

Abstract
Chorionic somatomammotropin (CSH) is a placenta-specific hormone associated with fetal growth, and fetal and maternal metabolism in both humans and sheep. We hypothesized that CSH deficiency could impact sheep fetal liver glucose utilization. To generate CSH-deficient pregnancies, day 9 hatched blastocysts were infected with lentiviral particles expressing CSH-specific shRNA (RNAi) or scramble control shRNA (SC) and transferred to synchronized recipients. CSH RNAi generated two distinct phenotypes at 135 days of gestational age (dGA); pregnancies with IUGR (RNAi-IUGR) or with normal fetal weight (RNAi-NW). Fetal body, fetal liver and placental weights were reduced (P<0.05) only in RNAi-IUGR pregnancies compared to SC. Umbilical artery plasma insulin and insulin-like growth factor 1 (IGF1) concentrations were decreased, whereas insulin receptor beta (IRβ) concentration in fetal liver was increased (P<0.05) in both RNAi phenotypes. The mRNA concentrations of IGF1, IGF2, IGF binding protein 2 (IGFBP2) and IGFBP3 were decreased (P<0.05) in fetal livers from both RNAi phenotypes. Fetal liver glycogen concentration and glycogen synthase 1 (GYS1) concentration was increased (P<0.05), whereas fetal liver phosphorylated-GYS (inactive GYS) concentration was reduced (P<0.05) in both RNAi phenotypes. Lactate dehydrogenase B (LDHB) concentration was increased (P<0.05) and IGF2 concentration was decreased (P<0.05) in RNAi-IUGR fetal livers only. Our findings suggest that fetal liver glucose utilization is impacted by CSH RNAi, independent of IUGR, and is likely tied to enhanced fetal liver insulin sensitivity in both RNAi phenotypes. Determining the physiological ramifications of both phenotypes, may help to differentiate direct effect of CSH deficiency or its indirect effect through IUGR.
Keywords: Chorionic Somatomammotropin, glycogen, insulin receptor, glycogen synthase
Introduction
Chorionic somatomammotropin (CSH), also known as placental lactogen (PL), is a peptide hormone produced by the placenta and secreted into both fetal and maternal circulation. The source of CSH is the syncytiotrophoblast in humans, trophoblast giant cells in mice and chorionic binucleate cells in sheep (Gootwine 2004; Sibiak et al. 2020). CSH binding has been identified in several maternal and fetal sheep tissues, including skeletal muscle, pancreas and liver (Chan et al. 1978). CSH has high affinity for prolactin (PRL) receptors as well as growth hormone (GH) receptors (Handwerger & Freemark 2000), although a distinct CSH receptor may exist in fetal liver (Freemark et al. 1987; Hill et al. 1988; Pratt et al. 1995). While CSH is produced by the placenta, it is found in high concentrations in both maternal and fetal blood (Handwerger & Freemark 2000), indicating that it might have widespread roles in different physiological processes in the mother and fetus.
Ovine CSH has long been thought to play an important role in fetal growth (Hurley et al. 1977; Min et al. 1996), and reduced CSH in maternal circulation has been associated with intrauterine growth restriction (IUGR) in both humans and sheep pregnancies (Spellacy et al. 1976; Lea et al. 2007). There are conflicting reports about the role of human CSH in fetal growth. Some studies have demonstrated that CSH deficiency or deletion of CSH locus in humans results in severe intrauterine growth restriction (Trapp et al. 1987; Rygaard et al. 1998) however other studies show that CSH deficiency or gene deletion does not affect fetal growth and results in otherwise uneventful pregnancies (Wurzel et al. 1982; Giampietro et al. 1984; Barbieri et al. 1986; Simon et al. 1986). Other than its effect on fetal growth, none of these studies investigated the possible physiological ramification of CSH deficiency. We recently reported (Baker et al. 2016) that CSH deficiency generates two distinct phenotypes; pregnancies affected by IUGR (CSH RNAi-IUGR) and pregnancies with normal fetal weight (CSH RNAi-NW). However, Baker et al. (2016) reported little on the characteristics of the CSH RNAi-NW (“non responders”) pregnancies. While correlations between CSH concentrations and fetal growth has been examined, the effect of CSH on glucose utilization in the fetal liver is not clear.
Glucose is the principal source of energy for the fetus, and optimal glucose utilization is crucial for normal fetal growth (Hay 2006). Due to high insulin sensitivity, glucose utilization in the fetal liver is primarily regulated by insulin (Hay 2006). In humans, the fetal liver plays a vital role in the maintenance of glucose metabolism and has all the necessary enzymes for gluconeogenesis and glycogen synthesis by as early as 8 weeks of gestation (Grijalva & Vakili 2013). IUGR pregnancies often exhibit an increase in fetal blood glucagon and fetal liver gluconeogenic enzymes, suggesting an early initiation of gluconeogenesis (Thorn et al. 2009; Culpepper et al. 2016; Wesolowski & Hay 2016). One such gluconeogenic enzyme is lactate dehydrogenase (LDHB) which coverts lactate to pyruvate, the first substrate for gluconeogenesis (Garrett & Grisham 2001). Similarly, phosphoenolpyruvate carboxykinase 2 (PCK2) is also a key gluconeogenic enzyme which converts oxaloacetate to phosphoenolpyruvate (Yang et al. 2009).
CSH is believed to regulate glycogen synthesis in fetal tissues, as it stimulates glycogen synthesis and storage in fetal rat hepatocytes (Freemark & Handwerger 1984a, 1985), and infusion of CSH increased fetal sheep liver glycogen content (Schoknecht et al. 1996). The glycogenic action of CSH in fetal liver is thought to be regulated through insulin signaling (Freemark & Handwerger 1984b). CSH is also believed to support fetal growth by stimulating insulin-like growth factor (IGF) production and by mobilizing maternal glucose and amino acids for transfer from the mother to the fetus (Handwerger & Freemark 2000). The fetal liver abundantly expresses insulin like growth factor 1 and 2 (IGF1 and IGF2) and insulin receptor beta (IRβ) (Liang et al. 2010). IGF2 regulates several biological functions including glycogen synthesis through its interaction with the type 1 IGF receptor and insulin receptor, and IGF2 and insulin receptor are main regulators of glycogen synthesis in fetal liver (Liang et al. 2010). Prenatal glycogen synthesis and storage is crucial to meet the requirements of glucose during the immediate perinatal period. Glycogen synthase (GYS), as the key enzyme involved in glycogen synthesis, is regulated by GYS kinase-3 (GSK3), which phosphorylates and inactivates GYS to p-GYS. Through activation of the insulin receptor signaling pathway, protein kinase B (PKB) phosphorylates GSK3, thereby inactivating GSK3 and hence reducing the proportion of inactivated p-GYS (Fang et al. 2000).
Previously, our laboratory reported a novel model of CSH-deficient pregnancies, using lentiviral-mediated trophectoderm-specific RNA interference (RNAi) (Baker et al. 2016). Using these pregnancies, we investigated the effect of CSH RNAi on fetal liver glucose utilization, to test the hypothesize that CSH regulates fetal liver glucose utilization by regulating insulin signaling.
Materials and methods
All procedures involving use of animals or lentivirus were approved by the Colorado State University Institutional Animal Care and Use Committee (Protocol 14-5257A) and the Institutional Biosafety Committee (Protocols 11–034B and 13–043B).
Generation of CSH-deficient pregnancies
We have previously reported the detailed procedures for the generation of these CSH-deficient pregnancies (Baker et al. 2016). Briefly, lentiviral particles were generated using a packaging plasmid (psPAX2), envelope plasmid (pMD2.G) and the shRNA vector (with SC or CSH specific tg6 shRNA), and the resulting lentiviral titer was calculated by infecting human embryonic kidney cells (HEK293) and counting green fluorescent protein (GFP)-positive cells or by TCID50 assay. Day 9 hatched, and fully expanded blastocysts were collected from naturally mated/non-superovulated donor ewes, and infected with 100,000 transducing units in a 100μL drop of CDM-2 medium for 4–6 hours. After infecting with lentiviral particles, blastocysts were surgically transferred to estrus synchronized ewes. One blastocyst was transferred in each recipient ewe. Terminal surgeries were conducted at 135 dGA and blood samples were collected from umbilical artery, umbilical vein, uterine artery and uterine vein from 8 SC and 16 CSH RNAi pregnancies. Fetal liver tissue and fetal cotyledons were collected and snap frozen in liquid nitrogen. The tissue was later pulverized using a mortar and pestle and stored in at −80°C until further use.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from 135 dGA pulverized fetal cotyledons and fetal liver tissue using RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. The detailed procedure and qRT-PCR primers have been previously described (Baker et al. 2016).
