

Abstract
Purpose:
A Phase I clinical trial (GOG-9929) examined the safety and efficacy of adjuvant immune modulation therapy with the checkpoint inhibitor ipilimumab (anti-CTLA4) following chemoradiation therapy (CRT) for newly diagnosed node-positive human papillomavirus (HPV)-related cervical cancer. To better understand the mechanism of action and to identify predictive biomarkers, immunological and viral correlates were assessed before, during, and after treatment.
Experimental Design:
Twenty-one patients who received CRT and ≥2 doses of ipilimumab and 5 patients who received CRT only were evaluable for translational endpoints. Circulating T cell subsets were evaluated by multi-parameter flow cytometry. Cytokines were evaluated by multiplex ELISA. HPV-specific T cells were evaluated in a subset of patients by IFNγ ELISpot.
Results:
Expression of the activation markers ICOS and PD-1 significantly increased on T cell subsets following CRT and were sustained or increased following ipilimumab treatment. Combined CRT/ipilimumab treatment resulted in a significant expansion of both central and effector memory T cell populations. Genotype-specific E6/E7-specific T cell responses increased post-CRT in 1/8 HPV16+ patients and in 2/3 HPV18+ patients. Elevation in levels of tumor-promoting circulating cytokines (TNFα, IL-6, IL-8) post-CRT were significantly associated with worse progression-free survival.
Conclusions:
Our data indicate that CRT alone and combined with ipilimumab immunotherapy show immune modulating activity in women with locally advanced cervical cancer and may be a promising therapeutic option for the enhancement of anti-tumor immune cell function after primary CRT for this population at high-risk for recurrence and metastasis. Several key immune biomarkers were identified that were associated with clinical response.
Trial registration #:
Keywords: cervical cancer, ipilimumab, cytotoxic T lymphocyte associated antigen-4 (CTLA-4), chemoradiation, immune checkpoint blockade, human papillomavirus (HPV)
Introduction
Cervical cancer (CC) affects an estimated 13,800 women and accounts for 4,290 deaths annually in the United States (1). Although the concurrent use of cytotoxic systemic chemotherapy with radiation therapy (CRT) increases the disease free and overall survival, patients who present with stage II-IV disease have a low 4-year progression free (PFS) and overall survival (OS) of 51% and 55%, respectively, and frequently succumb to their disease and/or related complications (2). Therefore, identifying a durable therapy to address the profound CC-related morbidity and mortality for patients with high risk, node-positive locally advanced CC continues to represent an important unmet clinical need.
There is continued interest across gynecological cancers to combine chemotherapy, radiation, and immunotherapy for synergistic effects (3). These combination therapies may impact host immune regulation, anti-tumor response, or enhance response to radiation therapy (RT) (4). For example, viral and mutated self-neoantigen release is stimulated by CRT, which results in antigen presentation to the immune system to initiate an anti-tumor response, a process termed immunogenic tumor cell death (3). Immunotherapy, such as the use of anti-CTLA4 blockade therapy may further enhance this immunogenic process. The E6 and E7 oncoproteins of high-risk human papillomaviruses are constitutively expressed in the majority of cervical tumor cells, are non-self-proteins, and are therefore attractive targets for T cell mediated immunotherapy. Early data suggest that immune cells, in particular CD8+ T cells, play a key role in tumor cell death within a radiation field (5). RT causes neoantigen release and migration of dendritic cells, which can result in T-cell activation and proliferation (3). Furthermore, RT increases the density of tumor infiltrating lymphocytes (TIL) into a tumor likely by activation of local chemokine release and extravasation of TIL into the extracellular space of tumors (3,6). However, RT has also been reported to increase PD-1 expression in the tumor microenvironment leading to immune exhaustion (7). A key understanding of the underlying immune-related mechanisms contributing to CC will allow for a more tailored therapy approach and selection of the optimal patient population for immune-modulatory treatment.
Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is an inhibitory co-receptor, also called a checkpoint inhibitor, that is upregulated on activated T cells and which functions to counteract stimulation by the CD28 costimulatory molecule to attenuate the immune response (8). In vivo blockade of CTLA-4 induces regression of established tumors and enhanced antitumor immune responses in several murine tumor models (9). Ipilimumab, a humanized IgG1κ monoclonal antibody (mAb) targeting human CTLA-4, is an immune checkpoint mAb. In late 2011, the NRG Oncology (legacy Gynecologic Oncology Group (GOG)) designed the GOG-9929 study to examine the safety and tolerability of ipilimumab in patients with HPV-related newly diagnosed locally advanced CC after undergoing CRT. Since the biological mechanism of CTLA-4 blockade is to confer sustained anti-tumor T cell activation, we hypothesized that priming of the immune system to CC tumor antigens, including HPV antigens, as a result of chemoradiation could be sustained by adjuvant ipilimumab therapy.
Because reliable biomarkers or immune-related early response indicators remain lacking, we conducted targeted immune monitoring of patients in GOG-9929 in order to gain insight of the patient’s immune regulation over the course of CC during CRT and during subsequent immune checkpoint blockade (ICB). We sought to identify those at greatest risk of recurrence by examining baseline, on-treatment, and post-treatment immune changes and regulation in the peripheral blood. Here we report on the immune and viral translational correlates that were analyzed to identify potential biomarkers of response to ipilimumab following CRT as well as predictive biomarkers for clinical outcome in this high-risk population.
Methods
Ethics approval and consent to participate
The GOG-9929 clinical trial was approved by the research ethics board at each participating center. Institutional approval for correlative analyses of clinical specimens was approved by the University of Southern California Institutional Review Board (IRB# HS-13-00777). Patients provided written informed consent in compliance with institutional, state, and federal guidelines. Studies were conducted in accordance with ethical principles outlined in The Belmont Report and the Report of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research.
Study design and patients
NRG Oncology/GOG designed and conducted this 3+3 design phase I dose escalation study. Eligible patients were women from 29 sites in the U.S. with newly diagnosed, histologically confirmed locally advanced CC FIGO stages IB2/IIA with positive para-aortic lymph nodes (PALN) or FIGO stages IIB/IIIB/ IVA with positive pelvic- and/or PALN (10). Nodal status was confirmed by PET/CT scan, fine needle biopsy, extraperitoneal biopsy, laparoscopic biopsy, or lymphadenectomy. Per standard of care for this population, patients were treated with 6 weekly doses of platinum-based chemotherapy (cisplatin, intravenous, 40 mg/m2) concurrent with extended field RT (EFRT), an involved nodal RT boost, and intracavitary brachytherapy. Two to 6 weeks after completing CRT, patients with no evidence of disease progression on diagnostic CT of the chest, abdomen, and pelvis initiated ipilimumab (Yervoy®, provided by Bristol-Myers Squibb) intravenously every 21 days for 4 doses. Two dose levels of ipilimumab, 3mg/kg (DL1) and 10mg/kg (DL2), were studied to identify the maximum tolerated dose (MTD). No reductions in DL were necessary and 10mg/kg was advanced to the expansion cohort as the MTD. Clinical responses were defined by the immune Response Evaluation Criteria in Solid Tumours (irRECIST version 1.1) (11), as evaluated by exam and by PET/CT or CT 3 months post adjuvant immunotherapy. Feasibility, safety, and clinical efficacy outcomes are reported elsewhere (12).
