

Abstract
Introduction:
TANK-binding kinase 1 (TBK1) is a Ser/Thr kinase with a central role in coordinating the cellular response to invading pathogens and regulating key inflammatory signaling cascades. While intact TBK1 signaling is required for successful anti-viral signaling, dysregulated TBK1 signaling has been linked to a variety of pathophysiologic conditions, including cancer. Several lines of evidence support a role for TBK1 in cancer pathogenesis, but the specific roles and regulation of TBK1 remain incompletely understood. A key challenge is the diversity of cellular processes that are regulated by TBK1, including inflammation, cell cycle, autophagy, energy homeostasis, and cell death. Nevertheless, evidence from pre-clinical cancer models suggest that targeting TBK1 may be an effective strategy for anti-cancer therapy in specific settings.
Areas covered:
This review provides an overview of the roles and regulation of TBK1 with a focus on cancer pathogenesis and drug targeting of TBK1 as an anti-cancer strategy. Relevant literature was derived from a PubMed search encompassing studies from 1999 to 2020.
Expert Opinion:
TBK1 is emerging as a potential target for anti-cancer therapy. Inhibition of TBK1 alone may be insufficient to restrain the growth of most cancers, hence combination strategies will likely be necessary. Improved understanding of tumor-intrinsic and tumor-extrinsic TBK1 signaling will inform novel therapeutic strategies.
Keywords: TBK1, cancer, immunotherapy, innate immunity, targeted therapy
1. Introduction
Tumor necrosis factor (TNF) receptor-associated factor (TRAF) family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1), is a serine/threonine-protein kinase that regulates innate immune responses by coordinating activation of key transcription factors including interferon regulatory factor 3 (IRF3) and NF-κB pathways [1–4]. TBK1 is a member of the IκB kinase (IKK) protein kinase family members that include canonical IKKs such as IKKα, IKKβ, and IKKγ (NEMO) and the noncanonical IKK members such as IKKε (IKBKE) and TBK1, with established roles coordinating innate immune response by inducing type I interferon (IFN) and modulating NFκB signaling [2]. Recognition of cytoplasmic nucleic acids by DNA/RNA sensors activates TBK1 leading to phosphorylation and activation of interferon regulatory factor 3 and 7 (IRF3/7), key transcription factors regulating expression of type I interferons (IFNs; IFN-α/β). Type I IFNs can induce the expression of numerous antiviral genes called interferon-stimulated genes (ISGs) to build an antiviral state and limit viral replication [1].
In addition to its well-established role in coordinating innate immune signaling, additional roles for TBK1 have been described, including regulation of inflammation, autophagy, cell cycle, cell death, and metabolism. Dysregulated TBK1 signaling has been implicated in several human disease states, although there has been an increasing interest in the role of TBK1 in cancer pathogenesis [5–10]. Despite the growing interest in studying the role(s) and regulation of TBK1 in cancer, the precise molecular mechanisms governing TBK1 signaling and its subsequent impact on cancer biology remain incompletely understood. In this article, we will review the structure and function of TBK1, including its cellular roles, mechanisms of regulation, role in cancer pathogenesis, and therapeutic targeting in cancer. We searched pubmed.ncbi.nlm.nih.gov for the following search terms with various combinations: TBK1, IKK, inhibitors, cancer, tumor microenvironment. We reviewed relevant papers from 1999 to 2020.
2. TBK1: From gene to enzyme
The gene encoding human TBK1 (TBK1), also referred to as NF-κB-activating kinase (NAK) or T2K, and is located at 12q14.1, contains 21 exons, giving rise to a 729 amino acid (84 kDa) protein [11–13] (Fig. 1). TBK1 is constitutively expressed across all tissues, whereas its homolog IKKε (IKBKE, also called IKK-i) is preferentially expressed in immune cells and is induced during inflammation [14,15]. Additionally, higher TBK1 expression has been observed in fibroblasts, cells in the CNS (central nervous system), skin, adipocytes, and cancer cells/tumors [5–7,16]. An alternatively spliced form of TBK1 (named TBK1s) that lacks exons 3-6 has been described in human and mouse cells and is suggested to inhibit pathways normally activated by full length TBK1 [17]. Additional splice variants have been described in zebrafish (TBK1_tv1 and TBK1_tv2) that also lack portion of the kinase domain and the ubiquitin-like domain (see section 2.2) were shown to negatively regulate TBK1-mediated innate immune signaling [18].
Fig. 1. TBK1 protein structure and post-translational modifications.
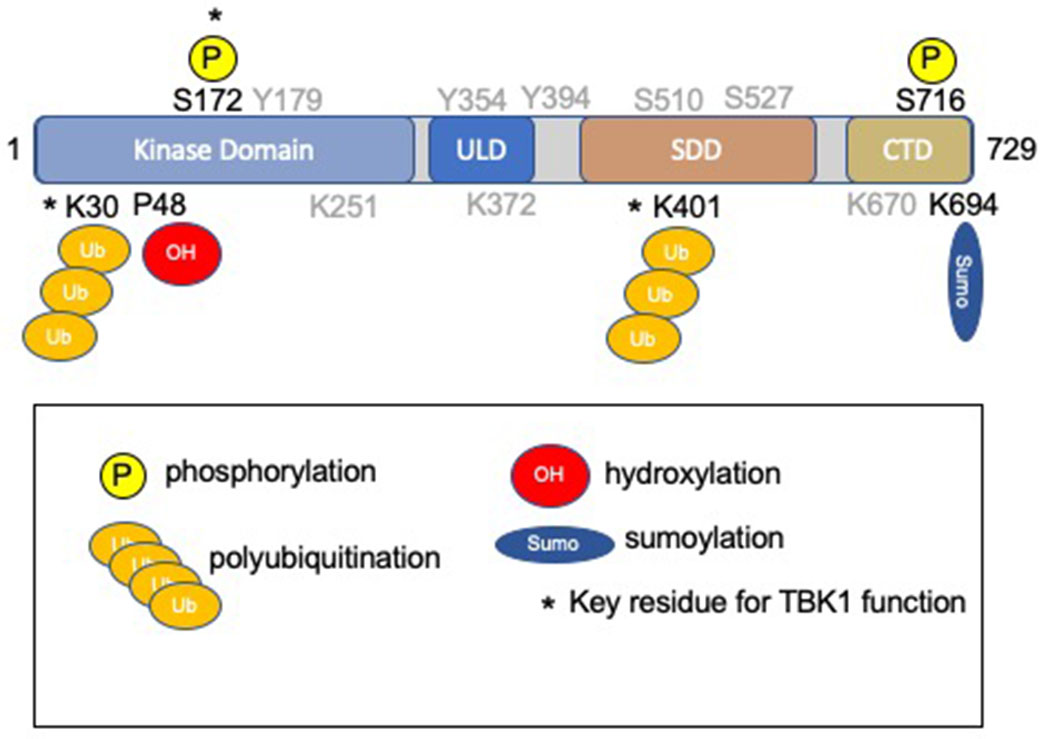
TBK1 is a 729 amino acids in length and is comprised of four domains: the kinase domain (a.a. 1-307), the ubiquitin-like domain (a.a. 310-385), the scaffold/dimerization domain (a.a. 407-657), and the C-terminal domain (a.a. 657-729) [2,19,48]. The kinase domain contains the activation loop (Leu164-Gly199), including Ser172. Key post-translational modifications required for enzyme function [19] are indicated (*). Additional residues subject to post-translational modification are indicated in gray and are described and reviewed elsewhere [2]. Hydrophobic residues (Leu 316, Ile 353, and Val 382) within the ULD have been implicated in protein-protein interactions.
