

Abstract
The natural products combretastatin A-1 (CA1) and combretastatin A-4 (CA4) function as potent inhibitors of tubulin polymerization and as selective vascular disrupting agents (VDAs) in tumors. Bioreductively activatable prodrug conjugates (BAPCs) can enhance selectivity by serving as substrates for reductase enzymes specifically in hypoxic regions of tumors. A series of CA1-BAPCs incorporating nor-methyl, mono-methyl, and gem-dimethyl nitrothiophene triggers were synthesized together with corresponding CA4-BAPCs, previously reported by Davis (Mol. Cancer Ther. 2006, 5 (11), 2886), for comparison. The CA4-gem-dimethylnitrothiophene BAPC (45) proved exemplary in comparison to its nor-methyl (43) and mono-methyl (44) congeners. It was stable in phosphate buffer (pH 7.4, 24 h), was cleaved (25%, 90 min) by NADPH-cytochrome P450 oxidoreductase (POR), was inactive (desirable prodrug attribute) as an inhibitor of tubulin polymerization (IC50 > 20 μM), and demonstrated hypoxia-selective activation in the A549 cell line [hypoxia cytotoxicity ratio (HCR) = 41.5]. The related CA1-gem-dimethylnitrothiophene BAPC (41) was also promising (HCR = 12.5) with complete cleavage (90 min) upon treatment with POR. In a preliminary in vivo dynamic bioluminescence imaging (BLI) study, BAPC 45 (180 mg/kg, IP) induced a decrease (within 4 h) in light emission in a 4T1 syngeneic mouse breast tumor model, implying activation and vascular disruption.
Graphical Abstract
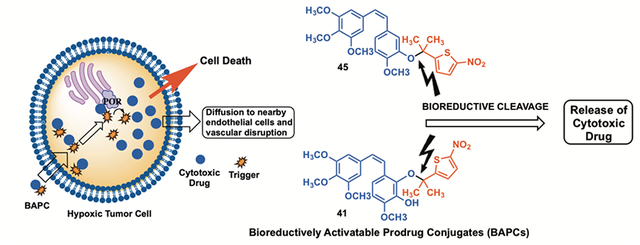
The tumor microenvironment exhibits unique attributes, notably associated with vascular architecture and associated blood flow dynamics.1–3 Solid tumors, once they reach approximately 2–5 mm3 in size, must establish their own vascular network in order to meet their rapidly accelerating demand for oxygen and nutrients.4–10 This rapid angiogenic development of tumor-associated vasculature results in disorganized, fragile, and leaky vessels lacking pericyte support, thus providing a target for therapeutic intervention.1–3,11–13 Vascular disrupting agents (VDAs) selectively damage established tumor-associated vasculature, thus denying necessary oxygen and nutrients, resulting in tumor necrosis.2,14–16 VDAs are mechanistically distinct from the established angiogenesis inhibiting agents (AIAs) such as bevacizumab (Avastin™).17,18 The natural products combretastatin A-1 (CA1, Figure 1) and combretastatin A-4 (CA4, Figure 1), isolated from the bark of the African bush willow tree Combretum caffrum (Combretacae) by Pettit and co-workers, are potent inhibitors of tubulin polymerization (binding at the colchicine site) that function biologically as antiproliferative agents and as VDAs.19–22 These agents cause rapid morphological changes to the endothelial cells lining tumor-associated blood vessels, resulting in irreversible vascular damage.1,23,24 The corresponding water-soluble phosphate prodrugs of CA1 and CA4 [referred to as CA1P (or OXi4503) and CA4P (fosbretabulin) respectively, Figure 1)] have advanced through preclinical evaluation and clinical trials.14,21,25–32
Figure 1.
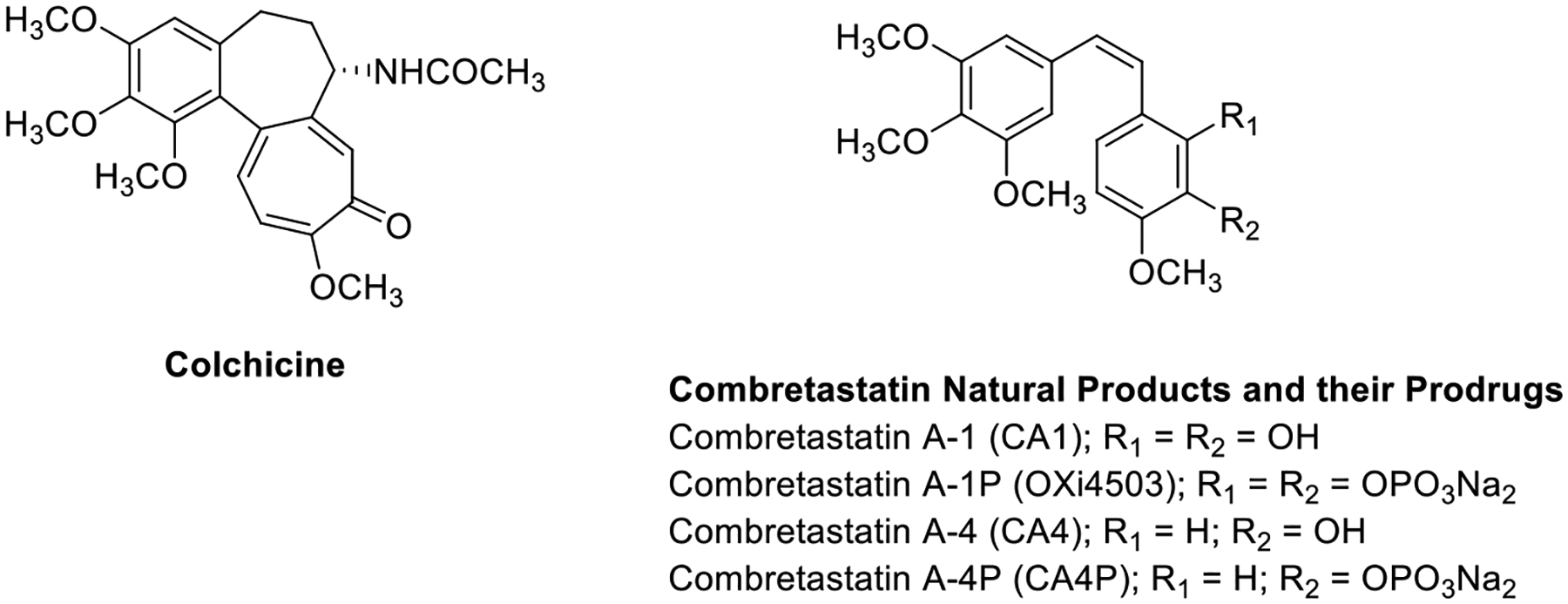
Colchicine and combretastatin natural products and phosphate prodrug salts.
In addition to an aberrant vascular network and elevated interstitial pressure due to immature and leaky vasculature, the tumor microenvironment is often characterized by regions of hypoxia and a pH gradient, with cells distant from blood vessels being in an acidic environment.3,33–37 Hypoxia represents a characteristic uniquely inherent to many solid tumors that does not naturally occur in normal tissue,3 thus offering a specific target based on selective activation of potent anticancer agents or their selective delivery achieved through appropriate prodrug strategies.3,38–41 Hypoxia-selective prodrugs undergo activation through either one- or two-electron reductase enzymes.3,34
One type of BAPC incorporates a bioreductive trigger covalently attached to an appropriate therapeutic agent, which is designed to undergo enzyme-mediated cleavage (to release the parent anticancer agent) under hypoxic conditions (Scheme 1).3,30,34 Davis and co-workers synthesized a series of nor-, mono-, and gem-dimethyl-nitrothienyl BAPCs (Figure S3, Supporting information) that incorporated CA4 and evaluated their efficacy through the generation of radical anions by pulse radiolysis and determination (spectrophotometrically) of their stability and fragmentation.42 They also evaluated these compounds in the presence of NADPH-cytochrome P450 oxidoreductase (POR) and further assessed their efficacy by determining their cytotoxicity (normoxia versus hypoxia) in the A549 human lung cancer cell line.42 It was determined that the gem-dimethyl-nitrothiophene trigger CA4 prodrug (Scheme 1) was the most resistant to aerobic metabolism (in comparison to the nor- and mono-methyl-nitrothiophene trigger CA4 prodrugs), and the gem-dimethyl CA4-BAPC remained intact in high oxygen environments.42 While the gem- and mono-substituted CA4 BAPCs were effective (as evidenced by release of CA4 in the presence of supersomal P450R) across a range of oxygen concentrations, the unsubstituted (nor-methyl) was only effective under extreme hypoxia (<0.01% O2).42
Scheme 1.
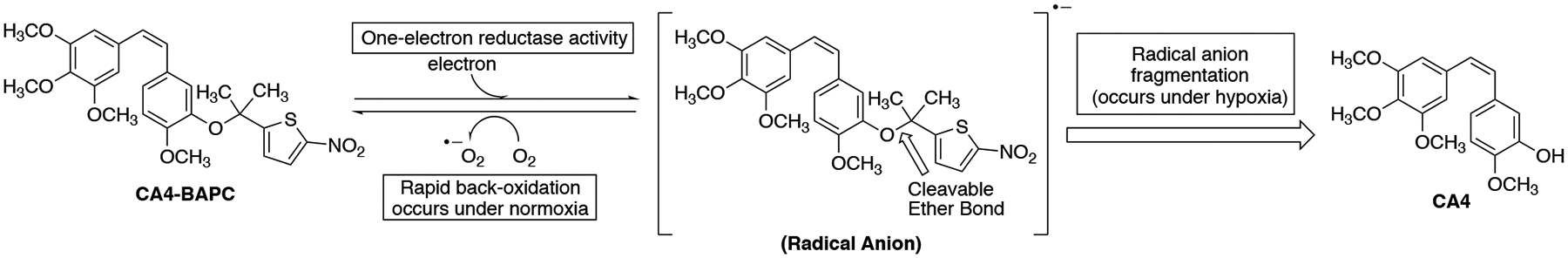
Proposed mechanism for the biological reduction and cleavage of CA4 gemdimethyl-nitrothiophene trigger releasing CA4.42
Inspired by the premise of targeting tumor hypoxia for the selective delivery of tubulin-active VDAs, and building on the encouraging results reported for the CA4-BAPCs, we designed and synthesized a series of BAPCs that incorporate the natural product CA1 and evaluated them in preliminary studies to determine their efficacy as therapeutic agents. A regioselective protecting group strategy (incorporating tert-butyldimethylsilyl, isopropyl, and tosyl groups), which we previously developed as part of a separate synthetic campaign,43,44 was utilized to differentiate the catechol functionality (C-2 and C-3 positions) inherent to CA1. The nitrothiophene triggers previously described by Davis and co-workers42 were synthesized using a revised synthetic strategy.41 The synthesized CA1-BAPCs were evaluated for their ability to inhibit tubulin polymerization and to function as substrates for the reductase enzyme POR. In addition, differential cytotoxicity studies (normoxia versus hypoxia) assessed cell-based (A549 lung cancer) hypoxia-selective activation (evidenced by enhanced cytotoxicity). Collectively these studies were designed to guide the potential therapeutic advancement of the most promising CA1-BAPCs as hypoxia-selective anticancer agents.
RESULTS AND DISCUSSION
Synthesis.
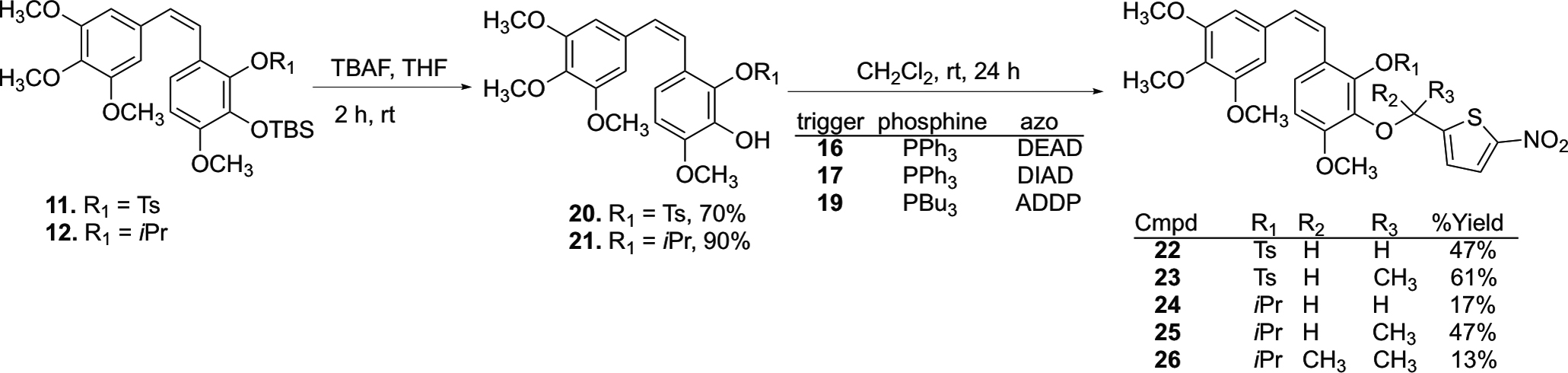
The CA1-BAPCs were synthesized utilizing two key reactions, a Wittig olefination to generate regioselectively protected CA1 followed by reaction between the tosyl, isopropyl, and tert-butyldimethylsilyl protected CA1 analogues (20, 21, 27, and 28 respectively, Schemes 3 and 4) and the nitrothienyl triggers (16, 17 and 19, Scheme S1, Supporting information) under Mitsunobu conditions.45,46 Synthesis of the regioselectively protected Z-CA1 analogues (11–13, Scheme 2) was facilitated by a Wittig olefination reaction between aldehydes 5–7 (Scheme 2) and triphenyl phosphonium salt 10.46 While the Wittig reaction produced a mixture of Z- and E-stilbene isomers, the Z-isomer was formed preferentially (Scheme 2).43,44,46,47
Scheme 3.
Scheme 4.
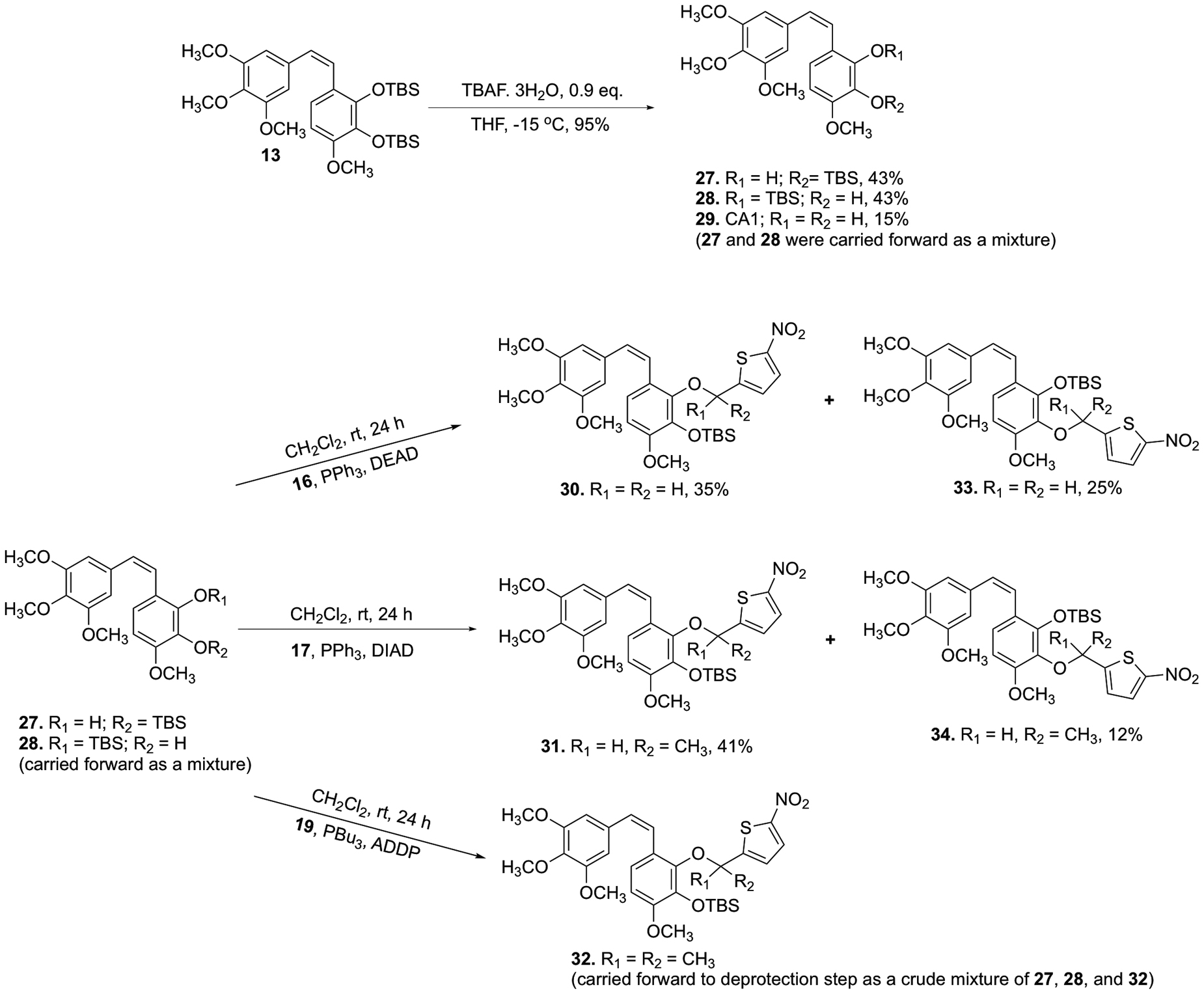
Synthesis of TBS-protected CA1-BAPCs 30–34.
Scheme 2.
