

Abstract
Repair of DNA double strand breaks (DSB) is performed by two major pathways: homology-dependent repair and classical non-homologous end joining. Recent studies have identified a third pathway: microhomology-mediated end-joining (MMEJ). MMEJ has similarities to homology-dependent repair in that repair is initiated with end resection, leading to single-stranded 3’ ends which require microhomology upstream and downstream of the DSB. Importantly, the MMEJ pathway is commonly upregulated in cancers, especially in homologous recombination-deficient cancers which display a distinctive mutational signature. Here we review the molecular process of MMEJ as well as new targets and approaches exploiting the MMEJ pathway in cancer therapy.
Keywords: MMEJ, DNA repair, cancer, POLQ
INTRODUCTION
DNA Damage and Double-Stranded Breaks
Maintenance of genome integrity is of upmost importance for cellular survival (1). Loss of genomic integrity results in permanent changes to DNA sequence and is the source of many human diseases, notably cancer (2–7). Genome integrity is achieved by DNA repair pathways, collectively known as the DNA damage response (DDR) (8). Double-stranded breaks (DSBs) are the most deleterious form of DNA damage (1). DSBs are generated either exogenously, for example, from exposure to ionizing radiation, or endogenously, for example, from reactive oxygen species secondary to aerobic metabolism or from errors during DNA replication and meiosis (9–11). DSBs were traditionally thought to be repaired by either of two distinct pathways, distinguished by the presence or absence of homology: homologous recombination repair (HR) and classical non-homologous end-joining (NHEJ), respectively (Figures 1A–B).
FIGURE 1: DSB Repair Pathways.
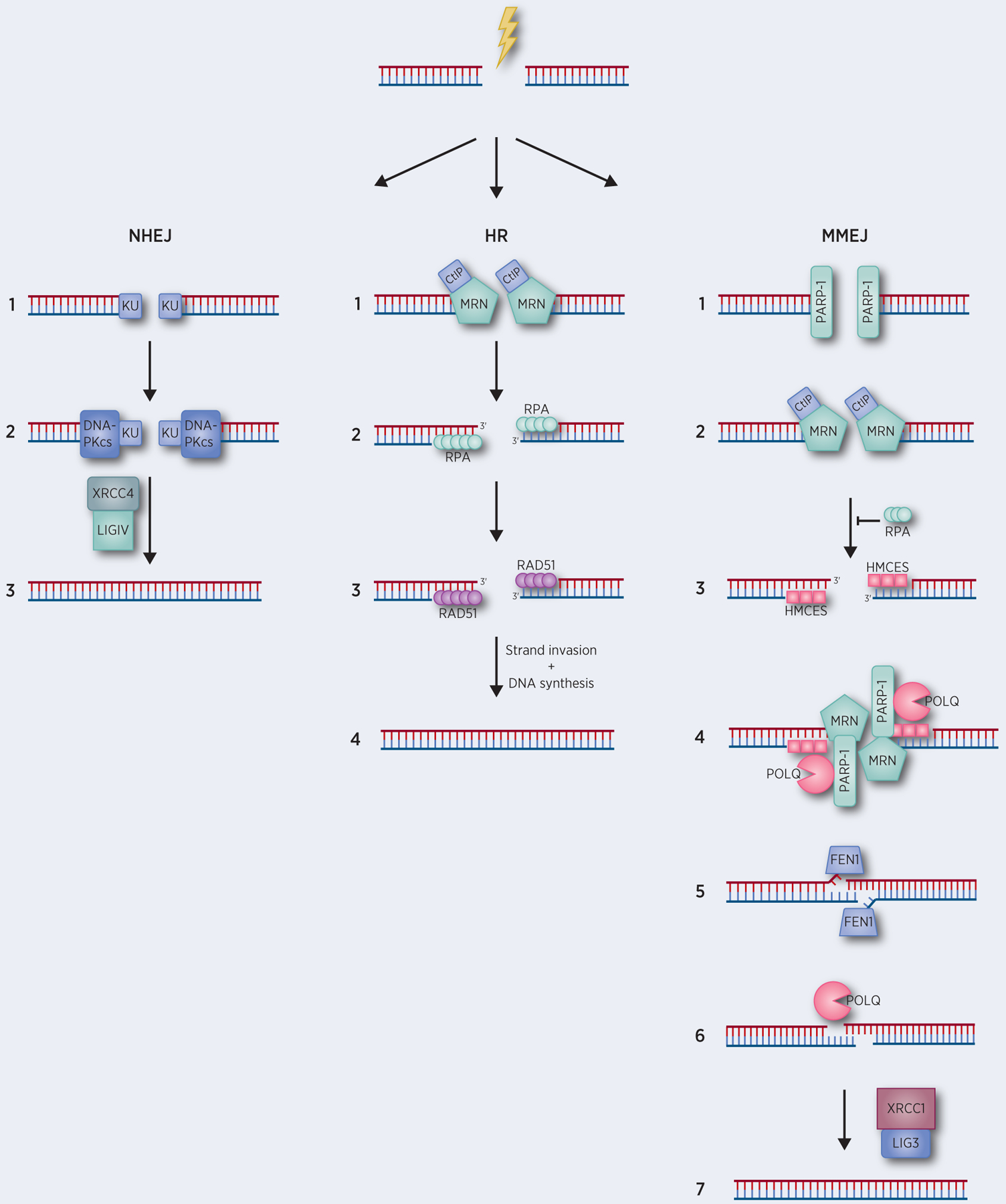
Repair of DSBs is a stepwise pathway. (A) NHEJ. (1) Ku70-Ku80 (Ku) heterodimer recognizes DSB end. (2) Recruitment of DNA-PKcs to Ku70-Ku80 heterodimer-DSB ends. If incompatible ends, further processing is required. (3) Ligation of DNA ends by XRCC4-DNA ligase IV. (B) HR. (1) MRN, stimulated by CtIP performs end-resection. (2) ssDNA is bound by RPA. (3) RAD51 replaces RPA, forming a RAD51-ssDNA nucleoprotein filament. (4) RAD51-ssDNA nucleoprotein filament performs strand invasion of the sister chromatid, leading to template-dependent strand extension followed by strand annealing. (C) MMEJ. (1) PARP-1 competes with Ku70–80 to bind DSBs ends, recruiting MRN and CtIP. (2) End-resection is initiated by MRN, stimulated by CtIP, in a stepwise manner first with the creation of a 3’ single-strand region endonucleolytically, subsequently followed by 3’ to 5’ exonucleolytic digestion. (3) Competition for binding of single-stranded DNA by RPA or HMCES with HMCES-ssDNA coating favored by MMEJ. (4) End-bridging and microhomology alignment. End-bridging is mediated by a combination of PARP-1, MRN, and POLQ. (5) Flap processing. Non-homologous 3’ tails are removed by ERCC1/XPF, FEN1, and likely as-yet identified endonucleases. Non-homologous 5’ tails are removed by FEN1. (6) POLQ gap filling. POLQ can uniquely prime DNA synthesis from non-optimal base-pairing leading distinctive templated insertions. (7) Ligation of ends by LIG3/XRCC1. See text for more details.
