

Summary
Herpes virus entry mediator (HVEM) regulates positive and negative signals for T-cell activation through co-signaling pathways. Dysfunction of the HVEM co-signaling network is associated with multiple pathologies related to autoimmunity, infectious disease and cancer, making the associated molecules biologically and therapeutically attractive targets. HVEM interacts with three ligands from two different superfamilies using two different binding interfaces. The engagement with ligands CD160 and B- and T- lymphocyte attenuator (BTLA), members of immunoglobulin superfamily, is associated with inhibitory signals, whereas inflammatory responses are regulated through the interaction with LIGHT from the TNF superfamily. We computationally redesigned the HVEM recognition interfaces using a residue-specific pharmacophore approach, ProtLID, to achieve switchable binding specificity. In subsequent cell-based binding assays the new interfaces, designed with only single or double mutations, exhibited selective binding to only one or two out of the three cognate ligands.
Keywords: Residue-specific pharmacophores, ProtLID, HVEM, Computational interface design, Switchable-binding selectivity
Graphical Abstract
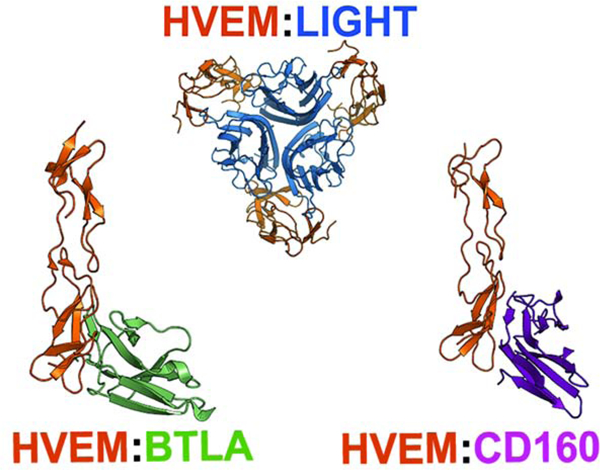
eTOC blurb
Shrestha et al. uses a residue-based pharmacophore a computational approach to design mutations for the interface of HVEM to make it selective to one or two out of its three cognate ligands. In cell assay experiments 15 of the 25 designed single and double point mutants proved to introduce statistically significant selectivity.
Introduction
Herpes virus entry mediator (HVEM) provides stimulatory and inhibitory co-signaling pathways (signal 2) for host immune responses (Murphy et al., 2006; Ward-Kavanagh et al., 2016) subsequent to T-cell receptor (TCR) engagement with the peptide/MHC complex (signal 1) (Bretscher and Cohn, 1970; Lafferty and Cunningham, 1975). HVEM interacts with multiple ligands from the tumor necrosis factor (TNF) and Immunoglobulin (Ig) superfamilies to provide functionally orthogonal signals. HVEM transmits a co-inhibitory signal to the T-cell upon binding CD160 (Cai et al., 2008) or B- and T- lymphocyte attenuator (BTLA) (Sedy et al., 2005) from the Ig superfamily, but elicits a co-stimulatory signal upon interaction with LIGHT or lymphotoxin α (LTα) from the TNF superfamily (Mauri et al., 1998). The actual functional role of HVEM (Cai et al., 2008; Cai and Freeman, 2009) depends on the identity of the ligands engaged, expression patterns on different cell types, and the trans or cis configuration of molecular interactions.
The extracellular domain of HVEM, a type I membrane protein, is composed of four cysteine-rich domains (CRDs); CRD1-CRD4, of which only CRD1–3 have been structurally characterized (Bodmer et al., 2002). The ectodomain of HVEM has two unique binding sites, with CRD1 of HVEM containing the binding sites for BTLA (Sedy et al., 2005) and CD160 (Cai et al., 2008), and CRD2 and CRD3 providing the binding sites for LIGHT and LTα (del Rio et al., 2010; Ware, 2008). Ligands in different superfamilies exhibited modestly cooperative interactions, as demonstrated by the twofold enhancement of BTLA and CD160 binding affinity when LIGHT is also present (Cai et al., 2008; Cai and Freeman, 2009). Co-expression of HVEM and BTLA or CD160 leads to the formation of a cis complex that inhibits binding of other ligands (LIGHT, BTLA, and CD160) interacting in trans (Cheung et al., 2009a). Engagement of membrane LIGHT with HVEM presumably hinders trans HVEM:BTLA interaction due to membrane restriction. In contrast, glycosylphosphatidylinositol (GPI) linked CD160 can form ternary complexes with membrane bound HVEM:LIGHT (Steinberg et al., 2011).
LIGHT, a member of TNF superfamily, is a type II transmembrane protein that forms homotrimers on the cell surface (Mauri et al., 1998). LIGHT has two alternatively spliced isoforms, a cytosolic and a membrane-bound form that lacks an intracellular domain for signaling. Interaction with the trimeric TNF-ligands, either LIGHT or LTα, directs clustering of HVEM and results in the recruitment of cytosolic TNFR-associated factor (TRAF) adaptor family of ubiquitin E3 ligases for cellular activation, differentiation, and survival signaling (Cheung et al., 2009b). Formation of these multimeric assemblies involving LIGHT and HVEM provides a co-stimulatory signal. LIGHT acts as a regulator of its receptor HVEM, the up-regulation of LIGHT during T-cell activation or DC maturation is inversely affecting HVEM expression (Morel et al., 2000; Tamada et al., 2000).
BTLA shares functional and structural similarity with other checkpoint receptors, such as CTLA-4 and PD-1 (Watanabe et al., 2003). BTLA is composed of an Ig domain with a cytoplasmic tail for signaling. BTLA delivers an inhibitory signal to immune cells (Cheung et al., 2009b; Steinberg et al., 2011). Engagement of BTLA with HVEM induces phosphorylation of the BTLA immunoreceptor tyrosine-based inhibitory motif, resulting in recruitment of tyrosine phosphatases SHP1 and SHP2, which reduces cellular activation and growth (Gavrieli et al., 2003; Sedy et al., 2005; Watanabe et al., 2003). The outcome of HVEM:BTLA interaction also impacts LIGHT engagement – membrane bound LIGHT disrupts cis HVEM:BTLA interaction by promoting HVEM activation, but soluble LIGHT promotes HVEM:BTLA inhibitory interactions (Steinberg et al., 2011).
Human CD160 was originally described as a cell surface antigen tethered through a GPI link (Maiza et al., 1993); however, subsequent reports also identified an alternative isoform containing transmembrane and signaling domains, which activate the ERK1/2 signaling pathway (Giustiniani et al., 2009). In both isoforms, the extracellular domain belonging to IgSF engages ligands that include MHC molecules (Agrawal et al., 1999; Barakonyi et al., 2004; Fons et al., 2006; Le Bouteiller et al., 2011) and induces NK and T cell activity. CD160 interacts with HVEM competitively at the same binding site as BTLA (Cai et al., 2008). The function of CD160:HVEM interaction varies on different cell types. Trans CD160 and HVEM interaction delivers a co-inhibitory signal in T lymphocytes to suppress T cell proliferation and cytokine production (Cai et al., 2008; Vigano et al., 2014), whereas due to the co-stimulatory signals in NK cells, it induces effector activity of NK cell in conjunction with cytokines and enhances lytic activity (Sedy et al., 2013; Tu et al., 2015).