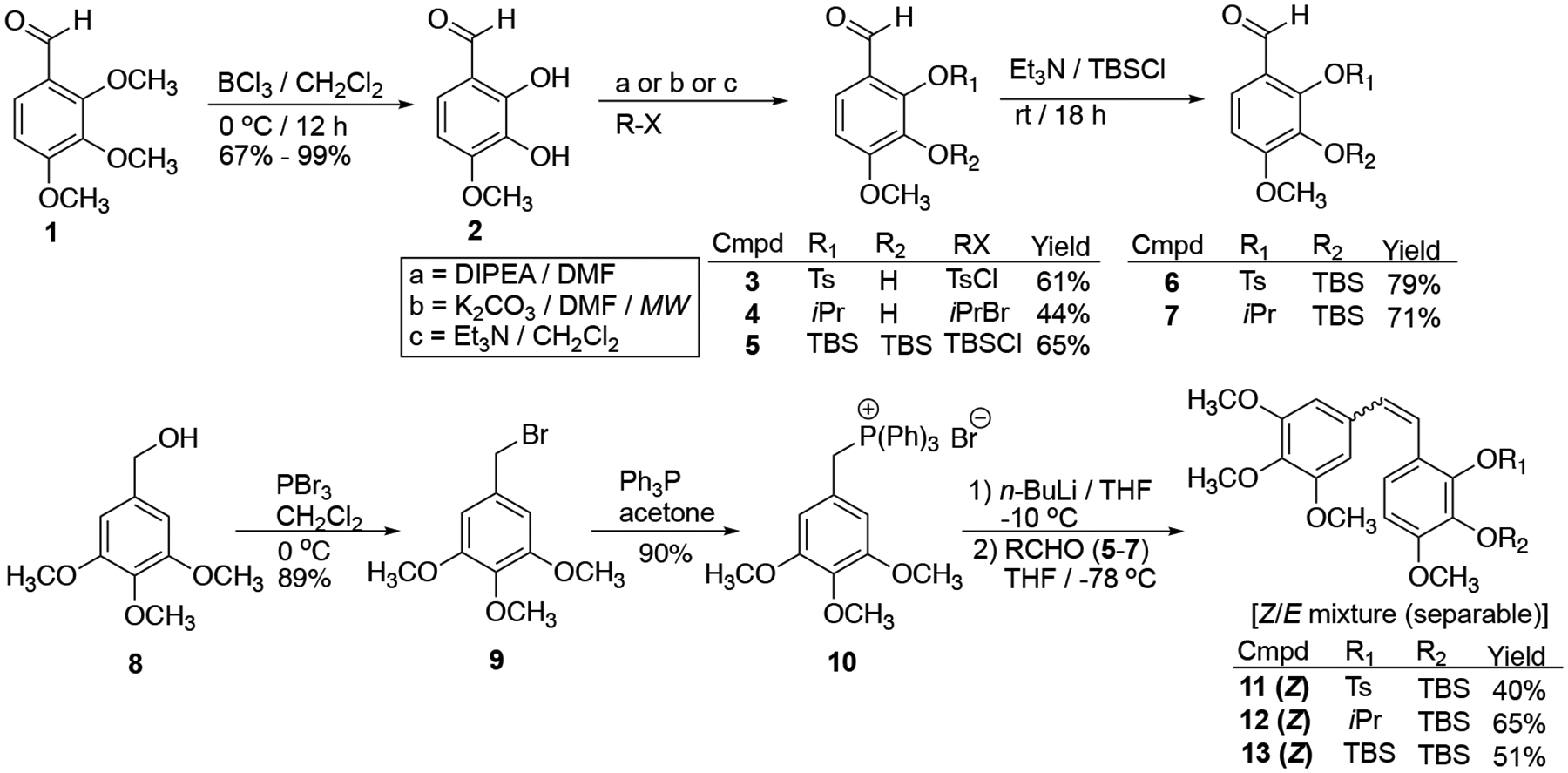
Synthesis of regioselectively protected CA1 derivatives 11–13.32,43
Selective demethylation of aldehyde 1 using BCl3 yielded catechol 2, which was subsequently converted to selectively protected aldehydes 3–7 (Scheme 2) using a previously reported synthetic strategy.43,44,46 Phosphonium salt 10 was prepared upon bromination of benzyl alcohol 8 using PBr3, followed by a reaction with triphenyl phosphine. A Wittig reaction between suitably protected aldehydes (5–7) and Wittig salt 10 yielded a mixture of Z- and E-stilbene isomers (11–13, favoring the Z-isomer), which were separated by flash column chromatography.
Synthesis of the three nitrothiophene triggers (16, 17, 19) utilized in the Mitsunobu reactions was achieved as previously described (Scheme S1, Supporting Information).41,42 Deprotection of CA1 analogues 11 and 12 using TBAF yielded the corresponding phenols 20 and 21, respectively, which were subjected to Mitsunobu conditions that utilized nitrothiophene triggers (16, 17 and 19), phosphine reagents (PPh3 or PBu3), and azo compounds [diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD) or 1,1′-(azodicarbonyl)-dipiperdine (ADDP)] to generate BAPCs 22–26 (Scheme 3).
Attempted deprotection of compounds 22 and 23 with NaOH (2 M) under either microwave or reflux conditions did not yield the desired product, but instead cleaved the nitrothiophene trigger from the starting material to regenerate compound 20 (Scheme S2, Supporting Information). Similarly, compound 24 regenerated compound 21 upon attempted deprotection using AlCl3 (Scheme S2, Supporting Information). In an effort to solve this problem, we attempted to partially cleave the bis-TBS-protected CA1 13 using a deficiency of TBAF, but this resulted in a mixture of regioisomers 27 and 28 (Scheme 4), which proved inseparable by flash column chromatography. This mixture of regioisomers 27 and 28 was further functionalized to incorporate nitrothienyl triggers under Mitsunobu conditions to synthesize regioisomeric TBS/trigger analogues 30–34. The parent natural product CA1 (29) proved unreactive under analogous reaction conditions. The protected CA1-BAPC 32 proved difficult to purify by column chromatography, so the crude product was taken to the next step. Interestingly, the conventional TBS-deprotection of compounds 30 and 31 using TBAF yielded ring-cyclized products 35 and 36 (proposed structures based on analysis of NMR and HRMS data) without producing any other discernable side products (Scheme 5). While a plausible mechanistic explanation for this cyclization has yet to be established, we place a high degree of confidence in the structural assignment for cyclic compounds 35 and 36 based on a combination of 1H-NMR, 13C-NMR, and mass spectrometry data (see Supporting Information for further details). Intrigued by this unusual cyclization (that produced 35 and 36), we investigated whether exposure of phenolic compounds 37 and 38 to strong base would also facilitate a similar cyclization reaction, and this indeed proved to be the case (Scheme 5).
Scheme 5.
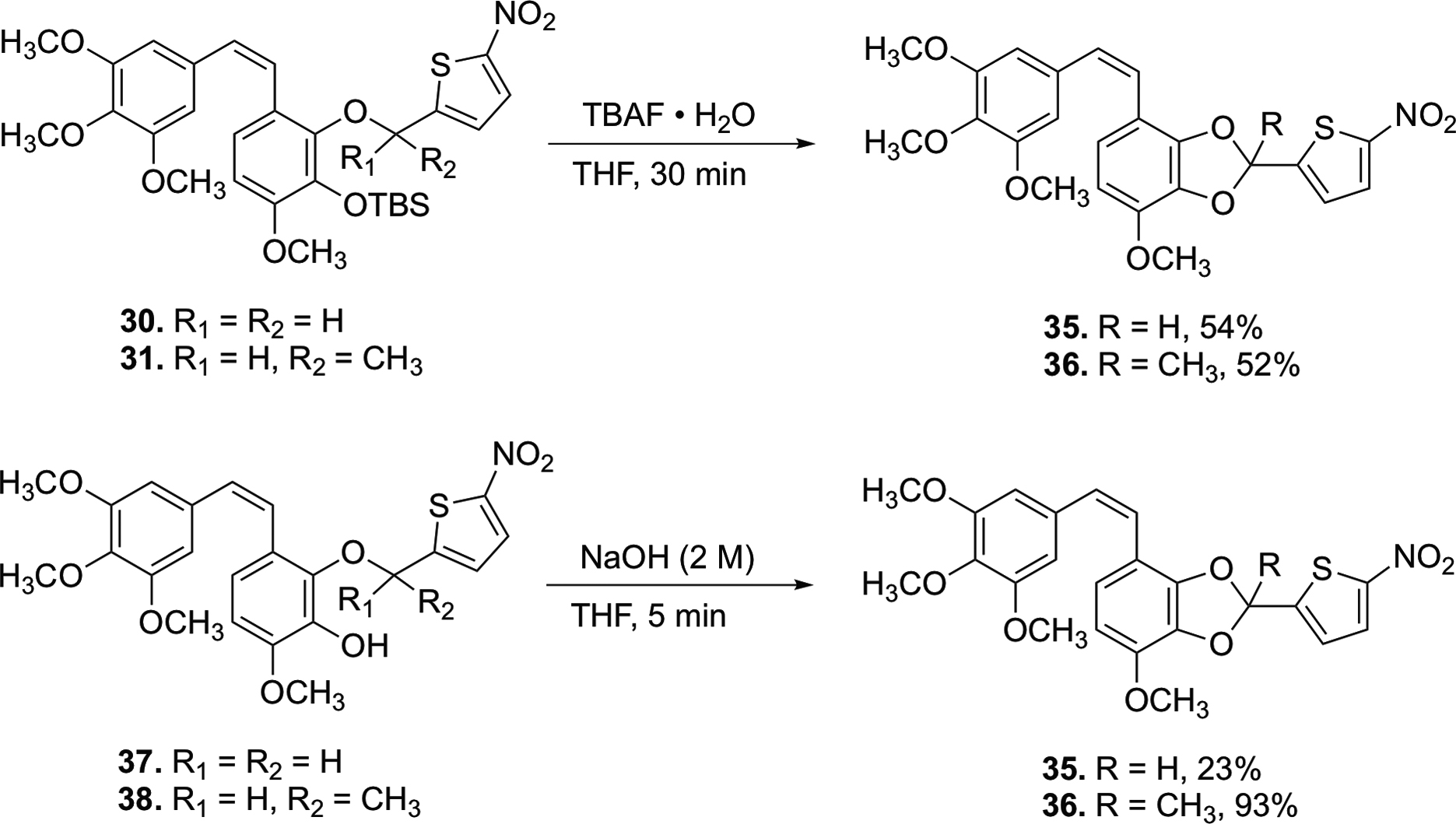
Generation of cyclized analogues 35 and 36.
Modification (Scheme 6) of the deprotection conditions [HCl (2 M)/AcOH, instead of TBAF], afforded the desired CA1-BAPCs 37–40 (regiochemistry was determined through 1D NOE NMR).
Scheme 6.
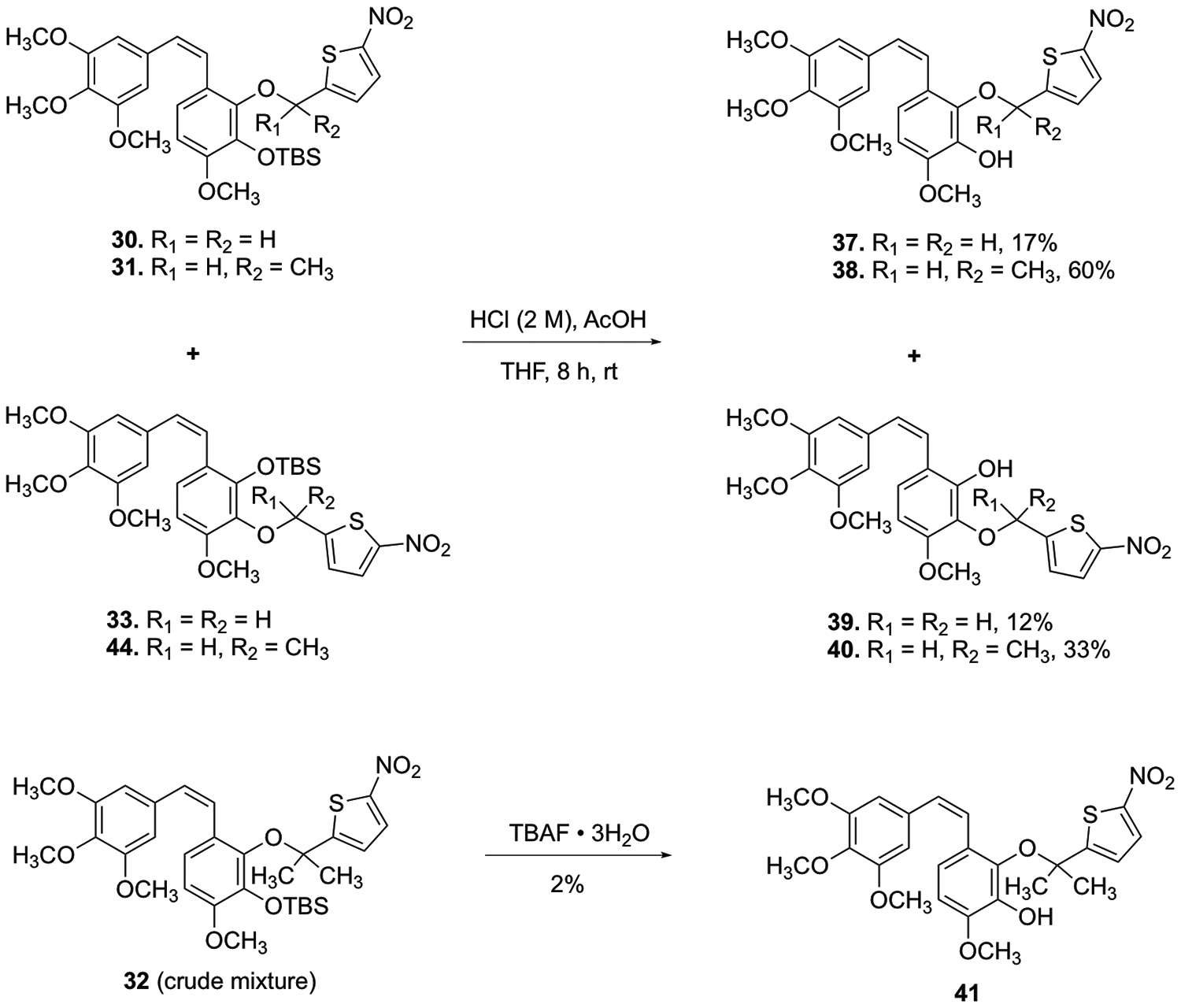
Synthesis of CA1 BAPCs 37–41.
Purification of the TBS-protected gem-dimethyl CA1 BAPC 32 by column chromatography did not result in the requisite level of purity necessary for meaningful biological evaluation. Thus, the crude mixture (containing 32, 27, and 28) underwent deprotection (Scheme 6) prior to purification by column chromatography. Regioisomeric assignment was confirmed by 1D NOE NMR. While the overall yield for this deprotection was quite low, the remaining material consisted only of starting material (crude mixture of 32, 27, and 28).
The known CA4-BAPCs were synthesized (as comparative compounds) under conditions similar to those previously reported by Davis et al., but with several useful modifications (Scheme 7).42 CA4-BAPC 43 was synthesized through a Mitsunobu reaction heated to 50 °C with nitrothiophene 16.42 CA4 and nitrothiophene 17 were reacted with DIAD and triphenylphosphine to generate BAPC 44.42 The gem-dimethyl CA4 BAPC 45 was synthesized from CA4, ADDP, nitrothiophene 19, and tributylphosphine.42 In order to improve the yield for the gem-dimethyl CA4 BAPC 45, subsequent Mitsunobu reactions were performed in toluene.42 While the overall yield was improved, the new method required a more extensive purification procedure to remove the remaining CA4 and gem-dimethyl thiophene trigger, both of which had nearly identical chromatographic retention times to the desired CA4-BAPC 45. Accordingly, the reaction mixture was subjected to chemical modification to facilitate chromatographic separation during purification. The phenolic moiety of CA4 was converted to its corresponding silyl ether (TBS), and the unreacted gem-dimethyl trigger was subsequently acetylated, allowing both of these compounds to be successfully separated chromatographically from the desired CA4 gem-dimethyl-nitrothiophene BAPC 45.
Scheme 7.
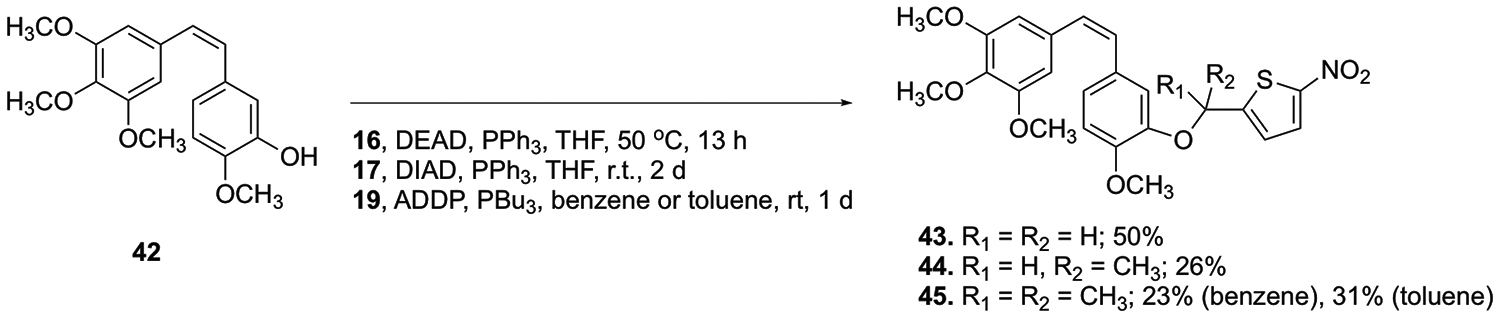
Synthesis of CA4-BAPCs.42
Biological Evaluation.
Inhibition of Tubulin Polymerization and Colchicine Binding,
The BAPCs and their parent anticancer agents (CA4 and CA1) were evaluated for their ability to inhibit tubulin polymerization and colchicine binding (Table 1). The parent anticancer agents [CA4, CA1, tosyl-protected CA1 (20), and isopropyl-protected CA1 (21)] utilized in this study were potent inhibitors of tubulin polymerization (IC50 = 0.64, 1.9, 0.84, and 0.82 μM, respectively) and strongly inhibited colchicine binding. The TBS-protected CA1 analogue 26 was only moderately active as an inhibitor of tubulin polymerization (IC50 = 9.5 μM). Ideally, the BAPCs prepared from these parent anticancer agents would be protected from binding to tubulin until cleaved (in vivo) to generate their corresponding anticancer agents. Considering the collective group of fifteen BAPCs synthesized for this study, seven BAPCs (22, 23, 25, 36, 43, 44, 45) were inactive (IC50 > 20 μM) as inhibitors of tubulin polymerization while four BAPCs (24, 26, 39, 40) were moderately active inhibitors (IC50 > 3 μM but < 20 μM) and four BAPCs (35, 37, 38, 41) proved to be potent inhibitors (IC50 < 3 μM).
Table 1.
Inhibition of Tubulin Polymerization and Colchicine Binding
| compound | inhibition of tubulin polymerizationa IC50 | inhibition of colchicine bindingb % inhibition ± SD | |
|---|---|---|---|
| (μM)±SD | 1 μM | 5 μM | |
| 29 (CA1)c | 1.9 | NDd | 99.6±0.7 |
| 42 (CA4) | 0.64±0.01 | 84±2 | 97±0.7 |
| 20 | 0.84±0.1 | 50±5 | 84±1 |
| 21 | 0.82±0.04 | 72±4 | 94± 0.7 |
| 22 | >20 | ||
| 23 | >20 | ||
| 24 | 12±1 | ||
| 25 | >20 | ||
| 26 | 9.5±0.9 | ||
| 35 | 1.7±0.2 | 25±3 | |
| 36 | >20 | ||
| 37 | 1.7±0.01 | 53±3 | 92±0.5 |
| 38 | 0.84±0.1 | 34±3 | 90±0.7 |
| 39 | 4.3±0.4 | 58±4 | |
| 40 | 6.2±0.3 | 72±3 | |
| 41 | 1.3±0.08 | 43±4 | |
| 43 | >20 | 26±1 | |
| 44 | >20 | 15±5 | |
| 45 | >20 | 33±3 | |
Enzyme-Mediated Cleavage of BAPCs and Stability in Phosphate Buffer.
In preliminary studies, the CA4-BAPCs were treated with POR supersomes (Table 2). Complete cleavage was observed for compound 45 (24 h, anaerobic conditions). Therefore, a shorter assay time period (90 min) was selected for compound comparison, and under these conditions compounds 43 and 44 underwent minimal cleavage (2.7% and 4.1% respectively) while 45 was more extensively cleaved (25.4%). These results are in accordance with the previously reported results from Davis and co-workers that utilized supersomal POR with compounds 43, 44, and 45 and demonstrated that 45 was cleaved more readily (to release CA4) than 43 and 44.42 This trend in cleavage (from gem-dimethyl to mono-methyl to nor-methyl) was also observed when the CA1-BAPCs were exposed to POR. The gem-dimethyl CA1-BAPC (41) and the isopropyl-protected gem-dimethyl BAPC (27) were cleaved more extensively in comparison to their corresponding mono-methyl and nor-methyl BAPCs. The mono-methyl and nor-methyl CA1-BAPCs (37, 38, 39 and 40) were cleaved by POR to different extents, depending on the position of the nitrothiophene side chain (bioreductive trigger) and the hydroxyl group. It should be noted that under these assay conditions, these four BAPCs (37, 38, 39, and 40) underwent cyclization to generate their corresponding cyclized analogues 35 or 36. While the mechanism of this cyclization is unknown, it is noteworthy that under these assay conditions cyclization that incorporates the bioreductive trigger was more favorable than the desired cleavage of the prodrug trigger. BAPC 41 was the only gem-dimethyl BAPC that was fully cleaved (100%) by POR (90 min) in this study, and thus its stability was further evaluated in the pH 7.4 buffer. BAPC 41 showed no apparent spontaneous hydrolysis for the first 150 min, but it was mostly hydrolyzed if incubated in the buffer for 24–48 h. Therefore, the cleavage (100%) of BAPC 41 by POR (90 min) was not due to spontaneous hydrolysis in buffer.
