

SUMMARY
CellMiner-SCLC (https://discover.nci.nih.gov/SclcCellMinerCDB/) integrates drug sensitivity and genomic data, including high-resolution methylome and transcriptome from 118 patient-derived small cell lung cancer (SCLC) cell lines, providing a resource for research into this “recalcitrant cancer.” We demonstrate the reproducibility and stability of data from multiple sources and validate the SCLC consensus nomenclature on the basis of expression of master transcription factors NEUROD1, ASCL1, POU2F3, and YAP1. Our analyses reveal transcription networks linking SCLC subtypes with MYC and its paralogs and the NOTCH and HIPPO pathways. SCLC subsets express specific surface markers, providing potential opportunities for antibody-based targeted therapies. YAP1-driven SCLCs are notable for differential expression of the NOTCH pathway, epithelial-mesenchymal transition (EMT), and antigen-presenting machinery (APM) genes and sensitivity to mTOR and AKT inhibitors. These analyses provide insights into SCLC biology and a framework for future investigations into subtype-specific SCLC vulnerabilities.
In Brief
Tlemsani et al. provide a unique resource, SCLC-CellMiner, integrating drug sensitivity and multi-omics data from 118 small cell lung cancer (SCLC) cell lines. They demonstrate that SCLCs have differential transcriptional networks driven by lineage-specific transcription factors (NEUROD1, ASCL1, POU2F3, and YAP1). Furthermore, YAP1-driven SCLCs have distinct drug sensitivity profiles.
Graphical Abstract
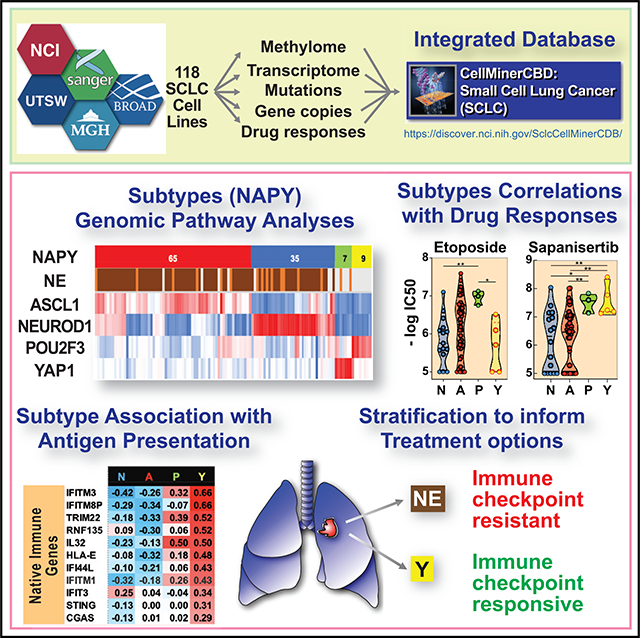
INTRODUCTION
Although small cell lung cancer (SCLC) represents only 15% of all lung cancers, it accounts for more than 30,000 cases/year in the United States, with most patients presenting with widely metastatic disease. Unlike the increasingly personalized treatment approaches for non-small cell lung cancer (NSCLC), SCLC is currently treated as a homogeneous disease (Rudin et al., 2019; Thomas and Pommier, 2016). The typical short life expectancy and the therapeutic options, which have not changed for decades (platinum-etoposide combination as first-line therapy and topotecan at relapse), caused the National Cancer Institute (NCI) to categorize SCLC as a “recalcitrant” cancer.
SCLC tumors are usually characterized by their neuroendocrine (NE) differentiation, which is immuno-histochemically visualized with markers including synaptophysin (SYP) and chromogranin A (CHGA) (Gazdar et al., 2017; McColl et al., 2017). Yet a small subset of SCLCs express low levels of these NE markers (“non-NE”) (McColl et al., 2017; Zhang et al., 2018). Hence, SCLCs have been historically defined as “classic” (NE) or “variant” (non-NE) (Zhang et al., 2018). Gazdar and colleagues proposed a classification (“NE score”) on the basis of the expression of 50 genes (25 with increased and 25 with decreased expression) for NE SCLC, including the transcription factors ASCL1 (achaete-scute homolog 1) and NEUROD1 (neurogenic differentiation factor 1), which are highly expressed in NE SCLC (Zhang et al., 2018). A consensus nomenclature for molecular subtypes has been recently proposed on the basis of differential expression of two additional transcription factors, YAP1 (Yes-associated protein 1) and POU2F3 (POU class 2 homeodomain box 3) for the non-NE SCLC subtypes (Rudin et al., 2019). POU2F3 encodes a POU domain transcription factor normally expressed in chemosensory cells of the intestinal and lung epithelium (Huang et al., 2018). YAP1, a key mediator of the Hippo signaling pathway, is reciprocally expressed relative to the NE marker INSM1 (McColl et al., 2017). Hence, SCLCs can be classified into four groups on the basis of the expression of NEUROD1, ASCL1, POU2F3, and YAP1 (Rudin et al., 2019). For brevity, we refer to this classification as “NAPY” (N for NEUROD1, A for ASCL1, P for POU2F3, and Y for YAP1).
Genomic initiatives spearheaded by The Cancer Genome Atlas (TCGA) consortium have accelerated the pace of discovery for many cancers. Yet TCGA was not extended to SCLC, because of a lack of readily accessible and adequate tumor tissue, as most patients are diagnosed by fine-needle aspiration. Nevertheless, SCLC research has benefited from the systematic collection of a large number of tumor cell lines, most of them developed at the NCI in the NCI-VA and NCI-Navy Medical Oncology Branches (Mulshine et al., 2019). This collection has been distributed widely and included in the cancer drug genomic databases of the NCI, Broad Institute/MIT, and Sanger/Massachusetts General Hospital (MGH) (Barretina et al., 2012; Garnett et al., 2012; Iorio et al., 2016; Polley et al., 2016). However, the data were until now accessible only from individual platforms, making it challenging to translate genomic knowledge of SCLC tumor biology and therapeutic possibilities. Additionally, a number of SCLC cell lines generated by the Minna-Gazdar group at UT Southwestern (UTSW) Medical Center (McMillan et al., 2018) had not been integrated in the NCI (NCI-SCLC), Broad Institute (Cancer Cell Line Encyclopedia [CCLE]/Cancer Therapeutics Response Portal [CTRP]), and Sanger/MGH (Genomics of Drug Sensitivity in Cancer [GDSC]) databases.
To extend our understanding of the genomics of SCLC, we performed genome-wide promoter methylation on the NCI set of 66 SCLC cell lines and whole-genome RNA sequencing (RNA-seq) for 72 cell lines of the UTSW set. We integrated those data in a global drug and genomic database (SCLC-Global) encompassing 118 SCLC lines from 115 individual patients. The integrated data, SCLC-CellMiner-CrossDataBase (SCLC-Cell-Miner), are available from a web-based tool (https://discover.nci.nih.gov/SclcCellMinerCDB/) derived from our CellMiner cross-database (CDB) web application (Rajapakse et al., 2018).
RESULTS
SCLC-CellMiner Resource
SCLC-CellMiner integrates genomic and drug activity data for 118 molecularly characterized SCLC cell lines, all of which have DNA fingerprints establishing their provenance (Figures 1A and 1C): 68 from the NCI collection (Polley et al., 2016), 74 from the GDSC (Garnett et al., 2012), 53 from the CCLE, 39 from the CTRP (Barretina et al., 2012), and 73 from UTSW (Gazdar et al., 2010). Seventeen cell lines (14%) are in all five data sources, 20 (17%) are in four data sources, 23 (20%) in three data sources, 15 (13%) in two data sources, and 43 (36%) in only one data source (Figure 1A; Table S1).
Figure 1. Summary of the Data Included in SCLC-CellMiner and Resources.

(A) Cell line overlap between the data sources. Cell lines in red are from the NCI database (n = 68), dark blue from CTRP (n = 39), light blue from CCLE (n = 53),orange from GDSC (n = 74), and green from UTSW (n = 73). Cell line details are provided in Table S1.
(B) Summary of the genomic and drug activities data in SCLC-CellMiner (https://discover.nci.nih.gov/SclcCellMinerCDB/). For microarray, mutations, copy number, and promoter methylation data, the numbers indicate the number of genes. For RNA-seq data, the numbers indicate the number of transcripts. The bottom row shows the total number of cell lines (N = 118) integrated in SCLC-CellMiner. New data analyses are highlighted in yellow.
(C) Cell line overlap between data sources (see Table S1 for details).
(D) Drug overlap between data sources.
Our integrated resource includes new analyses for high-resolution methylome (Krushkal et al., 2020) and copy number for 66 NCI cell lines and RNA-seq for 72 UTSW cell lines (Figure 1B). SCLC-CellMiner also makes accessible whole-exome mutation data for 12,537 genes across 72 cell lines of the UTSW SCLC database in addition to the previously released exome sequencing data for 52 cell lines from CCLE and 62 cell lines from GSDC.
Tested clinical drugs and investigational compounds in each dataset and across data sources are summarized in Figure 1D. The NCI dataset provides the largest number (n = 526), followed by the CTRP (n = 481), GDSC (n = 297), and CCLE (n = 224).
SCLC-CellMiner allows multiple analyses (Table 1): confirming cell line reproducibility and identity across datasets, drug activity reproducibility, determinants of gene expression (on the basis of DNA copy number, promoter methylation, and microRNA expression), exploration and validation of genomic networks, classification of the cell lines on the basis of metadata such as the NAPY, epithelial-mesenchymal transition (EMT) and antigen-presenting machinery (APM) scores, and validation and discovery of drug response determinants.