Microhomology-Mediated End-Joining as a Third Pathway for DSB Repair
Evidence for a third pathway that could repair DSBs first emerged following the observation that NHEJ-deficient yeast and hamster cells retained some degree of end-joining activity (12,13). Because this pathway was first reported in NHEJ-deficient cells, it was initially termed alternative end-joining and viewed as a backup mechanism if HR and NHEJ were unavailable. Subsequent work, especially in mammalian cells, has suggested a more substantial role for this pathway, now more commonly referred to as microhomology-mediated end-joining (MMEJ) (14,15).
CURRENT UNDERSTANDING OF DSB REPAIR BY MMEJ
The MMEJ pathway is a stepwise pathway (Figure 1C). It is initiated by end-resection near the DSB exposing short regions of complementary sequences ranging from 2 to 20 nucleotides (microhomologies). These microhomologies are used to align the DNA ends with the occurrence of end bridging. Next, the resultant 5’ flaps, created following alignment, are processed. A polymerase then fills in any gaps, followed ultimately by ligation (16).
End-Resection
The MMEJ pathway is initiated by end-resection. Following a DSB, poly (ADP-ribose) polymerase-1 (PARP-1) competes with Ku70-Ku80 for binding to DSBs ends, facilitating, though not essential for, recruitment of end-resection factors (17,18). As in HR, the initial end-resection in MMEJ is performed by the MRN complex (MRE11, RAD50, and NBS1) stimulated by C-terminal binding protein interacting protein (CtIP), with the initial creation of a 3’ single-strand region (19–21). If the microhomologies are distant, studies have suggested that further resection is performed by Bloom helicase-DNA2 helicase/nuclease and exonuclease 1 (EXO1); however, their roles have not been genetically confirmed. Furthermore, opposing data suggests that long range resection by EXO1 decreases MMEJ (14,19,20,22–27).
Normally, single-stranded DNA (ssDNA) is bound by Replication protein A (RPA) to stabilize the secondary structure. In yeast, RPA functions as a negative regulator of MMEJ. MMEJ is dramatically increased following end resection while HR remains relatively stable when a hypomorphic mutant allele of one of components of yeast RPA is expressed (28). Recently, Shukla et al. demonstrated that the 5-hydroxymethylcytosine binding, ESC-specific protein (HMCES), previously shown to protect stalled replication forks, can bind ssDNA and promote MMEJ repair (29,30). Furthermore, HMCES binds class switch regions and protects ssDNA overhangs to promote MMEJ, and mice deficient in HMCES have significant defects in class switch recombination, similar to mice deficient in the specialized DNA polymerase of the MMEJ pathway, polymerase theta (POLQ) (30,31). Taken together, these studies suggest that RPA is the critical ssDNA binding protein in HR repair, while HMCES is the critical ssDNA binding protein in MMEJ (Figures 1B–C).
End-Bridging and Alignment
End-bridging and alignment of microhomology sequences are next accomplished through the activities of PARP-1, the MRN complex, and POLQ. Recent in vitro studies suggest that PARP-1 may compete with Ku70–80 for binding to DSBs and may exhibit end-bridging activity through an unknown mechanism (32). PARP-1 also appears to have a role in the recruitment of POLQ to sites of DNA damage. Regarding the MRN complex, in vitro studies have shown that cohesive ends inhibit MRE11-mediated degradation. If a region of microhomology is revealed during resection, MRE11 stalls, thereby stabilizing the junction site (33,34). In addition, scanning force microscopy revealed a complex of MRE11 and RAD50 which can tether linear DNA molecules (35). These findings suggest that the MRN complex, while performing nucleolytic degradation, can facilitate DNA end-bridging and microhomology alignment. POLQ contains a N-terminal helicase domain, a long, unstructured central region, and a C-terminal polymerase domain (36). The crystal structure of the POLQ helicase domain revealed a tetrameric organization, suggesting a role of microhomology alignment and strand annealing in preparing a substrate for the polymerase domain (37). Indeed, both domains are required for MMEJ, and mutation of a conserved residue in the helicase domain reduces MMEJ (38,39).
Tail and Flap Processing
The processes of DNA end-bridging and alignment result in the generation of nonhomologous 3’ tails. These tails must be removed in order to complete the repair process. In some DNA repair processes, such as single-strand annealing, nonhomologous 3’ tails are removed by the heterodimeric structure-specific endonuclease ERCC1/XPF (Excision repair cross-complementing group 1 / Xeroderma pigmentosum, complementation group F) (40). ERCC1/XPF is also employed in nucleotide excision repair, in the Fanconi Anemia interstrand crosslink repair, and in base excision repair (41–43). Consequently, it was originally hypothesized that ERCC1/XPF was the endonuclease complex that removes nonhomologous 3’ flaps in MMEJ. However, murine ERCC1 −/− cells demonstrate only a minor MMEJ defect (44). These studies suggest that another functionally-distinct flap endonuclease, such as flap endonuclease 1 (FEN1), might be involved in the removal of nonhomologous 3’ tails in the MMEJ pathway.
