


Abstract
Cells rely on stress response pathways to uphold cellular homeostasis and limit the negative effects of harmful environmental stimuli. The stress- and mitogen-activated protein (MAP) kinases, p38 and JNK, are at the nexus of numerous stress responses, among these the ribotoxic stress response (RSR). Ribosomal impairment is detrimental to cell function as it disrupts protein synthesis, increase inflammatory signaling and, if unresolved, lead to cell death. In this review, we offer a general overview of the three main translation surveillance pathways; the RSR, Ribosome-associated Quality Control (RQC) and the Integrated Stress Response (ISR). We highlight recent advances made in defining activation mechanisms for these pathways and discuss their commonalities and differences. Finally, we reflect on the physiological role of the RSR and consider the therapeutic potential of targeting the sensing kinase ZAKα for treatment of ribotoxin exposure.
INTRODUCTION
Stress responses are evolutionarily conserved signaling pathways that have evolved to cope with detrimental cellular perturbations. As such, the cell's ability to rapidly sense and adequately respond to extra- and intra-cellular signals is crucial for maintaining cellular integrity. Such signals include hormones and growth factors, which are important for regulating cell physiology, but also agents of a more harmful nature such as toxins, infections, byproducts of metabolism, and ultra violet (UV)-radiation. Depending on the nature, duration and severity of stress signals, cells initiate stress responses that serve to curtail the negative consequences of disrupted homeostasis.
MAP kinase cascades are key signal transduction pathways involved in the coordination of numerous stress responses (Figure 1). This large kinase network can regulate a plethora of biological processes in response to intra- and extracellular stimuli. In general, MAP kinases are organized with a three-tier architecture starting with MAP kinase kinase kinases (MAP3Ks) that activate MAP kinase kinases (MAP2Ks) that in turn activate MAP kinases. This molecular hierarchy is highly conserved throughout evolution, and many of the kinases have homologs in other organisms, demonstrating their importance in maintaining homeostasis on a cellular level.
Figure 1.

The MAP kinase signaling network. (A) A vast array of stimuli can activate the MAP kinase network, including environmental stressors, inflammatory cytokines, growth factors and hormones. The canonical activation pattern of MAPK cascades begins with the stimulus-specific activation of a MAP kinase kinase kinase (MAP3K, green) that activates MAP kinase kinases (MAP2K, blue) that activates MAP kinases (MAPK, yellow). The cellular effects of MAP kinase signaling is mediated through the phosphorylation of a vast amount of effector proteins.
The proto-typical MAP kinases comprise three groups; Extracellular signaling-Regulated Kinase (ERK1 and 2), c-Jun N-terminal Kinase (JNK1, 2 and 3) and p38 (p38α, β, γ and δ). In addition, several atypical MAP kinases exist, denoted ERK3,4,5 and 7 and Nemo-Like Kinase (NLK). The three classes of typical MAP kinases respond to multiple cellular cues, and all require dual phosphorylation in their activation loop to achieve maximum enzymatic activity. The relevant region contains a Thr-X-Tyr motif, where both threonine and tyrosine must be phosphorylated for activation. X denotes Pro, Glu and Gly for JNKs, ERKs, and p38s respectively (1).
Members of the ERK MAP kinase family primarily respond to cytokines, hormones and growth factors. In contrast, the p38 and JNK signaling pathways are often referred to as the Stress-Activated Protein (SAP) kinases as they are activated in response to various exogenous and endogenous stress stimuli. These include environmental stressors, such as reactive oxygen species (ROS), osmolarity changes, toxins and UV-radiation, but also proinflammatory cytokines and bacterial lipopolysaccharide (LPS). Activation of the SAP kinases mediates cell-fate decisions, promoting both pro-survival and pro-apoptotic signaling, and modulate immune responses by controlling the expression of inflammatory cytokines (2,3). The importance of an appropriate activation of these kinases under diverse stresses is highlighted by the fact that dysregulation of SAP kinase signaling is associated with numerous pathologies, including neurodegenerative disorders, inflammatory diseases and cancer (2,4–6)
In this review, we summarize our knowledge on how p38 and JNK are activated upon ribosomal impairment, and the ensuing biological consequences. Ribosomal poisons in the form of ribotoxins, antibiotics and UV radiation interrupt translational elongation and elicit the RSR. In contrast to other ribosomal surveillance mechanisms such as Ribosomal Quality Control (RQC) and the Integrated Stress Response (ISR), the RSR elicits a proinflammatory cascade controlled by MAP kinase signaling.
THE p38 SIGNALING PATHWAY
There are four members of the p38 family, p38α/MAPK14, p38β/MAPK11, p38γ/MAPK12 and p38δ/MAPK13. They are encoded by four separate genes that give rise to a number of alternatively spliced constructs, and can further be divided into two groups based on their sequence homology: p38α/β and p38γ/δ (7). p38α and p38β are both ubiquitously expressed, whereas p38γ and p38δ are expressed in a cell type and tissue-specific manner, possibly reflecting a more specialized function. p38γ is mainly found in skeletal muscle, while p38δ is significantly expressed in pancreas, kidney, testis and small intestine (7). Knockout of p38α is embryonic lethal due to placental defects (8–10), but p38β-, p38γ- and p38δ-deficient mice are viable and fertile and present with minimal phenotypes (7). As an example, knockout of p38γ revealed that this isoform is important for endurance exercise-induced biogenesis of mitochondria, consistent with its marked overrepresentation in skeletal muscle (11). Furthermore, and in accordance with p38δ’s tissue-restricted expression pattern, p38δ knockout mice display a higher glucose tolerance due to increased insulin secretion from pancreatic β-cells (12).
The canonical activation mechanism for p38 is by dual phosphorylation of the Thr-Gly-Tyr motif by the MAP2Ks MKK3 and MKK6 (13–15). Additionally, the JNK-activating MAP2K MKK4 is also able to activate p38α under certain conditions (16). MKK3 and MKK6 appear to serve functionally redundant roles, and mice deficient for either MKK3 or MKK6 are viable and fertile although displaying reduced cytokine production (17–19). On the other hand, inactivation of both genes confers embryonic lethality, and embryos present with many of the same symptoms as p38α knockouts, including placental defects (16). Upstream of the MAP2K layer, several MAP3Ks exert control over p38 activation, and this subject is well treated in other reviews (20,21). Non-canonical modes of p38 activation have also been reported. Here, TAB1 (transforming growth factor-β-activated protein kinase 1-binding protein) mediates activation of p38α in the absence of upstream kinase signaling by promoting cis-autophosphorylation of the activation loop (22).
p38 was first identified as a protein with a critical role in production of the proinflammatory cytokines IL-1 and TNFα in response to LPS-stimulation of monocytes (23). It has since become apparent that p38 plays a broad role in immune signaling, as its activation results in stabilization and translation of multiple cytokine mRNA (7). Studies employing chemical inhibitors and genetic inactivation have led to the identification of numerous physiological targets of p38. These include components of the AP-1 transcription factors complex (e.g. ATF2, Fos), Ser/Thr kinases (e.g. MK2) and RNA binding proteins (e.g. HuR), as well an expanding list of regulatory, membrane and structural proteins (24). p38 is a proline-directed kinase, the substrate specificity of which is influenced by interactions between docking sites on the substrate and interaction motifs on the kinase. Albeit the four members of the p38 family have many overlapping targets and some functional redundancy, there are notable differences. For instance, while MK2 is a prominent substrate for p38α/β, this kinase is phosphorylated to a lesser degree by p38γ/δ (25,26). Additionally, small molecule kinase inhibitors such as SB203580 and SB202190, which have contributed greatly to our understanding of p38 signaling pathways, only inhibit p38α and p38β, while others like BIRB796 inhibit all four isoforms (27).
THE JNK SIGNALING PATHWAY
Three genes encode JNK kinases, JNK1/MAPK8, JNK2/MAPK9 and JNK3/MAPK10, collectively giving rise to at least 10 different isoforms by alternative splicing (28). While JNK1 and JNK2 are universally expressed, JNK3 is largely restricted to testis and neuronal tissue (29). Mice lacking any of the three isoforms are viable, as is also the case for double knockout of JNK1/3 and JNK2/3 (29–31). However, simultaneous knockout of JNK1 and JNK2 confers early embryonic lethality due to defective neural tube closing and dysregulated apoptosis in the brain (30,31). Such data suggest that JNK1 and JNK2 are largely functionally redundant kinases and highlights the importance of JNK signaling in the nervous system.
JNK kinases are activated by many of the same cellular stresses and MAP3Ks as the p38 module, but instead of MKK3 and MKK6, JNK activity is controlled by the MAP2Ks, MKK4 and MKK7 (32). Unlike the case for MKK3 and MKK6, these two MAP2Ks exhibit non-redundant roles, as MKK4 preferentially phosphorylates Tyr, whereas MKK7 generally phosphorylates Thr in JNK’s Thr-Pro-Tyr activation loop (32). Since JNK as a typical MAP kinase requires dual phosphorylation on Tyr and Thr for activation, it has been suggested that MKK4 and MKK7 work synergistically to achieve downstream kinase activation. In support of this, mice lacking either MKK4 or MKK7 succumb before birth presumably due to the inability of these kinases to functionally compensate for each other (29,33–35). Nevertheless, such a synergistic relationship is not fully consistent with the fact that MKK4 and MKK7 exhibit a distinct pattern of tissue enrichment and subcellular localization (36,37), and by the fact that mkk4-/- and mkk7-/- embryos display different phenotypic anomalies (33,34,38,39). Binary interaction between MAP3Ks and the corresponding MAP2Ks and MAPKs represent one determinant of selective MAPK pathway activation. Further specificity can be conferred by scaffolding and adaptor proteins. Such proteins stabilize and spatially restrict the direct interactions between MAP kinase signaling components. Examples of well-studied MAP kinase scaffold factors are the JNK-interacting proteins (JIPs) which are required for optimal JNK activation in a variety of settings (40).
JNK was originally identified in 1993 as the kinase that phosphorylated the transcription factor c-Jun in response to UV light and expression of transforming oncogenes (41,42). Phosphorylated c-Jun promotes formation of the AP-1 transcription factor complex and thus activates genes containing AP-1 binding sites. These include cytokine genes and genes that control cell cycle, survival and apoptosis (43–45). Substrates for JNKs also include other transcription factors, microtubule-associated proteins (MAPs) and members of the apoptosis-regulating BCL2 protein family (e.g. BAD and BAX) (46).