Table 1.
Examples of SCLC-CellMiner Capabilities
| SCLC-CellMiner Explores and Validates | Method | Examples | Examples of Findings | |
|---|---|---|---|---|
| 1 | cell line reproducibility and consistency | “Univariate Analyses: Plot Data:” expression of the same gene “across different datasets” (X and Y) | Figure 2 | cell lines are highly reproducible across datasets |
| 2 | omic data robustness and reproducibility | “Univariate Analyses: Plot Data:” expression, copy number variation, promoter methylation, mutations for the same gene “across datasets” (X and Y) | Figures 1B, 1C, and 2 | transcripts, promoter methylation, and gene copy number are highly reproducible across datasets |
| 3 | drug data robustness and reproducibility | “Univariate Analyses: Plot Data:” activity of the same drug “across datasets” (X and Y) | Figures 2E and 2F | warning: not all drugs are consistent across dataset |
| 4 | integrates all the SCLC cell line genomic datasets under SCLC-Global (NCI, GDSC, CCLE, CTRP, UTSW) | use the pull-down tabs for “Cell Line Sets” and choose “SCLC-Global” | Figures 4D, 6H, S2G, S2H, and S3C | the 119 SCLC cell lines can be classified in the four groups of NAPY; development of NAPY genomic signatures |
| 5 | integration with CellMinerCDB | open in parallel: https://discover.nci.nih.gov/cellminercdb | Figures 2, 4, and 5 | POU2F3 is selective for SCLC; YAP1 is expressed widely beyond SCLC; ASCL1 is co-expressed with NEUROD1 |
| 6 | select and compare subsets of cell lines based on tissue of origin or metadata: NAPY, TNBC, NSCLC | “Univariate Analyses:” select y axis: “Select Tissue/s of Origin” or “Select Tissues to Color” (NEUROD1, ASCL1, POU2F3, YAP1, NE) | Figures 2H, 5F, S3, and S6 | NEUROD1 and ASCL1 are also selectively expressed in CNS cancer cell lines |
| 7 | test phenotypic data (mda): NE, APM, EMT | “Univariate Analyses:” select “Data Type mda: NE, APM, EMT;” additional selection can be done for subset (see #6) | Figures 4H and 6 | NE cell lines have low antigen-presenting machinery (APM) score |
| 8 | tissue- or subset type-specific analyses (NAPY; NE) | “Select Tissue/s of Origin” or “Select Tissues to Color” | Figures 5, 6, and S4–S6 | YAP1 cell lines have lower replication and highest APM score |
| 9 | epigenetics: promoter methylation for any given gene | “Univariate Analyses: Plot Data:” expression of a given gene versus its methylation (X and Y “Data Type”) within a given “Cell Line Set” or across datasets (independent datasets can be tested for missing “Data Type” and confirmation) | Figure S1 | promoter methylation is a driver for gene expression (NAPY genes; SLFN11; MGMT; SMARCA1; CGAS) |
| 10 | gene amplification and deletions for any given gene | “Univariate Analyses: Plot Data:” expression of a given gene versus copy number (X and Y “Data Type”) within a given “Cell Line Set” or across datasets (independent datasets can be tested for validation and missing “Data Type”) | Figures 1, 3, and S1 | MYC genes and other oncogenes are often driven by copy number variation (CNV) |
| 11 | integrate and complement different datasets for common cell lines | “Univariate Analyses: Plot Data:” plot different parameters (“Data Type for” genomic or drug response) across “Cell Line Sets” (X and Y) to counter missing data in one dataset | Figures 1, 2, and 6 | drug response data in one dataset can be correlated with genomics of another dataset |
| 12 | genomic pathway discovery (coregulated genes and microRNAs) | “Univariate Analyses: Plot Data:” expression of a given gene (X or Y “Data Type”) within a given dataset or across datasets; also use the “Compare Patterns” tab | Figures 5, 6, S2, and S3 | ASCL1 and YAP1 are integrated in tight genomic networks connected with the NOTCH pathway |
| 13 | discover determinants of drug response and targeted drug delivery | “Univariate Analyses: Plot Data: Compare Patterns:” coregulated genes for a given gene (X or Y) within a given “dataset” (independent datasets can be tested for confirmation) | Figures 6 and S6 | resistance of YAP1 cell lines to chemotherapy and potential response to mTOR and immune checkpoint inhibitors; NAPY-specific antigen cell surface biomarkers |
| 14 | validate genomic determinant of drug response | “Univariate Analyses: Plot Data: Compare Patterns:” plot genomic parameter versus drug (X or Y “Data Type”) | Figure 6 | validation of SLFN11 for DNA damaging chemotherapy |
| 15 | examine drug correlations: COMPARE analyses | “Univariate Analyses: Plot Data: Data Type:” drug versus drug (X or Y); also select “Compare Patterns” to identify drug-drug correlations | Figure S1 | cell lines sensitive to etoposide are cross-sensitive to topotecan |
| 16 | multivariate models of drug response and genomic features | “Multivariate Analyses: Cell Line Set; Response Data Type; Predictor Data Type/s; Predictor Identifier:” enter drug and genomic parameters to be tested as identifier or use “LASSO” to discover additional non-redundant determinants of response | Figures 5B and 5D; Figure S3E | discover independent omic or drug parameters to build a molecular signature for drug response or gene expression |
| 17 | data download | “Univariate Analyses: View Data: Download” tabs or “Multivariate Analyses: Download” tab | Figure 6 | allow further in-depth analyses and data download in Excel |
| 18 | drug identifier conversion | not applicable | Figures 2E and 2F | allow drug identification across different sources |
Set off in quotation marks are the option tabs of SCLC-CellMiner (https://discover.nci.nih.gov/SclcCellMinerCDB/).
Data Validation, CDB Analyses, and CellMiner Univariate Analyses
Cross-comparison for matched cell lines was used to validate the new NCI-SCLC methylome (850K Illumina array) (Krushkal et al., 2020) by comparison with the published SCLC data of GDSC (450K array) (Rajapakse et al., 2018). The comparison yields high overall correlation for promoter methylation (Reinhold et al., 2017), with a median of 0.90 for 9,015 common genes with a wide expression range for the 43 common cell lines (Figures 2A and S1). Cross-correlation of the new RNA-seq data from UTSW with other gene expression data (microarray and RNA-seq) is also highly significant (Figures 2A and S1). This demonstrates the high reproducibility and stability of the key molecular characteristics in SCLC lines grown in tissue culture for widely divergent passages at different institutions and analyzed independently with different technical platforms (RNA-seq versus microarray, 850K versus 450K methylome arrays).
Figure 2. Validation and Reproducibility of the SCLC-CellMiner Data and Snapshots of Representative Outputs of SCLC-CellMiner (https://discover.nci.nih.gov/SclcCellMinerCDB/).

(A) Reproducibility between data sources. Pearson’s correlations are indicated above violin plots.
(B) Snapshot showing the reproducibility of SLFN11 gene expression across the 41 common cell lines (AffyArray for NCI/DTP on the x axis versus RNA-seq for UTSW). Each dot is a cell line. The data can also be readily displayed in tabular form and downloaded in tab-delimited format by clicking on the “View Data” tab to the right of the default “Plot Data” tab.
(C) Snapshot showing the reproducibility of SLFN11 promoter methylation across the 43 common cell lines independently of the methods used (850K Illumina Infinium MethylationEPIC BeadChip array for NCI/DTP versus Illumina HumanMethylation 450K BeadChip array for GDSC).
(D) Highly significant correlation between MYC copy number (NCI/DTP) and MYC expression (CCLE) for the 36 common SCLC cell lines.
(E–G) Examples of drug activity across databases for the common cell lines.
(H) High proliferation signature of SCLC cell lines on the basis of high PCNA and MYC expression. Snapshot shows that SCLC (green) overexpress PCNA and fall into two groups with respect to MYC.
Reproducibility across datasets can be tested with CellMinerCDB by plotting the same gene (expression, copy number, or promoter methylation), drug, or microRNA on the x and the y axes. For instance, Schlafen 11 (SLFN11), whose expression is highly predictive of response to a broad range of frontline treatments of SCLC (etoposide, topotecan, cis- and carboplatin) as well as drugs under investigation such as the poly(ADP-ribose polymerase) inhibitors (Farago et al., 2019; Gardner et al., 2017; Murai et al., 2019; Zoppoli et al., 2012) measured by RNA-seq in the UTSW database, shows a 0.92 Pearson correlation with its measured values by Affymetrix microarray in the NCI database (Figure 2B). SLFN11 promoter DNA methylation in the NCI database also shows a Pearson correlation of 0.9 with its value in the GDSC (Figure 2C).
CDB analyses are shown in Figure 2 for MYC, which is commonly amplified and drives proliferation of SCLC (Ireland et al., 2020), for BCL2, which encodes a canonical antiapoptotic protein targeted by navitoclax (ABT-263) (Rudin et al., 2012), and for two SCLC drugs, etoposide and topotecan. MYC amplification (in NCI) is correlated with its overexpression (by RNA-seq in CCLE) (Figure 2D). Navitoclax activity is correlated with BCL2 expression (Figure 2E). Response to etoposide is correlated in the NCI and CTRP despite different assays; cells responding to etoposide overlap for topotecan (Figures 2F and 2G).
