

Abstract
Despite significant advances, skin cutaneous melanoma (SKCM) is a common life-threatening cancer worldwide. Recently, pseudogenes have been discovered to be functional in many physiological processes and the pathogenesis of various diseases, including cancer. However, their relevance to SKCM remains largely unknown. In this study, seven upregulated pseudogenes were identified based on TCGA data. Among them, MTND4P12 was negatively correlated with the overall survival of SKCM patients. After constructing a pseudogene-miRNA-mRNA regulatory network, MTND4P12 was found to regulate the expression of oncogene AURKB by serving as a ceRNA. Both genetic and chemical inhibition of AURKB reduced viability and induced apoptosis of melanoma cells. Interestingly, DNA repair pathway seems to be involved in the anti-tumor effect of AURKB inhibition. Indeed, a synergistic therapeutic effect of AURKB inhibition and PARP inhibitor was confirmed both in vitro and in vivo. In conclusion, AURKB plays an oncogenic role and is a novel therapeutic target in SKCM. The combination of AURKB inhibition and PARP inhibitor has a synergistic effect, representing a promising treatment for SKCM.
Keywords: ceRNA, pseudogene, AURKB, PARPi, skin cutaneous melanoma
Introduction
Skin cutaneous melanoma (SKCM) is a common malignant tumor worldwide, with an estimated age-standardized incidence of 2.8-3.1 per 100,000 [1]. Its occurrence is still rapidly increasing annually throughout the world, faster than many other malignancies [2]. The prognosis for advanced melanoma remains poor, with a 5-year survival rate of less than 10% for patients with distant metastases [3]. Melanoma originates from neural-crest derived pigmented melanocytes [4], and the malignant transformation of melanocytes is a complex process with multigenic etiology. Ultraviolet radiation exposure and severe sunburns are certainly significant risk factors for the pathogenesis of melanoma [5]. Despite considerable progress in treating SKCM patients, better therapies are still urgently needed, considering the high mortality for advanced patients.
Competing endogenous RNAs (ceRNAs), first proposed by Salmena and colleagues [6], refer to transcripts containing the same miRNA response element that can regulate each other at the post-transcription level by competing for shared miRNAs. CeRNA networks link the function of protein-coding mRNAs with that of non-coding RNAs (ncRNAs) such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), pseudogenic RNAs and circular RNAs (circRNAs). An increase in the ncRNA level can function as RNA sponges for miRNAs and release repression on target genes, leading to an increase in target gene expression.
Pseudogenes are defined as defunct copies of protein-coding genes that emerge during the evolutionary process from random replications and mutations [7], which have been considered as “junk DNAs” for a long time [8]. However, growing evidence shows that pseudogenes play a significant role in the initiation and progression of cancers by serving as ceRNAs, affecting both their cognate and unrelated genes [9]. For instance, pseudogene PTENP1 functions as miR-499-5p sponge to manipulate the expression of parental gene PTEN in mice [10]. Pseudogene OCT4-pg4 regulates OCT4 expression by sponging miR-145 to promote hepatocarcinogenesis [11].
In this study, we identified MTND4P12 to be an oncogenic pseudogene upregulated in SKCM. It upregulates the expression of oncogene AURKB by serving as a ceRNA. In vitro study indicates that AURKB is a promising target for SKCM treatment, and the DNA repair pathway plays a significant intermediary role in the anti-tumor effect of AURKB inhibition. AURKB inhibitor and PARP inhibitor have a synergistic therapeutic effect on SKCM cells, thus providing a new strategy for SKCM treatment.
Materials and methods
Identification of differentially expressed pseudogenes
The screen process can be found in Figure 1. RNA-Seq expression data of pseudogenes in SKCM were downloaded from the dreamBase database [12]. The cut-off for differentially expressed pseudogenes in SKCM was set at |log2 Fold Change| > 6.0, P < 0.05.
Figure 1.
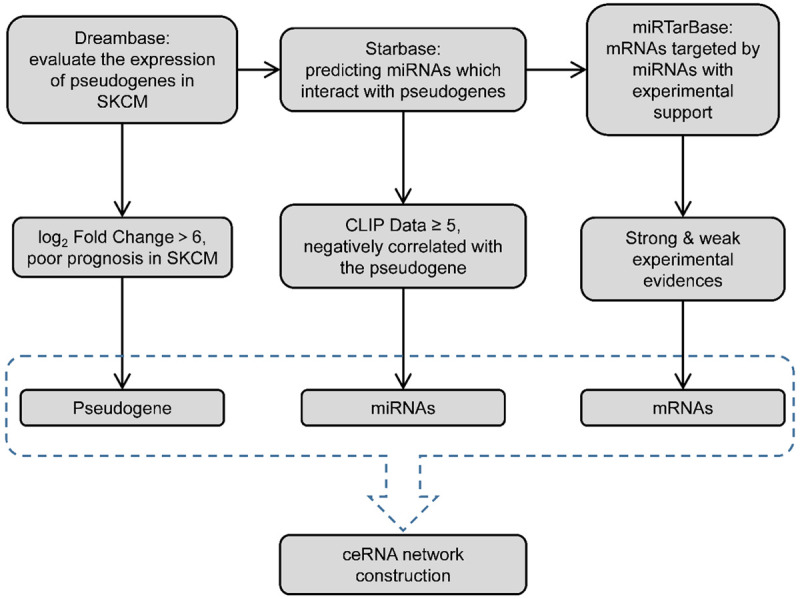
The screening flowchart.
Construction of the pseudogene-miRNA-mRNA regulatory network
miRNAs binding to pseudogenes were identified using starBase v2.0 [13]. Target genes of identified miRNA were retrieved from miRTarBase [14]. Pseudogene-miRNA-mRNA ceRNA network was visualized using Cytoscape v3.8.0 [15].