Table 2.
Stability of BAPCs and Their Cleavage by NADPH-Cytochrome P450 Oxidoreductase
| compound | percent BAPC hydrolysi s/cl eavage (non-enzymatic) in phosphate buffer (pH 7.4, 48 h) | percent BAPC cleavage by POR (90 min) |
|---|---|---|
| 22 | 0.25 | NCa |
| 23 | 0.84 | 13.5 |
| 24 | 1.59 | 1.1 |
| 25 | 0.69 | 3.8 |
| 26 | 4.03 | 7.6 |
| 35 | 0 | NC |
| 36 | 0 | NC |
| 37 | 0b | 14.2 |
| (24% cyclization of 37 to 35) | ||
| 38 | 0.5c | 5.6 |
| (48% cyclization of 38 to 36) | ||
| 39 | 0b | 17.9 |
| (46% cyclization of 39 to 35) | ||
| 40 | 0c | 25.5 |
| (35% cyclization of 40 to 36) | ||
| 41 | 100 | 100 |
| 43 | 0.35 | 2.7 |
| 44 | ND | 4.1 |
| 45 | 0.69 | 25.4 |
NC= no cleavage observed.
significant cyclization to 35.
significant cyclization to 36.
Hypoxia Cytotoxicity Ratio (HCR) Determined in A549 Lung Cancer Cell Line.
The initial cytotoxicity data for the CA1 and CA4 BAPCs showed promise for differential activity between oxic and hypoxic environments (Table 3), with several BAPCs demonstrating a positive hypoxia cytotoxicity ratio (HCR). A number of prodrugs stood out, notably compounds 24, 41, and 45. The most active prodrugs in the series were the gem-dimethyl BAPCs of CA1 (41) and CA4 (45) with HCRs of 12.5 and 41.5, respectively, consistent with previous studies by Davis and co-workers that demonstrated that the gem-dimethyl CA4-BAPC had greater resistance to cleavage in oxic environments, releasing the parent anticancer agent (CA4) selectively under hypoxic conditions.42 The hypoxia activated compounds, tirapazamine and RB6145 were included as positive controls. The lower HCR for tirapazamine (Table 3) compared to literature values was a result of modification of the assay conditions. The drug removal step after anoxic (or oxic) exposure was omitted in order to detect the antimitotic activity of the parent compounds CA1 and CA4 released from their corresponding BAPCs. (See Table S7 in Supporting Information for additional data). Under these conditions, tirapazamine gave an HCR of 9.2 in contrast to HCRs > 62 in assays in which it was washed out after 4 h.
Table 3.
In Vitro Potency and Hypoxia Cytotoxicity Ratio (HCR) of the CA4 and CA1 BAPCs in the A549 Human Cancer Cell Line
| compound | GI50 [oxic]a,b (μM)±SEM | GI50 [anoxic]a,b (μM)±SEM | HCR |
|---|---|---|---|
| RB6145 | >89 | 24±6.7 | >3.7 |
| Tirapazamine | 63±5.7 | 6.8±0.39 | 9.2 |
| 29 (CA1) | 1.2±0.48 | 0.82±0.12 | n/ac |
| 42 (CA4) | 0.0047±0.00021 | 0.0061±0.00048 | n/ac |
| 20 | 0.065±0.0030 | 0.19±0.015 | n/ac |
| 21 | 0.063±0.0087 | 0.046±0.0047 | n/ac |
| 22 | 0.44±0.022 | 0.52±0.060 | 0.85 |
| 23 | 4.6±0.14 | 3.3±0.36 | 1.4 |
| 24 | 1.9±0.66 | 0.30±0.047 | 6.3 |
| 25 | 2.9±0.22 | 1.2±0.20 | 2.4 |
| 26 | 1.5±0.39 | 0.49±0.033 | 3.1 |
| 35 | 0.046±0.0037 | 0.033±0.0012 | 1.4 |
| 36 | 0.55±0.025 | 0.72±0.17 | 0.76 |
| 37 | 0.17±0.040 | 0.28±0.082 | 0.61 |
| 38 | 0.18±0.061 | 0.26±0.077 | 0.69 |
| 39 | 0.32±0.014 | 0.69±0.031 | 0.46 |
| 40 | 0.31±0.035 | 0.051±0.0041 | 6.1 |
| 41 | 6.0±2.4 | 0.48±0.054 | 12.5 |
| 43 | 0.11±0.031 | 0.032±0.0073 | 3.4 |
| 44 | 0.15±0.027 | 0.038±0.0059 | 4.0 |
| 45 | 2.2±0.64 | 0.053±0.0088 | 41.5 |
Average of n ≥ 3 independent determinations
Incubation involved 4 h (oxic or anoxic) followed by 48 h oxic exposure
n/a (not applicable)
Preliminary In Vivo Assessment with BLI.
In line with ARRIVE guidelines, animal investigations were conducted in accordance with State and Federal guidelines and approved by the Institutional Animal Care and Use Committee of UT Southwestern under protocol APN#2017–102169. A preliminary in vivo study was performed using orthotopic 4T1-luc breast tumors growing in the left frontal mammary fat pad of syngeneic BALB/c mice treated with CA4-BAPC 45 [single dose (180 mg/kg at a concentration of 30 mg/mL), IP] to gauge initial tolerability and efficacy of this agent. 4T1 is a murine mammary tumor that arose spontaneously in an ageing BALB/C mouse and is considered to replicate many of the characteristics of human breast cancer.49 It is widely used in studies of chemotherapy, and several reports have used luciferase transfected clones to facilitate imaging of therapeutic response and metastasis.50–52 We are only aware of one previous report of evaluation of a VDA in 4T1, specifically OXi4503.53
Since BAPC 45 was insoluble in buffered saline or water, it was necessary to develop a suitable vehicle to solubilize this agent for in vivo use. While BAPC 45 proved soluble in DMSO, there are limits (in terms of volume tolerability) associated with the use of neat DMSO in mice. A solubilization study identified 10% DMSO / 55% sesame oil / 35% PEG 400 (hereafter referred to as DSP) as a suitable vehicle (see Supporting Information for further details). An initial evaluation of tolerability of vehicle alone (without added BAPC) led to the conclusion that 150 μL DSP (administered IP) approached the maximum usable volume. The solubility constraints (of BAPC 45 in this vehicle) limited the maximum single dose for injection. BLI was performed on a group of five mice at baseline. BLI was repeated again 4 h post administration of BAPC 45 (180 mg/kg, IP) in three mice, while two mice served as controls, with one mouse treated with vehicle alone and another mouse treated with CA4P54 (120 mg/kg, IP), a benchmark VDA.
Bioluminescence images are shown for the group of five mice at various time points (baseline to 48 h in Figure 3). At baseline each tumor showed an integrated light intensity of about 5×109 photons/s. Four hours following administration of BAPC 45 (180 mg/kg, IP), two of three mice showed a dramatic decrease in light emission (>80%) following administration of fresh luciferin (Figures 3 and 4). At 24 h these two tumors remained >75% depressed, but showed substantial recovery by 48 h. By comparison, CA4P caused a >99% drop in signal within 4 h, which remained reduced by >90% up to 72 h. The control mouse receiving vehicle alone showed relative stability up to 72 h. One mouse died during the night prior to BLI at 72 h, and two mice died under anesthesia during BLI at 72 h following treatment. We attribute this to the stress of anesthesia accompanying the high tumor burden. Tumors from four of the mice were harvested at 72 h or following additional imaging at 96 h and were stained with hematoxylin and eosin (H&E). Whole mount sections (Figure 5) showed substantial necrosis in all tumors including the vehicle control (46% necrosis), as also reported by others for this tumor type.50,53 The tumor showing a strong BLI signal response to BAPC 45 showed more necrosis than the unresponsive one (55 vs. 47% respectively). Likewise, the tumor on the mouse receiving CA4P was highly necrotic (70%). At higher magnification, extensive hemorrhage was apparent in several tumors together with congested blood vessels (see Figure S9, Supporting Information). These BLI and histology results are potentially indicative of in vivo cleavage by POR and subsequent vascular disruption by the released parent anticancer agent CA4. The differential response is not unexpected since 4T1 tumors show highly variable levels of hypoxia (see Figure S10, Supporting Information).
Figure 3.
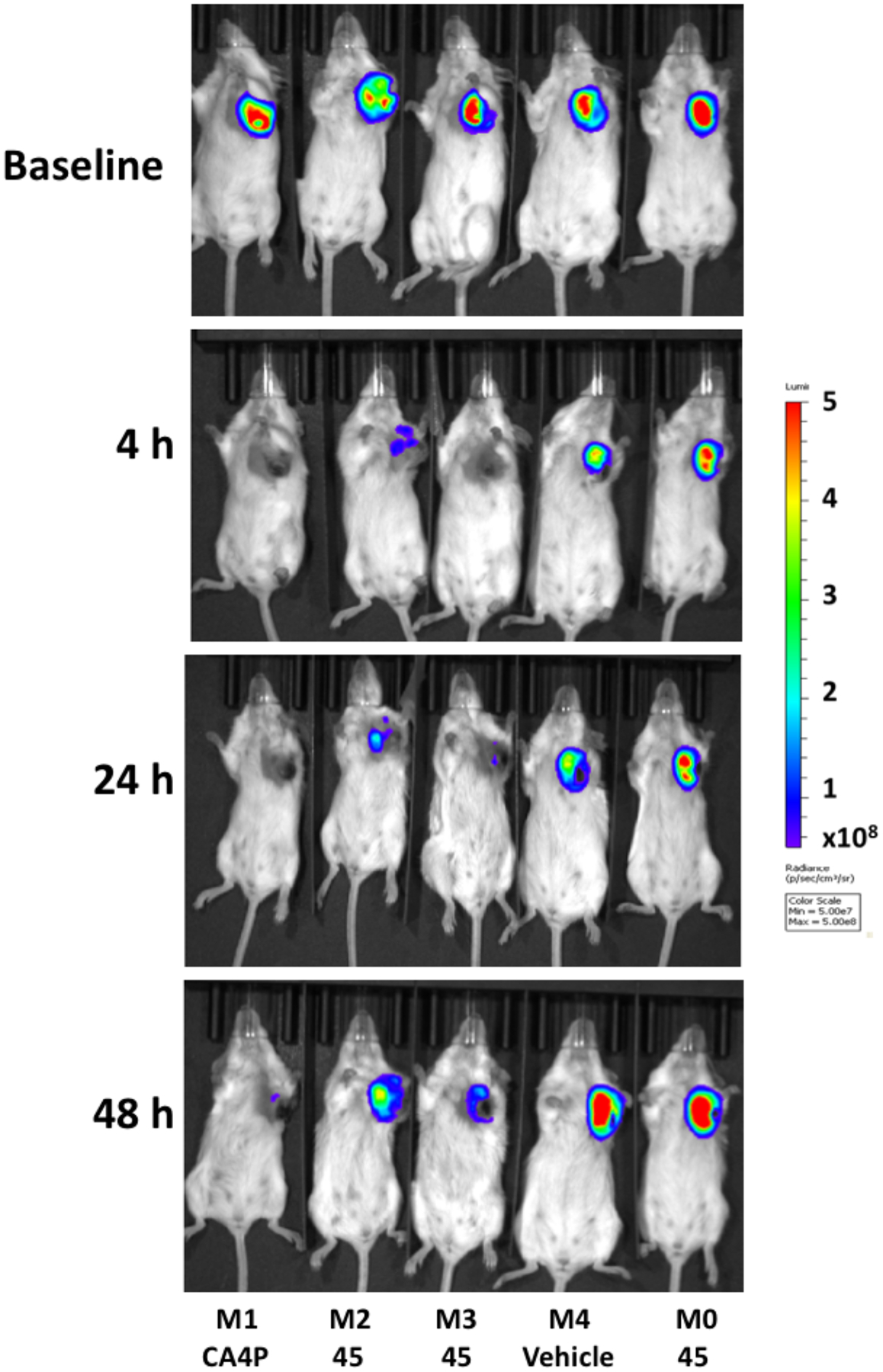
Bioluminescence images of 4T1-luc tumor-bearing BALB/c mice at various times following VDA administration. Baseline shows mice at 20 min following administration of 120 mg/kg luciferin subcutaneously in the foreback region of five BALB/c mice bearing orthotopic syngeneic 4T1-luc tumors growing in a frontal upper mammary fat pad. Immediately following baseline BLI, mice M2, M3 and M0 were injected IP with 180 mg/kg BAPC 45 dissolved in DPS. M1 received 120 mg/kg CA4P IP and M4 received DPS (vehicle alone). Four, 24 and 48 h later BLI was repeated following administration of fresh luciferin on each occasion. Light emission time courses are presented in Figure 4.
Figure 4.
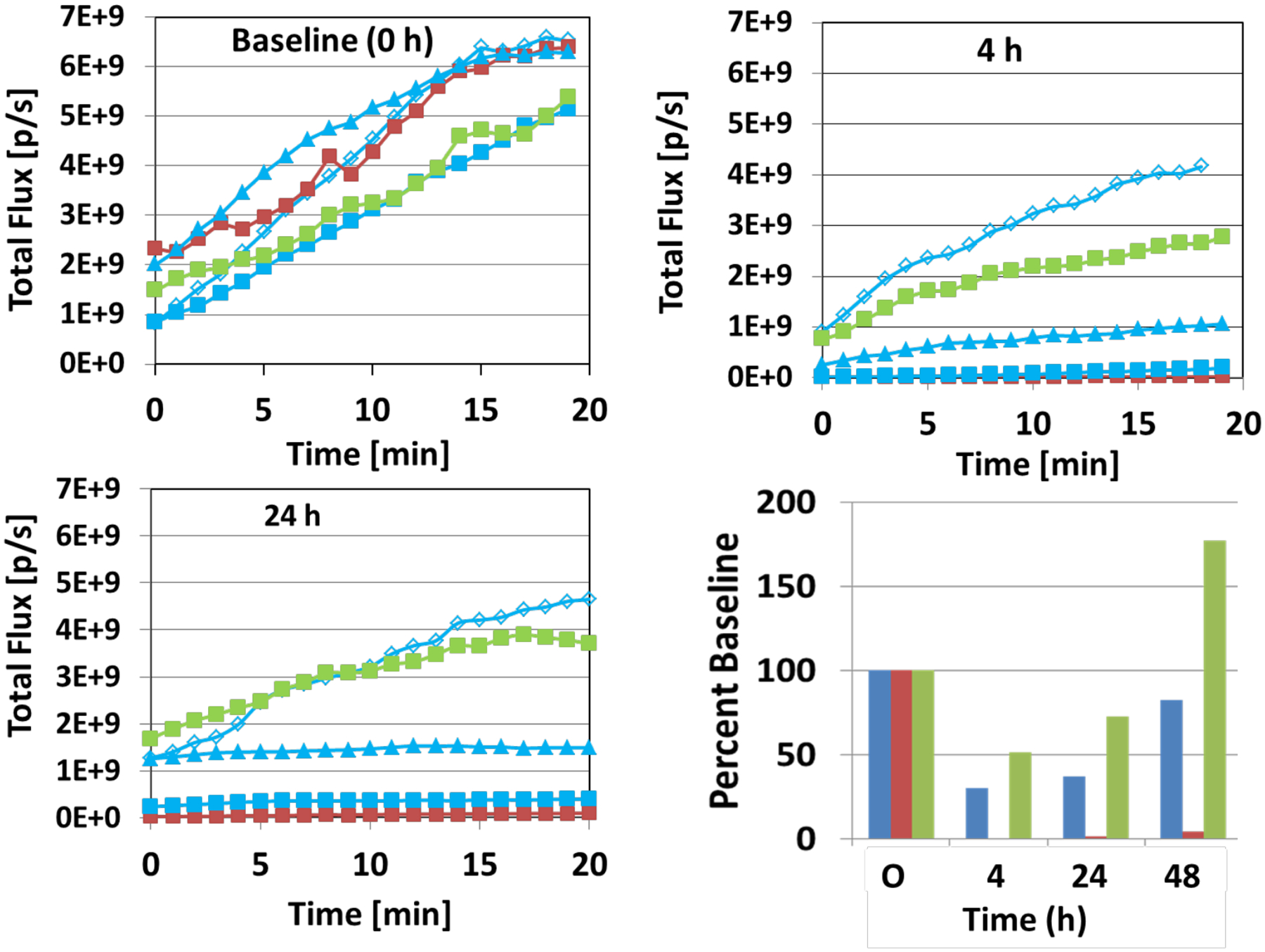
Dynamic light emission time courses with respect to vascular disruption. Following administration of luciferin, BLI was performed over a period of about 20 min for the group of mice shown in Figure 3, and variation of signal intensity is shown at baseline, 4 h and 24 h. The mouse represented by the red line received CA4P (M1); by the green line received vehicle (M4) and by the blue lines each received BAPC 45 (triangles, M2; squares, M3; open diamonds, M0). At baseline, all tumors showed similar light emission kinetics (upper left panel). Four hours later, two of three tumors receiving BAPC 45 and the CA4P treated tumor showed substantially reduced signals, while one BAPC 45 treated tumor and the control tumor were relatively unchanged (upper right panel). Data for 24 h are shown in the lower left panel. The results over 48 h are summarized for the tumors in the lower right panel: red bars (CA4P); green bars (vehicle) and blue bars [mean of three tumors receiving BAPC 45)].
Figure 5.
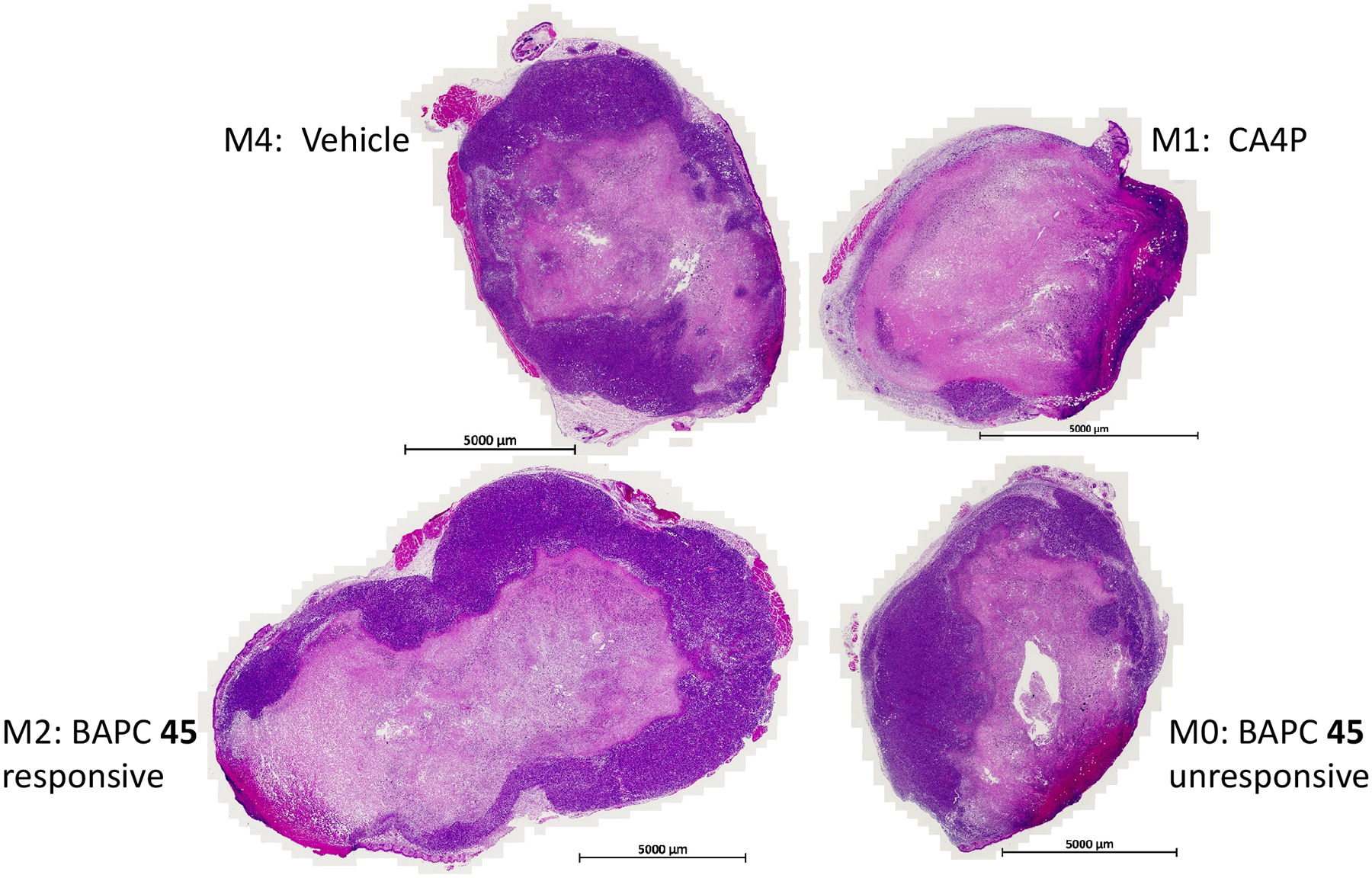
Histology of 4T1-luc tumors. H&E staining revealed substantial necrosis in all tumors, including the vehicle control. However, necrosis is particularly evident following CA4P (upper right) and in M2, which showed a strong response to BAPC 45. Expanded views of histology are presented in Figure S9 (Supporting Information).