HVEM mutations are linked to multiple pathologies - autoimmunity, infectious disease, and cancer (Boice et al., 2016; Coenen et al., 2009; del Rio et al., 2010; Ward-Kavanagh et al., 2016); therefore, HVEM has generated significant attention as a therapeutic target. Due to the bi-directional effects of HVEM in the immune system, careful and selective modulation of HVEM function is necessary to regulate the balance of the immune response. Blocking the co-stimulatory activity of HVEM:LIGHT interactions with specific biologics such as mABs or fusion proteins (del Rio et al., 2010) can modulate the immune system and control inflammatory and autoimmune pathology in transgenic mouse models (Shaikh et al., 2001). The immuno-suppressive role of HVEM:BTLA interaction was shown to counterbalance LIGHT-activated inflammation (Murphy and Murphy, 2010) in infectious diseases (Breloer et al., 2015). BTLA-specific checkpoint inhibitors (Sedy et al., 2014) are employed for anti-tumor treatment (Derre et al., 2010). Although CD160 performs cell-specific biological functions (Ward-Kavanagh et al., 2016), negative signaling through HVEM:CD160 engagement might also be useful in attenuating autoimmune diseases.
Computational approaches have supported remarkable achievements for the design of novel folds (Kuhlman et al., 2003), optimization of existing binders for the realization of modified or new enzymatic functions (Rothlisberger et al., 2008), and novel binding functions (Looger et al., 2003). Computational design methods facilitate the assessment of large-scale sequence variation to reduce the number of designs required for subsequent experimental validation (Lippow et al., 2007; Mandell and Kortemme, 2009). One important application of computational interface design includes the alteration of binding specificity. Interface design for selectivity can consider both positive and negative design elements to jointly optimize the desired interactions and reduce unwanted interactions (Bolon et al., 2005; Havranek and Harbury, 2003; Shrestha et al., 2019).
Here, we describe the application of ProtLID (Protein Ligand Interface Design) (Shrestha et al., 2019; Yap and Fiser, 2016) to computationally redesigned the HVEM binding interface for enhanced selectivity. ProtLID generates residue-based pharmacophores (rs-pharmacophore) over the binding interfaces of HVEM for LIGHT, CD160, and BTLA. These rs-pharmacophores identify positions for the introduction of mutations predicted to alter selectivity. A number of variants were experimentally validated using cell-based binding assays. We discovered single and double mutant variants of HVEM that support a selective recognition of its cognate ligands. These new constructs can serve as selective reagents or possible starting points in drug development applications.
Results
Generation of residue-specific pharmacophores
HVEM has multiple interacting ligands (LIGHT, BTLA, CD160, LTα ). In addition to ligand engagement, the functionality of HVEM pathway is sensitive to other variables – molecular form, expression pattern, and trans and cis interaction configuration. Understanding the context-dependent molecular interactions of HVEM with its cognate partners can reveal underlying mechanistic details and facilitate development of novel strategies to modulate its function. Engineered variants of HVEM that can selectively engage subsets of its ligands can facilitate this discovery process (Fig. 1.).
Figure 1.

Interfaces in the HVEM:LIGHT:BTLA:CD160 complex and schema of design process. Three rows refer to binary complexes of HVEM with LIGHT, BTLA, and CD160m, respectively. (A) Crystal structures of complexes of LIGHT, CD160, and BTLA with HVEM. (B) Corresponding cartoon representation: yellow eclipse represents cysteine rich domains of HVEM and rectangle-like shape represent ligand monomers. (C) Interface residues from both proteins are represented using circles within the white area and their corresponding interactions are shown in black line. (D) rs-pharmacophore residue preference as calculated by ProtLID (E) Rs-pharmacophore guided design of HVEM interface. (F) Effective mutations for modulating HVEM specificity in six different ways.
The recognition surfaces of BTLA and CD160 substantially overlap on CRD1 of HVEM, as defined by their respective complexes - HVEM:BTLA (PDB code: 2AW2) (Compaan et al., 2005) and HVEM:CD160 (6NG3)(Liu et al., 2019). (Equivalent positions of HVEM interface residues among these PDB files are listed in Suppl. Table 6.) The binding interface of the HVEM:BTLA complex is formed by interactions between 15 residues from BTLA and 12 residues from HVEM (Supp. Table 1), whereas the CD160:HVEM binding interface contains CD160 and HVEM, each contribute 10 residues. In the present work, we excluded Cys residues from the interface definition as they are critical for maintaining the CRD fold (Cys Rich Domains with three disulfide bonds). Although BTLA and CD160 both belong to the Immunoglobulin Superfamily (IgSF) and exhibit the same topology, they share only 16% sequence identity. Despite their limited homology, BTLA and CD160 share eight common residues that interact with HVEM (Supp. Table 1). Furthermore, some residues on the HVEM interface (D45, T71, G72, and V74) interact with physio-chemically similar residues, including G66, N28, I29, and L123 on CD160 and G76, Q37, L38, and I124 on BTLA (Supp. Table 1). Post-translational modifications currently cannot be included in the rs-pharmacophopre calculation, and these modifications can make major difference in binding affinity and specificity. N28 of CD160 is one such possible site, which is glycosylated.
BTLA- and CD160-specific rs-pharmacophores were generated using our ProtLID program(Yap and Fiser, 2016). Rs-pharmacophore describes the spatial positions and residue preferences of a hypothetical complementary interface for a given target binding site that are inferred from extensive molecular dynamics simulations of single amino acid probes. ProtLID calculation progresses through four major steps: (1) identifying interface residues using the CSU program (Sobolev et al., 1999). Qualifying residue interactions between the receptor and ligand must satisfy CSU classification for legitimate interactions and must be within 4.0 Å distance (Sobolev et al., 1999) and at least 1 Å2 accessible surface area; (2) A hypothetical mesh is generated over interface residues using a 1 Å distance and probe radius over the solvent accessible region of the interface residues, which serves as starting points for subsequent MD simulations; (3) performing extensive molecular dynamics (MD) simulations with single residue probes starting from each mesh point using AMBER (Case et al., 2005) with seven replicas, each with different starting orientations; (4) a surface normalized interacting residue preference is determined over the mesh points and assigned to interface residues.
The differences observed between the ligand specific rs-pharmacophores and the wild type interface can be utilized to guide the selection of positions and types of mutations to achieve a desired specificity. The patches obtained from rs-pharmacophore constructed on CD160 and BTLA interfaces were structurally aligned with the wild type HVEM interface (Supp. Table 1). Next, the rs-pharmacophores were used to select positions and residue types for designing interface selectivity. On average, the algorithm suggested eight variants per position for CD160 and BTLA (Suppl. Table 2). Positions with well defined, one-to-one interacting residue contacts usually accept a smaller number of variants such as G66 in CD160 that interacts only with D45 in HVEM and for which F and I residues are suggested by the CD160 specific rs-pharmacophore. Whereas residues that participate in a network of interactions, such as Q124 in CD160 that interacts with K56, G72, S58, T73 in HVEM has a lager variety of hypothetical contacts predicted by the rs-pharmacophore (DEHKNPQRSTWY) (Suppl. Table 1 and 2). On average, two contacts per residue were identified for the CD160:HVEM and BTLA:HVEM interface residues (Suppl. Table 2).