Protein extraction and western blotting
For protein extraction, pulverized fetal liver tissue was homogenized in RIPA buffer (20 mM Tris, 137 mM NaCl, 10% glycerol, 1% nonidet P-40, 3.5 mM SDS, 1.2 mM sodium deoxycholate, 1.6 mM EDTA, pH 8) containing 1x protease/phosphate inhibitor cocktail, and incubated for 5 minutes on ice. Homogenized samples were sonicated using a Bioruptor Sonication System (VWR Scientific) for 5 cycles of 30 seconds “ON” and 30 seconds “OFF”. The sonicated tissue lysate was centrifuged at 14,000 g for 5 minutes to remove tissue debris. Protein concentration in tissue lysates was measured using BCA protein assay kit (ThermoFisher, Waltham, USA), and 55–100μg of protein was electrophoresed through Mini-Protein 4–15% Tris-Glycine stain-free gels (Bio-Rad Laboratories, Hercules, USA) at 90 volts for 15 minutes and 200 volts for 30 minutes. The stain-free gels were activated for 1 minute using the ChemiDoc XRS+ chemiluminescence system (Bio-Rad Laboratories, Hercules, USA) and then transferred to 0.2 μm pore size nitrocellulose membrane at 100 volts for 2 hours. The membranes were imaged using the stain-free settings on the ChemiDoc XRS+ chemiluminescence system and then blocked in 5% non-fat dry milk solution in TBST (50 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.6) for 1 hour at room temperature. After blocking, the membranes were washed 3 times with 1x TBST for 5 min each, and then incubated at 4°C overnight with specific primary antibody. After overnight incubation, the membranes were washed 3 times with 1x TBST for 5 min each. After washing, the membranes were incubated with appropriate secondary antibody conjugated to horseradish peroxidase for 1 hour at room temperature. After removing the secondary antibody, the membranes were washed following the same procedure and developed using Super Signal WestDura Extended Duration Substrate and imaged using ChemiDoc XRS+ chemiluminescence system. The images were quantified using Image-Lab software. Protein quantification data were normalized to total protein quantity in each lane transferred to the nitrocellulose membrane. The antibodies used and their dilutions are listed in supplementary table 1.
Glycogen Assay
Glycogen quantity in fetal liver samples was measured using glycogen colorimetric/ fluorometric assay kit (BioVision) following the manufacturer’s protocol. Briefly, 10 mg of pulverized fetal liver tissue was homogenized in 200μL dH2O, boiled for 10 minutes to inactivate enzymes, and centrifuged to remove insoluble materials within the homogenate. Using a 96 well flat-bottom plate, 5 μL of tissue extract was added in two replicate wells and final volume was brought up to 50 μL using hydrolysis buffer. One well was used to measure glycogen while the second well was used to measure glucose background of each sample. A standard curve for glycogen was made by loading different glycogen quantities (0, 0.4, 0.8, 1.2, 1.6, and 2.0 μg per well) and final volume was brought to 50 μL using hydrolysis buffer. Two μL of Hydrolysis Enzyme Mix was added in all wells except the wells to be used to measure glucose background and incubated at room temperature for 30 minutes. Fifty μL of development reaction mix for each well was prepared by mixing 46 μL development buffer, 2 μL development enzyme and 2 μL OxiRed reagent. After incubation, 50 μl of development reaction mix was added in each well and incubated for 30 minutes at room temperature. Absorbance (OD 570nm) was measured using a BioTek Syngergy 2 Microplate Reader (BioTek, Winooski, USA) and glycogen quantity in each sample was determined from the standard curve.
Radioimmunoassay
The serum concentrations of insulin and IGF-1 were measured using radioimmunoassay as previously reported (Baker et al. 2016).
Serum Glucose Measurement
Maternal and fetal serum glucose concentrations were measured using the YSI 2700 SELECT Biochemistry Analyzer (YSI, Yellow Springs, OH) following the manufacturer’s protocol, as previously reported (Baker et al., 2016).
Statistical Analysis
As described in Baker et al. (2016), the determination of “responder” (RNAi-IUGR) pregnancies versus “nonresponder” (RNAi-NW) was done by determining which pregnancies had placental weights that fell two standard deviations below the mean of the SC pregnancies. Subsequently, all data were subjected to analysis of variance with treatment, fetal sex and sire as the dependent variables, and included all interactions. There were no treatment by fetal sex or treatment by sire interactions, therefore statistical analysis of the data presented herein, from both RNAi phenotypes, were compared with SC using one-way analysis of variance followed by Tukey’s HSD post-hoc test. Statistical significance was set at P ≤ 0.05 while statistical tendency was set at P≤ 0.10. The data figures are presented as scatter plots, with the horizontal line representing the mean, and the capped vertical lines representing the standard error of the mean (SEM).
Results
Trophectoderm-specific CSH RNAi generates two distinct phenotypes
As previously reported (Baker et al., 2016), trophectoderm-specific CSH RNAi generated two distinct phenotypes, as assessed by determining which CSH RNAi (tg6) pregnancies resulted in placental weights that fell two standard deviations below the mean of the SC pregnancies. As such, some fetuses were considered to be intrauterine growth restricted (RNAi-IUGR; n=8) and some had normal weight (RNAi-NW; n=8) compared to SC (n=8) (Figure 1A). A 52% reduction (P<0.05) in placental weight and a 40% reduction (P<0.05) in fetal liver weight was observed in RNAi-IUGR pregnancies compared to SC, whereas there was no significant change in placental weight and fetal liver weight in RNAi-NW pregnancies (Figure 1B–C). CSH mRNA was reduced by 50% (P<0.05) in RNAi-IUGR and by 47% (P<0.05) in RNAi-NW fetal cotyledons compared to SC (Figure 1D). Differences in cotyledonary CSH concentration for these pregnancies was previously reported (Baker et al. 2016).
Figure 1.
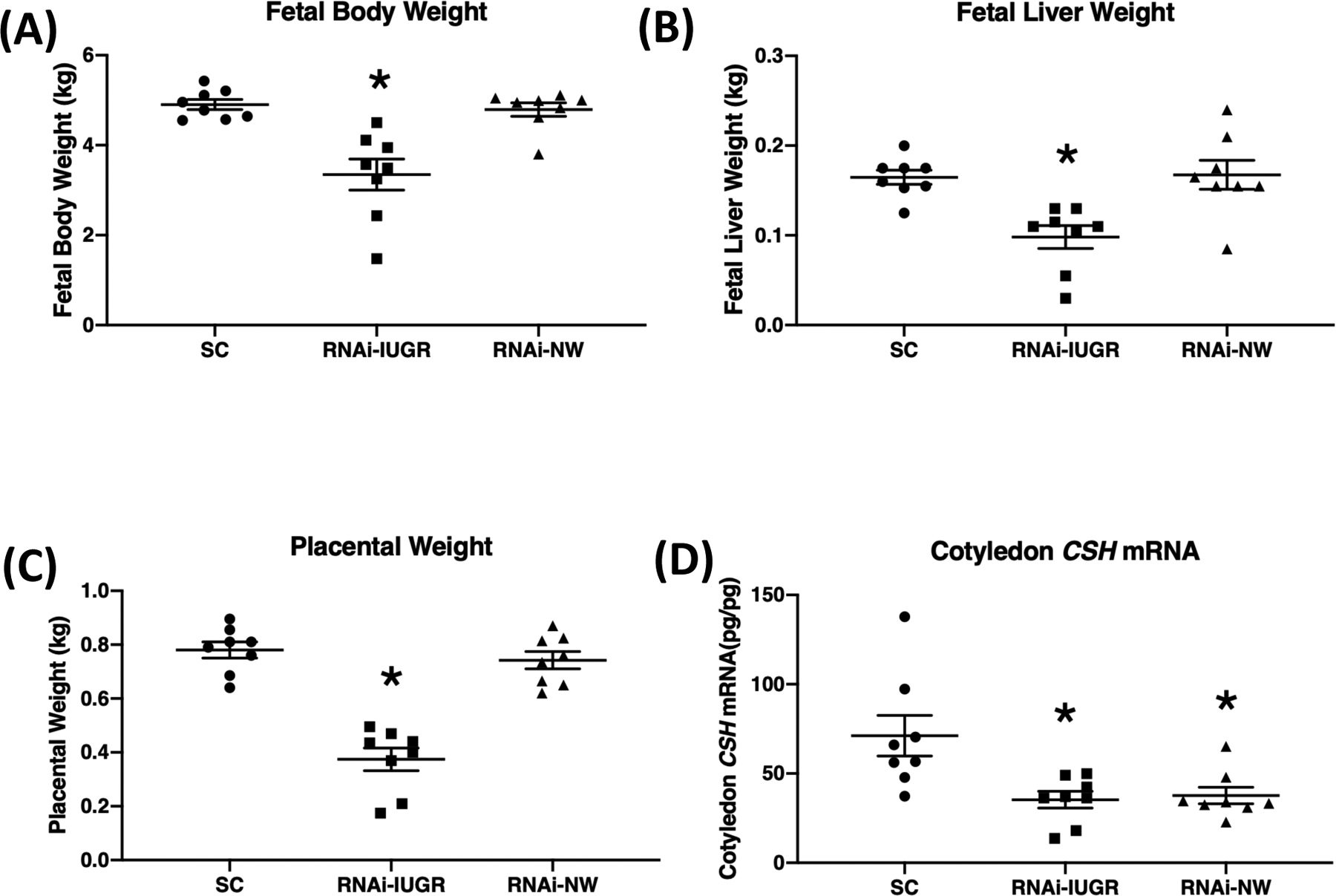
(A-C) Fetal body weight, liver weight and placental weight at 135 dGA in SC, RNAi-IUGR and RNAi-NW pregnancies. (n=8/treatment). SC and RNAi-IUGR data in this figure were previously presented (Baker et al. 2016) and is incorporated here to build a comparison. (D) Cotyledon CSH mRNA concentration in fetal cotyledons from SC, RNAi-IUGR and RNAi-NW pregnancies. (n=8/treatment), *p<0.05.