Protein-protein interaction network construction and hub genes analysis
The protein-protein interaction network of miRNA target genes was analyzed using STRING v11.0 and visualized by Cytoscape v3.8.0 [15]. Genes with top ten degree were identified as hub genes using Centiscape 2.2 plugin of Cytoscape v3.8.0 [16].
Kaplan-Meier survival analysis
For pseudogenes, Kaplan-Meier overall survival analysis and log-rank test were used to evaluate the statistical significance of survival differences between the two groups using GEPIA [17]. The cut-off value between the two groups was ‘Median’.
Gene expression analysis
The relative expression levels of target genes in SKCM were validated by GEPIA [17], using |logFC| ≥ 1 and P ≤ 0.01 as the cut-off criteria. The results are presented as box plots.
Gene ontology & KEGG pathway enrichment analysis
Pathway enrichment analysis for enriched target genes was conducted using ClueGO v2.5.7 plugin [18] of Cytoscape v3.8.0. GO items and KEGG pathways with P < 0.05 were considered significant.
Differential expression analysis and functional enrichment of AZD1152 treatment and control samples
The raw data were obtained from the GSE38466 dataset deposited in the GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38466). The dataset includes 42 human melanoma cell samples, including 38 treatment samples and 4 control samples. The platform is GPL10558 Illumina HumanHT-12 V4.0 expression beadchip. The DEGs were identified using the GEO2R tool [19]. Genes with P < 0.05 and |log2 Fold Change (FC)| > 1 were considered as DEGs between the AZD1152 treatment and control groups. For functional analysis of the identified DEGs, the DEGs were imported into the metascape [20] for enrichment and visualization. GO functional enrichment analysis and KEGG pathway enrichment analysis were conducted [21]. P < 0.05 and P < 0.01 were considered significant in GO and KEGG analysis.
GSEA analysis
Genome-wide expression profiles at the level of gene sets, which include genes with the common biological function, were analyzed by GSEA (http://software.broadinstitute.org/gsea/index.jsp).
Cell culture and drug treatment
B16 and A375 melanoma cells were cultured in DMEM medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin at 37°C and 5% CO2 in a humidified incubator. Olaparib and AZD2811 (Selleck) were dissolved in dimethyl sulfoxide (DMSO). The final concentrations of the drugs and duration of treatments are indicated in the figure legends.
Cell viability assay
The cells were treated with different concentrations of AZD2811 (0, 3.125, 6.25, 12.5, 25 and 50 μmol/L) for 72 h and 96 h. The cell viability of B16 and A375 melanoma cells was measured by the MTS cell proliferation assay.
Small interfering RNA transfection
The knockdown of AURKB was performed by small interfering RNA transfection. B16 melanoma cells were transfected with small interfering RNA (siRNA) targeting AURKB (#1: 5’-CCUUUGGGCAAAGGCAAAUTT-3’, #2: 5’-CCACAAGAAGAAGGUAAUUTT-3’) by Lipofectamine2000 (Invitrogen), according to the manufacturer’s protocol.
Western blot analysis
Cells were washed with ice-cold PBS, scraped, lysed, and sonicated in radioimmunoprecipitation (RIPA) buffer supplemented with 1 mM phenylmethylsulfonyl fluoride (Beyotime) and phosphatase inhibitor cocktail (Roche, Basel, Switzerland). The protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Reinach, Switzerland). The proteins were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then electrotransferred to a polyvinylidene fluoride membrane (Millipore, Burlington, MA, USA) at 100 V for 2 h. The membranes were blocked in 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) at room temperature for 2 h. After washing with TBST, the membranes were incubated for 8 h at 4°C with primary antibodies. The membranes were incubated in horseradish peroxidase-labeled goat antirabbit secondary antibody (1:5000; Proteintech) for 1 h at room temperature. The signal was detected by enhanced chemiluminescence (Millipore).
Flow cytometry
Annexin V-FITC/PI labeling was performed to detect apoptosis according to the manufacturer’s recommendations (Invitrogen). At least 10,000 cells were analyzed by flow cytometry to determine the percentage of apoptotic cells. The results are presented as the percentage of apoptotic cells.
Drug combination study in vitro
For drug combination study, approximately 3000 cells per well were seeded in the 96-well plates, and then treated with a single AZD2811, olaparib or with the combination of the two, and each compound for another 72 hours. Cell viability was measured using the MTS assay. The combination index (CI) and fraction affected (Fa) values were calculated using Compusyn software [22].
SKCM xenograft model and drug treatment
Six-week-old female nu/nu mice and male C57BL/6 mice were obtained and maintained in a specific pathologic-free environment. 12×106 and 4.1×106 viable B16 cells were inoculated subcutaneously in nude mice and C57BL/6 mice, and tumor incidence and growth were monitored after inoculation. Mice bearing xenograft tumors were randomly divided into four subgroups, which were scheduled to receive the following treatments: 50 mg/kg olaparib, 50 mg/kg AZD2811, a combination of both agents and vehicle as control. Tumor volume was measured after each treatment. The tumor volume of nu/nu mice was measured at day 1, day 5, day 7, day 9, day 12 and day 14 after B16 cells inoculation and the tumor volume of C57BL/6 mice was measured at day 7, day 10, day 12, day 14, day 17, day 19, day 21 and day 24 after B16 cells inoculation.
Results
Identification of upregulated pseudogenes in SKCM
To investigate the potential roles of functional pseudogenes in SKCM, differentially expressed pseudogenes in SKCM compared with normal tissues were identified using dreamBase [23]. Based on the cut-off criteria (|log2FC| > 6.0, P < 0.05), seven upregulated pseudogenes were identified in SKCM (Table 1). Among these pseudogenes, only high expression of MTND4P12 was identified to be significantly correlated with worse survival rate in SKCM (Figure 2), suggesting that MTND4P12 may play an essential role in the pathogenesis of SKCM.