TBK1 is highly conserved in mammals, with the human TBK1 polypeptide sharing 99% homology with its mouse ortholog [12]. TBK1 is composed of an N-terminal kinase domain (KD, residues 1–307) that houses its catalytic activity and three accessory/regulatory elements, along with a ubiquitin-like domain (ULD, 310-385), the scaffold/dimerization domain (SDD, 407-657), and a C-terminal domain (CTD, 657-729) involved in adaptor-binding [19] (Fig.1). The KD of TBK1 is comprised of two lobes termed N-terminal and C-terminal lobes, with the active site situated between these two lobes. The KD also contains an activation loop (Leu164-Gly199) which includes Ser172 that can be phosphorylated and induce TBK1 activation [20,21]. Phosphorylation of Ser172 results in a conformational change of the activation loop. The activation loop retracts towards and interacts with its own KD allowing substrate binding. Ser172 can be phosphorylated by upstream signaling or by autophosphorylation from the dimerization kinase [21]. While TBK1 integrates responses from a variety of extracellular and intracellular stimuli, the role of potential upstream kinases remains unresolved [22]. The dimerization domain can form a homodimer or a heterodimer with other IKKs [20]. As a result, there is transphosphorylation of Ser172 and activation of the kinase [21]. Proteins phosphatases (e.g. SHP1/2, PP4, Cdc25a, PPM1A, and PPM1B [23–27] limit TBK1 activity by removing the activating phosphorylation on Ser172. Additional phosphorylation events have been described for TBK1 [2]. Most recently, phosphorylation of TBK1 on Ser716 by protein kinase C θ (PKCθ) was demonstrated in response to growth factor signaling [28]. Lysine 38 is a conserved residue within the kinase domain (shared with IKKα, IKKβ, and IKKε), and when mutated to methionine (K38M) or alanine (K38A) rendering a kinase-deficient mutant of TBK1 that has been used in structural and functional studies to assess the TBK1 activity [11,12,14,19].
K63-linked polyubiquitination of TBK1 on residues Lys30 and Lys401 is a posttranslational modification that is essential for Ser172 phosphorylation, activation, and downstream signaling [19]. Several TRAF family E3 ubiquitin ligases, including MIB1, MIB2 RNF128, RNF144B, and RNF41/Nrdp1, have been shown to promote K63-polyubiquitination of TBK1 [29–32]. Thus, CYLD (cylindromatosis deubiquitinase) and ubiquitin-specific protease 2b (USP2b) have been shown to limit TBK1 activation by removing K63-linked ubiquitin chains [33,34]. In contrast, K48-linked polyubiquitination promotes TBK1 degradation serving as a mechanism to limit TBK1 signaling, providing a negative feedback loop on TBK1 activation [35–37]. Additional regulatory factors influencing TBK1 ubiquitination have been described [38], as well as SUMO-ylation on Lys694 which appears to contribute to anti-viral signaling [39]. A recent study suggested that TBK1 itself can serve as an E3-ligase and induce its own ubiquitination in vitro [40], although the significance of this finding is unknown and requires further exploration.
The ULD is a regulatory component of TBK1 and is involved in the control of kinase activation, substrate presentation, and downstream signaling pathways [19]. Various adaptor proteins can bind TBK1, regulating its localization, activation and participation in different signaling pathways [41–43]. Three residues in TBK1, Leu316, Ile353, and Val382, are suspected to be involved in these protein-protein interactions, as well as, the hydrophobic patch of the ubiquitin residues. For example, the adaptor proteins NAP1/AZI2, TANK, and Sintbad/TBKBP1 can bind the adaptor motif of TBK1 in a mutually exclusive manner and direct different subcellular localization downstream pathway [42]. NAP1/AZI2 and Sintbad/TBKBP1 are localized diffusely throughout the cytoplasm, and their association leads towards autophagy, whereas TANK is punctate in the perinuclear region, and its binding results in the induction of IFNα and IFNβ [32,41]. Along with NAP1/AZI2, TANK, and Sintbad/TBKBP1 [44], DDX3 can regulate type I IFN production by direct interaction with TBK and IKKε [45]. A variety of other direct protein-protein interactions with TBK1 have been described to influence TBK1 activity (positively or negatively), as well as, TBK1 localization, most of which have been characterized in the setting of anti-viral signaling and type I IFN [2]. Some of the negative regulators appear to have a physiologic role in completing a negative feedback loop to limit excess type I IFN signaling [46], or integrating cellular stress response with anti-viral responses [47]. Specific substrates of TBK1 and additional interactions are expertly reviewed elsewhere [2,48]. Below we will review specific roles and regulation of TBK1 in the context of cellular processes implicated in cancer pathogenesis and/or anti-cancer activity related to TBK1 inhibition.
3 -. Cellular Roles of TBK1
3.1. Anti-viral and innate immune response.
TBK1 is a key signaling kinase in regulating the innate immune response to invading pathogens, especially viruses [1]. Pattern recognition receptors (PRRs) recognize specific components of invading pathogens to initiate the innate immune response (Figure 2) [49]. Following recognition of double stranded (ds)-RNA (via TLR3-TRIF), LPS (via TLR4-TRIF), viral RNA (via RIG-I-MAVS), and dsDNA (via cGAS-STING) [50], TBK1 promotes activation of key transcription factors, namely IRF3/IRF7 [51] and NF-κB [16]. The mechanism of TBK1 activation in response to cytoplasmic dsDNA has been extensively characterized and involves generation of the cyclic dinucleotide 2′3′-cGAMP, a second messenger generated by cyclic GMP-AMP synthase (cGAS) [52,53]. Stimulator of interferon genes (STING, TMEM173) is an ER-resident adaptor protein that lacks kinase activity and oligomerizes following binding of cGAS-derived 2′3′-cGAMP [54], and then serves as a platform for the recruitment and activation of TBK1 and IRF3 [50,55]. After direct binding of STING to TBK1, TBK1 phosphorylates STING on Ser366, in a process that appears to requires oligomerization of both proteins [55], and enhances binding to TBK1 via STING’s C-terminal domain [54]. TBK1 (and IKKε) directly phosphorylate and activate IRF3, promoting dimerization and translocation into the nucleus to increase transcription of type I IFN [43,56]. Recent studies have suggested that activation of IRF3 signaling by TBK1 requires translocation of TBK1 to the Golgi in response to viral sensing promotes transphosphorylation and activation [57]. The K30/K401-ubiquitinated and S172-phosphorylated TBK1 localizes to the Golgi apparatus after the stimulation of RIG-I-like receptors or TLR3. Relocalization of K38/K401-ubiquitinated TBK1 requires the ubiquitin-binding protein optineurin (OPTN) following RLR or TLR3 stimulation. Golgi proximal OPTN foci appear to regulate innate immune signaling and cytokine production via interactions with TBK1 and the linear ubiquitin assembly complex (LUBAC) [58]. Of note, these OPTN foci also contain ATG9A, a transmembrane protein involved in autophagy that cycles between the Golgi and the endocytic pathway.
Fig. 2. TBK1 and Innate Immune Signaling.
