (1:1000; Rockland, #600-401-379), Click-iT EdU Cell Proliferation Kit for Imaging Alexa 647 (Invitrogen, #C10340), Sox9 (1:200; Chemicon, #AB5535), Muc2 (1:300; Santa Cruz, #sc-15334), WGA (1 ug/ml; Invitrogen, #W32464), Cleaved Caspase3 (Asp175) (1:300; CST, #9661), GFP (1:300; abcam, #13970) and Ki67 (1:300; abcam, #15580). Representative images are shown for each staining.
Imaging and quantification of nuclear Sox9
Sox9, β-catenin, WGA and DAPI stained histological sections were imaged using point scanning confocal Nikon Eclipse Ti microscope with a Plan Apo 20x dry NA=0.75 water immersion objective and the NIS-Elements AR 5.02.01 software (Nikon). Using ImageJ software, the imaged stack was collapsed (Sum intensity) and a region of interest (ROI) was manually drawn around the nucleus based on the DNA stain (DAPI). Apc-mutant cells were identified based on strong nuclear β-catenin staining, and the remaining cells within that crypt were considered wild-type. Nuclear Sox9 intensity was measured as mean fluorescent intensity (MFI) in wild-type ISC and TA-cells. Cells next to a WGA+ Paneth cell were classified as ISCs and other WGA- cells were considered as TA cells. Average of the nuclear Sox9 MFI from all ISCs within a single crypt was compared to the average Sox9 MFI of TA cells in the same crypt.
Statistical analyses
The smallest sample size that could give a significant difference was chosen in accordance with the 3Rs. Given the robust phenotype of the Apcfl/fl mice and assuming no overlap in the control versus the experimental group, the minimum sample size was 3 animals per group. For analysis of organoid cultures, investigators were blinded when possible. Investigators performed all histological quantification blinded to genotype or treatment. For RNAseq analysis, statistical significance was determined using the DESeq function in R, which uses a Wald test (two-tailed) to test for significance of the coefficients after fitting a negative binomial GLM to the data. For all other data, statistical analysis was performed with GraphPad Prism v8.0.0 for Windows (GraphPad Software) using two-tailed Mann–Whitney U-tests unless otherwise stated. Statistical comparisons of survival data were performed using the log-rank (Mantel-Cox) test. For individual value plots, data are displayed as mean ± standard error of the mean (s.e.m.). P-values ≤ 0.05 were considered significant. Statistical tests and corresponding P-values are indicated in the figure legends and figures, respectively.
Extended Data
Extended Data Figure 1. Notum secreted by Apc-mutant clones inhibits wild-type organoid growth.

a, Volcano plot of significantly differentially expressed Wnt-target genes (red dots) in Apc-mutant (VilCreER;Apcfl/+) tumour tissue compared to wild-type small intestine (n=3 WT mice, n=5 VilCreER;Apcfl/+mice). This is the same dataset as in Fig. 1a but with different Wnt-target genes highlighted (green dots) using Wald test (two-tailed). b, Notum-ISH shows high levels of Notum expression in the intestine of multiple Wnt-driven tumour models of the indicated genotypes (Ctnnb1Ex3/+, 30 days; Apc1322T/+, 98 days; ApcMin/+, 125 days). Sections from n=4 mice per genotype were stained. Scale bar, 200 μm. c, ISH of serial en face sections of intestinal tissue from Lgr5CreER;Apcfl/fl mice 10 days post tamoxifen-induction. BaseScope ISH for recombined Apc (ApcEx14-ISH) and Notum (Notum-ISH) shows exclusive expression of Notum in cells that have recombined Apc (cells lacking pink RNA dots in right panel). Boxed areas show close-up of Notum+ Apc-mutant crypts, demarcated by a dashed line. Sections from n=4 mice per genotype were stained. Scale bar, 20 μm. d, Notum-ISH timecourse (days 6, 28, 60, and >100) post tamoxifen-induction in Lgr5CreER;Apcfl/fl mice shows specific Notum expression in progressively dysplastic epithelium. Sections from n=4 mice per genotype were stained. Scale bar, 100 μm. e, Quantification of the number of crypt domains per WT organoid following indicated treatments. Each data point represents a single mouse, n=6 mice. f, Relative WT organoid viability measured at passage 3 following treatments as indicated. n=5 mice/condition. g, Top, schematic illustrating mouse breeding scheme to generate Lgr5CreER;Apcfl/fl;Notumfl/fl mice for treatment of WT organoids with Apc-/-;Notum-/- conditioned medium (Apc-/-;Notum-/- CM). Bottom, representative images of WT organoids grown in Apc-/-;Notum-/- CM for 5 days and supplemented with recombinant NOTUM. Treatments were repeated twice on WT organoids derived from n=3 mice. Scale bar, 200 μm. Data are mean ± s.e.m. In e, f, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
Extended Data Figure 2. Apc-mutant cells upregulate negative regulators of Wnt signalling.