Table 1.
Information of upregulated pseudogenes with log2 Fold Change (FC) more than six in skin cutaneous melanoma
| Pseudogene name | Ensembl ID | Genome location | Transcript biotype | Parent gene | Fold Change | Hazard Ratio | P-value |
|---|---|---|---|---|---|---|---|
| SRP9P1 | ENST00000445874 | chr10:91,806,043-91,807,533 | Processed pseudogene | SRP9 | 11.2 | 1.2 | 0.26 |
| RP4-800G7.1 | ENST00000461005 | chr7:149,191,043-149,191,223 | Processed pseudogene | RPL36A | 7.79 | 1 | 0.91 |
| CH17-264B6.3 | ENST00000275546 | chr7:73,065,529-73,077,353 | Unprocessed pseudogene | PMS2 | 6.93 | 0.91 | 0.49 |
| C1DP1 | ENST00000426049 | chr10:32,510,628-32,511,819 | Processed pseudogene | C1D | 6.7 | 0.79 | 0.081 |
| MTND4P12 | ENST00000498999 | chr5:134,926,660-134,928,036 | Processed pseudogene | MTND4 | 6.27 | 1.3 | 0.039 |
| LDHAP3 | ENST00000448312 | chr2:41,819,648-41,821,649 | Processed pseudogene | LDHA | 6.25 | 0.9 | 0.42 |
| RP11-359E7.3 | ENST00000634073 | chr10:121,557,729-121,587,837 | Processed pseudogene | VMA21 | 6.04 | 0.86 | 0.25 |
Figure 2.

The expression and survival analysis of MTND4P12 in skin cutaneous melanoma. Overall survival (A), disease free survival analyses (B) and the expression of MTND4P12 in skin cutaneous melanoma (C) were performed using the GEPIA online platform. The solid line represents the survival curve and the dotted line represents the 95% confidence interval. Patients with expression above the median are indicated by red lines, and patients with expression below the median are indicated by blue lines. Log-rank P < 0.05 was considered to indicate a statistically significant difference. HR: hazard ratio. Box plots derived from gene expression data in GEPIA comparing expression of MTND4P12 in skin cutaneous melanoma tissue and normal tissues, *P < 0.05.
Construction of pseudogene-miRNA-mRNA regulatory network
Since pseudogenes mainly function as ceRNAs, we screened potential miRNAs binding to pseudogene MTND4P12 and negatively correlated with the expression of MTND4P12 using starBase [13]. Hsa-let-7e-5p and hsa-miR-1193 were finally identified to be candidate miRNAs (Figure 3; Supplementary Table 1). Target genes of hsa-let-7e-5p and hsa-miR-1193 were further explored by strong and weak experimental method using miRTarBase [14]. In total, 697 genes were identified to be direct targets of candidate miRNAs (Supplementary Table 2). The pseudogene-miRNA-mRNA regulatory network was then visualized using Cytoscape [15] (Figure 4A). Thus, MTND4P12 may exert biological functions through regulating these target genes.
Figure 3.

Identification of hsa-let-7e-5p and hsa-miR-1193 as candidate miRNAs regulated by MTND4P12. A. Co-Expression Analysis for hsa-let-7e-5p and MTND4P12. B. Co-Expression Analysis for hsa-miR-1193 and MTND4P12.
Figure 4.

Bioinformatics analysis of miRNA target genes. (A) Top 100 Target genes for these miRNAs were retrieved from miRtarbase. Pseudogene-miRNA-mRNA interaction network was visualized using Cytoscape v3.8.0. (B-D) Gene ontology (GO) enrichment analysis of miRNA target genes. Colors represent the GO groups, each node is a GO biological process (B), GO Cellular component (C), GO Molecular function (D), the edges show the connectivity between each node based on the connection of the genes and the size of nodes depends on the number of genes that are grouped. (E) KEGG pathway enrichment analysis of miRNA target genes. Colors represent the KEGG groups and the size of nodes depends on the number of genes that are grouped.
Functional enrichment analysis of miRNA target genes
To investigate the potential functions of MTND4P12 in cancer development, we performed GO and KEGG pathway enrichment analysis using ClueGo [18]. Several GO terms were enriched such as DNA binding, RNA binding, gene expression and mitotic cell cycle (Figure 4B-D). KEGG pathway analysis indicated that miRNA target genes were enriched in several pathways such as pathways in cancer, FoxO signaling pathway, p53 signaling pathway and proteoglycans in cancer (Figure 4E). These findings indicate that MTND4P12 is involved in carcinogenesis.
AURKB was identified as a target gene of MTND4P12
To further analyze the functional roles of MTND4P12, a protein-protein interaction network was generated using STRING [24], and hub genes were selected using Centiscape 2.2 [16] (Figure 5A). Genes with top ten degree (MYC, CDC5L, STAT3, CCND1, PLK1, PPP2R1A, CREBBP, EZH2, AURKB and RPSA) were identified as hub genes (Table 2). Prognostic outcomes of ten hub genes in SKCM were analyzed using GEPIA [17] (Table 3). It was found that high mRNA expression level of STAT3 was significantly associated with good overall survival (P=0.034; Table 3), high mRNA expression levels of PLK1, PPP2R1A, CREBBP and AURKB were significantly associated with poor overall survival (P=0.00041, P=0.0029, P=0.0012, P=0.0047, respectively; Table 3). Pearson correlation analysis between MTND4P12 and ten hub genes expressions in SKCM showed that only the correlation between AURKB and MTND4P12 expression levels was significantly positive (Table 2). AURKB mRNA expression levels were further validated higher in SKCM compared with normal tissues in GEPIA (Figure 5B). These findings indicate that MTND4P12 regulates AURKB expression by serving as ceRNA in SKCM.