Gap filling in MMEJ is performed by POLQ, through the process of strand displacement and DNA synthesis, and this process creates displaced 5’ ssDNA flaps requiring subsequent removal (45). FEN1 is a structure-specific endonuclease that recognizes and cleaves these 5’ ssDNA flaps (46). Using a cell-free system created from rat testes and thymus to study proteins involved in MMEJ, Sharma et al. demonstrated that siRNA-mediated depletion of FEN1 strongly reduces MMEJ activity (47). More recently, genetic screens to identify genes required for the upregulation of MMEJ and the survival of BRCA2-deficient cells identified FEN1, and specifically the 5’ flap endonuclease activity of FEN1 (48). Furthermore, siRNA-mediated depletion of FEN1 resulted in a defect in MMEJ repair in a cell-based MMEJ reporter assay, confirming that FEN1 is an essential enzyme in MMEJ, and suggesting that FEN1 may be the critical endonuclease which cleaves 5’ flaps created by POLQ.
Polymerase Theta (POLQ) Gap Filling
Once the DNA microhomology regions are aligned and the 3’ nonhomologous tails have been removed, there will be gaps between the duplex DNA. The specialized DNA polymerase involved in MMEJ, POLQ, was first identified from studies in Drosophila by using DSBs induced by P-element transposition or sequence-specific endonuclease (38,49). Subsequent studies have shown that POLQ is essential for MMEJ in all mammalian species examined to date (50). POLQ is a 290 kDA A-family DNA polymerase with a unique structure containing a N-terminal helicase domain, a long, unstructured central region, and a C-terminal polymerase domain (36).
The N-terminal domain of POLQ contains a superfamily 2 Hel308-typeS helicase domain. The helicase domain has both ATPase and DNA unwinding activities, which can act independently of each other. The ATPase activity is required for both the suppression of HR by binding RAD51 and inhibiting its assembly along ssDNA, and the survival of HR deficient cells (36,51). Expression of POLQ mutated at ATPase catalytic residues did not reduce RAD51 foci formation, nor decrease recombination frequency (51). The ATPase activity also facilitates removal of RPA from ssDNA to allow MMEJ and inhibit HR (52). Conceivably, with removal of RPA from ssDNA, HMCES could bind, protecting ssDNA overhangs and promoting MMEJ (30). In vitro, the POLQ helicase domain can unwind DNA, thereby facilitating displacement synthesis by the POLQ polymerase domain. Further studies are required to understand the coordination of these domains in vivo (53).
The long, unstructured central region of POLQ contains a RAD51 interaction motif. Protein interaction studies of POLQ demonstrated that this site directly interacts with RAD51 via three distinct motifs, though only one region (amino acids 847 to 894) was both necessary and sufficient for RAD51 binding. Interaction of the central domain of POLQ with RAD51 inhibited RAD51-ssDNA nucleoprotein filament assembly. In this way, POLQ acts as an anti-recombinase capable of inhibiting HR and promoting MMEJ (51). This process appears to mirror that of the POLQ helicase domain which can displace RPA, inhibit HR, and promote MMEJ (52). These interactions of the POLQ helicase domain with RPA and the POLQ central domain with RAD51, respectively, demonstrate the competitive nature between MMEJ and HR, occurring at the level of ssDNA binding. In addition, the central domain of POLQ regulates POLQ multimerization (54).
The C-terminal domain of POLQ contains an A-family DNA polymerase domain that performs gap filling. In vitro and cellular experiments have elucidated POLQ’s gap filling mechanism. POLQ anneals to short regions of ssDNA microhomology of 2 to 6 base-pairs (37,55,56). Because POLQ can uniquely prime DNA synthesis from non-optimal base-pairing leading to the introduction of insertions at the break sites, with the 3’ flaps acting as a template, it is highly error-prone (45,57,58). In addition, the POLQ polymerase is proofreading deficient and quite promiscuous because of its robust terminal transferase activity (59). Biochemically, which domains of POLQ are necessary for DNA synthesis depend on the substrate (54). Purified POLQ polymerase domain alone is active on short ssDNA and short 3’ overhangs. Longer ssDNA substrates require both the POLQ N-terminal helicase domain and the C-terminal polymerase domain, and it is the helicase domain’s DNA annealing ability that is required rather than its helicase activity. Further, extension of long ssDNA substrates requires multimers of POLQ, and multimerization requires the central region (54).
End Ligation
DSB repair by MMEJ is completed upon ligation of the DNA ends. In mammalian cells, three DNA ligases are encoded: DNA ligase I, DNA ligase III, and DNA ligase IV (60). Ligase IV functions in NHEJ and its deficiency does not impact MMEJ (61). Substantial literature has demonstrated that Ligase III is the major ligase in MMEJ. First, in vitro biochemical and plasmid-rejoining studies identified Ligase III (32,62). Second, these studies were confirmed in vivo, where mouse cells deficient in Ligase III demonstrated reduced frequency of chromosomal translocations. In this case, the few remaining translocations lacked areas of microhomology commonly seen in MMEJ (63). Third, sequential depletion of Ligase III followed by Ligase I depletion was additive, suggesting the latter is a backup ligase (63). Ligase III forms a stable complex with the scaffold protein, X-ray repair cross-complementing protein 1 (XRCC1), and both proteins in the complex interact with PARP-1 (64,65). This interaction with PARP-1 is not required for recruitment of Ligase III/XRCC1 to DSBs as there is no reduction in recruitment in the presence of PARP inhibition (15). Rather, it appears that the MRN complex might recruit Ligase III/XRCC1 to DSBs during repair by MMEJ (66).