Integrating the CellMinerCDB database of more than 1,000 cell lines of all lineages, which includes 74 and 53 SCLC cell lines in GDSC and CCLE (Figures 1A and 1C) (Rajapakse et al., 2018), allows comparisons among tissue of origin. For instance, MYC expression is correlated with the replication processivity factor PCNA (proliferating cell nuclear antigen) in SCLC versus other tissues, including NSCLC, consistent with the replicative genotype of SCLC and high PCNA expression compared with NSCLC (Figure 2H).
The SCLC Methylome
Two prior studies described the promoter methylation profiles of SCLC with limited data for cell lines; 18 were examined by Kalari et al. (2013) and 7 by Poirier et al. (2015) together with primary tumors and patient-derived xenograft (PDX) samples. Here we analyzed the methylome of the 66 cell lines of the NCI and processed the methylome of the whole 985 GDSC cancer cell line dataset, including its 61 SCLC cell lines. Individual probe analysis for the Illumina 850K platform in the NCI SCLC cell lines is reported in a parallel publication (Krushkal et al., 2020), while SCLC-CellMiner provides promoter methylation score (Reinhold et al., 2017). The promoter methylation data are highly reproducible between the NCI and GDSC datasets for the 43 common cell lines despite the different Illumina platforms (850K versus 450K) (Figures 2A and 2C). Thus, SCLC-CellMiner provides promoter methylation for a total of 84 individual SCLC cell lines (43 common + 23 specific to NCI-SCLC + 18 specific to GDSC).
Low Global Methylation and Promoter Methylome of SCLC Cell Lines
Global methylation levels show marked differences between the SCLC and the other cancer cell lines from different histologies across the GDSC, with SCLC showing the lowest median level of global methylation among 21 cancer subtypes (Figure 3A).
Figure 3. Methylation Profile of SCLC Cell Lines.

(A) Global hypomethylation in SCLC cell lines. Each point represents the median methylation level of individual cell lines for the total set of 17,559 genes. Twenty-one cancer subtypes from GDSC are ranked according their global methylation levels. SCLC cell lines are in red (NCI) and green (GDSC).
(B) Comparison of promoter methylation profiles for 287 cell lines including SCLC (NCI and GDSC), NSCLC (GDSC and NCI-60), and non-lung cancer cell lines from the NCI-60. The heatmap displays the levels of methylation of 1,813 genes with high dynamic range. Examples of genes are indicated at right and details provided in Table S3. Clusters a, b, and c include 68, 117, and 102 cell lines, respectively.
(C) Pathway analysis.
(D) Functional categories with significant correlation between gene expression and promoter methylation for the NCI-SCLC cell lines (n = 66). Median values transcript expression versus DNA methylation level correlations of 20 functional groups including 17,144 genes (Table S5).
(E) Correlations between gene expression and predictive values of DNA copy number. R values of −1 and +1 indicate perfect negative and positive predictive power, respectively. Each point represents 1 of a total of 14,046 genes analyzed. Oncogenes and tumor suppressor genes (highlighted in purple and in blue, respectively) are driven primarily by copy number. Histone genes (red) and epithelial genes (green) are driven primarily by DNA methylation (Table S5). SCLC key genes (ASCL1, NEUROD1, POU2F3, and YAP1) are also labeled.
To assess the distinctiveness of the methylome of the SCLC cell lines, we compared 61 cell lines from GDSC and 66 cell lines from NCI with the 75 NSCLC cell lines of the GDSC and the 60 cell lines of the NCI-60, which include nine NSCLC cell lines. We selected 1,813 genes with the highest methylation range (SD > 0.25). Hierarchical clustering (Figure 3B) shows that SCLC cell lines come together (cluster b), except for nine cell lines (one in cluster a, eight in cluster c), which are all SCLCs not expressing NE features (“non-NE” SCLCs). Of the five NSCLC cell lines in the SCLC cluster (b), three are large cell lung cancers and one is a carcinoid (Table S2). This demonstrates a promoter methylation signature for SCLC cell lines associated with NE phenotype.
Genes clustered as (1) hypomethylated in SCLC (clusters 1–3), including ASCL1, NEUROD1, INSM1, and CHGA (Figure S2); (2) hypermethylated in SCLC (cluster 5); and (3) variably methylated independently of tissue of origin (cluster 4) (Table S2). Pathway analysis of the 1,082 hypomethylated genes (clusters 1–3) shows enrichment of neurological as well as extracellular matrix (ECM) pathways (Figure 3C; Table S2), consistent with the NE and aggregation features of classic SCLC cell lines. Many genes involved in EMT (Kohn et al., 2014) also tend to be hypomethylated in SCLC cell lines, including ZEB1, CLDN7, and ESRP2.
Histone and Epithelial Genes Are Driven by Methylation in SCLC Cell Lines
To determine the influence of promoter methylation on gene expression, we selected gene categories on the basis of our previously established Development Therapeutics Branch (DTB) gene sets (Table S3) (Reinhold et al., 2017). Epithelial and histone genes stood out (Figure 3D, with median correlation of −0.53 and −0.50, respectively). Canonical histones showed the highest negative correlation between expression and methylation (Figure 3E), suggesting that epigenetic regulation of canonical histones is a feature of SCLC carcinogenesis.
We also performed gene set enrichment analyses (GSEAs) looking at Gene Ontology (GO) and functional gene set collections (MSigDB Hallmark gene set, C2 curated pathway gene set, and C5 GO gene set, as well as our DTB functional gene sets; Table S3). They confirmed the high significance of the histones and epithelial genes as well as additional GO categories, including protein modifications, microtubule cytoskeleton, mitotic cell cycle, and cellular responses to DNA damage (Table S4).
SCLC DNA Copy Number versus Methylome as Drivers of Gene Expression
To evaluate the relative importance of promoter methylation and gene copy number, we derived copy number data from the Illumina 850K methylome array and correlated the expression of each gene with DNA copy number and methylation in the NCI-SCLC dataset (Figure 3E) (Reinhold et al., 2017). Correlations for individual genes can be readily displayed with SCLC-CellMiner (https://discover.nci.nih.gov/SclcCellMinerCDB/), and snapshots of genes involved in SCLC carcinogenesis and driven by methylation (NEUROD1, ASCL1, POU2F3, YAP1, and SLFN11) are presented in Figure S1.
Unlike the histone and epithelial genes, the expression of SCLC growth-driving genes, such as the oncogenes (MYC, MYCL, MYCN, and AKT1), tumor suppressor genes (CDKN2A, BAP1, and VHL), and chromatin remodeler genes (EP300 and CREBBP), are driven primarily by copy number alterations (Figure 3E; Table S5). CellMinerCDB snapshots showing increased (MYC, MYCL, and MYCN) or decreased (BAP1 and VHL) copy number variation are provided in Figure S1.
SCLC-Global Integrates Transcriptomes and Molecular and Phenotypic Data for 116 Cell Lines
To integrate expression data from microarray and/or RNA-seq across the five data sources (Figure 1), we created the “SCLC-Global” expression set by regrouping all datasets by Z score normalization, which enables CDB analyses of gene expression (and also other genomic, epigenomic, and phenotypic drug response information). Principal-component and correlation analyses (Circos- and CAT-plots) validated the approach (Figures S2A–S2D and S2F). The “SCLC-Global” data are available in the pull-down tabs “x-Axis Cell Line Set” and “y-Axis Cell Line Set” (https://discover.nci.nih.gov/SclcCellMinerCDB/). The “SCLC-Global” mRNA dataset shows very high correlation with each dataset (NCI-SCLC, GDSC, CCLE, and UTSW) (Figure S2E). For example, ASCL1 expression in SCLC-Global versus SCLC NCI/DTP is highly correlated (r = 0.99, p = 1.9e-55). SCLC-Global offers many other features, including cross-correlation with other databases for DNA methylation, DNA copy number, DNA mutation, microRNA expression, and drug activity.
SCLC-Global can also be used to retrieve all the genes correlated with the expression of any given gene. For instance, for MYCN, the top correlate (p = 0.967) is MYCNOS (Figures S2G–S2I), the MYCN Opposite Strand antisense RNA. The data for individual cell lines can also be visualized by plotting MYCNOS against MYCN in the SCLC-Global database (Figure S2H). Plotting MYCN versus MYCNOS in the CCLE database using CellMinerCDB extends the finding that MYCN is co-expressed with its antisense RNA in both SCLC and brain tumor cell lines (Figure S2I).
NE, NAPY, MYC, and EMT Molecular Signatures
Ranking of the 116 cell lines of SCLC-Global on the basis of their NE scores (Zhang et al., 2018) shows the expected high correlation with SYP, CHGA, NCAM1, and INSM1 expression (Figure 4A). To explore the selectivity of those genes for SCLC, we examined the GDSC and CCLE human tumor cell line collections with CellMinerCDB (Rajapakse et al., 2018). CHGA, INSM1, and SYP are selective for SCLC and brain tumors, consistent with the neuronal differentiation of SCLC (Figures S3A and S3B). The SCLC cell lines with high NE scores, which can be readily labeled in SCLC-CellMinerCDB under the “Select Tissues to Color” tab, have significantly higher levels of expression of CHGA and SYP than cell lines with low NE score (Figure S3C).
Figure 4. SCLC Genomic Molecular Classifications.

(A) NE classification. Cell lines with high and low NE score are in dark brown and gray, respectively (n = 116 cell lines; CellMiner-Global). CHGA, SYP, and INSM1 expression after Z score normalization.
(B) NAPY classification for the 116 SCLC cell lines. Expression values across the five data sources were obtained after normalization by Z score (Table S3).
(C) NEUROD1 and ASCL1 expression are specific for both SCLC and brain tumor cell lines (GDSC database; each point is a cell line; n = 986).