HPV and HLA genotyping
Cervical cells were collected via cervical swab prior to any treatment using the Digene Cervical sampler (Qiagen, Valencia, CA). HPV genotyping on genomic DNA was performed using the INNO-LiPA HPV Genotyping Extra kit (Innogenetics, Seguin, TX). Low-resolution DNA typing for HLA-A2 was performed for all patients using standard endpoint PCR or by flow cytometry of isolated white blood cells using a fluorochrome conjugated anti-HLA-A2 antibody (BD Biosciences, Franklin Lakes, NJ). For patients testing positive for HLA-A2, high-resolution genotyping was performed by PCR to provide allele level typing at the HLA-A2 locus with the A*02 SSP UniTray Kit (Life Technologies, Carlsbad, CA).
Immune subset and functional marker analysis
Whole blood was drawn by venipuncture into ACD-A-containing blood tubes prior to CRT, 2-6 weeks post-CRT when CRT-associated toxicities resolved but pre-ipilimumab treatment, and 2 weeks post-ipilimumab treatment. Blood from each participating center was shipped overnight (on the day of sample collection) to a central lab (USC Beckman Center for Immune Monitoring) for processing, storage, and immunoassay testing. Peripheral blood mononuclear cells (PBMC) were isolated over a Ficoll-paque density gradient, cryopreserved, and stored in vapor phase liquid nitrogen prior to batch testing. After thawing, PBMC were stained with antibodies for human CD3 (OKT3), CD4 (OKT4), CD8 (RPA-T8), CD25 (BC96), CD45RO (UCHL1), CD62L (DREG-56), CD127/IL-7Rα (A019D5), CD137/4-1BB (4B4-1), CD197/CCR7 (G043H7), CD278/ICOS (C398.4A), FoxP3 (206D), CD152/CTLA4 (L3D10), CD279/PD-1 (EH12.2H7), and Zombie Aqua live/dead exclusion dye (all from Biolegend, San Diego, CA) for multiparameter flow cytometric analysis. Data were acquired on a BD FACS Canto II cytometer and analyzed with FlowJo software version 10.5 (BD Biosciences). Live cells in the lymphocyte gate (by forward/side scatter) were included in the analysis. T cell subsets were quantified as a percentage of the total lymphocyte population. Activation marker expression was quantified as the percentage of cells expressing a particular marker within a certain T cell subset (CD4+ or CD8+ T cells). Appropriate isotype control antibodies and fluorescence-minus-one control stains were used to set flow gating strategies.
IFNγ Enzyme-linked immunospot (ELISpot) assay
Fifteen amino acid long peptides overlapping by eleven spanning the entire open reading frames of HPV16 E6/E7 and HPV18 E6/E7 were purchased from New England Peptide (Gardner, MA). Peptides were purified by HPLC to >80%. Cryopreserved PBMC were quickly thawed and resuspended in RPMI-1640 supplemented with 10% pre-qualified fetal bovine serum (Omega Scientific, Tarzana, CA), 2 mM L-Glutamine, 50 μg/ml gentamycin, 1mM sodium pyruvate, 2mM non-essential amino acids, 50 μM 2-mercaptoethanol, and 10 U/mL Benzonase nuclease (Millipore Sigma). Cells were “rested” in 37°C, 5% CO2 incubator for 2 hrs to remove dead or apoptotic cells, then transferred to a 24-well plate in the presence or absence of 5 μg/mL of HPV E6 or HPV E7 peptides combined in pools. After 4 days, cells were harvested, washed, rested, counted, and plated in four replicate wells of a sterile 96 well plate pre-coated human IFNγ ELISpotPLUS plate (Mabtech 3420-4APT-10). PHA-L (Sigma, St. Louis, MO) was added at a final concentration of 5μg/mL to positive control wells. After 18 h incubation (37°C, 5% CO2), cells were removed by washing and assay performed per the manufacturer’s recommendations. Spots were imaged and counted using the automated computer-assisted video-imaging KS ELISPOT analysis system (Carl Zeiss, Thornwood, NY). The number of peptide-specific spots was calculated by subtracting the mean number of spots from medium control wells from the mean number of spots from experimental wells and normalizing to spots per million PBMC. Positive response wells fit the pre-specified cut-point of 50 IFNγ-secreting cells per million PBMC. A treatment-induced antigen-specific response was defined as a 2-fold increase over the baseline response.
Cytokine studies
Plasma was separated from whole blood within 2 hours of receipt and stored at −80°C until measurement with no freeze-thaw cycles. Circulating cytokine measurements were assayed using the Luminex multiplexing cytokine suspension array technology. Measurements of plasma interleukin (IL)-1β , IL-2, IL-4, IL-5, IL-6, IL-7, IL-8/CXCL8, IL-10, IL-12p70, IL-13, tumor necrosis factor alpha (TNFα), IFNγ, and GM-CSF were analyzed using a MilliPlex MAP human high sensitivity cytokine 13-plex panel (MilliporeSigma, St. Louis, MO). Transforming Growth Factor (TGF) β1 was measured using a MillipPlex MAP TGFβ 1,2,3 magnetic bead kit (MilliporeSigma). Plates were read on a Bio-Plex 200 System using Bio-Plex Manager software version 6.0 (BioRad, Hercules, CA). Five parameter logistic regression algorithms were used to determine cytokine concentrations based on provided cytokine standards and averages from triplicate wells.
Statistical analyses
Statistical tests for laboratory-generated data used a 0.05 two-sided significance level, adjusted for multiple comparisons where appropriate, using GraphPad Prism software v8.2.0 (GraphPad Software Inc., San Diego, CA). Immune subset flow cytometry and cytokine data were analyzed using a Mixed-effects analysis, with the Geisser-Greenhouse correction for any missing data, followed by the Tukey’s multiple comparisons test, with individual variances computed for each comparison. Data correlated to objective tumor responses were analyzed using an unpaired Mann-Whitney U test per individual comparison. Changes in biomarker expression relative to PFS and OS were analyzed using Cox proportional hazards regression models adjusted for baseline values (SAS software version 9.4, Cary, NC), and effect size was expressed with hazard ratio (HR) and 95% confidence interval (CI).
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its supplemental files).
Results
Patient characteristics
From December 2012 to September 2016, 34 patients were enrolled; the safety, efficacy, and adverse events were previously reported (12). Twenty-six patients with biospecimens from ≥2 time points were evaluable for translational endpoints with 20 patients having specimens at all three time points. Twenty-one patients (81%) were treated according to protocol and received at least 2 cycles of sequential ipilimumab and were therefore evaluable for all study endpoints. Five patients who were not allocated to receive ipilimumab, but who received CRT and from whom we were able to collect a blood sample post-CRT are included in the intent-to-treat (ITT) cohort. Patients in the ITT cohort were not eligible to receive ipilimumab if CRT-related toxicities did not resolve to ≤ grade 1 within the allowable 2-6 week window or if they experienced disease progression. Demographic and clinical data of patients are shown in Table 1.
Table 1.