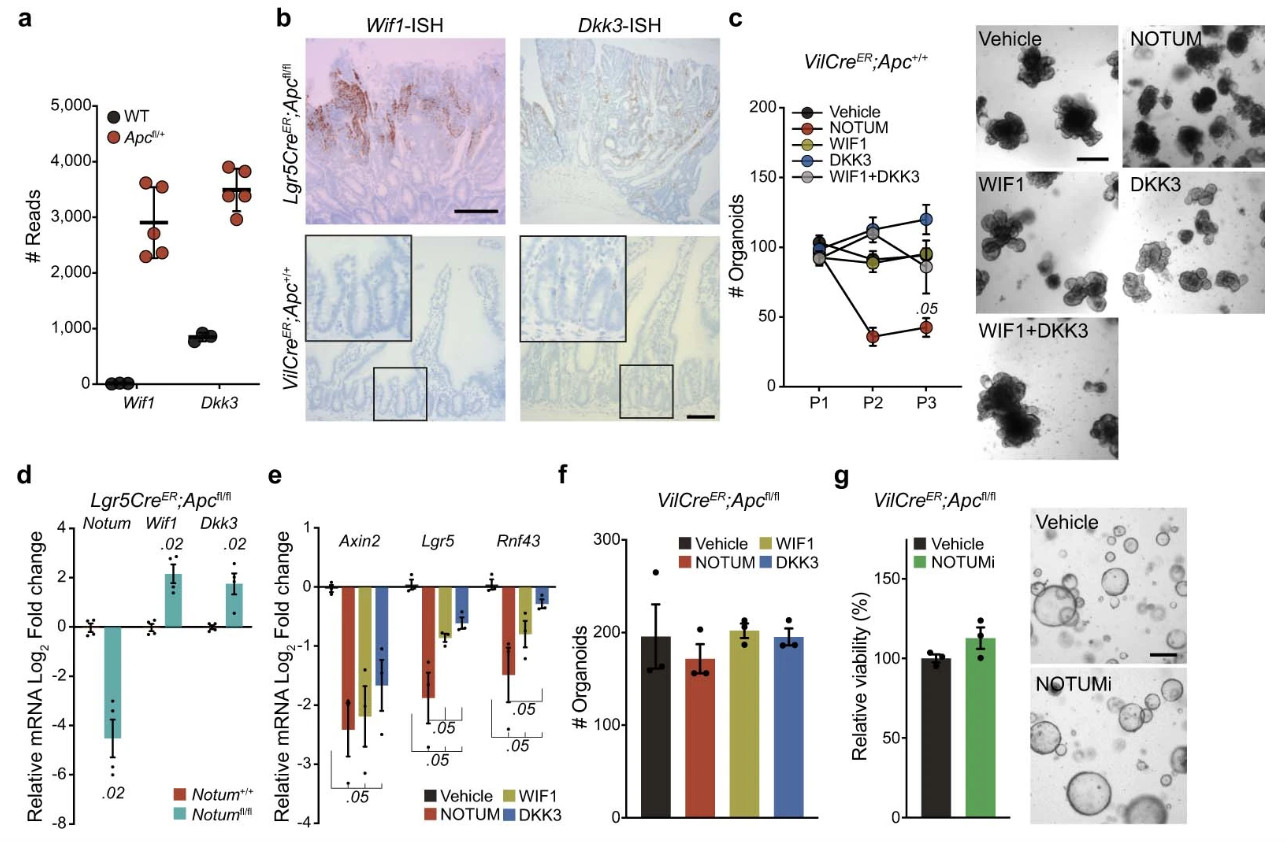
a, Raw sequence reads for transcripts encoding Wnt-negative regulators, Wif1 and Dkk3, in Apc-mutant (VilCreER;Apcfl/+) tumour tissue compared to wild-type (WT) small intestine (n=3 WT mice, n=5 VilCreER;Apcfl/+mice). b, Representative ISH for Wif1 and Dkk3 in Lgr5CreER;Apcfl/fl tumour (top panels) and WT small intestine (bottom panels). n=3 mice. Images of mice shown aged between 3–4 months. Boxed areas show close-up of WT crypts. Scale bar, 200 μm. For fluorescence-activated cell sorting (FACS) gating strategy, see Supplementary Fig. 1. c, Quantification and representative images of WT organoids, formed over multiple passages (P1, P2, and P3), during culture supplemented with recombinant WIF1, DKK3, and NOTUM. WT organoids treated with recombinant proteins were derived from n=3 mice. Images taken at P2. Mann–Whitney one-tailed U-test. Scale bar, 200 μm. d, qPCR for Wnt antagonists expressed by tamoxifen-induced Lgr5CreER;Apcfl/fl (Notum+/+) and Lgr5CreER;Apcfl/fl;Notumfl/fl (Notumfl/fl) small intestinal organoids. n=4 mice per genotype. e, qPCR for Wnt targets expressed in WT organoids 3 days following treatment with indicated recombinant Wnt antagonists. n=3 mice per treatment. Mann–Whitney one-tailed U-test. f, Quantification of VilCreER;Apcfl/fl organoids 3 days after culture with recombinant WIF1, DKK3, and NOTUM. n=3 mice per