EXPERIMENTAL SECTION
General Experimental Procedures.
Acetic acid (AcOH), acetic anhydride, acetonitrile, CH2Cl2, dimethylformamide (DMF), ethanol, methanol, hexanes, nitric acid, sulfuric acid, ethyl acetate (EtOAc), and tetrahydrofuran (THF) were used in their anhydrous forms or as obtained from the chemical suppliers. Reactions were performed under N2 gas. Thin-layer chromatography (TLC) plates (precoated glass plates with silica gel 60 F254, 0.25 mm thickness) were used to monitor reactions. Purification of intermediates and products was carried out with a Biotage Isolera or Teledyne Combiflash flash purification system using silica gel (200–400 mesh, 60 Å) or RP-18 prepacked columns or was performed manually in glass columns. Intermediates and products synthesized were characterized on the basis of their 1H NMR (600 or 500 MHz), 13C NMR (150, 125 or 90 MHz) and 31P NMR (240 MHz) spectroscopic data using a Varian VNMRS 500 MHz, a Bruker DRX 600 MHz, or a Bruker DPX 360 MHz instrument. Spectra were recorded in CDCl3 or (CD3)2CO. All chemical shifts are expressed in ppm (δ), and peak patterns are reported as broad (br), singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), sextet (sext), septet (sept), double doublet (dd), double double doublet (ddd), and multiplet (m). Purity of the final compounds was further analyzed at 25 °C using an Agilent 1200 HPLC system with a diode-array detector (λ = 190–400 nm), a Zorbax XDB-C18 HPLC column (150 mm, 5 μm), and a Zorbax reliance cartridge guard-column; solvent A acetonitrile, solvent B H2O; Method A: H2O; gradient, 10% A/90% B to 100% A/0% B over 0 to 40 min; post-time 10 min, Method B: H2O; gradient, 50% A/50% B to 90% A/10% B over 0 to 30 min; post-time 10 min; flow rate 1.0 mL/min; injection volume 20 μL; monitored at wavelengths of 210, 230, 254, 280, and 320 nm. Mass spectrometry was carried out under either positive or negative ESI (electrospray ionization) or positive or negative APCI/APPI (atmospheric pressure chemical ionization/atmospheric pressure photoionization) using a Thermo Scientific LTQ Orbitrap Discovery instrument.
2,3-Dihydroxy-4-methoxybenzaldehyde (2)43,44.
2,3,4-Trimethoxybenzaldehyde (4.00 g, 20.4 mmol) was added to dry CH2Cl2 (80 mL) in an ice bath (0 °C). BCl3 (45 mL, 45 mmol, 1.0 M) was added dropwise to the reaction mixture, and it was stirred for 18 h. The reaction was then quenched with NaHCO3 and acidified to pH 2 with concentrated HCl. The reaction mixture was extracted with EtOAc, and the organic phase was dried with Na2SO4 and evaporated under reduced pressure. The crude mixture was then filtered through silica gel in a frit funnel with CH2Cl2, and the solvent was evaporated under reduced pressure. Flash chromatography of the crude product using a prepacked 100 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 10% A/90% B over 1.19 min (1 CV), 10% A/90% B → 69% A/31% B over 13.12 min (10 CV), 69% A/31% B over 2.38 min (2 CV); flow rate 50.0 mL/min; monitored at 254 and 280 nm] yielded 2,3-dihydroxy-4-methoxybenzaldehyde (2) (2.64 g, 15.7 mmol, 77%) as a yellow solid: 1H NMR (500 MHz, CDCl3) δ 11.12 (1H, s, OH), 9.76 (1H, s, CHO), 7.15 (1H, d, J = 8.5 Hz, ArH), 6.63 (1H, d, J = 8.5 Hz, ArH), 5.46 (1H, s, OH), 3.99 (3H, s, OCH3); 13C NMR (125 MHz, CDCl3) δ 195.2, 153.0, 149.0, 133.0, 126.1, 116.1, 103.6, 56.4.
6-Formyl-2-hydroxy-3-methoxyphenyl 4-methylbenzenesulfonate (3)43,44.
To a solution of aldehyde 2 (1.15 g, 6.76 mmol) and DIPEA (2.50 mL, 14.3 mmol) in anhydrous DMF (10 mL), p-TSCl (1.29g, 6.73 mmol) was added in portions while stirring at room temperature. After stirring for 5 h, the reaction mixture was quenched with H2O (20 mL), and extracted with EtOAc (3 × 25 mL). The combined organic phase was washed with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. Flash chromatography of the residue using a prepacked 50 g silica column [eluents; solvent A, EtOAc, solvent B, hexanes; gradient, 40% A/60% B over 1.19 min (1 CV), 40% A/60% B →100% A/0% B over 16.3 min (10 CV), 100% A/0% B over 3.18 min (2 CV); flow rate 40.0 mL/min; monitored at 254 and 280 nm] afforded aldehyde 3 (1.33 g, 4.3 mmol, 61% yield) as a white solid: 1H NMR (600 MHz, CDCl3) δ 9.85 (1H, s, CHO), 7.87 (2H, d, J = 8.3 Hz, ArH), 7.50 (1H, d, J = 8.6 Hz, ArH), 7.36 (2H, d, J = 8.0 Hz, ArH), 6.90 (1H, d, J = 8.6 Hz, ArH), 5.91 (1H, s, OH), 3.97 (3H, s, OCH3), 2.47 (3H, s, CH3); 13C NMR (151 MHz, CDCl3) δ 187.0, 153.2, 146.2, 139.2, 138.2, 132.0, 130.0, 128.7, 124.1, 120.6, 109.2, 56.7, 21.8.
3-Hydroxy-2-isopropoxy-4-methoxybenzaldehyde (4)43,44
2,3-Dihydroxy-4-methoxybenzaldehyde (0.400 g, 2.34 mmol), K2CO3 (0.330 g, 2.38 mmol), and 2-bromopropane (0.21 mL, 2.3 mmol) were dissolved in dry DMF (5mL) in a 5 mL Biotage microwave vial. The reaction was run in a Biotage microwave reactor (2 h, 90 °C, normal absorbance). The reaction was then quenched with water, and the reaction mixture was extracted with EtOAc. The organic phase was washed with water and brine, dried with Na2SO4, and evaporated under reduced pressure. Flash column chromatography of the crude product using a prepacked 50 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 10% A/90% B over 1.19 min (1 CV), 10% A/90% B → 54% A/46% B over 13.12 min (10 CV), 54% A/46% B over 2.38 min (2 CV); flow rate 40.0 mL/min; monitored at 254 and 280 nm] yielded 3-hydroxy-2-isopropoxy-4-methoxybenzaldehyde (4) (0.220 g, 1.05 mmol, 44%) as a tan solid: 1H NMR (CDCl3, 600 MHz) δ 10.24 (1H, s, CHO), 7.41 (1H, d, J = 8.7 Hz, ArH), 6.71 (1H, d, J = 8.7 Hz, ArH), 5.77 (1H, d, J = 4.8 Hz, OH), 4.67 (1H, sept, J = 6.1 Hz, CH), 3.94 (3H, s, OCH3), 1.34 (6H, d, J = 6.2 Hz, C(CH3)2); 13C NMR (151 MHz, CDCl3) δ 189.68, 152.75, 147.90, 138.62, 124.34, 120.36, 106.22, 77.00, 56.44, 22.43.
2,3-Bis((tert-butyldimethylsilyl)oxy)-4-methoxybenzaldehyde (5)43,44.
To a solution of 2,3-dihydroxy-4-methoxybenzaldehyde (1.00 g, 5.95 mmol), Et3N (2.00 mL, 14.3 mmol), and DMAP (0.025 g, 0.200 mmol) in CH2Cl2 (30 mL), was added dropwise TBSCl (2.10 g, 13.9 mmol) dissolved in DMF. The reaction mixture was stirred for 12 h at room temperature. H2O was used to quench the reaction, and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] affording 2,3-bis((tert-butyldimethylsilyl)oxy)-4-methoxybenzaldehyde (0.650 g, 1.64 mmol, 65%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 10.22 (1H, s, CHO), 7.48 (1H, d, J = 8.8 Hz, ArH), 6.62 (1H, d, J = 8.8 Hz, ArH), 3.84 (3H, s, OCH3), 1.04 (9H, s, C(CH3)3), 0.99 (9H, s, C(CH3)3), 0.13 (12H, s, Si (CH3)2); 13C NMR (151 MHz, CDCl3) δ 189.64, 157.88, 151.32, 137.10, 123.64, 121.69, 105.73, 55.53, 26.51, 26.36, 19.07, 18.89, −3.51.
2-((tert-Butyldimethylsilyl)oxy)-6-formyl-3-methoxyphenyl 4-methylbenzenesulfonate (6)43,44.
Aldehyde 3 (0.501 g, 1.77 mmol), Et3N (2.00 mL, 14.3 mmol), and DMAP (0.035 g, 0.28 mmol) were dissolved in dry CH2Cl2 (45 mL). TBSCl (0.327 g, 2.17 mmol) was added, and the reaction mixture was stirred for 18 h. The reaction was quenched with water and extracted with diethyl ether. The organic phase was washed with water and brine, dried with Na2SO4, and evaporated under reduced pressure. Flash column chromatography of the residue using a prepacked 50 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 12% A/88% B over 1.19 min (1 CV), 12% A/88% B → 54% A/46% B over 13.12 min (10 CV), 54% A/46% B over 2.38 min (2 CV); flow rate 35.0 mL/min; monitored at 254 and 280 nm] yielded aldehyde 6 (0.610 g, 1.40 mmol, 79%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 9.60 (1H, d, J = 0.47 Hz, CHO), 7.71 (2H, d, J = 8.34 Hz, ArH), 7.52 (1H, d, J = 8.70 Hz, ArH), 7.32 (2H, d, J = 8.05 Hz, ArH), 6.87 (1H, d, J = 8.63 Hz, ArH), 3.87 (3H, s, OCH3), 2.45 (3H, s, CH3), 0.97 (9H, s, C(CH3)3), 0.10 (6H, s, Si (CH3)2); 13C NMR (126 MHz, CDCl3) δ 186.7, 157.3, 145.9, 143.0, 138.9, 132.1, 129.9, 128.5, 124.0, 121.3, 109.8, 55.6, 25.7, 21.7, 18.6, −4.4.
3-((tert-Butyldimethylsilyl)oxy)-2-isopropoxy-4-methoxybenzaldehyde (7)43,44.
Aldehyde 4 (1.39 g, 6.61 mmol), Et3N (1.40 mL, 9.91 mmol), and DMAP (0.050 g, 0.40 mmol) were dissolved in dry CH2Cl2 (50 mL). TBSCl (1.50 g, 9.95 mmol) was added, and the reaction mixture was stirred for 18 h. The reaction was quenched with water and extracted with diethyl ether. The organic phase was washed with water and brine, dried with Na2SO4, and evaporated under reduced pressure. Flash column chromatography of the residue using a prepacked 50 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 12% A/88% B over 1.19 min (1 CV), 12% A/88% B → 54% A/46% B over 13.12 min (10 CV), 54% A/46% B over 2.38 min (2 CV); flow rate 35.0 mL/min; monitored at 254 and 280 nm] yielded aldehyde 7 (1.53 g, 4.71 mmol, 71%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 10.11 (1H, s, CHO), 7.35 (1H, d, J = 8.7 Hz, ArH), 6.56 (1H, d, J = 8.7 Hz, ArH), 4.60 – 4.45 (1H, m, CH), 3.71 (3H, s, OCH3), 1.10 (6H, d, J = 6.2 Hz, C(CH3)2), 0.86 (9H, d, J = 2.0 Hz, C(CH3)3), 0.00 (6H, s, Si(CH3)2); 13C NMR (151 MHz, CDCl3) δ 190.0, 157.4, 152.7, 138.4, 125.2, 121.4, 106.9, 75.5, 55.5, 25.9, 22.3, 18.7, −4.3.
3,4,5-Trimethoxybenzylbromide (9)43,44.
A mixture of 3,4,5-trimethoxybenzylalcohol (20.1g, 101.4 mmol) and PBr3 (4.8 mL, 50.7 mmol) in anhydrous CH2Cl2 was stirred for 1 h at 0 °C under N2. Water (10 mL) was added, and the organic layer was separated and extracted with CH2Cl2 (2 × 100 mL). The combined organic layer was washed with brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. After the recrystallization of the crude solid from 10% (EtOAc/hexanes), the off-white solid of bromide 9 (23.6 g, 90.3 mmol, 89% yield) was obtained and needed no further purification: 1H NMR (500 MHz, CDCl3) δ 6.62 (2H, s, ArH), 4.47 (2H, s, CH2), 3.87 (6H, s, OCH3), 3.85 (3H, s, OCH3); 13C NMR (125 MHz, CDCl3) δ 153.3, 138.2, 133.2, 106.1, 60.9, 56.1, 34.3.
3,4,5-Trimethoxybenzyltriphenylphosphonium Bromide (10)43,44.
A mixture of bromide 9 (11.00 g, 42.1 mmol) and PPh3 (12.1 g, 46.3 mmol) in acetone (100 mL, anhydrous) was stirred in a flask under N2. After 5 h, the resulting suspension was filtered through a Buchner funnel, and the solid was washed with acetone (100 mL) and hexanes (50 mL) to afford an off-white solid. The solid was dried in vacuo to obtain the phosphonium salt 10 (20.3 g, 38.2 mmol, 92% yield) as a white solid: 1H NMR (600 MHz, CDCl3) δ 7.74 – 7.64 (9H, m, ArH), 7.58 – 7.50 (6H, m, ArH), 6.43 (2H, d, J = 2.6 Hz, ArH), 5.29 (2H, d, J = 14.1 Hz, CH2), 3.70 (3H, d, J = 3.4 Hz, OCH3), 3.43 (6H, d, J = 3.7 Hz, OCH3); 13C NMR (125 MHz, CDCl3) δ 153.0, 137.6, 134.8, 134.6, 130.0, 122.4, 117.8, 108.8, 60.8, 56.2, 30.8; 31P NMR (243 MHz, CDCl3) δ 23.2.
(Z)-2-((tert-Butyldimethylsilyl)oxy)-3-methoxy-6-(3,4,5-trimethoxystyryl)phenyl 4-methylbenzenesulfonate (11)43,44.
Triphenyl(3,4,5-trimethoxybenzyl)phosphonium bromide (3.25 g, 6.20 mmol) was dissolved in dry THF (90 mL) in an ice/salt bath (−10 °C). n-Butyllithium (2.4 mL, 6.0 mmol, 2.5 M) was added dropwise, and the reaction mixture was stirred for 30 min. The aldehyde 6 (2.01 g, 4.60 mmol) was dissolved in dry THF (30 mL) and added dropwise to the reaction mixture, which was stirred for 5 h. The reaction was quenched with water, and the THF was evaporated under reduced pressure. The mixture was extracted with EtOAc, and the organic phase was washed with water and brine, dried with Na2SO4, and evaporated under reduced pressure. Flash chromatography of the residue using a prepacked 100 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 10% A/90% B over 1.19 min (1 CV), 10% A/90% B → 80% A/20% B over 13.12 min (10 CV), 80% A/20% B over 2.38 min (2 CV); flow rate 35.0 mL/min; monitored at 254 and 280 nm] yielded Z-isomer 11 (1.11 g, 1.84 mmol, 40%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.82 (2H, d, J = 8.5 Hz, ArH), 7.25 (2H, d, J = 8 Hz, ArH), 6.77 (1H, d, J = 8.5 Hz, ArH), 6.61 (1H, d, 8.5 Hz, ArH), 6.44 (2H, s, ArH), 6.19 (1H, d, J = 12 Hz, CH), 6.16 (1H, d, J = 12 Hz, CH), 3.82 (3H, s, OCH3), 3.76 (3H, s, OCH3), 3.67 (6H, s, OCH3), 0.95 (9H, s, C(CH3)3), 0.04 (6H, s, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 152.6, 151.3, 144.8, 140.2, 139.1, 134.5, 132.2, 130.4, 129.5, 128.4, 125.3, 124.7, 122.1, 109.5, 106.1, 60.8, 55.8, 55.4, 25.8, 25.7, 25.6, 21.6, 18.7, −4.5.
(Z)-tert-Butyl(2-isopropoxy-6-methoxy-3-(3,4,5-trimethoxystyryl)phenoxy)-dimethylsilane (12)43,44.
Triphenyl(3,4,5-trimethoxybenzyl)phosphonium bromide (1.94 g, 3.70 mmol) was dissolved in dry THF (50 mL) and cooled to −15 °C. n-Butyllithium (2.5 M in hexanes, 1.78 mL, 4.44 mmol, 2.5 M) was added dropwise, and the reaction mixture was stirred for 25 min. The reaction mixture was cooled to −78 °C, and a solution of aldehyde 7 in THF (30 mL) was added dropwise. The reaction mixture was stirred for 5 h. The reaction was quenched with water, and the THF was evaporated under reduced pressure. The mixture was extracted with EtOAc, and the organic phase was washed with water and brine, dried with Na2SO4, and evaporated under reduced pressure. The crude product was purified using flash column chromatography to yield the Z-isomer (resolved from the E-isomer) (0.982 g, 2.01 mmol, 65%) as a reddish-white solid: 1H NMR (600 MHz, CDCl3) δ 6.83 (1H, d, J = 8.6 Hz, ArH), 6.62 (1H, d, J = 12.1 Hz, ArH), 6.52 (2H, s, ArH), 6.45 (1H, d, J = 8.6 Hz, CH), 6.41 (1H, d, J = 12.1 Hz, CH), 4.61 (1H, sept, J = 6.1 Hz, CH), 3.82 (3H, s, OCH3), 3.76 (3H, s, OCH3), 3.65 (6H, s, OCH3), 1.27 (6H, d, J = 6.1 Hz, CH3), 1.02 (9H, s, C(CH3)3), 0.14 (6H, s, Si(CH3)2); 13C NMR (151 MHz, CDCl3) δ 152.7, 151.4, 148.0, 138.6, 136.8, 132.9, 128.5, 126.9, 125.1, 122.4, 106.0, 105.9, 74.2, 60.9, 55.8, 55.2, 25.9, 22.3, 18.7, −4.4.