Patient characteristics for immune and viral correlate analysis
| Characteristic | Per Protocol (PP) (N=21)a |
Intent To Treat (ITT) (N=5)b |
Total Evaluable (N=26) |
P-valuesc |
|---|---|---|---|---|
| Median age, years (range) | 48 (34-62) | 51 (26-56) | 49 (26-62) | 0.79 |
| Race, N (%) | ||||
| White | 14 (67) | 4 (80) | 18 (69) | 1.00 |
| African American | 4 (19) | 1 (20) | 5 (19) | |
| Asian | 2 (9) | 0 (0) | 2 (8) | |
| Other | 1 (5) | 0 (0) | 1 (4) | |
| Ethnicity, N (%) | ||||
| Non-Hispanic | 14 (67) | 4 (80) | 18 (69) | 1.00 |
| Hispanic | 7 (33) | 1 (20) | 8 (31) | |
| Histology, N (%) | ||||
| Squamous cell carcinoma | 19 (90) | 4 (80) | 23 (88) | 0.49 |
| Adenosquamous | 1 (5) | 0 (0) | 1 (4) | |
| Adenocarcinoma | 1 (5) | 1 (20) | 2 (8) | |
| Lymph node involvement, N (%) | ||||
| Pelvic | 15 (71) | 3 (60) | 18 (69) | 0.63 |
| Pelvic and Para-aortic | 6 (29) | 2 (40) | 8 (31) | |
| FIGOd Stage, N (%) | ||||
| 1B | 3 (14) | 1 (20) | 4 (15) | 0.69 |
| IIA | 1 (5) | 1 (20) | 2 (8) | |
| IIB | 11 (52) | 2 (40) | 13 (50) | |
| IIIB | 5 (24) | 1 (20) | 6 (23) | |
| IVA | 1 (5) | 0 (0) | 1 (4) | |
| HLA-A*0201 status, N (%) | ||||
| Positive | 8 (38) | 1 (20) | 9 (35) | 0.63 |
| Negative | 13 (62) | 4 (80) | 17 (65) | |
| Ipilimumab Dose Level, N (%) | ||||
| 3 mg/kg | 3 (14) | 0 (0) | 3 (12) | 1.00 |
| 10 mg/kg | 18 (86) | 5 (100) | 23 (88) | |
| Ipilimumab Doses, N (%) | ||||
| 2 | 3 (14) | 0 (0) | 3 (14) | <0.001 |
| 4 | 18 (86) | 0 (0) | 18 (86) |
PP patients received CRT and ≥ 2 doses ipilimumab
ITT patients received CRT only
Comparing PP patients vs. ITT patients, using the Wilcoxon rank sum test and Fisher’s exact test where appropriate
FIGO, International Federation of Gynecology and Obstetrics
HPV genotype and HLA-A*0201 status
The HLA-A2 allele family is the largest and most frequently found allele in many ethnic groups, predominated by HLA-A*0201 (13). The HLA-A*0201-restricted immune response is the most commonly studied subtype for which defined CD8+ T cell MHC epitopes have been defined for HPV16 and HPV18 E6 and E7 (14-16). Thirty-five percent of overall patients expressed HLA-A*0201 (Table 1), consistent with the prevalence of this allele in various ethnic populations, ranging from 35-50% (13). HPV16 was found in 13 (50%) patients and HPV18 was found in 4 (15%) patients (Figure 1). Other high-risk HPV genotypes were found in the remaining 9 patients. In 7 patients, more than one high-risk HPV was detected, as might be expected from a cervical swab sampling (17). These type attributions are consistent with the genotype prevalence in CC reported in other studies (18).
Figure 1. Represented high-risk HPV types in patients enrolled in GOG9929.
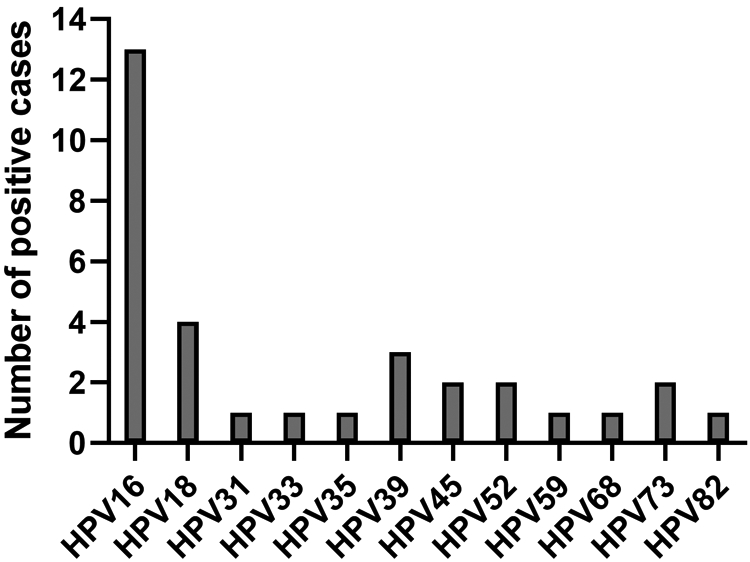
Sampling of the cervix of each subject at baseline was performed to determine probable HPV genotype of the cervical cancer. Isolated genomic DNA was tested in the INNO-LiPA HPV Genotyping Extra assay. Shown is the number of cases detected for the listed high-risk HPV genotypes. Seven patient samples were positive for more than one HPV type. HPV16 and HPV18 were the most frequently detected HPV genotypes in this patient population.
CD4 and CD8 T cell activation following chemoradiation therapy and ipilimumab treatment
To investigate the pharmacodynamic effects of CRT and sequential ipilimumab on the frequency and activation of T cell subsets, PBMC from pre-treatment, post-CRT, and post-ipilimumab treatment were analyzed by flow cytometry. Frequencies of CD8+ T cells, CD4+ T cells, Tregs, and expression of the T cell activation/exhaustion markers inducible T cell costimulatory (ICOS), CD137 (4-1BB), and programmed cell death 1 (PD-1) were enumerated. There was a significant decline in the frequency of CD4+ T cells post-CRT compared to baseline, which did not completely recover post-ipilimumab treatment (Figure 2a). Although the frequencies of CD8+ T cells appeared to increase post-CRT in some patients and return to baseline, the median differences were non-significant (Figure 2a). The frequency of Tregs increased marginally over the course of CRT and iplimumab treatment, although the ratios of CD8+ T cells to Tregs at each time point did not change significantly (Figure 2b).
Figure 2. Circulating T cell subsets and expression of immune activation markers.

PBMC subsets and expression of T cell activation markers were assessed in all evaluable patients (PP and ITT) with a baseline (pre-CRT) sample, a post-CRT sample, and a post-ipilimumab sample (in PP patients) by multi-parameter flow cytometry. (A) Percentages of CD4 and CD8 T cells are shown. (B) Percentage of Tregs and ratio of CD8:Tregs is shown. (C) For activation marker expression, the percentage change from baseline is shown. Response is color coded for recurrence-free censored patients (black symbols) versus patients with disease recurrence during the follow-up period (red symbols). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns, non-significant (Mixed-effects analysis followed by the Tukey’s multiple comparisons test).