Figure 5.

Identification of AURKB as the hub gene of interest. (A) The protein-protein interaction network of miRNA target genes was constructed. Genes (yellow nodes) with top 10 degree value were identified as hub genes. The expression (B) and overall survival (C) of AURKB in skin cutaneous melanoma were performed using the GEPIA online platform. Box plots derived from gene expression data comparing expression of AURKB in skin cutaneous melanoma tissue and normal tissues, *P < 0.05. The solid line represents the survival curve and the dotted line represents the 95% confidence interval. Patients with expression above the median are indicated by red lines, and patients with expression below the median are indicated by blue lines. Log-rank P < 0.05 was considered to indicate a statistically significant difference. HR: hazard ratio.
Table 2.
Ten hub genes identified by Centiscape 2.2
| Gene names | Degree | Closeness | Betweenness | Gene descriptions | R* | P-value |
|---|---|---|---|---|---|---|
| MYC | 92 | 0.000726 | 42223.56959 | MYC Proto-Oncogene, BHLH Transcription Factor | -0.05 | 2.81E-01 |
| CDC5L | 60 | 0.000672 | 27760.35872 | Cell Division Cycle 5 Like | -0.149 | 1.20E-03 |
| STAT3 | 58 | 0.000663 | 15201.15151 | Signal Transducer and Activator of Transcription 3 | -0.05 | 2.81E-01 |
| CCND1 | 56 | 0.000664 | 9247.362858 | Cyclin D1 | -0.018 | 7.02E-01 |
| PLK1 | 54 | 0.000667 | 14629.18024 | Polo Like Kinase 1 | 0.071 | 1.22E-01 |
| PPP2R1A | 53 | 0.000663 | 16111.20213 | Protein Phosphatase 2 Scaffold Subunit Aalpha | 0.024 | 6.03E-01 |
| CREBBP | 50 | 0.000646 | 16371.64887 | CREB Binding Protein | -0.106 | 2.14E-02 |
| EZH2 | 47 | 0.000629 | 7417.059528 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit | -0.014 | 7.68E-01 |
| AURKB | 43 | 0.000643 | 7336.007453 | Aurora Kinase B | 0.104 | 2.36E-02 |
| RPSA | 42 | 0.000625 | 6433.209 | Ribosomal Protein SA | -0.046 | 3.19E-01 |
Pearson correlation analysis between MTND4P12 and ten hub genes expressions in skin cutaneous melanoma.
Table 3.
Prognostic values of ten hub genes in skin cutaneous melanoma
| Gene names | RNAseq IDs | HRs with 95% CIs | Prognostic outcomes | P-value |
|---|---|---|---|---|
| MYC | 4609 | 1.3 | Poor | 0.08 |
| CDC5L | 988 | 1 | No difference | 0.74 |
| STAT3 | 6774 | 0.75 | Good | 0.034 |
| CCND1 | 595 | 1.1 | Poor | 0.33 |
| PLK1 | 5347 | 1.6 | Poor | 0.00041 |
| PPP2R1A | 5518 | 1.5 | Poor | 0.0029 |
| CREBBP | 1387 | 1.6 | Poor | 0.0012 |
| EZH2 | 2146 | 1.1 | Poor | 0.52 |
| AURKB | 9212 | 1.5 | Poor | 0.0047 |
| RPSA | 3921 | 1.1 | Poor | 0.7 |
AURKB inhibition reduced viability and induced apoptosis of B16 melanoma cells in vitro
To validate the biological relevance of AURKB in melanoma, we treated melanoma cells with AURKB inhibitor, AZD2811, for 72 h and 96 h, respectively. It was observed that AZD2811 significantly inhibited cell viability in a time- and dose-dependent manner (Figure 6A). Moreover, AURKB siRNA treatment significantly increased cleaved-CASP3 and cleaved-PARP1 protein expression in melanoma cells compared with the control (Figure 6B). In addition, flow cytometric analysis of early and late stage apoptosis revealed that AURKB siRNA treatment significantly increased apoptosis of melanoma cells compared with the control (Figure 6C). These findings suggest that the inhibition of AURKB could reduce viability and induce apoptosis of melanoma cells in vitro.
Figure 6.

Inhibition of AURKB induced apoptosis of B16 melanoma cells. A. The cell viability of B16 cells by MTS cell proliferation assay at 72 h and 98 h after the treatment of AZD2811. B. siRNA-mediated AURKB inhibition potentiates apoptosis. B16 cell lysates were subjected to western blotting for the detection of cleaved caspase-3 and PARP-1 activation. Tubulin expression was utilized for normalization. C. Flow cytometry was used to detect the effect of si-AURKB on apoptosis. B16 cells were stained with PE-CF594-A/FITC, and the total apoptosis rates were examined by flow cytometry, including the early (Q4) and late (Q2) apoptosis rates. Data represent means ± SD. ***P < 0.0005.
DNA repair pathway played an essential role in mediating the anti-tumor effect of AURKB inhibition
To further explore the mechanism by which AURKB regulates the viability of melanoma cells, we performed gene expression comparisons of human melanoma cells treated with AURKB inhibitor, AZD2511, for 48 h and the control group, based on the GSE38466 dataset from GEO database. The results revealed that 111 and 83 genes were upregulated and downregulated, respectively (Supplementary Table 3). GO enrichment analysis showed the predominance of genes related to DNA replication, DNA repair and inter-strand cross-link repair (Figure 7A). Besides, several KEGG pathways were significantly enriched, such as DNA replication, Fanconi anemia pathway and homologous recombination (Figure 7B). These data highlighted the importance of AURKB in DNA repair pathway. To give further insight into the relationship between the DNA repair and AURKB, we conducted the Gene set enrichment analysis (GSEA). It was found that the “Regulation of Double Strand Break Repair via Homologous Recombination” gene set was significantly enriched in control group with higher expression of AURKB (Figure 7C). Thus, we speculate that AURKB regulates SKCM cells viability through DNA repair pathways.