(D) POU2F3 is selectively expressed in SCLC but not in brain tumor cell lines (GDSC; n = 986).
(E) YAP1 shows a high range of expression across different cell line subtypes (GDSC; n = 986). (C)–(E) are snapshots (https://discover.nci.nih.gov/cellminercdb).
(F) Co-expression of NEUROD1 and ASCL1 in SCLC-Global.
(G) Subtypes of cell lines in GDSC.
(H) EMT signature and NAPY classification in CellMiner-Global.
(I) Classification based on expression of the three MYC genes in 106 SCLC cell lines across the five data sources after Z score normalization.
Next we tested the lineage transcription factor molecular classification on the basis of the expression of NEUROD1 and ASCL1 for NE and YAP1 and POU2F3 for non-NE SCLC (Rudin et al., 2019) and found clear separation (Figure 4B; Table S6). Comparison with other tissues showed selective expression of NEUROD1 and ASCL1 in SCLC and brain tumors (Figure 4C), while POU2F3 was expressed only in a subset of SCLC cell lines (Figure 4D). In contrast, YAP1 is not exclusive to SCLC and is expressed in a wide range of cancer types (except blood and lymphoid tumors) (Figure 4E), consistent with its broad role in carcinogenesis (Ma et al., 2019). We also noted a significant fraction of NE-SCLC cells with dual expression of ASCL1 and NEUROD1 (Figures 4B and 4F).
The three MYC genes (MYC, MYCL, and MYCN) play key roles in SCLC carcinogenesis (Johnson et al., 1987; Little et al., 1983; Nau et al., 1985, 1986). With SCLC-Global, ~80% of the SCLC cell lines highly express one of the MYC genes, and MYC and MYCL are most prevalent (Figure 4H). Expression of the MYC genes is mutually exclusive (Ireland et al., 2020; Mollaoglu et al., 2017), with the non-NE cell lines (Y and P) expressing MYC and the NE cell lines expressing MYCL and MYCN (Figures 4H and S3).
The EMT status (Rajapakse et al., 2018) derived from the expression of 37 genes (Kohn et al., 2014) showed that the SCLC-P cell lines are consistently epithelial, while the SCLC-Y cell lines have a mesenchymal signature (Figure 4I), except for NCI-H1607, expressing both YAP1 and POU2F3 (Figure 4B, left). The SCLC-NE cells form two subgroups, one mesenchymal and the other intermediate (Figure 4I).
SCLC Transcriptional Networks for the ASCL1, YAP/TAZ, and NOTCH Pathways
As a pioneer transcription factor, ASCL1 binds E-box motifs (as does NEUROD1) to promote chromatin opening and activation of neuronal genes. Figure 5A summarizes the ASCL1-NOTCH network on the basis of our molecular interaction map (MIM) conventions (https://discover.nci.nih.gov/mim/index.jsp) (Kohn et al., 2006). Notably both NKX2.1 and PROX1 transcription factors are highly significantly co-expressed with ASCL1, suggesting that they function together (Pozo et al., 2020). This co-expression is not due to the location of those genes on the same chromosomes (Figure 5A), indicating upstream regulatory transcriptional control with the likely implication of super-enhancers. As expected, the transcriptional targets of ASCL1 were co-expressed with ASCL1 (Figures 5A and 5B). One of those, BCL2, is positively correlated not only with ASCL1 but also with POU2F3, whereas BCL2 expression is negatively correlated with NEUROD1 expression (Figures S3H and S3I). Expression of the cancer-driving genes RET, SOX1, SOX2, FOXA1, and FOXA2 is also highly correlated with ASCL1 (Figure 5A). Expression of DLL3, a known inhibitor of the NOTCH pathway and direct target of ASCL1, was found to be significantly correlated with ASCL1 (r = 0.61, p = 4.05e-13; Figure 5A).
Figure 5. Integration of the Transcriptional Networks of the SCLC-A and SCLC-Y Cell Lines with the NOTCH Pathway for the 116 Cell Lines Derived from SCLC-Global Analyses.

(A) Highly significant correlations between ASCL1 expression and NKX2–1 and PROX1 and downstream transcriptional targets (bayonet arrows). Numbers to the right indicate the significantly positive Pearson’s correlations coefficients (red) (https://discover.nci.nih.gov/SclcCellMinerCDB/) irrespective of chromosome locations (black in parenthesis). The NOTCH receptor network with its transcriptional target REST (yellow box) shows significant negative Pearson’s correlations (blue).
(B) Correlations between the expression of ASCL1 and the genes shown in (A) (snapshot from the multivariate analysis tool of SCLC-CellMiner).
(C and D) Same as (A) and (B) except for YAP1.
(E) Correlations between the NOTCH receptors and ligands genes and ASCL1 versus YAP1. Pearson’s correlation coefficients are indicated in parenthesis.
(F) Correlation between NOTCH1 and NOTCH2 expression. YAP1 cells show significantly high expression of both NOTCH1 and NOTCH2.
(G) Correlation between NOTCH1 and NOTCH2 expression across the 1,036 cell lines of the CCLE. SCLC-Y cells have highest expression.
(H) SCLC-Y cells have significantly fewer RB1 mutations.
(I) t-Distributed stochastic neighbor embedding clustering plot using gene expression data of 60 SCLC and 100 NSCLC cell lines (microarray; GDSC data source).
Analysis of the NOTCH pathway whose inactivation is crucial in NE-SCLC (Gazdar et al., 2017; Leonetti et al., 2019; Ouadah et al., 2019) showed that NOTCH1, NOTCH2, and NOTCH3 are jointly downregulated in the SCLC-A cell lines (Figures 5A and 5B). Functional downregulation of the NOTCH pathway is consistent with the negative correlation (r = −0.545, p = 2.45e-10) between ASCL1 and REST, the transcriptional target of NOTCH (Figure 5A). The NEUROD1 subset of NE-SCLC (SCLC-N) did not show significant correlation between NEUROD1 and DLL3 expression (r = −0.18, p = NS) (Figures S3J and S3K), questioning whether DLL3 downregulates the NOTCH pathway in SCLC-N cell lines.
Of the 116 SCLC cell lines in SCLC-CellMiner, 9 belong to the YAP subset (Figure 4). Because expression of YAP1 is a feature in a wide variety of solid tumors (Figure 4E), we explored the YAP transcriptional network (Figure 5C). The first notable finding is that YAP1 expression is highly correlated with the expression of its heterodimeric partner TAZ (encoded by the WWTR1/TAZ gene) both in the SCLC-Global dataset (Figures 5C and 5D) and across the 986 cell lines of the GDSC (Figure S4), suggesting a master transcriptional regulator upstream of both genes, or YAP1 acting as super-enhancer (Figure 5C).
YAP/TAZ functions as a direct activator of the TEAD transcription factors (encoded by TEAD2/TEAD3/TEAD4), whose expressions are highly significantly coregulated with YAP1 (Figure 5C). As expected, known transcriptional targets of the TEADs are also significantly correlated with YAP1 expression (Figure 5C). Others can readily be revealed with the “Compare Patterns” feature of SCLC-CellMiner using TEAD or YAP1 as “seeds.” Among those are the cancer- and growth-related SMAD3 and SMAD5 genes, CCN1/CYR61 (encoding a growth factor interacting with integrins and heparan sulfate), and VGLL4 (Figures 5C and 5D).
Next, we explored the Hippo pathway, which acts as a negative regulator of YAP/TAZ and is commonly inactivated in solid tumors (Dasgupta and McCollum, 2019; Ma et al., 2019; Totaro et al., 2018). Expression of LATS2 and LATS1, which encode the core kinase of the Hippo pathway and negatively regulate YAP by sequestering phosphorylated YAP in the cytoplasm, are significantly positively correlated with YAP1 expression (Figures 5C and 5D). Similarly, the transcripts of MOB1A and MOB1B, the cofactors of LATS1/2, are positively correlated with YAP1 (Figures 5C and 5D). Moreover, the transcripts of the negative regulators of YAP, AMOT and AMOTL2, which are released by depolymerized F-actin and sequester YAP from its nuclear translocation, are also significantly positively coregulated with YAP1 (Figures 5C and 5D) (Dasgupta and McCollum, 2019; Wang et al., 2019). Together, these results demonstrate that the SCLC-Y cell lines co-express both YAP/TAZ and its negative regulator genes driving the Hippo pathway, suggesting an equilibrium (“metastable”) state in which the Hippo pathway remains active to potentially negatively regulate YAP/TAZ in SCLC-Y cells.
Consistent with the NOTCH pathway as transcriptional target of YAP/TAZ and the TEADs (Totaro et al., 2018), YAP1 expression is highly correlated with NOTCH1, NOTCH2, NOTCH3, and REST (Figures 5C–5E). In contrast, expression of the NOTCH ligand DLL3, which acts as negative regulator of the NOTCH receptors (Andersson et al., 2011), is negatively correlated with YAP1 (Figure 5E). These results support the conclusion that the NOTCH pathway is “on” in the SCLC-Y cells. In contrast, in the SCLC-A cells, the opposite is observed (Figures 5E and S4C). The SCLC-P cells also show a positive correlation between the NOTCH receptor and REST effector transcripts and POU2F3 expression (Figures 5F, S4C, and S4F). These analyses demonstrate a difference between NE and non-NE SCLC with respect to NOTCH, with the pathway “off” in the NE subset (N and A) and “on” in the non-NE subset (P and Y).