ICOS, PD-1, and C137 expression are all inducible upon T cell activation. ICOS is a protein homologous to CD28 and CTLA-4, which functions to co-stimulate T cell proliferation and cytokine production (19). There were significant increases in ICOS expression on CD4+ T cells from baseline to post-CRT, with further increases post-ipilimumab (Figure 2c). Furthermore, ICOS expression of CD8+ T cells increased significantly post-ipilimumab compared to baseline. PD-1 expression was similarly significantly increased on CD4+ and CD8+ T cells after CRT and high levels were sustained post-ipilimumab (Figure 2c). A high percentage of the T cells expressing ICOS also co-expressed PD-1, an inhibitory molecule which provides a regulatory feedback mechanism to control the immune response. Notably, the increased expression of immune activation markers on T cells prior to any ipilimumab suggests that CRT alone has a significant effect on lymphocyte activation resulting in priming of the immune system. CD137, a TNF-receptor superfamily member, also known as 4-1BB, is induced upon T cell activation and provides signals for proliferation, enhanced IL-2 production and inhibition of apoptosis (20). Changes in CD137 expression, although increased in some patients following CRT, were not consistently upregulated among all patients (Figure 2c).
Changes in frequencies of T cell subsets and expression markers from baseline to post-CRT were compared among patients who experienced an objective tumor response during the study observation period, separating patients into two response groups: complete response (CR) and partial response (PR) vs. stable disease (SD) and progressive disease (PD). Changes in the overall frequencies of CD4+ and CD8+ T cells from baseline to post-CRT visits were not associated with objective tumor response (Supplementary Figure S1a). Changes in expression of ICOS, PD-1, and CD137 on CD4+ T cells and CD8+ T cells were also not significantly associated with objective tumor response, although ICOS expression on CD4+ T cells did appear to trend higher in patients with a CR/PR (Supplementary Figure S1b, c). There were no significant differences noted in immune markers among patients who received ipilimumab at the MTD of 10 mg/kg for 4 cycles versus those with reduced dose intensity (3 mg/kg or < 4 cycles) (Supplementary Figure S2).
In addition to the above selected immunological biomarkers, we also measured CD4+ and CD8+ naïve, effector, and memory T cell subsets. Expansion of CD4+ effector memory (Tem; CD62L-/CD45RO+) and central memory (Tcm; CD62L+/CD45RO+) T cells was significantly increased post-CRT compared to baseline with further expansion post-ipilimumab treatment, and a concomitant decrease in the naïve (CD62L+/CD45RO-) CD4+ T cell population (Figure 3a). Similar expansion of the CD8+ Tem and Tcm subsets and decrease in naïve CD8+ T cells were observed (Figure 3b). Changes in median overall effector T cell populations (Teff; CD62L-/CD45RO-) were not significant between time points. These variables failed to show statistically significant associations with objective tumor response, although we did observe increases in the CD4+ Tem population and greater decreases in naïve T cells in the clinical responders (Supplementary Figure S3). Collectively, these results indicate that CRT by itself stimulates T cell activation in patients with CC, even as absolute numbers of leukocytes decrease from cisplatin-based therapy, and this immune activation is sustained throughout adjuvant CTLA-4 blockade.
Figure 3. Changes in circulating T cell memory subsets post CRT and ipilimumab treatment.

(A) Percentage change from baseline in CD4+ T cell memory subsets. (B) Percentage change from baseline in CD8+ T cell memory subsets. Percentages were calculated from CD3+CD4+ or CD3+CD8+ gated lymphocytes. Response is color coded for recurrence-free censored patients (black symbols) versus patients with disease recurrence (red symbols). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns, non-significant (Mixed-effects analysis followed by the Tukey’s multiple comparisons test).
Each measured immune biomarker parameter at each time point was correlated to both PFS and OS using adjusted Cox proportional hazards models. Additionally, two change variables were considered to characterize the kinetics of the immune response with the different treatment modalities: (i) the change from baseline to post-CRT value, and (ii) the change from post-CRT to post-ipilimumab value. Only significant associations are reported. There was significant correlation of increases in activated CD4+ T cells expressing ICOS and PD-1 with PFS (HR 0.806, 95% CI 0.650-0.999, p=0.049; Figure 4) and similarly with OS (HR 0.705, 95% CI 0.508-0.978, p=0.036; Supplementary Figure S4) over the 1-year observation period, suggesting that greater immune activation is associated with lower risk of progression and death over time.
Figure 4. Association of changes in immune biomarkers with progression-free survival.
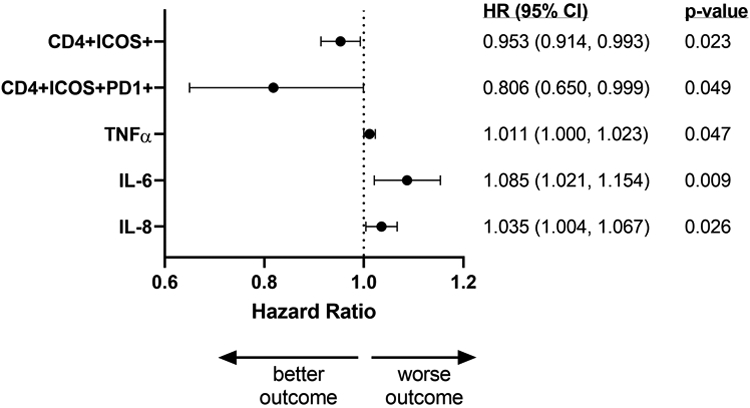
Increased changes (baseline to post-CRT values) in immune parameters were related to PFS using adjusted Cox proportional hazards models, with each variable fit in individual model adjusted for baseline value. Figure shows hazard ratios with 95% confidence intervals (lower limit, upper limit) and associated p-values for statistically significant associations found for immune activation markers and plasma cytokines. Expansion of the CD4+ICOS+ and CD4+ICOS+PD-1+ subsets post-CRT are associated with lower risk of progression while increases in inflammatory cytokines TNFα, IL-6, and IL-8 post-CRT are associated with higher risk of tumor progression. Nineteen PP and five ITT patients were included in immune phenotyping analysis. Twenty-one PP and five ITT patients were included in cytokine analysis.
Circulating cytokine response to therapy
Cytokines produced by immune cells are key players in controlling immune responses to cancer through both tumor-promoting and/or tumor-suppressing properties. To investigate whether CRT and sequential ipilimumab-induced changes in circulating cytokines, we evaluated a set of 14 cytokines comprising different functional groups using a Luminex-based multiplexing immunoassay. Cytokines tested were growth factors (GM-CSF, IL-7), proinflammatory cytokines (TNFα, IL-1β, IL-6, IL-8), CD4+ T helper type 1 (Th1)-associated cytokines (IL-2, IL-12p70, IFNγ), CD4+ T helper type 2 (Th2)-associated cytokines (IL-4, IL-5, IL-13), and immunosuppressive cytokines (IL-10, TGFβ1). Significant decreases were observed in GM-CSF, IL-2, IL-4, IL-5, IL-7 and TGFβ1 levels between baseline and post-CRT (Supplementary Figure S5), likely due to myelosuppression as a result of CRT. Changes in median values of IFNγ, TNFα, IL-1β, IL-6, IL-8, IL-10, IL-12p70, and IL-13 were not statistically different between baseline, post-CRT, and post-ipilimumab treatment. Absolute levels of baseline and post-CRT cytokines did not correlate with objective tumor response (Supplementary Figure S6). TGFβ1 levels were high in all patients whether they exhibited an objective tumor response or not. However, increased levels of the inflammatory cytokines TNFα, IL-6, and IL-8 from baseline to post-CRT were significantly and independently correlated to PFS (Figure 4) and OS (Supplementary Figure S4) with high IL-6 levels contributing the highest risk, suggesting that greater increases in systemic inflammation post-CRT are associated with risk of progression and death over time.
