

Abstract
The cyclooxygenase (COX) metabolic pathway regulates immune responses and inflammation. The effect of the COX pathway on innate pulmonary inflammation induced by protease-containing fungal allergens such as Alternaria alternata is not fully defined. In this study, we tested the hypothesis that COX inhibition augments Alternaria-induced pulmonary group 2 innate lymphoid cell (ILC2) responses and IL-33 release. Mice were treated with the COX inhibitor indomethacin, flurbiprofen, or vehicle and challenged intranasally with Alternaria extract for four consecutive days to induce innate lung inflammation. We found that indomethacin and flurbiprofen significantly increased the numbers of ILC2, and IL-5 and IL-13 expression by ILC2 in the lung. Indomethacin also increased ILC2 proliferation, the percentages of eosinophils and mucus production in the lung. Both indomethacin and flurbiprofen augmented the release of IL-33 in bronchoalveolar lavage fluid after Alternaria challenge, suggesting that more IL-33 was available for ILC2 activation and that a COX product(s) inhibited IL-33 release. This is supported by the in vitro finding that the COX product PGE2 and the PGI2 analogs cicaprost decreased Alternaria extract-induced IL-33 release by human bronchial epithelial cells. Although contrasting effects of PGD2, PGE2, and PGI2 on ILC2 responses have been previously reported, the overall effect of the COX pathway on ILC2 function is inhibitory in Alternaria-induced innate airway inflammation. (Word count: Abstract 211; Manuscript: 5109)
INTRODUCTION
Group 2 innate lymphoid cells (ILC2) are critical in innate airway inflammation and promote adaptive type 2 inflammatory and immune responses in allergic diseases. The cytokines IL-33, IL-25 and TSLP activate ILC2 in a tissue-specific manner. IL-33 activates lung ILC2 that express IL-33 receptor ST2, while IL-25 and TSLP play more prominent roles in activating gut- and skin-associated ILC2, respectively.(1–8) IL-33 is constitutively expressed mainly by epithelial cells and endothelial cells, and is sequestered in the nucleus.(9) IL-33 can be released into the extracellular space during cell injury and necrosis, and acts as an alarmin to activate ILC2 for IL-5 and IL-13 expression. The release of IL-33 into the airway has been reported in mouse models of fungal allergen-induced lung inflammation. Challenging mice with Alternaria alternata extract (Alt), house dust mite (HDM) and papain in the lung resulted in IL-33 release in bronchoalveolar lavage fluid (BALF).(5, 6, 10) Alt caused epithelial damage in vivo and induced epithelial cell necrosis, suggesting that Alt-induced cell damage and death is a mechanism of IL-33 release in mouse lungs.(11) Other mechanisms proposed for Alt-induced IL-33 release include the critical role of epithelial cell dual oxidase 1 activity, elevated extracellular secretion of ATP, augmented intracellular calcium concentrations, and generation of reactive oxygen species (ROS).(12–14) The importance of IL-33 in ILC2 function and innate type 2 inflammation is supported by diminished ILC2 activation in IL-33 KO mice in Alt-challenged mouse lungs.(14)
Bioactive lipid molecules formed in arachidonic acid metabolism have regulatory functions on physiological processes and cell signaling in mice and humans. They also influence immune responses and inflammation. Arachidonic acid can be metabolized through the cyclooxygenase (COX) pathway for production of prostaglandin (PG) D2, PGE2, PGF2α, and PGI2 and thromboxane A2, or through the lipoxygenase pathways for leukotriene and lipoxin formation. Cysteinyl leukotrienes (CysLTs) formed in the 5-lipoxygenase (5-LO) pathway potently stimulated ILC2 production of IL-4, IL-5 and IL-13 in vitro and enhanced Alt-induced ILC2 activation and IL-5 production in mouse lungs.(15–18) In contrast, lipoxin A4 suppressed ILC2 responses.(19) In the COX pathway, PGD2 promoted type 2 inflammation and the PGD2 receptor DP2 is selectively expressed on ILC2.(20) PGD2 induced chemotaxis of human peripheral blood ILC2 and mouse ILC2, and potentiated mouse ILC2 for IL-33- and IL-25-induced type 2 cytokine responses.(20–22) Mouse and human ILC2 also express the PGI2 receptor IP and PGE2 receptors (EP1, EP2, EP3, and EP4). PGI2 markedly inhibited IL-33-induced ILC2 IL-5 and IL-13 expression in vitro in an IP-specific manner.(7, 23) IP-deficient mice had greater numbers of IL-5- and IL-13-expressing ILC2 and elevated eosinophilia and inflammation in Alt-challenged lungs.(7) Similarly, PGE2 suppressed IL-5 and IL-13 production by mouse ILC2 and decreased Alt-induced mouse and human lung ILC2 responses through the EP2 signaling pathway.(24, 25) Due to the diverse or opposite effects of individual PGs on ILC2, the overall impact of the COX pathway on ILC2-mediated immune responses has not been fully defined. Considering that COX inhibiting drugs are the most commonly used pain-relieving and fever-reducing drugs,(26) studies on the effect of COX inhibitors on ILC2 responses could provide insights into ILC2-mediated innate type 2 inflammation and ILC2-enhanced adaptive allergic immune responses.
In this study, we hypothesized that COX inhibition increases protease-containing fungal allergen-induced ILC2 responses and type 2 cytokine production. We tested the hypothesis by using the mouse model of Alt-induced and ILC2-mediated innate lung inflammation. WT and IL-33 KO mice were treated with the non-selective COX inhibitor indomethacin (Indo), flurbiprofen, or vehcle. COX inhibition increased Alt-induced ILC2 responses and IL-5 and IL-13 production. This elevated ILC2 response was associated with greater percentages of eosinophils in BALF and airway mucus in WT mice. Furthermore, COX inhibition increased Alt-induced ROS production and IL-33 release in the lung independent of 5-lipoxygenase, and the COX products PGI2 and PGE2 decreased IL-33 release and cell damage of human bronchial epithelial cells (hBE33 cells) in vitro. In IL-33 KO mice, COX inhibition did not increase IL-5 and IL-13 production compared to the vehicle controls. Interestingly, COX inhibition also increased ILC2 responses in mice challenged with recombinant mouse IL-33 in the lung.
MATERIALS AND METHODS
Mice
Wild-type (WT) BALB/c mice were obtained from The Charles River Laboratories (Raleigh, NC). Il33Cit/Cit functional Il33 knockout (IL-33 KO) mice on a BALB/c genetic background were a gift from Dr. Andrew N.J. McKenzie (MRC Laboratory of Molecular Biology, Hills Road, Cambridge, UK).(27) 5-LO KO mice on a 129 genetic background were purchased from The Jackson Laboratory and backcrossed 10 generations to BALB/c genetic background. Age-matched female WT, IL-33 KO, and 5-LO KO mice were used at 8–12 weeks old. Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at Vanderbilt University, and were conducted according to the guidelines for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council.
Indomethacin, flurbiprofen, and BrdU administration and Alternaria challenge
Wild type (WT), IL-33 knockout (IL-33 KO), and 5-LO KO mice on BALB/c background were used at 8–12 weeks of age. The mice were treated with the non-selective COX inhibitor indomethacin (Indo, 30 μg/ml, Sigma), flurbiprofen (75 μg/ml, Sigma), or vehicle (Veh, ethanol) in the drinking water for 3 days followed by intranasally challenging mice daily for 4 days with Alt (Greer Laboratories, 5 µg in 100 µl PBS per mouse) or heat-inactivated (on 110 °C heat block for 2 h) Alternaria extract (HIA). The mice were continuously treated with Indo, flurbiprofen, or Veh throughout the experiment protocol. In some experiments, mice were intranasally challenged daily for 4 days with recombinant mouse IL-33 (rIL-33, PeproTech Inc., 500 ng in 100 µl PBS per mouse) or PBS. Mice were sacrificed by an intraperitoneal injection of Fatal-Plus solution (pentobarbital sodium) 24 h after the last challenge, and BALF and lungs were harvested for analyses. For IL-33 release, the mice were treated with Indo or Veh for 3 days and challenged with one dose of Alt or HIA. BALF was harvested 1 h after Alt or HIA challenge. The one-hour time point was chosen based on published findings that IL-33 release peaked at 1h after Alt challenge.(14, 28) In some experiments, 1 mg BrdU (BD Biosciences) was injected intraperitoneally after each Alt challenge for analyses of ILC2 proliferation at 24 h after the last Alt/BrdU injections.
Measurement of prostaglandins and cysteinyl leukotrienes
Urine PGE2 and the PGI2 metabolite 6-keto-PGF1α in mouse urine samples were determined by PGE2 Express ELISA kit and 6-keto-PGF1α EIA kit (Cayman Chemical). CysLTs in mouse BALF harvested 1 h after Alt challenge from Indo- or Veh-treated mice were measured by CysLT ELISA kit (Cayman chemicals). Creatinine levels in the urine were determined by Creatinine Colorimetric Assay Kit (Cayman Chemical). For mouse urine collection, WT BALB/c mice were placed in metabolic cages, three mice per cage, and treated with drinking water containing either Indo or EtOH as described above. After the mice were treated with either Indo or EtOH for 3 days, they were challenged with Alt or HIA daily for 2 consecutive days. Urine was collected daily from each cage before the first and second challenges and at 24 h after the second challenge for the measurement of PGE2, 6-keto-PGF1α and CysLTs .