CSH deficiency affects insulin signaling in fetal liver
There was a 48% reduction (P=0.07) in umbilical artery insulin in RNAi-IUGR pregnancies and a 36% reduction (P=0.15) in RNAi-NW pregnancies compared to SC (table 1), but umbilical artery glucose concentrations were not impacted. Insulin and glucose concentrations in the uterine artery were not impacted in either RNAi-IUGR and RNAi-NW pregnancies (table 1), however, there was a significant increase in the uterine artery to uterine vein glucose gradient (P<0.05) in both RNAi-IUGR and RNAi-NW phenotypes (table 1). No significant difference was observed in IGF1 concentration in uterine arterial blood in either RNAi-IUGR and RNAi-NW pregnancies, however, umbilical artery IGF-1 concentration was reduced (P<0.05) in both RNAi phenotypes compared to SC (Figure 2). The mRNA concentrations of IGF1, IGF2, IGFBP2 and IGFBP3 were reduced (P<0.05) in fetal livers from both RNAi phenotypes compared to SC (table 2). Moreover, mRNA concentration of IGFBP1 did not change in RNAi-IUGR fetal livers but tended (P=0.08) to be reduced in RNAi-NW fetal livers compared to SC (table 2). Analysis of IRβ concentrations revealed a 192% (P<0.05) increase in RNAi-IUGR fetal livers and 106% (P<0.05) in RNAi-NW fetal livers, compared to SC (Figure 3).
Table 1.
Fetal and maternal serum measurements
| Serum Measurements | SC (Mean ± SEM) | RNAi-IUGR (Mean ± SEM) | P value (SC vs RNAi-IUGR) | RNAi-NW (Mean ± SEM) | P value (SC vs RNAi-NW) |
|---|---|---|---|---|---|
| UtA insulin (ng/ml) | 0.40 ± 0.08 | 0.94 ± 0.31 | 0.14 | 0.40 ± 0.10 | 0.99 |
| UtA gluocse (mmol/L) | 3.39 ± 0.56 | 3.70 ± 0.54 | 0.69 | 3.05 ± 0.16 | 0.54 |
| UtA insulin:glucose | 0.12 ± 0.02 | 0.24 ± 0.06 | 0.08 | 0.13 ± 0.03 | 0.78 |
| UmbA insulin (ng/ml) | 0.56 ± 0.13 | 0.29 ± 0.06 | 0.07 | 0.36 ± 0.04 | 0.15 |
| UmbA glucose (mmol/L) | 0.80 ± 0.31 | 0.53 ± 0.21 | 0.48 | 0.61 ± 0.11 | 0.57 |
| UmbA insulin:glucose | 1.21 ± 0.27 | 1.77 ± 0.78 | 0.51 | 0.62 ± 0.11 | 0.08 |
| UtV glucose (mmol/L) | 3.06 ± 0.50 | 3.34 ± 0.56 | 0.71 | 2.64 ± 0.17 | 0.43 |
| UtA-UtV glucose(mmol/L) | 0.23 ± 0.04 | 0.36 ± 0.03 | 0.02 | 0.42 ± 0.06 | 0.02 |
| UmbV Glucose (mmol/L) | 0.97 ± 0.33 | 0.77 ± 0.22 | 0.62 | 0.76 ± 0.10 | 0.56 |
| UmbV-UmbA glucose (mmol/L) | 0.17 ± 0.04 | 0.24 ± 0.07 | 0.42 | 0.15 ± 0.03 | 0.66 |
Serum concentrations are mean ± SEM. UtA, uterine artery; UtV, uterine vein; UmbA, umbilical artery; UmbV, umbilical vein. SC and RNAi-IUGR data in this table were previously presented (Baker et al. 2016) and is incorporated here to build a comparison. (n=8/treatment). P ≤ 0.10 indicates a trending difference from controls. P ≤ 0.05 indicates a significant difference from controls.
Figure 2.
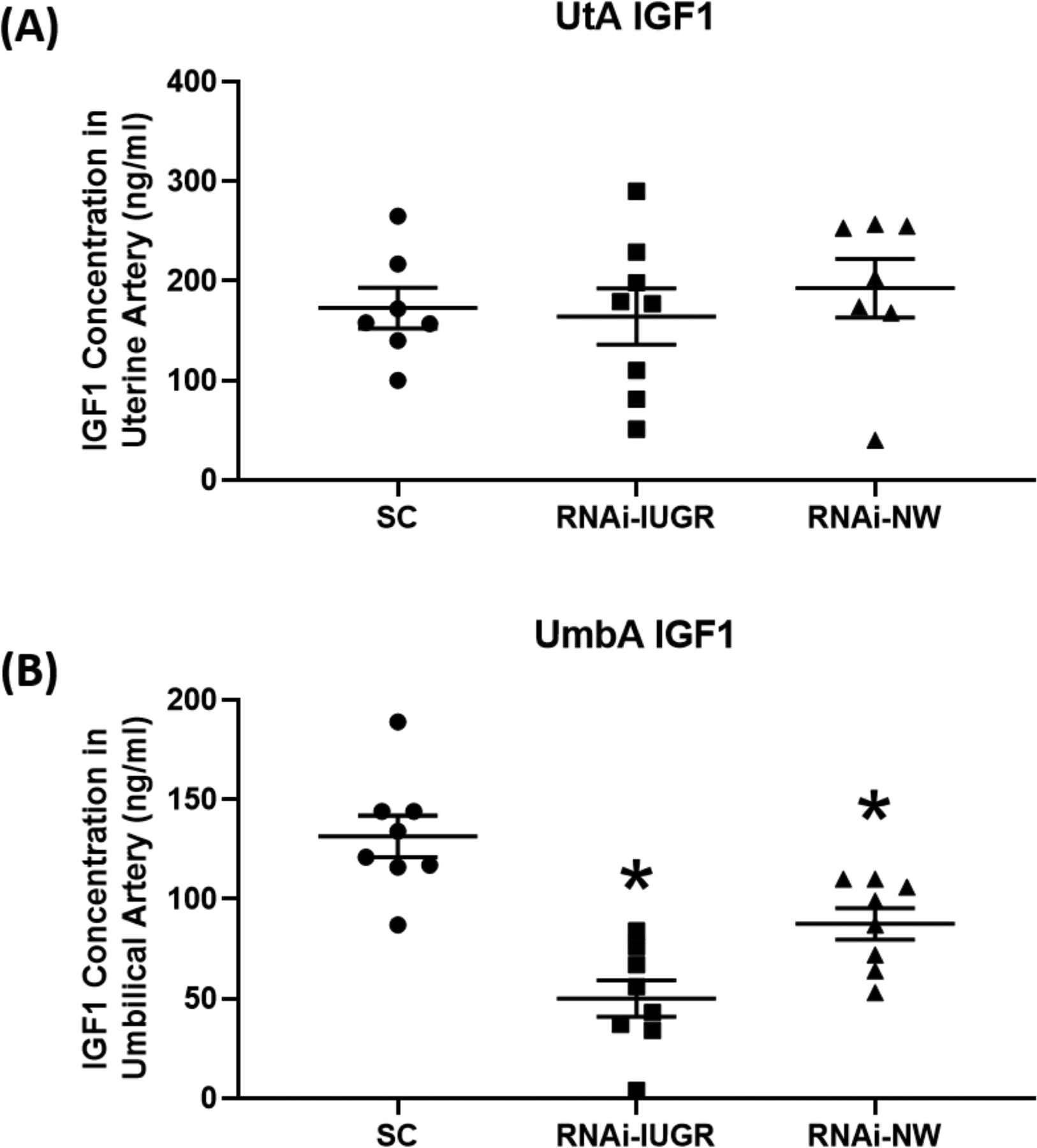
IGF-1 concentration in uterine artery (UtA; n=7 for SC and RNAi-IUGR, and n=8 for RNAi-NW) and umbilical artery (UmbA; n=8/treatment)) blood. SC and RNAi-IUGR data in this figure were previously presented (Baker et al. 2016) and is incorporated here to build a comparison. (n=8/treatment), *p<0.05.
Table 2.
Fetal liver insulin-like growth factor mRNA concentrations
| Gene | SC (Mean ± SEM) | RNAi-IUGR (Mean ± SEM) | P value (SC vs RNAi-IUGR) | RNAi-NW (Mean ± SEM) | P value (SC vs RNAi-NW) |
|---|---|---|---|---|---|
| IGF1 (pg/pg) | 0.04 ± 0.01 | 0.01 ± 0.002 | 0.0001 | 0.02 ± 0.003 | 0.0019 |
| IGF2 (pg/pg) | 0.49 ± 0.08 | 0.14 ± 0.04 | 0.001 | 0.22 ± 0.04 | 0.01 |
| IGFBP1 (pg/pg) | 0.72 ± 0.16 | 0.64 ± 0.31 | 0.813 | 0.37 ± 0.10 | 0.08 |
| IGFBP2 (pg/pg) | 0.18 ± 0.04 | 0.05 ± 0.03 | 0.022 | 0.03 ± 0.01 | 0.006 |
| IGFBP3 (pg/pg) | 0.09 ± 0.01 | 0.02 ± 0.006 | 0.0002 | 0.02 ± 0.003 | 0.0002 |
mRNA concentrations are mean ± SEM for the starting quantity of mRNA of interest (pg) divided by geomean of starting quantity (pg) for the housekeeping mRNA. SC and RNAi-IUGR data in this table were previously presented (Baker et al. 2016) and is incorporated here to build a comparison. (n=8/treatment), P ≤ 0.10 indicates a trending difference from controls. P ≤ 0.05 indicates a significant difference from controls.