Detection of HPV genotype-specific T cell responses
After observing the significant effect that CRT and CTLA-4 blockade had on activating both CD4+ and CD8+ T cells and expanding the Tcm and Tem memory subsets, we next examined whether we could detect increases in functional HPV-specific T cell responses either post-CRT or post-ipilimumab treatment. We hypothesized that CRT-induced tumor cell death would release HPV tumor antigens, namely the E6 and E7 oncoproteins, into the tumor environment, which in combination with immune danger signals would prime and activate tumor antigen-specific T cells. Long overlapping peptide pools representing the E6/E7 antigens from the HPV16 or HPV18 genotypes were used to stimulate PBMC samples at each time point from patients who were HPV16- or HPV18-positive at baseline. Functional E6/E7 antigen-specific T cells were enumerated through an IFNγ ELISpot assay. The presence of HPV-specific T cells of each patient varied with respect to presence and magnitude of response. Among the eight HPV16+ patients from whom sufficient PBMC were available for the assay, only one patient (patient no. 15) demonstrated pre-existing HPV16-specific T cells at baseline (Figure 5). There was a small, but non-statistically significant increase post-CRT which was not sustained during the CTLA-4 blockade. No other HPV16+ patients demonstrated E6/E7-specific T cell responses at any of the time points. In the three HPV18+ patients tested, two showed a pre-existing response to HPV18 E6/E7 antigens (Figure 5). The third patient did not have sufficient cell recovery at baseline to test. Two of the HPV18+ patients showed high levels of HPV18-specific T cell responses post-CRT (in one patient at least 2-fold over baseline). The small patient numbers in this study preclude any reliable correlation of HPV-specific responses to clinical outcomes. However, it is noteworthy that the two HPV18+ high responding patients demonstrated positive objective tumor responses, whereas the patient with the low T cell response (patient no. 7) developed progressive disease. As none of the patients with positive HPV-specific responses were also HLA-A*0201+, we were unable to explore T cell responses to known HLA-A*0201-restricted epitopes in more detail to specifically examine HPV-specific CD8+ T cell activation and exhaustion cell marker expression over the course of treatment.
Figure 5. HPV16 or HPV18-specific T cell responses detected by IFNγ ELISpot assay.
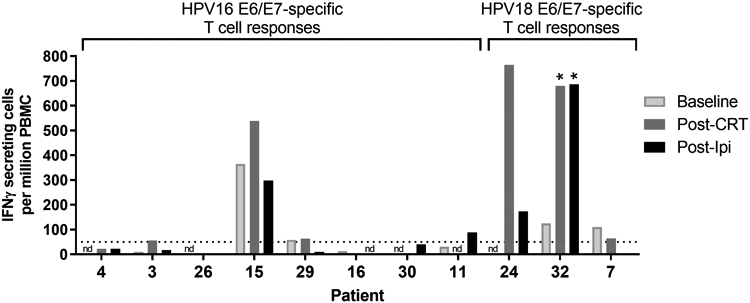
HPV-specific T cell responses were detected at baseline, post-CRT and post-ipilimumab from a subset of evaluable patients who tested positive for either HPV16 or HPV18 at baseline. Shown is the number of IFNγ-secreting protein-specific T cells (in spots per million PBMC) responding to either E6 or E7 peptides from each HPV genotype. Sample time points labeled “nd” were not tested due to insufficient cell numbers or quality recovered post-PBMC thaw. The dotted horizontal line indicates the threshold for a positive response (> 50 IFNγ-secreting cells per million PBMC) based on assay sensitivity. Samples labeled with an asterisk indicate significant increases in HPV-specific T cells ≥ 2-fold over baseline.
Discussion
Virtually all cervical cancers arise from persistent infection by high-risk HPVs with cellular transformation occurring as a consequence of E6 and E7oncogene expression (21). There is abundant evidence that HPV infection can be controlled by the immune system even with its multiple immune evasion strategies, and further, that HPV-transformed cells can be targeted and cleared by HPV oncogene-specific cytotoxic T lymphocytes (CTLs) (22-24). As a virally-driven cancer, there is significant interest in the role of immunotherapy and ICB as new treatment paradigms for CC (25,26). To our knowledge, this is the first longitudinal description of clinical immune biomarkers over the course of multimodality treatment including ipilimumab ICB for treatment-naïve, high risk locally advanced CC. We report on the HPV-specific response and immune activation status at baseline prior to any treatment, after CRT, and after ipilimumab in order to help inform how CLTA-4 blockade may be used with standard of care treatment or in future CTLA-4 immune therapy combinations.
The ability to longitudinally assess immune biomarkers to predict which patients are more likely to recur after standard of care treatment would represent a significant clinical advance. Prior reports have indicated that analysis of peripheral immune cell subsets prior to initiation of immunotherapy has the potential to predict, with statistical significance, those patients most likely to achieve clinical benefit (27-29). Upregulation of the T cell-costimulatory molecule ICOS has been shown to be a consistent pharmacodynamic marker of CTLA-4 blockade in pre-clinical models and in patient clinical trials (30-33). In the absence of ICOS/ICOSL pathway stimulation, efficacy of CTLA-4 blockade is severely diminished (34). Consistent with previous studies, we found ICOS expression to be significantly upregulated in our patient population treated with ipilimumab, supporting its known biological mechanism of action. Interestingly, ICOS and PD-1 expression were induced by CRT alone, supporting the view that radiotherapy induced immunogenic cell death in these patients results in the potential activation of tumor-directed T cells. Previous reports with chemotherapy alone have shown little effect on immune parameters, including ICOS and PD-1 expression, in PBMCs (33). In the CD4+ T cell population, ipilimumab further increased and sustained ICOS expression 14-20 weeks after CRT was administered when the final blood samples were taken. We also noted significant expansions in both the Tem and Tcm memory T cell populations in both CD4+ and CD8+ subsets despite the fact that overall frequencies of CD4+ and CD8+ T cell subsets either remained stable or even decreased over the course of treatment.
Several studies report that the CD4+ICOShigh subset is most significantly increased and possibly associated with clinical benefit after ipilimumab administration (31,35). In this study, baseline to post-CRT increases in the CD4+ICOS+ and CD4+ICOS+PD-1+ subsets were significantly correlated to overall PFS and OS over the 1-year observation period, confirming an association with clinical benefit as seen in other cancers. Unlike findings reported by Tang et. al. who observed clinical benefit associations with CD8+ T cells expressing CD137 (4-1BB) and PD-1 after combined ipilimumab and stereotactic ablative radiation (36), we did not see any clinical associations with upregulated CD137 or PD-1 expression on either CD4+ or CD8+ T cells.