Global analyses of the NOTCH pathway across 1,036 cell lines from the 22 different tissue types of CCLE (Figures 5G, S4D, and S4E) show that NOTCH2 and NOTCH3 are co-expressed in many tumor types and that the NE-SCLC cell lines are characterized by low NOTCH expression (Figures 5G and S4D). In contrast, the SCLC-Y- and -P cells are among the highest NOTCH-expressing cells.
The SCLC-Y Transcriptome Clusters with NSCLC Cell Lines
Next, we examined the relationship between the SCLC-Y and the NSCLC cell lines (Figure 5I). tSNE (t-distributed stochastic neighbor embedding) is a method to highlight strong patterns by reducing the dimensionality of a dataset while preserving as much “variability” as possible. tSNE analysis using gene expression data between NSCLC (n = 100) and SCLC (n = 60) cell lines from the GDSC grouped the SCLC-Y with the NSCLC cell lines. Among the few NSCLC cancer cell lines clustering with the NE-SCLC were carcinoids and large cell lung cancers (Figure 3B; Table S2). Our analysis supports that SCLC-Y cell lines are distinct among the SCLC subtypes with transcriptome similarity to NSCLC.
Another characteristic of the SCLC-Y cell lines is their low number of RB1 mutations (only one cell line among nine shows RB1 mutation; Figure 5H). However, several of the SCLC-Y lines (NCI-H196, NCI-H841, NCI-H1339, and NCI-H1607) do not express RB1 protein (Modi et al., 2000). The SCLC-Y cell lines also show reduced replication transcriptional network with lowest PCNA, MCM2, and RNASEH2A expression (Figure S5). Additionally, the SCLC-Y cells express the mesenchymal marker VIM, the cytoskeleton component and regulators CNN2 (actomyosin and F-actin component), and the AMOT genes, which regulate cell migration and actin stress fiber assembly (Figure 5C) (Dasgupta and McCollum, 2019).
Global Drug Activity Profiling Suggests Transcription Elongation Pathways as General Drug Response Determinants and Hypersensitivity of the SCLC-P Cell Lines
To explore connections between the NAPY classification and drug responses, we analyzed the drug responses of the 66 SCLC-NCI cell lines using 134 compounds with the broadest activity range (Polley et al., 2016). Unsupervised hierarchical clustering generated two groups of cell lines: those globally drug resistant and those globally drug sensitive, with a bimodal distribution (Figure 6A). Although the NE cell lines (SCLC-N and SCLC-A) and SCLC-Y were distributed in both clusters, the SCLC-P cell lines clustered among the most drug sensitive.
Figure 6. Predictive Biomarkers for SCLC Responses.

(A) Global response of the NCI-SCLC cell lines (NAPY classification to the left).
(B) SCLC-P cells are the most sensitive to etoposide and talazoparib. SCLC-Y cell lines are the most resistant.
(C) Selective activity of the BCL2-BCL-XL inhibitor in a subset of the SCLC-A cells and highly significant correlation with BCL2 expression (right).
(D) Activity of mTOR/AKT inhibitors in a subset of non-NE cells.
(E) Activity of the PI3K inhibitors in non-NE SCLC cells.
(F) SLFN11 expression across the 116 SCLC cells exhibits bimodal distribution in all four SCLC subsets and is predictive of response to DNA damaging chemotherapeutics (Figure S6).
(G) Selective expression of native immune pathway genes in SCLC-Y (correlations between each of the NAPY genes and the listed native immune response genes. Significantly positive and negative correlations are in red and blue, respectively.
(H) Snapshot from SCLC-CellMiner illustrating the correlation between the YAP1 and IFITM3 transcripts across the 116 cell lines of SCLC-Global (Figure S6).
(I) Selective expression of the DLL3 and CEACAM5 (Figure S6).
(J) Potential surface biomarker targets for NE-SCLC and SCLC-P cells.
(K) Potential surface biomarkers for SCLC-Y cells.
Data in (A)–(E) and (I)–(K) are from the 66 cell lines from the NCI-DTP drug and genomic database.
Differential gene expression followed by enrichment pathway analyses (Figures S6A and S6B) showed the ribosomal and EIF2 signaling pathway selectively activated in the sensitive cell lines. EIF2 (eukaryotic translation initiation factor 2A) catalyzes the first regulated step of protein synthesis initiation, promoting the binding of the initiator tRNA to 40S ribosomal subunits. EIF2 factors are also downstream effectors of the PI3K-AKT-mTOR and RAS-RAF-MAPK pathways. These results suggest that global drug response in SCLC is associated with active protein synthesis.
Drug Activity Profiling in Relationship with the NAPY Classification
Both the SCLC-A and N subgroups showed a broad range of response to etoposide, topotecan, and cisplatin, as well as to the PARP inhibitor talazoparib (Figures 6B and S6C). The most significant genomic predictor of response for this NE subgroup is SLFN11 expression (Figure S6C), consistent with analyses in other tissue types (Barretina et al., 2012; Rajapakse et al., 2018; Zoppoli et al., 2012). The potential value of SLFN11 expression as a predictive biomarker is borne out by its highly dynamic and bimodal expression pattern (Figure 6F). Approximately 40% of the 116 SCLC cell lines of SCLC-Global do not express SLFN11 (Figure S6D) and are predicted to be DNA damaging agent resistant.
Methylguanine methyltransferase (MGMT) is a predictive biomarker of drug response is for temozolomide (TMZ). Cancer cells (typically glioblastomas) with MGMT inactivation are selectively sensitive to TMZ. Analyses of SCLC-Global reveals lack of MGMT expression in 33% (N = 38) of the cell lines (Figure S6D). Notably, the non-NE SCLC cell lines all express MGMT, indicating that the SCLC-P- and -Y cancer cells are predicted to be poor candidates for TMZ-based therapies (Farago et al., 2019).
The SCLC-Y cell lines show the greatest resistance to the standard-of-care drugs (etoposide, cisplatin, and topotecan) (Figure 6B). This result is not limited to SCLC, as a highly significant drug resistance phenotype is observed between YAP1 expression and response to etoposide and camptothecin across the whole database of the CCLE-CTRP (Figure S6E).
To determine whether the NAPY classification predicts sensitivity to drugs not commonly used as standard of care for SCLC, we analyzed 526 compounds of the NCI database (Polley et al., 2016) (Table S7). Eighteen drugs were highly subtype specific (p < 0.01, Kruskal-Willis test). Although the BCL2 inhibitor ABT-737 was selective of the SCLC-A cells, seven PI3K-AKT-mTOR inhibitors showed high activity in the non-NE cell lines (SCLC-Y and SCLC-P) (Figures 6D and 6E). The SCLC-P and -Y cell lines are also more sensitive to multi-kinase inhibitors, including dasatinib and ponatinib.
Immune Pathways Are Selectively Expressed in the YAP1 Subgroup of SCLC
Although immune checkpoints inhibitors have been approved for SCLC, the benefit in an unselected patient population is modest with approximately 2-month improvement in median overall survival when immunotherapy is added to first-line platinum and etoposide.
To explore the immune pathways in the 116 cell lines of SCLC-Global and the potential value of the NAPY classification for selecting SCLC patients likely to respond to immune checkpoint inhibitors, we explored the transcriptome of a subset of established native immune response and antigen-presenting genes. Figures 6G and 6H shows that the SCLC-Y cell lines are the only subset expressing innate immune response genes. Expression of the innate immune effector genes CGAS and STING, HLA-E and interferon-inducible genes (IFIT3, IFITM1, IFI44L, IFIT, IFITM8P, and IFITM3) are positively correlated with YAP1 expression. In contrast, the NE subtype shows negative correlation between NEUROD1 and ASCL1 expression for those same immune genes (Figure 6G).
On the basis of the study of Wang et al. (2019) reporting a novel APM transcription signature score yielding a high prediction index for tumor response to immune checkpoint inhibitors, we tested the APM score in the SCLC cell lines (Figure S6). The APM score showed a high correlation with PD-L1 expression, which is notable as PD-L1 is not included among the 13 genes constituting the APM score. Interestingly, the SCLC-Y subtype showed the highest APM score (Figure S6K).
Cell Surface Biomarkers for Targeted Therapy in Relation with the NAPY Classification
Antibody-targeted therapies including antibody-drug conjugates (ADC) represent a promising approach for specific homing, increased uptake, and drug retention at tumor sites while reducing drug exposure to normal tissues and the associated dose-limiting side effects (Coats et al., 2019).
A primary criterion for efficient drug delivery is to choose an exclusively or overexpressed target for the cancer cells. Figures 6I and S6 show the expression of two receptors of clinical ADCs in the SCLC cell lines: DLL3 (used for SCLCs as rovalpituzumab tesirine [Rova-T]; Morgensztern et al., 2019; Rudin et al., 2017) and the carcinoembryonic antigen CEACAMC5 (used in other clinical indications as labetuzumab govitecan; Das, 2017). DLL3 expression is highly correlated with ASCL1 expression (p = 0.62), suggesting that targeting DLL3 could be selective toward SCLC-A tumors. CEACAM5 is highly expressed in only a subset of SCLC-A cell lines that may be potentially sensitive to labetuzumab govitecan (IMMU-130). Both DLL3 and CEACAM5 show highest expression in SCLC among all GDSC tissue types (Figure S6). Expression of TACSTD2 (TROP2), which is used as target for sacituzumab govitecan (IMMU-132) in patients with triple-negative breast cancer (TNBC), exhibits a low expression level in all SCLC cell lines, suggesting that TACSTD2 as a targeted receptor may not be efficient in SCLC (Figure S6).