Figure 7.
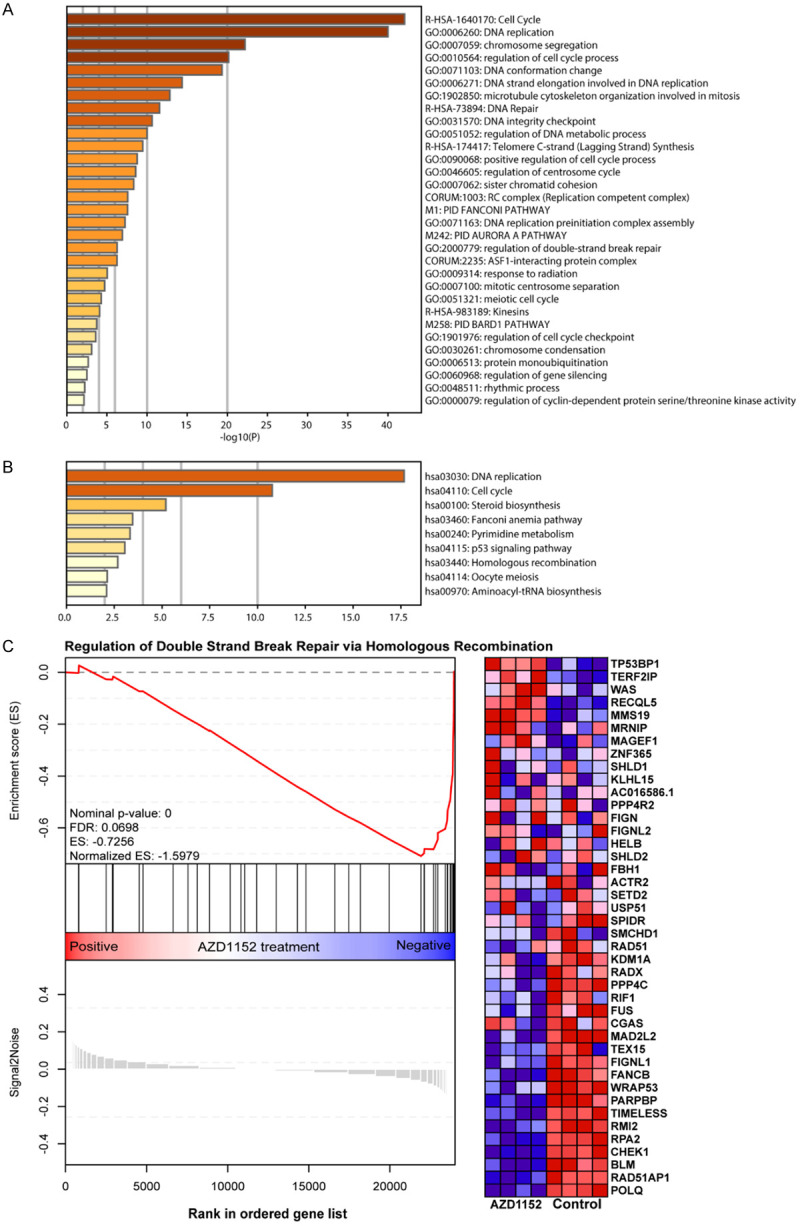
GO enrichment analysis, KEGG pathway enrichment analysis and Gene Set Enrichment Analysis (GSEA) of target genes upon AURKB inhibition. GO enrichment analysis (A) and KEGG pathway enrichment analysis (B) of target genes upon AURKB inhibition in human melanoma cells. The color intensity of bars indicated the P-value of the corresponding term. GSEA of target genes upon AURKB inhibition in human melanoma cells (C). GSEA plot for gene set GO_REGULATION_OF_DOUBLE_STRAND_BREAK_REPAIR_VIA_HOMOLOGOUS_RECOMBINATION. Heatmap of core enrichment genes for gene set GO_REGULATION_OF_DOUBLE_STRAND_BREAK_REPAIR_VIA_HOMOLOGOUS_RECOMBINATION. “AZD1152” stands for AZD1152 treatment for 48 h.
AURKB inhibitor and PAPR inhibitor synergistically promoted apoptosis of melanoma cells
Inspired by these findings, we sought to explore the therapeutic potential of the combination of PARP inhibitor and AURKB inhibitor against melanoma. Our data from the drug combination tests revealed that olaparib, a PARP inhibitor, had a strong synergistic anti-growth effect with AURKB inhibitor (Figure 8A). After treating melanoma cells with AZD2811 and olaparib alone or in combination, cleaved caspase-3 and cleaved PARP1 were increased, accompanied by the increase of DNA damage and repair marker, γ-H2AX (Figure 8B). In agreement with these results, flow cytometry data showed that the combination of olaparib and AZD2811 resulted in much more cells undergoing apoptosis as compared to single agent alone, as evidenced by 20.25% apoptosis ratio in combination group while 2.36% and 5.2% in olaparib or AZD2811 group, respectively (Figure 8C and 8D). In addition, the similar effect can be observed in another melanoma cell line A375, confirming that the synergy between AURKB inhibitor and PARP inhibition is not cell line specific (Supplementary Figure 1A, 1B). Collectively, these findings reveal that PARP inhibitor and AURKB inhibitor synergistically promote melanoma cell apoptosis.
Figure 8.