The presence of increased frequencies of Tregs has been associated with poor outcomes in patients with HPV-related cancers, including CC (37). In preclinical models, optimal efficacy of CTLA-4 blockade was achieved through the synergistic effects of enhanced effector T cell function and blocking Treg activity (38). Although the median percentage of Tregs increased post-CRT in this study, the ratio of CD8+ T cells:Tregs did not change significantly. As a result, the significance of greater increases in Tregs in patient responders is unclear, given that they also likely had expansions in the CD8+ population. Studies to date have reported inconsistent ipilimumab- or tremelimumab-induced changes in peripheral Tregs, supporting the notion that intratumoral Tregs may be more biologically relevant (35). Indeed, frequencies of Tregs during CTLA-4 blockade may be less important than their function, as it has been shown that in tremelimumab-treated melanoma patients, CD4+CD45RO+ memory T cells developed a resistance to the suppressive function of Tregs (39). Differences between our observations and others could also be attributed to the cancer type, the sequencing and type of treatment (neoadjuvant vs. concurrent vs. adjuvant therapy), or the timing of peripheral sampling.
We hypothesized that those patients with measurable pre-treatment levels of HPV-specific T cells may represent a patient subgroup that has an immune system primed with already-activated tumor-specific T cells. In addition, we also hypothesized that the magnitude of change in the levels of HPV-specific T cells between the pre-treatment time point and the pre-ipilimumab time point (when CRT has been on-going) may predict an increased likelihood of response to ipilimumab treatment due to a rigorous activation of HPV-specific T cells from CRT alone. We were able to detect HPV-specific T cells above our cutoff threshold from two patients prior to any treatment. These occasional responses correlate with earlier observations in patients with HPV16+ cervical lesions (40). Both of these patients experienced an expansion in those cells as a result of CRT. A third patient displayed a high level of HPV-specific T cells post-CRT; however, we were not able to examine her baseline sample due to poor sample quality. After ipilimumab treatment, the frequency of HPV-specific T cells declined in two of three patients, which could either be explained by the natural immune contraction of this population or alternatively, cells may have left the periphery and trafficked to sites of residual tumor. Future analysis of T cell expansion to CC neoantigens may also prove to be informative, since both viral and non-viral antigens can be important for T cell-mediated regression of CC (24). Information from these types of studies may be used not only retrospectively, but more importantly, carry the possibility of determining, prior to immune-modulating therapy, who may best respond to such therapeutic strategies.
Soluble immune mediators, such as cytokines, have been shown to be important players in both the initiation and progression of cancer through their tumor-promoting properties (41). Elevated levels of circulating TNFα, IL-1β, IL-6, IL-8, IL-10, and TGFβ in cancer patients are associated with more aggressive cancers and poorer outcomes (42). In our study, we found that elevated levels of TNFα, IL-1β, IL-6, and IL-8 post-CRT were associated with worse clinical outcomes (PFS and OS), perhaps suggesting that sustained high levels of these tumor-promoting cytokines could indicate risk of progression or recurrence after primary treatment. ITT patients were included in the analysis since at the time of post-CRT sampling, neither ITT nor PP patients had received ipilimumab.
There are several limitations to our study. Evaluation of clinical efficacy and translational biomarkers was meant to be descriptive and hypothesis-generating given the small number of patients on this phase I trial with follow-up restricted to just 1 year. Although all 34 patients had a baseline blood sample drawn, we limited the detailed immunological analysis to the 26 patients from which we had at least one follow-up blood sample. Our overall attrition rate for the study was 38% (13 of 34 who did not continue to ipilimumab), but all of the attrition was due to reasons unrelated to ipilimumab toxicity, such as not meeting inclusion criteria, not adhering to treatment, cisplatin allergy, unresolved toxicity following CRT, withdrawal of consent, or progression of disease after CRT (12). Patients who were ineligible to receive ipilimumab went off study and were not followed given the primary objectives of the study nor asked to provide follow-up blood samples for exploratory endpoints at the same time point as patients who received ipilimumab. Therefore, we were unable to assess post-CRT outcomes in ipilimumab-untreated patients to attribute any long term immune related changes in those treated with CRT alone vs. those patients treated with CRT and ipilimumab combined. In addition, RT can change the tumor microenvironment, and it would have been interesting to determine the acute and late effects of RT on the TME (3). While we report here associations of immune biomarkers with PFS and OS that were found to be significant, we acknowledge that our small sample size limits the precision of the Cox models due to limited events within our short evaluation time frame. Clinical variables were not adjusted for in the analysis, also due to limited number of events. There were 7 PFS events and 4 OS events within our study population (PP and ITT combined). Inclusion of the ITT group allowed us to look at more events when evaluating the immunological change from baseline to post-CRT, which was valuable in itself to investigate the effects of CRT since neither group had received any ipilimumab yet. Based on Kaplan-Meier estimates for the PP patients only, 12-month PFS was 81% and 12-month OS was 90% in our PP group; medians of neither end point were reached as previously reported (12). As with most phase I studies, a larger and ideally randomized trial would be needed to further explore immunotherapy and CRT in the definite setting and confirm our findings presented here.
The optimal timing of administration of ipilimumab immunotherapy with RT or CRT remains unanswered. In order to promote synergistic effects of CRT and ICB, these therapeutic modalities need to be given in a sequence relative to their proposed biological action. To reduce the potential cumulative toxicities of CRT with concurrent ICB, ipilimumab was administered post-CRT to not interfere with standard therapy. The decision to administer ipilimumab post-CRT was also made due to the significant lymphopenia that was expected as a result of the CRT as used in this study. Although absolute lymphocyte count (ALC) and myeloid cell counts were not collected as an outcome measure in this study, no patients receiving the 3 mg/kg ipilimumab dose had grade 3 toxicity of ALC or neutrophil count (12). In the 10 mg/kg dose group, 2 patients experienced a grade 3 ALC decrease and 1 patient experienced a grade 3 neutrophil count decrease (12). Recent data suggests that CTLA-4 blockade works more effectively at the time of antigen presentation and T cell priming that occurs in lymphoid tissues. This is in contrast to blocking the PD-1/PD-L1 axis which involves inhibiting tumor-induced immunoregulation of infiltrating T cells where PD-L1 is expressed in peripheral tissues (43). The presence and composition of immune infiltrate or “immunoscore” in tumors has been shown in some cases to be an important predictor of clinical benefit regardless of the treatment modality (44,45). In this study, pre-treatment biopsies were not required, therefore we were unable to examine tumor PD-L1 expression or other tumor-associated biomarkers that might have proven to be informative.
Our study results are relevant to clinical trial design in locally advanced CC. Our data suggests that multimodality therapy can indeed increase immunogenicity. Due to the observation that CRT increases PD-1 expression on CD4+ and CD8+ T cells, immunotherapy such as checkpoint blockade that targets the PD-1/PDL-1 axis may further enhance the response in patients who already have favorable outcomes. Clinical trials in metastatic and recurrent CC have shown that PD-1/PD-L1 blockade is well-tolerated and shows anti-tumor effects. Keynote-028 evaluated pembrolizumab at a dose of 10 mg/kg and demonstrated an overall response rate of 17% (95% CI: 5 to 37%) (46). Keynote-158 evaluated pembrolizumab at a dose of 200 mg/kg and demonstrated an overall response rate of 14.6% (95% CI: 7.8 to 24.2%) in patients with PD-L1-positive tumors (47). On the latter trial’s basis, pembrolizumab was granted accelerated FDA approval to patients with advanced PD-L1-positive CC whose disease progressed during or after chemotherapy. The Checkmate-358 study evaluated nivolumab at a dose of 240 mg every two weeks in recurrent/metastatic cervical, vaginal, or vulvar cancers and showed an overall response rate of 26.3% (95% CI: 9.1 to 51.2%) for CC (48). Ipilimumab has been tested in women with metastatic CC with disease recurrence after ≥ 1 line of prior chemotherapy. In that study, Lheureux et. al. showed that ipilimumab for patients with metastatic and recurrent CC was safe and toxicities were manageable, however the study did not meet its primary endpoint and ipilimumab had limited single agent activity (49). Improved response rates in CC could be achieved if ipilimumab is combined with a PD-1/PD-L1 mAb, as has been shown in advanced melanoma where combination checkpoint inhibitors showed better long-term efficacy compared to each single-agent group (50).