In sum, a plethora of intra- and extracellular signals trigger activation of the SAP kinases p38 and JNK. The context-specific dosing of downstream phosphorylation events determine the balance between cellular outcomes such as survival, cell death, differentiation and inflammatory signaling.
MAP KINASE ACTIVATION IN RESPONSE TO RIBOTOXIC STRESS
Given the essential role of protein synthesis (Figure 2A) in all living cells, it is not surprising that stress response pathways have evolved to detect and counteract physiological and pathological obstructions to translation. One such pathway is the RSR, which is a MAP kinase-mediated proinflammatory signaling cascade that is mounted in response to different ribosomal insults (Figure 2B). The term ‘ribotoxic stress response’ was coined in 1997 by Iordanov et al. The authors demonstrated that p38 and JNK was activated by drugs and toxins that interfere with the region of 28S rRNA critical for aa-tRNA binding, peptidyl transfer and translocation (47). Since then additional triggers of the RSR has been identified, and they can roughly be subdivided into translation inhibitors (e.g. anisomycin and cycloheximide), ribotoxins (e.g. ricin, Shiga toxin and α-sarcin), chemotherapeutics (e.g. doxorubicin) and UV-radiation (Figure 2C, Table 1). These agents all cause structural damage to rRNA and impair ribosome function, and lead to strong activation of p38 and JNK. Such signaling initially triggers a proinflammatory response, but a sufficient duration or magnitude of RSR signaling will eventually cause cell death (48–51).
Figure 2.

The translation elongation cycle and the ribotoxic stress response. (A) The ribosome can decode messenger RNA (mRNA) and translate it into protein in three sequential events; initiation, elongation and termination. Elongation is a three-step cycle that is repeated until the protein is fully synthesized, and the stop codon is reached. The key processive reactions are (i) the capture of amino acid loaded tRNA, (ii) transfer of the amino acid to the growing polypeptide chain and (iii) the release of vacant tRNA, which occur in the ribosomal A-, P- and E-sites, respectively. (B) Ribotoxic stress is sensed by the MAP3K ZAKα leading to activation of MAPKs p38 and JNK and inflammatory signaling. In case of strong and sustained signaling the cell will undergo regulated cell death. (C) Overview of ribosomal insults known to activate the RSR – see also Table 1. (D) Alternative splicing of the ZAK gene results in two different MAP3Ks, α and β, that share the first 11 exons followed by unique exons. ZAKα is an 800 amino acid protein containing a kinase domain, a leucine zipper (LZ), a sterile-alpha motif (SAM) and two ribosome-binding domains (RBDs). ZAKβ is a 455 amino acid protein that is identical to ZAKα in its N-terminus but has a unique C-terminus.
Table 1.
Known ribotoxic stress agents and their mechanism of action
| Ribosomal target | Cellular effect | |
|---|---|---|
| Translation inhibitors | ||
| Anisomycin | Prevents peptide bond formation at PTC, 60S A-site (52) Interacts with domain V of 28S rRNA | p38 and JNK activation (47) in a ZAKα dependent manner (66,70,72) and leads to transcription and secretion of IL-8 via ZAK/p38 signaling axis (48,71) |
| Cycloheximide | Inhibits translocation via interactions with the 60S E-site | p38 and JNK activation in a ZAKα dependent manner (47,72) |
| Lactimidomycin | Blocks initiation by inhibiting first round of translocation at the 60S E-site (52) | p38 and JNK activation in a ZAKα dependent manner (72) |
| Harringtonine | Blocks initiation by inhibiting first peptide bond formation at PTC, 60S A-site (153) | p38 and JNK activation in a ZAKα dependent manner (72) |
| Deoxynivalenol / trichothecene toxins | Prevents peptide bond formation at PTC, 60S A-site | p38 and JNK activation (154) |
| Onnamide A | Inhibits protein translation measured by [3H]-leucine incorporation (62) | p38 and JNK activation (62) |
| Theopederin B | Inhibits protein translation measured by [3H]-leucine incorporation (62) | p38 and JNK activation (62) |
| Ribotoxins | ||
| Ricin Ricinus communis | Depurination of adenine in the sarcin/ricin loop located in domain VI of 28S rRNA (155) | p38 and JNK activation and IL8 production in a ZAKα dependent manner (47,71,72,77) Ricin-induced cell death is mediated by ZAK (71) Zak-/- mice have lower duodenal pathology score than wt mice following oral ricin intoxication (77) |
| Shiga toxin Shigella dysenteriae Escherichia Coli | Depurination of adenine in the sarcin/ricin loop located in domain VI of 28S rRNA (155) | p38 and JNK activation (50) in a ZAK dependent manner (71) N-glycosidase activity of Stx1 is required for p38 and JNK activation (49) Stx at high doses induce cell death via p38 and ZAK (49,71) |
| α-sarcin Aspergillus giganteus | Cleavage of phosphodiester bond in the sarcin/ricin loop (53) | p38 and JNK activation (47) |
| Chemotherapeutics | ||
| Doxorubicin | Inhibits protein translation measured by [3H]-leucine incorporation (61) | p38 and JNK activation and cell death in a ZAK dependent manner (61,94) Upregulation of transcription and secretion of inflammatory cytokines IL-6 and CXCL1 via p38/ZAK (58,94) |
| Daunorubicin | Structurally similar to doxorubicin (61) | p38 and JNK activation and cell death in a ZAK dependent manner (61) |
| UV-radiation | ||
| UV-B and UV-C | Induce rRNA lesions at domain V and VI of 28S rRNA (63) | p38 and JNK activation (63) in a ZAKα dependent manner (66,70) Sorafenib, a multi-kinase inhibitor, repress UV-induced apoptosis via ZAK and JNK (76) |
| UV-damaged mRNA | Activation of ZAKα, p38 and JNK in cells after transfection with UV-irradiated mRNA (66) |
Ribotoxic stress-inducing agents
Anisomycin is an antibiotic that targets the peptidyl transferase center (PTC), where the peptide bonds are formed during protein synthesis (52). Other translation inhibitors, like cycloheximide, occupy the ribosomal E-site and blocks the ribosomal translocation step of elongation (52). In support of their hypothesis that interference with 28S rRNA is the trigger of the RSR, Iordanov et al. also achieved strong activation of the RSR by administration of ribotoxins (Shiga, ricin and α-sarcin). These enzymes all exert their cytotoxic function through modification of the sarcin/ricin loop (SRL) located on the 28S rRNA (47). Shiga is produced by the dysentery-causing bacterium Shigella dysenteriae and Shiga-like toxins (Stx) are produced by some pathogenic strains of Escherichia coli. Ricin is a plant-derived glycoprotein isolated from castor beans (Ricinus communis), while α-sarcin is an RNA endonuclease from the fungus Aspergillus giganteus. Ricin and Shiga toxins both hydrolyze the N-glycosidic bond of a conserved adenine residue in the SRL, whereas α-sarcin cleaves the phosphodiester bond adjacent to this adenine residue (53,54). The SRL is critical for delivery of amino acid-bound tRNA and elongation factors to the translational pocket of the ribosome. In addition, the SRL plays a critical role in stimulating the GTPase activity and subsequent release of elongation factors from the ribosome (55–57).
MAP kinase-regulated production of proinflammatory cytokines are common responses to a range of chemotherapeutics (58,59) Such signaling is widely believed to be a significant cause of detrimental side effects to cancer treatment. Doxorubicin is an anthracycline widely used in the treatment of hematological and solid tumors, and it is known to induce p38 and JNK activation (58,60). Sauter et al. reported that treatment of cells with high doses of doxorubicin activates the RSR in keratinocytes leading to inflammatory signaling and apoptosis (61). An argument for the ribosome being the relevant target was supported by the finding that doxorubicin at the same doses strongly inhibited protein translation. In addition, pre-treatment of cells with the ribosome inhibitor emetine, which by itself does not activate RSR, blocked doxorubicin-induced activation of p38 and JNK. Since then, several other chemotherapeutic agents have been shown to inhibit the ribosome and activate RSR signaling. These include daunorubicin, another anthracycline-class drug (61), and Onnamide A and Theopederin B, two natural compounds related to mycalamides (62).
Finally, UV light has also been shown to be a ribotoxic stress-inducing agent (63). Even in the absence of DNA, UV-light is a potent inducer of MAPK activation. Studies with enucleated HeLa cells demonstrated a nucleus-independent activation of JNK after UV irradiation (64). This extranuclear response has been suggested to be mediated by UV-induced inactivation of protein tyrosine phosphatases and the resulting increase in tyrosine kinase activities (65) or by ribotoxic stress (47,63). In support of the latter hypothesis, Iordonov et al. concluded that UV-irradiation introduces ribosomal photodamage and inhibits ribosome function (63). In primer extension reactions, the authors detected UV-induced nucleotide damage in the 3′end of 28S rRNA. These lesions occurred mainly at adjacent pyrimidine nucleotides, suggesting that they consist of pyrimidine dimers, similar to photoproducts induced by UV light in DNA. Some of these lesions affected two critical regions of 28S rRNA; domain V, containing the peptidyl transferase ring, and domain VI, containing the SRL (63). As previously mentioned, these rRNA structures are also the relevant targets of several ribotoxic stress agents, including anisomycin and ribotoxic enzymes (47). More recent work has shown that the RSR is also elicited by translation arrest on a UV-damaged mRNA template (66). Whether it is a combination of damage to rRNA and mRNA, or only the latter, that activates the RSR after UV-irradiation remains to be clarified.
Activation of the ribotoxic stress response
The RSR is a MAP kinase signaling cascade mounted in response to defective ribosomes. Several upstream kinases have been implicated in the activation of this response including the double-stranded RNA-dependent Protein Kinase (PKR) (67,68), HCK (69) and the sterile-motif alpha and leucine zipper kinase (ZAK) (70–72). One group found that PKR is partially needed for complete MAPK activation in response to the RSR activators anisomycin, deoxynivalenol and ricin (67,68). However, a study using MEFs from PKR knockout mice did not support a role for PKR in activation of p38 and JNK in response to anisomycin or UV (73). Similarly, evidence for HCK playing an important role in the response is limited.