MMEJ’S ROLE IN HUMAN DISEASE
Because of the mutagenic nature of POLQ, it is likely that POLQ and hence MMEJ are only activated under rare circumstances. Indeed, POLQ protein expression is highly limited in normal tissue (51,67). These rare circumstances might include the following. First, as POLQ expression greatly increases in HR-deficient cancers, the presence of active HR could inhibit expression of POLQ to prevent a more mutagenic DSB repair (51,68). Perhaps the BRCA1 protein, which has some transcriptional functions, reduces the transcription of the POLQ gene (69). Second, embryonic stem cells and somatic cells rely on different DSB repair pathways (70). MMEJ might play a greater role during gametogenesis, embryogenesis, or in hematopoietic stem cells as expression of POLQ is greatest in the testis, human placental tissue, and hematopoietic stem cells (36,71). Third, POLQ appears to have a prominent role in the repair of four-stranded guanine rich structures (G-quadruplex) (72–74). These structures can have both beneficial and deleterious functions. On the one hand, G-quadruplexes can protect telomeres, regulate transcription, and promote immunoglobulin gene recombination. On the other hand, these structures are prone to genomic instability and can inhibit DNA replication through fork stalling (75). Indeed, mice deficient in POLQ have increased genomic instability (76). Finally, POLQ might play a preferred role in the repair of repetitive DNA, as HR could be a deleterious option given the possibility of recombination between repetitive elements.
MMEJ and Cancer
One of the hallmarks of cancer is genomic instability (77). The repair of DSBs by MMEJ is an intrinsically mutagenic pathway. The creation of templated insertions at DSB junction sites is characteristic of MMEJ (38,51,78,79). Templated insertions are small insertions, typically, 3 to 30 bps, synthesized from DNA flanking the DSB junction into sites of microhomology deletions (38). Templated insertions, are abundant in the ClinVar database, raising the question of MMEJ’s contribution to malignancy (80). Microhomology signatures have been observed at sites of oncogenic chromosomal translocation breakpoints in primary human cancer cells, raising the possibility that MMEJ could be the causative mechanism of these translocation (23,26). Further, review of The Cancer Genome Atlas (TCGA) demonstrates that POLQ is overexpressed in breast, lung, bladder, colorectal, gastric, glioblastoma, pancreatic, prostate, melanoma, and uterine cancers, correlating with poor prognosis (67,81). The elevated POLQ expression in these tumors may result, at least in part, from an underlying HR deficiency.
Cancers deficient in HR up-regulate the expression of POLQ as a survival strategy. Aberrant use of MMEJ could create more mutations in cancers, promoting both cancer growth and resistance (82). These cancers become addicted to POLQ expression, routing the repair of DSBs to MMEJ, as evidenced by the accumulation of a distinct pattern of MMEJ-mediated templated insertions (51,68,83). This mutation signature, initially termed mutation signature 3, was identified by whole genome sequence analysis of HR-deficient tumor and thought to typify tumors that were potentially HR-deficient (84). In actuality, this signature better represents up-regulation and addiction to POLQ rather than a specific signature for HR deficiency (85). Indeed, in a recent revision of mutational signatures in cancers, signature 3 has been further resolved. The MMEJ specific signature, now defined as insertion-deletion signature 6, is characterized predominantly by greater than 5bp deletions with overlapping microhomology at deletion boundaries and is correlated with single-base substitution signature 3 (86).
THERAPEUTIC APPLICATION OF MMEJ FOR CANCER TREATMENT
Loss of DNA repair pathways is a common feature of cancers and likely provides a growth advantage (87). Concomitantly, there is increased dependence on the remaining DNA repair pathways. This dependence renders the cancer vulnerable to inhibition of the remaining DNA repair pathways and is the basis of the synthetic lethality. This concept of synthetic lethality has been successfully exploited by the use of PARP inhibitors in breast and ovarian cancers harboring inactivating mutations in BRCA1 or BRCA2, and could similarly be employed with MMEJ inhibition (88,89). Because PARP-1 is also a component of MMEJ (see section 2), PARP inhibitors also inhibit MMEJ (15). Indeed, inhibition of MMEJ appears to be a major mechanism of synthetic lethality of PARP inhibitors. Loss of expression of POLQ or other MMEJ genes in HR-deficient cells is synthetically lethal (51,68). Some cancers also have mutations in NHEJ genes, and these cancers exhibit synthetically lethality from depletion of POLQ (58,90). Importantly, some HR-deficient tumors acquire PARP inhibitor resistance by downregulating NHEJ. This offers the opportunity for using a POLQ inhibitor, since NHEJ-deficient cells are also dependent on MMEJ. Taken together these observations provide a strong rationale for targeting MMEJ in the treatment of cancers, including PARP inhibitor resistant cancers (Figure 2) (88,89).