In the era of personalized medicine, emphasis is placed on determining which patients will derive maximal benefit from therapy to decrease non-responder toxicity and futile treatments. The data from this study contribute to the understanding of the immune regulation of CC, response to CRT, and immunotherapy. Knowledge of the immunotherapeutic effects of ICB has the potential to identify relevant biomarkers that may either predict response or enrich the patient population for those who are most likely to benefit from future combined immunotherapy strategies.
Supplementary Material
Translational Relevance.
Systematic monitoring of select immune biomarkers may offer insight into complementary mechanisms of action of immunotherapies and traditional treatments (e.g., chemoradiation, CRT), and may guide how best to design rational immune-modulating combinations. This study provides detailed immune profiling in newly diagnosed women with locally advanced cervical cancer who received CRT and sequential ipilimumab. Our findings demonstrate that CRT is by itself immune stimulating and induces T cell activation, including in some cases, of HPV oncogene-specific T cells. Addition of ipilimumab was able to sustain and, in the CD4+ T cell population, enhance T cell activation including expression of ICOS and PD-1. Our data suggest that ipilimumab after CRT may be able to strengthen the antitumor response to cervical cancer. Together with PD-1/PD-L1 inhibitors, this potential combination may provide a desirable immunological boost to patients at high risk for disease recurrence.
Acknowledgements
This study was supported by National Institutes of Health National Cancer Institute grants to the NRG Biospecimen Bank-Columbus (U24 CA196067), the NRG Oncology Operations Center (U10 CA180868) (H.A. Lankes), and the NRG Oncology Statistics and Data Management Center (U10 CA180822) (DM Enserro). Translational research was supported by grants from the Cancer Research Institute Clinic and Laboratory Integration Program (WM Kast, DM Da Silva), the University of Southern California Norris Comprehensive Cancer Center Women’s Auxilliary Club (YG Lin, DM Da Silva), National Cancer Institute R01 CA074397 (WM Kast, JG Skeate, DM Da Silva), and the University of Southern California Norris Comprehensive Cancer Center Support Grant Award P30CA014089 (DM Da Silva, WM Kast).
We thank the following NRG Oncology/Gynecologic Oncology Group member institutions who participated in this study: Augusta University Medical Center, University of Oklahoma Health Sciences Center, University of Southern California, Thomas Jefferson University Hospital, University of California at Davis, and Virginia Commonwealth University. We are grateful to Kim Lühen, Suma Gudipati, Heike Brand, and Elena Chavez-Juan for technical assistance in the USC Immune Monitoring Core Laboratory.
Footnotes
Disclaimer
This study was designed by the investigators, who oversaw all data collection and interpretation. Representatives from the National Institutes of Health, Bristol Myers Squibb, and Cancer Research Institute had no role in the design, accrual, management or analysis of the data, interpretation of the results, or writing of the article.
Disclosure of Potential Conflicts of Interest
DMD, DME, JGS, HQP, HAL, and WMK have no conflicts to disclose. JSM is a consultant for Varian Medical Systems and reports receiving honoraria and travel reimbursement from AstraZeneca. KM reports receiving honoraria from Chugai, textbook editorial support from Springer, and travel reimbursement from VBL Therapeutics. KMM is a consultant for Tessa Therapeutics and Clovis Oncology. SAG is a consultant for Seattle Genetics; reports receiving research grants from GlaxoSmithKline, Merck, Genentech-Roche, Takeda, Seattle Genetics, Advaxis, Bristol-Meyers Squibb, Abbvie and Tesaro; received travel reimbursement from Intuitive Surgical and Merck; participates in Speakers’ Bureau for Tesaro/GlaxoSmithKline. YGL is an employee of Genentech-Roche; has stock ownership in Genentech-Roche; is a consultant for Venn Biosciences Corporation. RJS is a consultant for Celsion, Incyte, Flatiron Health, Immunogen, and Clovis Oncology. MJB is a consultant for Genentech-Roche, Sanofi, Acceleron Pharma, Merrimack, Oxigene, Threshold Pharmaceuticals, Immunogen, Clovis Oncology, Tesaro, and AstraZeneca.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: a cancer journal for clinicians 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Chemoradiotherapy for Cervical Cancer Meta-Analysis C. Reducing uncertainties about the effects of chemoradiotherapy for cervical cancer: a systematic review and meta-analysis of individual patient data from 18 randomized trials. J Clin Oncol 2008;26(35):5802–12 doi 10.1200/JCO.2008.16.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol 2015;16(13):e498–509 doi 10.1016/S1470-2045(15)00007-8. [DOI] [PubMed] [Google Scholar]
- 4.Dyer BA, Zamarin D, Eskandar RN, Mayadev JM. Role of Immunotherapy in the Management of Locally Advanced and Recurrent/Metastatic Cervical Cancer. J Natl Compr Canc Netw 2019;17(1):91–7 doi 10.6004/jnccn.2018.7108. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 2009;114(3):589–95 doi 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallahan D, Kuchibhotla J, Wyble C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer research 1996;56(22):5150–5. [PubMed] [Google Scholar]
- 7.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. The Journal of clinical investigation 2014;124(2):687–95 doi 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182(2):459–65 doi 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271(5256):1734–6 doi 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 10.Pecorelli S Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet 2009;105(2):103–4 doi 10.1016/j.ijgo.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 11.Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017;18(3):e143–e52 doi 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayadev JS, Enserro D, Lin YG, Da Silva DM, Lankes HA, Aghajanian C, et al. Sequential Ipilimumab After Chemoradiotherapy in Curative-Intent Treatment of Patients With Node-Positive Cervical Cancer. JAMA Oncol 2020;6(1):92–9 doi 10.1001/jamaoncol.2019.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis JM, Henson V, Slack R, Ng J, Hartzman RJ, Katovich Hurley C. Frequencies of HLA-A2 alleles in five U.S. population groups. Predominance Of A*02011 and identification of HLA-A*0231. Human immunology 2000;61(3):334–40. [DOI] [PubMed] [Google Scholar]
- 14.Kast WM, Brandt RM, Sidney J, Drijfhout JW, Kubo RT, Grey HM, et al. Role of HLA-A motifs in identification of potential CTL epitopes in human papillomavirus type 16 E6 and E7 proteins. J Immunol 1994;152(8):3904–12. [PubMed] [Google Scholar]
- 15.Ressing ME A Sette, R. M. Brandt, J. Ruppert, P. A. Wentworth, M. Hartman, , C. Oseroff HMG, C. J. Melief, and W. M. Kast. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. The Journal of Immunology 1995;154:5934–43. [PubMed] [Google Scholar]
- 16.Rudolf MP, Man S, Melief CJ, Sette A, Kast WM. Human T-cell responses to HLA-A-restricted high binding affinity peptides of human papillomavirus type 18 proteins E6 and E7. Clin Cancer Res 2001;7(3 Suppl):788s–95s. [PubMed] [Google Scholar]