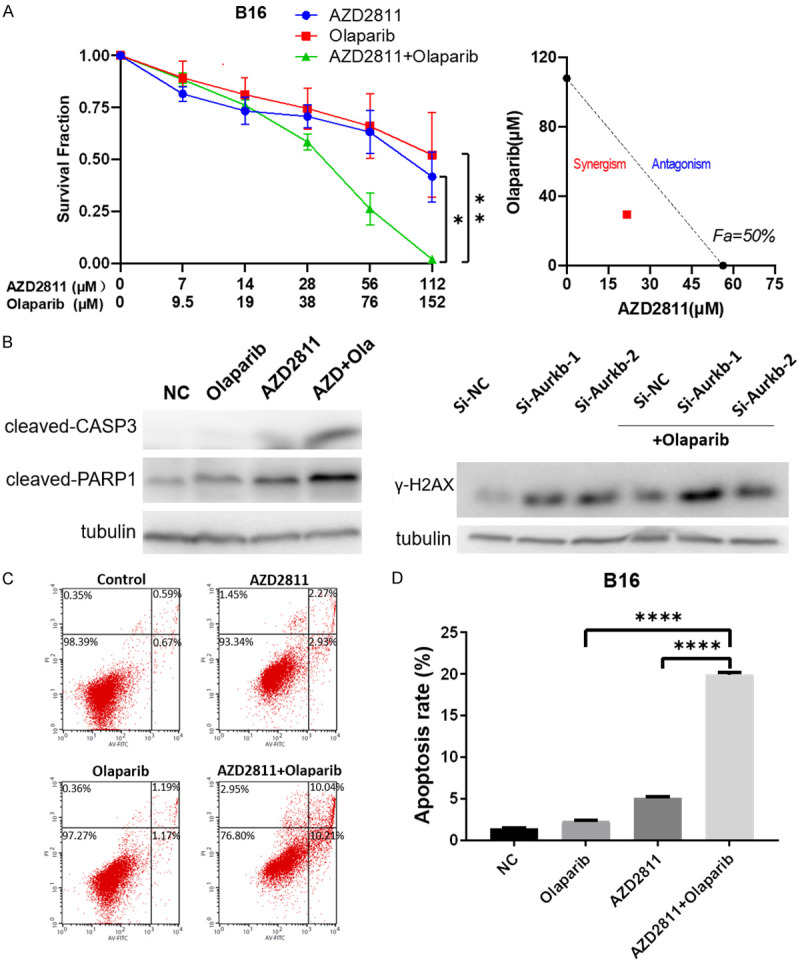
Drug combination identifies PARPi acting synergistically with AURKB inhibition in melanoma cells. A. Sensitivity of B16 to AZD2811, Olaparib alone or AZD2811 plus Olaparib. Survival fraction (left) and Isobologram analysis (right) are shown. The diagonal, dotted line indicates additivity, and the red symbol shows dose requirements to achieve 50% cancer cell inhibition. Data points below the line of additivity indicate synergy, data points above denote antagonism. B. Western blotting analysis (left) showing the effects of AZD2811 (28 μM), Olaparib (38 μM), and AZD2811+Olaparib on the expression of cleaved caspase-3 and PARP1. Western blotting analysis (right) showing γH2AX expression in si-AURKB transfected B16 before and after the treatment of Olaparib. Tubulin as a loading control. C. Flow cytometry analysis of early and late apoptosis in treated B16 cells with AZD2811 (28 μM), Olaparib (38 μM) and AZD2811+Olaparib. D. Percentage of apoptotic cells are shown. Data represent means ± SD. *P < 0.05, **P < 0.005, ****P < 0.0001.
Combinational treatment of olaparib and AZD2811 impaired tumor growth in vivo
We next exploited xenograft mouse models to validate the therapeutic effects of olaparib and AZD2811 in vivo. It was found that treatment with olaparib or AZD2811 alone partially inhibited tumor growth, whereas the combination of the olaparib and AZD2811 significantly inhibited tumor growth in C57BL/6 mice and nude mice, with approximately 80% tumor volume reduction relative to control in C57BL/6 mice (Figure 9A and 9B, Supplementary Figure 1C). Taken together, these findings from the preclinical SKCM animal models indicate that olaparib and AZD2811 can synergistically inhibit growth of melanoma in vivo.
Figure 9.
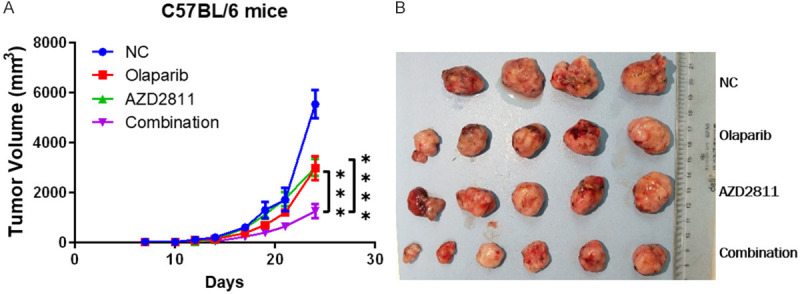
Combinational treatment of olaparib and AZD2811 impaired tumor growth in vivo. A. The combination of PARPi and AURKB inhibition synergistically suppressed the growth of B16 melanoma in C57BL/6 mice. Data represent means ± SD. ***P < 0.0005. ****P < 0.0001. B. The representative tumors of tumor-bearing mouse in the control group and the medicated groups are shown. Saline solution (control), AZD2811 only, Olaparib only and AZD2811+Olaparib were administered to C57BL/6 mice with skin cutaneous melanoma.