(Z)-((3-Methoxy-6-(3,4,5-trimethoxystyryl)-1,2-phenylene)bis(oxy))bis(tert-butyldimethylsilane) (13)43,44.
n-Butyllithium (11.4 mL, 2.5 M) was added to a solution of phosphonium salt (11.2 g, 21.4 mmol) in THF (350 mL). The resulting solution was stirred for 15 min at −78 °C. Aldehyde 5 (5.66 g, 14.3 mmol) was dissolved in THF and added dropwise using a dropping funnel. The reaction mixture was stirred for 5 h. H2O was used to quench the reaction, and the residue was extracted with Et2O. The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using a prepacked 340 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 85 mL/min; monitored at 254 and 280 nm] affording compound 13 (2.89 g, 5.15 mmol, 51%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 6.91 (1H, d, J=8.6 Hz, ArH), 6.62 (2H, s, ArH), 6.58 (1H, d, J = 12.2 Hz, CH), 6.37 (1H, d, J = 9.2 Hz, ArH), 6.37 (1H, d, J = 12 Hz, CH), 3.83 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.67 (6H, s, OCH3), 1.04 (9H, s, C(CH3)3), 1.00 (9H, s, C(CH3)3), 0.19 (6H, s, Si(CH3)2), 0.10 (6H, s, Si(CH3)2); 13C NMR (151 MHz, CDCl3) δ 153.0, 152.0, 146.5, 137.1, 133.1, 128.0, 127.7, 123.5, 122.5, 106.2, 104.5, 61.2, 56.1, 55.3, 26.7, 26.4, 19.1, −2.9, −3.6.
(Z)-2-Hydroxy-3-methoxy-6-(3,4,5-trimethoxystyryl)phenyl 4-methylbenzene-sulfonate (20)43,44.
To a solution of Z-stilbene 11 (0.754 g, 1.26 mmol) in dry THF (40 mL) at −15° C, a solution of TBAF·3H2O (3.8 mL, 3.8 mmol) dissolved in THF (10 mL) was added drop-wise. The reaction was stirred for 12 h. H2O (40 mL) was used to quench the reaction, THF was removed by evaporation, and the residue was extracted with EtOAc (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 82%A / 18%B (10 CV), 82%A / 18%B (2 CV); flow rate: 35 mL/min; monitored at 254 and 280 nm] affording compound 20 (0.429 g, 0.882 mmol, 70%) as a dark green solid: 1H NMR (500 MHz, CDCl3) δ 7.91 (2H, d, J = 8.1 Hz, ArH), 7.29 (2H, d, J = 8.0 Hz, ArH), 6.71 (1H, d, J = 8.6 Hz, ArH), 6.62 (1H, d, J = 8.6 Hz, ArH), 6.42 (2H, s, ArH), 6.36 (1H, d, J = 12.0 Hz, CH), 6.32 (1H, d, J = 12.0 Hz, CH), 5.89 (1H, s, OH), 3.86 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.66 (6H, s, OCH3), 2.42 (3H, s, CH3); 13C NMR (126 MHz, CDCl3) δ 151.9, 146.6, 144.5, 138.5, 136.4, 134.5, 132.7, 131.2, 130.5, 128.7, 127.7, 124.9, 123.4, 119.9, 108.3, 105.4, 75.9, 60.0, 55.6, 55.0, 20.9.
(Z)-2-Isopropoxy-6-methoxy-3-(3,4,5-trimethoxystyryl)phenol (21)43,44.
To a solution of compound 12 (0.150 g, 0.251 mmol) in THF (5 mL) at room temperature, TBAF· 3H2O (0.0952 g, 0.302 mmol) dissolved in THF was added dropwise. The reaction was stirred for 0.5 h. H2O (5 mL) was used to quench the reaction, THF was removed by evaporation, and the residue was extracted with EtOAc (3 × 10 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (13 CV), 60%A / 40%B (2 CV); flow rate: 8 mL/min; monitored at 254 and 280 nm] affording compound 21 (0.135 g, 0.361 mmol, 90%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 6.75 (1H, d, J = 8.6 Hz, ArH), 6.59 (1H, d, J = 12.1 Hz, CH), 6.51 (1H, d, J = 8.7 Hz, ArH), 6.51 (2H, s, ArH), 6.46 (1H, d, J = 12.1 Hz, CH), 5.60 (1H, s, OH), 4.56 (1H, sept, J = 6.1 Hz, CH), 3.86 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.66 (6H, s, OCH3), 1.32 (6H, d, J = 6.2 Hz, CH(CH3)2); 13C NMR (126 MHz, CDCl3) δ 152.7, 146.9, 143.2, 138.9, 137.1, 132.5, 129.4, 125.8, 124.1, 120.5, 106.3, 106.0, 75.7, 60.9, 56.2, 55.8, 22.5; HRMS m/z 397.1713 [M+Na]+ (calcd for NaC21H26O6+, 397.1713); HPLC (Method A) 14.7 min, 98%.
(Z)-3-Methoxy-2-((5-nitrothiophen-2-yl)methoxy)-6-(3,4,5-trimethoxystyryl)phenyl 4-methylbenzenesulfonate (22).
To a solution of compound 20 (0.700 g, 1.44 mmol), nor-methyl trigger 16 (0.191 g, 1.20 mmol), and DIAD (0.32 mL) in CH2Cl2 (10 mL), PPh3 (0.610 g, 2.33 mmol) dissolved in CH2Cl2 was added dropwise. The reaction mixture was stirred for 12 h at room temperature. The reaction mixture was then quenched with H2O and extracted with CH2Cl2 (3 × 30 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording tosyl-protected CA1 nor-methyl BAPC 22 (0.125 g, 0.236 mmol, 47%) as a tan-white solid: 1H NMR (600 MHz, CDCl3) δ 7.84 (2H, d, J = 8.2 Hz, ArH), 7.77 (1H, d, J = 4.1 Hz, ArH), 7.24 (2H, d, J = 8.1 Hz, ArH), 6.94 (1H, d, J = 8.7 Hz, ArH), 6.90 (1H, d, J = 4.1 Hz, ArH), 6.69 (1H, d, J = 8.8 Hz, ArH), 6.46 (2H, s, ArH), 6.40 (1H, d, J = 11.9 Hz, CH), 6.33 (1H, d, J = 11.9 Hz, CH), 5.06 (2H, s, CH2), 3.85 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.68 (6H, s, OCH3), 2.40 (3H, s, CH3); 13C NMR (151 MHz, CDCl3) δ 152.8, 152.5, 151.8, 147.9, 145.2, 141.8, 140.3, 137.2, 134.3, 132.0, 131.7, 129.6, 128.3, 128.1, 126.1, 125.9, 125.8, 124.0, 110.4, 106.1, 69.0, 60.9, 56.2, 55.9, 21.7; HRMS m/z 650.1120 [M+Na]+ (calcd for NaC30H29NO10S2+, 650.1125); HPLC (Method A) 18.5 min, 97%, 1.3% parent (Ts-protected CA1 analogue 20), 0% CA1.
(Z)-3-Methoxy-2-(2-(5-nitrothiophen-2-yl)propoxy)-6-(3,4,5-trimethoxystyryl)-phenyl-4-methylbenzenesulfonate (23).
To a solution of compound 20 (0.200 g, 0.411 mmol), DIAD (0.100 g, 0.495 mmol), and 1-(5-nitrothiophen-2-yl) ethanol (0.059 g, 0.34 mmol) in CH2Cl2 (25 mL), triphenylphosphine (0.216 g, 0.822 mmol) was added, and the reaction mixture was stirred for 24 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic phase was dried with Na2SO4 and evaporated under reduced pressure. Flash chromatography of the residue using a prepacked 25 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 12% A/88% B over 1.19 min (1 CV), 12% A/88% B → 100% A/0% B over 13.12 min (10 CV), 100% A/0% B over 2.38 min (2 CV); flow rate 25.0 mL/min; monitored at 254 and 280 nm] yielded (Z)-3-methoxy-2-(2-(5-nitrothiophen-2-yl)propoxy)-6-(3,4,5-trimethoxystyryl)phenyl-4-methylbenzenesulfonate (23) (0.160 g, 0.249 mmol, 61%) as a yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.90 (1H, d, J = 4.3 Hz, ArH), 7.86 (2H, d, J = 8.3 Hz, ArH), 7.44 (2H, d, J = 8.3 Hz, ArH), 7.01 (1H, d, J = 8.8 Hz, ArH), 6.99 (2H, m, ArH), 6.57 (2H, s, ArH), 6.51 (1H, d, J = 12.0 Hz, CH), 6.44 (1H, d, J = 11.9 Hz, CH), 5.47 (1H, q, J = 6.5 Hz, CH), 3.90 (3H, s, OCH3), 3.73 (3H, s, OCH3), 3.66 (6H, s, OCH3), 2.44 (3H, s, CH3), 1.43 (3H, d, J = 6.5 Hz, CH3); 13C NMR (125 MHz, CDCl3) δ 159.6, 158.2, 158.1, 150.5, 147.2, 144.2, 139.9, 137.2, 136.8, 134.9, 133.5, 131.1, 130.9, 129.5, 129.1, 116.0, 111.7, 80.7, 73.4, 64.8, 60.9, 60.5, 26.5, 26.0, 25.8; HRMS m/z 642.1465 [M+H]+ (calcd for C31H32NO10S2+, 642.1462); HPLC (Method A) 18.2 min, 99%, 1.0% parent (Ts-protected CA1 analogue 20), 0% CA1.
(Z)-2-((2-Isopropoxy-6-methoxy-3-(3,4,5-trimethoxystyryl)phenoxy)methyl)-5-nitrothiophene (24).
To a solution of isopropyl-protected CA1 21 (0.350 g, 0.843 mmol), nor-methyl trigger 16 (0.162 g, 1.02 mmol), and DEAD (0.220 mL) in CH2Cl2 (10 mL), PPh3 (0.430 g, 1.64 mmol) dissolved in CH2Cl2 was added dropwise. The reaction mixture was stirred for 24 h at room temperature. H2O (40 mL) was added to quench the reaction, and the resultant liquid mixture was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 17 mL/min; monitored at 254 and 280 nm] affording CA1-BAPC 24 (0.0600 g, 0.116 mmol, 17%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.82 (1H, d, J = 4.1 Hz, ArH), 7.02 (1H, d, J = 8.8 Hz, ArH), 7.00 (1H, d, J = 4.2 Hz, ArH), 6.60 (1H, d, J = 12.1 Hz, CH), 6.53 (1H, d, J = 8.7 Hz, ArH), 6.50 (2H, s, ArH), 6.47 (1H, d, J = 12.1 Hz, CH), 5.17 (2H, s), 4.60 (1H, quint, J = 6.2 Hz, CH), 3.83 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.67 (6H, s, OCH3), 1.32 (3H, s, CH3), 1.31 (3H, s, CH3); 13C NMR (126 MHz, CDCl3) δ 152.8, 152.8, 151.7, 149.9, 149.0, 140.6, 137.1, 132.6, 129.3, 128.2, 125.9, 125.7, 125.3, 125.1, 106.8, 106.0, 76.0, 69.2, 60.9, 55.9, 55.8, 22.6; HRMS m/z 538.1506 [M+Na]+ (calcd for NaC26H29NO8S+, 538.1506); HPLC (Method A) 14.7 min, 93%, 2.1% parent (isopropyl-protected CA1 analogue 21), <1% (trace) CA1.
(Z)-2-(1-(2-Isopropoxy-6-methoxy-3-(3,4,5-trimethoxystyryl)phenoxy)ethyl)-5-nitrothiophene (25)45.
To a solution of isopropyl-protected CA1 21 (0.267 g, 0.715 mmol), mono-methyl trigger 17 (0.136 g, 0.785 mmol), and DIAD (0.190 mL) in CH2Cl2 (10 mL), PPh3 (0.364 g, 1.39 mmol) dissolved in CH2Cl2 was added dropwise. The reaction mixture was stirred for 12 h at room temperature. H2O was added to quench the reaction, and the resultant liquid mixture was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] yielding CA1-BAPC 25 (0.125 g, 0.236 mmol, 47%) as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.78 (1H, d, J = 4.2 Hz, ArH), 6.99 (1H, d, J = 8.7 Hz, ArH), 6.91 (1H, d, J = 4.1 Hz, ArH), 6.59 (1H, d, J = 12.1 Hz, CH), 6.49 (1H, d, J = 8.6 Hz, ArH), 6.47 (2H, s, ArH), 6.45 (1H, d, J = 12.2 Hz, CH), 5.49 (1H, q, J = 6.4 Hz, CH), 4.61 (1H, sept, J = 6.1 Hz, CH), 3.82 (3H, s, OCH3), 3.75 (3H, s, OCH3), 3.65 (6H, s, OCH3), 1.66 (3H, d, J = 6.5 Hz, CH3), 1.30 (3H, d, J = 6.1 Hz, CH3), 1.26 (3H, d, J = 6.1 Hz, CH3); 13C NMR (151 MHz, CDCl3) δ 155.2, 153.1, 152.8, 151.0, 150.2, 139.2, 137.0, 132.6, 129.2, 128.1, 125.9, 125.9, 125.3, 123.5, 106.6, 105.9, 75.7, 75.4, 60.9, 55.9, 55.8, 22.6, 22.5, 22.2; HRMS m/z 552.1660 [M+Na]+ (calcd for NaC27H31NO8S+, 552.1663); HPLC (Method B) 20.5 min, 95%, 0.3% parent (isopropyl-protected CA1 analogue 21), <0.5% (trace) CA1.
(Z)-2-(2-(2-Isopropoxy-6-methoxy-3-(3,4,5-trimethoxystyryl)phenoxy)propan-2-yl)-5-nitrothiophene (26)45
To a solution of isopropyl-protected CA1 21 (0.150 g, 0.402 mmol), gem-dimethyl trigger 19 (0.091 g, 0.486 mmol), and ADDP (0.137 g, 0.543 mmol) in CH2Cl2 (10 mL), PBu3 (0.199 mL) was added dropwise. The reaction was stirred for 24 h at room temperature. H2O was added to quench the reaction, and the resultant liquid mixture was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm] yielding CA1-BAPC 26 (0.020 g, 0.037 mmol, 13%) as an orange oil: 1H NMR (600 MHz, CDCl3) δ 7.80 (1H, d, J = 4.2 Hz, ArH), 7.01 (1H, d, J = 8.7 Hz, ArH), 6.93 (1H, d, J = 4.3 Hz, ArH), 6.58 (1H, d, J = 12.1 Hz, CH), 6.49 (2H, s, ArH), 6.47 (1H, d, J = 8.6 Hz, ArH), 6.45 (1H, d, J = 12.1 Hz, CH), 4.60 (1H, sept, J = 6.0 Hz, CH), 3.82 (3H, s, OCH3), 3.67 (3H, s, OCH3), 3.66 (6H, s, OCH3), 1.71 (6H, s, CH3), 1.23 (6H, d, J = 6.1 Hz, CH3); 13C NMR (151 MHz, CDCl3) δ 161.4, 154.6, 152.8, 151.6, 150.4, 137.1, 137.0, 132.7, 129.0, 128.1, 126.4, 126.2, 125.3, 122.1, 106.4, 105.9, 81.7, 75.1, 60.9, 55.8, 55.5, 28.8, 22.4; HRMS m/z 566.1819 [M+Na]+ (calcd for NaC28H33NO8S+, 566.1819); HPLC (Method B) 22.3 min, 91%, 4.9% parent (isopropyl-protected CA1 analogue 21), 0% CA1.
Synthesis of Compounds 27, 28 and 29.
Deprotection of the TBS group of compound 13 using TBAF (0.9 eq.) yielded an inseparable mixture of compounds 27 and 28. At the same time, about 15% CA1 (compound 29) was also isolated.
(Z)-2-((tert-Butyldimethylsilyl)oxy)-3-methoxy-6-(3,4,5-trimethoxystyryl)phenol (27) and (Z)-2-((tert-butyldimethylsilyl)oxy)-6-methoxy-3-(3,4,5-trimethoxystyryl)phenol (28)43,44.
To a solution of di-TBS CA1 13 (2.00 g, 3.57 mmol) in THF (150 mL) at −15 °C, TBAF· 3H2O (1.01 g, 3.20 mmol) dissolved in THF (10 mL) was added dropwise. The reaction was stirred for 0.5 h. H2O was used to quench the reaction, THF was removed by evaporation, and the residue was extracted with EtOAc (3 × 30 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 70%A / 30%B (13 CV), 70%A / 30%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] affording a mixture of compounds 27 and 28 (0.860 g, 2.59 mmol, 43%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 6.80 (1H, d, J = 8.7 Hz, ArH), 6.71 (1H, d, J = 8.7 Hz, ArH), 6.58 (2H, d, J = 12.0 Hz, CH), 6.52 (4H, s, ArH), 6.47 (1H, d, J = 12.1 Hz, CH), 6.41 (1H, d, J = 12.2 Hz, CH), 6.36 (1H, d, J = 8.5 Hz, ArH), 6.30 (1H, d, J = 8.6 Hz, ArH), 5.66 (1H, s, OH), 5.45 (1H, s, OH), 3.81 (6H, s, OCH3), 3.78 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.64 (12H, d, J = 2.2 Hz, OCH3), 1.01 (9H, d, J = 5.2 Hz, C(CH3)3), 1.00 (9H, s, C(CH3)3), 0.22 (6H, s, Si(CH3)2), 0.19 (6H, s, Si(CH3)2); 13C NMR (126 MHz, CDCl3) δ 152.7, 152.7, 149.3, 146.9, 145.9, 141.2, 137.0, 137.0, 136.8, 132.9, 132.8, 131.6, 129.6, 129.0, 126.8, 124.5, 123.2, 122.0, 120.1, 117.1, 106.1, 106.0, 103.8, 103.0, 60.9, 60.8, 56.1, 55.8, 55.7, 55.2, 26.0, 26.0, 18.6, 18.6, −3.9, −4.4.
(Z)-3-methoxy-6-(3,4,5-trimethoxystyryl)benzene-1,2-diol (29).
Combretastatin A-1 (CA1) 29 (0.179 mg, 0.538 mmol, 15%) was isolated as a white solid: 1H NMR (500 MHz, CDCl3) δ 6.76 (1H, d, J = 8.6 Hz, ArH), 6.59 (1H, d, J = 12.1 Hz, CH), 6.54 (1H, d, J = 11.9 Hz, CH), 6.52 (2H, s, ArH), 6.39 (1H, d, J = 8.6 Hz, ArH), 5.39 (2H, s, OH), 3.86 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.67 (6H, s, OCH3); 13C NMR (126 MHz, CDCl3) δ 152.9, 146.5, 141.7, 137.4, 132.7, 132.6, 130.5, 124.2, 120.5, 118.0, 106.1, 103.1, 76.9, 61.0, 56.3, 56.0; HRMS m/z 355.1154 [M+Na]+ (calcd for NaC18H20O6+, 355.1152); HPLC (Method A) 11.3 min, 99%.
Synthesis of Compounds 30 and 33.
To a mixture of compounds 27 and 28 (1.00 g, 2.24 mmol), nor-methyl trigger 16 (0.428 g, 2.69 mmol), and DIAD (0.867 mL) in CH2Cl2 (50 mL), PPh3 (1.47 g, 5.60 mmol) was added dropwise. The reaction mixture was stirred (24 h) at room temperature. H2O (40 mL) was added to quench the reaction, and the resultant liquid mixture was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 75 mL/min; monitored at 254 and 280 nm].
(Z)-tert-Butyl(6-methoxy-2-((5-nitrothiophen-2-yl)methoxy)-3-(3,4,5-trimethoxystyryl)-phenoxy)dimethylsilane (30).