- 17.Wentzensen N, Schiffman M, Dunn T, Zuna RE, Gold MA, Allen RA, et al. Multiple human papillomavirus genotype infections in cervical cancer progression in the study to understand cervical cancer early endpoints and determinants. International journal of cancer 2009;125(9):2151–8 doi 10.1002/ijc.24528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Sanjose S, Quint WG, Alemany L, Geraets DT, Klaustermeier JE, Lloveras B, et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol 2010;11(11):1048–56 doi 10.1016/S1470-2045(10)70230-8. [DOI] [PubMed] [Google Scholar]
- 19.Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999;397(6716):263–6 doi 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Paulete AR, Labiano S, Rodriguez-Ruiz ME, Azpilikueta A, Etxeberria I, Bolanos E, et al. Deciphering CD137 (4–1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur J Immunol 2016;46(3):513–22 doi 10.1002/eji.201445388. [DOI] [PubMed] [Google Scholar]
- 21.Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, et al. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2016;2:16086 doi 10.1038/nrdp.2016.86. [DOI] [PubMed] [Google Scholar]
- 22.Smola S, Trimble C, Stern PL. Human papillomavirus-driven immune deviation: challenge and novel opportunity for immunotherapy. Ther Adv Vaccines 2017;5(3):69–82 doi 10.1177/2051013617717914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Draper LM, Kwong ML, Gros A, Stevanovic S, Tran E, Kerkar S, et al. Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed against E6. Clin Cancer Res 2015;21(19):4431–9 doi 10.1158/1078-0432.CCR-14-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevanovic S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol 2015;33(14):1543–50 doi 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otter SJ, Chatterjee J, Stewart AJ, Michael A. The Role of Biomarkers for the Prediction of Response to Checkpoint Immunotherapy and the Rationale for the Use of Checkpoint Immunotherapy in Cervical Cancer. Clin Oncol (R Coll Radiol) 2019. doi 10.1016/j.clon.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Minion LE, Tewari KS. Cervical cancer - State of the science: From angiogenesis blockade to checkpoint inhibition. Gynecologic oncology 2018;148(3):609–21 doi 10.1016/j.ygyno.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farsaci B, Donahue RN, Grenga I, Lepone LM, Kim PS, Dempsey B, et al. Analyses of Pretherapy Peripheral Immunoscore and Response to Vaccine Therapy. Cancer Immunol Res 2016;4(9):755–65 doi 10.1158/2326-6066.CIR-16-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czystowska M, Gooding W, Szczepanski MJ, Lopez-Abaitero A, Ferris RL, Johnson JT, et al. The immune signature of CD8(+)CCR7(+) T cells in the peripheral circulation associates with disease recurrence in patients with HNSCC. Clin Cancer Res 2013;19(4):889–99 doi 10.1158/1078-0432.CCR-12-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 2014;9(2):e87705 doi 10.1371/journal.pone.0087705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proceedings of the National Academy of Sciences of the United States of America 2008;105(39):14987–92 doi 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res 2010;16(10):2861–71 doi 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res 2013;1(4):229–34 doi 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yi JS, Ready N, Healy P, Dumbauld C, Osborne R, Berry M, et al. Immune Activation in Early-Stage Non-Small Cell Lung Cancer Patients Receiving Neoadjuvant Chemotherapy Plus Ipilimumab. Clin Cancer Res 2017;23(24):7474–82 doi 10.1158/1078-0432.CCR-17-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res 2011;71(16):5445–54 doi 10.1158/0008-5472.CAN-11-1138. [DOI] [PubMed] [Google Scholar]
- 35.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol 2010;37(5):473–84 doi 10.1053/j.seminoncol.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang C, Welsh JW, de Groot P, Massarelli E, Chang JY, Hess KR, et al. Ipilimumab with Stereotactic Ablative Radiation Therapy: Phase I Results and Immunologic Correlates from Peripheral T Cells. Clin Cancer Res 2017;23(6):1388–96 doi 10.1158/1078-0432.CCR-16-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ou Y, Cannon MJ, Nakagawa M. Regulatory T Cells in Gynecologic Cancer. MOJ Immunol 2018;6(2):34–42. [PMC free article] [PubMed] [Google Scholar]
- 38.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 2009;206(8):1717–25 doi 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menard C, Ghiringhelli F, Roux S, Chaput N, Mateus C, Grohmann U, et al. Ctla-4 blockade confers lymphocyte resistance to regulatory T-cells in advanced melanoma: surrogate marker of efficacy of tremelimumab? Clin Cancer Res 2008;14(16):5242–9 doi 10.1158/1078-0432.CCR-07-4797. [DOI] [PubMed] [Google Scholar]
- 40.Ressing ME, van Driel WJ, Celis E, Sette A, Brandt MP, Hartman M, et al. Occasional memory cytotoxic T-cell responses of patients with human papillomavirus type 16-positive cervical lesions against a human leukocyte antigen-A *0201-restricted E7-encoded epitope. Cancer Res 1996;56(3):582–8. [PubMed] [Google Scholar]
- 41.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. The Journal of clinical investigation 2007;117(5):1175–83 doi 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Visconti L, Nelissen K, Deckx L, van den Akker M, Adriaensen W, Daniels L, et al. Prognostic value of circulating cytokines on overall survival and disease-free survival in cancer patients. Biomark Med 2014;8(2):297–306 doi 10.2217/bmm.13.122. [DOI] [PubMed] [Google Scholar]
- 43.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017;170(6):1120–33 e17 doi 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313(5795):1960–4 doi 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 45.Angell HK, Bruni D, Barrett JC, Herbst R, Galon J. The Immunoscore: Colon Cancer and Beyond. Clin Cancer Res 2019. doi 10.1158/1078-0432.CCR-18-1851. [DOI] [PubMed] [Google Scholar]
- 46.Frenel JS, Le Tourneau C, O’Neil B, Ott PA, Piha-Paul SA, Gomez-Roca C, et al. Safety and Efficacy of Pembrolizumab in Advanced, Programmed Death Ligand 1-Positive Cervical Cancer: Results From the Phase Ib KEYNOTE-028 Trial. J Clin Oncol 2017;35(36):4035–41 doi 10.1200/JCO.2017.74.5471. [DOI] [PubMed] [Google Scholar]
- 47.Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 2019;37(17):1470–8 doi 10.1200/JCO.18.01265. [DOI] [PubMed] [Google Scholar]
- 48.Naumann RW, Hollebecque A, Meyer T, Devlin MJ, Oaknin A, Kerger J, et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J Clin Oncol 2019;37(31):2825–34 doi 10.1200/JCO.19.00739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lheureux S, Butler MO, Clarke B, Cristea MC, Martin LP, Tonkin K, et al. Association of Ipilimumab With Safety and Antitumor Activity in Women With Metastatic or Recurrent Human Papillomavirus-Related Cervical Carcinoma. JAMA Oncol 2018;4(7):e173776 doi 10.1001/jamaoncol.2017.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19(11):1480–92 doi 10.1016/S1470-2045(18)30700-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplemental files).