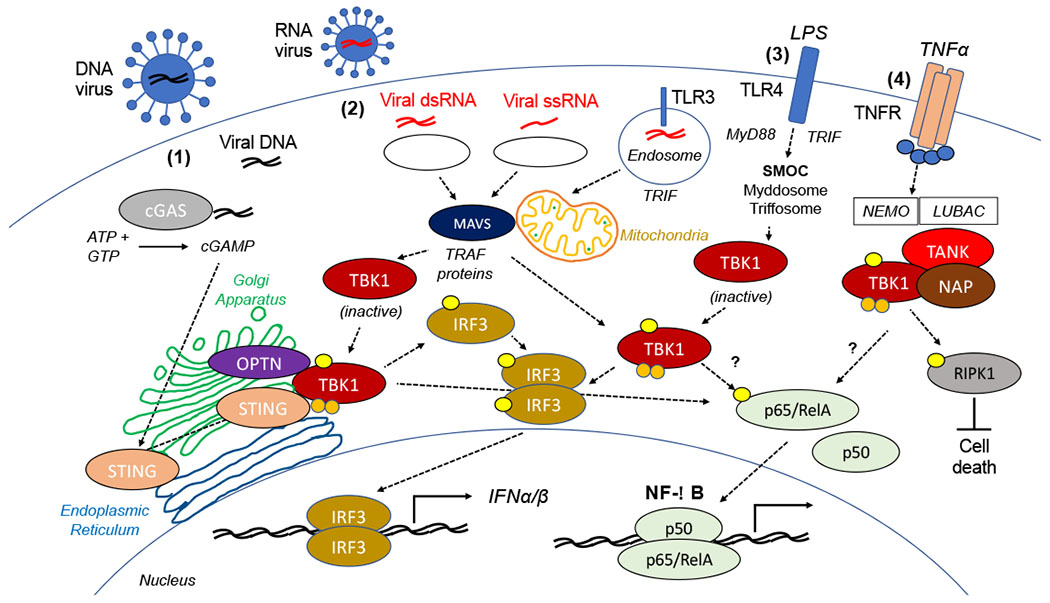
In response to invading pathogens, (1) Cytosolic DNA (e.g. viral dsRNA) is recognized by cyclic GMP-AMP synthase (cGAS) which converts ATP and GTP to the second messenger, cyclic GMP- AMP (cGAMP). cGAMP activates STING (TMEM173) promoting recruitment of TBK1. STING promotes activation of TBK1 and subsequent phosphorylation and dimerization of IRF3. Translocation of IRF3 to the nucleus promotes transcription of type I inteferons (IFNα/β). (2) In response to a viral infection, cytosolic ssRNA or dsRNA are recognized by RIG-I like receptors (RLRs) and Toll-like receptor 3 (TLR3). Signals from retinoic acid-inducible gene I (RIG-I, DDX58) and melanoma differentiation-associated protein 5 (MDA5, IFIH1) are transduced through mitochondrial antiviral signaling protein (MAVS; a.k.a. IPS1, VISA, CARDIF) to activation IRF3 and NF-κB signaling through TBK1 and TRAF proteins [204]. Translocation of K38/K401-polyubiquitinated and pSer172 phosphorylated TBK1 to the Golgi apparatus following RLR or TLR3 stimulation requires the ubiquitin-binding protein optineurin (OPTN) to promote IRF3/IFN type I signaling [57]. (3) In response to LPS, Toll-like receptor 4 (TLR4) is regulated by two supramolecular organizing complexes (SMOCs), the myddosome (containing MyD88) and the triffosome (containing Trif) [113]. This pathway can active IRF3 and NF-κB signaling, as well as promote metabolic changes (e.g. regulation of glycolysis). (4) TBK1 (and IKKε) limit cell death following tumor necrosis factor α (TNFα) treatment by promoting the phosphorylation of RIPK1 [97]. This requires NEMO (IKKγ), the linear-ubiquitin chain assembly complex (LUBAC), and the known TBK1/IKKε interacting proteins NAP1 and TANK. TANK-dependent TBK1 signaling has been shown to promote NF-κB activation [11], although the precise role of TBK1 in regulation NF-κB signaling remains unclear.
Viruses have evolved a variety of strategies to directly or indirectly inhibit the function of TBK1 and its downstream target IRF3 to blunt the production of key anti-viral cytokines, namely the type I interferons (IFNα/β) [1,59]. Bluetongue virus NS3 protein binds OPTN at the Golgi apparatus, neutralizing its activity and thereby decreasing TBK1 activation and downstream signaling, and impairing TBK1 targeting to the Golgi apparatus [57]. Proteases of foot-and-mouth disease virus (FMDV) and mouse hepatitis virus A59 (MHV-A59) inhibit ubiquitination of TBK1 [3,4]. Others like herpes simplex viruses (HSV), hepatitis C virus (HCV) , vaccinia virus (VACV), severe acute respiratory syndrome (SARS), coronavirus and hantavirus suppress TBK1-containing complex formation and downstream signaling [60–64], whereas Borna disease virus (BDV) P protein is phosphorylated by TBK1 potentially acting as a decoy substrate [65].
Impaired type I interferon responses have been observed in patients with COVID-19 [66,67], and recent studies have confirmed impaired activation of TBK1 and blunted IFNβ production following SARS-CoV-2 infection [68]. Evaluation of host protein interaction networks for SARS-CoV-2 open reading frames (ORFs) identified an interaction between non-structural protein 13 (nsp13) and TBK1 [69]. Interestingly, multiple SARS-CoV-1 viral proteins (M protein, papain-like protease, helicase) are capable of disrupting TBK1-IRF3-IFN type I signaling [62,70,71]. As a key signaling molecule in the crossroads of anti-viral response and induction of inflammatory cytokines [72], disruption of TBK1 signaling may be an important part of the pathophysiology of severe COVID-19 and efforts to restore type I IFN signaling may prove useful [73,74]. Additionally, the role of host variations in TBK1 function may impact disease severity, as has been demonstrated in childhood herpes simplex encephalitis (HSE) in which heterozygous mutations in TBK1 has been observed [75].
3.2. Autophagy.
Autophagy promotes clearance of damaged cellular proteins and organelles and is a key source of energy and survival for cells under stress [76]. Autophagy is part of the cell maintenance mechanism but is also involved in various pathological conditions, including cancer [77]. TBK1-null cells exhibit basal dysregulation of autophagy, exhibiting increased levels of the autophagic marker ubiquitin-like microtubule-associated protein light chain 3 (LC3) [10,78]. Stimulated autophagy in response to invading bacteria [79] or mycobacteria [10] is also defective in TBK1-null cells. TBK1 has been shown to interact with several autophagy receptors and adaptor proteins, including optineurin (OPTN) [79], p62 (SQSTM1) [10], NDP52, and TAX1BP1 [80]. OPTN is phosphorylated by TBK1 on Ser177 [79] leading to enhanced interaction of OPTN with the family of the ATG family (Atg8) that are essential for autophagy. In response to bacterial lipopolysaccharide (LPS), TBK1 activates to autophagy pathway by interacting with optineurin (OPTN) to promote effective bacterial clearance [59]. TBK1 has also been shown to sequester the autophagy/xenophagy cargo receptor Ndp52 [80] and promote phosphorylation of p62/SQSTM1 on Ser403 upon mitochondrial damage or bacterial attack to promote autophagy [81]. More recently, TBK1 was shown to phosphorylate syntaxin 17 (Stx17) on S202 to promote the formation of the pre-autophagosomal structures [82].
TBK1 is also involved in mitophagy, a form of autophagy that targets damaged mitochondria for degradation [83]. Removal of dysfunctional mitochondria involves the polyubiquitination of damaged mitochondria and targeting for autophagosomal degradation [83]. PINK1 and Parkin are established mediators of mitophagy, that promote the translocation and activation of TBK1 to damaged mitochondria. Following assembly of ubiquitin chains on mitochondria, adapter proteins recruit and activate TBK1 in a complex containing NDP52, OPTN, and SQSTM1 (p62) [84]. Upon mitochondrial damage or depolarization, TBK1 can be recruited to the mitochondria by NDP52 to promote mitophagy [85]. Recently, TBK1 was shown to promote activation of the PINK1-PARKIN pathway via phosphorylation of RAB7A on Ser72 [86]. PINK1/Parkin-mediated activation of TBK1 at the mitochondria during mitophagy was shown to induce a block in mitosis due to the sequestration of TBK1 from its physiological role at centrosomes [87]. It is also reported that Zika virus (ZIKV) infection of neuro-epithelial stem cells and radial glial cells induces mitochondrial translocation of TBK1 associated with loss of centrosomal localization [88].