ZAK, also known as MAP3K20 or MLTK, is a MAP3K belonging to the MLK subfamily. Two major splice variants of ZAK exist; ZAKα (800 amino acids) and ZAKβ (455 amino acids). Originally, overexpression of these proteins were found to promote apoptosis in a hepatoma cell line (74), and ZAK was also reported to be activated by osmotic stress, causing disruption of actin fibers and altered cell morphology (75). Since then multiple functions of ZAK have been proposed, including the activation p38 and JNK in response to ribotoxic stress (61,70,72,76). This was first described in 2005 by Wang et al. through use of the ZAK inhibitor dihydro-pyrrolopyrazole quinoline (DHP-2) and by siRNA-mediated knockdown (70). These authors further showed that ZAK mediates p38 and JNK activation in COS-7 cells exposed to anisomycin and UV, but not the inflammatory cytokines TNFα and IL-1β.
An activating role for ZAK also in ribotoxin-induced RSR was demonstrated in 2008 by Jandhyala et al. They showed that both chemical inhibition and siRNA-mediated knockdown of ZAK prevented MAP kinase activation in response to ricin and Stx (71), which was later corroborated in ZAK knockout cells (72). Later, the same group showed that bone-marrow derived macrophages established from zak-/- mice were completely deficient for p38 and JNK activation upon ricin exposure (77). ZAK also appears to be the relevant upstream MAP3K when cells encounter other sources of ribotoxic stress. Thus, ablation of ZAK activity, both through chemical inhibition with sorafenib and nilotinib and siRNA-mediated knockdown suppressed doxorubicin-induced activation of p38 and JNK (61). In addition, knockdown of ZAK suppressed UV-induced MAP kinase activation (70), which was recently validated in ZAK knockout cells (66). Taken together, an overwhelming body of literature using diverse sources of ribotoxic stress places ZAK at the top of the MAP kinase signaling cascade that constitutes the RSR.
Sensing ribotoxic insults
Despite the emergence of ZAK as a key upstream regulator of RSR signaling, it is not fully understood how cells sense and communicate the presence of structurally diverse ribotoxic insults towards p38 and JNK activation. The underlying activation signal(s) does not appear to constitute mere translation arrest, as several translation inhibitors, such as emetine, pactamycin, and puromycin, are unable to (or only weakly) trigger the response (47). Additionally, toxins that inactivate the elongation factor EEF2 do not initiate RSR (47,78). A recent study by our group showed that the long isoform of ZAK, ZAKα, constitutes a molecular sensor of ribotoxic stress by virtue of two C-terminal ribosome binding domains (72) (Figure 2D). The shorter ZAKβ isoform does not contain such domains and does not support RSR signaling. Instead its C-terminus is encoded for by an alternative exon, and this protein is likely to mediate MAP kinase activation in response to other and presently unknown cellular insults. By inserting its flexible C terminus into the inter-subunit space, ZAKα is placed in a unique position to probe structural conformations of the translationally active pocket of the ribosome (72). This direct communication with the ribosome allows ZAKα kinase activation in the presence of functionally diverse ribosome inhibitors, structural rRNA damage induced by ribotoxins and presumably UV-induced photo-products. ZAKα’s signature Sterile Alpha Motif (SAM) domain likely plays a key role in harnessing kinase activity, as mutation of this domain was shown to confer constitutive activation (72) and cause a developmental bone/limb disorder in humans and mice (79).
Pathological implications of the Ribotoxic Stress Response
The primary outcome of ZAKα-induced RSR signaling elicits activation of SAPKs p38 and JNK. In addition to controlling cell-fate decisions such as cell death and differentiation, these kinases activate inflammatory signaling by virtue of their control over cytokine expression (3,6). In addition, it has been shown that RSR signaling in concert with the Unfolded Protein Response (UPR) regulates Bcl-2 to balance apoptosis and cell survival in leukocytes during immune responses (80–82). The ability of the RSR to activate both pro-inflammatory and pro-apoptotic pathways has motivated the proposal of pathway inhibition as a therapeutic strategy to combat ribotoxin intoxication (83,84). It is likely that RSR signaling contributes to the pathology of the gastrointestinal and renal complications associated with infection with Stx-producing E. coli (STEC). This group of food-borne pathogens can cause severe disease in humans including hemorrhagic colitis and, in susceptible patients, hemolytic uremic syndrome (HUS). HUS is a triad of hemolytic anemia, thrombocytopenia, and acute kidney failure. In support of this notion, mice that are unable to produce TNFα from macrophages survive Stx2 intoxication longer compared to WT mice (85). Additionally, injection of TNFα increases the mortality of mice intoxicated with sub-lethal doses of Stx1. This enhanced phenotype was associated with exacerbated glomerular damage, decreased kidney function, and increased renal apoptosis (86). Several studies also support that RSR-driven MAP kinase signaling boosts cytokine production upon Stx exposure (49,71,87,88).
p38 and JNK are both attractive drug targets for a number of diseases, including the potentially lethal Stx-induced gastrointestinal and renal damage caused by STEC infection. However, the many and pleiotropic roles of these kinases have so far prevented successful clinical trials with chemical inhibitors (89,90). Instead, inhibition of upstream and signal-specific activator kinases may represent a viable approach (83,89). This strategy has already been pursued with some success with inhibition of the MAP3K ASK1 for the treatment of nonalcoholic steatohepatitis (91). In the case of ribotoxin exposure, inhibition of ZAK could similarly represent a promising therapeutic strategy. Indeed, a couple of studies suggest that RSR signaling contributes to the pathogeneses observed after ribotoxin exposure (77,83). Jandhyala et al. reported that ZAK knockout mice suffer less duodenal damage upon ricin exposure, likely due to attenuation of cytokine-mediated tissue destruction (77). Other supporting evidence was obtained in rabbits exposed to Stx (83). Here, ZAK inhibition by the multiple-kinase inhibitor imatinib decreased heterophil infiltration and colonic inflammation (83). In spite of these encouragements, attempts to exploit our knowledge of RSR signaling therapeutically have so far been very limited. Further pre-clinical studies are required to determine how much of the detrimental organismal effects of ribotoxin exposure can be attributed to RSR signaling (84,92).
The RSR is also activated by a subset of often used anti-cancer drugs. This signaling may be linked to some of the detrimental side effects of chemotherapy including heart failure (59). For example, ribosome inhibition and ensuing p38 signaling has been suggested to be involved in doxorubicin-induced cardiomyopathy (60,61). MAP kinase inhibition has been shown to effectively reduce apoptosis of cardiomyocytes after doxorubicin and daunorubicin treatment (60,93). Inhibition of p38 in macrophages was also shown to reduce doxorubicin-induced proinflammatory responses, without compromising the anti-proliferative effects of the drug in a cancer cell line (58). Doxorubicin caused activation of p38 and JNK in a ZAK-dependent manner in both keratinocytes and monocytes (61,94), and in keratinocytes ZAK inhibition also effectively blocked apoptosis. Notably, this effect was not observed for cancerous HeLa cells (61). Time will tell whether there is a rationale for the use of ZAK inhibition in combination with RSR-activating chemotherapy (61,94).
In cultured cells, UV-irradiation is another potent trigger of RSR activation. This does not appear to be dependent on DNA damage, and human skin fibroblasts exhibit an almost 7-fold higher amount of oxidized RNA compared to DNA (95). It intuitively follows that the RSR could play an important physiological role in the skin's response to UV light. Melanoma patients treated with the BRAF inhibitor sorafenib are in high risk of developing UV-induced squamous cell carcinoma during the course of their treatment. (96,97). Sorafenib is known to have strong off-target effect against ZAK (61), and was reported to suppress UV-induced and ZAK-dependent apoptosis in cultured keratinocytes (76). These results suggest that ZAK and RSR signaling has an anti-tumorigenic role in skin. Defining the role(s) of the RSR in UV-irradiated skin will be an important future challenge, both with regards to elucidation of the physiological roles of the RSR and potential new insights into skin cancer biology.
OTHER RIBOSOMAL SURVEILLANCE PATHWAYS
The RSR is only one of several signaling pathways that originate from distressed ribosomes. During translation, ribosomes can encounter a range of obstacles on the mRNA, such as nucleotide damage, secondary structures and lack of a stop codon. Ribosomes may also struggle with inefficient decoding due to a shortage of amino acids (98). Such events will slow down elongation and may in the most extreme case cause translational arrest of individual ribosomes. Transient pausing of the elongation cycle generally allows time for resolving such issues, however if not sufficient, quality control and stress response mechanisms are required to salvage the affected ribosomes (98–100).
Ribosome-associated quality control
Irreversible ribosome stalling induces active splitting and rescue of ribosome subunits, and degradation of the incomplete protein product (Figure 3). Under these conditions, the ASC-1 helicase complex (Slh1/Rqt2 in yeast) or the recycling factors Pelota, HBS1L (or GTPBP2) and ABCE1 (Dom34, Hbs1 and Rli1 in yeast) split the stalled ribosome into its constituent 60S and 40S subunits (101,102). Subsequently, the released mRNA is degraded by the 5′-3′ exoribonuclease Xrn1 and the exosome complex to prevent the aberrant mRNA from recruiting a new ribosome. The released 40S subunit is recycled (reviewed in (98,100)), which likely requires the action of the recycling factor ABCE1 (103,104). The 60S subunit must first be relieved of the trapped tRNA-conjugated polypeptide chain (105).
Figure 3.