FIGURE 2: Therapeutic Application of MMEJ for Cancer Treatment.
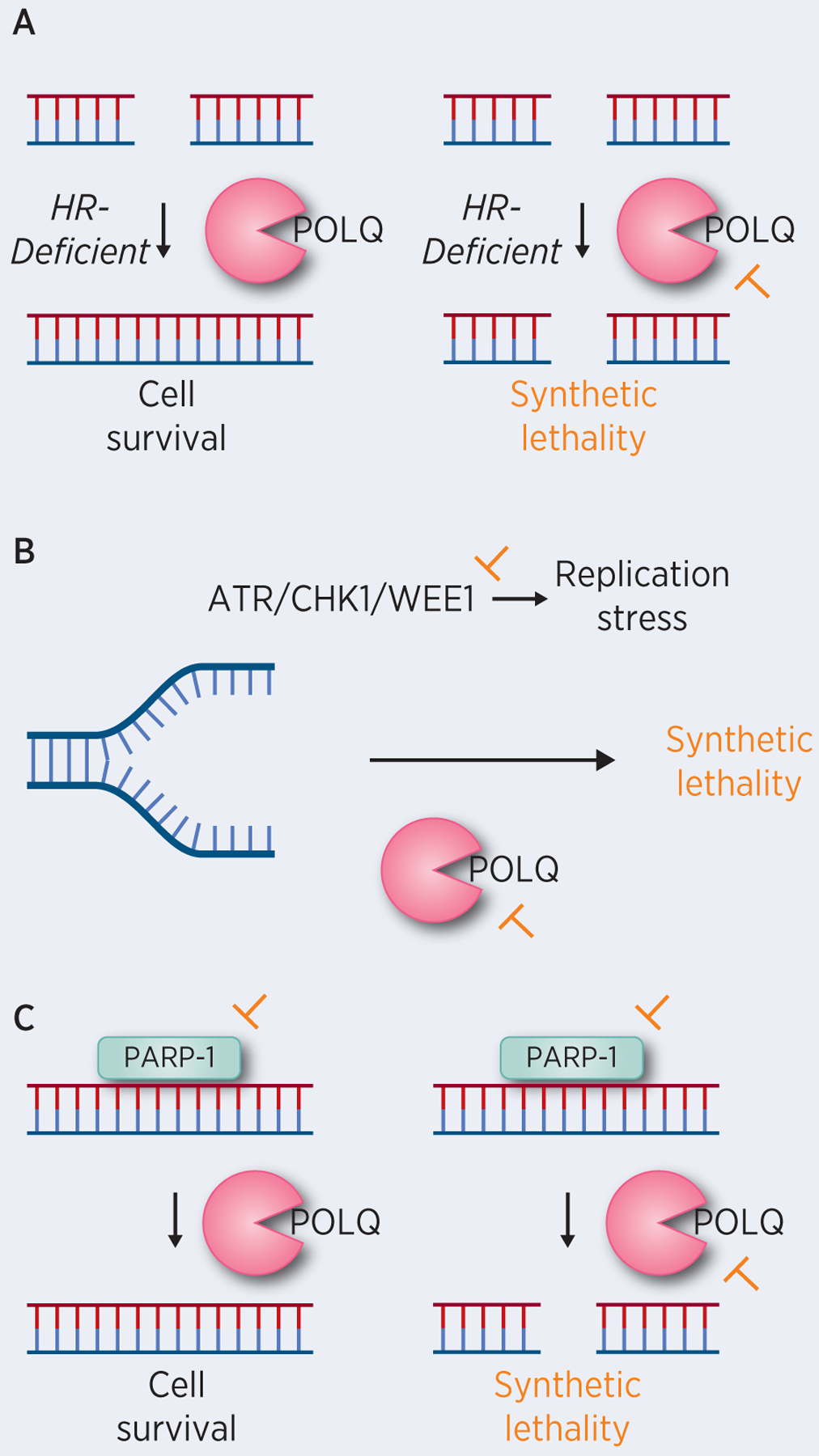
(A) HR-deficient cancers require MMEJ to repair DSBs. Inhibition of POLQ results in synthetic lethality in HR-deficient cancers. (B) POLQ deficiency is synthetically lethal with ATR inhibition. Inhibition of the ATR-CHK1-WEE1 signaling cascade leading to replication stress in combination with POLQ inhibition would result in synthetic lethality. (C) PARP inhibition results in DSBs which can be repaired by MMEJ. Inhibition of both PARP and POLQ (MMEJ) induces synthetic lethality.
Synthetic Lethality: Targeting MMEJ and HR
POLQ has limited protein expression in normal tissue but is overexpressed in wide variety of cancers (51,67,81). This overexpression is increased with concomitant p53 loss (91). These cancers may be addicted to POLQ expression because of the need to repair DSBs (51,68,83). Furthermore, POLQ, via its ATPase activity and RAD51 binding ability, can prevent the accumulation of toxic RAD51 on resected ssDNA, thereby enhancing cancer cell survival. Loss of POLQ in these tumors results in cell death by impairing MMEJ-mediated of DSBs and promoting the buildup of toxic RAD51 (51,68). Thus, POLQ is an attractive target for drug development in HR-deficient cancers (Figure 2A) (92).
The POLQ protein has three domains: a N-terminal helicase domain, a long, unstructured central region, and a C-terminal polymerase domain (36). POLQ inhibitors could be designed to target either the N-terminal helicase domain or the C-terminal polymerase domain. Targeting the polymerase domain would inhibit repair of resected DSBs at the gap-filling step in HR-deficient cancers. Targeting the helicase domain, however, might be a better option, as MMEJ repair of DSBs would be inhibited and toxic RAD51 foci would accumulate (51). POLQ inhibitors are currently undergoing active pre-clinical development. Indeed, Zhou et al. have recently identified the antibiotic Novobiocin as a specific POLQ helicase domain inhibitor that selectively kills HR-deficient cancer cells in vitro and in vivo (93).
In addition to POLQ, FEN1 is an essential enzyme in MMEJ, required at least for removal of the 5’ flap and potentially the 3’ flap (47,48). Depletion of FEN1 results in a defect in MMEJ repair and is also synthetically lethal with HR deficiency, recapitulating the synthetic lethality observed with POLQ inhibition (48,51,68). Hydroxyurea based FEN1 inhibitors have been developed that induce synthetic lethality and did not induce toxicity as a monotherapy (94). Importantly, FEN1 is not exclusive to the MMEJ pathway. It is also involved in long-patch base excision repair, thereby adding to its value as a target for anti-cancer drug development.
Synthetic Lethality: Targeting More Than HR-Deficiency
Synthetic lethality occurs not only in HR-deficient cells but also in cells deficient in NHEJ, where depletion of POLQ decreases cell survival (58). There are only a few cancers with underlying defects in NHEJ (90). However, DNA-dependent protein kinase (DNA-PK) is a key component of the NHEJ pathway, and DNA-PK inhibitors are currently under development because they block NHEJ and enhance the activity of DNA-damaging agents (95,96). DNA-PK inhibition results in a significant increase in MMEJ-mediated templated insertions and, coupled with genetic downregulation of POLQ, results in significantly reduced viability (91). These results provide evidence for the application of POLQ inhibition beyond HR-deficient cancers and for combination treatment strategies (Figures 2B–C).
Cell cycle DNA damage checkpoints allow time for repair by the DDR. The protein kinases, ataxia-telangiectasia-mutated (ATM) and ataxia-telangiectasia and Rad3-related protein (ATR), play central roles in these DNA damage checkpoints, creating an opportunity for synthetic lethality (97). ATM is activated by DSBs and initiates a DNA damage response via a signaling cascade of phosphorylation with activation of checkpoint kinase 2 (CHK2) and ultimately p53, as well as activation of the HR pathway. ATR, in contrast, is activated by replication stress. Replication stress is common in oncogene-driven cancer cells, but not in normal tissue (98). Through its kinase activity, ATR activates checkpoint kinase 1 (CHK1) and ultimately Wee1-like protein kinase (WEE1), leading to prolongation of the S/G2 cell cycle phase (99).