In addition to these reports which show a role for TBK1 in the regulation of autophagy/mitophagy, there is evidence to suggest that autophagy also regulates TBK1 as part of a negative feedback loop. Yang and colleagues demonstrated that cells lacking ATG5 exhibited enhanced activation of TBK1 and increased production of inflammatory cytokines following challenge with interleukin-1β [89]. Interestingly, turnover of upstream stimulators of TBK1 activation such as STING are also subject to regulation by autophagy [90]. Following challenges with dsDNA, STING levels decrease in a matter of hours before returning to baseline levels. Decrease in protein levels results from targeted degradation of ubiquitinated STING in a p62-dependent manner. In TBK1- or IRF3-null cells, p62 is not activated and STING levels remain constant. These findings suggest that these mechanisms are normally subject to tight feedback regulation. Dysregulation of such feedback mechanisms may underlie certain disease states, such as neurodegenerative diseases, metabolic diseases, aging and cancer [91–93].
3.3. Cell cycle.
TBK1 is induced at mitosis and directly phosphorylates the mitotic kinase, Polo-like kinase 1 (PLK1) and its upstream regulator AKT [42]. In line with that, TBK1 activating phosphorylation on pSer172, was increased during mitosis, and loss of TBK1 blocked mitotic-associated phosphorylation of PLK1. Other substrates of mitosis including CEP170 and NUMA, that colocalize with TBK1 in centrosomes [94]. TBK1 is necessary for CEP170 centrosomal localization and binding to the microtubule depolymerase Kif2b, as well as, for NUMA binding to the microtubule motor dynein. Inhibition of TBK1 binding to CEP170 results in microtubule instability and defects in mitosis. Optineurin and the PINK1/Parkin pathway regulate mitophagy via TBK1 in a cell-cycle dependent manner [87,95], providing an intriguing connection between autophagy/mitophagy, the cell cycle, and innate immune signaling. This biology may have implications for cancer cell growth, as recruitment of TBK1 to the mitochondria (by PINK1/Parkin) results in a G2/M block due to loss of TBK1 at centrosomes and mitotic spindles. In A549 cancer cells, inhibition of TBK1 led to an increase in tetraploid cells indicating a role for TBK1 in mitotic progression [94] and suggesting that impaired TBK1 signaling in cancer cells may impact successful chromosome segregation. The impact of TBK1 deletion or inhibition on accumulated chromosomal alterations, both acutely and over time, as well as the upstream signals promoting TBK1 activation before entering mitosis, remain unclear and will require further investigation.
3.4. Cell death.
TBK1 and IKKε are now known to restrict cell death in multiple settings [96], particularly following challenge with TNFα [97]. Initial clues linking TBK1 to cell death came from studies characterizing TBK1-knockout mice which die at day 13.5-14.5 owing to massive hepatocyte cell death [13], a phenotype shared by mice lacking other key components of TNF signaling, including p65/RelA, IKKβ, and IKKγ/NEMO [98]. TBK1 was initially characterized as a component of a multi-protein signaling complex (along with TANK and TRAF2) downstream of the TNF receptor (TNFR) [11] capable of activating NF-κB [11,12]. TBK1 is recruited to TNF receptor 1 (TNFR1) following TNFα stimulation [99], leading to formation of a membrane-associated signaling complex (complex I) that contains several adaptor proteins, kinases (e.g. RIPK1, TAK1), and ubiquitin E3 ligases (cIAP1, cIAP2, LUBAC) [96]. TNF-induced apoptosis is restrained by activation of the IκB kinase (IKK) signaling complex and subsequent activation of NF-κB and upregulation of anti-apoptotic and inflammatory signaling pathways [100,101]. Following LUBAC-mediated activation, TBK1 phosphorylates RIPK1 in the TNFR1 signaling complex, thereby preventing RIPK1-dependent cell death [97]. TNF/TNFR1-dependent embryonic lethality of Tbk1-null mice [13,102,103], can also be blocked by inactivating mutations in RIPK1 [102]. Therefore, in response to TNFα challenge, cells lacking TBK1 fail to phosphorylate RIPK1 and the TNFR-associated signaling complex I is unable to assemble [96]. Instead, TNFR signaling promotes formation secondary cell death signaling complex (complex II), leading to caspase 8-dependent apoptosis or RIPK3-dependent necroptosis [104,105]. These findings suggest that targeting TBK1 (and/or IKKε) sensitizes cells to undergo necroptosis, specifically following exposure to TNFα. There are clear implications for TBK1-targeted therapeutic development in cancer, but also to the pathophysiology of certain neurodegenerative conditions. Heterozygous loss-of-function mutations in TBK1 have been described in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [85,106,107], and dysregulated TBK1 signaling may underlie promote neuroinflammation and cell death [108]. The role of kinase-independent functions of TBK1 in modulating cell death signaling remains poorly understood, although embryonic lethality of TBK1-null mice could be rescued using a truncated allele (TBK1Δ/Δ) which lacks kinase activity [12].
3.5. Metabolism.
Several studies have demonstrated a role for TBK1 in the regulation of cellular metabolism and energy homeostasis in both normal and cancer cells. In normal adipocytes, expression and activation of TBK1 is increased in mice fed with a high fat diet (HFD mice) [6,109]. Mice expressing a mutant version of TBK1, lacking enzymatic activity [109], and mice with adipose-specific TBK1 deletion were resistant to HFD induced obesity [6]. TBK1 loss promoted insulin resistance and glucose intolerance via increased NF-κB driven inflammation.
Tumor cells maintain rapid growth by switching to glycolysis for a needed supply of energy. In this process, glucose uptake is elevated in the cancer cells and this appears to be controlled by increased expression of glucose transporters [110]. It was reported that upon activation of RalA, downstream of RAS, TBK1 phosphorylates the exocyst protein Exo84, which leads to translocation of the GLUT4 glucose transporter to the cell membrane [111]. TBK1 also phosphorylates the insulin receptor (thereby blocking its activity) potentially contributing to insulin-resistance [112]. Alterations in tumor cell glycolysis have also been described following epidermal growth factor (EGF) stimulation of cancer cells [28].
The connection(s) between cellular metabolism and inflammation remains incompletely understood, but recent insights into tunable supramolecular organizing centers (SMOCs) may offer a potential explanation. SMOCs are the signaling organelles of the innate immune system that consist of oligomeric protein complexes, including myddosomes (containing MyD88), triffosomes (containing TRIF), and inflammasomes (containing NLRP4), which induce transcription-dependent and -independent inflammatory responses [113]. MyD88-TBK1 signaling in myddosomes was necessary to stimulate glycolysis in response to TLR4 activation with lipopolysaccharide (LPS), but not NF-κB activation.
Another connection between TBK1 and metabolism involves regulation of mammalian target of rapamycin (mTOR), the master regulator of cell growth [114]. Several studies have described context-dependent activation or inhibition of mTOR signaling by TBK1 [115–117]. Recently, TBK1 was shown to restrict mTORC1 activity by directly phosphorylating Raptor on Ser877 [118]. TBK1 has also been shown to promote mTORC1 signaling via phosphorylation of Akt1 [119] and mTOR itself [115], suggesting complex and context-dependent signaling. As mTOR signaling regulates inflammation, autophagy, and metabolism, the role of TBK1 activating (and inhibiting) mTOR signaling may offer further insights into the complex roles and regulation of TBK1 signaling.