Figure 3.
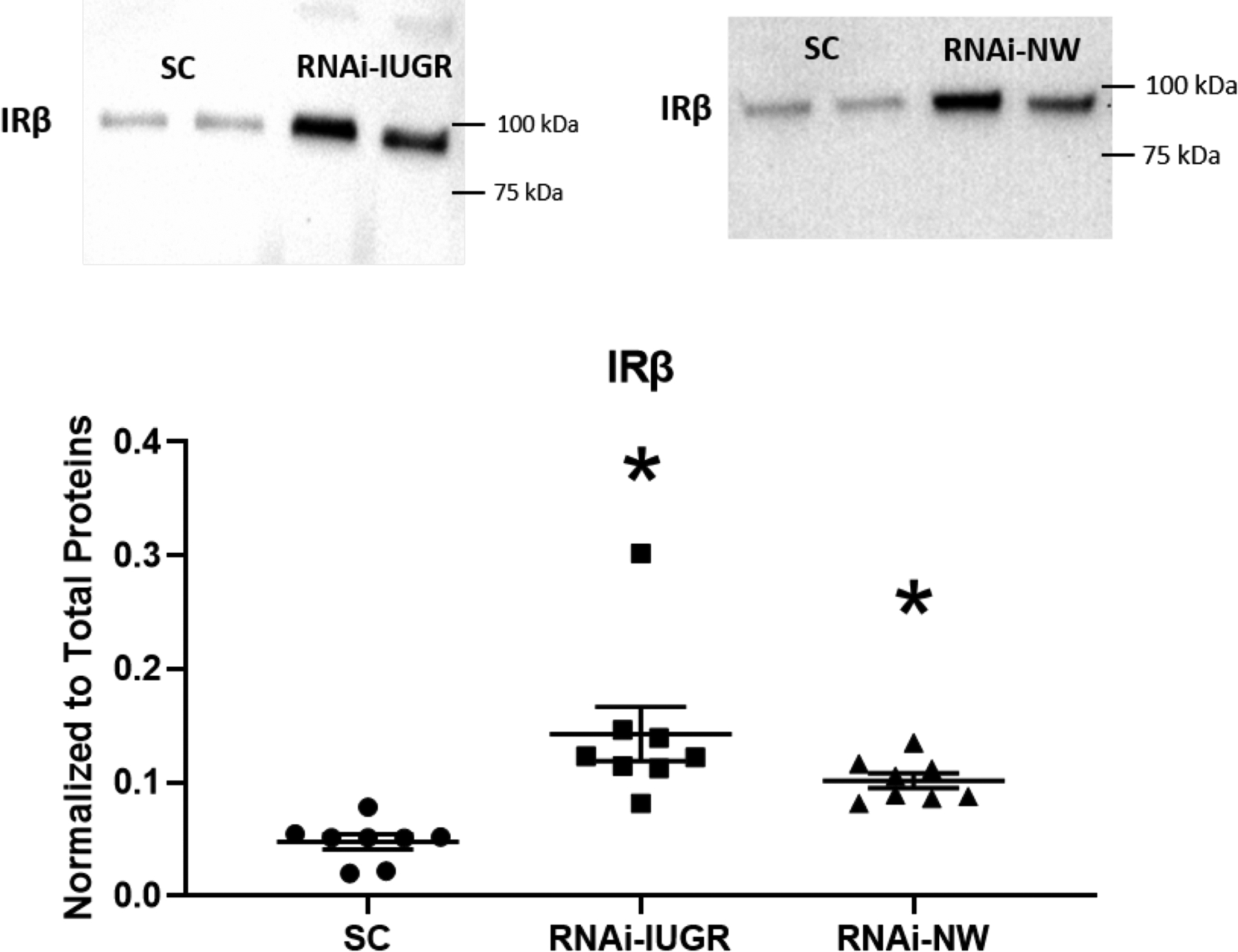
Representative immunoblots and densitometric analysis for IRβ protein in fetal liver from SC, RNAi-IUGR and RNAi-NW pregnancies. (n=8/treatment), *p<0.05.
CSH deficiency leads to increased glycogen storage in fetal liver
Glycogen concentration in fetal livers increased by 80% (P<0.05) in RNAi-IUGR pregnancies and by 89% (P<0.05) in RNAi-NW pregnancies compared to SC (Figure 4). Glycogen synthase 1 (GYS1), glycogen synthase 2 (GYS2) and phosphorylated glycogen synthase (p-GYS) were also assessed. GYS1 concentration in fetal liver increased 92% (P<0.05) in RNAi-IUGR pregnancies and 47% (P<0.05) in RNAi-NW pregnancies compared to SC (Figure 5A). There was no statistical difference in GYS2 concentration in fetal livers in either RNAi-IUGR and RNAi-NW pregnancies as compared to SC (Figure 5B). The inactive form of glycogen synthase, p-GYS, was reduced in fetal livers by 34% (P<0.05) in RNAi-IUGR pregnancies and 30% (P<0.05) in RNAi-NW pregnancies compared to SC (Figure 5C).
Figure 4.
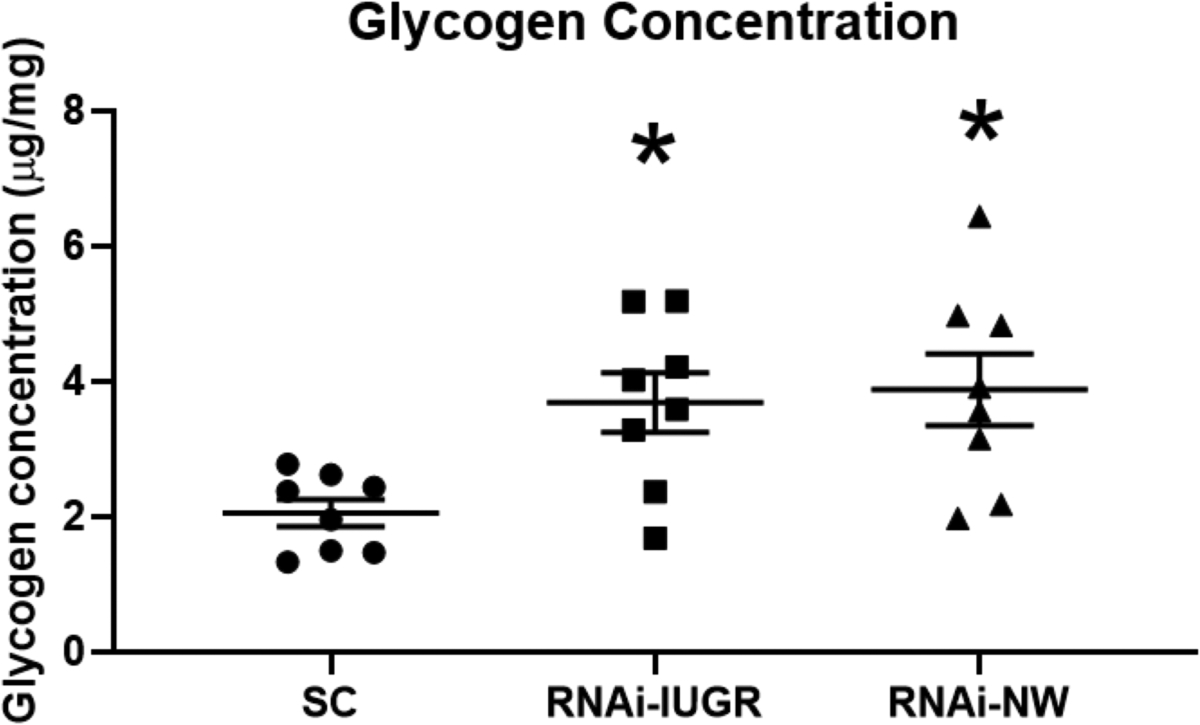
Glycogen concentration in fetal liver from SC, RNAi-IUGR and RNAi-NW pregnancies. (n=8/treatment), *p<0.05.
Figure 5.