condition. g, Relative organoid viability and representative images of VilCreER;Apcfl/fl intestinal organoids treated with vehicle or NOTUMi for 3 days. n=3 mice/condition. Scale bar, 100 μm. Data are mean ± s.e.m. In d, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
Extended Data Figure 3. Notum is required for Apc-mutant cells to form intestinal tumours.

a, Survival plot for Lgr5CreER;Apcfl/fl;Notum+/+ (Notum+/+) and Lgr5CreER;Apcfl/fl;Notumfl/fl (Notumfl/fl) mice aged until clinical endpoint following induction with 0.15 mg tamoxifen. (n=10 Notum+/+ mice, n=10 Notumfl/fl mice). P=0.12, log-rank test. b, Total small intestinal tumour burden (area) per mouse from mice in a, (n=10 Notum+/+ mice, n=9 Notumfl/fl mice). c, Small intestinal tumour number per mouse from mice in a, (n=10 Notum+/+ mice, n=10 Notumfl/fl mice). d, Representative H&E and Notum-ISH staining on serial sections from Notum+/+ and Notumfl/fl mice in a. Asterisks denote intestinal adenomas. Boxed areas are close-up of adenomas stained for Notum. Note that adenomas grow out as Notum-positive lesions in Notumfl/fl mice suggesting that retaining Notum confers a survival advantage during adenoma development. Scale bars, 200 μm. Data are mean ± s.e.m. In, b, c, Mann–Whitney U-test; P values are shown in the corresponding panels.
Extended Data Figure 4. Generation and characterisation of novel Notum conditional knockout.