4. TBK1 in cancer pathogenesis
TBK1 signaling has been implicated in several hallmarks of tumor progression, including evading growth suppression, evading immune destruction, tumor-promoting inflammation, inducing angiogenesis, genomic instability, resisting apoptosis, and reprogramming energy metabolism [120]. The mechanisms by which TBK1 is activated in tumor cells are still not fully understood, although there appears to be a clear role for tumor cell autonomous mechanisms. In 2006, Chien and colleagues showed that TBK1 is activated downstream of the RAS effector RalB in NSCLC (non-small cell lung carcinoma) to inhibit apoptotic pathways, and demonstrated that TBK1 was necessary for RAS-induced oncogenic transformation of mouse embryonic fibroblasts [121]. Subsequently, a synthetic lethality screen found TBK1 essential for survival of KRAS mutant lung cancer cells, by NF-κB activation that promotes anti-apoptotic signals [122]. Further study of TBK1 signaling in KRAS mutant lung cancer has demonstrated a key role for autocrine cytokine signaling in this pathway [123]. Recently, Zhu and colleagues confirmed activation of TBK1 downstream by growth factor receptor signaling and identified a key role for TBKBP1 (SINTBAD) in promoting mTORC1 activation and subsequent tumorigenesis [28], confirming a role for TBK1 in KRAS mutant NSCLC. Furthermore, TBK1 was shown to directly phosphorylate the mitosis substrates CEP170 and NUMA independent of KRAS mutational status [42], as well as directly localize to the centrosomes during mitosis [94].
TBK1 also promotes cancer cell growth via activation of AKT [116,119] which in turn regulates mTORC1 that controls metabolic processes and cell survival (see section 3.5) [124]. TBK1 regulation of mTOR seems to be a fine-tuning mechanism, or cell dependent, as in prostate cancer TBK1 can promote cancer dormancy by mTORC1 inhibition [125]. Consistent with these overall findings, TBK1 localizes with components of the mTORC1 complex [125]. As discussed above (section 3.5), the relationship between TBK1 and mTOR signaling appears to be both complex and context dependent. TBK1 regulation of autophagy in cancer is similarly complex, as blocking of autophagy, by Atg5 deletion in pancreatic cancer, enhanced activation of TBK1 in vivo, increased neutrophil and T-cell tumor infiltration and caused PD-L1 upregulation, while blocking of TBK1 resulted in blocking of autophagy [89]. This suggests a negative feedback mechanism between TBK1 and autophagy, whereby active TBK1 promotes basal autophagy in PDA cells and is then degraded in the autophagic process to limit over-activation of autophagy.
Cellular de-differentiation via epithelial-to-mesenchymal transition (EMT) appears to influence sensitivity to TBK1 inhibition [126], and may account for some differences in context-dependent TBK1 signaling. Loss of TBK1 disrupts KRAS-driven, AXL-mediated EMT in pancreatic adenocarcinoma [127], and was similarly required for platelet-induced EMT in breast cancer cells [128]. Pharmacologic inhibition of TBK1 was capable of reversing EMT in prostate cancer cells [129], and EMT in response to radiation in lung cancer cells [130]. De-differentiated melanoma cells were shown to be inherently more resistant to MAPK pathway inhibition, but more sensitive to TBK1 inhibition, and was associated with a de-differentiation and activation of Akt and YAP signaling [131]. Interestingly, this TBK1i-sensitive/MAPK-resistant cell state could be recapitulated by inhibition of the H3K27 histone methyl transferase, EZH2 [132]. NSCLC cells with a mesenchymal gene signature exhibited increased sensitivity to TBK1 inhibition, which was correlated with activation of Akt and mTORC1 (mechanistic target of rapamycin complex 1) [116]. However, loss of TBK1 has been shown to promote EMT in estrogen receptor-positive breast cancer cells [133], suggesting a more complex connection between TBK1 and EMT.
Recently, Hu and colleagues described a novel relationship between TBK1 and VHL [134]. The von Hippel-Lindau (VHL) gene product, is a ubiquitin E3 ligase with established tumor suppressor function that is absent in clear cell renal cell carcinoma (ccRCC), resulting in accumulation of hypoxia inducible factor α (HIFα) and induction of hypoxia-induced gene programs which promote angiogenesis and glycolysis [135]. Under hypoxic conditions, TBK1 (and not IKKε), was shown to be hyperactivated (increased pSer172) leading to phosphorylation of p62/SQSTM1 on Ser366 and increases its stability thereby promoting cancer cell growth [134]. Hydroxylation of proline 48 (see Fig. 1) on TBK1 by EGLN1 promoted binding of VHL and PP1MB, thereby enhancing TBK1 dephosphorylation [134,136]. These findings suggest synthetic lethal relationship and identify another role for TBK1 in cancer, independent from its role regulating innate immune responses.
5. TBK1 in the tumor immune microenvironment
Tumor cells co-opt normal stromal cells to escape detection by the immune system and to facilitate tumor growth [120]. Secreted cytokines, onco-metabolites, and recruitment of immune suppressive cell types all contribute to an immune suppressive tumor immune microenvironment (TIME) [137]. Cancer cells have the ability to activate different immune checkpoint pathways that harbor immunosuppressive functions [138–140]. Given its ability to influence secretion of cytokines and chemokines, tumor cell autonomous TBK1 signaling can also regulate cancer progression by regulating the tumor microenvironment (TME) [123,141], including endothelial cell proliferation and angiogenesis [142,143]. Interestingly, the function of TBK1 in the TIME may extend beyond cancer cells (Figure 3). Dendritic cell-specific deletion of Tbk1 causes T cell activation and autoimmune symptoms, as well as, enhances antitumor immunity in animal models of melanoma and thymoma [144], suggesting a role for TBK1 in restraining tumor immune attack. TBK1 is required for PRR signaling in macrophages and loss of TBK1 results in impaired inflammatory signaling with reduced elaboration of inflammatory cytokines, including TNFα, IL-6, IFNβ, and CXCL10 (IP-10) [145]. TBK1 deletion has been shown to influence T cell function in murine models of neuroinflammation [146], and in vitro pharmacologic inhibition of TBK1/IKKε enhanced production of T cell cytokines, IL-2 and IFNγ [147].
Fig. 3. Targeting TBK1 in the Tumor Microenvironment.
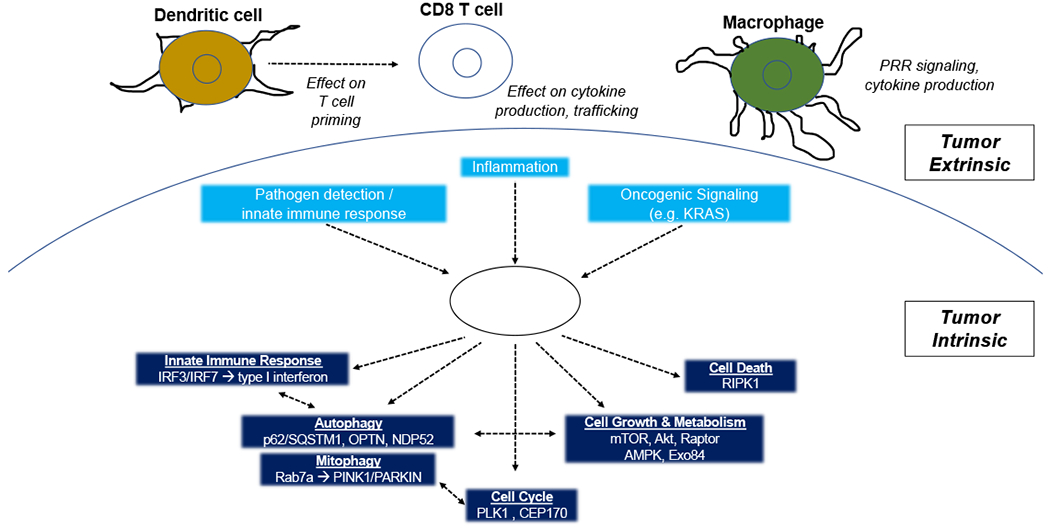
Tumor intrinsic TBK1 signaling in the setting of innate immune response, inflammation, and oncogenesis have been described. Subsequent roles for TBK1 have been established for innate immune signaling (e.g. IRF3/IFN type 1), autophagy/mitophagy, cell cycle, cell growth/metabolism, and cell death, with some evidence of crosstalk between these pathways. Tumor extrinsic TBK1 signaling has been suggested with potential roles for dendritic cells, cytotoxic T lymphocytes, and macrophages.