This isomer 30 (0.350 g, 0.739 mmol, 35%) was isolated as a brownish-yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.76 (1H, d, J = 4.1 Hz, ArH), 6.91 (1H, d, J = 4.1 Hz, ArH), 6.87 (1H, dd, J = 8.6, 0.8 Hz, ArH), 6.57 (1H, d, J = 8.6 Hz, ArH), 6.50 (1H, d, J = 12.0 Hz, CH), 6.45 (1H, d, J = 12.2 Hz, CH), 6.44 (2H, s, ArH), 5.12 (2H, s, CH2), 3.82 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.65 (6H, s, OCH3), 0.99 (9H, s, C(CH3)3), 0.13 (6H, s, Si(CH3)2); 13C NMR (126 MHz, CDCl3) δ 152.7, 151.6, 151.4, 148.6, 147.6, 138.4, 137.1, 132.4, 130.4, 128.2, 125.1, 124.9, 124.4, 122.3, 107.5, 105.9, 68.5, 60.9, 55.8, 55.4, 25.8, 18.6, −4.6.
(Z)-tert-Butyl(3-methoxy-2-((5-nitrothiophen-2-yl)methoxy)-6-(3,4,5-trimethoxystyryl)phenoxy)dimethylsilane (33).
This isomer 33 (0.250 g, 0.425 mmol, 25%) was isolated as a brownish-yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.81 (1H, d, J = 4.1 Hz, ArH), 7.00 (1H, d, J = 8.7 Hz, ArH), 6.96 (1H, d, J = 4.1 Hz, ArH), 6.56 (1H, d, J = 12.2 Hz, CH), 6.52 (2H, s, ArH), 6.44 (1H, d, J = 11.6 Hz, CH), 6.42 (1H, d, J = 8.5 Hz, ArH), 5.30 (2H, s, CH2), 3.83 (3H, s, OCH3), 3.80 (3H, s, OCH3), 3.67 (6H, s, OCH3), 1.01 (9H, s, C(CH3)3), 0.18 (6H, s, Si(CH3)2); 13C NMR (151 MHz, CDCl3) δ 153.4, 152.8, 152.7, 148.8, 147.8, 138.4, 137.0, 132.6, 129.1, 128.2, 126.3, 125.9, 125.4, 123.3, 105.9, 104.9, 68.8, 60.9, 55.9, 55.8, 26.1, 18.6, −3.9.
Synthesis of Compounds 31 and 34.
Mono TBS CA1 [mixture of 27 and 28, (0.680 g, 1.52 mmol)], diisopropylazodicarboxylate (0.415 g, 2.05 mmol), and 1-(5-nitrothiophen-2-yl)ethan-1-ol (0.317 g, 1.82 mmol) were dissolved in THF (50 mL). Triphenylphosphine (0.793 g, 3.04 mmol) was added, and the reaction mixture was stirred (3 d). The reaction mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 40%A / 60%B (13 CV), 40%A / 60%B (2 CV); flow rate: 80 mL/min; monitored at 254 and 280 nm].
(Z)-tert-butyl(6-methoxy-2-(1-(5-nitrothiophen-2-yl)ethoxy)-3-(3,4,5-trimethoxystyryl)phenoxy)dimethylsilane (31).
Isomer 31 (0.375 g, 0.623 mmol, 41%) was isolated as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.60 (1H, d, J = 4.2 Hz, ArH), 6.76 – 6.69 (2H, m, ArH), 6.41 (1H, d, J = 8.2 Hz, ArH), 6.39 (1H, d, J = 11.9 Hz, CH), 6.32 (2H, s, ArH), 6.29 (1H, d, J = 12.1 Hz, CH), 5.58 (1H, q, J = 6.4 Hz, CH), 3.69 (3H, s, OCH3), 3.65 (3H, s, OCH3), 3.51 (6H, s, OCH3), 1.48 (3H, d, J = 6.4 Hz, CH3), 0.85 (9H, s, C(CH3)3), 0.00 (3H, s, Si(CH3)), −0.02 (3H, s, Si(CH3)); 13C NMR (151 MHz, CDCl3) δ 155.0, 152.7, 151.4, 150.9, 146.1, 138.5, 137.2, 132.4, 129.8, 128.1, 125.6, 124.9, 123.4, 122.5, 107.0, 106.0, 74.4, 60.9, 55.8, 55.3, 25.8, 22.3, 18.6, −4.3, −4.4.
(Z)-tert-butyl(3-methoxy-2-(1-(5-nitrothiophen-2-yl)ethoxy)-6-(3,4,5-trimethoxystyryl)phenoxy)dimethylsilane (34).
Isomer 34 (0.073 g, 0.122 mmol, 12%) was isolated as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.61 (1H, d, J = 4.2 Hz, ArH), 6.84 (1H, d, J = 8.7 Hz, ArH), 6.71 (1H, d, J = 4.2 Hz, ArH), 6.38 (1H, d, J = 12.0 Hz, CH), 6.37 (2H, s, ArH), 6.26 (1H, d, J = 12.3 Hz, CH), 6.24 (1H, d, J = 8.6 Hz, ArH), 5.26 (1H, q, J = 6.5 Hz, CH), 3.67 (3H, s, OCH3), 3.61 (3H, s, OCH3), 3.50 (6H, s, OCH3), 1.47 (3H, d, J = 6.5 Hz, CH3), 0.84 (9H, s, C(CH3)3), 0.03 (3H, s, Si(CH3)), 0.00 (3H, s, Si(CH3)); 13C NMR (151 MHz, CDCl3) δ 153.3, 151.1, 150.9, 149.1, 146.1, 135.5, 135.2, 130.7, 126.9, 126.2, 124.6, 123.7, 121.5, 121.5, 104.1, 103.0, 73.1, 59.0, 53.9, 24.2, 24.2, 19.8, 16.7, −5.2, −5.7.
(Z)-tert-Butyl(6-methoxy-2-((2-(5-nitrothiophen-2-yl)propan-2-yl)oxy)-3-(3,4,5-trimethoxystyryl)phenoxy)dimethylsilane (32).
Mono TBS CA1 [mixture of 27 and 28, (1.07 g, 2.40 mmol)], gem-dimethyl trigger 19 (0.540 g, 2.88 mmol), and ADDP (0.832 g, 3.30 mmol) were dissolved in CH2Cl2 (100 mL). Tributylphosphine (1.26 mL, 5.04 mmol) was added dropwise, and the reaction mixture was stirred (2d). The reaction mixture was then concentrated under reduced pressure. Flash chromatography yielded the crude product which was taken to the next step for deprotection.
(Z)-4-Methoxy-2-(5-nitrothiophen-2-yl)-7-(3,4,5-trimethoxystyryl)-benzo[d][1,3]dioxole (35).
To a solution of 30 (0.095 g, 0.162 mmol) in THF (10 mL) at 0 °C, TBAF· 3H2O (0.0672 g, 0.213 mmol) dissolved in THF (10 mL) was added dropwise. The reaction mixture was stirred (30 min), and H2O (5 mL) was added. THF was removed by evaporation, and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude organic product was purified by flash column chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (13 CV), 60%A / 40%B (2 CV); flow rate: 20 mL/min; monitored at 254 and 280 nm] affording compound 35 (0.0510 g, 0.108 mmol, 54%) as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.83 (1H, d, J = 4.2 Hz, ArH), 7.15 (1H, d, J = 4.2 Hz, ArH), 7.07 (1H, s, CH), 6.86 (1H, d, J = 8.8 Hz, ArH), 6.56 (1H, d, J = 12.0 Hz, CH), 6.50 (2H, s, ArH), 6.48 (1H, d, J = 8.8 Hz, ArH), 6.44 (1H, d, J = 12.0 Hz, CH), 3.90 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.69 (6H, s, OCH3); 13C NMR (151 MHz, CDCl3) δ 152.9, 146.0, 145.2, 143.2, 137.3, 133.8, 132.6, 131.2, 128.1, 126.0, 123.4, 121.7, 113.5, 107.8, 105.7, 105.6, 105.2, 60.9, 56.6, 55.9; 13C NMR DEPT (CDCl3, 151 MHz) δ 131.2, 128.1, 126.0, 123.4, 121.7, 107.8, 105.6, 105.2, 60.9, 56.6, 55.9; HRMS m/z 494.0881 [M+Na]+ (calcd for NaC23H21NO8S+, 494.0880); HPLC (Method A) 17.2 min, 98%, 0% parent (CA1).
(Z)-4-Methoxy-2-(5-nitrothiophen-2-yl)-7-(3,4,5-trimethoxystyryl)-benzo[d][1,3]dioxole (35) [Base cyclization method].
Compound 37 (0.0380 g, 0.0803 mmol) was dissolved in THF (5 mL) at room temperature. NaOH (1 mL, 2 M) was added dropwise, and the reaction mixture was then stirred (5 min). THF was removed by evaporation, and the residue was extracted with CH2Cl2 (3 × 10 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 36 mL/min; monitored at 254 and 280 nm] affording compound 35 (0.0090 g, 0.019 mmol, 23%) as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.83 (1H, d, J = 4.2 Hz, ArH), 7.15 (1H, d, J = 4.2 Hz, ArH), 7.07 (1H, s, CH), 6.86 (1H, d, J = 8.8 Hz, ArH), 6.56 (1H, d, J = 12.0 Hz, CH), 6.50 (2H, s, ArH), 6.48 (1H, d, J = 8.8 Hz, ArH), 6.44 (1H, d, J = 12.0 Hz, CH), 3.90 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.69 (6H, s, OCH3); 13C NMR (151 MHz, CDCl3) δ 152.9, 146.0, 145.2, 143.2, 137.3, 133.8, 132.6, 131.2, 128.1, 126.0, 123.4, 121.7, 113.5, 107.8, 105.7, 105.6, 105.2, 60.9, 56.6, 55.9.
(Z)-4-Methoxy-2-methyl-2-(5-nitrothiophen-2-yl)-7-(3,4,5-trimethoxystyryl)-benzo[d][1,3]dioxole (36).
Compound 31 (0.105 g, 0.174 mmol) was dissolved in CH2Cl2 (20 mL) at −10 °C. Tert-butylammonium fluoride trihydrate (0.0620 g, 0.191 mmol) was dissolved in CH2Cl2 (2 mL) and slowly added dropwise to the reaction mixture, which was then stirred (18 min). H2O (5 mL) was used to quench the reaction, and the layers were partitioned. The residue was extracted with CH2Cl2 (3 × 10 mL), washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 12%A / 88%B (1 CV), 12%A / 88%B → 100%A / 0%B (13 CV), 100%A / 0%B (2 CV); flow rate: 10 mL/min; monitored at 254 and 280 nm] affording compound 36 (0.044 g, 0.0906 mmol, 52%) as a yellow oil: 1H NMR (500 MHz, acetone) δ 7.76 (1H, d, J = 4.3 Hz, ArH), 7.03 (1H, d, J = 4.2 Hz, ArH), 6.81 (1H, d, J = 8.8 Hz, ArH), 6.56 (1H, d, J = 12.0 Hz, CH), 6.48 (2H, s, ArH), 6.45 (1H, d, J = 8.8 Hz, ArH), 6.42 (1H, d, J = 11.9 Hz, CH), 3.88 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.67 (6H, s, OCH3), 2.02 (3H, s, CH3); 13C NMR (126 MHz, acetone) δ 152.8, 151.5, 145.1, 143.1, 137.2, 133.8, 132.7, 131.1, 128.3, 124.0, 123.0, 121.9, 113.8, 113.4, 107.4, 105.7, 60.9, 56.5, 55.9, 26.6; HRMS m/z 486.1219 [M+H]+ (calcd for C24H23NO8S+, 486.1217). HPLC (Method A) 14.9 min, 96%, 0% parent (CA1).
(Z)-4-Methoxy-2-methyl-2-(5-nitrothiophen-2-yl)-7-(3,4,5-trimethoxystyryl)-benzo[d][1,3]dioxole (36) [Base cyclization method]
Compound 38 (0.0500 g, 0.103 mmol) was dissolved in THF (5 mL) at room temperature. NaOH (1 mL, 2 M) was added dropwise, and the reaction mixture was then stirred (5 min). THF was removed by evaporation, and the residue was extracted with CH2Cl2 (3 × 10 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 36 mL/min; monitored at 254 and 280 nm] affording compound 36 (0.0470 g, 0.0964 mmol, 93%) as a yellow oil: 1H NMR (500 MHz, acetone) δ 7.76 (1H, d, J = 4.3 Hz, ArH), 7.03 (1H, d, J = 4.2 Hz, ArH), 6.81 (1H, d, J = 8.8 Hz, ArH), 6.56 (1H, d, J = 12.0 Hz, CH), 6.48 (2H, s, ArH), 6.45 (1H, d, J = 8.8 Hz, ArH), 6.42 (1H, d, J = 11.9 Hz, CH), 3.88 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.67 (6H, s, OCH3), 2.02 (3H, s, CH3); 13C NMR (126 MHz, acetone) δ 152.8, 151.5, 145.1, 143.1, 137.2, 133.8, 132.7, 131.1, 128.3, 124.0, 123.0, 121.9, 113.8, 113.4, 107.4, 105.7, 60.9, 56.5, 55.9, 26.6.
(Z)-6-Methoxy-2-((5-nitrothiophen-2-yl)methoxy)-3-(3,4,5-trimethoxystyryl)phenol (37).
AcOH (7 mL) and HCl (5 mL, 2 M) were added dropwise to a solution of compound 30 (0.115 g, 0.196 mmol) in THF (30 mL). The reaction mixture was stirred for 8 h at room temperature. H2O (40 mL) was used to quench the reaction, THF was removed by evaporation, and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 20 mL/min; monitored at 254 and 280 nm] affording compound 37 (0.020 g, 0.0422 mmol, 17%) as a brown oil: 1H NMR (600 MHz, acetone) δ 8.15 (1H, s, OH), 7.93 (1H, d, J = 4.2 Hz, ArH), 7.21 (1H, d, J = 4.1 Hz, ArH), 6.95 (1H, d, J = 8.7 Hz, ArH), 6.58 (2H, s, ArH), 6.54 (1H, d, J = 12.2 Hz, CH), 6.49 (1H, d, J = 8.7 Hz, ArH), 6.44 (1H, d, J = 12.2 Hz, CH), 5.29 (2H, s, CH2), 3.86 (3H, s, OCH3), 3.68 (3H, s, OCH3), 3.62 (6H, s, OCH3); 13C NMR (151 MHz, acetone) δ 153.1, 152.3, 149.2, 148.6, 137.4, 134.1, 132.7, 129.0, 128.6, 126.5, 125.1, 124.5, 124.5, 117.9, 106.2, 103.0, 68.5, 59.6, 55.4, 55.2; 13C NMR DEPT (151 MHz, acetone) δ 129.0, 128.6, 126.5, 125.1, 124.5, 106.2, 103.0, 68.5, 59.6, 55.4, 55.2; HRMS m/z 496.1034 [M+Na]+ (calcd for NaC23H23NO8S+, 496.1037); HPLC (Method B) 10.0 min, 95%, 0% parent (CA1).
(Z)-6-Methoxy-2-(1-(5-nitrothiophen-2-yl)ethoxy)-3-(3,4,5-trimethoxystyryl)phenol (38).
Compound 31 (0.200 g, 0.333 mmol) was dissolved in THF (5 mL). Glacial acetic acid (7 mL) and HCl (2 M, 4 mL) were added dropwise, and the reaction mixture was stirred (30 min). Glacial acetic acid (4 mL) and HCl (2 M, 2.5 mL) were added dropwise, and the reaction mixture was stirred (8 h). H2O (30 mL) was used to quench the reaction, and the reaction mixture was concentrated under reduced pressure. The residue was extracted with CH2Cl2 (3 × 30 mL), and the combined organic phase was washed multiple times with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A/ 90%B → 80%A / 20%B (13 CV), 80%A / 20%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] affording compound 38 (0.094 g, 0.199 mmol, 60%) as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.74 (1H, d, J = 4.2 Hz, ArH), 6.93 (1H, d, J = 4.2 Hz, ArH), 6.80 (1H, d, J = 8.5 Hz, ArH), 6.55 (1H, d, J = 8.6 Hz, ArH), 6.53 (1H, d, J = 12.5 Hz, CH), 6.47 (2H s, ArH), 6.45 (1H, d, J = 12.2 Hz, CH), 5.71 (1H, q, J = 6.4 Hz, CH), 3.87 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.66 (6H, s, OCH3), 1.71 (3H, d, J = 6.4 Hz, CH3); 13C NMR (126 MHz, CDCl3) δ 153.6, 152.8, 151.4, 147.5, 137.1, 132.6, 132.2, 130.2, 128.2, 125.4, 123.8, 123.8, 117.5, 105.8, 103.4, 75.2, 60.9, 55.9, 55.8, 29.7, 21.7; HRMS m/z 488.1363 [M+H]+ (calcd for C24H25NO8S+, 488.1374); HPLC (Method B) 10.1 min, 96%, 4% parent (CA1).
(Z)-3-Methoxy-2-((5-nitrothiophen-2-yl)methoxy)-6-(3,4,5-trimethoxystyryl)phenol (39).
AcOH (10 mL) and HCl (10 mL, 2 M) were added dropwise to a solution of compound 33 (0.250 g, 0.425 mmol) in THF (25 mL). The reaction mixture was stirred for 8 h at room temperature. H2O (40 mL) was used to quench the reaction, THF was removed by evaporation, and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 20 mL/min; monitored at 254 and 280 nm] affording compound 33 (0.030 g, 0.0634 mmol, 12%) as a brown oil: 1H NMR (CDCl3, 600 MHz) δ 7.77 (1H, d, J = 4.1 Hz, ArH), 6.97 (1H, d, J = 4.1 Hz, ArH), 6.79 (1H, d, J = 8.4 Hz, ArH), 6.56 (1H, d, J = 8.4 Hz, ArH), 6.55 (1H, d, J = 12.2 Hz, CH), 6.50 (1H, d, J = 12.2 Hz, CH), 6.45 (2H, s, ArH), 5.59 (1H, s, OH), 5.24 (2H, s, CH2), 3.88 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.65 (6H, s, OCH3); 13C NMR (CDCl3, 151 MHz) δ 152.8, 151.8, 148.5, 147.0, 142.6, 138.4, 137.2, 132.4, 130.7, 128.2, 125.5, 124.6, 124.3, 120.5, 106.6, 106.0, 68.7, 60.9, 56.4, 55.8; HRMS m/z 496.1033 [M+Na]+ (calcd for NaC23H23NO8S+, 496.1037). HPLC (Method B): 12.5 min, 96%, 0% parent (CA1).
(Z)-3-Methoxy-2-(1-(5-nitrothiophen-2-yl)ethoxy)-6-(3,4,5-trimethoxystyryl)phenol (40).
Compound 34 (0.100 g, 0.167 mmol) was dissolved in THF (3 mL). Glacial acetic acid (5.6 mL) and HCl (2 M, 3.3 mL) were added dropwise, and the reaction mixture was stirred (8 h). H2O (20 mL) was used to quench the reaction, and the reaction mixture was concentrated under reduced pressure. The residue was extracted with CH2Cl2 (3 × 20 mL), and the combined organic phase was washed multiple times with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 80%A / 20%B (13 CV), 80%A / 20%B (2 CV); flow rate: 100 mL/min; monitored at 254 and 280 nm] affording compound 40 (0.026 g, 0.055 mmol, 33%) as a yellow oil: 1H NMR (600 MHz, CDCl3) δ 7.79 (1H, d, J = 4.2 Hz, ArH), 6.96 (1H, d, J = 8.7 Hz, ArH), 6.93 (1H, d, J = 4.2 Hz, ArH), 6.53 (2H, d, J = 12.4 Hz, CH), 6.49 (2H, s, ArH), 6.37 (1H, d, J = 8.8 Hz, ArH), 5.56 (1H, q, J = 6.5 Hz, CH), 3.84 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.67 (6H, s, OCH3), 1.73 (3H, d, J = 6.5 Hz, CH3); 13C NMR (126 MHz, CDCl3) δ 153.6, 152.8, 151.4, 147.5, 132.6, 132.2, 130.2, 128.2, 125.3, 123.8, 123.7, 117.5, 105.8, 103.4, 103.3, 75.2, 60.9, 55.9, 55.8, 29.7, 21.7; HRMS m/z 488.1373 [M+H]+ (calcd for C23H23NO8S+), 488.1374; HPLC (Method B) 11.1 min, 93%, 1% parent (CA1).