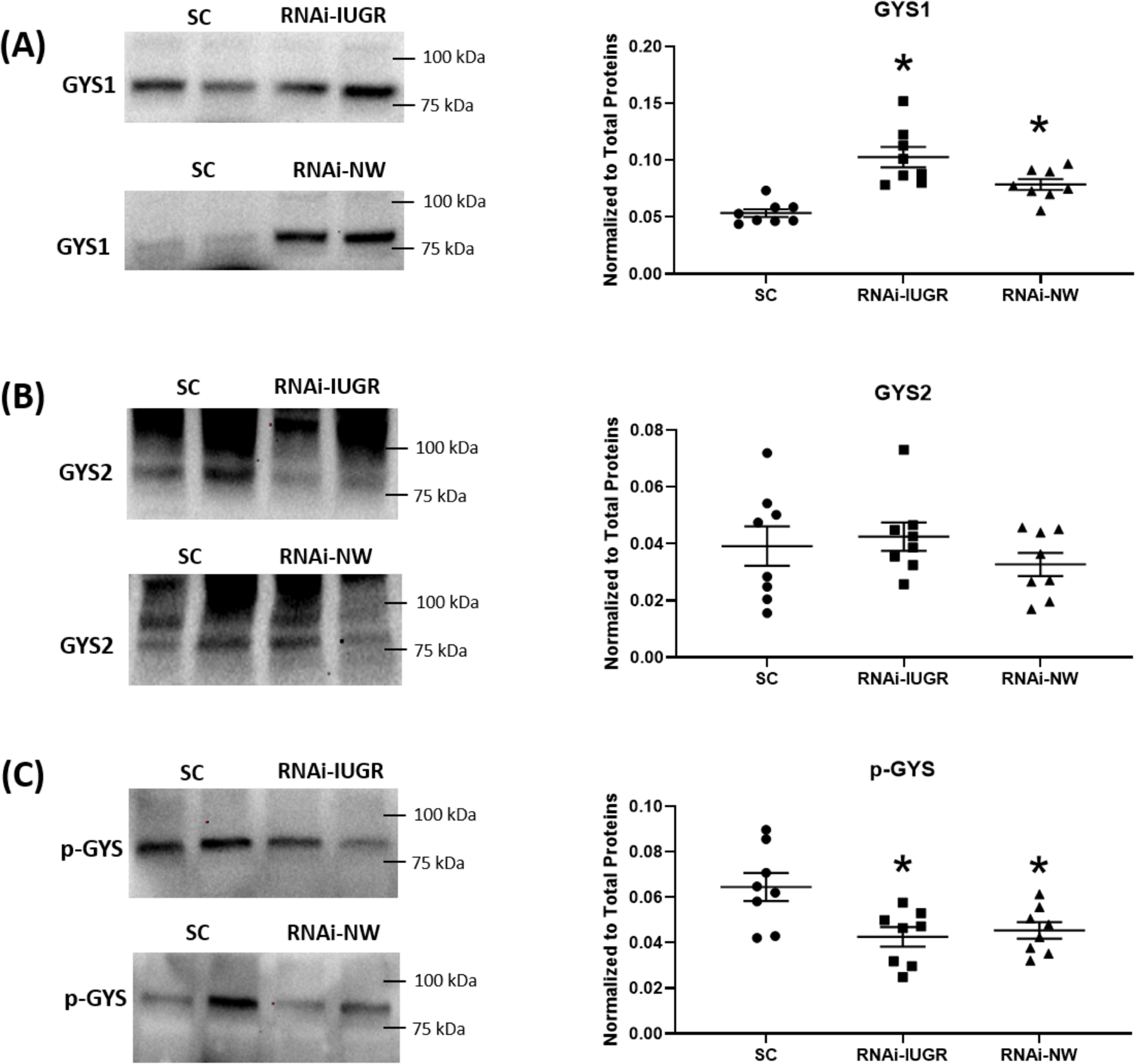
Representative immunoblots and densitometric analysis for GYS1, GYS2 and p-GYS in fetal liver from SC, RNAi-IUGR and RNAi-NW pregnancies. (n=8/treatment), *p<0.05.
CSH deficiency affects the expression of genes with known role in glucose metabolism
Analysis of LDHB concentrations in fetal liver revealed a 130% (P<0.05) increase in RNAi-IUGR pregnancies, whereas no significant change was observed in fetal liver from RNAi-NW pregnancies, compared to SC (Figure 6A). There was no significant difference in fetal liver PCK2 concentrations in fetal livers from either RNAi-IUGR and RNAi-NW pregnancies, compared to SC (Figure 6B). However, IGF2 concentration was reduced by 23% (P<0.05) in RNAi-IUGR fetal liver, whereas no change was observed in RNAi-NW fetal liver, as compared to SC (Figure 6C).
Figure 6.
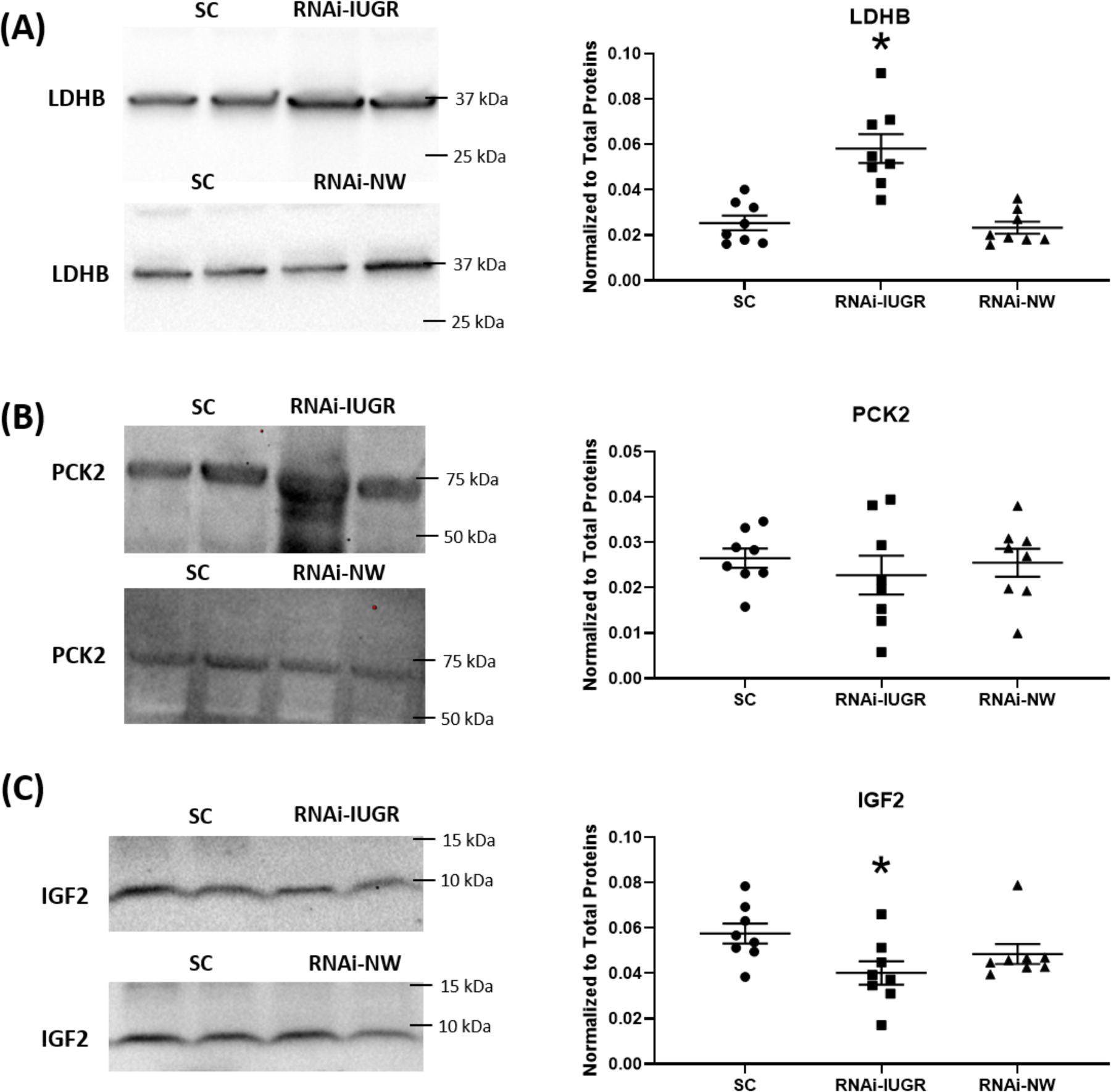
Representative immunoblots and densitometric analysis for LDHB, PCK2 and IGF2 in fetal liver from SC, RNAi-IUGR and RNAi-NW pregnancies. (n=8/treatment), *p<0.05.
Discussion
In sheep, chorionic somatomammotropin (CSH) is produced by chorionic binucleate cells and is secreted into both fetal and maternal circulation (Gootwine 2004; Sibiak et al. 2020). CSH has been previously studied for its potential to modulate fetal growth (Spellacy et al. 1976; Freemark 2010; Baker et al. 2016; Jeckel et al. 2018). Recently, we generated CSH-deficient pregnancies by trophectoderm-specific RNA interference (RNAi) of CSH (Baker et al. 2016; Jeckel et al. 2018). In this unique approach, hatched day 9 sheep blastocysts were incubated with shRNA-expressing lentiviral particles which infect specifically the trophectoderm and leads to placenta specific modifications without affecting the inner cell mass (Baker et al. 2016). We reported that CSH deficiency in sheep resulted in two distinct phenotypes at 135 dGA; pregnancies with IUGR (RNAi-IUGR) or pregnancies with normal placental and fetal weights (RNAi-NW). CSH mRNA in fetal cotyledons was significantly reduced in both RNAi-IUGR and RNAi-NW phenotypes (Figure 1D), whereas cotyledonary CSH was only significantly reduced in RNAi-IUGR pregnancies (Baker et al. 2016). Despite comparable reductions in CSH mRNA, variation existed on the impact of fetal and placental growth between the CSH RNAi phenotypes. Jeckel et al., (2018) reported that trophectoderm-specific RNAi in sheep causes fetal growth restriction at 50 dGA, in the absence of significant reductions in cotyledonary CSH mRNA and protein. Both in our study and Jeckel et al., (2018) study, the placental tissue samples were collected after terminating the pregnancies and CSH concentrations were determined at only one timepoint in gestation. However, the efficiency of RNAi is affected by several factors including stabilization of target mRNA by binding proteins, target mRNA turnover rate, abundance of target transcript, efficiency of lentivirus transduction, epigenetic modifications and shRNA-driving promoter efficiency (Ramezani et al. 2000; Holen et al. 2002; Hong et al. 2014). These factors can affect the degree of CSH RNAi at different stages of gestation starting from day of infection (day 9) till the termination of pregnancy. However, it is not possible to access the variability in RNAi in placenta across gestation, without terminating the pregnancies. These factors can vary from animal to animal, and there is possibility that, in some animals, RNAi efficiency is low at a critical time of fetal and placental development and vice versa, which will affect the phenotype of that pregnancy. Several studies in humans have also reported contrasting results regarding CSH’s role in fetal growth. Some reports have shown that CSH deficiency or deletion of the CSH locus in humans causes severe fetal growth restriction (Trapp et al. 1987; Rygaard et al. 1998), however in some other cases it has no impact on fetal growth (Wurzel et al. 1982; Giampietro et al. 1984; Barbieri et al. 1986; Simon et al. 1986). Absence of growth restriction in CSH-deficient pregnancies suggests that there might be some compensatory mechanisms supporting fetal growth in these pregnancies (Freemark 2010), which are yet to be explored.