However, questions remain about the impact of targeting TBK1 in immune cells versus tumor cells. Activating the STING-TBK1 pathway using STING agonists appears to effectively reduce tumor growth alone or in combination with cancer immunotherapy [148]. Using transplantable mouse tumor models, there is evidence that cGAS and STING are required for effective generation of CD8 T cells, presumably by enhancing dendritic cell (DC) priming via enhanced cross-presentation of tumor-associated antigens [149,150]. Thus, disrupting TBK1 early in tumorigenesis might inhibit CD8 T cell priming similar to the phenotype observed in STING-null mice. However, a tumor cell autonomous role for cGAS-STING signaling has been demonstrated, including a role for ‘self’ DNA derived from chromosomal instability (CIN) promoting metastasis [151]. Another recent study found that IFI16 (Interferon-inducible 16) activates STING-TBK1 pathway, which leads to increased PD-L1 expression and immunosuppressive TME [152]. Further study is needed to clarify the impact of STING agonism versus TBK1 inhibition with particular attention cell type specific signaling (e.g. tumor versus immune).
6. Therapeutic targeting of TBK1 in cancer
As TBK1 has emerged as a druggable target for multiple illnesses, several inhibitors (TBK1i) have been developed (see Table 1) [153]. TBK1i were shown to be effective in restricting tumor growth in various cancers [116,154–158], inhibiting metastatic potential [129,158], as well as, improve response to immunotherapy in mice and patient-derived 3D tumor spheroids [147]. In breast cancer, TBK1 was shown to phosphorylate estrogen receptor α (ERα) on Ser-305 thereby promoting resistance of the cells to tamoxifen [159]. Moreover, TBK1 expression in breast cancer correlated with poor response to tamoxifen. In a murine breast cancer model, a TBK1/IKKε inhibitor combined with chemotherapy (docetaxel) reduced metastasis and improved survival owing to reduced cancer cell growth and impaired migration [158]. TBK1/IKKε inhibition was also effective in a murine model of Her2-mutant breast cancer and demonstrated cooperativity with the lapatinib [160]. In addition, both glioblastoma and lung cancer, IKKε inhibition increased the response to chemotherapy, suggesting that IKKs inhibition can serve as a combined therapy with chemotherapy as well [161].
Table 1 – Small Molecules and PROTACs Targeting TBK1.
Select references indicating cancer-related and non-cancer related studies, data on cross reactivity against other kinases, and IC50 (inhibitors) or DC50 (for proteolysis targeting chimeras).
| TBK1i | Non-cancer related | Cancer-related | Cross reactivity | IC50 / DC50 |
|---|---|---|---|---|
| Amlexanox | Inflammatory diseases, obesity, asthma, allergies [188,189] | breast cancer [158], colorectal cancer [157], prostate cancer [129,190], glioblastoma [156], leukemia | TBK1/IKKε Binds also to some S100 proteins and alters cell signaling and cytoskelston organization [191,192] |
0.85uM 4.7uM 1-300 μg/ml for cell culture assays [193] |
| BX795 | Herpes Simplex infections [194] | Lung cancer [116], bladder cancer [155], melanoma [131,167], squamous cell carcinoma [154] | TBK1/IKKε PDK1 Aurora B, ERK 8 And more [22] |
1nM 1nM 1.3 nM 2 nM Working concentrations 100 nM - 10 μM |
| MRT67307 | rheumatoid arthritis | T cell Leukemia [195] | TBK1/IKKε ULK1/2 [196] SIK [197] |
19 nM 160 nM 40nM 1-20 μM for cell culture assays |
| CYT387 (Momelotinib) [198] | Myelofibrosis [199] | Lymphoma [200]; AML [162]; PDAC [171]; NSCLC [123,170], |
JAK1 JAK2 TBK1/IKKε |
11nM 18nM 58 nM / 42 nM |
| Cmpd1 | N/A | Colon cancer (mouse model) [147]; Renal cell carcinoma [134] | TBK1/IKKε (less potent against JAK1/2/3) [147] |
1nM (TBK1) / 5.6 nM (IKKε) |
| GSK8612 | N/A | N/A | TBK1 [201] | N/A |
| Compound II | Autoimmune diseases [202,203] | melanoma [131] | TBK1/IKKε [203] |
N/A |
| Compound 3i | N/A | KRAS mutant lung cancer [166] | Selective for TBK1 | DC50 = 12 nM |
| UNC6587 | N/A | VHL-mutant renal cell carcinoma [134] | Selective for TBK1 | N/A |
Most of the established TBK1 inhibitors are potent with in vitro IC50 in the mid-low nM range, and EC50 in the low μM to high nM range [153]. Unfortunately, many of these inhibitors are not selective, and are cross reactive with multiple kinases with high potency [78]. It is worth noting that all TBK1 inhibitors to date also effectively inhibit IKKε. While IKKε (IKBKE) is regulated transcriptionally and exhibits distinct tissue expression, predominately in immune cells, compensatory upregulation of IKKε in response to TBK1 silencing has been observed in some cell lines [162], and serves as a reminder that genetic confirmation is necessary to establish the specificity of small molecule inhibitors. While there appears to be some degree of redundancy between TBK1 and IKKε in several settings [97,163], additional studies have demonstrated a clear role for IKKε and not TBK1 in cancer cell growth [164,165]. Possible explanations for non-overlapping roles of TBK1 and IKKε include potential differences in upstream regulators, such as RALA/B Kras-mutant in pancreatic cancer [165] and VHL status in renal cell carcinoma which affected TBK1 but not IKKε [134]. In addition, selectivity of specific downstream targets (e.g. p62, in RCC [134]). The roles of IKKε in cancer are reviewed elsewhere in greater detail [161]. Due to the lack of specificity of small molecules other approaches were taken to develop TBK1 specific inhibitors, such as PROTACs (proteolysis targeting chimeras), which are small molecules that binds the target protein and an E3 ligase and promoting the target protein for ubiquitination mediated degradation. This type of inhibitor is highly specific towards TBK1 versus IKKε and can be used for application when only TBK1 has to targeted [166].
Genetic, epigenetic, and/or environmental factors have been shown to influence tumor-intrinsic sensitivity to TBK1 loss. Initial studies suggested certain RAS-mutant cancers exhibited a specific vulnerability to TBK1 inhibition [122,123,167]. David Barbie’s lab studied addition of other drugs (e.g. MEK inhibitors) to enhance response to TBK1 inhibition [123], but epigenetic, tumor-intrinsic resistance mechanisms limited these combinations as well. Triple therapy with TBK1 inhibition, trametinib (MEKi), and a BET inhibitor (JQ1) was required to overcome resistance to TBK1i +/− MEKi [141]. Altered transcriptional cell state appears to influence intrinsic activity of the STING-TBK1-IRF3 pathway in KRAS-mutant NSCLC cell lines based on co-mutations in TP53 versus STK11 (LKB1). Kitajima and colleagues demonstrated that cells with loss-of-function mutations in LBK1 undergo epigenetic silencing of TMEM173 (encoding STING) resulting in impaired sensing of cytoplasmic dsDNA [168]. Given the poor response to cancer immunotherapy with PD-1 blockade observed in patients with KRas/LKB1 mutant (“KL”) NSCLC [169], it has been suggested that mechanisms to restore STING expression may enhanced response to immunotherapy in these patients.