Discussion
Our results reveal that the pseudogene MTND4P12, associated with poor prognosis in SKCM, can regulate the expression of oncogene AURKB through the ceRNA mechanism. Notably, the combination of AURKB inhibitor and PARP inhibitor robustly inhibited tumor growth and promoted apoptosis both in vitro and in vivo. These findings offer a strong rationale for the future clinical application of AURKB inhibitors for patients with SKCM combined with PARP inhibitors that has been approved for the treatment of various cancers including SKCM.
The oncogenic role of AURKB has attracted tremendous researches in recent years. AURKB belongs to the highly conserved Aurora family of mitotic kinases, encoding serine/threonine kinase [25]. AURKB overexpression can result in aneuploidy, probably contributing to cancer initiation or progression [26]. AURKB is also involved in promoting cell cycle by regulating cycle-related targets, thus promoting tumor cell survival [27]. In line with our study, anti-proliferative effects of AURKB inhibition have been observed in animal tumor models of several human cancers, including breast, ovarian, pancreatic, colorectal, lung cancers and hepatocellular carcinoma [28-32]. These findings strongly indicate a possible therapeutic role of AURKB inhibition in cancer patients. Currently, AURKB inhibitors have already been evaluated for their potential in the clinical management of cancers (https://clinicaltrials.gov/show/NCT01118611).
Synthetic lethality is a concept in which the simultaneous loss of function of two different genes results in cell death while the loss of just one gene continues to be compatible with cell viability [33]. For example, PARP inhibitors (PARPi) display synthetic lethality in cells with impaired homologous recombination (HR)-mediated DNA repair function, such as tumors with BRCA1/2 mutation [34]. Unfortunately, BRCA1/2 mutation-occur in solely a small percentage of tumors, significantly restricting the clinical efficacy of PARPi-based therapies [35,36]. Therefore, the development of new paradigms to elicit PARPi synthetic lethality in HR-competent cancers remains an intriguing treatment strategy. Herein, we found that DNA repair pathways were involved in the AURKB regulation process by bioinformatics approaches. Given this finding, we conducted the drug combination test of AURKB inhibitor and PARP inhibitor in SKCM cells. We found significant synergistic effects, which might due to increased DNA damage after AURKB inhibition. A similar conclusion was obtained when L.R. Mitchell et al. investigated the effects of AURKB inhibitor, AZD1152, on radio-sensitization of prostate cancer cells [37]. Their results demonstrated that more AZD1152-treated cells sustained DNA damage than irradiated controls 30 minutes post-radiation [37]. It has been reported that AURKB can phosphorylate p53 to accelerate its degradation through polyubiquitination-proteasome pathway [38], and p53 can inhibit homologous recombination repair by its interaction with RAD51 and RAD54 [39]. The inhibition of AURKB, as a result, will lead to the impairment of homologous recombination dependent DNA repair due to the accumulation of p53, resulting in increased DNA damage and conferring synthetic lethality to PARP inhibition. Another study showed that inhibition of AURKB sensitized cells to both cisplatin and oxaliplatin [38]. Therefore, it is conceivable that a rationally designed combination based on AURKB inhibitor and other anti-cancer drugs, especially the new generation drug PARP inhibitor, may be a reasonable and feasible strategy for enhancing therapeutic efficacy and reducing undesirable side effects in melanoma patients.
In summary, the present study indicates that the pseudogene MTND4P12 can regulate the expression of oncogene AURKB through the ceRNA mechanism in SKCM. Our findings also reveal the potent and synergistic therapeutic effects of AURKB inhibitor and PARP inhibitor, offering a novel promising strategy for SKCM treatment.
Acknowledgements
This work is supported by grants from National Natural Science Foundation of China (81761138047), Natural Science Foundation of Zhejiang (Q18H160015 and D21H160001).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Ali Z, Yousaf N, Larkin J. Melanoma epidemiology, biology and prognosis. EJC Suppl. 2013;11:81–91. doi: 10.1016/j.ejcsup.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trinh VA. Current management of metastatic melanoma. Am J Health Syst Pharm. 2008;65(Suppl 9):S3–8. doi: 10.2146/ajhp080460. [DOI] [PubMed] [Google Scholar]
- 4.Prasad P, Vasas A, Hohmann J, Bishayee A, Sinha D. Cirsiliol suppressed epithelial to mesenchymal transition in B16F10 malignant melanoma cells through alteration of the PI3K/Akt/NF-κB signaling pathway. Int J Mol Sci. 2019;20:608. doi: 10.3390/ijms20030608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leiter U, Garbe C. Epidemiology of melanoma and nonmelanoma skin cancer--the role of sunlight. Adv Exp Med Biol. 2008;624:89–103. doi: 10.1007/978-0-387-77574-6_8. [DOI] [PubMed] [Google Scholar]
- 6.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanin EF. Processed pseudogenes: characteristics and evolution. Annu Rev Genet. 1985;19:253–272. doi: 10.1146/annurev.ge.19.120185.001345. [DOI] [PubMed] [Google Scholar]