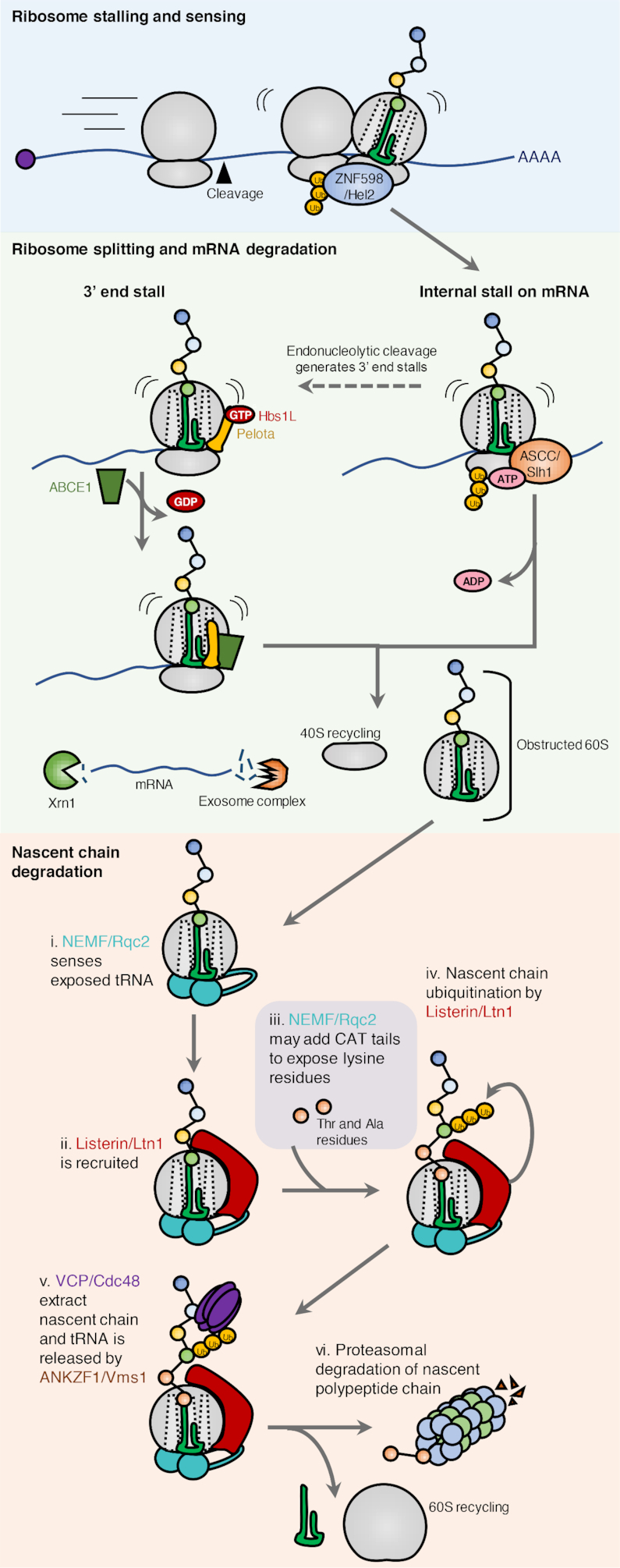
Ribosome collision and ribosome-associated quality control. (A) Translational stalling causes collision of ribosomes. This unique structure is recognized by the E3 ubiquitin ligase ZNF598 that promotes ubiquitination of the ribosomal proteins uS10, eS10 and uS3. Stalling on internal mRNA sequences is recognized by the ASC complex (Slh1 in yeast) which liberates the leading ribosome. The trailing ribosomes can then resume translation. Under certain circumstances, endonucleases cleave the mRNA between ribosomes resulting in ribosomes stalled on 3′end of the mRNA. This makes them accessible for splitting by the recycling factors Pelota, HBS1L and ABCE1 (Dom34, Hbs1 and Rli1 in yeast). The released mRNA is degraded by the 5′-3′ exoribonuclease Xrn1 and the exosome complex to prevent the aberrant mRNA from being translated again. While the 40S subunit is directly ready for recycling, the peptidyl-tRNA remains associated with the large ribosomal subunit. The obstructed 60S subunit is recognized by the RQC component NEMF (Rqc2 in yeast) that recruits the E3 ubiquitin ligase Listerin (Ltn1 in yeast) to the native peptide chain. NEMF/Rqc2 may employ ‘CAT-tailing’ to expose ribosome-buried and ubiquitinatable lysine residues in the native chain. Ubiquitination by listerin/Ltn1 recruits the ATPase VCP (Cdc48 in yeast), and once the nascent chain has been released from the tRNA by ANKZF1 (Vms1 in yeast), VCP can deliver the polypeptide to the proteasome, where it is degraded.
The molecular signal for initiation of the RQC pathway involves collision of ribosomes which occurs when a trailing ribosome collides with a slowed-down or blocked leading ribosome. Such perturbations may be pathological in nature, but can also occur naturally. An example of the latter is when membrane domain insertion and/or folding fails. Decreasing the translation rate in response to inefficient folding of a transmembrane protein, can protect the cell from potentially toxic aberrant proteins (106). Collision creates a ‘disome’ with an extensive 40S–40S interface that can be recognized by different quality control factors (107–110). Recently, the very abundant EDF1 protein was reported to be the first factor to engage disomes (111,112). If collisions persist, EDF1 stabilizes the recruitment of the GIGYF2–4EHP complex and the E3 ubiquitin ligase ZNF598 (Hel2 in yeast). Both pathways aim to reduce collision events: GIGYF2–4EHP by preventing re-initiation on problematic messages, and ZNF598 by resolving the internally stalled ribosome and initiating RQC (101,113,114). ZNF598 can selectively recognize and ubiquitinate collided ribosomes, whereas GIGYF2–4EHP is dependent on EDF1 for recruitment (111). The longer stalling persist, the greater the likelihood of 40S ribosome ubiquitination by ZNF598 (110,115). It has been speculated that 40S ubiquitination signifies a ubiquitin code that distinguishes physiologically paused ribosomes from pathological structures (116). Indeed, a new study propose that the splitting factor ASCC irreversibly commits ribosomes to RQC by liberating the leading ribosome—a process that requires eS10 and uS10 ubiquitination by ZNF598 (101). In the absence of ASCC, or if the pathway is oversaturated, internally stalled ribosomes become substrates for endonuclease-mediated mRNA cleavage, likely via ubiquitin-dependent nuclease recruitment (109,117). These endonucleases sever the mRNA in between collided ribosomes, thus generating a 3′ end stall, which can be rescued by Pelota-Hbs1L-ABCE1. Consequently, absence of ubiquitination permits a certain degree of translation of aberrant mRNAs due to the lack of ASCC recruitment and inefficient mRNA cleavage (118,119).
RQC constitutes a cellular pathway for detecting and splitting stalled ribosomes, resolve the peptidyl-tRNA–60S complex and promote degradation of the nascent polypeptide chain (100,120). Resolution of the obstructed 60S subunit is initiated by NEMF (Rqc2/Tae2 in yeast), a protein that recognizes the exposed peptidyl-tRNA (121). Binding of NEMF prevents re-association of the two ribosomal subunits, as it occupies the 40S binding interphase (121). In addition, NEMF recruits the E3 ubiquitin ligase Listerin/LTN1 (Ltn1 in yeast) to ubiquitinate the nascent chain protruding from the 60S exit tunnel (118,122,123). In cases where the exposed peptide chain does not contain any lysines amenable for ubiquitination, non-canonical elongation with alanines and threonines is catalyzed by NEMF/Rqc2. This process, coined CAT-tailing, is likely to expose lysines from the part of the nascent chain that was buried within the 60S exit tunnel (124,125). Subsequently, ANKZF1 (Vms1 in yeast) releases the polypeptide from the conjugated tRNA. ANKZF1 was originally believed to be a tRNA hydrolase that attacks the peptide chain, but recent studies suggest that this enzyme instead processes the tRNA through its endonuclease activity (126,127). In the final step of RQC, the ubiquitinated nascent peptide is extracted from 60S through the ubiquitin segregase activity of p97/VCP and degraded by the proteasome (123,128).
The physiological importance of functional RQC is underscored by the fact that knockout of LTN1 is embryonically lethal. Mice carrying hypomorphic LTN1 mutations that support life suffer from early onset neurodegeneration (129). This phenotype is likely to be a consequence of compromised RQC, as mutation of the RQC factor GTPBP2, a binding partner of the ribosome recycling factor Pelota, also causes neurodegeneration in mice (130). It thus appears that RQC is an essential process that when impaired results in proteotoxic stress, including truncated and misfolded protein products, sequestration of inactivated 60S subunits, protein aggregate formation and organelle dysfunction. This will in turn contribute to proteome imbalance which is one of the hallmarks of neurodegeneration (100,131,132).
Integrated stress response
The integrated stress response is a convergence of several stress responses that respond to cellular imbalances (Figure 4). The ISR can be activated by four distinct molecular stress signals, each of which activates a specific protein kinase. Amino acid deficiency is sensed by general control non-depressible protein 2 (GCN2), heme deprivation by heme-regulated eIF2α kinase (HRI), viral infection by double stranded RNA-dependent protein kinase (PKR) and ER stress by PKR-like ER kinase (PERK) (133). Upon activation, all of these kinases phosphorylate and inhibit the function of eukaryotic translation initiation factor eIF2α (133). This phosphorylation event interferes with most forms of translation initiation, but allows the ribosome to selectively translate messages of critical stress-combatting genes, regulated by the transcription factor ATF4 (134,135). The ISR thus attempts to restore cellular homeostasis in the face of cell stress or biochemical imbalances. The ISR can also activate cell death pathways via selective translation of the pro-apoptotic transcription factors CHOP and ATF3 (133).
Figure 4.
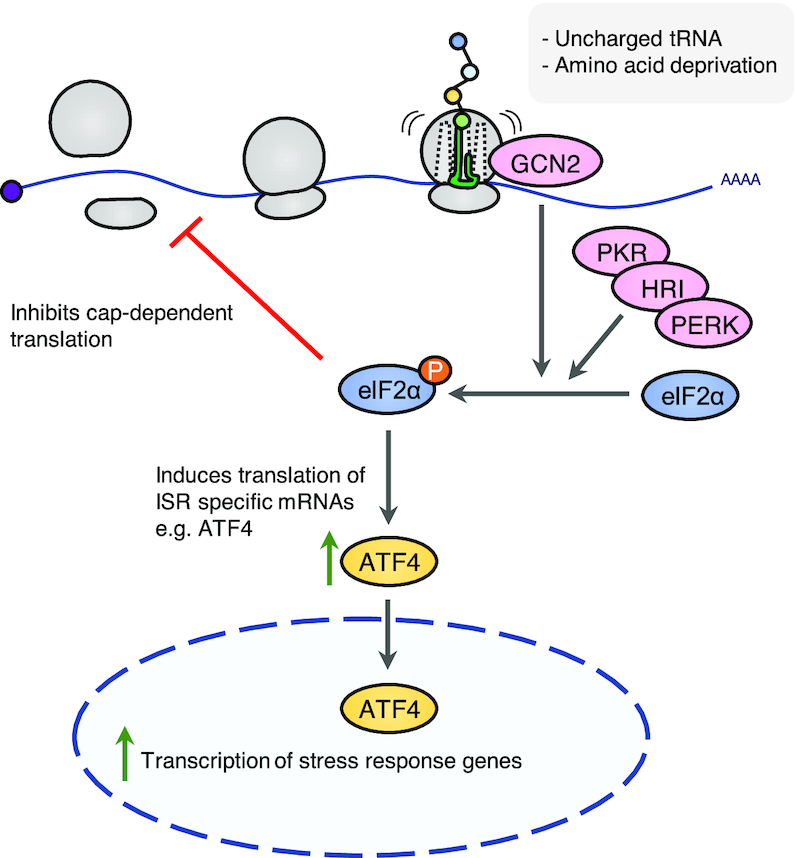
The integrated stress response. (A) The integrated stress response (ISR) converges on phosphorylation of eIF2α, resulting in global inhibition of cap-dependent mRNA translation. GCN2 is the relevant ISR-kinase upon translational arrest. Cap-independent pathways allow for the selective translation of ISR-specific mRNAs including ATF4. ATF4 is a transcription factor and effector of ISR that controls expression of stress response genes.