Several initial studies demonstrated modest single-agent activity of TBK1/IKKε inhibitors, prompting combination studies often with MEK inhibitors [123,141,167]. In clinical trials momelotinib (MMB, CYT387), a multi-targeted inhibitor of JAK/TBK1/IKKε, was tested in combination with either chemotherapy or trametinib (MEKi) for metastatic KRAS mutant NSCLC and PDAC (NCT02258607, NCT02244489, NCT02101021). Results of the Phase 1B trial assessing MMB + trametinib in KRAS-mutant NSCLC were disappointing, with no patients showing an objective response [170]. MMB with chemotherapy in pancreatic cancer was well tolerated with 28% partial response rate although given the lack of survival benefit over chemotherapy alone, there was insufficient evidence to support further evaluation of this regimen as a first-line treatment in patients with PDAC [171].
7. Conclusions
TBK1 is a multi-functional Ser/Thr kinase with well-established roles in regulating the innate immune responses in response to invading pathogens [1], with additional roles in autophagy/mitophagy/xenophagy, cellular metabolism, cell cycle, and cell death signaling [2,9,48]. TBK1 signaling is subject to multiple layers of regulation, including post-translational modifications, dynamic protein-protein interactions, and alterations in subcellular localization. Such complexity is required for a single enzyme to exhibit such influence in so many different biological processes, and likely underlies the context-dependent impact of TBK1 deletion/inhibition [116,131]. Improved understanding of the roles and regulation of TBK1 has begun to reveal novel insights and connections [87,172], although the precise signaling pathway(s) relevant to cancer pathogenesis are incompletely understood.
8. Expert opinion
Over the last decade, TBK1 has emerged as a potential therapeutic target in cancer based on genetic and pharmacologic loss-of-function studies [5,78]. Despite promising pre-clinical data in certain cancers and cancer sub-types, early phase clinical trials of TBK1 inhibitors +/− chemotherapeutic agents or small molecules (e.g. MEK inhibitors) were not promising. Possible explanations for the poor clinical performance include issues with the chosen drug itself (e.g. potency, specificity), the patient population in which the drug was evaluated, lack of predictive biomarkers, the combination therapy strategy, and lack of appropriate pre-clinical models that recapitulate key features of the TME to perform drug sensitivity testing.
There are several TBK1 inhibitors that have been described in the literature (Table 1), although only amlexanox is FDA-approved (aphthous ulcers) and only momelotinib has undergone early phase clinical testing for cancer. Amlexanox has been shown to improve glucose control in patients with diabetes and was generally well tolerated [173], but has not been evaluated in patients with malignancy. Newer more selective TBK1 inhibitors, as well as TBK1 PROTACs, are still in pre-clinical development, and currently there are no clinical trials evaluating TBK1 inhibitors that are enrolling patients (clinicaltrials.gov), with the exception of momelotinib which remains under investigation in myelofibrosis largely due its activity as a JAK inhibitor. Several drug companies have developed molecules that inhibit TBK1 in the low nM range [153], a handful of which have been published [131,147]. Key to the future of TBK1-directed therapy in cancer will be improved understanding of the cellular roles and molecular mechanisms governing TBK1 activity, as well as improved understanding of the inter- and intra-patient heterogeneity of TBK1 signaling.
Even with the development of a highly selective and potent TBK1 inhibitor, the right drug must be used in the appropriate therapeutic context. Precision medicine broadly refers to approaches to match the “right drug(s) with the right patient,” although this has largely been synonymous with genomic approaches to identify druggable driver mutations. Despite great success with this approach, FDA-approved drugs are available for a small subset of cancer patients based on “targetable” mutations identified from next-generation DNA sequencing [174], and there is an unmet need for new and complementary approaches to evaluate novel therapies and therapeutic combinations. Initial studies suggested genomic features (e.g. RAS mutations) indicated sensitivity to TBK1 inhibition, although more recent evaluation of larger numbers of cell lines suggests that transcriptional cell state may better predict sensitivity to TBK1 inhibition than specific genomic features. Michael White’s group performed pharmacogenomic analyses of melanoma [131] and NSCLC [116] cell lines and identified a gradient of sensitivity to TBK1 inhibition. The TBK1i-sensitive transcriptional cell state in both studies is more mesenchymal and de-differentiated, suggesting a connection between transcriptional cell state and innate immune signaling. Single-cell RNA-sequencing has offered unprecedented insights into the evolution of cancer cell programs and tumor cell states that can be therapeutically targeted [175,176], and may prove useful identifying specific cell states that are uniquely sensitive to TBK1 inhibition. The development of adaptive resistance to cancer therapies gives rise to distinct transcriptional cell states [175,177] that also may exhibit unique response profiles to TBK1-directed therapies. Recent studies also suggest development of adaptive mutability in response to targeted cancer therapies [178], which should also be considered with respect to TBK1-directed therapeutic strategies. Deep characterization and understanding of the molecular adaptations underlying response (and resistance) to TBK1-directed therapies are also expected to improve our understanding of the complex roles and regulation of TBK1 in cancer.
While TBK1 inhibition has shown some activity as a monotherapy, most pre-clinical studies have evaluated TBK1 inhibitors in combination with other anti-cancer agents. Given the clinical success of cancer immunotherapy with immune checkpoint blockade, there has been early interest in improving the response to PD-1 blockade by TBK1i [147], as TBK1 has been shown to promote immune suppression in murine cancer models [28]. As it expressed both in tumor and immune tissues further studies are required to confirm tumor-intrinsic and extrinsic roles of TBK1 signaling. A particularly intriguing observation arising from several studies is that cancer cells sensitive to TBK1 inhibition exhibit activation of innate immune signaling pathways [116,131]. Whether TBK1 inhibition in such a setting would enhance response to cancer immunotherapy is unknown but is a subject of active investigation. As several CRISPR screens have nominated TBK1 as a promising candidate to sensitize tumor cells to immune attack [179–181], the tumor-intrinsic role of TBK1 may be particularly relevant. Inhibiting TBK1 may also prime anti-tumor immune responses [144,145,147,179,181], suggesting that targeting TBK1 in the TME may have greater efficacy than tumor-specific targeting. As there is a growing interest in developing strategies to normalize the TME [182], targeting TBK1 signaling may prove particularly effective in combination in certain tumor types depending on tumor cell transcriptional state, innate immune signaling, as well as immune contexture.
In the coming years, we anticipate TBK1 will re-emerge as a therapeutic target in cancer, especially as part of combination strategies directed at the TME. More selective small molecule inhibitors and PROTACs targeting TBK1 will provide the appropriate tools for further pre-clinical and clinical development. Further research is needed to facilitate development of rational TBK1-directed therapeutic strategies as well as predictive biomarkers to determine which patients are most likely to respond to treatment. One of the greatest challenges facing the development of TBK1-directed cancer therapeutics is the use of appropriate pre-clinical model systems that faithfully recapitulate key features of the TME. Newer patient-derived tumor model systems (e.g. organoids, organotypic tumor spheroids) [147,183] may enable testing of combination strategies prior to first-in-human clinical trials. Such personalized cancer models may not only facilitate pre-clinical evaluation using clinically relevant patient-derived tumor models, but may also serve as a functional precision medicine tools to guide patient selection as part of early phase clinical testing [184–187].
Article Highlights.
TBK1 is a multi-functional serine/threonine kinase with well-established roles in regulating the innate immune responses in response to invading pathogens
Regulation of TBK1 signaling involves post-translational modifications, dynamic protein-protein interactions, and alterations in subcellular localization
Role of TBK1 in cancer include governing innate immune signaling, inflammation, autophagy, cellular metabolism, cell cycle, and cell death signaling
Tumor-intrinsic and tumor-extrinsic roles of TBK1 in cancer have been described
TBK1 has emerged as a potential therapeutic target in cancer based on genetic and pharmacologic loss-of-function studies.