ProtLID generated rs-pharmacophores for the HVEM:LIGHT interface, which is formed by the contribution of 14 and 12 residues from HVEM and LIGHT, respectively (Suppl. Table 3). These interfacial residues are distributed over two adjacent LIGHT protomers and interact with the CRD2 and CRD3 domains of HVEM. The rs-pharmacophore suggested an average of 9 variants per residue for the exploration of LIGHT-specific HVEM interface design (Suppl. Table 4).
The predicted rs-pharmacophore for CD160, BTLA and LIGHT recapitulated the wild type residues at 45%, 62% and 75%, respectively, of the positions contributing to the HVEM interface (Suppl. Tables 2 and 4). With the exception of CD160, this represents a favorable enrichment of wild type residues within the calculated pharmacophores, as compared with the randomly expected 58%, 24% and 7%, respectively. The random model calculates the probability of matching the wild type interface residue from the same number of choices as the number of residues suggested by the rs-pharmacophore. In addition, in the case of LIGHT, the five positions where the wild type residues are not recapitulated include A85 and G89, which are excluded from the pharmacophore calculation due to the lack of functionally discriminative side-chain atoms.
Selection of variants for interface redesigning
Two distinct surfaces on the HVEM molecule function to recognize ligands within two different superfamilies (i.e., the IgSF and TNFSF). The BTLA and CD160 interfaces overlap on the surface of HVEM CRD1. Comparison of the rs-pharmacophores generated for CD160 and BTLA, with the corresponding wild type interface of HVEM predicted mutations for achieving the desired selectivity (Suppl. Table 1 and 2). As part of the design process, positions with the same residue types in both rs-pharmacophores and that also matches the wild type HVEM interface residue are eliminated from further consideration, as these are not likely to provide discrimination (Suppl. Fig. 2). Priority was given to rs-pharmacophore differences at equivalent structural positions that were predicted to either enhance or disrupt the interface.
A total of six mutations distributed over five positions, were selected to re-engineer the LIGHT specific interface (Suppl. Table 3). Most of these mutations were selected to disrupt the targeted interaction; therefore, amino acids that were excluded from the rs-pharmacophore were chosen for experimental validation. For instance, for D100 in HVEM, only a single interacting residue was identified in LIGHT, R226 (Suppl. Tables 3 and 4) and these form a hydrogen bond or salt bridge interaction, therefore we selected R and K residues to replace D100 in HVEM, which were excluded from the preferred list of residues for this position in the rs-pharmacophore (DEHNPQSTY) in order to introduce a disruptive electrostatic interaction.
To modulate the HVEM recognition surface shared by BTLA and CD160, we followed a selection strategy similar to that employed for engineering PD-1 variants that selectively bind to only one of its ligand, PD-L1 (Shrestha et al., 2019). First, we focused on positions proximal to only one ligand. For instance, E52 of HVEM is near R42 of BTLA, but the binding interface for CD160 does not include an interaction with E52 (Suppl. Table 1 and Fig. 6). We sought to introduce a disrupting mutation at this position, which would reduce the strength of the HVEM:BTLA, while not impacting the HVEM:CD160 interaction. Positively charged residues (R or K) were selected to mutate E52 in HVEM, as these were not listed in rs-pharmacophore generated for BTLA (DENPQSTY) (Suppl. Table 2).
Figure 6.

Structural environment of some of the selective mutants. (A) E52K for HVEM:BTLA, (B) S58Q for HVEM:CD160. Upper and lower panels are for wild type and mutant residue. HVEM, CD160, and BTLA are colored with magenta, green, and cyan, respectively.
Next, we focused on interface residues involved in a complex network of interactions with multiple contact residues. Based on the calculated rs-pharmacophores, these residues are predicted to tolerate a more diverse set of substitutions, therefore providing greater flexibility for exploration. This situation is the case for Q124 of CD160 and E125 of BTLA (Suppl. Table 1). These two residues from the two ligands are in close proximity to the same three residues (S58, G72, and T73) on HVEM (Suppl. Table 1). The calculated rs-pharmacophores for CD160 has a larger set of preferred residues than BTLA. The additional and unique rs-pharmacophore residues (DEHPS) for Q124 in CD160 appear to compensate hydrogen-bond donor or acceptor properties (excluding proline), and these are absent for E125 in BTLA, presumably to avoid the unfavorable contacts between same charges. Shared residue preferences at these positions include positively charged (RK), polar (QNT) and aromatic (WY) residues (Suppl. Table 1). We chose S58 for mutation on HVEM because it interacts with three key residues of CD160 (D67, R122, and Q124) and is proximal to E125 of BTLA, as well. A total of four variants (S58L, S58R, S58K, and S58Q) were selected for experimental testing.
Finally, we also explored residues that were modestly distal to the interface, which did not satisfy the default distance criteria required for binding (i.e., at least one atom from the interacting residues being within 4 Å of each other). However, considering the dynamics of the interface structures and especially if a mutation from a smaller to a larger sidechain takes place, these residues can participate in the interface. These positions, which include L87F/W/Y and L70D in HVEM, were identified by relaxed criteria (increasing distance thresholds) for interface definition.
Synergy of double mutants
After a first round of experimental testing of 18 single point mutations, we considered combinations of eight single mutants to generate seven double mutants with the aim of achieving higher selectivity through additivity or synergy (Suppl. Table 5).
For instance, when focusing to establish a BTLA-selective HVEM variant, single point mutations H86I and D100R on HVEM were effective in reducing HVEM binding to LIGHT, but binding to CD160 was only moderately reduced. Therefore, double mutants of D100R with three others, H86I, M103K and G89I were constructed to explore a possible synergy. Among these newly constructed double mutants, D100R with M103K achieved the highest level of BTLA-selective binding, with almost complete elimination of binding to both LIGHT and CD160 (Fig. 2B). We also combined D100R with G89I, in order to reduce HVEM binding to CD160. Both single mutants diminished LIGHT binding and when combined showed reduced LIGHT and CD160 binding, thus generating an HVEM variant selective for BTLA binding.
Figure 2.