a, Schematic of Notum locus and recombined Notumc allele with relevant genome editing sites indicated. b, Southern blot analysis of embryonic stem (ES) cell Notumc clones and wild-type genomic DNA showing successful recombination at the Notum locus (4.7 kb product). The 13.6- and 4.7 kb bands represent endogenous and recombined alleles, respectively (arrows). c, Top, schematic of tamoxifen treatment regimen and tissue analysis 7 and 14 d.p.i. of Lgr5CreER;Apcfl/fl;Notum+/+ (NotumWT) and Lgr5CreER;Apcfl/fl;Notumc/c (NotumcKO) mice. Bottom, representative images of small intestinal sections stained for β-catenin from NotumWT and NotumcKO mice 14 days following induction with 120 mg/kg (3 mg) tamoxifen. Arrows indicate dysplastic crypts with nuclear β-catenin (magenta border). Boxed area shows close-up of β-catenin+ crypts. Sections from n=4 mice per genotype were stained. Scale bar, 50 μm. d, Representative agarose gel electrophoresis of products from conventional PCR showing relative recombination of Apcfl and Notumc alleles 5 days post tamoxifen-induction. The 250- and 513 bp bands represent recombined Apc and Notum, respectively. For gel source data, see Supplementary Fig. 1. n=3 mice. e, Quantification of small intestinal β-catenin+ lesions in NotumWT and NotumcKO mice, induced with 3 mg tamoxifen, and sampled at 7 and 14 d.p.i. n=4 per genotype at 7 d.p.i; n=18 NotumWT mice and n=12 NotumcKO mice at 14 d.p.i. f, Quantification of large intestinal β-catenin+ lesions at 14 d.p.i (n=18 NotumWT mice, n=12 NotumcKO mice). g, Left, representative images of β-catenin IHC depicting fully and partially fixed Apc-mutant crypts in NotumWT and NotumcKO mice, respectively. Mice were induced with 3 mg tamoxifen and sampled at 14 d.p.i (n=4 per genotype). Right, ratio of fully-to-partially fixed crypts in NotumWT and NotumcKO mice. Scale bar, 100 μm. h, Relative percentages of clonal crypt classification (clonal crypt phenotype) from mice described in g. i, Analysis of NotumWT and NotumcKO adenomas at 14 d.p.i. Left, representative confocal images. Ki67 (magenta), Lgr5-EGFP (green), nuclei (cyan), β-catenin (white), adenomas (yellow dashed line). Middle, quantification of Lgr5+ cell frequency within β-catenin+ Apc-mutant clones. Right, proliferation of Lgr5+ and Lgr5- Apc-mutant cells (β-catenin+). n=5 mice/genotype. Scale bar, 20 μm. In box plots, the line represents median, the box shows interquartile range and whiskers represent the range. Data are mean ± s.e.m. In e, f, g, i, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
Extended Data Figure 5. Deletion of Notum does not disrupt intestinal homeostasis.
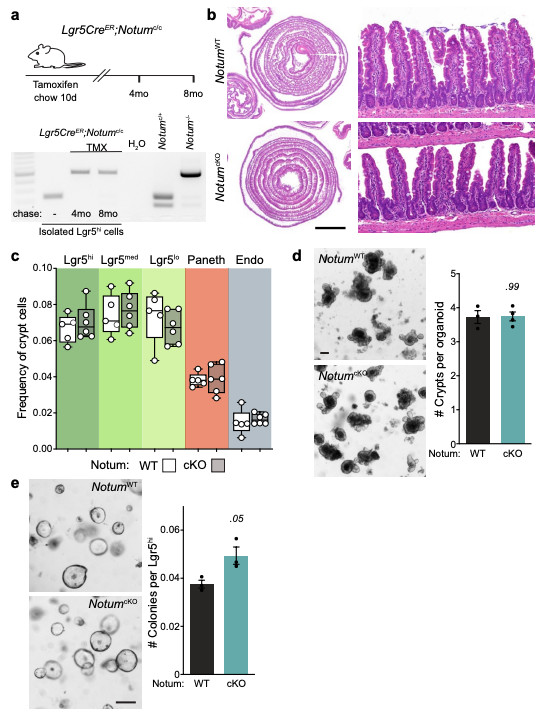
a, Top, schematic of the tamoxifen (TMX) treatment regimen and analysis of tissues from Lgr5CreER;Notumc/c mice. Bottom, representative agarose gel electrophoresis of products from conventional PCR detecting alleles for non-recombined and recombined Notumc (238 and 513 bp, respectively) in Lgr5hi cells isolated from Lgr5CreER;Notumc/c mice with or without tamoxifen-induction. Cells were isolated from n=3 mice/genotype per timepoint. b, H&E staining of WT (NotumWT) and NotumcKO tissue harvested 8 months post tamoxifen-induction. Right panels show regular crypt-villus architecture in both cohorts. Sections from n=4 mice per genotype were stained. Scale bar, 5 mm. c, Cellular frequencies of crypt cells, analysed by flow cytometry, remain unchanged after Notum deletion (WT, n=5; NotumcKO, n=6). ISCs (Lgr5hi), transit-amplifying cells (Lgr5med and Lgr5lo), Paneth cells, and enteroendocrine cells (Endo). For fluorescence-activated cell sorting (FACS) gating strategy, see Supplementary Fig. 2. In box plots, the line represents median, the box shows interquartile range and whiskers represent the range. d, Representative images and quantification of organoid regeneration (shown as number of crypt domains per organoid) of WT and NotumcKO (cKO) organoid cultures, (WT, n=3; NotumcKO, n=4). Scale bar, 100 μm. e, Clonogenic growth of isolated Lgr5hi cells is increased in NotumcKO (cKO) compared to WT. Colonies were quantified 7 days post seeding, n=3 independent organoid lines per genotype. Mann–Whitney one-tailed U-test. Scale bar, 100 μm. Data are mean ± s.e.m. In d, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels. Representative images taken at day 6 (d) and 7 (e) of culture. For gel source data, see Supplementary Fig. 1.