BALF collection and differential cell counting
BALF collection was performed by instilling 800 µl saline through a tracheostomy tube and then withdrawing the fluid with gentle suction via a syringe. Total white blood cells in BALF were counted on a hemocytometer. Cytospin slides were prepared with 100 µl BALF and stained using Diff Quik (American Scientific Products). Differential cell counts were based on counts of 200 cells using standard morphological criteria to classify the cells as eosinophils, lymphocytes, neutrophils, or macrophages.
Histopathology of Lungs
Lungs were harvested and fixed in 10% buffered formalin overnight. The lungs were transferred to 70% EtOH and then embedded in paraffin blocks. Tissue sections (5 μm) were stained with periodic acid-Schiff (PAS) to assess mucus production. Slides were examined and scored by a pathologist who was blinded to the experimental groups. PAS scoring criteria: 0 = PAS positive cell are not observed in the examined sections; 1 = Less than 10% of cells in medium and small airways are PAS positive; 2 = 10–20% PAS positive cells in medium and small airways; 3 = Greater than 20% cells in medium and small airways are PAS positive and hyperplasia of PAS positive cells is observed; 4= Greater than 20% cells in medium and small airways are PAS positive, hyperplasia of PAS positive cells and mucus plugging of airways are observed.
Flow cytometry
Lungs were disrupted by mechanical mincing and enzymatic digestion with 1 mg/ml collagenase (Sigma) and 0.02 mg/ml DNase I (Sigma) in RPMI with 5% FBS for 30 min at 37°C as described.(7) EDTA was added to quench the enzymatic reaction, and the tissue was ground and filtered through a 70-µm plastic strainer. RBCs were lysed and the lung cells were cultured with phorbol 12-myristate 13-acetate (PMA,10 nM, Sigma), ionomycin (1 µg/ml, Sigma), and GolgiStop (BD Biosciences) for 4–6 h. The cells were stained with Ghost Dye™ Violet 450 dead cell dye (Tonbo Biosciences) and fixed with IC fixation buffer (eBiosciences). After cell permeabilization with eBiosciences™ Perm Buffer, the cells were blocked with anti-CD16/CD32 (BD Biosciences) and stained for the surface markers with specific Abs for CD3 (BD Biosciences), CD90.2 (BD Biosciences), KLRG1 (BioLegend), ST2 (BD Biosciences), ICOS (BD Biosciences), CD25 (Invitrogen), lineage cell markers (Ab cocktail, Miltenyi); and the intracellular cytokines IL-5 (BD Biosciences) and IL-13 (eBiosciences). The lineage marker Ab cocktail contains antibodies against CD5, CD45R (B220), CD11b, Gr-1 (Ly-6G/C), 7–4, and Ter-119. 123count eBeads (Invitrogen) were added for cell counting by flow cytometry. Samples were analyzed on a BD LSR II flow cytometer and data were analyzed by FlowJo software version X. ILC2 were identified as Lin−CD90.2+KLRG1+ST2+ cells except that ILC2 were identified as Lin−CD90.2+ICOS+CD25+ cells in Supplemental Fig. 3. For ILC2 proliferation assay, lung cells were stained with APC BrdU Flow Kit (Catalogue # 552598) following the manufacturer’s protocol. The proliferating ILC2 were identified as BrdU+Lin−CD90.2+KLRG1+ST2+ cells.
Cytokine and LDH quantitation
The levels of mouse IL-5, IL-13, IL-33 and TSLP in the lung tissue, IL-33 in BALF, and human IL-33 in hBE33 culture supernatant were measured with Quantikine ELISA kits (R&D Systems). HMGB1 in BALF was measured with HMG1/HMGB1 ELISA kit (LifeSpan Biosciences). LDH in BALF and in hBE33 culture supernatant was determined with LDH Cytotoxicity assay kits (Thermo Fisher Scientific).
Protease Assay
The protease activity in Alt was measured by Protease Fluorescent Detection Kit (Sigma). Alt was mixed with reduced L-glutathione (GSH, Sigma) or N-acetyl-L-cysteine (NAC, Sigma) at various concentrations and incubated at r.t. for 10 min followed by protease activity determination.
In vivo ROS quantification
Alt-induced ROS was determined by using L-012, a chemiluminescent probe that reacts with ROS and generates luminescence.(29) A bioluminescence imager for small animals (PerkinElmer IVIS Spectrum) was used to detect in vivo luminescent signal and IVIS SpectrumCT software was used for the quantification of signal intensity. WT BALB/c mice were treated with Indo or Veh in drinking water for 3 days and challenged intranasally with one dose of 5 µg Alt or HIA in 100 µl PBS. Sixty min after challenge, the mice were injected with 400 µg L-012 in 100 µl PBS retro-orbitally and imaged at 10 min after L-012 injection.
hBE33 cell culture
The immortalized human epithelial cell line (hBE33) that stably express high levels of human IL-33 in the nuclei (12) was seeded in 24 well plates and cultured in BronchiaLife Epithelial Basal Media (LifeLine cell technology). At 80–90% cell confluence, the cells were treated with cicaprost, PGE2, the cAMP-elevating agent forskolin, or the respective vehicles for 30 min. Alt or HIA was then added at 30 µg/ml and the cell culture supernatant was collected at 15 min after Alt or HIA addition for human IL-33 ELISA and lactic acid dehydrogenase (LDH) assay (Thermo Fisher Scientific). In some experiments, hBE33 cells were treated with Indo or Veh for 4 h followed by challenging with HIA or Alt at 30 µg/ml for 15 min.
Statistical analysis
Data were graphed by using GraphPad Prism software and analyzed with standard one-way ANOVA with Sadik’s multiple comparisons tests. For experiments with significant differences in SDs among treatment groups, the data were analyzed with Brown-Forsythe and Welch ANOVA with Dunnett T3 multiple comparisons tests. The results were presented as mean ± SEM. Differences were considered significant if p<0.05.
RESULTS
COX inhibition decreased the levels of PGE2 and PGI2 production in the urine.
To study the effect of COX inhibition on Alt-induced innate lung inflammation, we determined whether the COX inhibitor Indo suppresses prostaglandin formation in the mouse Alt model. We found that Alt challenge significantly increased PGE2 and PGI2 production and treating mice with Indo in the drinking water significantly decreased the levels of PGE2 and the PGI2 metabolite 6-keto-PGF1α in the urine, compared with the vehicle treatment (Supplemental Fig. 1). Therefore, this model provides the opportunity to test the effect of COX inhibition on Alt-induced innate airway responses.
COX inhibition increased Alt-induced ILC2 responses.
Alt induced lung ILC2 responses in an IL-33-dependent manner.(28) We and others have previously shown that ILC2 are the predominant producers of type 2 cytokines in the model of Alt-induced innate lung inflammation.(24, 30) To test the hypothesis that COX inhibition augments Alt-induced lung ILC2 responses, we treated mice with Indo or Veh and challenged mice intranasally with Alt or HIA daily for 4 days and analyzed ILC2 numbers and IL-5 and IL-13 expression in the lung by flow cytometry (Supplemental Fig. 2A for flow gating strategy). We found that COX inhibition with Indo significantly increased the total number of ILC2 in Alt-challenged lungs compared to Veh-Alt group (Fig. 1A). COX inhibition resulted in greater percentages of IL-13 single positive ILC2 cells (Fig. 1A) than Veh-Alt control, but similar percentages of IL-5/IL-13 double positive cells and IL-5 single positive cells in the lung to those in Veh-Alt group (Complemental Fig. 2C and 2D). The total numbers of IL-5−IL-13+ ILC2, IL-5+IL-13+ ILC2, and IL-5+IL-13− ILC2 were all significantly greater in Indo group than in Veh group (Fig. 1E–1G). In vivo ILC2 proliferation analyses of BrdU incorporation indicated that Indo significantly increased the numbers of BrdU+ILC2 cells (Fig. 1H), but not BrdU+CD4 T cells (Fig. 1I) in Alt-challenged lungs. Indo did not change the total numbers of live cells (Supplemental Fig. 2B), CD4 T cells (Supplemental Fig. 2A for gating strategy and 2E), and CD4 T cells that expressed IL-5 and/or IL-13 (Supplemental Fig. 2F–2H), indicating that in this acute innate inflammation model, CD4 T cells were not activated. These results indicate that COX inhibition significantly enhanced Alt-induced ILC2 proliferation and ILC2 type 2 cytokine responses in the lung.
Figure 1.