It has long been thought that, other than fetal growth, CSH also regulates glucose metabolism. According to Freemark and Handwerger (1984), ovine CSH has insulin-like effects and stimulates incorporation of glucose into glycogen in fetal rat hepatocytes in a dose dependent manner (Freemark & Handwerger 1984a). They also reported that, other than stimulating glycogen synthesis, ovine CSH inhibits glucagon-induced glycogen degradation in fetal rat hepatocytes (Freemark & Handwerger 1985). CSH receptors have also been identified in many maternal and fetal tissues including liver suggesting a role in regulating liver function (Freemark et al. 1986, 1987, 1992; Hill et al. 1988; Pratt et al. 1995). The expression of CSH receptors in maternal and fetal sheep liver is positively correlated with glucose and insulin concentrations in maternal and fetal blood (Freemark et al. 1990, 1992). However, the effect of CSH deficiency on glucose utilization by sheep fetal liver has never been examined. As little was reported on the CSH RNAi-NW (non-responders) in Baker et al. (2016), here we further report on both phenotypes (RNAi-IUGR and RNAi-NW) and extend the investigation into the fetal liver glucose utilization.
According to Schoknecht et al., (1996), intravenous infusion of 1.2 mg/day of ovine CSH into sheep fetal circulation starting at 122 dGA increases hepatic glycogen deposition without affecting fetal weight (Schoknecht et al. 1996). Therefore, it would be expected that reduced placental CSH would result in a decrease in liver glycogen stores. Glycogen, however, was found to be significantly increased in fetal liver from both RNAi-IUGR and RNAi-NW pregnancies, which is contradictory to former findings (Freemark & Handwerger 1984a; Schoknecht et al. 1996). One potential reason for this contradiction is that previous studies established a correlation between CSH and glycogen synthesis by CSH infusion into fetus (Schoknecht et al. 1996) or stimulating rat hepatocytes with ovine CSH (Freemark & Handwerger 1984a) whereas we investigated this correlation using a CSH deficiency model. We suggest that, compared to CSH infusion or stimulation, CSH deficiency could modulate more pathways involved in glucose utilization. Another possible explanation is that the tissues in our study were collected after terminal surgeries performed under general anesthesia and after ~16 h of fasting. Although fasting might not impact the plasma concentrations of CSH (Butler et al. 1987; Bauer et al. 1995), the stress of the surgical procedure and general anesthesia can increase CSH concentrations in fetal and maternal plasma (Taylor et al. 1980). Although increased CSH concentration due to surgery or anesthesia can affect the glucose utilization by the fetal liver, it is possible that an increase in CSH concentration during the surgical procedure (~1 h), could drive enhanced glycogen concentration in fetal liver.
Several studies have demonstrated a correlation between CSH and insulin. Bennet et al., showed that injection of human CSH in hypophysectomized rats increases pancreatic insulin secretion (Bennett et al. 1976). Handwerger et al., reported that injecting CSH in pregnant or non-pregnant ewes resulted in a decrease in insulin after 1hr of CSH injection which then increases above the baseline (Handwerger et al. 1976). Treatment of human fetal pancreatic explants with human CSH in the presence of glucose causes a significant increase in insulin synthesis and release (Swenne et al. 1987). In our study, while there were reductions in umbilical artery insulin concentrations in both RNAi-IUGR and RNAi-NW pregnancies, neither reached statistical significance (P<0.05), and there were no changes in umbilical glucose concentrations either. However, insulin receptor beta (IRβ) concentration was significantly increased in fetal livers from both RNAi-IUGR and RNAi-NW pregnancies. Increased IRβ concentration in the fetal liver might be a compensatory response to reduced insulin in fetal blood, thus increasing fetal liver insulin sensitivity (Beale 2013; Boucher et al. 2014). Increased insulin sensitivity could explain the increased glycogen concentration in fetal livers from both RNAi-IUGR and RNAi-NW pregnancies. Hypoinsulinemia and increased insulin sensitivity is a hallmark of human IUGR pregnancies (Gagnon 2003), however, the existence of enhanced insulin sensitivity in the absence of IUGR has not been reported.
CSH is thought to support fetal growth by stimulating the production of fetal insulin-like growth factors (IGF) and insulin (Handwerger 1991; Schoknecht et al. 1996). Thorn et al., reported that placental-insufficiency-based IUGR in sheep resulted in a 60% reduction in fetal liver weight compared to control fetuses at 134 dGA (Thorn et al. 2009). They further reported that IUGR fetuses had significant reductions in plasma insulin and IGF1 concentrations and 80% increase in insulin receptor in fetal skeletal muscle, however insulin receptor concentration in the fetal liver was not reported (Thorn et al. 2009). In our study, we saw a significant reduction in IGF1 in umbilical artery blood from both RNAi-IUGR and RNAi-NW groups. Moreover, fetal livers from both RNAi-IUGR and RNAi-NW pregnancies had a significant reduction in mRNA encoding IGFI, IGF2, IGF-binding protein 2 (IGFBP2) and IGFBP3. IGF1 and IGF2 stimulate glycogen synthesis by interacting with insulin receptors (Liang et al. 2010; Muhič et al. 2015). IGFBPs bind IGFs and reduced their bioavailability, and nearly 75% of IGFs are bound to IGFBP3 (Ding & Wu 2018). Based on these findings we suggest that, other than increased insulin sensitivity, increased bioavailability of IGFs due to reduced IGFBPs could also contribute in glycogen synthesis resulting in increased glycogen in fetal liver from CSH-deficient pregnancies in our study. We also measured tissue IGF2 concentrations in the fetal liver, which was significantly reduced in RNAi-IUGR pregnancies, with no change in fetal livers from RNAi-NW pregnancies. IGF2 is a key determinant of fetal growth and IGF2 infusion in mouse increases fetal growth and body weight (White et al. 2018). Therefore, reduced IGF2 quantity only in RNAi-IUGR fetal liver might be driving the growth restriction in this group.
By its interaction with IRβ, insulin initiates a signaling cascade that results in enhanced glycogen synthesis (Bouskila et al. 2010). One major step in this cascade is the increase in the ratio of active to inactive GYS in response to interaction of insulin with IRβ (Bouskila et al. 2010). In humans, glycogen synthase has two paralogous isozymes; GYS1 known as the muscle isozyme and GYS2 known as the liver isozyme (Browner et al. 1989; Westphal & Nuttall 1992). However, we found that sheep fetal liver has high expression of both GYS1 and GYS2, which are active forms of glycogen synthase. GYS1 concentration was significantly increased in fetal livers from RNAi-IUGR and RNAi-NW pregnancies, whereas there was no change in GYS2 concentrations. The quantity of p-GYS (inactive form of GYS) was significantly reduced in both RNAi-IUGR and RNAi-NW pregnancies, indicating overall increased proportion of active GYS. Other studies have also shown that during late gestation in rats, insulin increases the proportion of active glycogen synthase leading to glycogen deposition in rat fetal liver (Eisen et al. 1973; Beurel et al. 2015). Our data suggests that increased insulin sensitivity in fetal liver in both RNAi-IUGR and RNAi-NW pregnancies lead to increased active GYS, and hence increased glycogen synthesis and storage.
Normally, gluconeogenesis does not occur in utero until the immediate perinatal period (Grijalva & Vakili 2013), however some studies suggest that gluconeogenic activity is initiated earlier in IUGR pregnancies as an adaptive mechanism to meet the demand of glucose utilization within the fetus (Hay 2006; Limesand et al. 2007; Thorn et al. 2009; Brown et al. 2015). Lactate dehydrogenase B (LDHB) and phosphoenolpyruvate carboxykinase 2 (PCK2) are key gluconeogenic enzymes. LDHB converts lactate into pyruvate which is then converted to oxaloacetate through a series of reactions. With the aid of PCK2, the oxaloacetate is shuttled across the mitochondrial membrane to be converted into phosphoenolpyruvate, a substrate for gluconeogenesis (Garrett & Grisham 2001; Yang et al. 2009). The CSH-deficient RNAi-IUGR pregnancies had significantly increased in LDHB concentrations, but there was no significant change in PCK2 concentrations. Increased LDHB might initiate gluconeogenesis but unchanged PCK2 suggests otherwise. There was no significant change in LDHB and PCK2 quantity in RNAi-NW fetal liver. From these results we suggest that increased glycogen storage in RNAi-IUGR and RNAi-NW fetal liver is primality due to altered insulin signaling rather than gluconeogenesis.