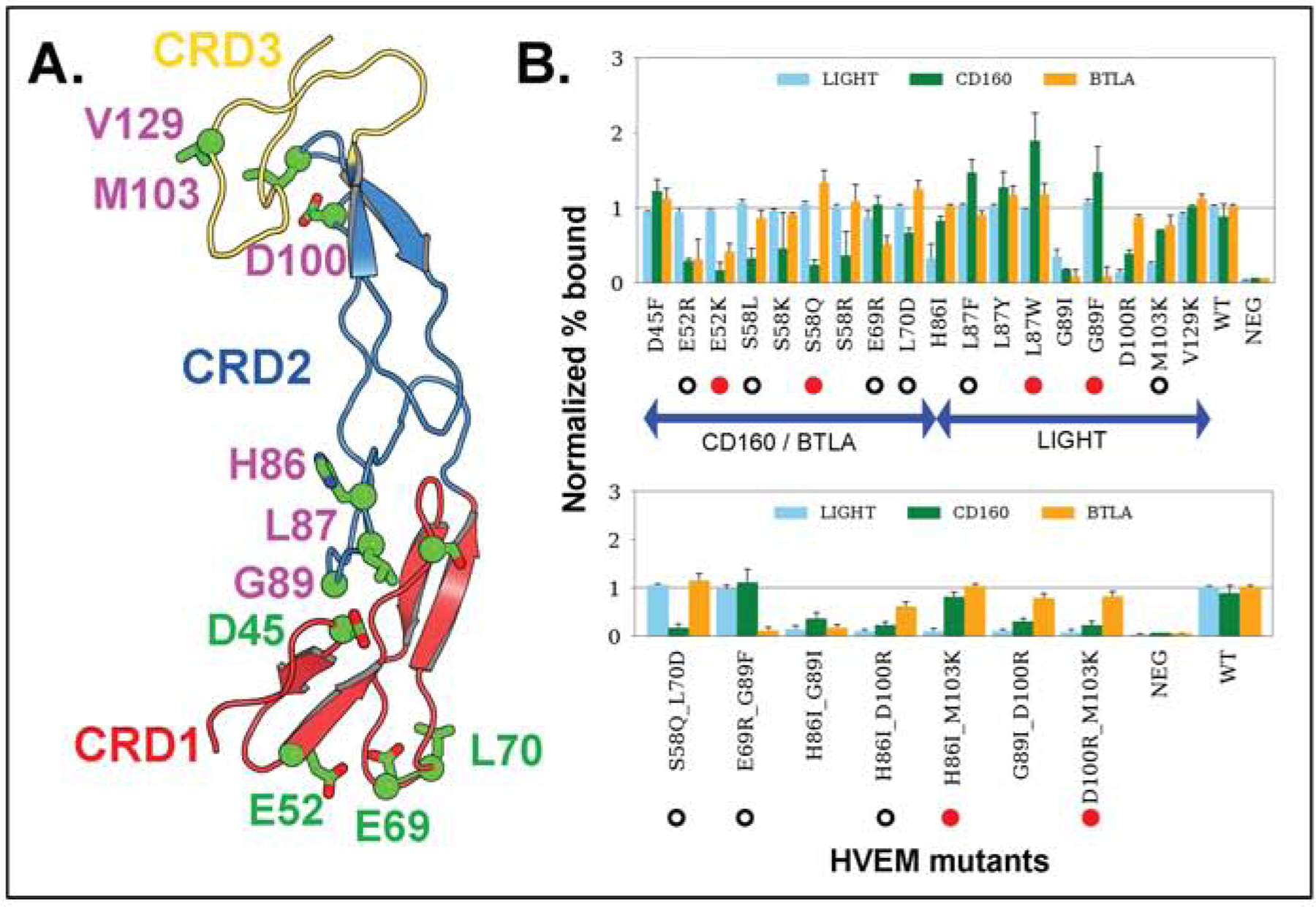
Engineered positions in HVEM and their binding affinity. (A) Cartoon representation of HVEM domains - CRD1 (red), CRD2 (blue), and CRD3 (yellow). Targeted residue positions are in stick and ball model and highlighted in colors. Residues with green color represent the mutational sites for BTLA or CD160 and magenta color shows residues binding with LIGHT. (B) Experimentally observed Normalized % Bound measures for three ligands - LIGHT, CD160, and BTLA. Single and double point mutations are in the upper and lower panels, respectively. Mutations represented by red circles are the most effective for switching the binding specificities according to their p-values obtained from Welch’s t-test (Suppl. Table 5). Other mutations with statistically significant modulation of binding selectivity are marked with hollow circles.
Redesign the interface to switch binding specificity
We studied the HVEM interaction network with its three ligands through the design of 18 single mutants (Fig. 2A) and subsequently created seven double mutants. Site directed mutagenesis was performed, and all point mutations were sequence validated and expressed as GFP fusions presented on the surface of suspension adapted HEK-293 cells. Protein expression levels of mutants were statistically indistinguishable from WT HVEM (Suppl. Fig.3). Analysis of the HVEM mutants binding to its ligands was performed by high-throughput flow cytometry. The percent of HVEM-expressing cells bound to its ligands was determined and the data normalized to the highest ligand concentration for wild-type HVEM binding.
Out of 25 HVEM mutants examined experimentally, we identified 15 mutants, including five double mutants, covering a total of nine positions on HVEM (Fig. 2B), the most effective of which are delineated in Table 1. EC50 and Bmax values for all 15 mutants are listed in Fig 3. and shown in Suppl. 4.The corresponding concentration-dependent binding of ligands for the six most effective mutants are shown in Fig. 4. Concentration dependent titration curves for the remaining 9 variants are shown in Suppl. Fig. 6. Corresponding raw FACS plots of binding are shown for the most effective HVEM mutants and the wild type protein in Suppl. Figure 5.
Table1:
List of identified HVEM mutations for binding selectivity to it three ligands. Statistical p-values of pairwise comparisons are marked as: BTLA:LIGHT (italic), CD160:LIGHT (regular), and BTLA:CD160 (Bold).
| Mutations | TNFSF | IgSF | p-value (Welch’s t-test) | |
|---|---|---|---|---|
| LIGHT | CD160 | BTLA | ||
| E52K | + | − | − | 9.62E-03, 3.56E-03 |
| L87W | − | + | − | 6.58E-02, 5.15E-02 |
| D100R:M103K | − | − | + | 1.44E-03, 1.00E-03 |
| H86I:M103K | − | + | + | 1.22E-03, 7.31E-05 |
| G89F | + | + | − | 1.17E-02, 3.21E-03 |
| S58Q | + | − | + | 3.17E-03, 3.63E-04 |
Figure 3.
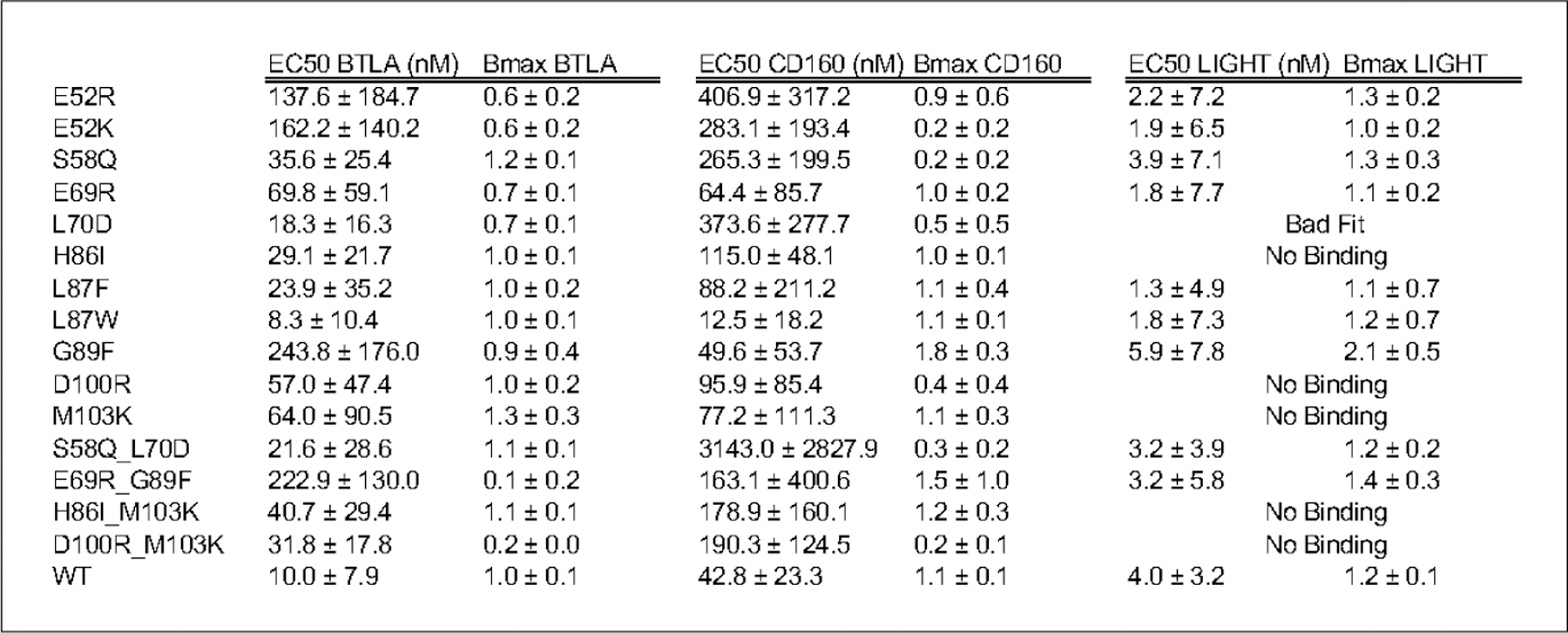
EC50 and Bmax values of binding of the hHVEM mutants to ligands BTLA, CD160 and LIGHT.