Replication stress activates the ATR pathway to prevent replication fork collapse and allow DNA replication to continue (99). By creating single-strand breaks at replication forks, using either a Cas9 nickase or a topoisomerase inhibitor, Wang and colleagues demonstrated synthetic lethality with POLQ deficiency, demonstrating a role for MMEJ in replication stress tolerance (100). Inhibition of the ATR-CHK1-WEE1 signaling cascade is likely to prove useful in combination with POLQ inhibition (Figure 2B). Inhibitors of ATR, CHK1, or WEE1 are currently under investigation in clinical trials and have activity through their ability to promote replication stress. Gemcitabine is another agent which can promote replication stress. A randomized phase II clinical trial of the ATR inhibitor M6620 plus gemcitabine demonstrated efficacy through the induction of replication stress in cancer cells (101). Thus, combination treatment strategies involving induction of replication stress with gemcitabine coupled with POLQ inhibition provides a strong rationale for future trials. Finally, a targeted CRISPR screen of 309 murine genes known to be involved in DDR uncovered 140 genes synthetically lethal with POLQ deficiency with the majority of these identified genes being previously unknown. It is likely that whole genome wide CRISPR screening to identify genetic vulnerabilities that interact with POLQ will reveal new combination therapy targets.
Targeting MMEJ to Overcome Therapy Resistance
The success of PARP inhibitors (PARPi) in the treatment of breast and ovarian cancers harboring inactivating mutations in BRCA1 or BRCA2 has demonstrated the power of synthetic lethality (88,89). Unfortunately, these cancers ultimately acquire PARPi resistance, with the primary mechanisms of resistance being either restoration of HR repair or replication fork stabilization (102). Reactivation of HR in PARPi resistant cancers would be predicted to alleviate dependence on MMEJ for DSB repair. However, POLQ via its RAD51 binding ability, prevents accumulation of toxic RAD51 secondary to abortive HR. Though HR is often reactivated in PARPi resistant cancers, through somatic reversion of BRCA1/2 mutations or via downregulation of NHEJ, inhibition of POLQ could still result in accumulation of toxic RAD51, resulting in cellular toxicity (51). Furthermore, POLQ and the MMEJ are required for coping with replication stress (100). Inhibition of POLQ in PARPi resistant cancers would still result in increased replication stress and cellular toxicity. Indeed, PARPi resistant tumors are sensitive to the specific POLQ inhibitor, Novobiocin, both in vitro and in vivo (93). Targeting MMEJ by inhibition of POLQ might help prevent the development of PARPi resistance or re-sensitize PARP inhibitor resistant cancers (Figure 2C).
Cancer cells often lose expression of p53 or express a mutant version (103). Loss of p53 function in cancer cells increases resistance to ionizing radiation, a common treatment modality for cancer (104,105). Given POLQ is overexpressed in p53-deficient cancers, this raises the question of whether POLQ inhibition might re-sensitize p53-deficient cancers to ionizing radiation or if POLQ inhibition could be used as an adjuvant with radiation treatment (91). Indeed, treatment of bone marrow stromal cells derived from POLQ deficient mice demonstrated increased sensitivity to ionizing radiation (106).
Biomarker-Guided Development of MMEJ Inhibitors
The development of predictive biomarkers to guide the application of MMEJ inhibition in cancers is crucial. There are at least four potential MMEJ/POLQ biomarkers. First, since POLQ is overexpressed in HR-deficient cancers or in p53-deficient cancers, dependence on POLQ activity would be predicted by upregulation of POLQ protein expression (51,68,83,91). Assessment of POLQ expression in cancers, via immunohistochemistry or RT-PCR, could serve as a predictive biomarker for response to POLQ inhibition. Second, depletion of POLQ in HR-deficient cancers leads to RAD51 accumulation (51). Assessing RAD51 accumulation could serve as a pharmacodynamic biomarker for POLQ inhibition. Third, the MMEJ pathway is required for alleviation of replication stress and is synthetically lethal with ATR inhibition (100). Replication stress activates the ATR pathway (99). Phosphorylation of RPA is downstream of ATR activation and is an immunohistochemical marker of replication stress (107). Thus, the assessment of phosphorylated-RPA could be a biomarker of the effectiveness of an MMEJ inhibitor. Finally, templated insertions are a biomarker of ongoing (or previous) MMEJ activity in the tumor cells and are a potential indicator of POLQ inhibitor efficacy (84,85). The development of biomarkers will be extremely useful in future clinical trials of POLQ inhibitors.
SUMMARY
DNA is under constant threat from both endogenous and exogenous damage, and the maintenance of genomic integrity is critical for cell survival (1). The most deleterious type of DNA damage is DSBs. Mammalian cells have three main pathways to repair DSBs: NHEJ, HR, and MMEJ. Initially, NHEJ and HR were the best studied DSB repair pathways and were thought to be the dominant pathways, with MMEJ as a weaker alternative pathway. However, evidence has since emerged to support MMEJ as a more substantial third pathway given the functional relevance of the pathway to both DSB repair and human disease. Indeed, the prevalence of a distinctive MMEJ mutational signature of templated insertions in up to 20% of all human cancers provides evidence of the widespread relevance of this pathway. Given the success of targeting DDR dependencies as a therapeutic approach to cancers, for example with PARP inhibitors in HR-deficient ovarian and breast cancers, targeting MMEJ is especially enticing. Furthermore, targeting MMEJ could be helpful in the setting of acquired drug resistance to prior therapies.
ACKNOWLEDGEMENTS
This This research was supported by grants from the U.S. National Institutes of Health (R37HL052725, P01HL048546), the U.S. Department of Defense (BM110181, BC151331P1), the Breast Cancer Research Foundation, and the Fanconi Anemia Research Fund (to A.D.D.). A.D.D is also supported by a Pancreatic Cancer Collective New Therapies Challenge Grant, an initiative of the Lustgarten Foundation and Stand Up To Cancer, Grant Number SU2C-AACR-PCC-02-18. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The research grant is administered by the American Association for Cancer Research, the Scientific Partner of SU2C. We thank Connor Clairmont, Wei-Chi Tsai, and Jia Zhou for critical reading of the manuscript.