Among potential new targets for the development of ADCs, the specific NE markers NCAM1, CD24, CADM1, and ALCAM are highly expressed in non-YAP1 SCLC (Figure 6J), suggesting the potential of developing ADCs targeting such surface receptors for NE-SCLC and SCLC-P patients. In contrast, the non-NE surface markers CD151 and EPH2 are highly expressed in the YAP1 cell lines (Figure 6K), suggesting their potential for SCLC-Y cancers.
DISCUSSION
SCLC-CellMiner (https://discover.nci.nih.gov/SclcCellMinerCDB/) provides a unique and first-of-its-kind resource of patient-derived SCLC cell lines characterized comprehensively using multi-omics and drug sensitivity. It also includes new high-resolution methylome, detailed in a complementary publication (Krushkal et al., 2020). SCLC-CellMiner enables interrogation of different databases. The data are highly reproducible across databases, which allowed us to build an integrated platform (“SCLC-Global”) to examine genomic characteristics and drug sensitivities across 116 SCLC cell lines.
Patient-derived cancer cell lines remain the most widely used models and the primary basis to study the biology of cancers. They enable high-throughput testing of new drugs and determinant-of-response hypotheses. The database of 116 SCLC cell lines reported here models the genetic and molecular diversity of SCLC, as exemplified by their stratification across the four recently proposed subgroups (NAPY classification) (Rudin et al., 2019).
Several studies of human cancer cell lines have revealed a drift at the transcriptomic level for individual cell lines over multiple passages, or passages in different laboratories. This raised the concern that cancer cell lines bear more resemblance to each other, regardless of the tissue of origin, than to the clinical samples that they model. However, several other studies have come to the opposite conclusion, demonstrating the need for human cancer cell line panels (Barretina et al., 2012; Neve et al., 2006; Reinhold et al., 2019; Wang et al., 2006; Weinstein, 2012; Zoppoli et al., 2012). For lung cancer cell lines, it has been shown that the genomic drift during culture life is not a dominant feature (Wistuba et al., 1999). The recent analyses across SCLC cell lines, PDX models, and human tissues reported by Rudin et al. (2019) and our present analyses provide strong evidence that the molecular features of SCLC are stable.
SCLC is highly proliferative and under replication stress (Thomas and Pommier, 2016). SCLC-CellMiner confirms that genes involved in DNA replication such as PCNA and MKI67 are highly expressed in SCLC (Figure S7). We also found evidence of chromatin adaptation in SCLC. Not only are many core histone gene promoters hypermethylated (Figure 3) but also H2AFY, a non-canonical histone encoding macroH2A.1, is highly expressed in SCLC cell lines. Two H2AFY splice variants have been identified and SCLC cell lines predominantly express the macroH2A1.2 variant, known to promote homologous recombination and proliferation (Kim et al., 2018). In the context of chromatin and histone genes, ACTL6B, which encodes a subunit of the BAF (BRG1/brm-associated factor) complex, is highly expressed in the SCLC cell lines (Figure S7). The BAF complex is functionally related to SWI/SNF complexes that facilitate transcriptional activation of specific genes by antagonizing chromatin-mediated transcriptional repression. ACTL6B expression is specific to SCLC and brain tumor cell lines and highly correlated with the expression of other chromatin genes, including HMGN2, KDM4B, and SMARCA4 (Figure S7). Only the NE cells express ACTL6B, while the non-NE cells express lowest KDM4B and SMARCA4. These results suggest that this specific BAF complex subunit may be critical in determining the cell fate of NE cells.
Supporting the importance of epigenetics in SCLC carcinogenesis, SCLC cell lines exhibit distinct promoter methylation profile. First, they are globally hypomethylated, suggesting their plasticity. Second, they exhibit a distinct epigenetic profile compared with NSCLC (Figure 3B). Most genes with low methylation are involved in neuronal pathways, suggesting that NE differentiation is driven by promoter methylation. In contrast, Poirier et al. (2015) reported that SCLCs tend to have high methylation levels. The apparent discrepancy could be due to the inclusion of PDX and tumor samples in their study. Also, they did not measure promoter methylation but the proportion of highly variable CpGs, leading them to conclude that high methylation instability is consistent with the plasticity of SCLC (Poirier et al., 2015).
SCLC-CellMiner validates the NAPY classification (Rudin et al., 2019) and provides insights into the coordinated network regulated by each lineage transcription factor. Potential upstream regulators (super-enhancers, microRNAs, or non-coding RNAs) may explain the co-expression of ASCL1 with NKX2-1 and PROX1 and YAP1 with TAZ and warrants further investigations, which can be facilitated by SCLC-CellMiner. Consistent with the results of Rudin et al. (2019), the NAPY classification shows that the cell lines driven by ASCL1 and NEUROD1 often overlap and share common features (Figures 4 and 6). Yet they differ in their relationship with respect to the NOTCH pathway, with the SCLC-A cells showing strong negative correlation with NOTCH genes expression, consistent with NOTCH acting as negative regulator of ASCL1 (George et al., 2015) (Figure 5).
Transcriptome and drug response analyses highlight the distinguishing features of the SCLC-Y. In contrast to ASCL1, NEUROD1, and POU2F3, YAP1 is expressed widely across different tissue types (Figure 4) (Ma et al., 2019), and transcriptome analyses cluster the SCLC-Y cell lines with NSCLC (Figure 5F). SCLC-Y cells also express the NOTCH pathway, in contrast to SCLC-A. This feature could be related to the direct transcriptional activation of the NOTCH pathway by YAP/TAZ (Figure 5C) (Yimlamai et al., 2014). In addition, SCLC-Y cells do not express MYCL or MYCN but rather MYC (Figure 4) (McColl et al., 2017; Mollaoglu et al., 2017). They tend to be RB1 wild-type (Figure 5H) and have lower expression of replication and proliferation genes than the other SCLC subtypes (Figures S5 and S7). SCLC-Y cells were also often derived from non-smoking patients (Table S1; Figure S5). In total, our data suggest that SCLC-Y cell lines are probably derived from a different cell type compared with the NE and SCLC-P subgroups. Our findings of differential drug sensitivities on the basis of transcriptional subtypes support this notion (Figures 6 and S6) and are consistent with recent studies showing that non-NE and MYC-driven SCLC cell lines are sensitive to PI3K-AKT-mTOR, AURKA, and HSP90 inhibitors (Chalishazar et al., 2019; Wooten et al., 2019).
Overall, our data suggest that targeted therapies in patient subgroups selected on the basis of NAPY stratification may be beneficial. Additional therapeutic insights can be derived from our study. First, although SCLC is among the cancer types with the lowest expression of immune-related genes, the SCLC-Y cells notably demonstrate high presenting and native immune predisposition (Figures 6G, 6H, and S6). If verified in clinical cohorts of immunotherapy-treated patients, this finding might enable patient selection. Second, we highlight potential surface markers that could be targeted on the basis of the NAPY subgroups. For example, SCLC-Y cells express neither the therapeutically relevant surface epitopes DLL3 or CEACAM5 (Das, 2017; Morgensztern et al., 2019; Rudin et al., 2017), which tend to be specific for the SCLC-A (and N). However, SCLC-Y express CD151 and EPHA2 (Figure 6K) and might respond to the YAP1 and NOTCH inhibitors in clinical development (Crawford et al., 2018; Leonetti et al., 2019).
Our analyses demonstrate the value of cancer cell line databases and imply that updating drug testing with new clinical drug candidates shall provide valuable information to guide clinical trials. Our results also suggest the potential value of using the NAPY classification to select patients for targeted therapies. It is likely that genomic signatures based on transcriptome and promoter DNA methylation will have to be developed to build reliable tools to assign samples to each of the NAPY subgroups and determine their prognostic and therapeutic value.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by Lead Contact Yves Pommier (pommier@nih.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The scripts and data used for the analysis can be obtained at https://zenodo.org/record/3959142.
All newly generated methylation datasets are available from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) under the accession number GEO: GSE145156.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
SCLC-CellMiner is a dedicated CellminerCDB version for SCLC cell lines (Reinhold et al., 2012, 2014, 2017, 2019) https://discover.nci.nih.gov/cellminercdb/).
The cell line sets included in SCLC-CellMiner Cross-Data-Base (CDB) currently are from the National Cancer Institute SCLC cell lines from the Developmental Therapeutics Program Small Cell Lung Cancer Project (SCLC NCI-DTP), Cancer Cell Line Encyclopedia (CCLE), Genomics and Drug Sensitivity in Cancer (GDSC), Cancer Therapeutics Response Portal (CTRP), the University of Texas SouthWestern (UTSW) and a new resource SCLC-Global. The data source details are described in “Help” section of the SCLC-Cell-Miner website.
Most of the data including drug activity and genomics experiments were processed at the institute of origin and were downloaded from their website or provided from their principal investigator. The genomic data from CTRP and CCLE are common for the overlapping cell lines. However, methylation, mutation and copy number data were processed at Development Therapeutics Branch (DTB), CCR, NCI to generate a gene level summary as described previously (Barretina et al., 2012; Garnett et al., 2012; Krushkal et al., 2020; McMillan et al., 2018; Polley et al., 2016). The new Global expression (SCLC-Global) was developed at DTB by merging the gene expression of all the data sources.