As reported previously, CSH deficiency generated by lentiviral-mediated RNAi can result in significant fetal growth restriction (Baker et al., 2016; Jeckel et al., 2018), but at least near-term (Baker et al., 2016) there is a cohort of CSH RNAi pregnancies that do not exhibit IUGR. While there are a number of potential explanations for these two phenotypes (e.g., variability in RNAi efficiency, response to fasting and anesthesia, potential compensatory mechanisms, etc.), our results complement the existing case reports of human pregnancies in which the CSH locus is disrupted/deleted. As mentioned earlier, in some human cases severe fetal growth restriction (Trapp et al. 1987; Rygaard et al. 1998) occurred, whereas in some other cases there was no apparent impact on fetal growth (Wurzel et al. 1982; Giampietro et al. 1984; Barbieri et al. 1986; Simon et al. 1986). Based on our results, it could be argued that the offspring of a CSH-deficient pregnancy may well be impacted by CSH deficiency, even in the absence of IUGR. The pregnancies that we studied were harvested at a single gestational age, and the samples collected reflect a single-point in time. As such, there is clearly a need to investigate both CSH-deficient phenotypes under steady-state non-anesthetized/non-stressed conditions, as these types of stressors (fasting and/or anesthesia) impact the acute physiological status of both the mother and fetus, in order to fully define the impact of CSH deficiency on fetal physiology.
Supplementary Material
Funding
This work was supported by NIH-NICHD HD093701 and HD094952, NIH-NIDDK DK08813, and Agriculture and Food Research Initiative Grant nos. 2012-67015-30215 from the United State Department of Agriculture.
Footnotes
Declaration of interest
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- Baker CM, Goetzmann LN, Cantlon JD, Jeckel KM, Winger QA & Anthony RV 2016. Development of ovine chorionic somatomammotropin hormone-deficient pregnancies. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology 310 R837–R846. (doi: 10.1152/ajpregu.00311.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri F, Botticelli A, Consarino R, Genazzani AR & Volpe A 1986. Failure of placenta to produce hPL in an otherwise uneventful pregnancy: a case report. Biological Research in Pregnancy and Perinatology 7 131–133. [PubMed] [Google Scholar]
- Bauer MK, Breier BH, Harding JE, Veldhuis JD & Gluckman PD 1995. The fetal somatotropic axis during long term maternal undernutrition in sheep: evidence for nutritional regulation in utero. Endocrinology 136 1250–1257. (doi: 10.1210/endo.136.3.7867579) [DOI] [PubMed] [Google Scholar]
- Beale EG 2013. Insulin Signaling And Insulin Resistance. Journal of Investigative Medicine : The Official Publication of the American Federation for Clinical Research 61 11–14. (doi: 10.231/JIM.0b013e3182746f95) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett LL, Curry DL & Li CH 1976. Enhancement of insulin secretion by human chorionic somatomammotropin and related hormones. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine (New York, N.Y.) 152 281–283. (doi: 10.3181/00379727-152-39379) [DOI] [PubMed] [Google Scholar]
- Beurel E, Grieco SF & Jope RS 2015. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacology & Therapeutics 148 114–131. (doi: 10.1016/j.pharmthera.2014.11.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J, Kleinridders A & Kahn CR 2014. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harbor Perspectives in Biology 6. (doi: 10.1101/cshperspect.a009191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskila M, Hunter RW, Ibrahim AFM, Delattre L, Peggie M, van Diepen JA, Voshol PJ, Jensen J & Sakamoto K 2010. Allosteric Regulation of Glycogen Synthase Controls Glycogen Synthesis in Muscle. Cell Metabolism 12 456–466. (doi: 10.1016/j.cmet.2010.10.006) [DOI] [PubMed] [Google Scholar]
- Brown LD, Rozance PJ, Bruce JL, Friedman JE, Hay WW & Wesolowski SR 2015. Limited capacity for glucose oxidation in fetal sheep with intrauterine growth restriction. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 309 R920–928. (doi: 10.1152/ajpregu.00197.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browner MF, Nakano K, Bang AG & Fletterick RJ 1989. Human muscle glycogen synthase cDNA sequence: a negatively charged protein with an asymmetric charge distribution. Proceedings of the National Academy of Sciences 86 1443–1447. (doi: 10.1073/pnas.86.5.1443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler WR, Huyler SE, Grandis AS & Handwerger S 1987. Failure of fasting and changes in plasma metabolites to affect spontaneous fluctuations in plasma concentrations of ovine placental lactogen. The Journal of Endocrinology 114 391–397. (doi: 10.1677/joe.0.1140391) [DOI] [PubMed] [Google Scholar]
- Chan JS, Robertson HA & Friesen HG 1978. Distribution of binding sites for ovine placental lactogen in the sheep. Endocrinology 102 632–640. (doi: 10.1210/endo-102-2-632) [DOI] [PubMed] [Google Scholar]
- Culpepper C, Wesolowski SR, Benjamin J, Bruce JL, Brown LD, Jonker SS, Wilkening RB, Hay WW & Rozance PJ 2016. Chronic anemic hypoxemia increases plasma glucagon and hepatic PCK1 mRNA in late-gestation fetal sheep. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 311 R200–208. (doi: 10.1152/ajpregu.00037.2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H & Wu T 2018. Insulin-Like Growth Factor Binding Proteins in Autoimmune Diseases. Frontiers in Endocrinology 9. (doi: 10.3389/fendo.2018.00499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen HJ, Goldfine ID & Glinsmann WH 1973. Regulation of Hepatic Glycogen Synthesis During Fetal Development: Roles of Hydrocortisone, Insulin, and Insulin Receptors. Proceedings of the National Academy of Sciences of the United States of America 70 3454–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Yu SX, Lu Y, Bast RC, Woodgett JR & Mills GB 2000. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proceedings of the National Academy of Sciences 97 11960–11965. (doi: 10.1073/pnas.220413597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemark M 2010. Placental Hormones and the Control of Fetal Growth. The Journal of Clinical Endocrinology & Metabolism 95 2054–2057. (doi: 10.1210/jc.2010-0517) [DOI] [PubMed] [Google Scholar]
- Freemark M & Handwerger S 1984a. Ovine placental lactogen stimulates glycogen synthesis in fetal rat hepatocytes. The American Journal of Physiology 246 E21–24. (doi: 10.1152/ajpendo.1984.246.1.E21) [DOI] [PubMed] [Google Scholar]
- Freemark M & Handwerger S 1984b. Synergistic effects of oPL and insulin on glycogen metabolism in fetal rat hepatocytes. The American Journal of Physiology 247 E714–718. (doi: 10.1152/ajpendo.1984.247.6.E714) [DOI] [PubMed] [Google Scholar]
- Freemark M & Handwerger S 1985. Ovine placental lactogen inhibits glucagon-induced glycogenolysis in fetal rat hepatocytes. Endocrinology 116 1275–1280. (doi: 10.1210/endo-116-4-1275) [DOI] [PubMed] [Google Scholar]
- Freemark M, Comer M & Handwerger S 1986. Placental lactogen and GH receptors in sheep liver: striking differences in ontogeny and function. The American Journal of Physiology 251 E328–333. (doi: 10.1152/ajpendo.1986.251.3.E328) [DOI] [PubMed] [Google Scholar]
- Freemark M, Comer M, Korner G & Handwerger S 1987. A unique placental lactogen receptor: implications for fetal growth. Endocrinology 120 1865–1872. (doi: 10.1210/endo-120-5-1865) [DOI] [PubMed] [Google Scholar]
- Freemark M, Comer M, Mularoni T, D’Ercole AJ, Grandis A & Kodack L 1990. Placental lactogen receptors in maternal sheep liver: effects of fasting and refeeding. The American Journal of Physiology 258 E338–346. (doi: 10.1152/ajpendo.1990.258.2.E338) [DOI] [PubMed] [Google Scholar]
- Freemark M, Keen A, Fowlkes J, Mularoni T, Comer M, Grandis A & Kodack L 1992. The placental lactogen receptor in maternal and fetal sheep liver: regulation by glucose and role in the pathogenesis of fasting during pregnancy. Endocrinology 130 1063–1070. (doi: 10.1210/endo.130.2.1310275) [DOI] [PubMed] [Google Scholar]
- Gagnon R 2003. Placental insufficiency and its consequences. European Journal of Obstetrics, Gynecology, and Reproductive Biology 110 Suppl 1 S99–107. (doi: 10.1016/s0301-2115(03)00179-9) [DOI] [PubMed] [Google Scholar]
- Garrett RH & Grisham CM 2001. Principles of Biochemistry: With a Human Focus. Harcourt College Publishers. [Google Scholar]