Extended Data Figure 6. Notum drives the elimination of wild-type cells from the crypt independent of apoptosis.

a, Quantification and representative confocal imaging of cleaved caspase-3+ cells (CC3) within clonal crypts and the surrounding non-mutant epithelium in NotumWT and NotumcKO mice at 14 d.p.i. n=4 per genotype. Crypts adjacent (Adj.) to or remote from (Rem.) Apc-mutant (clone) crypts were scored as non-mutant epithelia. Red arrows indicate CC3+ cells (green) in areas of Apc-mutant clones (purple border) and surrounding epithelia. Scale bar, 50 μm. Data are mean ± s.e.m; Mann–Whitney two-tailed U-test.
Extended Data Figure 7. Cells that escape Notum deletion upregulate Wif1, but not Dkk3.
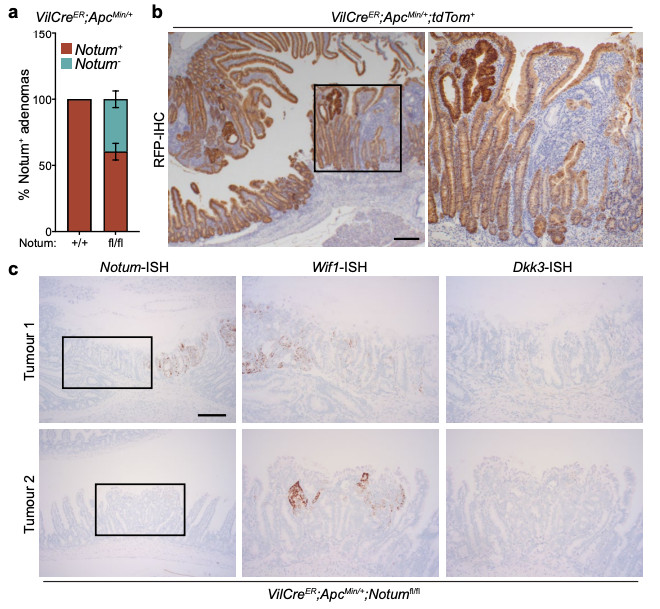
a, Quantification of the relative percentage of Notum+ adenomas (as detected via Notum-ISH) in VilCreER;ApcMin/+;Notum+/+ (+/+) and VilCreER;ApcMin/+;Notumfl/fl (fl/fl) mice aged 85 days. Mice were induced with 2 mg tamoxifen at 6 and 8 weeks of age. n=11 Notum+/+ and n=8 Notumfl/fl mice. b, Representative RFP-IHC of VilCreER;ApcMin/+;tdTom+ tumour tissue to show recombination efficiency at 85 days of age. Mice were induced with 2 mg tamoxifen at 6 and 8 weeks of age. Boxed area shows close-up of recombined (dark brown cells) and non-recombined (light brown cells) tumour epithelium from a single VilCreER;ApcMin/+;tdTom+ animal. Sections from n=4 mice were stained. Scale bar, 10 mm. c, Serial sections of intestinal tumour tissue stained via ISH for Notum, Wif1, and Dkk3 from two separate VilCreER;ApcMin/+;Notumfl/fl mice described in a. Note, Wif1 is upregulated in Notum-negative epithelial cells (boxed area). Scale bar, 50 μm. Data are mean ± s.e.m.
Extended Data Figure 8. Notum is expressed by ligand-independent and not ligand-dependent tumours.