METHOD DETAILS
DNA methylation data
Gene-level methylation using the 850K Illumina Infinium MethylationEPIC BeadChip array was summarized based on (Reinhold et al., 2017). In short, methylation data were normalized using the minfi package using default parameters, where probe-level beta-values and detection p values were calculated for each probe. This provided 866,091 methylation probe measurements. Methylation probe beta-values for individual cell lines with detection p values > = 10–3 were set to missing. Also probes with median p value > = 10–6 were set to missing for all cells and removed from the analysis. Probe locations on the human genome (hg19 version) defined by Illumina was used for the analysis, annotating proximal gene transcripts and CpG islands. Probes were designated as category “1” or “2,” with category “1” considered to be most informative. Category “1” probes overlapped CpG islands and they overlapped either the TSS region within a 1.5kb distance, the first exon or 5′-UTR region. Additionally, probes on the upstream shore of a CpG island with a maximal distance of 200bp from the TSS were also included as category “1” probes. Category “2” probes were positioned either in the upstream- or downstream shore of a CpG island and overlapping the first exon, or on the downstream shore of CpG islands overlapping a 200bp region from the TSS, or in 5′-UTR. In case of genes with multiple transcript start sites, the transcript methylation with the most negative correlation to the gene level expression was used. The analysis resulted in gene-level methylation values for 23,202 genes.
Copy number
Genome wide copy number for the cell lines was estimated from the methylation array data using the Chip Analysis Methylation Pipeline (ChAMP) (Tian et al., 2017) package. ChAMP returns lists of genomic segments with putative copy number estimates. However, the estimate is not valid for regions with high methylation detection p values. For this reason, regions spanning more than 1kb with at least 5 probes with high detection p values (p > 0.05) were filtered out. The copy number estimates were set to missing for those areas. Gene level copy number (for n = 25,568 genes) was calculated for each gene individually, by calculating the average estimate between the transcription start sites and transcription end sites.
RNaseq data
The RNA-seq gene expression data from UTSW SCLC were obtained from analyses based on McMillan et al. (2018). The raw data have been previously submitted to dbGaP (accession phs001823.v1.p1). The paired-end RNA-seq reads from the 70 UTSW SCLC cell lines were aligned to the human reference genome GRCh38 using STAR aligner (version 2.7), FPKM expression values were generated with cufflinks (version 2.2.1) (Bullard et al., 2010) and log-transformation.
Global expression data
We generate a new Global SCLC dataset (SCLC-Global) using all combined cell line resources: NCI SCLC, CCLE, CTRP, GDSC and UTSW. The data sources have a mixture of microarray and RNA-seq gene expression. For each experiment, genes were scaled across all cell lines to create a z-score normalized dataset. The SCLC-Global expression was calculated by averaging the z-scored gene expressions from all sources. To test for removal of batch effects by gene scaling (z-score normalization), we clustered the cell lines based on gene expression using the raw data (Figure S2A) and the normalized data (Figure S2B) in R using the hclust() for clustering, and the ape package (version 5.3) to create the clustering dendrograms.
QUANTIFICATION AND STATISTICAL ANALYSES
Pathway level correlation of expression and DNA methylation
The correlation between methylation and gene expression for multiple functional categories was calculated based on genes in Table S4 using R programming language. For each category, the median correlation of the related genes was calculated to identify potential categories of interest.
Predictive power of DNA copy number and methylation on transcript expression
Testing the predictive power of DNA copy number and methylation on transcript expression was performed with linear regression analysis (as seen in Figure 3E. For each of the 15,798 genes with all three forms of data available (transcript, methylation, and copy number levels) a linear regression model was fit, with both copy number and methylation as independent variables and transcript expression as the dependent variable. The model provided coefficients for the copy number and methylation that gave the lowest squared error between fitted values and true expression. We separated individual contributions of these two factors for gene expression prediction using the method of relative importance (Gromping, 2006), using the lmg method (Bacher, 1980) from the R package relaimpo to compute individual R2 values. Total (or combined) R2 is the summation of these two. Square roots of the R2 values were multiplied by the sign of the coefficients of the factors in the combined model to get the value of R.
Methylome cluster analysis
The methylation cluster analysis was performed using the methylation data from the NCI-SCLC cell lines, GDSC lung cancer (SCLC and NSCLC) cell lines and the NCI-60 cell lines. Genes with high standard deviation (> 0.25) in the GDSC lung cancer cell lines were selected for the analysis. The number of reported clusters was selected based on the cutreeDynamic() function of the dynamicTree-Cut R package (v1.63–1), which split genes into 5 main clusters and cells into 3 main clusters (as reported in the figure). The methylation heatmap was created with the ComplexHeatmap (Gu et al., 2016) R package (version 1.20.0).
SCLC subtypes and heatmaps
The SCLC cell lines were classified into the NAPY subtypes using the expression of NEUROD1, ASCL1, POU2F3 and YAP1 with the SCLC-Global expression dataset. Clustering was performed using distance matrix based on Euclidean distance and “ward.D” clustering using the hclust() function in R programming language.
SCLC neuroendocrine score
Cell line neuroendocrine score was calculated based the method reported in Zhang et al. (2018) that uses a gene set of 25 neuroendocrine and 25 non-neuroendocrine genes for classification. For each cell line, the expression values of genes were correlated with the expression averages of neuroendocrine [NE] cells and non-neuroendocrine [non-NE] cells from Zhang et al. (2018). The NE score was calculated with the following formula:
where Xi denotes the gene expression values of cell line i, [NE] is the mean expression of genes in neuroendocrine cells from Zhang et al. (2018) and [nonNE] is the mean expression of genes in non-neuroendocrine cells from Zhang et al. (2018). The R script that calculates the NE score from the SCLC-Global expression data is available in the supplementary materials.
t-SNE clustering of GDSC lung cell lines using gene expression
SCLC and NSCLC cell line grouping was performed with the gene expression data from the GDSC microarray dataset using the t-SNE algorithm in R (v3.5.1). The random seed was set to 1, the Euclidean distance of genes was calculated with the dist() function with default settings. The t-SNE grouping was calculated using the Rtsne() function from the Rtsne (van der Maaten, 2014) package (v0.15) using the calculated distance matrix, with perplexity set to 10, and 5k maximum iterations.
Clustering drug data of NCI-SCLC cell lines
SCLC cell line expression heatmaps for the SCLC markers, NAPY genes and MYC genes were done using the ComplexHeatmap (Gu et al., 2016) R package (version 1.20.0).
The NCI SCLC drug activity heatmap was generated using R. First, drugs with coefficient of variation less or equal to 0.09 were filtered out. Then the remaining data for the selected 134 drugs (from originally 527) across the 66 SCLC lines were clustered using the hierarchical method based on Euclidean distance and complete linkage.
Gene set enrichment analysis and GSEA analysis
A preranked gene set enrichment analysis was run in R using the clusterProfiler (Yu et al., 2012) and ReactomePA (Yu and He, 2016) packages. Pathways with an adjusted p value below 0.05 were considered as significantly enriched. Single sample gene set enrichment score (APM score) was computed using the R package GSVA (version 1.28.0).
A pre-ranked gene set enrichment analysis (GSEA version 4.0.3) was performed for the correlation between the gene expression and methylation across all the NCI SCLC cell lines. The score was 1/p value if correlation was positive and −1/p value otherwise. The gene sets included our DTB 21 gene sets with the Hallmark, C2 (pathways) and C5 (GO) GSEA signatures. The analysis was done using the classic enrichment statistic with a minimum gene set size of 15 and a maximum of 1000.
Statistical methods
Correlations, heatmaps, and histograms were generated mostly using The R Project for Statistical Computing. Some plots and analysis (such as the Kruskal Willis test) were generated using Partek Genomics suite v7.17.1222 (https://www.partek.com/partek-genomics-suite/) or using SCLC-CellMiner and CellMinerCDB (https://discover.nci.nih.gov/cellminercdb).
Wilcoxon rank-sum tests were used to test the difference between continuous variables such as drug sensitivity and gene expression according NAPY classification. We considered changes significant if p values were below 0.05. In the figures, p values below 0.00005 were summarized with four asterisks, p values below 0.0005 were summarized with three asterisks, p values below 0.005 were summarized with two asterisks and p values below 0.05 were summarized with one asterisk. The scripts and data used for the analysis can be obtained at https://zenodo.org/record/3959142.
Supplementary Material
KEY RESOURCES TABLE.
Highlights.
SCLC-CellMiner is an extensive cell line genomic and pharmacology resource
SCLC cell lines show a methylome consistent with their plasticity and lineage
Transcriptome analyses reveal lineage transcriptional networks and drug predictions
SCLC-Y cells differ from other subgroups by transcriptome and potential therapeutics
ACKNOWLEDGMENTS
C.T., L.P., F.E., N.R., S.V., V.N.R., R.S., M.I.A., W.C.R., A.T., and Y.P. are supported by the Center for Cancer Research, CCR-NCI-NIH (Z01-BC-006150). P.M. is supported by the Center for Cancer Research, CCR-NCI-NIH (ZIA BC 011091). J.K. and B.A.T. are supported by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis (DCTD)-NCI-NIH (HHSN261200800001E). L.G., K.E.H., and J.D.M. are supported by P50 CA070907, U24 CA213274, U01 CA2113338, and Cancer Prevention and Research Institute of Texas (CPRIT) RP110708. We acknowledge the role of the late Adi Gazdar in generating many of the SCLC lines.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108296.