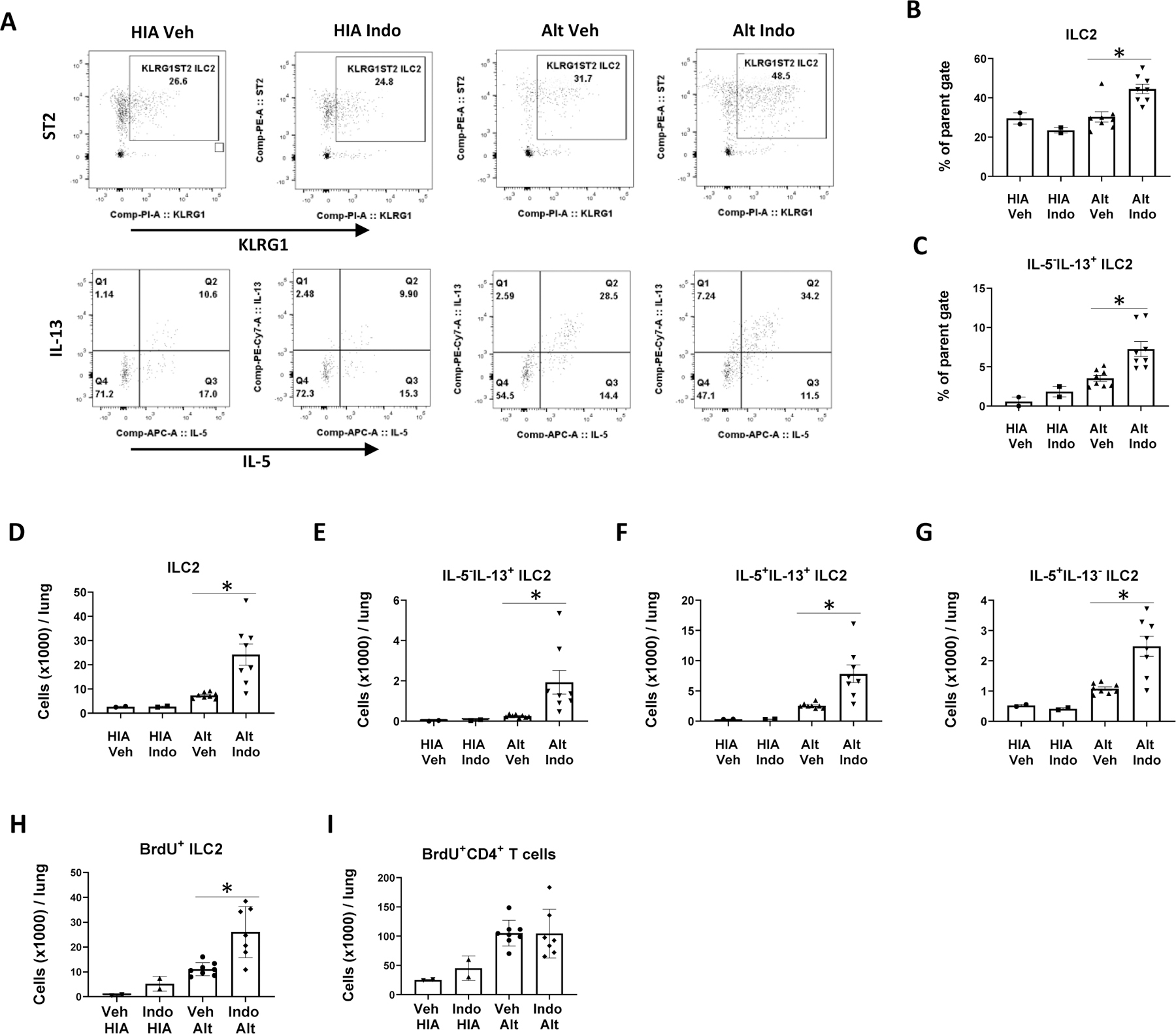
COX inhibition increased Alternaria-induced ILC2 responses in the lung. WT BALB/c mice were treated with indomethacin (Indo, 30 μg/ml) or vehicle (Veh) in drinking water and challenged intranasally with heat-inactivated Alternaria extract (HIA) or Alternaria extract (Alt) daily for 4 days. Mouse lungs were harvested 24 h after the last Alt challenge for flow cytometry. (A) Representative flow plots showing the percentages of ILC2 (Lin−CD90+KLRG1+ST2+ cells) and IL-5+ and/or IL-13+ ILC2. (B) Percentages of ILC2. (C) Percentages of IL-5−IL-13+ ILC2. (D)–(G) Total numbers of ILC2, IL-5−IL-13+ ILC2, IL-5+IL-13+ ILC2, and IL-5+IL-13− ILC2 in the lung. (H)-(I) BrdU was injected intraperitoneally after each Alt challenge. The numbers of (H) BrdU+ ILC2 and (I) BrdU+ CD4 T cells 24 h after the last Alt/BrdU injections were determined by flow cytometry. * p<0.05, n=2 mice in HIA groups and 8 mice in Alt groups.
Figure 2.
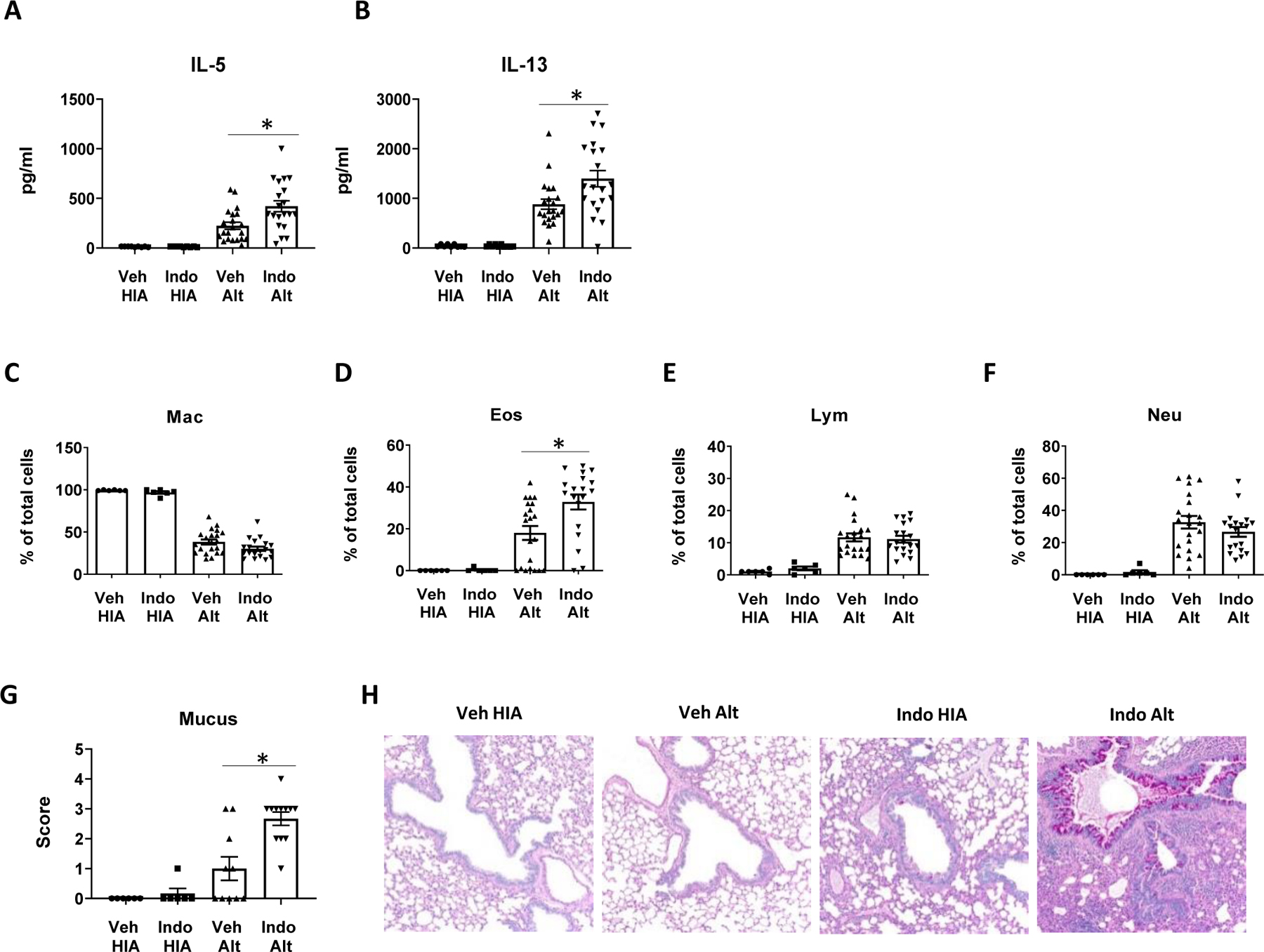
COX inhibition increased IL-5 and IL-13 responses and type 2 inflammation. WT BALB/c mice were treated with Indo or Veh and challenged intranasally with HIA or Alt daily for 4 days. Bronchoalveolar lavage fluid (BALF) and lungs were harvested 24h after the last Alt challenge. (A and B) IL-5 and IL-13 levels in the lung homogenate. (C–F) Percentages of macrophage, eosinophils, lymphocytes and neutrophils in BALF. (G and H) Mucus score (PAS staining) and images of PAS stains of the lung tissue. Combined data of 3 (A–F) or 2 (G) independent experiments were shown. * p<0.05, n=9–22 mice (A–B), 6–21 mice (C–F), and 6–11 mice (G).
In the experiments described above, ILC2 were defined as Lin−CD90+KLRG1+ST2+ cells. We also performed COX inhibition and Alt-challenge experiments in that ILC2 were identified as Lin−CD90+ICOS+CD25+ cells. We found similar results that Indo significantly increased total numbers of ILC2 and IL-5+ and/or IL-13+ ILC2 in the lung (Supplemental Fig. 3). Therefore, COX inhibition increased Alt-induced ILC2 responses.
COX inhibition increased Alt-induced airway eosinophilia and remodeling.
To test the hypothesis that COX inhibition increases Alt-induced innate type 2 lung inflammation, mice were treated with Indo or Veh and challenged with Alt daily for 4 days and lungs and lungs were harvested for cytokine measurements and differential cell counts. We found that COX inhibition with Indo significantly augmented IL-5 and IL-13 responses in the lung (Fig. 2A, 2B). Indo significantly increased the percentage of eosinophils, but not the percentages of macrophages, lymphocytes, and neutrophils in the BALF (Fig. 2C–2F). Indo also significantly augmented airway mucus production (Fig. 2G, 2H). These results demonstrated that COX inhibition has an overall stimulatory effect on Alt-induced innate type 2 cytokine responses and eosinophilia. Indo decreased the numbers of total cells, macrophages, lymphocytes, and neutrophils in BALF, but not eosinophils (Supplemental Fig. 4A–4E), also supporting that COX inhibition preferentially promoted innate type 2 responses.