Shortage of amino acids and ribosome pausing are sensed by GCN2, and this is one of the triggers of the ISR (136,137). Earlier models of GCN2 activation centered around sensing of deacylated tRNAs caused by amino acid depletion. Such tRNAs were shown to be recognized by the regulatory HisRS-like domain of GCN2, located C-terminal to the kinase domain (138). In its inactive form, GCN2 is a dimer where the C-terminal segment and the HisRS-like domain engages in autoinhibitory interactions with the kinase domain in trans. Binding of uncharged tRNA to the HisRS-like domain in vitro relieves this inhibition and allows GCN2 to phosphorylate its main substrate eIF2α (139,140). In vivo studies in higher species have challenged this view and point towards alternative or complementary activation mechanisms for GCN2. Ishimura et al. observed that GCN2 is activated in brains of mice prone to widespread ribosomal stalling due to deficiency of just a single arginine tRNA isoacceptor and the ribosome rescue factor GTPBP2. The authors propose that ribosome stalling is sufficient to activate a GCN2-dependent ISR, independently of the presence of deacylated tRNAs (136).
Other recent studies lend further support and mechanistic insight into a ribosome-templated activation mechanism for GCN2 (141,142). Inglis et al. showed that GCN2 binds directly to ribosomes, an interaction that strongly stimulates GCN2 kinase activity in vitro. Binding involved both the pseudokinase domain, C-terminal Domain (CTD) and HisRS-like domains of GCN2, and was mediated by the ribosomal P-stalk. In eukaryotes, this flexible structure is a pentameric complex consisting of one subunit of the 60S ribosomal protein RPLP0 and two subunits of both RPLP1 and RPLP2. The P-stalk is situated adjacent to the ribosomal A-site and together with the SRL it forms the main ribosomal interaction site for translation elongation factors such as EEF1A and EEF2 (143). Studies in archaea show that the flexible acidic tails of P-stalk proteins RPLP1 and RPLP2 play an important role in capturing these elongation factors and direct them towards their ribosomal site of action (144,145). A potential role for the P-stalk was highlighted several years ago in yeast, where recombinant P-stalk complexes were shown to activate GCN2 in vitro (146). Besides corroborating that both intact ribosomes and the P-stalk activate GCN2 in vitro, Harding et al. also isolated mammalian ribosome-mutated cell lines that supported basal ribosome function, but did not allow for GCN2 activation by amino acid starvation in vivo. These mutations affected the P-stalk proteins, and the ability of these ribosomes to activate GCN2 in vitro was strongly attenuated (142). In spite of this recent insight, sensing of tRNAs may still play an important role during GCN2 activation, and it has not been clarified if the P-stalk carries a specific activating signal upon amino acid starvation or merely constitutes a structural platform on which GCN2 is activated by other signals. An attractive possibility is that the elongation factors compete with GCN2 for P-stalk binding, and thus represses GCN2 activation when the ribosome is actively translating mRNAs. This traffic is naturally interrupted by lack of amino acids and may expose the latent capacity of the P-stalk to bind and activate GCN2 (142).
GCN2 knockout mice are overall normal and present mainly with mild metabolic phenotypes, probably related to an altered response to nutrient availability. A physiological role of GCN2 in responding to amino acid deprivation was elegantly shown by Zhang et al., who mated GCN2 KO female mice with heterozygous males. The genotypes of the offspring followed the expected mendelian distribution, but under conditions mimicking amino acid starvation, significantly fewer knockout pups were born and these also had a higher perinatal morbidity (147). Dysregulation of ISR signaling has been linked to tumor progression (133,148,149), and therapeutic targeting of the ISR for cancer treatment has been proposed (150,151).
DISCUSSION
Protein translation is essential for cell survival and must be tightly controlled with respect to activity, selectivity and fidelity. The Ribotoxic Stress Response (RSR), the Ribosome-associated Quality Control (RQC) and the Integrated Stress Response (ISR) constitute a palette of surveillance pathways that monitor and respond to ribosomal dysfunction. Compared to the last two of these pathways, we know precious little about the physiological relevance of the RSR. Unlike RQC and ISR, the RSR pathway does not appear to be conserved from yeast, and the vast majority of studies on the subject address only mammalian cell lines exposed to exotic sources of ribotoxic stressors. These include ribotoxic enzymes and microbial ribosome inhibitors that cells are highly unlikely to become exposed to. It thus transpires that there must exist a more pertinent evolutionary rationale for the appearance of this stress response in multicellular organisms. Potential phenotypes of ZAK knockout mice have not yet been elucidated, and only rudimentary experiments in C. elegans has confirmed that this pathway supports survival and adaptation upon organismal stress (72). Resolving these issues remain pressing and is an obvious avenue for future research.
While the recent realization that ZAKα is a ribosome-binding and ribotoxic stress-sensing kinase is an important step forward (66,72), it also raises several unanswered questions. How can two sensing domains in a single protein accommodate the diverse structural conformations associated with the broad spectrum of ribotoxic lesions? And how are these interactions channeled into ZAKα kinase activation? The solution to this sensor conundrum does not necessarily lie in recognition of the actual lesions, but could equally well be dependent on interaction with a translation-relevant intermediate of the elongation cycle. In this scenario, ZAKα would only be competent for activation when bound to the ribosome, but kept inactive when satisfied by simultaneous probing of a non-pathological structure in the translation pocket. Recent work from the Green lab suggests that one direct, and potentially universal, activation signal for both RSR and ISR is brought about by collision of ribosomes and that a ‘disome’ structure is key for activation of ZAKα and GCN2 kinases (66) (Figure 5). In yeast, where the RSR pathway did not evolved, Gcn2 was also proposed to be activated by collision of ribosomes (66,152). This is a very interesting concept that unifies the activating signal for the three ribosomal stress responses dealt with in this review. It also suggests that RQC, RSR and ISR are physiologically interconnected, and represents a three-pronged defence against common and stochastic translational perturbations. At present, this model does not fully explain the lack of direct correlation between the potency of ribotoxic stressors with respect to inhibiting translation and activating the RSR. Nor can it account for the strongly biased activation of the RSR vs. the other stress signaling pathways by specific translation-perturbing agents (72). An attractive possibility is that collided ribosomes can present themselves in several forms that are differentially sensed by sensing factors. An alternative explanation that would satisfy the collision model is that ZAKα simultaneously monitors disome formation and other signals associated with translational arrest. Thus, while ZAKα appears to be the universal sensor of ribotoxic stress, the exact identity and full scope of ribosomal signal(s) that it senses, and how the recognition of these signals translates into kinase activation, is not clear at present.
Figure 5.

Sensing mechanisms and outcomes of ribosomal stress-surveillance pathways. Local translational arrest leads to collision of ribosomes, which is a signal for recruitment and activation of the RQC pathway sensor ZNF598. The RSR sensor ZAKα and the ISR sensor GCN2 may also be activated by recognition of this structure, or alternatively by sensing signals directly on stalled ribosomes. Once activated, the RQC pathway will attempt to rescue and recycle stalled ribosomes. The ISR pathway activates stress responses by inhibiting cap-dependent translation and facilitating selective translation of stress response proteins. The RSR pathway activates MAP kinase-driven stress responses and inflammatory signaling and may also mediate apoptotic signaling.
CONCLUDING REMARKS AND FUTURE PERSPECTIVE
We have reviewed the current knowledge on ribosomal surveillance systems with an emphasis of the roles and underlying mechanisms of RSR-associated MAP kinase signaling. Among the pathways described (RSR, RQC and ISR), our knowledge of the significance of the RSR is lacking behind. Future work is required to elucidate the physiologically relevant signals that activate this pathway and determine the full scope of its physiological roles. Given the already established connections between dysregulated RSR and pathology, such knowledge is likely to inform new therapeutic strategies for combating a range of human diseases.
Contributor Information
Anna Constance Vind, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, Blegdamsvej 3B, DK-2200 Copenhagen, Denmark.
Aitana Victoria Genzor, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, Blegdamsvej 3B, DK-2200 Copenhagen, Denmark.
Simon Bekker-Jensen, Center for Healthy Aging, Department of Cellular and Molecular Medicine, University of Copenhagen, Blegdamsvej 3B, DK-2200 Copenhagen, Denmark.