Figure 4.
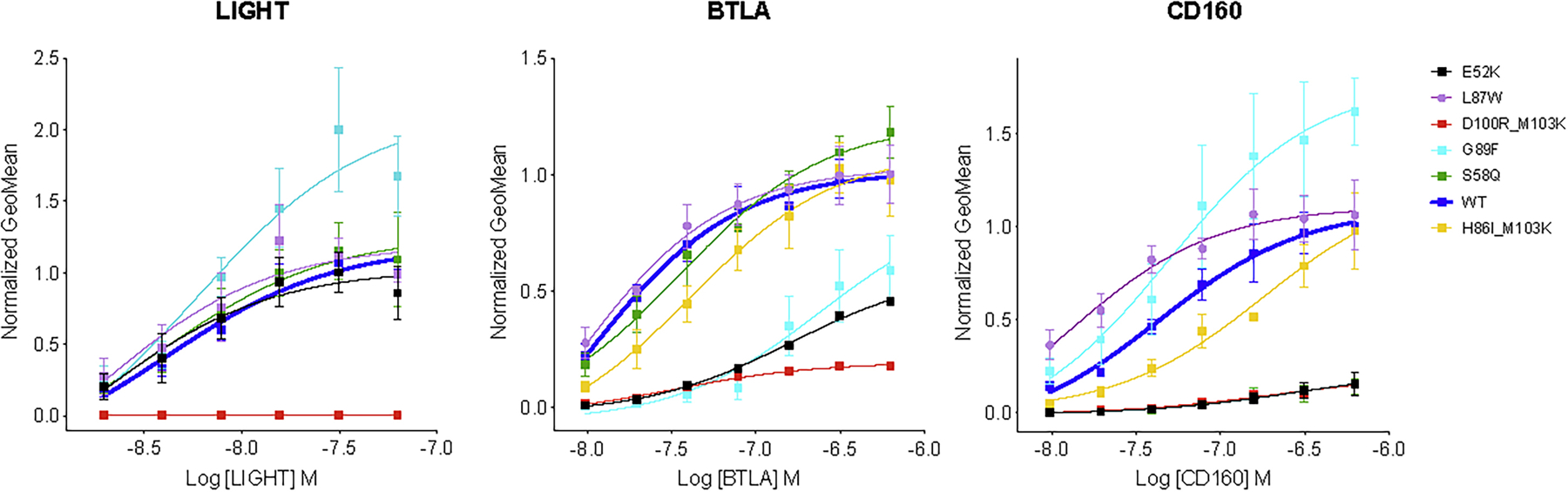
Geometric mean of percent bound ligand as a function of concentration. Panels show HVEM binding of ligands LIGHT, BTLA and CD160, respectively. Different colors of lines refer to different mutant variants of HVEM: black: E52K, purple: L87W, red: D100R_M103K, light blue: G89F, green: S58K and yellow H86I_M103K. Blue line corresponds to the wild type HVEM.
The overlapping binding sites of BTLA and CD160 on HVEM create competition for binding, and selectivity can be achieved either by positive or negative design. We also screened for designs that either preserved or eliminated binding to LIGHT. The S58Q single mutant (Table 1, Fig. 4) and the S58Q:L70D double mutant of HVEM (Suppl. Table 5, Fig. 3) exhibited the greatest selectivity for BTLA, while still preserving LIGHT binding. Specifically, these mutations slightly enhanced the binding specificity for BTLA and drastically decreased it for CD160, while leaving the interaction with LIGHT unchanged (Fig. 2B). Another double mutant (D100R:M103K) resulted in BTLA binding close to wild type, with substantial reductions in binding to both LIGHT and CD160 (Table 1).
Binding selectivity for CD160 relative to BTLA, while retaining close to wild type LIGHT binding, was achieved through the single HVEM G89F mutant using positive design principles, where HVEM binding to CD160 increased significantly. The G89F:E69R double mutant resulted in similar selectivity by drastically reducing BTLA binding, while LIGHT and CD160 binding remained close to wild type levels (Table1, Fig. 3). The HVEM L87W or L87F single mutants exhibited increased selectivity for CD160 through positive design, as binding to CD160 increased, while LIGHT and BTLA binding stayed close to wild type (Suppl. Table 5, Fig 3). LIGHT-selective binding was exhibited by the E52R or E52K single mutants, which reduced binding to both BTLA and CD160. Finally, the HVEM H86I:M103K double mutant reduced LIGHT binding, while preserving CD160 and BTLA binding at close to wild type levels (Table 1, Fig. 4).
One of the variables associated with bi-directional HVEM signaling (co-stimulatory or co-inhibitory) to T-cells is ligand engagement. The binding of LIGHT to HVEM signals T cell activation, whereas CD160 or BTLA results in down-regulation. Perturbing the binding site for CD160 and BTLA allows for exclusive binding of LIGHT to HVEM, which elicits the associated co-stimulatory signal; however, the overall effect of HVEM upon the concomitant engagement of LIGHT and BTLA/CD160 results in a co-inhibitory signal (Cai and Freeman, 2009). Achieving ligand selectivity with HVEM is primarily important for balancing the signal; therefore, our design efforts include an HVEM interface selective for LIGHT. LIGHT selective mutations such as E52K or E52R maintain wild type binding affinity for LIGHT, but disrupts the binding to CD160 (p-value 0.01) and BTLA (p-value 0.006) (Table. 1 and Fig. 2B, Fig 4).
Structural environment of selective mutants
We generated comparative models of the complexes with Modeller (Sali and Blundell, 1993) to examine the predicted interactions between interfacial residues. The E52K mutation in HVEM CRD1 experimentally reduces binding with BTLA and CD160. One speculative explanation is that this mutation introduces contacts unfavorable for CD160 binding because of the presence of H126 in CD160, which prefers K52 over E52 in HVEM (Fig 5A and 6A). Similarly, E52K (HVEM) could also be disruptive due to unfavorable electrostatic with R42 on BTLA (Fig. 5A and 6AA). In contrast, there are no contacts between E52 and any residues from LIGHT, consistent with the preserved wild type binding activity with LIGHT.
Figure 5.

Structural insight for selected mutations using homology models. Upper panel is for wild type residue positions – (A) E52K, (B) S58Q, (C) H86I/M103K whereas lower panel represents models of corresponding mutant structures. HVEM, CD160, BTLA, and LIGHT are colored with magenta, green, cyan, and yellow, respectively.