(Z)-6-Methoxy-2-((2-(5-nitrothiophen-2-yl)propan-2-yl)oxy)-3-(3,4,5-trimethoxystyryl)phenol (41).
To a solution of compound 32 [as a mixture of 32, 27, and 28, (2.35 g, 3.82 mmol)] in THF (250 mL) at −15 °C, TBAF· 3H2O (1.32 g, 4.19 mmol) dissolved in THF (10 mL) was added dropwise. The reaction was stirred for 1 h. H2O (40 mL) was used to quench the reaction, THF was removed by evaporation, and the residue was extracted with CH2Cl2 (3 × 20 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a prepacked 10 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (13 CV), 60%A / 40%B (2 CV); flow rate: 20 mL/min; monitored at 254 and 280 nm] affording compound 41 (0.050 g, 0.980 mmol, 2%) as a brownish-yellow solid: 1H NMR (CDCl3, 600 MHz) δ 7.77 (1H, d, J = 4.2 Hz, ArH), 6.93 (1H, d, J = 4.2 Hz, ArH), 6.84 (1H, d, J = 8.6 Hz, ArH), 6.56 (1H, d, J = 8.6 Hz, ArH), 6.52 (2H, s, ArH), 6.48 (1H, d, J = 12.2 Hz, CH), 6.30 (1H, d, J = 12.2 Hz, CH), 5.47 (1H, s, OH), 3.86 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.67 (6H, s, OCH3), 1.79 (6H, s, CH3); 13C NMR (CDCl3, 151 MHz) δ 161.5, 152.9, 150.6, 147.1, 140.6, 140.2, 137.3, 132.5, 129.3, 128.4, 127.2, 126.6, 122.2, 120.6, 106.9, 106.0, 81.9, 61.1, 56.4, 56.0, 29.5; HRMS m/z 524.1352 [M+Na]+ (calcd for NaC25H27NO8S+, 524.1350); HPLC (Method B) 12.3 min, 98%, 0% parent (CA1).
[(Z)-2-((2-Methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)methyl)-5-nitrothiophene (43)42.
(5-Nitrothiophen-2-yl)methanol (0.100 g, 0.628 mmol), triphenylphosphine (0.336 g, 1.28 mmol), and combretastatin A-4 (0.396 g, 1.25 mmol) were dissolved in tetrahydrofuran (2 mL). DEAD (0.218 g, 1.25 mmol) was added, and the reaction mixture was stirred for 4 h at 50 °C. The reaction mixture solvent was then removed by evaporation under reduced pressure. The residue was extracted with EtOAc, and the solution was washed with water and brine, dried with Na2SO4, and evaporated under reduced pressure. Flash chromatography of the residue using a prepacked 100 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 10% A/90% B over 1.19 min (1 CV), 10% A/90% B → 67% A/33% B over 13.12 min (10 CV), 67% A/33% B over 2.38 min (2 CV); flow rate 50.0 mL/min; monitored at 254 and 280 nm] and recrystallization from EtOAc and hexanes yielded [(Z)-2-((2-methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)methyl)-5-nitrothiophene (43) (0.286 g, 0.625 mmol, 50%) as a yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.80 (1H, d, J = 4 Hz, ArH), 6.97 (1H, dd, J = 8 Hz, J = 1.5 Hz, ArH), 6.89 (1H, d, J = 4 Hz, ArH), 6.87 (1H, d, J = 2 Hz, ArH), 6.84 (1H, d, J = 8.5 Hz, ArH), 6.50 (2H, s, ArH), 6.48 (2H, d, J = 12 Hz, CH), 5.07 (2H, s, CH2), 3.89 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.72 (6H, s, OCH3); 13C NMR (125 MHz, CDCl3) δ 153.0, 149.1, 148.3, 146.4, 137.1, 132.9, 129.8, 129.2, 129.1, 128.4, 124.8, 124.0, 115.4, 111.7, 105.8, 66.4, 60.9, 56.0, 56.0; HRMS m/z 480.1088 [M+Na]+ (calcd for NaC23H23NO7S+, 480.1087); HPLC (Method A) 17.1 min, 97%, 1.5% parent (CA4).
(Z)-2-(1-(2-Methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)ethyl)-5-nitrothiophene (44)42.
Combretastatin A-4 (0.251 g, 0.79 mmol), triphenylphosphine (0.105 g, 0.400 mmol), and 1-(5-nitrothiophen-2-yl)ethanol (0.197 g, 1.14 mmol) were dissolved in dry THF (10 mL). DEAD (0.155 g, 0.890 mmol) was added dropwise, and the reaction mixture was stirred for 24 h. The reaction was quenched with water and partitioned, and the aqueous phase was extracted with EtOAc. The EtOAc phase was dried with Na2SO4 and evaporated under reduced pressure. Flash chromatography of the residue using a prepacked 25 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 15% A/85% B over 1.19 min (1 CV), 15% A/85% B → 100% A/0% B over 13.12 min (10 CV), 100% A/0% B over 2.38 min (2 CV); flow rate 25.0 mL/min; monitored at 254 and 280 nm] yielded (Z)-2-(1-(2-methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)ethyl)-5-nitrothiophene 44 (0.090 g, 0.19 mmol, 24%) as a yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.75 (1H, d, J = 4.5 Hz, ArH), 6.94 (1H, dd, J = 8 Hz, J = 2 Hz, ArH), 6.81 (3H, m, ArH), 6.46 (1H, d, J = 12.5 Hz, CH), 6.45 (2H, s, ArH), 6.44 (1H, d, J = 12 Hz, CH), 5.25 (1H, q, J = 6 Hz, CH), 3.86 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.69 (6H, s, OCH3), 1.63 (3H, d, J = 6.5 Hz, CH3); 13C NMR (125 MHz, CDCl3) δ 155.3, 153.0, 149.9, 145.7, 137.1, 132.9, 129.9, 129.2, 129.1, 128.4, 124.4, 123.0, 118.2, 112.0, 105.8, 73.8, 60.9, 55.9, 55.9, 23.1; HRMS m/z 472.1428 [M+H]+ (calcd for C24H25NO8S+, 472.1424); HPLC (Method A) 17.6 min, 98%, 0% parent (CA4).
[(Z)-2-(2-(2-Methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)propan-2-yl)-5-nitrothiophene (45)42.
Combretastatin A-4 (1.87 g, 5.91 mmol), 2-(5-nitrothiophen-2-yl)propan-2-ol (1.17 g, 6.25 mmol), and ADDP (1.46 g, 5.79 mmol) were dissolved in benzene (15 mL). Tributylphosphine (1.43 mL, 5.91 mmol) was added dropwise, and the reaction mixture was stirred for 24 h. The reaction was quenched with water, and the reaction mixture was extracted with EtOAc. The organic phase was dried with Na2SO4 and evaporated under reduced pressure. Flash chromatography of the residue using a prepacked 25 g silica column [eluents: solvent A, EtOAc; solvent B, hexanes; gradient, 15% A/85% B over 1.19 min (1 CV), 15% A/85% B → 100% A/0% B over 13.12 min (10 CV), 100% A/0% B over 2.38 min (2 CV); flow rate 25.0 mL/min; monitored at 254 and 280 nm] yielded (Z)-2-(2-(2-methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)propan-2-yl)-5-nitrothiophene (45) (0.670 g, 1.38 mmol, 23%) as a dark red oil; 1H NMR (500 MHz, CDCl3) δ 7.72 (1H, d, J = 4.2 Hz, ArH), 7.01 (1H, dd, J = 2.0 Hz, 8.4 Hz, ArH), 6.83 (1H, d, J = 8.4 Hz, ArH), 6.77 (1H, d, J = 4.2 Hz, ArH), 6.72 (1H, d, J = 2.0 Hz, ArH), 6.43 (4H, m, ArH and CH), 3.85 (3H, s, OCH3), 3.80 (3H, s, OCH3), 3.71 (6H, s, OCH3), 1.59 (6H, s, CH3); 13C NMR (125 MHz, CDCl3): δ 161.0, 152.9, 152.4, 150.3, 142.8, 136.8, 132.9, 129.5, 129.2, 129.1, 128.3, 125.8, 124.0, 122.2, 111.8, 105.7, 79.9, 60.7, 55.8, 55.6, 28.6; HRMS m/z 508.1399 [M+Na]+ (calcd for NaC25H27NO8S+, 508.1400); HPLC (Method A) 16.2 min, 97%, 0% parent (CA4); X-ray Crystallography Single crystal X-ray diffraction (CCDC 1502383) provided further structural characterization for compound 45 (see Supporting Information).
[(Z)-2-(2-(2-Methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)propan-2-yl)-5-nitrothiophene (45)42 (Alternate Purification Route).
Combretastatin A-4 (1.61 g, 5.09 mmol), 2-(5-nitrothiophen-2-yl)propan-2-ol (1.00 g, 5.34 mmol), and ADDP (1.35 g, 5.34 mmol) were dissolved in toluene (101 mL). Tributylphosphine (1.3 mL, 5.34 mmol) was added dropwise, and the reaction mixture was stirred for 20 h. The reaction mixture was diluted with EtOAc and quenched with water. The resulting mixture was extracted with EtOAc (30 mL X 3), and the organic phase was dried with Na2SO4 and evaporated under reduced pressure. The residue was purified by flash chromatography using 5–20% EtOAc-hexanes. The column-purified product contained free alcohol, CA4 and BAPC (product) in the ratio of 1.0:2.4:3.0. The amount of BAPC was 921 mg, 1.90 mmol (calculated from NMR) (theoretical yield: 2.469 g). The amount of free alcohol was 118 mg, 0.63 mmol. The amount of CA4 was 480 mg, 1.52 mmol. To a solution of these three compounds in CH2Cl2 (30.0 mL), imidazole (1.03 g, 15.2 mmol) was added, followed by TBSCl (2.52 g, 16.7 mmol). The solution was stirred at room temperature for 4 h. The reaction was quenched with water and extracted with EtOAc (30 mL X 3). The EtOAc phase was dried with Na2SO4 and evaporated under reduced pressure. The product was purified by flash chromatography using 5–20% EtOAc-hexanes. This resulted in removal of the CA4, leaving a mixture of BAPC and the free alcohol in a ratio of 3:1 by NMR (807 mg, 1.66 mmol BAPC and 103 mg, 0.55 mmol free alcohol). A mixture of BAPC, free alcohol, DMAP (67 mg, 0.55 mmol) and TEA (0.85 mL, 6.05 mmol) in CH2Cl2 (11 mL) was treated with acetic anhydride (0.52 mL, 5.5 mmol) and stirred at room temperature for 4.0 h. The reaction was quenched with water and extracted with EtOAc (30 mL X 3). The EtOAc phase was dried with Na2SO4 and evaporated under reduced pressure. The product was purified by flash chromatography using 5–15% EtOAc-hexanes. (Z)-2-(2-(2-methoxy-5-(3,4,5-trimethoxystyryl)phenoxy)propan-2-yl)-5-nitrothiophene (45) (0.760 g, 1.56 mmol, 31%) was isolated as a dark red oil. By NMR the free alcohol was no longer observed in the purified product; 1H NMR (500 MHz, CDCl3) δ 7.72 (1H, d, J = 4.2 Hz, ArH), 7.01 (1H, dd, J = 2.0 Hz, 8.4 Hz, ArH), 6.83 (1H, d, J = 8.4 Hz, ArH), 6.77 (1H, d, J = 4.2 Hz, ArH), 6.72 (1H, d, J = 2.0 Hz, ArH), 6.43 (4H, m, ArH and CH), 3.85 (3H, s, OCH3), 3.80 (3H, s, OCH3), 3.71 (6H, s, OCH3), 1.59 (6H, s, CH3); 13C NMR (125 MHz, CDCl3) δ 161.0, 152.9, 152.4, 150.3, 142.8, 136.8, 132.9, 129.5, 129.2, 129.1, 128.3, 125.8, 124.0, 122.2, 111.8, 105.7, 79.9, 60.7, 55.8, 55.6, 28.6.
Biological Evaluation
Cell culture
Non-small cell lung carcinoma A549 wt (ATCC) cells were grown and maintained in MEM-alpha media containing 10% fetal bovine serum (FBS), 17 mM D-glucose (Sigma-Aldrich), and 1% gentamycin sulfate (Teknova, Hollister, CA). These cells were maintained in a log growth phase in a humidified 37 °C incubator under 95% air plus 5% CO2.
Differential cytotoxicity.
The bioreductive assay used was modified from Jaffar et al.55 For the anoxic arm of the assay, A549 cells were introduced as a small sample into an anaerobic chamber (Coy Chamber), and then resuspended to the appropriate volume using medium that had been pre-conditioned for a minimum of 48 h in the anaerobic chamber. Cells were plated at 6000 cells/well in a 100 μL volume into plastic 96-well plates (Corning Costar) that had been degassed in the anaerobic chamber for a minimum of 48 h, with a maximum limit of 10 ppm oxygen. Cells were allowed to attach for 2 h. Serial dilutions were made from 10 mg/mL DMSO stock solutions to twice the required concentrations in preconditioned medium, and 100 μL was added per well in duplicate for each experiment. The highest final concentration of control drugs and test compounds was 50 μg/mL. Plates were incubated for 4 h under anaerobic conditions, and then for 48 h under aerobic conditions prior to SRB determination of cytotoxicity.56–59 The procedure for the normoxic arm of the assay was identical to that described above except that all manipulations were carried out under ambient conditions with drug exposure carried out under 95% air plus 5% CO2. Tirapazamine was used as a positive control as previously described.60–62
Inhibition of Tubulin Polymerization63
Tubulin assembly reaction mixtures (0.25 mL final volume)63 contained 1.0 mg/mL (10 μM) purified bovine brain tubulin, 0.8 M monosodium glutamate (pH 6.6 in a 2 M stock solution), 4% (v/v) DMSO, 0.4 mM GTP, and varying compound concentrations. Initially, all components except GTP were preincubated for 15 min at 30 °C in 0.24 mL. The reaction mixtures were placed on ice, and 10 μL of 0.01 M GTP was added. The reaction mixtures were transferred to cuvettes held at 0 °C in Beckman DU-7400 and DU-7500 spectrophotometers equipped with electronic temperature controllers. The temperature was jumped to 30 °C over about 30 s, and the polymerization reaction was followed turbidimetrically at 350 nM for 20 min. Each reaction set included a reaction mixture without compound, and the IC50 was defined as the concentration of compound that inhibited the extent of assembly by 50%.
Colchicine Binding Assay64
Inhibition of [3H]colchicine binding to tubulin was determined in reaction mixtures (100 μL) containing 1.0 μM tubulin, 5.0 μM [3H]colchicine (Perkin-Elmer), 5% (v/v) DMSO, potential inhibitors at the indicated concentrations and components demonstrated to stabilize the colchicine binding activity of tubulin64 (1.0 M monosodium glutamate [adjusted to pH 6.6 with HCl in a 2.0 M stock solution], 0.5 mg/mL bovine serum albumin, 0.1 M glucose-1-phosphate, 1.0 mM MgCl2, and 1.0 mM GTP). Only compounds showing significant activity as inhibitors of tubulin polymerization (defined as having IC50 values below 5 μM) were evaluated for inhibitory effects on colchicine binding. Incubation was for 10 min at 37 °C (at this time point the binding reaction in the control is about 50% complete). Reactions were stopped by adding 2.0 mL 0 °C water and chilling the samples to 0 °C. Each sample was poured onto a stack of two DEAE-cellulose filters, followed immediately by 6 mL of 0 °C water. The water was aspirated under a weak vacuum, and the filters were washed three times with 2 mL of water. Each set of 2 filters was placed into a scintillation vial containing 5 mL of Biosafe II scintillation cocktail. Samples were counted at least 18 h later in a Beckman scintillation counter. Samples with potential inhibitors were compared to controls with no inhibitor to determine percent inhibition. All samples were corrected for colchicine that bound to the filters in the absence of tubulin.
NADPH-Cytochrome P450 Oxidoreductase Cleavage Assay64,65
Rat NADPH-cytochrome P450 oxidoreductase (POR) supersomes and protocatechuate 3,4-dioxygenase (PCD) were purchased from Corning® and Sigma-Aldrich, respectively, and their enzymatic activities were determined in terms of enzyme units (U) with their corresponding substrates (Sigma-Aldrich), cytochrome c and procatechuate (PCA, 3,4-dihydroxybenzoic acid), respectively. All bioreductive prodrugs were dissolved in DMSO as 10 mM stock solutions. Aliquots of the compound stock solution were added to 200 mM pH 7.4 potassium phosphate buffer containing 0.1% Triton X-100 (to facilitate BAPC solubility) and 400 μM freshly dissolved protocatechuic acid (PCA). The components were fully mixed in a microvessel capped with a rubber septum stopper and subjected to three cycles of evacuation and flushing with N2 using a manifold, followed by sparging with N2 for an additional 20 min. PCD (0.08 units) was added, to remove remaining traces of O2 by PCA/PCD. POR stock (0.006 units) and NADPH (0.8 mM final concentration) were introduced sequentially. The anaerobic reaction mixture was incubated for designated times at 37 °C, cooled on ice and treated with a 2x volume of acetonitrile. After centrifugation and syringe filtration, the samples were analyzed by HPLC using a gradient of water/acetonitrile for elution. Solutions without POR were used as controls. Standard curves for CA4, CA1, and each BAPC were used for quantitation.
In Vivo Tumor Model.66
Murine 4T1-luc breast cancer cells that stably express firefly luciferase reporter66 were grown in RPMI1640 medium (Hyclone Laboratories, Logan, Utah) with 10% FBS (Sigma) under 5% CO2 at 37 °C. Induction of tumors was carried out by injecting 50 μL containing 106 cells in PBS mixed with 30% Matrigel™ (BD Biosciences, San Jose, CA) into the left upper ventral mammary fat pads of anesthetized adult female BALB/CJ mice (18–24.4 g, age 56 days, UTSW breeding colony). Mice were housed as a group of 5 in a shoe box cage with nestlet and free access to water and chow in a conventional vivarium with a 12 h light/dark cycle (6 AM / 6 PM). Tumors were allowed to grow to a size of 10–12 mm in diameter, determined by calipers (Carbon Fiber Composite Digital Caliper, Fisher Scientific), over 11 days before BLI.
In Vivo Bioluminescence Imaging (BLI).
BLI was carried out as described previously.67,68 Briefly, anesthetized, tumor bearing mice (O2, 2% isoflurane, Henry Schein Inc., Melville, NY) were injected subcutaneously in the fore-back neck region with 80 μL of a solution of luciferase substrate, D-luciferin (sodium salt, 120 mg/kg, in saline, Gold Biotechnology, St. Louis, MO). Mice were maintained under anesthesia (2% isoflurane in oxygen, 1 dm3/min) while baseline BLI was performed using a Caliper Xenogen IVIS® Spectrum (Perkin-Elmer, Alameda, CA). A series of BLI images was collected using the following settings: exposure time 0.5 to 2 s depending on signal intensity, f-stop = 2, field of view = D, binning = 4 (medium). Light intensity-time curves obtained from these images were analyzed using Living Image® software, and light emission was compared based on area under the light emission curve. Mice were injected intraperitoneally with either 120 μL of DPS vehicle, CA4P (120 mg/kg in saline), or BAPC 45 (180 mg/kg in DPS vehicle) immediately after baseline BLI was obtained. BLI was repeated, with new luciferin injections, 4, 24, and 48 h later. Surviving mice were sacrificed after 96 h by cervical dislocation under isoflurane anesthesia and tumors harvested for routine H&E histology.