Footnotes
CONFLICT OF INTEREST DISCLOSURE
A.D. D’Andrea reports receiving commercial research grants from Eli Lilly & Company, Sierra Oncology, and EMD Serono and is a consultant/advisory board member for Eli Lilly & Company, Sierra Oncology, and EMD Serono. J.P.F. reports no conflicts of interests. J.P.F. reports no conflicts of interests. J.P.F. reports no conflicts of interests.
REFERENCES
- 1.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016;26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–50. [DOI] [PubMed] [Google Scholar]
- 3.Heyer W-D, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. [DOI] [PubMed] [Google Scholar]
- 5.Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carvalho CMB, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilles N, Fahrenkrog B. Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudley DD, Chaudhuri J, Bassing CH, Alt FW. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol. 2005;86:43–112. [DOI] [PubMed] [Google Scholar]
- 10.Neale MJ, Keeney S. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature. 2006;442:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieber MR. NHEJ and its backup pathways in chromosomal translocations. Nat Struct Mol Biol. 2010;17:393–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boulton SJ, Jackson SP. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–103. [PMC free article] [PubMed] [Google Scholar]
- 13.Kabotyanski EB, Gomelsky L, Han JO, Stamato TD, Roth DB. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 1998;26:5333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Truong LN, Li Y, Shi LZ, Hwang PY-H, He J, Wang H, et al. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:7720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, Pandey A, et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017;45:2585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sallmyr A, Tomkinson AE. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem. 2018;293:10536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haince J-F, McDonald D, Rodrigue A, Déry U, Masson J-Y, Hendzel MJ, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283:1197–208. [DOI] [PubMed] [Google Scholar]
- 19.Garcia V, Phelps SEL, Gray S, Neale MJ. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011;479:241–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–5. [DOI] [PubMed] [Google Scholar]
- 21.Daley JM, Niu H, Miller AS, Sung P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair (Amst). 2015;32:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–24. [DOI] [PubMed] [Google Scholar]
- 24.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol. 2011;18:75–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng SK, Yin Y, Petes TD, Symington LS. Mre11-Sae2 and RPA Collaborate to Prevent Palindromic Gene Amplification. Mol Cell. 2015;60:500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng SK, Gibb B, de Almeida MJ, Greene EC, Symington LS. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, et al. HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell. 2019;176:144–153.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shukla V, Halabelian L, Balagere S, Samaniego-Castruita D, Feldman DE, Arrowsmith CH, et al. HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells. Mol Cell. 2020;77:384–394.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masuda K, Ouchida R, Takeuchi A, Saito T, Koseki H, Kawamura K, et al. DNA polymerase theta contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc Natl Acad Sci U S A. 2005;102:13986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–26. [DOI] [PubMed] [Google Scholar]
- 33.Paull TT, Gellert M. The 3’ to 5’ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–79. [DOI] [PubMed] [Google Scholar]
- 34.Paull TT, Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc Natl Acad Sci U S A. 2000;97:6409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jäger R, Gisslinger H, Passamonti F, Rumi E, Berg T, Gisslinger B, et al. Deletions of the transcription factor Ikaros in myeloproliferative neoplasms. Leuk Off J Leuk Soc Am Leuk Res Fund, UK. 2010;24:1290–8. [DOI] [PubMed] [Google Scholar]
- 36.Seki M, Marini F, Wood RD. POLQ (Pol theta), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003;31:6117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman JA, Cooper CDO, Aitkenhead H, Gileadi O. Structure of the Helicase Domain of DNA Polymerase Theta Reveals a Possible Role in the Microhomology-Mediated End-Joining Pathway. Structure. 2015;23:2319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan SH, Yu AM, McVey M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 2010;6:e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beagan K, Armstrong RL, Witsell A, Roy U, Renedo N, Baker AE, et al. Drosophila DNA polymerase theta utilizes both helicase-like and polymerase domains during microhomology-mediated end joining and interstrand crosslink repair. Symington LS, editor. PLOS Genet. 2017;13:e1006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Motycka TA, Bessho T, Post SM, Sung P, Tomkinson AE. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J Biol Chem. 2004;279:13634–9. [DOI] [PubMed] [Google Scholar]
- 41.Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, et al. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol. 2009;29:6427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gregg SQ, Robinson AR, Niedernhofer LJ. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair (Amst). 2011;10:781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodrick J, Gupta S, Camacho S, Parvathaneni S, Choudhury S, Cheema A, et al. A new sub-pathway of long-patch base excision repair involving 5’ gap formation. EMBO J. 2017;36:1605–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat Struct Mol Biol. 2015;22:230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou G-M, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008;7:2012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015;6:e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D’Andrea AD, et al. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol Cell. 2019;73:885–899.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010;38:5706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sfeir A, Symington LS. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci. 2015;40:701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mateos-Gomez PA, Kent T, Deng SK, McDevitt S, Kashkina E, Hoang TM, et al. The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat Struct Mol Biol. 2017;24:1116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozdemir AY, Rusanov T, Kent T, Siddique LA, Pomerantz RT. Polymerase θ-helicase efficiently unwinds DNA and RNA-DNA hybrids. J Biol Chem. 2018;293:5259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Black SJ, Ozdemir AY, Kashkina E, Kent T, Rusanov T, Ristic D, et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat Commun. 2019;10:4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hogg M, Sauer-Eriksson AE, Johansson E. Promiscuous DNA synthesis by human DNA polymerase θ. Nucleic Acids Res. 2012;40:2611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yousefzadeh MJ, Wyatt DW, Takata K-I, Mu Y, Hensley SC, Tomida J, et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014;10:e1004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, et al. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol Cell. 2016;63:662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zahn KE, Averill AM, Aller P, Wood RD, Doublié S. Human DNA polymerase θ grasps the primer terminus to mediate DNA repair. Nat Struct Mol Biol. 2015;22:304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2:477–84. [DOI] [PubMed] [Google Scholar]