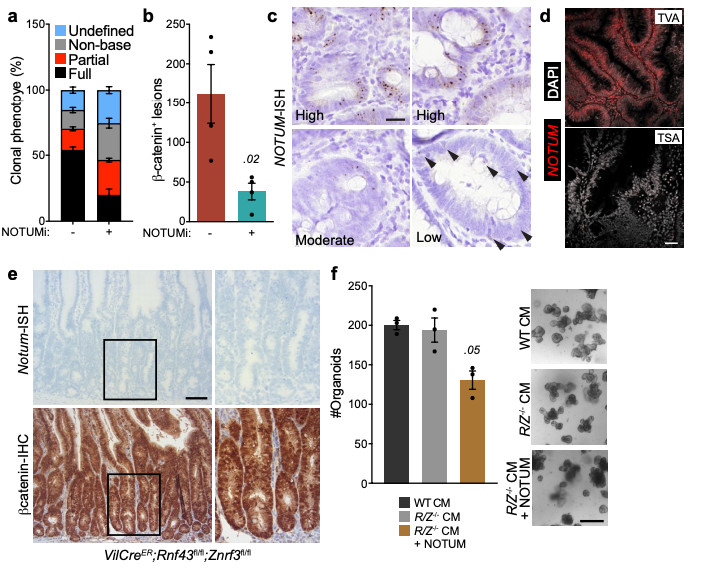
a, Relative percentages of clonal crypt classification (clonal crypt phenotype) from Lgr5CreER;Apcfl/fl mice induced with 0.15 mg tamoxifen followed by twice daily treatment with vehicle or NOTUMi (30 mg/kg) and sampled 21 days post induction. n=4 mice per treatment group. b, Quantification of β-catenin+ lesions from mice described in a. c, Representative examples of high, moderate and low NOTUM expression (as shown via ISH) within human colonic adenoma tissue. Arrows indicate single NOTUM positive cells. Staining was performed on >10 patient samples. Scale bar, 20 μm. d, Representative NOTUM expression, as shown by fluorescent ISH (FISH) on human colonic adenoma tissue. Tubulovillous adenoma (TVA), traditional serrated adenoma (TSA). Of note, NOTUM expression is minimally expressed in known RSPO1 fusion mutant adenoma tissue (TSA). Staining was performed on >10 patient samples. Scale bar, 50 μm. e, Representative Notum-ISH and β-catenin-IHC on VilCreER;Rnf43fl/fl;Znrf3fl/fl mice 14 d.p.i. (2 mg tamoxifen). Boxed areas show close-up of nuclear β-catenin+/Notum- epithelium (right panels). Sections from n=4 mice per genotype were stained. Scale bar, 50 μm. f, Quantification and representative images of WT organoids treated for 5 days with conditioned medium (CM) harvested from VilCreER;Rnf43fl/fl;Znrf3fl/fl organoids (R/Z-/- CM) ± recombinant NOTUM. R/Z-/- CM was collected from organoids derived from n=3 mice. WT organoids treated with CM ± NOTUM were derived from n=3 mice. Treatments were repeated twice. Mann-Whitney one-tailed U-test. Scale bar, 100 μm. Data are mean ± s.e.m. In b, Mann–Whitney two-tailed U-test; P values are shown in the corresponding panels.
Supplementary Material
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Acknowledgements
We thank the Core Services and Advanced Technologies at the Cancer Research UK Beatson Institute (C596/A17196 and A31287), and particularly the Biological Services Unit, Histology Service and Molecular Technologies. We are thankful to members of the Sansom and Katajisto labs for discussions of the data and manuscript. O.J.S. and his lab members were supported by Cancer Research UK (A28223, A21139, A12481 and A17196). D.J.F and M.C.H were supported by UK Medical Research Council (MR/R017247/1 and MR/J50032X/1, respectively). P.K and his lab members were supported by the Academy of Finland Centre of Excellence MetaStem (#266869, #304591 and #320185), ERC Starting Grant 677809, Swedish Research Council 2018-03078, Cancerfonden 190634, Jane and Aatos Erkko Foundation and Cancer Foundation Finland. N.P. was supported by the Finnish Cultural Foundation, Biomedicum Helsinki Foundation, Orion Research Foundation sr. and The Paulo Foundation. PVF was supported by Alzheimer’s Research UK (ARUK) and The Francis Crick Institute. The ARUK UCL Drug Discovery Institute is core funded by Alzheimer’s Research UK (520909). The Francis Crick Institute receives its core funding from Cancer Research UK (FC001002), the UK Medical Research Council (FC001002), and the Wellcome Trust (FC001002). We thank BRC Oxford for supplying patient material.