COX inhibition increased IL-33 and LDH release
To determine the mechanisms by which COX inhibition augmented Alt-induced innate pulmonary type 2 responses, we focused on IL-33, the major ILC2-activating cytokine in the lung tissue.(8) We hypothesized that COX inhibition increases Alt-induced IL-33 release and provides more IL-33 for ILC2 activation in the lung. We found that Alt induced IL-33 release in BALF while HIA was unable to induce IL-33 release (Fig. 3A). COX inhibition significantly increased the levels of IL-33 in BALF of Alt-challenged mice compared to vehicle control (Fig. 3A). In addition, COX inhibition increased the release of the cell damage marker LDH in BALF (Fig. 3B), suggesting a correlation between Alt-induced cell damage and IL-33 release. HMGB1 is a nuclear protein that binds to DNA as an architectural chromatin-binding factor and can be released from cells as an alarmin following cell damage.(31) HMGB1 enhanced IL-33 expression in an LPS stimulation model of epithelial cells.(32) We therefore measured HMGB1 levels in BALF harvested 1 h after Alt challenge. Indo decreased HMGB1 release in WT mice (Fig. 3C), indicating a negative correlation between HMGB1 release and Indo-augmented ILC2 responses. We also investigated the effect of COX inhibition on Alt-induced TSLP expression in the lung and found that Indo decreased TSLP expression at 6 h post Alt challenge (Fig. 3D), a time point at which we found the peak of TSLP expression occurs after Alt challenge. These results suggest that the increase of Alt-induced ILC2 responses in Indo-treated mice was related to the positive effect of Indo on IL-33 release, but not to the effects of Indo on either TSLP or HMGB1 expression.
Figure 3.
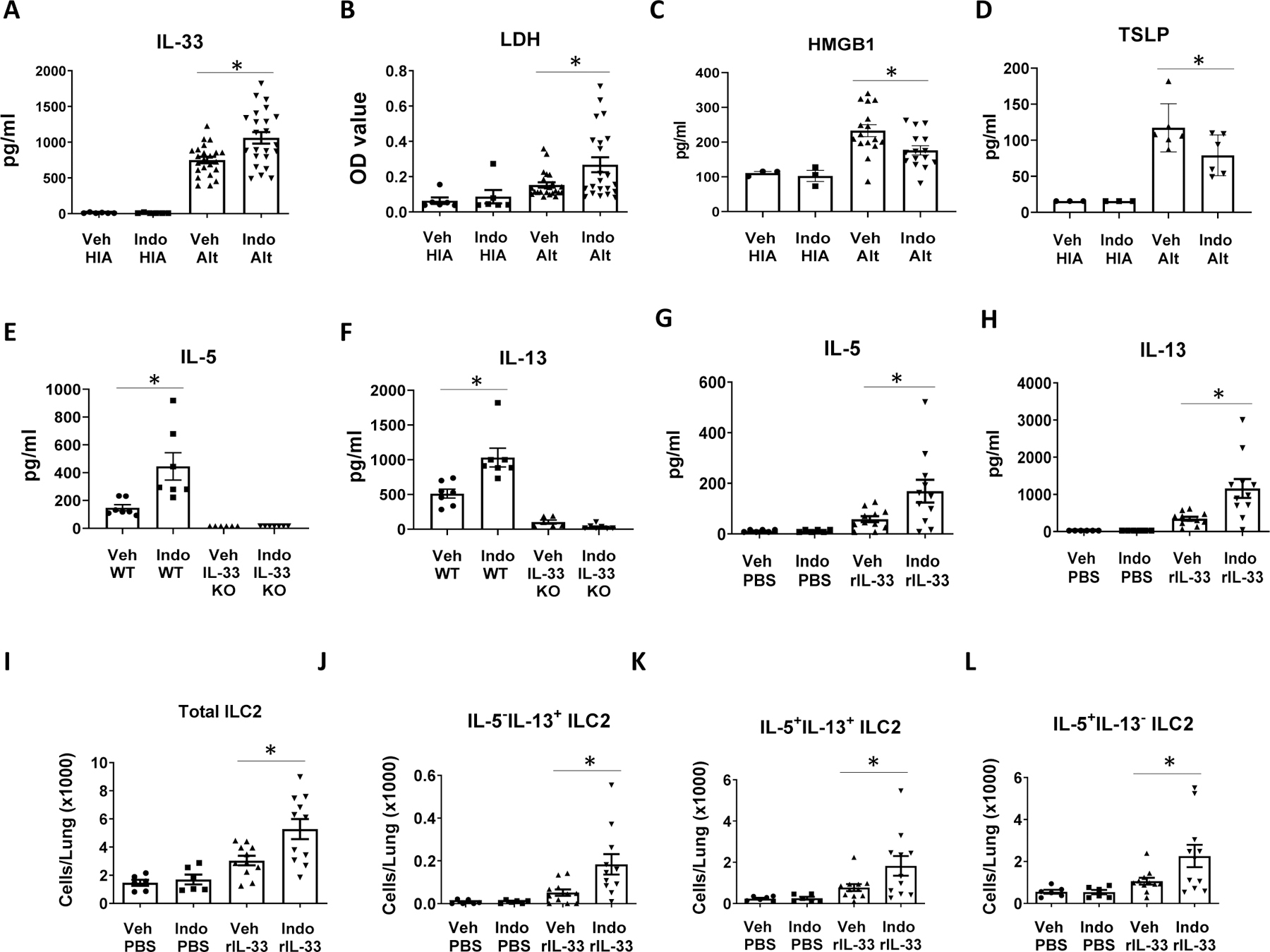
COX inhibition increased IL-33 release in BALF and IL-33 is required for the effect of COX inhibition on Alt-induced IL-5 and IL-13 responses. (A-D) WT BALB/c mice were treated with Indo or Veh and challenged intranasally with one dose of HIA or Alt. (A–C) Mouse BALF was harvested 1 h after the challenge. The levels of (A) IL-33, (B) LDH, and (C) HGBM1 in BALF. (D) The levels of TSLP in the lung homogenate at 6 h after Alt challenge. (E-F) WT mice and IL-33 KO mice were treated with Indo or Veh and challenged intranasally with Alt daily for 4 days. Mouse lungs were harvested 24 h after the last Alt challenge. The levels of (E) IL-5 and (F) IL-13. (G–H) WT BALB/c mice were treated with Indo or Veh and challenged intranasally with 500 ng rIL-33 or PBS daily for 4 days. Mouse lungs were harvested 24 h after the last rIL-33 challenge for ELISA and flow cytometry. The levels of (G) IL-5 and (H) IL-13 in the lung homogenate. (I–L) Flow cytometry data. The numbers of (I) total ILC2, (J) IL-5−IL-13+ ILC2, (K) IL-5+IL-13+ ILC2, and (L) IL-5+IL-13− ILC2 in the lung. Combined data of 2 independent experiments were shown. * p<0.05, n=6–23 mice (A–B), 3–11 mice (C), 3–6 mice (D), 6–7 mice (E–F), or 6–11 mice (G–K).
The stimulatory effect of COX inhibition on Alt-induced IL-5 and IL-13 responses is dependent on IL-33
Because IL-33 is critical for ILC2 activation, proliferation, and cytokine production in the lung,(33) we therefore sought to determine whether the stimulatory effect of COX inhibition on Alt-induced type 2 responses is dependent on the IL-33 signaling pathway. We used WT and IL-33 KO mice for COX inhibition and Alt challenges. We found that IL-33 KO mice did not respond to Alt challenge and produced minimum levels of IL-5 and IL-13 in the lung (Fig. 3E, 3F), indicating that Alt-induced lung type 2 cytokine responses were dependent on the presence of IL-33. There was no difference in IL-5 and IL-13 production in the lung between Indo group and Veh group in IL-33 KO mice (Fig. 3E, 3F), indicating that COX inhibition did not increase type 2 cytokine responses in the absence of IL-33.
COX inhibition increased recombinant IL-33-induced lung type 2 responses
To further investigate the role of IL-33 in COX inhibition-augmented lung type 2 responses, we treated BALB/c mice with Indo or Veh and challenged mice intranasally for 4 consecutive days with mouse rIL-33 to test the hypothesis that COX inhibition increases rIL-33-induced lung type 2 responses. We found that Indo significantly increased not only IL-33-induced IL-5 and IL-13 production (Fig. 3G, 3H), but also rIL-33-induced ILC2 numbers and IL-5 and IL-13 single positive and double positive cell numbers in the lung (Fig. 3I–3L). These data suggest that COX inhibition with Indo also increases type 2 cytokine responses and ILC2 functions by affecting the pathways downstream of IL-33 release.