- 62.Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–30. [DOI] [PubMed] [Google Scholar]
- 63.Simsek D, Brunet E, Wong SY-W, Katyal S, Gao Y, McKinnon PJ, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomkinson AE, Sallmyr A. Structure and function of the DNA ligases encoded by the mammalian LIG3 gene. Gene. 2013;531:150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okano S, Lan L, Tomkinson AE, Yasui A. Translocation of XRCC1 and DNA ligase IIIalpha from centrosomes to chromosomes in response to DNA damage in mitotic human cells. Nucleic Acids Res. 2005;33:422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Della-Maria J, Zhou Y, Tsai M-S, Kuhnlein J, Carney JP, Paull TT, et al. Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J Biol Chem. 2011;286:33845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lemée F, Bergoglio V, Fernandez-Vidal A, Machado-Silva A, Pillaire M-J, Bieth A, et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc Natl Acad Sci U S A. 2010;107:13390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kennedy RD, Gorski JJ, Quinn JE, Stewart GE, James CR, Moore S, et al. BRCA1 and c-Myc Associate to Transcriptionally Repress Psoriasin, a DNA Damage–Inducible Gene. Cancer Res. 2005;65:10265–72. [DOI] [PubMed] [Google Scholar]
- 70.Nagaria P, Robert C, Rassool FV. DNA double-strand break response in stem cells: mechanisms to maintain genomic integrity. Biochim Biophys Acta. 2013;1830:2345–53. [DOI] [PubMed] [Google Scholar]
- 71.Shima N, Munroe RJ, Schimenti JC. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol. 2004;24:10381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koole W, van Schendel R, Karambelas AE, van Heteren JT, Okihara KL, Tijsterman M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat Commun. 2014;5:3216. [DOI] [PubMed] [Google Scholar]
- 73.Lemmens B, van Schendel R, Tijsterman M. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat Commun. 2015;6:8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zyner KG, Mulhearn DS, Adhikari S, Martínez Cuesta S, Di Antonio M, Erard N, et al. Genetic interactions of G-quadruplexes in humans. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bryan TM. Mechanisms of DNA Replication and Repair: Insights from the Study of G-Quadruplexes. Molecules. 2019;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Soulier J, Leblanc T, Larghero J, Dastot H, Shimamura A, Guardiola P, et al. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood. 2005;105:1329–36. [DOI] [PubMed] [Google Scholar]
- 77.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 78.Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc Natl Acad Sci U S A. 2012;109:2473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wood RD, Doublié S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair (Amst). 2016;44:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schimmel J, van Schendel R, den Dunnen JT, Tijsterman M. Templated Insertions: A Smoking Gun for Polymerase Theta-Mediated End Joining. Trends Genet. 2019;35:632–44. [DOI] [PubMed] [Google Scholar]
- 81.Higgins GS, Harris AL, Prevo R, Helleday T, McKenna WG, Buffa FM. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget. 2010;1:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carvajal-Garcia J, Cho J-E, Carvajal-Garcia P, Feng W, Wood RD, Sekelsky J, et al. Mechanistic basis for microhomology identification and genome scarring by polymerase theta. Proc Natl Acad Sci U S A. 2020;117:8476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feng W, Simpson DA, Carvajal-Garcia J, Price BA, Kumar RJ, Mose LE, et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat Commun. 2019;10:4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kass EM, Moynahan ME, Jasin M. When Genome Maintenance Goes Badly Awry. Mol Cell. 2016;62:777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 89.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 90.Sishc BJ, Davis AJ. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers (Basel). 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumar RJ, Chao HX, Roberts VR, Sullivan AR, Shah SJ, Simpson DA, et al. Hyperactive end joining repair mediates resistance to DNA damaging therapy in p53-deficient cells. bioRxiv. 2020;2020.04.01.021253. [Google Scholar]
- 92.Higgins GS, Boulton SJ. Beyond PARP-POLθ as an anticancer target. Science. 2018;359:1217–8. [DOI] [PubMed] [Google Scholar]
- 93.Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. Polymerase Theta Inhibition Kills Homologous Recombination Deficient Tumors. bioRxiv. 2020;2020.05.23.111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Exell JC, Thompson MJ, Finger LD, Shaw SJ, Debreczeni J, Ward TA, et al. Cellularly active N-hydroxyurea FEN1 inhibitors block substrate entry to the active site. Nat Chem Biol. 2016;12:815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801–17. [DOI] [PubMed] [Google Scholar]
- 97.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–17. [DOI] [PubMed] [Google Scholar]
- 98.Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276–89. [DOI] [PubMed] [Google Scholar]
- 99.Dobbelstein M, Sørensen CS. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov. 2015;14:405–23. [DOI] [PubMed] [Google Scholar]
- 100.Wang Z, Song Y, Li S, Kurian S, Xiang R, Chiba T, et al. DNA polymerase θ (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J Biol Chem. 2019;294:3909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Konstantinopoulos PA, Cheng S-C, Hendrickson AEW, Penson RT, Schumer ST, Doyle LA, Lee EK, Kohn EC, Duska LR, Crispens MA. Randomized Phase 2 Study of the ATR inhibitor Berzosertib in Combination with Gemcitabine versus Gemcitabine alone in Platinum Resistant High Grade Serous Ovarian Cancer. Lancet Oncol. 2020;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–8. [DOI] [PubMed] [Google Scholar]
- 103.Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–8. [DOI] [PubMed] [Google Scholar]
- 104.Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A. 1993;90:5742–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McIlwrath AJ, Vasey PA, Ross GM, Brown R. Cell cycle arrests and radiosensitivity of human tumor cell lines: dependence on wild-type p53 for radiosensitivity. Cancer Res. 1994;54:3718–22. [PubMed] [Google Scholar]
- 106.Goff JP, Shields DS, Seki M, Choi S, Epperly MW, Dixon T, et al. Lack of DNA polymerase theta (POLQ) radiosensitizes bone marrow stromal cells in vitro and increases reticulocyte micronuclei after total-body irradiation. Radiat Res. 2009;172:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ, et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018;8:1404–21. [DOI] [PMC free article] [PubMed] [Google Scholar]