- 8.Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17:792–798. doi: 10.1261/rna.2658311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lou W, Ding B, Fu P. Pseudogene-derived lncRNAs and their miRNA sponging mechanism in human cancer. Front Cell Dev Biol. 2020;8:85. doi: 10.3389/fcell.2020.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Zhang N, Wang Z, Ai DM, Cao ZY, Pan HP. Pseudogene PTENP1 functions as a competing endogenous RNA (ceRNA) to regulate PTEN expression by sponging miR-499-5p. Biochemistry (Mosc) 2016;81:739–747. doi: 10.1134/S0006297916070105. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Guo ZY, Zhang R, Xin B, Chen R, Zhao J, Wang T, Wen WH, Jia LT, Yao LB, Yang AG. Pseudogene OCT4-pg4 functions as a natural micro RNA sponge to regulate OCT4 expression by competing for miR-145 in hepatocellular carcinoma. Carcinogenesis. 2013;34:1773–1781. doi: 10.1093/carcin/bgt139. [DOI] [PubMed] [Google Scholar]
- 12.Zheng LL, Zhou KR, Liu S, Zhang DY, Wang ZL, Chen ZR, Yang JH, Qu LH. dreamBase: DNA modification, RNA regulation and protein binding of expressed pseudogenes in human health and disease. Nucleic Acids Res. 2018;46:D85–D91. doi: 10.1093/nar/gkx972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, Chiew MY, Tai CS, Wei TY, Tsai TR, Huang HT, Wang CY, Wu HY, Ho SY, Chen PR, Chuang CH, Hsieh PJ, Wu YS, Chen WL, Li MJ, Wu YC, Huang XY, Ng FL, Buddhakosai W, Huang PC, Lan KC, Huang CY, Weng SL, Cheng YN, Liang C, Hsu WL, Huang HD. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46:D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25:2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2012;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou TC, Martin N. CompuSyn for drug combinations. PC software and user’s guide: a computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values. 2005 [Google Scholar]
- 23.Zheng LL, Zhou KR, Liu S, Zhang DY, Wang ZL, Chen ZR, Yang JH, Qu LH. dreamBase: DNA modification, RNA regulation and protein binding of expressed pseudogenes in human health and disease. Nucleic Acids Res. 2017;46:D85–D91. doi: 10.1093/nar/gkx972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 26.Sansregret L, Swanton C. The role of aneuploidy in cancer evolution. Cold Spring Harb Perspect Med. 2017;7:a028373. doi: 10.1101/cshperspect.a028373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.González-Loyola A, Fernández-Miranda G, Trakala M, Partida D, Samejima K, Ogawa H, Cañamero M, de Martino A, Martínez-Ramírez Á, de Cárcer G, Pérez de Castro I, Earnshaw WC, Malumbres M. Aurora B overexpression causes aneuploidy and p21Cip1 repression during tumor development. Mol Cell Biol. 2015;35:3566–3578. doi: 10.1128/MCB.01286-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schöffski P, Besse B, Gauler T, de Jonge MJ, Scambia G, Santoro A, Davite C, Jannuzzo MG, Petroccione A, Delord JP. Efficacy and safety of biweekly i.v. administrations of the Aurora kinase inhibitor danusertib hydrochloride in independent cohorts of patients with advanced or metastatic breast, ovarian, colorectal, pancreatic, small-cell and non-small-cell lung cancer: a multi-tumour, multi-institutional phase II study. Ann Oncol. 2015;26:598–607. doi: 10.1093/annonc/mdu566. [DOI] [PubMed] [Google Scholar]
- 29.Larsen SL, Yde CW, Laenkholm AV, Rasmussen BB, Duun-Henriksen AK, Bak M, Lykkesfeldt AE, Kirkegaard T. Aurora kinase B is important for antiestrogen resistant cell growth and a potential biomarker for tamoxifen resistant breast cancer. BMC Cancer. 2015;15:239. doi: 10.1186/s12885-015-1210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma YX, Li XZ. Effect of aurora kinase B inhibitor AZD1152 in the treatment of cisplatin-resistant ovarian carcinoma. Zhonghua Fu Chan Ke Za Zhi. 2013;48:46–50. [PubMed] [Google Scholar]
- 31.Aihara A, Tanaka S, Yasen M, Matsumura S, Mitsunori Y, Murakata A, Noguchi N, Kudo A, Nakamura N, Ito K, Arii S. The selective Aurora B kinase inhibitor AZD1152 as a novel treatment for hepatocellular carcinoma. J Hepatol. 2010;52:63–71. doi: 10.1016/j.jhep.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Azzariti A, Bocci G, Porcelli L, Fioravanti A, Sini P, Simone GM, Quatrale AE, Chiarappa P, Mangia A, Sebastian S, Del Bufalo D, Del Tacca M, Paradiso A. Aurora B kinase inhibitor AZD1152: determinants of action and ability to enhance chemotherapeutics effectiveness in pancreatic and colon cancer. Br J Cancer. 2011;104:769–780. doi: 10.1038/bjc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 34.Lee JM, Ledermann JA, Kohn EC. PARP inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25:32–40. doi: 10.1093/annonc/mdt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br J Cancer. 2000;83:1301–1308. doi: 10.1054/bjoc.2000.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malone KE, Daling JR, Neal C, Suter NM, O’Brien C, Cushing-Haugen K, Jonasdottir TJ, Thompson JD, Ostrander EA. Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer. 2000;88:1393–1402. doi: 10.1002/(sici)1097-0142(20000315)88:6<1393::aid-cncr17>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell LR, Kopsombut P, Li J, Kim K, Sun Y, Lu B. AZD1152, an aurora kinase b inhibitor, sensitizes prostate cancer cells through mitotic arrest and reduced DNA damage repair. Int J Radiat Oncol Biol Phys. 2009;75:S569. [Google Scholar]
- 38.Akiyama M, Izumi H, Wang KY, Yamaguchi T, Kuma A, Kitamura N, Harada Y, Oya R, Yamaguchi K, Iwai Y, Kohno K. Hypersensitivity to aurora kinase inhibitors in cells resistant against platinum- containing anticancer agents. Anticancer Agents Med Chem. 2014;14:1042–1050. doi: 10.2174/1871520614666140207154351. [DOI] [PubMed] [Google Scholar]
- 39.Menon V, Povirk L. Involvement of p53 in the repair of DNA double strand breaks: multifaceted roles of p53 in homologous recombination repair (HRR) and non-homologous end joining (NHEJ) Subcell Biochem. 2014;85:321–336. doi: 10.1007/978-94-017-9211-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.