- Giampietro O, Ferdeghini M & Scatena P 1984. Human placental lactogen (hPL) deficiency in a normal pregnancy. Postgraduate Medical Journal 60 689–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootwine E 2004. Placental hormones and fetal-placental development. Animal Reproduction Science 82–83 551–566. (doi: 10.1016/j.anireprosci.2004.04.008) [DOI] [PubMed] [Google Scholar]
- Grijalva J & Vakili K 2013. Neonatal liver physiology. Seminars in Pediatric Surgery 22 185–189. (doi: 10.1053/j.sempedsurg.2013.10.006) [DOI] [PubMed] [Google Scholar]
- Handwerger S 1991. Clinical counterpoint: the physiology of placental lactogen in human pregnancy. Endocrine Reviews 12 329–336. (doi: 10.1210/edrv-12-4-329) [DOI] [PubMed] [Google Scholar]
- Handwerger S & Freemark M 2000. The roles of placental growth hormone and placental lactogen in the regulation of human fetal growth and development. Journal of Pediatric Endocrinology & Metabolism: JPEM 13 343–356. (doi: 10.1515/jpem.2000.13.4.343) [DOI] [PubMed] [Google Scholar]
- Handwerger S, Fellows RE, Crenshaw MC, Hurley T, Barrett J & Maurer WF 1976. Ovine placental lactogen: acute effects on intermediary metabolism in pregnant and non-pregnant sheep. The Journal of Endocrinology 69 133–137. (doi: 10.1677/joe.0.0690133) [DOI] [PubMed] [Google Scholar]
- Hay WW 2006. Placental-Fetal Glucose Exchange and Fetal Glucose Metabolism. Transactions of the American Clinical and Climatological Association 117 321–340. [PMC free article] [PubMed] [Google Scholar]
- Hill DJ, Freemark M, Strain AJ, Handwerger S & Milner RD 1988. Placental lactogen and growth hormone receptors in human fetal tissues: relationship to fetal plasma human placental lactogen concentrations and fetal growth. The Journal of Clinical Endocrinology and Metabolism 66 1283–1290. (doi: 10.1210/jcem-66-6-1283) [DOI] [PubMed] [Google Scholar]
- Holen T, Amarzguioui M, Wiiger MT, Babaie E & Prydz H 2002. Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Research 30 1757–1766. (doi: 10.1093/nar/30.8.1757) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SW, Jiang Y, Kim S, Li CJ & Lee D 2014. Target Gene Abundance Contributes to the Efficiency of siRNA-Mediated Gene Silencing. Nucleic Acid Therapeutics 24 192–198. (doi: 10.1089/nat.2013.0466) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley TW, D’Ercole AJ, Handwerger S, Underwood LE, Furlanetto RW & Fellows RE 1977. Ovine placental lactogen induces somatomedin: a a possible role in fetal growth. Endocrinology 101 1635–1638. (doi: 10.1210/endo-101-5-1635) [DOI] [PubMed] [Google Scholar]
- Jeckel KJ, Boyarko AC, Bouma GJ, Winger QA & Anthony RV 2018. Chorionic Somatomammotropin Impacts Early Fetal Growth and Placental Gene Expression. The Journal of Endocrinology 237 301–310. (doi: 10.1530/JOE-18-0093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea RG, Wooding P, Stewart I, Hannah LT, Morton S, Wallace K, Aitken RP, Milne JS, Regnault TR, Anthony RV et al. 2007. The expression of ovine placental lactogen, StAR and progesterone-associated steroidogenic enzymes in placentae of overnourished growing adolescent ewes. Reproduction (Cambridge, England) 133 785–796. (doi: 10.1530/REP-06-0294) [DOI] [PubMed] [Google Scholar]
- Liang L, Guo WH, Esquiliano DR, Asai M, Rodriguez S, Giraud J, Kushner JA, White MF & Lopez MF 2010. Insulin-Like Growth Factor 2 and the Insulin Receptor, But Not Insulin, Regulate Fetal Hepatic Glycogen Synthesis. Endocrinology 151 741–747. (doi: 10.1210/en.2009-0705) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Smith D & Hay WW 2007. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. American Journal of Physiology. Endocrinology and Metabolism 293 E1716–1725. (doi: 10.1152/ajpendo.00459.2007) [DOI] [PubMed] [Google Scholar]
- Min SH, Mackenzie DD, Breier BH, McCutcheon SN & Gluckman PD 1996. Growth-promoting effects of ovine placental lactogen (oPL) in young lambs: comparison with bovine growth hormone provides evidence for a distinct effect of oPL on food intake. Growth Regulation 6 144–151. [PubMed] [Google Scholar]
- Muhič M, Vardjan N, Chowdhury HH, Zorec R & Kreft M 2015. Insulin and Insulin-like Growth Factor 1 (IGF-1) Modulate Cytoplasmic Glucose and Glycogen Levels but Not Glucose Transport across the Membrane in Astrocytes. The Journal of Biological Chemistry 290 11167–11176. (doi: 10.1074/jbc.M114.629063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt SL, Kappes SM & Anthony RV 1995. Ontogeny of a specific high-affinity binding site for ovine placental lactogen in fetal and postnatal liver. Domestic Animal Endocrinology 12 337–347. (doi: 10.1016/0739-7240(95)00030-i) [DOI] [PubMed] [Google Scholar]
- Ramezani A, Hawley TS & Hawley RG 2000. Lentiviral vectors for enhanced gene expression in human hematopoietic cells. Molecular Therapy: The Journal of the American Society of Gene Therapy 2 458–469. (doi: 10.1006/mthe.2000.0190) [DOI] [PubMed] [Google Scholar]
- Rygaard K, Revol A, Esquivel-Escobedo D, Beck BL & Barrera-Saldaña HA 1998. Absence of human placental lactogen and placental growth hormone (HGH-V) during pregnancy: PCR analysis of the deletion. Human Genetics 102 87–92. (doi: 10.1007/s004390050658) [DOI] [PubMed] [Google Scholar]
- Schoknecht PA, McGuire MA, Cohick WS, Currie WB & Bell AW 1996. Effect of chronic infusion of placental lactogen on ovine fetal growth in late gestation. Domestic Animal Endocrinology 13 519–528. (doi: 10.1016/s0739-7240(96)00090-2) [DOI] [PubMed] [Google Scholar]
- Sibiak R, Jankowski M, Gutaj P, Mozdziak P, Kempisty B & Wender-Ożegowska E 2020. Placental Lactogen as a Marker of Maternal Obesity, Diabetes, and Fetal Growth Abnormalities: Current Knowledge and Clinical Perspectives. Journal of Clinical Medicine 9. (doi: 10.3390/jcm9041142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon P, Decoster C, Brocas H, Schwers J & Vassart G 1986. Absence of human chorionic somatomammotropin during pregnancy associated with two types of gene deletion. Human Genetics 74 235–238. (doi: 10.1007/BF00282540) [DOI] [PubMed] [Google Scholar]
- Spellacy WN, Buhi WC & Birk SA 1976. Human placental lactogen and intrauterine growth retardation. Obstetrics and Gynecology 47 446–448. [PubMed] [Google Scholar]
- Swenne I, Hill DJ, Strain AJ & Milner RD 1987. Effects of human placental lactogen and growth hormone on the production of insulin and somatomedin C/insulin-like growth factor I by human fetal pancreas in tissue culture. The Journal of Endocrinology 113 297–303. (doi: 10.1677/joe.0.1130297) [DOI] [PubMed] [Google Scholar]
- Taylor MJ, Jenkin G, Robinson JS, Thorburn GD, Friesen H & Chan JS 1980. Concentrations of placental lactogen in chronically catheterized ewes and fetuses in late pregnancy. The Journal of Endocrinology 85 27–34. (doi: 10.1677/joe.0.0850027) [DOI] [PubMed] [Google Scholar]
- Thorn SR, Regnault TRH, Brown LD, Rozance PJ, Keng J, Roper M, Wilkening RB, Hay WW & Friedman JE 2009. Intrauterine Growth Restriction Increases Fetal Hepatic Gluconeogenic Capacity and Reduces Messenger Ribonucleic Acid Translation Initiation and Nutrient Sensing in Fetal Liver and Skeletal Muscle. Endocrinology 150 3021–3030. (doi: 10.1210/en.2008-1789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp M, De Wilde R, Holzgreve W, Stals HJ & Bohnet HG 1987. [A pregnancy without detectable human placental lactogen (hPL)]. Zentralblatt Fur Gynakologie 109 130–133. [PubMed] [Google Scholar]
- Wesolowski SR & Hay WW 2016. Role of placental insufficiency and intrauterine growth restriction on the activation of fetal hepatic glucose production. Molecular and Cellular Endocrinology 435 61–68. (doi: 10.1016/j.mce.2015.12.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal SA & Nuttall FQ 1992. Comparative characterization of human and rat liver glycogen synthase. Archives of Biochemistry and Biophysics 292 479–486. (doi: 10.1016/0003-9861(92)90019-S) [DOI] [PubMed] [Google Scholar]
- White V, Jawerbaum A, Mazzucco MB, Gauster M, Desoye G & Hiden U 2018. IGF2 stimulates fetal growth in a sex- and organ-dependent manner. Pediatric Research 83 183–189. (doi: 10.1038/pr.2017.221) [DOI] [PubMed] [Google Scholar]
- Wurzel JM, Parks JS, Herd JE & Nielsen PV 1982. A gene deletion is responsible for absence of human chorionic somatomammotropin. DNA (Mary Ann Liebert, Inc.) 1 251–257. (doi: 10.1089/dna.1.1982.1.251) [DOI] [PubMed] [Google Scholar]
- Yang J, Kalhan SC & Hanson RW 2009. What is the metabolic role of phosphoenolpyruvate carboxykinase? The Journal of Biological Chemistry 284 27025–27029. (doi: 10.1074/jbc.R109.040543) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.