Footnotes
Author contributions D.J.F, N.P, P.K and O.J.S designed and interpreted the results of all experiments. D.J.F, N.P, K.L, A.P.R, L.M, J.I.E, A.W, S.S, N.N, E.G, E.M, M.C.H and R.A.R performed all experiments and analysed the results. D.J.F performed and analysed organoid experiments. K.G, K.K and W.C processed and analysed the RNA-seq data. C.N performed ISH. N.S provided manuscript preparation advice. A.K.N, N.W, B.R, C.P, A.C.W, H.C, P.V.F, P.L, M.L, V.H, K.A, A.R, S.L, E.Y.J and J.P.V provided advice and reagents. D.J.F, N.S, N.P, P.K and O.J.S wrote the paper.
Competing interests
The authors have no competing interests to declare.
Additional information
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
RNA sequencing data generated in this study are publicly available through Gene Expression Omnibus (GEO) with accession code GSE167008. All other data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
- 1.Barker N, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 2.Huels DJ, et al. Wnt ligands influence tumour initiation by controlling the number of intestinal stem cells. Nature communications. 2018;9:1132. doi: 10.1038/s41467-018-03426-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blokzijl F, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee-Six H, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–537. doi: 10.1038/s41586-019-1672-7. [DOI] [PubMed] [Google Scholar]
- 5.Powell SM, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 6.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Vermeulen L, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 8.Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO reports. 2014;15:62–69. doi: 10.1002/embr.201337799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morin PJ, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 10.Cammareri P, et al. TGFbeta pathway limits dedifferentiation following WNT and MAPK pathway activation to suppress intestinal tumourigenesis. Cell Death Differ. 2017;24:1681–1693. doi: 10.1038/cdd.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 12.Kakugawa S, et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature. 2015;519:187–192. doi: 10.1038/nature14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pentinmikko N, et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature. 2019;571:398–402. doi: 10.1038/s41586-019-1383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 15.Vincent JP, Kolahgar G, Gagliardi M, Piddini E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev Cell. 2011;21:366–374. doi: 10.1016/j.devcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarver JE, Jr, et al. Stimulation of cortical bone formation with thienopyrimidine based inhibitors of Notum Pectinacetylesterase. Bioorg Med Chem Lett. 2016;26:1525–1528. doi: 10.1016/j.bmcl.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 17.Brommage R, et al. NOTUM inhibition increases endocortical bone formation and bone strength. Bone Res. 2019;7:2. doi: 10.1038/s41413-018-0038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canal F, et al. Generation of Mice with Hepatocyte-Specific Conditional Deletion of Notum. PLoS ONE. 2016;11:e0150997. doi: 10.1371/journal.pone.0150997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blache P, et al. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. The Journal of cell biology. 2004;166:37–47. doi: 10.1083/jcb.200311021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roche KC, et al. SOX9 maintains reserve stem cells and preserves radioresistance in mouse small intestine. Gastroenterology. 2015;149:1553–1563.:e1510. doi: 10.1053/j.gastro.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleeman SO, et al. Exploiting differential Wnt target gene expression to generate a molecular biomarker for colorectal cancer stratification. Gut. 2019 doi: 10.1136/gutjnl-2019-319126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao HX, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012;485:195–200. doi: 10.1038/nature11019. doi:nature11019 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Koo BK, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665–669. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- 24.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 25.el Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 26.Shibata H. Rapid Colorectal Adenoma Formation Initiated by Conditional Targeting of the Apc Gene. Science. 1997;278:120–123. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- 27.Pollard P, et al. The Apc 1322T mouse develops severe polyposis associated with submaximal nuclear beta-catenin expression. Gastroenterology. 2009;136:2204–2213. doi: 10.1053/j.gastro.2009.02.058. e2201-2213, doi:S0016-5085(09)00342-4 [pii] [DOI] [PubMed] [Google Scholar]
- 28.Harada N, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. Embo J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeuchi O, et al. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt S, et al. A MYC-GCN2-eIF2alpha negative feedback loop limits protein synthesis to prevent MYC-dependent apoptosis in colorectal cancer. Nature cell biology. 2019;21:1413–1424. doi: 10.1038/s41556-019-0408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gay DM, et al. Loss of BCL9/9l suppresses Wnt driven tumourigenesis in models that recapitulate human cancer. Nature communications. 2019;10:723. doi: 10.1038/s41467-019-08586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data generated in this study are publicly available through Gene Expression Omnibus (GEO) with accession code GSE167008. All other data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.