FUNDING
Lundbeck Foundation [R190-2014-4037]; NEYE Foundation; Danish Medical Research Council [9039-00007B]; European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program [863911-PHYRIST]. Funding for open access charge: Lundbeck Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cargnello M., Roux P.P.. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011; 75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wagner E.F., Nebreda Á.R.. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer. 2009; 9:537–549. [DOI] [PubMed] [Google Scholar]
- 3. Dong C., Davis R.J., Flavell R.A.. Map kinases in the immune response. Annu. Rev. Immunol. 2002; 20:55–72. [DOI] [PubMed] [Google Scholar]
- 4. Mazzitelli S., Xu P., Ferrer I., Davis R.J., Tournier C.. The loss of c-Jun N-terminal protein kinase activity prevents the amyloidogenic cleavage of amyloid precursor protein and the formation of amyloid plaques in vivo. J. Neurosci. 2011; 31:16969–16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Funding A.T., Johansen C., Kragballe K., Iversen L.. Mitogen- and stress-activated protein kinase 2 and cyclic AMP response element binding protein are activated in lesional psoriatic epidermis. J. Invest. Dermatol. 2007; 127:2012–2019. [DOI] [PubMed] [Google Scholar]
- 6. Hotamisligil G.S., Davis R.J.. Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 2016; 8:a006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cuenda A., Rousseau S.. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta. 2007; 1773:1358–1375. [DOI] [PubMed] [Google Scholar]
- 8. Tamura K., Sudo T., Senftleben U., Dadak A.M., Johnson R., Karin M.. Requirement for p38α in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000; 102:221–231. [DOI] [PubMed] [Google Scholar]
- 9. Adams R.H., Porras A., Alonso G., Jones M., Vintersten K., Panelli S., Valladares A., Perez L., Klein R., Nebreda A.R.. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell. 2000; 6:109–116. [PubMed] [Google Scholar]
- 10. Allen M., Svensson L., Roach M., Hambor J., McNeish J., Gabel C.A.. Deficiency of the stress kinase p38α results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J. Exp. Med. 2000; 191:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pogozelski A.R., Geng T., Li P., Yin X., Lira V.A., Zhang M., Chi J.T., Yan Z.. P38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One. 2009; 4:e7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sumara G., Formentini I., Collins S., Sumara I., Windak R., Bodenmiller B., Ramracheya R., Caille D., Jiang H., Platt K.A. et al.. Regulation of PKD by the MAPK p38δ in insulin secretion and glucose homeostasis. Cell. 2009; 136:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dérijard B., Raingeaud J., Barrett T., Wu I., Han J., Ulevitch R.J., Davis R.J., Derijard B., Raingeaud J., Barrett T. et al.. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995; 267:682–685. [DOI] [PubMed] [Google Scholar]
- 14. Raingeaud J., Whitmarsh A.J., Barrett T., Dérijard B., Davis R.J.. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 1996; 16:1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han J., Lee J.D., Jiang Y., Li Z., Feng L., Ulevitch R.J.. Characterization of the structure and function of a novel MAP kinase kinase (MKKG). J. Biol. Chem. 1996; 271:2886–2891. [DOI] [PubMed] [Google Scholar]
- 16. Brancho D., Tanaka N., Jaeschke A., Ventura J.J., Kelkar N., Tanaka Y., Kyuuma M., Takeshita T., Flavell R.A., Davis R.J.. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003; 17:1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka N., Kamanaka M., Enslen H., Dong C., Wysk M., Davis R.J., Flavell R.A.. Differential involvement of p38 mitogen-activated protein kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep. 2002; 3:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu H.-T. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3(Mkk3)-deficient mice. EMBO J. 1999; 18:1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wysk M., Yang D.D., Lu H.T., Flavell R.A., Davis R.J.. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:3763–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morrison D.K. MAP kinase pathways. Cold spring Harb. Perspect. Biol. 2012; 4:a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gallo K.A., Johnson G.L.. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 2002; 3:663–672. [DOI] [PubMed] [Google Scholar]
- 22. Ge B., Gram H., Di Padova F., Huang B., New L., Ulevitch R.J., Luo Y., Han J.. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science (80-.). 2002; 295:1291–1294. [DOI] [PubMed] [Google Scholar]
- 23. Lee J.C., Laydon J.T., McDonnell P.C., Gallagher T.F., Kumar S., Green D., McNulty D., Blumenthal M.J., Keys J.R., Land Vatter S.W. et al.. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994; 372:739–746. [DOI] [PubMed] [Google Scholar]
- 24. Borisova M.E., Voigt A., Tollenaere M.A.X., Sahu S.K., Juretschke T., Kreim N., Mailand N., Choudhary C., Bekker-Jensen S., Akutsu M. et al.. P38-MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage. Nat. Commun. 2018; 9:1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goedert M., Cuenda A., Craxton M., Jakes R., Cohen P.. Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); Comparison of its substrate specificity with that of other SAP kinases. EMBO J. 1997; 16:3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuenda A., Cohen P., Buée-Scherrer V., Goedert M.. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); Comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J. 1997; 16:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuma Y., Sabio G., Bain J., Shpiro N., Márquez R., Cuenda A.. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005; 280:19472–19479. [DOI] [PubMed] [Google Scholar]
- 28. Gupta S., Barrett T., Whitmarsh A.J., Cavanagh J., Sluss H.K., Dérijard B., Davis R.J.. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996; 15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 29. Yang D.D., Kuan C.-Y., Whitmarsh A.J., Rinco M., Zheng T.S., Davis R.J., Rakic P., Flavell R.A.. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997; 389:865–870. [DOI] [PubMed] [Google Scholar]
- 30. Sabapathy K., Jochum W., Hochedlinger K., Chang L., Karin M., Wagner E.F.. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech. Dev. 1999; 89:115–124. [DOI] [PubMed] [Google Scholar]
- 31. Kuan C.Y., Yang D.D., Samanta Roy D.R., Davis R.J., Rakic P., Flavell R.A.. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999; 22:667–676. [DOI] [PubMed] [Google Scholar]
- 32. Tournier C., Dong C., Turner T.K., Jones S.N., Flavell R.A., Davis R.J.. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001; 15:1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ganiatsas S., Kwee L., Fujiwara Y., Perkins A., Ikeda T., Labow M.A., Zon L.I.. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:6881–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nishina H., Vaz C., Billia P., Nghiem M., Sasaki T., De la Pompa J.L., Furlonger K., Paige C., Hui C.C., Fischer K.D. et al.. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK1/MKK4. Development. 1999; 126:505–516. [DOI] [PubMed] [Google Scholar]
- 35. Dong C., Yang D.D., Tournler C., Whitmarsh A.J., Xu J., Davis R.J., Flavell R.A.. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000; 405:91–94. [DOI] [PubMed] [Google Scholar]
- 36. Coffey E.T., Hongisto V., Dickens M., Davis R.J., Courtney M.J.. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J. Neurosci. 2000; 20:7602–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X., Destrument A., Tournier C.. Physiological roles of MKK4 and MKK7: Insights from animal models. Biochim. Biophys. Acta - Mol. Cell Res. 2007; 1773:1349–1357. [DOI] [PubMed] [Google Scholar]
- 38. Wada T., Joza N., Cheng H.M., Sasaki T., Kozieradzki I., Bachmaier K., Katada T., Schreiber M., Wagner E.F., Nishina H. et al.. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat. Cell Biol. 2004; 6:215–226. [DOI] [PubMed] [Google Scholar]
- 39. Yang D., Tournier C., Wysk M., Lu H.T., Xu J., Davis R.J., Flavell R.A.. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yasuda J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999; 19:7245–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hibi M., Lin A., Smeal T., Minden A., Karin M.. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993; 7:2135–2148. [DOI] [PubMed] [Google Scholar]
- 42. Dérijard B., Hibi M., Wu I.H., Barrett T., Su B., Deng T., Karin M., Davis R.J.. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994; 76:1025–1037. [DOI] [PubMed] [Google Scholar]
- 43. Lamb J.A., Ventura J.J., Hess P., Flavell R.A., Davis R.J.. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell. 2003; 11:1479–1489. [DOI] [PubMed] [Google Scholar]
- 44. Ventura J.-J., Kennedy N.J., Lamb J.A., Flavell R.A., Davis R.J.. c-Jun NH2-terminal kinase is essential for the regulation of AP-1 by tumor necrosis factor. Mol. Cell. Biol. 2003; 23:2871–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Read M.A., Whitley M.Z., Gupta S., Pierce J.W., Best J., Davis R.J., Collins T.. Tumor necrosis factor α-induced E-selectin expression is activated by the nuclear factor-κB and c-JUN N-terminal kinase/p38 mitogen-activated protein kinase pathways. J. Biol. Chem. 1997; 272:2753–2761. [DOI] [PubMed] [Google Scholar]
- 46. Zeke A., Misheva M., Reményi A., Bogoyevitch M.A.. JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev. 2016; 80:793–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iordanov M.S., Pribnow D., Magun J.L., Dinh T., Pearson J.A., Li-ye Chen S., Magun B.E.. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the a-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997; 17:3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thorpe C.M., Hurley B.P., Lincicome L.L., Jacewicz M.S., Keusch G.T., Acheson D.W.K.. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999; 67:5985–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith W.E., Kane A.V., Campbell S.T., Acheson D.W.K., Cochran B.H., Thorpe C.M.. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003; 71:1497–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foster G.H., Tesh V.L.. Shiga toxin 1-induced activation of c-Jun NH2-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-α. J. Leukoc. Biol. 2002; 71:107–114. [PubMed] [Google Scholar]
- 51. Korcheva V., Wong J., Corless C., Iordanov M., Magun B.. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and p38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am. J. Pathol. 2005; 166:323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garreau de Loubresse N., Prokhorova I., Holtkamp W., Rodnina M.V., Yusupova G., Yusupov M.. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014; 513:517–522. [DOI] [PubMed] [Google Scholar]
- 53. Endo Y., Tsurugi K.. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988; 263:8735–8739. [PubMed] [Google Scholar]
- 54. McCluskey A.J., Poon G.M.K., Bolewska-Pedyczak E., Srikumar T., Jeram S.M., Raught B., Gariépy J.. The catalytic subunit of shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol. 2008; 378:375–386. [DOI] [PubMed] [Google Scholar]