The S58Q mutant located on HVEM CRD1 (Fig. 5B and Fig. 6B) participates in interactions with BTLA and CD160 and results in different outcomes - it disrupts interaction with CD160 while significantly improving the binding with BTLA, with no appreciable effect on LIGHT binding . The S58Q mutation at the HVEM:BTLA interface could potentially create more favorable environment for E125 (OE2) with Q58 (NE2). But in case of CD160, the S58Q substitution in HVEM is unfavorable due to the orientation of hydrogen bond donor and acceptor atoms of R122 and Q124 (Fig. 6B). It presumably disrupts the contact observed in wild type between hydrogen bond acceptor S58 (HVEM) and hydrogen bond donor R122 (CD160). Another possible explanation is rearrangement of backbone structure upon mutation.
In another example, selective single point mutants were combined to enhance the binding selectivity of HVEM towards only BTLA and CD160, such as M103K and H86I. Structural modeling of the M103K mutation predicts unfavorable electrostatics with R226 in LIGHT. Likewise, H86I is predicted to disrupt favorable polar interactions between H86 and E175 of LIGHT (Fig. 5C). The non-proximal positions of these residues suggests that there is no synergy between these two mutations (M103K and H86I) when binding to LIGHT, and the resulting strong inhibition of the double mutant is the additive effect of the two individual mutations. For some mutations identified with the pharmacophore approach, including D100R, G89F, L87F, and L87W, the experimental outcomes did not agree with those predicted by the pharmacophore and may potentially require dynamic simulation to uncover the underlying structural mechanism, which may involve allosteric effects.
Discussion
HVEM is involved in a wide range of physiological immune responses, and is of central biomedical importance; for instance, it is frequently mutated in germinal center lymphomas(Cheung et al., 2010; Launay et al., 2012; Lohr et al., 2012), as loss of HVEM leads to B cell proliferation and drives germinal center lymphomas. Loss of HVEM supports tumor microenvironment. These effects are connected to the loss of inhibitory signals elicited by HVEM-BTLA engagement, which can be restored by administration of soluble HVEM ectodomain and promote tumor repression (Boice et al., 2016). HVEM is highly expressed in the gut epithelium and contributes to the pathogenesis of colitis (Boice et al., 2016). The mutations described in the current study were compared with the somatic mutations cataloged in the COSMIC database (Sondka et al., 2018). Of the 11 unique HVEM positions examined in this study, only three have a corresponding entry in the COSMIC database. D45F mutation was suggested in our study as a possible site to alter binding, but experimental evaluation did not show a statistically significant change in binding properties. The single somatic mutation with a clinical correlation was the D45G, which is associated with large intestinal cancer. The H86I mutation in our study statistically significantly reduces LIGHT binding and as might be predicted to cause T cell stimulation. A single corresponding sample entry in the COSMIC database reported a H86Y mutant and associated was it with malignant melanoma. Finally, the G89I or G89F mutations, which we demonstrate results in a complete abolishment of binding to all three ligands (G89I) or a sharp reduction in BTLA binding (G89F). The only corresponding entry in the COSMIC database reports a G89C mutation associated with lung carcinoma.
A recent study explored binding selectivity of HVEM using alanine scanning, saturation and combinatorial mutagenesis (Sedy et al., 2017). The authors found four positions to be important for BTLA binding selectivity, of which two were examined in our study. S58R was shown in both studies to reduce CD160 binding while maintaining or slightly increasing LIGHT and BTLA binding. The L70W mutation was reported to increase binding affinity for BTLA. We have not explored this specific mutation, but we did demonstrate that L70D exhibited a similar effect, increased binding to BTLA and a reduced affinity for CD160. However, this was not the most selective mutant we observed for this type of selectivity (i.e., the S58Q mutant was most effective) (Table 1, Fig. 2, 3 and 4).
In this work we employed a computational approach to identify mutant variants of HVEM that selectively bind to one or two of its three cognate ligands. A total of 25 mutations were proposed and 15 of those exhibited a statistically significant change in binding preferences. These variants will be useful for further dissecting the complex HVEM signaling network. The rs-pharmacophore based approach described in this work could serve as a paradigm for the rational design of binding specificities in biomedically important proteins and the development of new and enhanced biologics.
STAR Methods
Resource Availability
Lead Contact
Requests for further information or resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andras Fiser (andras.fiser@einsteinmed.org).
Materials Availability
Constructs generated in this study are available upon request.
Data and Code Availability
This study did not generate new code.
Experimental Models and Subject Details
Experimental models
We used the human embryonic kidney HEK 293 Freestyle (Invitrogen). These cells are maintained in serum free HEK Freestyle media (Invitrogen). Cells are grown at 37 °C in a humidified shaking incubator (Kuhner) with 5% CO2. The sex of the HEK 293 cells is Female.
Mutagenesis of human HVEM & binding to LIGHT, CD160, and BTLA
Site-directed mutagenesis was carried out using overlapping primer PCR with KOD Hot Start DNA polymerase and a full-length human HVEM GFP fusion construct as template. Double mutants were generated using single mutants as initial templates for PCR. Sequence-verified clones were transiently transfected into HEK 293 Freestyle suspension cells (Invitrogen) and checked by flow cytometry for GFP expression. Cell surface expressed hHVEM mutants (100K cells) were queried with recombinant 0.25µg BTLA, CD160 and LIGHT His-tagged proteins (R&D systems) in 1x Dulbecco’s phosphate buffered saline and 0.2% BSA (DPBS/BSA). After 1 hour incubation at room temperature shaking at 900rpm cells were washed two times with 1X DPBS/BSA by centrifugation at 500 rpm and subsequently incubated with 0.1 ug PE conjugated anti-6X HIS tag antibody (AbCam) for 30min. After secondary antibody incubation cells were washed two more time with DPBS/BSA and immediately analyzed by flow cytometry. Cells were gated using single color controls and untransfected cells. Percent bound was determined as the percentage of GFP positive cells that were positive for PE (double positive) and was normalized to wild-type for each protein queried. Titrations were carried out similarly except that all three proteins queried were added to 100K WT or mutant HVEM expressing cells at the indicated concentrations. For titration experiments flow data was gated for GFP positive cells (HVEM expression) and the GeoMean of PE fluorescence (protein bound) was plotted as a function of soluble protein concentration. Titration data was normalized to the GeoMean fluorescence value for WT binding at the highest concentration of query protein. Titration data was fit using the single-site binding equation Y = Bmax*X/(Kd + X) in the Graphpad Prism software and represents three independent experiments.
Method details
HVEM:LIGHT, HVEM:CD160, and HVEM:BTLA complexes
Rs-pharmacophore generation starts with determining the binding interface from known receptor-ligand complexes using the program CSU (Sobolev et al., 1999) (Supp. Fig1). The crystallographic structures of ectodomains (PDB codes and chain identifiers: BTLA: 2AW2.A; LIGHT: 4RSU.A and 4RSU.B; CD160: 6NG3.A; HVEM: 4RSU.D, 2AW2.B, and 6NG3.A) and complexes (HVEM:BTLA PDB code 2AW2; HVEM:LIGHT PDB code 4RSU; HVEM:CD160 PDB code 6NG3) were used for this step. Missing residues in the complexes were added using Modeller (Sali and Blundell, 1993).