COX inhibition increased Alt-induced ROS production in the lung
It has been previously reported that Alt-induced IL-33 release was dependent on oxidative stress in the lung.(12) We therefore hypothesized that COX inhibition increases ROS production in the lung and tested this hypothesis with L-012, a luminol-based chemiluminescent probe, to detect ROS in vivo. We found that mice in Alt-Veh group had significantly increased ROS signals than those in HIA-Veh group, indicating that Alt challenge induced ROS production in the lung (Fig. 4). Indo treatment significantly increased ROS levels in Alt-challenged lungs (Fig. 4, Veh-Alt vs. Indo-Alt), suggesting that COX inhibition augments IL-33 release by promoting ROS production. Uchida and colleagues reported that either L-glutathione (GSH) or N-acetyl-L-cysteine (NAC) mixed with Alt for mouse airway challenge decreased Alt-induced IL-33 release in BALF (12). We therefore sought to determine whether GSH and NAC can change the effect of COX inhibition on IL-33 release. Because GSH and cysteine may inactivate enzymes by their disulfide,(34) we hypothesized that GSH and NAC directly inactivate Alt proteases. We found that mixing Alt with either GSH or NAC dose-dependently decreased the protease activity of Alt (Supplemental Fig. 4F-4G), indicating direct inactivation of Alt protease by GSH or NAC. Because Alt proteases are essential for the induction of IL-33 release(28), we did not further use GSH or NAC as ROS scavengers to determine their effect on Alt-induced IL-33 release.
Figure 4.
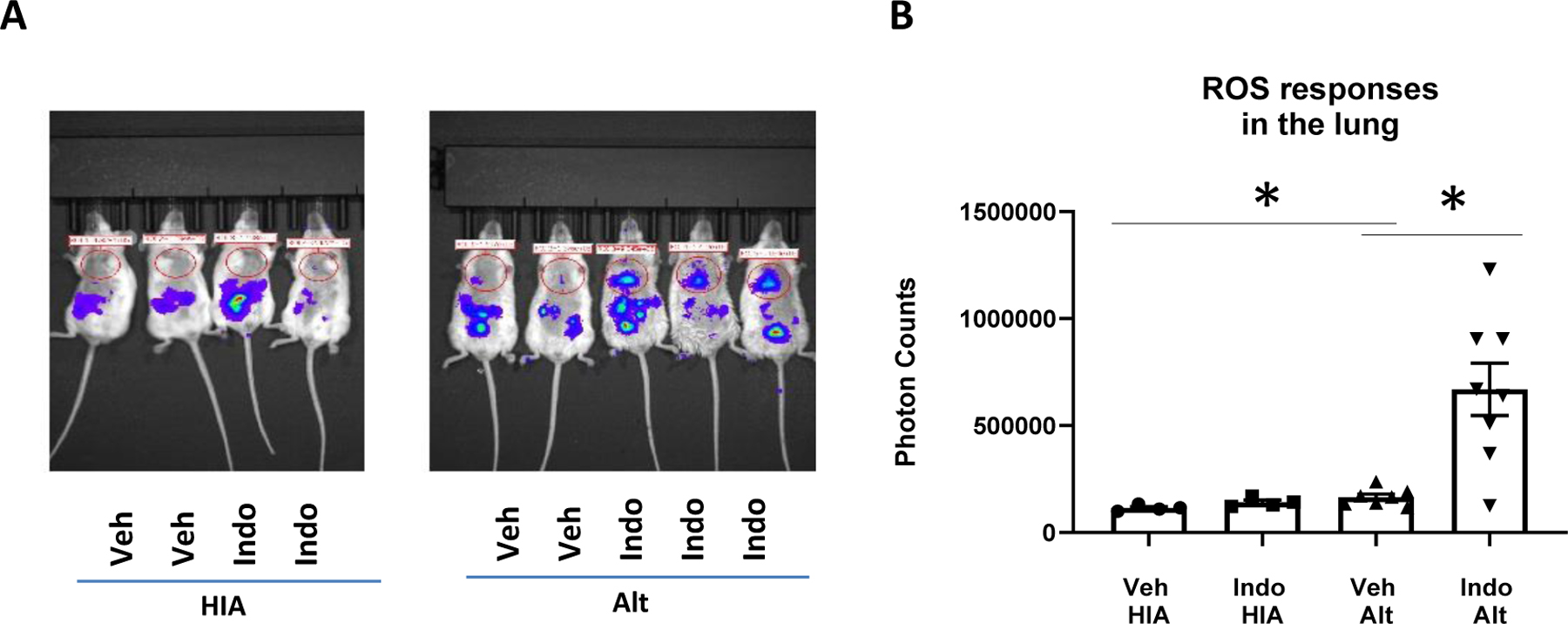
COX inhibition increased reactive oxygen species (ROS) production. WT BALB/c mice were treated with Indo or Veh and challenged intranasally with one dose of HIA or Alt. The mice were retro-orbitally injected with a chemiluminescent ROS prob, L-012, at 60 min after HIA or Alt challenge, and the ROS signal was captured and imaged with a small animal bioluminescence imager 10 min after L-012 injection. (A) Mouse images of ROS responses. (B) Quantitation of the ROS signals. Combined data of 2 independent experiments were shown. * p<0.05, n=4–8 mice.
COX inhibition increased Alt-induced IL-33 release independent of 5-LO expression
Inhibition of the COX pathway by COX inhibitors may cause increased activity of the 5-LO pathway and CysLTs production.(35) We therefore measured the levels of CysLTs in BALF harvested 1 h after Alt challenge. We found that Indo significantly increased CysLTs production in Alt-challenged mice (Fig. 5A). To determine the impact of the 5-LO pathway and the increase of CysLTs that occurred with Indo treatment on Alt-induced IL-33 release, we used WT and 5-LO KO mice and treated the mice with Indo or Veh in drinking water and challenged the mice with Alt for BALF harvest at 1 h after challenge. We found that Indo increased IL-33 release in both WT and 5-LO KO mice (Fig. 5B, 5C), indicating that the effect of Indo on Alt-induced IL-33 release was not dependent on the 5-LO pathway and leukotriene production.
Figure 5.

COX inhibition increased CysLT release in WT mice and augmented IL-33 release in WT and 5-LO KO mice. WT BALB/c and 5-LO KO mice were treated with Indo or Veh and challenged intranasally with one dose of HIA or Alt. Mouse BALF was harvested 1h after the challenge for CysLT and IL-33 ELISA. (A) The levels of CysLTs. (B) IL-33 levels in WT mice. (C) IL-33 levels in 5-LO KO mice. Combined data of 2 independent experiments were shown. * p<0.05, n=3–16 mice.
COX inhibition with flurbiprofen increased Alt-induced ILC2 responses
To further determine the effect of COX inhibition on Alt-induced lung ILC2 responses, we used the non-selective COX inhibitor flurbiprofen as a comparison with Indo for in vivo experiments. We found that, like Indo, flurbiprofen significantly increased IL-33 release to BALF at 1 h after Alt challenge (Fig. 6A) and elevated the levels of IL-5 and IL-13 production in the lung homogenate (Fig. 6B and 6C). Although neither Indo or flurbiprofen changed the total numbers of live cells in the lung (Fig. 6D), both COX inhibitors increased total numbers of ILC2, IL-13 single positive ILC2, and IL-5/IL-13 double positive ILC2 in the lung, compared to Veh control (Fig. 6E–6G). Taken together, these results indicate that COX inhibition promotes ILC2 responses and innate type 2 cytokine production in the lung.
Figure 6.
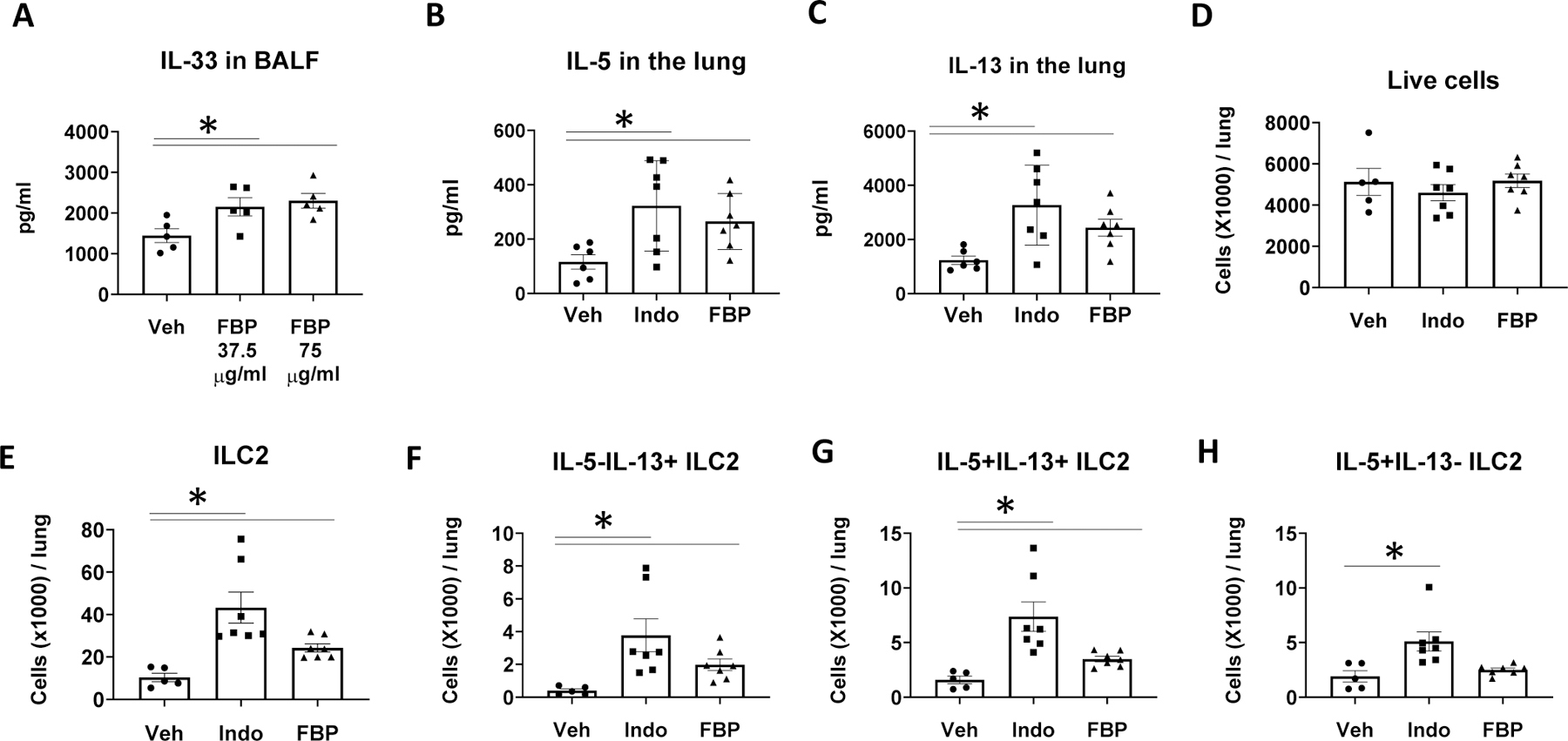
COX inhibition with flurbiprofen increased ILC2 and type 2 cytokine responses. WT BALB/c mice were treated with Indo, flurbiprofen (FBP, 75 μg/ml, B–H) or Veh in drinking water and challenged intranasally with Alt (A) once, or (B-H) daily for 4 days. Mouse lungs were harvested (A) 1 h after Alt challenge for ELISA, or (B–H) 24 h after the last Alt challenge for ELISA and flow cytometry. (A) IL-33 levels in BALF. (B)–(C) IL-5 and IL-13 levels in the lung homogenate. (D) Total numbers of live cells. (E–H) The numbers of ILC2 (Lin−CD90+KLRG1+ST2+ cells), IL-5−IL-13+ ILC2, IL-5+IL-13+ ILC2, and IL-5+IL-13− ILC2 in the lung. * p<0.05, n=5–7 mice per group.