- 55. Gao Y.-G., Selmer M., Dunham C.M., Weixlbaumer A., Kelley A.C., Ramakrishnan V.. The structure of the ribosome with elongation factor G trapped in the post-translocational state. Science. 2009; 326:694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmeing T.M., Voorhees R.M., Kelley A.C., Gao Y.-G., IV F.V.M., Weir J.R., Ramakrishnan V.. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009; 326:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Voorhees R.M., Schmeing T.M., Kelley A.C., Ramakrishnan V.. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010; 330:835–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Elsea C.R., Roberts D.A., Druker B.J., Wood L.J.. Inhibition of p38 MAPK suppresses inflammatory cytokine induction by etoposide, 5-fluorouracil, and doxorubicin without affecting tumoricidal activity. PLoS One. 2008; 3:e2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wood L.J., Nail L.M., Gilster A., Winters K.A., Elsea C.R.. Cancer chemotherapy-related symptoms: evidence to suggest a role for proinflammatory cytokines. Oncol. Nurs. Forum. 2006; 33:535–542. [DOI] [PubMed] [Google Scholar]
- 60. Kang Y.J., Zhou Z.X., Wang G.W., Buridi A., Klein J.B.. Suppression by metallothionein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J. Biol. Chem. 2000; 275:13690–13698. [DOI] [PubMed] [Google Scholar]
- 61. Sauter K.A.D., Magun E.A., Iordanov M.S., Magun B.E.. ZAK is required for doxorubicin, a novel ribotoxic stressor, to induce SAPK activation and apoptosis in HaCaT cells. Cancer Biol. Ther. 2010; 10:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee K.H., Nishimura S., Matsunaga S., Fusetani N., Horinouchi S., Yoshida M.. Inhibition of protein synthesis and activation of stress-activated protein kinases by onnamide A and theopederin B, antitumor marine natural products. Cancer Sci. 2005; 96:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Iordanov M.S., Pribnow D., Magun J.L., Dinh T.H., Pearson J.A., Magun B.E.. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 1998; 273:15794–15803. [DOI] [PubMed] [Google Scholar]
- 64. Devary Y., Rosette C., DiDonato J.A., Karin M.. NF-κB activation by ultraviolet light not dependent on a nuclear signal. Science. 1993; 261:1442–1445. [DOI] [PubMed] [Google Scholar]
- 65. Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M.. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005; 120:649–661. [DOI] [PubMed] [Google Scholar]
- 66. Wu C.C., Peterson A., Zinshteyn B., Regot S., Green R.. Ribosome collisions trigger general stress responses to regulate cell fate. Cell. 2020; 182:404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhou H.-R., He K., Landgraf J., Pan X., Pestka J.J.. Direct activation of ribosome-associated double-stranded RNA-dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: a new model for ribotoxic stress response induction. Toxins (Basel). 2014; 6:3406–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou H.-R., Lau A.S., Pestka J.J.. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003; 74:335–344. [DOI] [PubMed] [Google Scholar]
- 69. Zhou H.R., Jia Q., Pestka J.J.. Ribotoxic stress response to the trichothecene deoxynivalenol in the macrophage involves the Src family kinase Hck. Toxicol. Sci. 2005; 85:916–926. [DOI] [PubMed] [Google Scholar]
- 70. Wang X., Mader M.M., Toth J.E., Yu X., Jin N., Campbell R.M., Smallwood J.K., Christe M.E., Chatterjee A., Goodson T. et al.. Complete inhibition of anisomycin and UV radiation but not cytokine induced JNK and p38 activation by an aryl-substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J. Biol. Chem. 2005; 280:19298–19305. [DOI] [PubMed] [Google Scholar]
- 71. Jandhyala D.M., Ahluwalia A., Obrig T., Thorpe C.M.. ZAK: a MAP3 kinase that transduces Shiga toxin‐ and ricin‐induced proinflammatory cytokine expression. Cell. Microbiol. 2008; 10:1468–1477. [DOI] [PubMed] [Google Scholar]
- 72. Vind A.C., Snieckute G., Blasius M., Tiedje C., Krogh N., Bekker-Jensen D.B., Andersen K.L., Nordgaard C., Tollenaere M.A.X., Lund A.H. et al.. ZAKα recognizes stalled ribosomes through partially redundant sensor domains. Mol. Cell. 2020; 78:700–713. [DOI] [PubMed] [Google Scholar]
- 73. Goh K.C., DeVeer Mi., Williams R.G.. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000; 19:4292–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu T.C., Huang C., Chu Y., Wei C., Chou C., Chou M., C Y.. Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-[alpha] motif. Biochem. Biophys. Res. Commun. 2000; 274:811–816. [DOI] [PubMed] [Google Scholar]
- 75. Gotoh I., Adachi M., Nishida E.. Identification and characterization of a novel MAP kinase kinase kinase, MLTK*. J. Biol. Chem. 2001; 276:4276–4286. [DOI] [PubMed] [Google Scholar]
- 76. Vin H., Ching G., Ojeda S.S., Adelmann C.H., Chitsazzadeh V., Dwyer D.W., Ma H., Ehrenreiter K., Baccarini M., Ruggieri R. et al.. Sorafenib suppresses JNK-dependent apoptosis through inhibition of ZAK. Mol. Cancer Ther. 2013; 13:221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jandhyala D., Wong J., Mantis N., Magun B., Leong J., Thorpe C.. A novel Zak knockout mouse with a defective ribotoxic stress response. Toxins (Basel). 2016; 8:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Saenz J.B., Li J., Haslam D.B.. The MAP kinase-activated protein kinase 2 (MK2) contributes to the Shiga toxin-induced inflammatory response. Cell. Microbiol. 2010; 12:516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Spielmann M., Kakar N., Tayebi N., Leettola C., Nürnberg G., Sowada N., Lupiáñez D.G., Harabula I., Flöttmann R., Horn D. et al.. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res. 2016; 26:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee M.S., Cherla R.P., Leyva-Illades D., Tesh V.L.. Bcl-2 regulates the onset of shiga toxin 1-induced apoptosis in THP-1 cells. Infect. Immun. 2009; 77:5233–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee M.S., Koo S., Jeong D.G., Tesh V.L.. Shiga toxins as multi-functional proteins: induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins (Basel). 2016; 8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Deng X., Xiao L., Lang W., Gao F., Ruvolo P., May W.S.. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001; 276:23681–23688. [DOI] [PubMed] [Google Scholar]
- 83. Stone S.M., Thorpe C.M., Ahluwalia A., Rogers A.B., Obata F., Vozenilek A., Kolling G.L., Kane A.V., Magun B.E., Jandhyala D.M.. Shiga toxin 2-induced intestinal pathology in infant rabbits is A-subunit dependent and responsive to the tyrosine kinase and potential ZAK inhibitor imatinib. Front. Cell. Infect. Microbiol. 2012; 2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hall G., Kurosawa S., Stearns-Kurosawa D.J.. Shiga toxin therapeutics: beyond neutralization. Toxins (Basel). 2017; 9:291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Barrett T.J., Potter M.E., Strockbine N.A.. Evidence for participation of the macrophage in Shiga-like toxin II-induced lethality in mice. Microb. Pathog. 1990; 9:95–103. [DOI] [PubMed] [Google Scholar]
- 86. Lentz E.K., Cherla R.P., Jaspers V., Weeks B.R., Tesh V.L.. Role of tumor necrosis factor alpha in disease using a mouse model of Shiga toxin-mediated renal damage. Infect. Immun. 2010; 78:3689–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jandhyala D.M., Rogers T.J., Kane A., Paton A.W., Paton J.C., Thorpe C.M.. Shiga toxin 2 and flagellin from Shiga-toxigenic Escherichia coli superinduce interleukin-8 through synergistic effects on host stress-activated protein kinase activation. Infect. Immun. 2010; 78:2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Leyva-Illades D., Cherla R.P., Lee M.S., Tesh V.L.. Regulation of cytokine and chemokine expression by the ribotoxic stress response elicited by shiga toxin type 1 in human macrophage-like THP-1 cells. Infect. Immun. 2012; 80:2109–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009; 21:317–324. [DOI] [PubMed] [Google Scholar]
- 90. Coulthard L.R., White D.E., Jones D.L., Mcdermott M.F., Burchill S.A.. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009; 15:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Loomba R., Lawitz E., Mantry P.S., Jayakumar S., Caldwell S.H., Arnold H., Diehl A.M., Djedjos C.S., Han L., Myers R.P. et al.. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. 2018; 67:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Karpman D., Loos S., Tati R., Arvidsson I.. Haemolytic uraemic syndrome. J. Intern. Med. 2017; 281:123–148. [DOI] [PubMed] [Google Scholar]
- 93. Zhu W., Zou Y., Aikawa R., Harada K., Kudoh S., Uozumi H., Hayashi D., Gu Y., Yamazaki T., Nagai R. et al.. MAPK superfamily plays an important role in daunomycin-induced apoptosis of cardiac myocytes. Circulation. 1999; 100:2100–2107. [DOI] [PubMed] [Google Scholar]
- 94. Wong J., Smith L.B., Magun E.A., Engstrom T., Kelley-Howard K., Jandhyala D.M., Thorpe C.M., Magun B.E., Wood L.J.. Small molecule kinase inhibitors block the ZAK-dependent inflammatory effects of Doxorubicin. Cancer Biol. Ther. 2013; 14:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Warner W.G., Wei R.R.. In vitro photooxidation of nucleic acids by ultraviolet A radiation. Photochem. Photobiol. 1997; 65:560–563. [DOI] [PubMed] [Google Scholar]
- 96. Autier J., Escudier B., Wechsler J., Spatz A., Robert C.. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch. Dermatol. 2008; 144:886–892. [DOI] [PubMed] [Google Scholar]
- 97. Arnault J.P., Mateus C., Escudier B., Tomasic G., Wechsler J., Hollville E., Soria J.C., Malka D., Sarasin A., Larcher M. et al.. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin. Cancer Res. 2012; 18:263–272. [DOI] [PubMed] [Google Scholar]
- 98. Schuller A.P., Green R.. Roadblocks and resolutions in eukaryotic translation. Nat. Rev. Mol. Cell Biol. 2018; 19:526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yan L.L., Zaher H.S.. How do cells cope with RNA damage and its consequences. J. Biol. Chem. 2019; 294:15158–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Joazeiro C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019; 20:368–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Juszkiewicz S., Speldewinde S.H., Wan L., Svejstrup J.Q., Hegde R.S.. The ASC-1 complex disassembles collided ribosomes. Mol. Cell. 2020; 79:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Matsuo Y., Tesina P., Nakajima S., Mizuno M., Endo A., Buschauer R., Cheng J., Shounai O., Ikeuchi K., Saeki Y. et al.. RQT complex dissociates ribosomes collided on endogenous RQC substrate SDD1. Nat. Struct. Mol. Biol. 2020; 27:323–332. [DOI] [PubMed] [Google Scholar]