REFERENCES
- Andersson ER, Sandberg R, and Lendahl U (2011). Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612. [DOI] [PubMed] [Google Scholar]
- Bacher F (1980). Introduction to bivariate and multivariate-analysis - Lindman,Rh, Merenda,Pf, Gold,Rz. Ann Psychol 83, 265–266. [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, and Dudoit S (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalishazar MD, Wait SJ, Huang F, Ireland AS, Mukhopadhyay A, Lee Y, Schuman SS, Guthrie MR, Berrett KC, Vahrenkamp JM, et al. (2019). MYC-driven small-cell lung cancer is metabolically distinct and vulnerable to arginine depletion. Clin. Cancer Res 25, 5107–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coats S, Williams M, Kebble B, Dixit R, Tseng L, Yao NS, Tice DA, and Soria JC (2019). Antibody-drug conjugates: future directions in clinical and translational strategies to improve the therapeutic index. Clin. Cancer Res 25, 5441–5448. [DOI] [PubMed] [Google Scholar]
- Crawford JJ, Bronner SM, and Zbieg JR (2018). Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: a patent review. Expert Opin. Ther. Pat 28, 867–873. [DOI] [PubMed] [Google Scholar]
- Das M (2017). Labetuzumab govitecan in metastatic colorectal cancer. Lancet Oncol 18, e563. [DOI] [PubMed] [Google Scholar]
- Dasgupta I, and McCollum D (2019). Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem 294, 17693–17706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago AF, Yeap BY, Stanzione M, Hung YP, Heist RS, Marcoux JP, Zhong J, Rangachari D, Barbie DA, Phat S, et al. (2019). Combination olaparib and temozolomide in relapsed small-cell lung cancer. Cancer Discov 9, 1372–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, et al. (2017). Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 31, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. (2012). Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Girard L, Lockwood WW, Lam WL, and Minna JD (2010). Lung cancer cell lines as tools for biomedical discovery and research. J. Natl. Cancer Inst 102, 1310–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Bunn PA, and Minna JD (2017). Small-cell lung cancer: what we know, what we need to know and the path forward. Nat. Rev. Cancer 17, 725–737. [DOI] [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromping U (2006). Relative importance for linear regression in R: the package relaimpo. J. Stat. Softw 17, 1–27. [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE, et al. (2018). POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev 32, 915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. (2016). A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D, et al. (2020). MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell 38, 60–78.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Ihde DC, Makuch RW, Gazdar AF, Carney DN, Oie H, Russell E, Nau MM, and Minna JD (1987). myc family oncogene amplification in tumor cell lines established from small cell lung cancer patients and its relationship to clinical status and course. J. Clin. Invest 79, 1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalari S, Jung M, Kernstine KH, Takahashi T, and Pfeifer GP (2013). The DNA methylation landscape of small cell lung cancer suggests a differentiation defect of neuroendocrine cells. Oncogene 32, 3559–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Sturgill D, Sebastian R, Khurana S, Tran AD, Edwards GB, Kruswick A, Burkett S, Hosogane EK, Hannon WW, et al. (2018). Replication stress shapes a protective chromatin environment across fragile genomic regions. Mol. Cell 69, 36–47.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn KW, Aladjem MI, Weinstein JN, and Pommier Y (2006). Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol. Biol. Cell 17, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn KW, Zeeberg BM, Reinhold WC, and Pommier Y (2014). Gene expression correlations in human cancer cell lines define molecular interaction networks for epithelial phenotype. PLoS ONE 9, e99269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krushkal J, Silvers T, Reinhold WC, Sonkin D, Vural S, Connelly J, Varma S, Meltzer PS, Kunkel M, Rapisarda A, et al. (2020). Epigenome-wide DNA methylation analysis of small cell lung cancer cell lines suggests potential chemotherapy targets. Clin. Epigenetics 12, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, and Horvath S (2008). Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24, 719–720. [DOI] [PubMed] [Google Scholar]
- Leonetti A, Facchinetti F, Minari R, Cortellini A, Rolfo CD, Giovannetti E, and Tiseo M (2019). Notch pathway in small-cell lung cancer: from preclinical evidence to therapeutic challenges. Cell Oncol. (Dordr.) 42, 261–273. [DOI] [PubMed] [Google Scholar]
- Little CD, Nau MM, Carney DN, Gazdar AF, and Minna JD (1983). Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 306, 194–196. [DOI] [PubMed] [Google Scholar]
- Ma S, Meng Z, Chen R, and Guan KL (2019). The Hippo pathway: biology and pathophysiology. Annu. Rev. Biochem 88, 577–604. [DOI] [PubMed] [Google Scholar]
- McColl K, Wildey G, Sakre N, Lipka MB, Behtaj M, Kresak A, Chen Y, Yang M, Velcheti V, Fu P, and Dowlati A (2017). Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 8, 73745–73756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan EA, Ryu MJ, Diep CH, Mendiratta S, Clemenceau JR, Vaden RM, Kim JH, Motoyaji T, Covington KR, Peyton M, et al. (2018). Chemistry-first approach for nomination of personalized treatment in lung cancer. Cell 173, 864–878.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S, Kubo A, Oie H, Coxon AB, Rehmatulla A, and Kaye FJ (2000). Protein expression of the RB-related gene family and SV40 large T antigen in mesothelioma and lung cancer. Oncogene 19, 4632–4639. [DOI] [PubMed] [Google Scholar]
- Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al. (2017). MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 31, 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, Glisson BS, Farago AF, Dowlati A, Rudin CM, et al. (2019). Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: results from the phase II TRINITY study. Clin. Cancer Res 25, 6958–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulshine JL, Ujhazy P, Antman M, Burgess CM, Kuzmin I, Bunn PA Jr., Johnson BE, Roth JA, Pass HI, Ross SM, et al. (2019). From clinical specimens to human cancer preclinical models-a journey the NCI-cell line database-25 years later. J. Cell. Biochem 121, 3986–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Thomas A, Miettinen M, and Pommier Y (2019). Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther 201, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, McBride OW, Bertness V, Hollis GF, and Minna JD (1985). L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 318, 69–73. [DOI] [PubMed] [Google Scholar]
- Nau MM, Brooks BJ Jr., Carney DN, Gazdar AF, Battey JF, Sausville EA, and Minna JD (1986). Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc. Natl. Acad. Sci. U S A 83, 1092–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouadah Y, Rojas ER, Riordan DP, Capostagno S, Kuo CS, and Krasnow MA (2019). Rare pulmonary neuroendocrine cells are stem cells regulated by Rb, p53, and Notch. Cell 179, 403–416.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, and Strimmer K (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL, and Rudin CM (2015). DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 34, 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, Ogle C, Reinhart R, Selby M, Connelly J, et al. (2016). Small cell lung cancer screen of oncology drugs, investigational agents, and gene and microRNA expression. J. Natl. Cancer Inst 108, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Kolllpara RK, Rodarte K, Kelenis DP, Zhang X, Minna JD, and Johnson JE (2020). Lineage transcription factors ASCL1, NKX2–1, and PROX1 are enriched at super enhancers and co-regulate subtype-specific genes in small cell lung cancer . bioRxiv 10.1101/2020.08.13.249029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, Iorio F, Sousa FG, Elloumi F, Aladjem MI, et al. (2018). CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience 10, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, and Pommier Y (2012). CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res 72, 3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Varma S, Sousa F, Sunshine M, Abaan OD, Davis SR, Reinhold SW, Kohn KW, Morris J, Meltzer PS, et al. (2014). NCI-60 whole exome sequencing and pharmacological CellMiner analyses. PLoS ONE 9, e101670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Varma S, Sunshine M, Rajapakse V, Luna A, Kohn KW, Stevenson H, Wang Y, Heyn H, Nogales V, et al. (2017). The NCI-60 methylome and its integration into CellMiner. Cancer Res 77, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Varma S, Sunshine M, Elloumi F, Ofori-Atta K, Lee S, Trepel JB, Meltzer PS, Doroshow JH, and Pommier Y (2019). RNA sequencing of the NCI-60: integration into CellMiner and CellMiner CDB. Cancer Res 79, 3514–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, et al. (2012). Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res 18, 3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, Byers LA, Johnson ML, Burris HA 3rd, Robert F, et al. ; SCRX16–001 investigators (2017). Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 18, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D, et al. (2019). Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer 19, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, and Pommier Y (2016). Small cell lung cancer: time to revisit DNA-damaging chemotherapy. Sci. Transl. Med 8, 346fs12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, and Teschendorff AE (2017). ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Panciera T, and Piccolo S (2018). YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol 20, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L (2014). Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res 15, 3221–3245. [Google Scholar]
- Wang H, Huang S, Shou J, Su EW, Onyia JE, Liao B, and Li S (2006). Comparative analysis and integrative classification of NCI60 cell lines and primary tumors using gene expression profiling data. BMC Genomics 7, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, He Z, Wang X, Li H, and Liu X-S (2019). Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. eLife 8, e49020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JN (2012). Drug discovery: Cell lines battle cancer. Nature 483, 544–545. [DOI] [PubMed] [Google Scholar]
- Wistuba II, Bryant D, Behrens C, Milchgrub S, Virmani AK, Ashfaq R, Minna JD, and Gazdar AF (1999). Comparison of features of human lung cancer cell lines and their corresponding tumors. Clin. Cancer Res 5, 991–1000. [PubMed] [Google Scholar]
- Wooten DJ, Groves SM, Tyson DR, Liu Q, Lim JS, Albert R, Lopez CF, Sage J, and Quaranta V (2019). Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. PLoS Comput. Biol 15, e1007343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, and Camargo FD (2014). Hippo pathway activity influences liver cell fate. Cell 157, 1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, and He QY (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst 12, 477–479. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Girard L, Zhang YA, Haruki T, Papari-Zareei M, Stastny V, Ghayee HK, Pacak K, Oliver TG, Minna JD, and Gazdar AF (2018). Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl. Lung Cancer Res 7, 32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, Doroshow JH, and Pommier Y (2012). Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. U S A 109, 15030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scripts and data used for the analysis can be obtained at https://zenodo.org/record/3959142.
All newly generated methylation datasets are available from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) under the accession number GEO: GSE145156.