COX products inhibit Alt-induced human IL-33 release in vitro
Immortalized human epithelial cells (hBE33) that were genetically modified to express high levels of human IL-33 were used to study Alt-induced IL-33 release in vitro.(12) To study the mechanism by which COX inhibition increases IL-33 release, we treated hBE33 cells with Indo or Veh for 4 h followed by challenging the cells with Alt or HIA. Fifteen min after Alt challenge, we harvested the culture supernatant for IL-33 measurements. Indo did not significantly change Alt-induced IL-33 release by hBE33 and the levels of IL-33 were 996±129 pg/ml (Veh) and 883±220 pg/ml (Indo at 10 μM). We then hypothesized that exogenous COX products such as PGE2 and PGI2 suppress Alt-induced IL-33 release by hBE33 cells in vitro. We found that PGE2, the PGI2 analogs cicaprost and iloprost, and the cAMP-elevating agent forskolin dose-dependently decreased IL-33 release from Alt-treated hBE33 cells compared to vehicle treatments (Fig. 7A–7D). The decreased IL-33 release was correlated with the lower levels of LDH in the cell culture supernatant (Fig. 7E–7F), suggesting that PGE2, cicaprost, iloprost, and forskolin negatively affected IL-33 release by attenuating Alt-induced cell damage.
Figure 7.
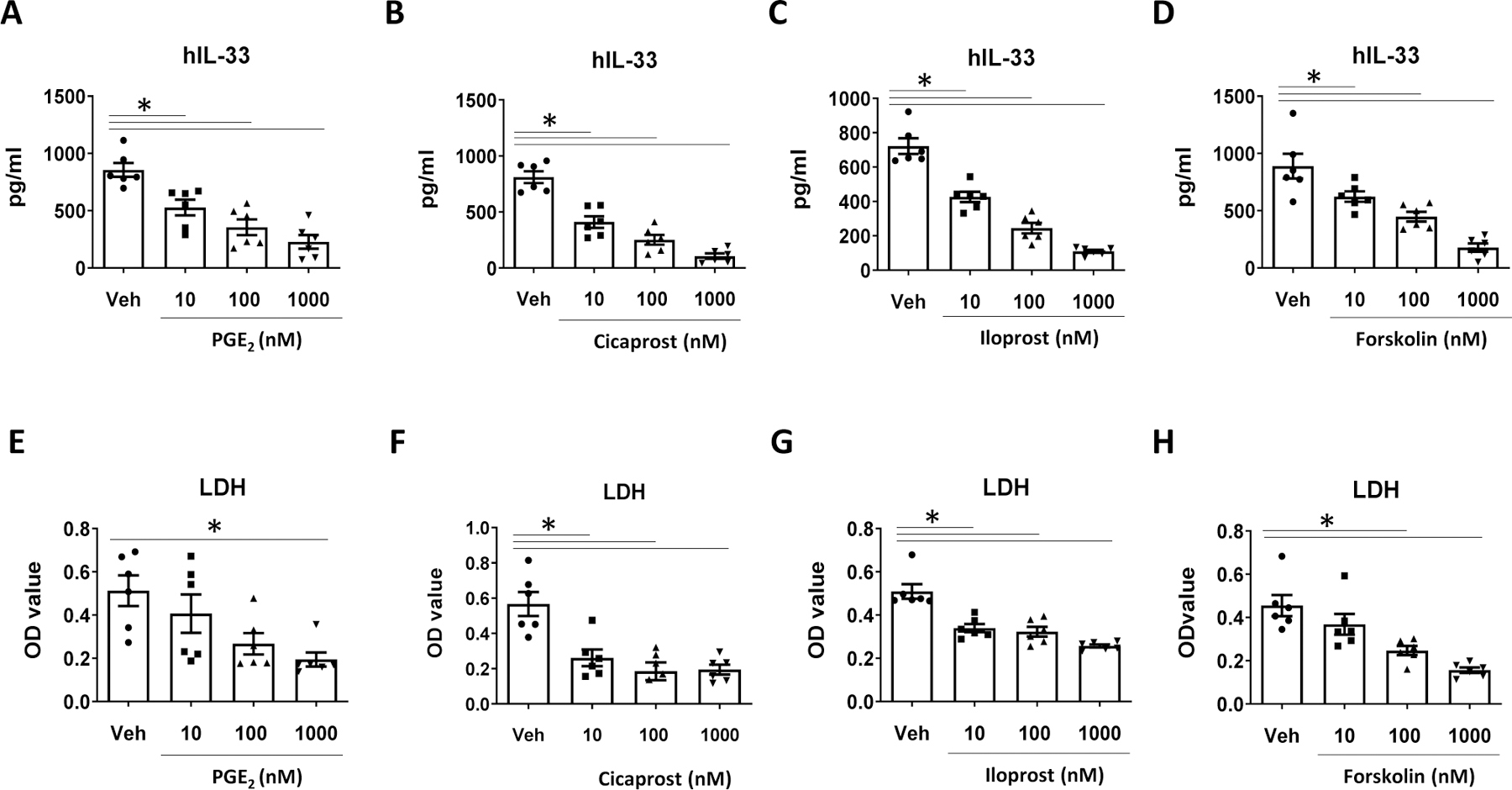
COX products inhibited IL-33 release from human bronchial epithelial (hBE33) cells. hBE33 cells were treated with PGE2, cicaprost, iloprost, and the cyclic AMP-elevating agent forskolin at 10 nM, 100 nM, or 1000 nM or respective vehicles for 30 min followed by treatment with Alt (30 µg/ml) for 15 min. The levels of (A–D) human IL-33 and (E–H) LDH in the culture supernatant. Combined data of 2 independent experiments were shown. * p<0.05, n=6 wells.
DISCUSSION
A number of studies have shown diverse or opposing effects of individual COX products (PGD2, PGE2, and PGI2) on ILC2 responses.(7, 15, 22–25) In this study, we show that nonselective COX inhibition with Indo augmented Alt-induced ILC2 proliferation, activation and IL-5 and IL-13 expression. The increased ILC2 type 2 cytokine responses correlated with increased eosinophil percentages in BALF and augmented airway mucus. Alt-induced type 2 cytokine responses and the effect of COX inhibition were absent in IL-33 KO mice, indicating an essential role of IL-33 in this model. We found that Indo-increased ILC2 responses correlated with the elevated levels of IL-33 release, increased cell damage, and heightened ROS production. These results suggest that Indo-augmented ILC2 responses are at least partially mediated by promoting Alt-induced epithelial cell damage, reactive oxidant stress, and IL-33 release for ILC2 activation and IL-5 and IL-13 production.
COX inhibition with Indo increased rIL-33-stimulated lung ILC2 activation and type 2 cytokine responses, suggesting that COX inhibition also acts on the biological processes downstream of IL-33 release to increase ILC2 responses. Our group and others showed that PGE2 and PGI2 signaling directly inhibited ILC2 responses in Alt-induced airway inflammation,(7, 24, 25) while PGD2 increased ILC2 responses by inducing ILC2 chemotaxis and priming ILC2 type 2 cytokine responses to IL-33 and IL-25.(20–22) By suppressing PGE2 and PGI2 production, COX inhibition could increase ILC2 activation and proliferation, and augment IL-5 and IL-13 production. We show that Alt challenge in the lung increased PGE2 and PGI2 levels in the mouse urine, indicating an activation of the COX pathway. Production of PGE2 and PGI2 was increased in Alt-challenged lungs, which is consistent with the expression of microsomal PGE2 synthase in epithelial and endothelial cells, and PGI synthase in endothelial cells.(36, 37) Conversely, Indo increased CysLT levels in the BALF, suggesting that COX inhibition alleviates PGE2-mediated suppression of 5-LO function.(38) COX inhibition increased IL-33 release in both WT and 5-LO KO mice, suggesting that Indo-augmented IL-33 release is not dependent on the 5-LO pathway and the downstream CysLT products. Since CysLTs promoted ILC2 chemotaxis and potentiated ILC2 for IL-33 responses, CysLTs may also play a role in Indo-augmented ILC2 responses in the four-day Alt challenge model.