- 103. Nürenberg‐Goloub E., Kratzat H., Heinemann H., Heuer A., Kötter P., Berninghausen O., Becker T., Tampé R., Beckmann R.. Molecular analysis of the ribosome recycling factor ABCE 1 bound to the 30S post‐splitting complex. EMBO J. 2020; 39:e103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kiosze-Becker K., Ori A., Gerovac M., Heuer A., Nürenberg-Goloub E., Rashid U.J., Becker T., Beckmann R., Beck M., Tampé R.. Structure of the ribosome post-recycling complex probed by chemical cross-linking and mass spectrometry. Nat. Commun. 2016; 7:13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shao S., Von der Malsburg K., Hegde R.S.. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol. Cell. 2013; 50:637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lakshminarayan R., Phillips B.P., Binnian I.L., Gomez-Navarro N., Escudero-Urquijo N., Warren A.J., Miller E.A.. Pre-emptive quality control of a misfolded membrane protein by ribosome-driven effects. Curr. Biol. 2020; 30:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Matsuo Y., Ikeuchi K., Saeki Y., Iwasaki S., Schmidt C., Udagawa T., Sato F., Tsuchiya H., Becker T., Tanaka K. et al.. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun. 2017; 8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Simms C.L., Yan L.L., Zaher H.S.. Ribosome collision Is critical for quality control during no-go decay. Mol. Cell. 2017; 68:361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ikeuchi K., Tesina P., Matsuo Y., Sugiyama T., Cheng J., Saeki Y., Tanaka K., Becker T., Beckmann R., Inada T.. Collided ribosomes form a unique structural interface to induce Hel2‐driven quality control pathways. EMBO J. 2019; 38:e100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Juszkiewicz S., Chandrasekaran V., Lin Z., Kraatz S., Ramakrishnan V., Hegde R.S.. ZNF598 is a quality control sensor of collided ribosomes. Mol. Cell. 2018; 72:469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Juszkiewicz S., Slodkowicz G., Lin Z., Freire-Pritchett P., Peak-Chew S.Y., Hegde R.S.. Ribosome collisions trigger cis-acting feedback inhibition of translation initiation. Elife. 2020; 9:e60038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sinha N.K., Ordureau A., Best K.M., Saba J.A., Zinshteyn B., Sundaramoorthy E., Fulzele A., Garshott D.M., Denk T., Thoms M. et al.. EDF1 coordinates cellular responses to ribosome collisions. Elife. 2020; 9:e58828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hickey K., Dickson K., Cogan Z., Replogle J., Schoof M., D’Orazio K., Sinha N., Frost A., Green R., Kostova K. et al.. GIGYF2 and 4EHP inhibit translation initiation of defective messenger RNAs to assist ribosome-associated quality control. Mol. Cell. 2019; doi:10.1101/792994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tollenaere M.A.X., Tiedje C., Rasmussen S., Nielsen J.C., Vind A.C., Blasius M., Batth T.S., Mailand N., Olsen J.V., Gaestel M. et al.. GIGYF1/2-driven cooperation between ZNF598 and TTP in posttranscriptional regulation of inflammatory signaling. Cell Rep. 2019; 26:3511–3521. [DOI] [PubMed] [Google Scholar]
- 115. Sundaramoorthy E., Leonard M., Mak R., Liao J., Fulzele A., Bennett E.J.. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol. Cell. 2017; 65:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Garshott D.M., Sundaramoorthy E., Leonard M., Bennett E.J.. Distinct regulatory ribosomal ubiquitylation events are reversible and hierarchically organized. Elife. 2020; 9:e54023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. D’Orazio K.N., Wu C.C.-C., Sinha N., Loll-Krippleber R., Brown G.W., Green R.. The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. Elife. 2019; 8:e49117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Brandman O., Stewart-Ornstein J., Wong D., Larson A., Williams C.C., Li G.W., Zhou S., King D., Shen P.S., Weibezahn J. et al.. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012; 151:1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Juszkiewicz S., Hegde R.S.. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol. Cell. 2017; 65:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Brandman O., Hegde R.S.. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016; 23:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Shao S., Brown A., Santhanam B., Hegde R.S.. Structure and assembly pathway of the ribosome quality control complex. Mol. Cell. 2015; 57:433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bengtson M.H., Joazeiro C.A.P.. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010; 467:470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Defenouillère Q., Yao Y., Mouaikel J., Namane A., Galopier A., Decourty L., Doyen A., Malabat C., Saveanu C., Jacquier A. et al.. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl Acad. Sci. U.S.A. 2013; 110:5046–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Shen P.S., Park J., Qin Y., Li X., Parsawar K., Larson M.H., Cox J., Cheng Y., Lambowitz A.M., Weissman J.S. et al.. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science. 2015; 347:75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zurita Rendón O., Fredrickson E.K., Howard C.J., Van Vranken J., Fogarty S., Tolley N.D., Kalia R., Osuna B.A., Shen P.S., Hill C.P. et al.. Vms1p is a release factor for the ribosome-associated quality control complex. Nat. Commun. 2018; 9:2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kuroha K., Zinoviev A., Hellen C.U.T., Pestova T.V.. Release of ubiquitinated and non-ubiquitinated Nascent chains from stalled mammalian ribosomal complexes by ANKZF1 and Ptrh1. Mol. Cell. 2018; 72:286–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Su T., Izawa T., Thoms M., Yamashita Y., Cheng J., Berninghausen O., Hartl F.U., Inada T., Neupert W., Beckmann R.. Structure and function of Vms1 and Arb1 in RQC and mitochondrial proteome homeostasis. Nature. 2019; 570:538–542. [DOI] [PubMed] [Google Scholar]
- 128. Verma R., Oania R.S., Kolawa N.J., Deshaies R.J.. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife. 2013; 2:e00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chu J., Hong N.A., Masuda C.A., Jenkins B.V., Nelms K.A., Goodnow C.C., Glynne R.J., Wu H., Masliah E., Joazeiro C.A.P. et al.. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:2097–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ishimura R., Nagy G., Dotu I., Zhou H., Yang X.L., Schimmel P., Senju S., Nishimura Y., Chuang J.H., Ackerman S.L.. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014; 345:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Choe Y.J., Park S.H., Hassemer T., Körner R., Vincenz-Donnelly L., Hayer-Hartl M., Hartl F.U.. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 2016; 531:191–195. [DOI] [PubMed] [Google Scholar]
- 132. Sweeney P., Park H., Baumann M., Dunlop J., Frydman J., Kopito R., McCampbell A., Leblanc G., Venkateswaran A., Nurmi A. et al.. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017; 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pakos‐Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M.. The integrated stress response. EMBO Rep. 2016; 17:1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Harding H.P., Zhang Y., Zeng H., Novoa I., Lu P.D., Calfon M., Sadri N., Yun C., Popko B., Paules R. et al.. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003; 11:619–633. [DOI] [PubMed] [Google Scholar]
- 135. Lu P.D., Harding H.P., Ron D.. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004; 167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ishimura R., Nagy G., Dotu I., Chuang J.H., Ackerman S.L.. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife. 2016; 5:e14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Darnell A.M., Subramaniam A.R., O’Shea E.K.. Translational control through differential ribosome pausing during amino acid limitation in mammalian cells Alicia. Mol. Cell. 2018; 71:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wek R.C., Jackson B.M., Hinnebusch A.G.. Juxtaposition of domains homologous to protein kinase and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:4579–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Dong J., Qiu H., Garcia-Barrio M., Anderson J., Hinnebusch A.G.. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell. 2000; 6:269–279. [DOI] [PubMed] [Google Scholar]
- 140. Wek S.A., Zhu S., Wek R.C.. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol. 1995; 15:4497–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Inglis A.J., Masson G.R., Shao S., Perisic O., McLaughlin S.H., Hegde R.S., Williams R.L.. Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:4946–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Harding H.P., Ordonez A., Allen F., Parts L., Inglis A.J., Williams R.L., Ron D.. The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. Elife. 2019; 8:e50149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gonzalo P., Reboud J.P.. The puzzling lateral flexible stalk of the ribosome. Biol. Cell. 2003; 95:179–193. [DOI] [PubMed] [Google Scholar]
- 144. Ito K., Honda T., Suzuki T., Miyoshi T., Murakami R., Yao M., Uchiumi T.. Molecular insights into the interaction of the ribosomal stalk protein with elongation factor 1α. Nucleic Acids Res. 2014; 42:14042–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Nomura N., Honda T., Baba K., Naganuma T., Tanzawa T., Arisaka F., Noda M., Uchiyama S., Tanaka I., Yao M. et al.. Archaeal ribosomal stalk protein interacts with translation factors in a nucleotide-independent manner via its conserved C terminus. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:3748–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Jiménez-Díaz A., Remacha M., Ballesta J.P.G., Berlanga J.J.. Phosphorylation of initiation factor eIF2 in response to stress conditions is mediated by acidic ribosomal P1/P2 proteins in saccharomyces cerevisiae. PLoS One. 2013; 8:e84219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhang P., McGrath B.C., Reinert J., Olsen D.S., Lei L., Gill S., Wek S.A., Vattem K.M., Wek R.C., Kimball S.R. et al.. The GCN2 eIF2α Kinase Is Required for Adaptation to Amino Acid Deprivation in Mice. Mol. Cell. Biol. 2002; 22:6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Denoyelle C., Abou-Rjaily G., Bezrookove V., Verhaegen M., Johnson T.M., Fullen D.R., Pointer J.N., Gruber S.B., Su L.D., Nikiforov M.A. et al.. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006; 8:1053–1063. [DOI] [PubMed] [Google Scholar]
- 149. Hart L.S., Cunningham J.T., Datta T., Dey S., Tameire F., Lehman S.L., Qiu B., Zhang H., Cerniglia G., Bi M. et al.. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 2012; 122:4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kline C.L.B., Heuvel A.P.J.V, Allen J.E., Prabhu V.V., Dicker D.T., El-Deiry W.S.. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci. Signal. 2016; 9:ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Axten J.M., Romeril S.P., Shu A., Ralph J., Medina J.R., Feng Y., Li W.H.H., Grant S.W., Heerding D.A., Minthorn E. et al.. Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 2013; 4:964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Meydan S., Guydosh N.R.. Disome and trisome profiling reveal genome-wide targets of ribosome quality control article disome and trisome profiling reveal genome-wide targets of ribosome quality control. Mol. Cell. 2020; 79:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Shifrin V.I., Anderson P.. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999; 274:13985–13992. [DOI] [PubMed] [Google Scholar]
- 155. Endo Y., Tsurugi K., Yutsudo T., Takeda Y., Ogasawara T., Igarashi K.. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes: RNA N‐glycosidase activity of the toxins. Eur. J. Biochem. 1988; 171:45–50. [DOI] [PubMed] [Google Scholar]