Supplementary Material
Figure 2.
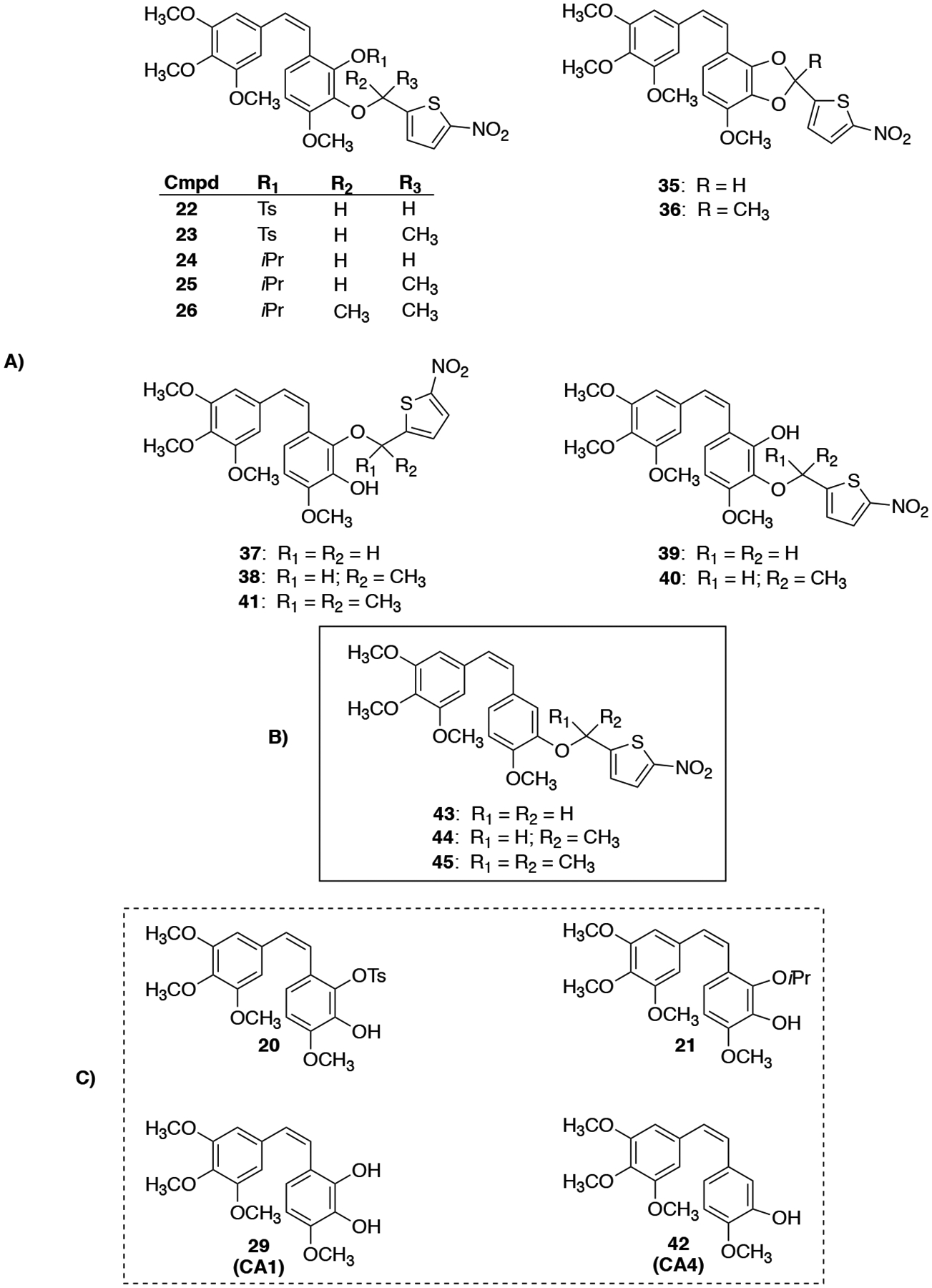
Compilation of parent anticancer agents and their corresponding BAPCs utilized in this study: A) CA1-BAPCs; B) CA4-BAPCs; C) Parent CA1 and CA4 Agents.
ACKNOWLEDGMENTS
The authors are grateful to the Cancer Prevention and Research Institute of Texas (CPRIT, Grant No. RP140399 to K.G.P., M.L.T., and R.P.M.), the National Cancer Institute of the National Institutes of Health (Grant No. 5R01CA140674 to K.G.P., M.L.T., and R.P.M.), Mateon Therapeutics, Inc. (grant to K.G.P. and M.L.T.), and the Undergraduate Research and Scholarly Activity Small Grant Program, the University Research Committee (URC), and the Vice Provost for Research at Baylor University (grants to M.L.T.) for their financial support of this project. BLI was facilitated by the Southwestern Small Animal Imaging Resource supported in part by the NIH Comprehensive Cancer Center Grant P30 CA142543 and a Shared Instrumentation Grant 1S10 RR024757. The authors also thank Dr. Michelle Nemec (Director) for the use of the shared Molecular Biosciences Center at Baylor University, the Mass Spectrometry Core Facility, Baylor University, and Dr. Kevin Klausmeyer (X-ray analysis, Baylor University). The authors are grateful to Mr. David Ross IV (Baylor University), Ms. Taylor Deushane (Baylor University), Mr. Benji Gomez (Baylor University), and Ms. Evelyn Le (Baylor University) for their contributions to the synthesis of advanced intermediates. We appreciate the technical support of Alex Winters and valuable discussions with Dr. Li Liu (UT Southwestern) and Dr. Edward Graves (Stanford University). Histology was performed by Dr. John Shelton in the Histo Pathology Core at UT Southwestern. We thank the Whole Brain Microscopy Facility (WBMF) in the UT Southwestern Department of Neurology and Neurotherapeutics for use of high resolution scanning equipment used to obtain histological images of the 4T1-luc tumors. The WBMF is supported by the Texas Institute for Brain Injury and Repair. This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 1H NMR and 13C NMR data for compounds 2–7, 10–13, 15–17, 19–31, 33–41, 43–45; HRMS and HPLC data for compounds 21–26, 29, 35–41, 43–45; X-ray crystallography data for compound 45; details regarding the synthesis of bioreductive triggers 16, 17, 19; NADPH-cytochrome P450 oxidoreductase cleavage assay HPLC traces; NOE analysis for compounds 37, 38, 41; details regarding a preliminary PK study; cytotoxicity data; and additional histology associated with BAPC 45.
One of the authors (KGP) previously served as a paid consultant with Mateon Therapeutics, Inc., and another author (DJC) served as a former Chief Scientific Officer (CSO) of Mateon Therapeutics, Inc.
DEDICATION
Dedicated to Professor George R. Pettit on the occasion of his most recent birthday and in celebration of his lifetime scientific accomplishments that continue to define and advance the fields of natural products chemistry, medicinal chemistry, and synthetic organic chemistry.
REFERENCES
- (1).Siemann DW Cancer Treat. Rev 2011, 37, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mason RP; Zhao D; Liu L; Trawick ML; Pinney KG Integr. Biol 2011, 3, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Siemann DW; Horsman MR Pharmacol. Ther 2015, 153, 107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brem S; Brem H; Folkman J; Finkelstein D; Patz A Cancer Res 1976, 36, 2807–2812. [PubMed] [Google Scholar]
- (5).Carmeliet P; Jain RK Nature 2000, 407, 249–257. [DOI] [PubMed] [Google Scholar]
- (6).Pralhad T; Madhusudan S; Rajendrakumar KJ Pharm. Pharmacol 2003, 55, 1045–1053. [DOI] [PubMed] [Google Scholar]
- (7).Siemann DW; Chaplin DJ; Horsman MR Cancer 2004, 100, 2491–2499. [DOI] [PubMed] [Google Scholar]
- (8).Horsman MR Int. J. Hyperth 2008, 24, 57–65. [DOI] [PubMed] [Google Scholar]
- (9).Kaur S; Bajwa P Clin. Transl. Oncol 2014, 16, 115–121. [DOI] [PubMed] [Google Scholar]
- (10).Prieto-Vila M; Yan T; Calle AS; Nair N; Hurley L; Kasai T; Kakuta H; Masuda J; Murakami H; Mizutani A; Seno M Am. J. Cancer Res 2016, 6, 1906–1921. [PMC free article] [PubMed] [Google Scholar]
- (11).Dark GG; Hill SA; Prise VE; Tozer GM; Pettit GR; Chaplin DJ Cancer Res 1997, 57, 1829–1834. [PubMed] [Google Scholar]
- (12).Tozer GM; Prise VE; Wilson J; Locke RJ; Vojnovic B; Stratford MR; Dennis MF; Chaplin DJ Cancer Res 1999, 59, 1626–1634. [PubMed] [Google Scholar]
- (13).Tozer GM; Akerman S; Cross NA; Barber PR; Björndahl MA; Greco O; Harris S; Hill SA; Honess DJ; Ireson CR; Pettyjohn KL; Prise VE; Reyes-Aldasoro CC; Ruhrberg C; Shima DT; Kanthou C Cancer Res 2008, 68, 2301–2311. [DOI] [PubMed] [Google Scholar]
- (14).Patterson DM; Rustin GJS Clin. Oncol 2007, 19, 443–456. [DOI] [PubMed] [Google Scholar]
- (15).Kingston DGI J. Nat. Prod 2009, 72, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pattillo CB Drugs Future 2011, 36, 385. [Google Scholar]
- (17).Shih T; Lindley C Clin. Ther 2006, 28, 1779–1802. [DOI] [PubMed] [Google Scholar]
- (18).Los M; Roodhart JML; Voest EE The Oncologist 2007, 12, 443–450. [DOI] [PubMed] [Google Scholar]
- (19).Pettit GR; Singh SB; Niven ML; Hamel E; Schmidt JM J. Nat. Prod 1987, 50, 119–131. [DOI] [PubMed] [Google Scholar]
- (20).Pettit GR; Singh SB; Hamel E; Lin CM; Alberts DS; Garcia-Kendal D Experientia 1989, 45, 209–211. [DOI] [PubMed] [Google Scholar]
- (21).Pettit GR; Temple C; Narayanan VL; Varma R; Simpson MJ; Boyd MR; Rener GA; Bansal N Anticancer. Drug Des 1995, 10, 299–309. [PubMed] [Google Scholar]
- (22).Pettit GR; Singh SB; Boyd MR; Hamel E; Pettit RK; Schmidt JM; Hogan FJ Med. Chem 1995, 38, 1666–1672. [DOI] [PubMed] [Google Scholar]
- (23).Tozer GM; Kanthou C; Baguley BC Nat. Rev. Cancer 2005, 5, 423–435. [DOI] [PubMed] [Google Scholar]
- (24).Pinney KG; Pettit GR; Trawick ML; Jelinek C; Chaplin DJ In Anticancer Agents from Natural Products; CRC Press, Taylor and Francis Group, 2011. [Google Scholar]
- (25).Pettit GR; Lippert JW Anticancer. Drug Des 2000, 15, 203–216. [PubMed] [Google Scholar]
- (26).Pettit GR; Grealish MP; Herald DL; Boyd MR; Hamel E; Pettit RK J. Med. Chem 2000, 43, 2731–2737. [DOI] [PubMed] [Google Scholar]
- (27).Patterson DM; Zweifel M; Middleton MR; Price PM; Folkes LK; Stratford MRL; Ross P; Halford S; Peters J; Balkissoon J; Chaplin DJ; Padhani AR; Rustin GJS Clin. Cancer Res 2012, 18, 1415–1425. [DOI] [PubMed] [Google Scholar]
- (28).Lin CM; Singh SB; Chu PS; Dempcy RO; Schmidt JM; Pettit GR; Hamel E Mol. Pharmacol 1988, 34, 200–208. [PubMed] [Google Scholar]
- (29).Pettit GR; Rhodes MR; Herald DL; Chaplin DJ; Stratford MR; Hamel E; Pettit RK; Chapuis JC; Oliva D Anticancer. Drug Des 1998, 13, 981–993. [PubMed] [Google Scholar]
- (30).Bevacizumab With or Without Fosbretabulin Tromethamine in Treating Patients With Recurrent or Persistent Ovarian Epithelial, Fallopian Tube, or Peritoneal Cavity Cancer - Full Text View - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01305213 (accessed Aug 16, 2018).
- (31).A Safety and Efficacy Study of Carboplatin, Paclitaxel, Bevacizumab and CA4P in Non-Small Cell Lung Cancer - Full Text View - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00653939 (accessed Aug 16, 2018).
- (32).Pettit GR; Rhodes MR Anticancer. Drug Des 1998, 13, 183–191. [PubMed] [Google Scholar]
- (33).Vaupel P Semin. Radiat. Oncol 2004, 14, 198–206. [DOI] [PubMed] [Google Scholar]
- (34).Wilson WR; Hay MP Nat. Rev. Cancer 2011, 11, 393–410. [DOI] [PubMed] [Google Scholar]
- (35).Chaplin DJ; Olive PL; Durand RE Cancer Res 1987, 47, 597–601. [PubMed] [Google Scholar]
- (36).Brown JM Br. J. Radiol 1979, 52, 650–656. [DOI] [PubMed] [Google Scholar]
- (37).Brown JM In Methods in Enzymology; Oxygen Biology and Hypoxia; Academic Press, 2007; Vol. 435, pp 295–321. [Google Scholar]
- (38).Horsman MR; Mortensen LS; Petersen JB; Busk M; Overgaard J Nat. Rev. Clin. Oncol 2012, 9, 674–687. [DOI] [PubMed] [Google Scholar]
- (39).von Pawel J; von Roemeling R; Gatzemeier U; Boyer M; Elisson LO; Clark P; Talbot D; Rey A; Butler TW; Hirsh V; Olver I; Bergman B; Ayoub J; Richardson G; Dunlop D; Arcenas A; Vescio R; Viallet J; Treat JJ Clin. Oncol 2000, 18, 1351–1359. [DOI] [PubMed] [Google Scholar]
- (40).Duan J-X; Jiao H; Kaizerman J; Stanton T; Evans JW; Lan L; Lorente G; Banica M; Jung D; Wang J; Ma H; Li X; Yang Z; Hoffman RM; Ammons WS; Hart CP; Matteucci MJ Med. Chem 2008, 51, 2412–2420. [DOI] [PubMed] [Google Scholar]
- (41).Winn BA; Shi Z; Carlson GJ; Wang Y; Nguyen BL; Kelly EM; Ross RD; Hamel E; Chaplin DJ; Trawick ML; Pinney KG Bioorg. Med. Chem. Lett 2017, 27, 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Thomson P; Naylor MA; Everett SA; Stratford MRL; Lewis G; Hill S; Patel KB; Wardman P; Davis PD Mol. Cancer Ther 2006, 5, 2886–2894. [DOI] [PubMed] [Google Scholar]
- (43).Tanpure RP; Strecker TE; Chaplin DJ; Siim BG; Trawick ML; Pinney KG J. Nat. Prod 2010, 73, 1093–1101. [DOI] [PubMed] [Google Scholar]
- (44).Tanpure RP; Nguyen BL; Strecker TE; Aguirre S; Sharma S; Chaplin DJ; Siim BG; Hamel E; Lippert JW; Pettit GR; Trawick ML; Pinney KG J. Nat. Prod 2011, 74, 1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mitsunobu O; Yamada M Bull. Chem. Soc. Jpn 1967, 40, 2380–2382. [Google Scholar]
- (46).Wittig G; Schöllkopf U Chem. Ber 1954, 87, 1318–1330. [Google Scholar]
- (47).Pettit GR; Toki B; Herald DL; Verdier-Pinard P; Boyd MR; Hamel E; Pettit RK J. Med. Chem 1998, 41, 1688–1695. [DOI] [PubMed] [Google Scholar]
- (48).Monk KA; Siles R; Hadimani MB; Mugabe BE; Ackley JF; Studerus SW; Edvardsen K; Trawick ML; Garner CM; Rhodes MR; Pettit G; Pinney KG Bioorg. Med. Chem 2006, 14, 3231–3244. [DOI] [PubMed] [Google Scholar]
- (49).Pulaski BA; Ostrand-Rosenberg S Cancer Res 1998, 58, 1486–1493. [PubMed] [Google Scholar]
- (50).Tao K; Fang M; Alroy J; Sahagian GG BMC Cancer 2008, 8, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhang Y; Hong H; Nayak TR; Valdovinos HF; Myklejord DV; Theuer CP; Barnhart TE; Cai W Angiogenesis 2013, 16, 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Baklaushev VP; Grinenko NF; Yusubalieva GM; Abakumov MA; Gubskii IL; Cherepanov SA; Kashparov IA; Burenkov MS; Rabinovich EZ; Ivanova NV; Antonova OM; Chekhonin VP Bull. Exp. Biol. Med 2015, 158, 581–588. [DOI] [PubMed] [Google Scholar]
- (53).Wankhede M; Dedeugd C; Siemann DW; Sorg BS Oncol. Rep 2010, 23, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhao D; Richer E; Antich PP; Mason RP FASEB J 2008, 22, 2445–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jaffar M; Phillips RM; Williams KJ; Mrema I; Cole C; Wind NS; Ward TH; Stratford IJ; Patterson AV Biochem. Pharmacol 2003, 66, 1199–1206. [DOI] [PubMed] [Google Scholar]
- (56).Siles R; Ackley JF; Hadimani MB; Hall JJ; Mugabe BE; Guddneppanavar R; Monk KA; Chapuis J-C; Pettit GR; Chaplin DJ; Edvardsen K; Trawick ML; Garner CM; Pinney KG J. Nat. Prod 2008, 71, 313–320. [DOI] [PubMed] [Google Scholar]
- (57).Monks A; Scudiero D; Skehan P; Shoemaker R; Paull K; Vistica D; Hose C; Langley J; Cronise P; Vaigro-Wolff AJ Natl. Cancer Inst 1991, 83, 757–766. [DOI] [PubMed] [Google Scholar]
- (58).Vichai V; Kirtikara K Nat. Protoc 2006, 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- (59).Voigt W Methods Mol. Med 2005, 110, 39–48. [DOI] [PubMed] [Google Scholar]
- (60).Saunders MP; Patterson AV; Chinje EC; Harris AL; Stratford IJ Br. J. Cancer 2000, 82, 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Siim BG; Pruijn FB; Sturman JR; Hogg A; Hay MP; Brown JM; Wilson WR Cancer Res 2004, 64, 736–742. [DOI] [PubMed] [Google Scholar]
- (62).Błaszczak-Świątkiewicz K; Mikiciuk-Olasik E; Błaszczak-Świątkiewicz K; Mikiciuk-Olasik E Molecules 2014, 19, 15361–15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hamel E Cell Biochem. Biophys 2003, 38, 1–22. [DOI] [PubMed] [Google Scholar]
- (64).Hamel E; Lin CM Biochim. Biophys. Acta BBA - Gen. Subj 1981, 675, 226–231. [DOI] [PubMed] [Google Scholar]
- (65).Guengerich FP; Martin MV; Sohl CD; Cheng Q Nat. Protoc 2009, 4, 1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Vilalta M; Rafat M; Giaccia AJ; Graves EE Cell Rep 2014, 8, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Liu L; Beck H; Wang X; Hsieh H-P; Mason RP; Liu X PLOS ONE 2012, 7, e43314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Strecker TE; Odutola SO; Lopez R; Cooper MS; Tidmore JK; Charlton-Sevcik AK; Li L; MacDonough MT; Hadimani MB; Ghatak A; Liu L; Chaplin DJ; Mason RP; Pinney KG; Trawick ML Cancer Lett 2015, 369, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Niu H; Strecker TE; Gerberich JL; Campbell JW; Saha D; Mondal D; Hamel E; Chaplin DJ; Mason RP; Trawick ML; Pinney KG J. Med. Chem 2019, 62, 5594–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.