COX inhibition promoted Alt-induced cell damage and IL-33 release, and the effect could be mediated by attenuating the cytoprotective effects of COX products. IL-33 release happens when IL-33-expressing cells undergo cellular damage or necrotic cell death. Airway exposure of mice with Alt caused epithelial damage and induced exfoliation of necrotic airway epithelial cells in BALF within 30 min (11) and IL-33 release peaking at 1 h in BALF.(14) It appears that Alt-induced cell damage is the major mechanism of IL-33 release and this is consistent with our data showing an association between the LDH levels and IL-33 release in vivo and in vitro. The cytoprotective effect of PGE2 and/or PGI2 has been documented in studies on gastric mucosal cells.(39) Mechanisms proposed for the cytoprotective effect of PGE2 and/or PGI2 include stimulation of mucin secretion and the sodium pump function, activation of adenylyl cyclase, and protection of gastric mucosal barrier.(39) PGE2 and PGI2 also had a barrier-protective function on pulmonary endothelium and the effect was mediated by PKA and Epac/Rap pathways.(40, 41) In our study, PGE2 and PGI2 decreased Alt-induced LDH release by hBE33 cells, indicating a cytoprotective effect of these PGs. The cAMP elevating agent forskolin had similar cytoprotective effect on hBE33 cells and decreased IL-33 release, indicating an important role of the cAMP pathway in Alt-induced IL-33 release. Therefore, we propose that by suppressing the production of PGE2 and PGI2, Indo attenuates the cytoprotective functions of the lipid molecules, and enhances pulmonary epithelial and endothelial cell damage and IL-33 release. Although TSLP can enhance IL-33-induced ILC2 responses,(42–45) COX inhibition caused a significant decrease of TSLP expression in the lung, suggesting that TSLP does not play a significant role in Indo-augmented ILC2 responses.
We demonstrated that COX inhibition increased ROS levels that were correlated with IL-33 release. Alt proteases induced ROS formation and ATP release, leading to IL-33 release from cultured epithelial cells in vitro.(12, 14, 28, 46) Alt-induced oxidative stress has been shown to be critical for IL-33 release by airway epithelium in vivo. Indo augmented Alt-induced ROS production in the lung, which is consistent with previously published results showing that Indo induced ROS generation and lipid peroxidation in gastric mucosa and inhibited the activity of glutathione peroxidase.(47, 48) Therefore, the augmented ROS production appears to be one of mechanisms responsible for the increased cell damage and IL-33 release in Indo-treated mice. Uchida and colleagues showed that ROS scavengers including GSH and NAC dose-dependently reduced IL-33 release when the mice were challenged with the mixed solution of either Alt and GSH or Alt and NAC.(12) In our study, when GSH and NAC were mixed with Alt, they caused direct inactivation of Alt proteases. Because Alt proteases are essential for the induction of IL-33 release, these results suggest that the reduction of IL-33 release in the mice challenged with GSH/Alt or NAC/Alt could be a result of direct inactivation of Alt protease by GSH or NAC, rather than a protective effect of GSH or NAC on airway epithelial cells.
Flurbiprofen is a non-selective COX inhibitor with similar therapeutic function as indomethacin on relieving pain and stiffness of rheumatoid arthritis.(49) We found that flurbiprofen promoted IL-33 release in BALF, augmented IL-5 and IL-13 production in the lung, and increased total number of ILC2 and IL-5 and IL-13 expression by ILC2 in Alt-induced lung inflammation. The similar effects of flurbiprofen and indomethacin strongly support that COX inhibition increases Alt-induced ILC2 responses and type 2 cytokine production in this mouse model of innate lung inflammation. In a study of human ILC2 culture in vitro, Maric and colleagues reported that flurbiprofen significantly inhibited IL-5 and IL-13 secretion from ILC2 activated by a cytokine mixture of IL-33, TSLP, and IL-25.(50) They demonstrated that human ILC2 produced PGD2 upon cytokine activation and the endogenously produced PGD2 was crucial for the cytokine-induced activation of freshly isolated blood and tonsillar ILC2.(50) The suppressive function of flurbiprofen on human ILC2 activation was mediated by inhibiting ILC2 PGD2 production.(50) The opposing effects of flurbiprofen on mouse lung ILC2 in vivo and human ILC2 in vitro suggest that the microenvironmental milieu of ILC2, especially the presence or absence of PGE2 and PGI2, plays a role in the effects of COX inhibition. Our findings are supported by a very recent publication by Maruyama and colleagues who published that NSAID treatment heightened innate-type allergic responses to inhalation of papain or rIL-33.(51)
In summary, we have shown that COX inhibition increased IL-33 release and augmented lung ILC2 responses and type 2 inflammation after repeated Alt exposure in mice. While PGD2 augmented ILC2 responses,(20) and both PGE2 and PGI2 suppressed ILC2,(7, 24) our findings suggest an overall restraining function of COX products on protease-containing allergen-induced airway type 2 responses. The stimulatory effect of COX inhibition on fungal allergen associated- and ILC2-mediated innate type 2 responses provides an important addition to our previous discoveries of COX inhibition-augmented allergen-specific adaptive immune responses. PGE2 and PGI2 that are FDA approved drugs for labor induction and pulmonary hypertension, respectively, appear to have potential as therapies for allergic diseases and asthma.
Supplementary Material
Key points:
COX inhibition increased Alternaria extract-incuded pulmonary ILC2 responses. COX inhibition promoted Alternaria extract-incuded IL-33 release in the lung. COX inhibition augmented recombinant IL-33-induced pulmonary ILC2 responses.
Acknowledgments
Grant supports:
This work was supported by the National Institute of Health (R01 AI145265, R01 AI124456, R21 AI145397, U19 AI 95227, R01 AI 111820, T32 GM 007347, F30 AI118376–01, R56 AI076411), and the Department of Veterans Affairs (I01BX004299).
Abbreviations used
- 5-LO
5-lipoxygenase
- Alt
Alternaria alternata extract
- BALF
Bronchoalveolar lavage fluid
- COX
Cyclooxygenase
- CysLTs
Cysteinyl leukotrienes
- GSH
Reduced L-glutathione
- hBE33
Immortalized human bronchial epithelial cells
- HIA
Heat inactivated Alternaria extract
- HMGB1
High mobility group box protein 1
- HDM
House dust mite
- ILC2
Group 2 innate lymphoid cells
- Indo
Indomethacin
- KO
Knockout
- L-012
A luminol-based chemiluminescent probe
- LDH
Lactate Dehydrogenase
- NAC
N-acetyl-L-cysteine
- ROS
Reactive oxygen species
- TSLP
Thymic stromal lymphopoietin
- WT
Wild type
REFERENCES
- 1.Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, Bucks C, Wu X, Kane CM, Neill DR, Flynn RJ, Sayers I, Hall IP, and McKenzie AN. 2013. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J.Allergy Clin.Immunol 132: 933–941. [DOI] [PubMed] [Google Scholar]
- 2.Gronke K, and Diefenbach A. 2016. Tuft cell-derived IL-25 activates and maintains ILC2. Immunol.Cell Biol 94: 221–223. [DOI] [PubMed] [Google Scholar]
- 3.Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, and Artis D. 2013. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci.Transl.Med 5: 170ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, and Takei F. 2014. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halim TY, Krauss RH, Sun AC, and Takei F. 2012. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 36: 451–463. [DOI] [PubMed] [Google Scholar]
- 6.Li BW, de Bruijn MJ, Tindemans I, Lukkes M, KleinJan A, Hoogsteden HC, and Hendriks RW. 2016. T cells are necessary for ILC2 activation in house dust mite-induced allergic airway inflammation in mice. Eur.J.Immunol 46: 1392–1403. [DOI] [PubMed] [Google Scholar]
- 7.Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, Dulek DE, Bloodworth MH, Stier MT, Polosuhkin V, Gangula RD, Mallal SA, Broide DH, and Peebles RS Jr. 2016. Prostaglandin I2 Signaling and Inhibition of Group 2 Innate Lymphoid Cell Responses. Am J Respir Crit Care Med 193: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, Vaka D, Eckalbar WL, Molofsky AB, Erle DJ, and Locksley RM. 2018. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol 19: 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, and Girard JP. 2007. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A 104: 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, and Takei F. 2014. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murai H, Qi H, Choudhury B, Wild J, Dharajiya N, Vaidya S, Kalita A, Bacsi A, Corry D, Kurosky A, Brasier A, Boldogh I, and Sur S. 2012. Alternaria-induced release of IL-18 from damaged airway epithelial cells: an NF-kappaB dependent mechanism of Th2 differentiation? PLoS.One 7: e30280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uchida M, Anderson EL, Squillace DL, Patil N, Maniak PJ, Iijima K, Kita H, and O’Grady SM. 2017. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy 72: 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, van d. V.. 2016. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J.Allergy Clin.Immunol 137: 1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, and Kita H. 2011. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J.Immunol 186: 4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, and Broide DH. 2013. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J.Allergy Clin.Immunol 132: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu T, Barrett NA, Kanaoka Y, Yoshimoto E, Garofalo D, Cirka H, Feng C, and Boyce JA. 2018. Type 2 Cysteinyl Leukotriene Receptors Drive IL-33-Dependent Type 2 Immunopathology and Aspirin Sensitivity. J Immunol 200: 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salimi M, Stöger L, Liu W, Go S, Pavord I, Klenerman P, Ogg G, and Xue L. 2017. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J Allergy Clin Immunol 140: 1090–1100.e1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lund SJ, Portillo A, Cavagnero K, Baum RE, Naji LH, Badrani JH, Mehta A, Croft M, Broide DH, and Doherty TA. 2017. Leukotriene C4 Potentiates IL-33-Induced Group 2 Innate Lymphoid Cell Activation and Lung Inflammation. J Immunol 199: 1096–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, Wechsler ME, Israel E, and Levy BD. 2013. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci.Transl.Med 5: 174ra126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, Willis C, Budelsky A, Farber DL, and Artis D. 2015. The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol 8: 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang JE, Doherty TA, Baum R, and Broide D. 2014. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J.Allergy Clin.Immunol 133: 899–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, Klenerman P, and Ogg G. 2014. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J.Allergy Clin.Immunol 133: 1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikutani M, Tsuneyama K, Kawaguchi M, Fukuoka J, Kudo F, Nakae S, Arita M, Nagai Y, Takaki S, and Takatsu K. 2017. Prolonged activation of IL-5-producing ILC2 causes pulmonary arterial hypertrophy. JCI Insight 2: e90721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Wang W, Zhao C, Wang Y, Wu H, Sun X, Guan Y, and Zhang Y. 2018. Prostaglandin E2 Inhibits Group 2 Innate Lymphoid Cell Activation and Allergic Airway Inflammation Through E-Prostanoid 4-Cyclic Adenosine Monophosphate Signaling. Front Immunol 9: 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maric J, Ravindran A, Mazzurana L, Björklund Å, Van Acker A, Rao A, Friberg D, Dahlén SE, Heinemann A, Konya V, and Mjösberg J. 2018. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J Allergy Clin Immunol 141: 1761–1773.e1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoenfeld P, Kimmey MB, Scheiman J, Bjorkman D, and Laine L. 1999. Review article: nonsteroidal anti-inflammatory drug-associated gastrointestinal complications--guidelines for prevention and treatment. Aliment Pharmacol Ther 13: 1273–1285. [DOI] [PubMed] [Google Scholar]
- 27.Hardman CS, Panova V, and McKenzie AN. 2013. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur.J.Immunol 43: 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snelgrove RJ, Gregory LG, Peiró T, Akthar S, Campbell GA, Walker SA, and Lloyd CM. 2014. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol 134: 583–592.e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kielland A, Blom T, Nandakumar KS, Holmdahl R, Blomhoff R, and Carlsen H. 2009. In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radic Biol Med 47: 760–766. [DOI] [PubMed] [Google Scholar]
- 30.Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, Pham A, Miller M, Croft M, and Broide DH. 2012. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. J Immunol 188: 2622–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Barrett NA, Kanaoka Y, Buchheit K, Laidlaw TM, Garofalo D, Lai J, Katz HR, Feng C, and Boyce JA. 2019. Cysteinyl leukotriene receptor 2 drives lung immunopathology through a platelet and high mobility box 1-dependent mechanism. Mucosal Immunol 12: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang J, Xia Y, Wasserloos K, Deng M, Blose KJ, Vorp DA, Turnquist HR, Billiar TR, Pitt BA, Zhang MZ, and Zhang LM. 2017. Cyclic stretch induced IL-33 production through HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells. PLoS One 12: e0184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, and Vivier E. 2013. Innate lymphoid cells--a proposal for uniform nomenclature. Nat.Rev.Immunol 13: 145–149. [DOI] [PubMed] [Google Scholar]
- 34.Offermann MK, McKay MJ, Marsh MW, and Bond JS. 1984. Glutathione disulfide inactivates, destabilizes, and enhances proteolytic susceptibility of fructose-1,6-bisphosphate aldolase. J Biol Chem 259: 8886–8891. [PubMed] [Google Scholar]
- 35.Peebles RS Jr., Hashimoto K, Morrow JD, Dworski R, Collins RD, Hashimoto Y, Christman JW, Kang KH, Jarzecka K, Furlong J, Mitchell DB, Talati M, Graham BS, and Sheller JR. 2002. Selective cyclooxygenase-1 and −2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am.J.Respir.Crit Care Med 165: 1154–1160. [DOI] [PubMed] [Google Scholar]
- 36.Jabbour HN, Milne SA, Williams AR, Anderson RA, and Boddy SC. 2001. Expression of COX-2 and PGE synthase and synthesis of PGE(2)in endometrial adenocarcinoma: a possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. Br.J.Cancer 85: 1023–1031. [DOI] [PubMed] [Google Scholar]
- 37.Camacho M, Rodriguez C, Salazar J, Martinez-Gonzalez J, Ribalta J, Escudero JR, Masana L, and Vila L. 2008. Retinoic acid induces PGI synthase expression in human endothelial cells. J.Lipid Res 49: 1707–1714. [DOI] [PubMed] [Google Scholar]
- 38.Liu T, Laidlaw TM, Katz HR, and Boyce JA. 2013. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A 110: 16987–16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robert A 1979. Cytoprotection by prostaglandins. Gastroenterology 77: 761–767. [PubMed] [Google Scholar]
- 40.Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, and Birukov KG. 2007. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp.Cell Res 313: 2504–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoshino T, Tsutsumi S, Tomisato W, Hwang HJ, Tsuchiya T, and Mizushima T. 2003. Prostaglandin E2 protects gastric mucosal cells from apoptosis via EP2 and EP4 receptor activation. J.Biol.Chem 278: 12752–12758. [DOI] [PubMed] [Google Scholar]
- 42.Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, Betsuyaku T, Koyasu S, and Asano K. 2013. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat Commun 4: 2675. [DOI] [PubMed] [Google Scholar]
- 43.Han M, Rajput C, Hong JY, Lei J, Hinde JL, Wu Q, Bentley JK, and Hershenson MB. 2017. The Innate Cytokines IL-25, IL-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J Immunol 199: 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, Good J, Ito Y, Chu H, Gorska MM, Martin RJ, and Alam R. 2018. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J Allergy Clin Immunol 141: 257–268.e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toki S, Goleniewska K, Zhang J, Zhou W, Newcomb DC, Zhou B, Kita H, Boyd KL, and Peebles RS. 2020. TSLP and IL-33 reciprocally promote each other’s lung protein expression and ILC2 receptor expression to enhance innate type-2 airway inflammation. Allergy DOI: 10.1111/all.14196 [DOI] [PMC free article] [PubMed]
- 46.Yee MC, Nichols HL, Polley D, Saifeddine M, Pal K, Lee K, Wilson EH, Daines MO, Hollenberg MD, Boitano S, and DeFea KA. 2018. Protease-activated receptor-2 signaling through β-arrestin-2 mediates Alternaria alkaline serine protease-induced airway inflammation. Am J Physiol Lung Cell Mol Physiol 315: L1042–L1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maity P, Bindu S, Dey S, Goyal M, Alam A, Pal C, Mitra K, and Bandyopadhyay U. 2009. Indomethacin, a non-steroidal anti-inflammatory drug, develops gastropathy by inducing reactive oxygen species-mediated mitochondrial pathology and associated apoptosis in gastric mucosa: a novel role of mitochondrial aconitase oxidation. J.Biol.Chem 284: 3058–3068. [DOI] [PubMed] [Google Scholar]
- 48.Yoshikawa T, Naito Y, Kishi A, Tomii T, Kaneko T, Iinuma S, Ichikawa H, Yasuda M, Takahashi S, and Kondo M. 1993. Role of active oxygen, lipid peroxidation, and antioxidants in the pathogenesis of gastric mucosal injury induced by indomethacin in rats. Gut 34: 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Moor M, and Ooghe R. 1981. A double-blind comparison of flurbiprofen and indomethacin suppositories in the treatment of osteoarthrosis and rheumatoid disease. J Int Med Res 9: 495–500. [DOI] [PubMed] [Google Scholar]
- 50.Maric J, Ravindran A, Mazzurana L, Van Acker A, Rao A, Kokkinou E, Ekoff M, Thomas D, Fauland A, Nilsson G, Wheelock CE, Dahlén SE, Ferreirós N, Geisslinger G, Friberg D, Heinemann A, Konya V, and Mjösberg J. 2019. Cytokine-induced endogenous production of prostaglandin D2 is essential for human group 2 innate lymphoid cell activation. J Allergy Clin Immunol 143: 2202–2214.e2205. [DOI] [PubMed] [Google Scholar]
- 51.Maruyama N, Takai T, Kamijo S, Suchiva P, Ohba M, Takeshige T, Suzuki M, Hara M, Matsuno K, Harada S, Harada N, Nakae S, Sudo K, Okuno T, Yokomizo T, Ogawa H, Okumura K, and Ikeda S. 2019. Cyclooxygenase inhibition in mice heightens adaptive- and innate-type responses against inhaled protease allergen and IL-33. Allergy 74: 2237–2240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.