Residue-specific pharmacophore generation
ProtLID (Protein Ligand Interface Design) originally developed to identify cognate ligand binding partners for given target proteins (Yap and Fiser, 2016) and subsequently, it was used to redesign the interface for enhancing the binding specificity in PD-1 pathway (Shrestha et al., 2019). ProtLID consists multiple steps of a hypothetical interface design - identifying interface residues, generating a hypothetical mesh to represent an interface, running of MD simulation of single amino acids probes, clustering of MD trajectories, and generating a rs-pharmacophore.
In this study, three distinct sets of rs-pharmacophores for LIGHT, BTLA, and CD160 were generated using ProtLID focusing on redesigning HVEM interface for selectivity (Fig. 1). HVEM has two spatially distinct binding sites for a member of TNF superfamily (LIGHT) and IgSF superfamily (BTLA and CD160). First, we ran ProtLID protocol on LIGHT interface extracted from LIGHT:HVEM complex (4RSU). The interface is a dimer of homotrimeric LIGHT interacting on residues of CRD2 and CRD3 region of HVEM. Next, we calculated rs-pharmacophore for the BTLA and CD160 interfaces that bind to HVEM on CRD1, approximately on the opposite of LIGHT binding site using their respective complexes (2AW2 for BTLA:HVEM and 6NG3 for CD160:HVEM ) .
Computational HVEM interface redesign
Resulting rs-pharmacophores were compared with one other and the wild type residues of HVEM interface as defined by the CSU program (Sobolev et al., 1999). A union of residue preferences suggested by the rs-pharmacophores was considered for residues with multiple contacts.
After assigning ligand specific rs-pharmacophores for HVEM residues, the selection of potential mutants followed various strategies. For example, LIGHT specific variants for HVEM were chosen from the rs-pharmacophore suggested pools of residues, as these are assumed to enhance compatibility of interface contacts (positive design). However, using similar concept, the variants not suggested in the rs-pharmacophore were included for negative design. This approach is straightforward since there was no competing molecule included. However, BTLA and CD160 share a binding interface on HVEM therefore the selection of mutants relied on exploiting the differences between the ligands specific pharmacophores.
Quantification and Statistical Analysis
For statistical analysis of data on Figure 2B, 3, and 4, we used Graphpad Prism (https://www.graphpad.com/scientific-software/prism/). Statistical t-tests in Table 1 and S5 were performed by SciPy (https://docs.scipy.org/doc/scipy/reference/stats.html) and StatModels (https://www.statsmodels.org/stable/index.html). For all experiments N = 3 and represents the number of replicate independent experiments. All data shows average values obtained from three independent experiments and the error bars show standard deviation. All titration data was fit using a standard single-site binding model Y = Bmax*X/(EC50 + X).
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-6X HIS Tag PE | AbCam | Cat# ab72467 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human CD160-His | R&D Systems | Cat# 6177-CD-050 |
| Human BTLA-His | R&D Systems | Cat# 9235-BT-050 |
| Human LIGHT (TNF14) His | R&D Systems | Cat# 664-LI-025 |
| Deposited Data | ||
| Human HVEM and BTLA complex | (Compaan, et al., 2005) | PDB: 2AW2 |
| Human HVEM and LIGHT complex | N/A | PDB: 4RSU |
| Human HVEM and CD160 complex | (Liu, et al., 2019) | PDB: 6NG3 |
| Experimental Models: Cell Lines | ||
| HEK 293-F Freestyle | Thermofisher | CAt# R79007 |
| Oligonucleotides | ||
| DNA primers | see Table S3 for all primers | N/A |
| Recombinant DNA | ||
| human HVEM (TNFRSF14) | GeneCopoeia | Cat# EX-M0900-M98 |
| Software and Algorithms | ||
| Modeller | (Sali and Blundell, 1993) | https://salilab.org/modeller/ |
| PyMOL | PyMOL version 1.7 | https://sourceforge.net/projects/pymol/files/pymol/1.7/ |
| Prism | Prism version 8.3.0 | https://www.graphpad.com/scientific-software/prism/ |
| INKSCAPE | INKSCAPE version 0.92 | https://inkscape.org |
| Python | Python version 3.7.5 | https://www.python.org |
| StatsModels | StatsModels version 0.10.2 | https://www.statsmodels.org/stable/index.html |
| pandas | pandas version 0.25.3 | https://pandas.pydata.org/index.html |
| NumPy | NumPy version 1.17.4 | http://www.numpy.org |
| SciPy | SciPy version 1.3.3 | https://docs.scipy.org/doc/scipy/reference/stats.html |
| Matplotlib | Matplotlib version 3.1.2 | https://matplotlib.org |
| ProtLID | (Yap and Fiser, 2016) | N/A |
Highlights.
A residue-based pharmacophore approach is used to design specific protein interfaces
Designed point mutations of HVEM introduced six-way selectivity
Cell based assays confirmed selectivity of mutants
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants GM118709, GM136357R01 and AI141816.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The Authors declare no conflict of interest.
Data and Software Availability
N/A
References
- Agrawal S, Marquet J, Freeman GJ, Tawab A, Le Bouteiller P, Roth P, Bolton W, Ogg G, Boumsell L, and Bensussan A (1999). Cutting edge: MHC class I triggering by a novel cell surface ligand costimulates proliferation of activated human T cells. Journal of Immunology 162, 1223–1226. [PubMed] [Google Scholar]
- Barakonyi A, Rabot M, Marie-Cardine A, Aguerre-Girr M, Polgar B, Schiavon V, Bensussan A, and Le Bouteiller P (2004). Cutting edge: Engagement of CD160 by its HLA-C physiological ligand triggers a unique cytokine profile secretion in the cytotoxic peripheral blood NK cell subset. Journal of Immunology 173, 5349–5354. [DOI] [PubMed] [Google Scholar]
- Bodmer JL, Schneider P, and Tschopp J (2002). The molecular architecture of the TNF superfamily. Trends Biochem Sci 27, 19–26. [DOI] [PubMed] [Google Scholar]
- Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. (2016). Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 167, 405–418 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon DN, Grant RA, Baker TA, and Sauer RT (2005). Specificity versus stability in computational protein design. Proc Natl Acad Sci U S A 102, 12724–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breloer M, Hartmann W, Blankenhaus B, Eschbach ML, Pfeffer K, and Jacobs T (2015). Cutting Edge: the BTLA-HVEM regulatory pathway interferes with protective immunity to intestinal Helminth infection. J Immunol 194, 1413–1416. [DOI] [PubMed] [Google Scholar]
- Bretscher P, and Cohn M (1970). A theory of self-nonself discrimination. Science 169, 1042–1049. [DOI] [PubMed] [Google Scholar]
- Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, and Freeman GJ (2008). CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol 9, 176–185. [DOI] [PubMed] [Google Scholar]
- Cai GF, and Freeman GJ (2009). The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunological Reviews 229, 244–258. [DOI] [PubMed] [Google Scholar]
- Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr., Onufriev A, Simmerling C, Wang B, and Woods RJ (2005). The Amber biomolecular simulation programs. J Comput Chem 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Johnson NA, Affleck JG, Severson T, Steidl C, Ben-Neriah S, Schein J, Morin RD, Moore R, Shah SP, et al. (2010). Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res 70, 9166–9174. [DOI] [PubMed] [Google Scholar]
- Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, et al. (2009a). T Cell Intrinsic Heterodimeric Complexes between HVEM and BTLA Determine Receptivity to the Surrounding Microenvironment. Journal of Immunology 183, 7286–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, et al. (2009b). Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci U S A 106, 6244–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen MJH, Trynka G, Heskamp S, Franke B, van Diemen CC, Smolonska J, van Leeuwen M, Brouwer E, Boezen MH, Postma DS, et al. (2009). Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum Mol Genet 18, 4195–4203. [DOI] [PubMed] [Google Scholar]
- Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, and Hymowitz SG (2005). Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. J Biol Chem 280, 39553–39561. [DOI] [PubMed] [Google Scholar]
- del Rio ML, Lucas CL, Buhler L, Rayat G, and Rodriguez-Barbosa JI (2010). HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukocyte Biol 87, 223–235. [DOI] [PubMed] [Google Scholar]
- Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, Michielin O, Olive D, and Speiser DE (2010). BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest 120, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fons P, Chabot S, Cartwright JE, Lenfant F, L’Faqihi F, Giustiniani J, Herault JP, Gueguen G, Bono F, Savi P, et al. (2006). Soluble HLA-G1 inhibits angiogenesis through an apoptotic pathway and by direct binding to CD160 receptor expressed by endothelial cells. Blood 108, 2608–2615. [DOI] [PubMed] [Google Scholar]
- Gavrieli M, Watanabe N, Loftin SK, Murphy TL, and Murphy KM (2003). Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Bioph Res Co 312, 1236–1243. [DOI] [PubMed] [Google Scholar]
- Giustiniani J, Bensussan A, and Marie-Cardine A (2009). Identification and Characterization of a Transmembrane Isoform of CD160 (CD160-TM), a Unique Activating Receptor Selectively Expressed upon Human NK Cell Activation. Journal of Immunology 182, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havranek JJ, and Harbury PB (2003). Automated design of specificity in molecular recognition. Nat Struct Biol 10, 45–52. [DOI] [PubMed] [Google Scholar]
- Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, and Baker D (2003). Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364. [DOI] [PubMed] [Google Scholar]
- Lafferty KJ, and Cunningham AJ (1975). A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci 53, 27–42. [DOI] [PubMed] [Google Scholar]
- Launay E, Pangault C, Bertrand P, Jardin F, Lamy T, Tilly H, Tarte K, Bastard C, and Fest T (2012). High rate of TNFRSF14 gene alterations related to 1p36 region in de novo follicular lymphoma and impact on prognosis. Leukemia 26, 559–562. [DOI] [PubMed] [Google Scholar]
- Le Bouteiller P, Tabiasco J, Polgar B, Kozma N, Giustiniani J, Siewiera J, Berrebi A, Aguerre-Girr M, Bensussan A, and Jabrane-Ferrat N (2011). CD160: A unique activating NK cell receptor. Immunology Letters 138, 93–96. [DOI] [PubMed] [Google Scholar]
- Lippow SM, Wittrup KD, and Tidor B (2007). Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol 25, 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Garrett SC, Fedorov EV, Ramagopal UA, Garforth SJ, Bonanno JB, and Almo SC (2019). Structural Basis of CD160:HVEM Recognition. Structure 27, 1286–1295 e1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. (2012). Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A 109, 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looger LL, Dwyer MA, Smith JJ, and Hellinga HW (2003). Computational design of receptor and sensor proteins with novel functions. Nature 423, 185. [DOI] [PubMed] [Google Scholar]
- Maiza H, Leca G, Mansur IG, Schiavon V, Boumsell L, and Bensussan A (1993). A novel 80-kD cell surface structure identifies human circulating lymphocytes with natural killer activity. J Exp Med 178, 1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DJ, and Kortemme T (2009). Computer-aided design of functional protein interactions. Nat Chem Biol 5, 797–807. [DOI] [PubMed] [Google Scholar]
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. (1998). LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8, 21–30. [DOI] [PubMed] [Google Scholar]
- Morel Y, Schiano de Colella JM, Harrop J, Deen KC, Holmes SD, Wattam TA, Khandekar SS, Truneh A, Sweet RW, Gastaut JA, et al. (2000). Reciprocal expression of the TNF family receptor herpes virus entry mediator and its ligand LIGHT on activated T cells: LIGHT down-regulates its own receptor. J Immunol 165, 4397–4404. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Nelson CA, and Sedy JR (2006). Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol 6, 671–681. [DOI] [PubMed] [Google Scholar]
- Murphy TL, and Murphy KM (2010). Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol 28, 389–411. [DOI] [PubMed] [Google Scholar]
- Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, et al. (2008). Kemp elimination catalysts by computational enzyme design. Nature 453, 190–195. [DOI] [PubMed] [Google Scholar]
- Sali A, and Blundell TL (1993). Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Sedy J, Bekiaris V, and Ware CF (2014). Tumor necrosis factor superfamily in innate immunity and inflammation. Cold Spring Harb Perspect Biol 7, a016279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedy JR, Balmert MO, Ware BC, Smith W, Nemcovicova I, Norris PS, Miller BR, Aivazian D, and Ware CF (2017). A herpesvirus entry mediator mutein with selective agonist action for the inhibitory receptor B and T lymphocyte attenuator. J Biol Chem 292, 21060–21070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedy JR, Bjordahl RL, Bekiaris V, Macauley MG, Ware BC, Norris PS, Lurain NS, Benedict CA, and Ware CF (2013). CD160 Activation by Herpesvirus Entry Mediator Augments Inflammatory Cytokine Production and Cytolytic Function by NK Cells. Journal of Immunology 191, 828–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, et al. (2005). B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nature Immunology 6, 90–98. [DOI] [PubMed] [Google Scholar]
- Shaikh RB, Santee S, Granger SW, Butrovich K, Cheung T, Kronenberg M, Cheroutre H, and Ware CF (2001). Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J Immunol 167, 6330–6337. [DOI] [PubMed] [Google Scholar]
- Shrestha R, Garrett SC, Almo SC, and Fiser A (2019). Computational Redesign of PD-1 Interface for PD-L1 Ligand Selectivity. Structure 27, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolev V, Sorokine A, Prilusky J, Abola EE, and Edelman M (1999). Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327–332. [DOI] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MW, Cheung TC, and Ware CF (2011). The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunological Reviews 244, 169–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, and Chen L (2000). LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol 164, 4105–4110. [DOI] [PubMed] [Google Scholar]
- Tu TC, Brown NK, Kim TJ, Wroblewska J, Yang XM, Guo XH, Lee SH, Kumar V, Lee KM, and Fu YX (2015). CD160 is essential for NK-mediated IFN-gamma production. Journal of Experimental Medicine 212, 415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigano S, Banga R, Bellanger F, Pellaton C, Farina A, Comte D, Harari A, and Perreau M (2014). CD160-Associated CD8 T-Cell Functional Impairment Is Independent of PD-1 Expression. Plos Pathog 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward-Kavanagh LK, Lin WW, Sedy JR, and Ware CF (2016). The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 44, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CF (2008). Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol Rev 223, 186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Gavrieli M, Sedy JR, Yang JF, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang XX, et al. (2003). BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nature Immunology 4, 670–679. [DOI] [PubMed] [Google Scholar]
- Yap EH, and Fiser A (2016). ProtLID, a Residue-Based Pharmacophore Approach to Identify Cognate Protein Ligands in the Immunoglobulin Superfamily. Structure 24, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new code.