The next generation of TBK1-directed cancer therapies will focus on the tumor microenvironment and will likely be evaluated in combination with cancer immunotherapy and/or molecular targeted therapy.
Acknowledgments
Funding
The research of the authors is funded by Karin Grunebaum Cancer Research Foundation X, Melanoma Research Alliance 621819, U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute 1K08CA226391.
Footnotes
Declaration of interest
R Jenkins is on the Advisory Board of XSphera Biosciences and has a financial interest in this company; he has also received research support from Monopteros Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Zhao C, Zhao W. TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin Ther Targets. 2019. May;23(5):437–446. [DOI] [PubMed] [Google Scholar]
- 2.Zhou R, Zhang Q, Xu P. TBK1, a central kinase in innate immune sensing of nucleic acids and beyond. Acta Biochim Biophys Sin (Shanghai). 2020. May 27. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Fang L, Li P, et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J Virol. 2011. April;85(8):3758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng D, Chen G, Guo B, et al. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008. November;18(11):1105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Durand JK, Zhang Q, Baldwin AS. Roles for the IKK-Related Kinases TBK1 and IKKepsilon in Cancer. Cells. 2018. September 15;7(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao P, Wong KI, Sun X, et al. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell. 2018. February 8;172(4):731–743 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmad L, Zhang SY, Casanova JL, et al. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol Med. 2016. June;22(6):511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shu C, Sankaran B, Chaton CT, et al. Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure. 2013. July 2;21(7):1137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louis C, Burns C, Wicks I. TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front Immunol. 2018;9:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012. August 24;37(2):223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999. December 1;18(23):6694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]; * first identification of TBK1.
- 12.Tojima Y, Fujimoto A, Delhase M, et al. NAK is an IkappaB kinase-activating kinase. Nature. 2000. April 13;404(6779):778–82. [DOI] [PubMed] [Google Scholar]
- 13.Bonnard M, Mirtsos C, Suzuki S, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000. September 15;19(18):4976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]; * initial demonstration of embryonic lethality of TBK1-null mice.
- 14.Shimada T, Kawai T, Takeda K, et al. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int Immunol. 1999. August;11(8):1357–62. [DOI] [PubMed] [Google Scholar]; * first identification of IKKε.
- 15.Peters RT, Liao SM, Maniatis T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol Cell. 2000. March;5(3):513–22. [DOI] [PubMed] [Google Scholar]
- 16.Shin CH, Choi DS. Essential Roles for the Non-Canonical IkappaB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells. 2019. February 19;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng W, Shi M, Han M, et al. Negative regulation of virus-triggered IFN-beta signaling pathway by alternative splicing of TBK1. J Biol Chem. 2008. December 19;283(51):35590–7. [DOI] [PubMed] [Google Scholar]
- 18.Hu YW, Zhang J, Wu XM, et al. TANK-Binding Kinase 1 (TBK1) Isoforms Negatively Regulate Type I Interferon Induction by Inhibiting TBK1-IRF3 Interaction and IRF3 Phosphorylation. Front Immunol. 2018;9:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tu D, Zhu Z, Zhou AY, et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013. March 28;3(3):747–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larabi A, Devos JM, Ng SL, et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep. 2013. March 28;3(3):734–46. [DOI] [PubMed] [Google Scholar]
- 21.Ma X, Helgason E, Phung QT, et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A. 2012. June 12;109(24):9378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark K, Plater L, Peggie M, et al. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009. May 22;284(21):14136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.An H, Zhao W, Hou J, et al. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity. 2006. December;25(6):919–28. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Liang L, Fan Y, et al. PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell Signal. 2012. November;24(11):2197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhan Z, Cao H, Xie X, et al. Phosphatase PP4 Negatively Regulates Type I IFN Production and Antiviral Innate Immunity by Dephosphorylating and Deactivating TBK1. J Immunol. 2015. October 15;195(8):3849–57. [DOI] [PubMed] [Google Scholar]
- 26.Qi D, Hu L, Jiao T, et al. Phosphatase Cdc25A Negatively Regulates the Antiviral Immune Response by Inhibiting TBK1 Activity. J Virol. 2018. October 1;92(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Liu G, Sun L, et al. PPM1A regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLoS Pathog. 2015. Mar;11(3):e1004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu L, Li Y, Xie X, et al. TBKBP1 and TBK1 form a growth factor signalling axis mediating immunosuppression and tumourigenesis. Nat Cell Biol. 2019. December;21(12):1604–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song G, Liu B, Li Z, et al. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat Immunol. 2016. December;17(12):1342–1351. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Zhang L, Wang B, et al. RNF144B inhibits LPS-induced inflammatory responses via binding TBK1. J Leukoc Biol. 2019. December;106(6):1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Chen T, Zhang J, et al. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat Immunol. 2009. July;10(7):744–52. [DOI] [PubMed] [Google Scholar]
- 32.Li S, Wang L, Berman M, et al. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity. 2011. September 23;35(3):426–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman CS, O’Donnell MA, Legarda-Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008. September;9(9):930–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Zhao X, Zhang M, et al. Ubiquitin-specific protease 2b negatively regulates IFN-beta production and antiviral activity by targeting TANK-binding kinase 1. J Immunol. 2014. September 1;193(5):2230–7. [DOI] [PubMed] [Google Scholar]
- 35.Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012. March 4;13(4):387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang M, Wang L, Zhao X, et al. TRAF-interacting protein (TRIP) negatively regulates IFN-beta production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J Exp Med. 2012. September 24;209(10):1703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Q, Hou J, Zhou Y, et al. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015. October;25(10):1121–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010. May 14;285(20):14999–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saul VV, Niedenthal R, Pich A, et al. SUMO modification of TBK1 at the adaptor-binding C-terminal coiled-coil domain contributes to its antiviral activity. Biochim Biophys Acta. 2015. January;1853(1):136–43. [DOI] [PubMed] [Google Scholar]
- 40.Li D, Yang W, Ren J, et al. The E3 Ubiquitin Ligase TBK1 Mediates the Degradation of Multiple Picornavirus VP3 Proteins by Phosphorylation and Ubiquitination. J Virol. 2019. December 1;93(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goncalves A, Burckstummer T, Dixit E, et al. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6(9):e23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim JY, Welsh EA, Oguz U, et al. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc Natl Acad Sci U S A. 2013. July 23;110(30):12414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003. May;4(5):491–6. [DOI] [PubMed] [Google Scholar]; * TBK1 and IKKε are central to innate immune response.
- 44.Ryzhakov G, Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007. July 11;26(13):3180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008. August 6;27(15):2147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Li C, Xue P, et al. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc Natl Acad Sci U S A. 2009. May 12;106(19):7945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Meng F, Chen S, et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat Cell Biol. 2017. April;19(4):362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013. April 17;587(8):1230–7. [DOI] [PubMed] [Google Scholar]
- 49.Self-regulation Cao X. and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016. January;16(1):35–50. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012. March 6;5(214):ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gasteiger G, D’Osualdo A, Schubert DA, et al. Cellular Innate Immunity: An Old Game with New Players. J Innate Immun. 2017;9(2):111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013. February 15;339(6121):786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013. February 15;339(6121):826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao B, Du F, Xu P, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. 2019. May;569(7758):718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang C, Shang G, Gui X, et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019. March;567(7748):394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma S, tenOever BR, Grandvaux N, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003. May 16;300(5622):1148–51. [DOI] [PubMed] [Google Scholar]; * TBK1 and IKKε are central to IRF3/IRF7 activation as part of antiviral response.
- 57.Pourcelot M, Zemirli N, Silva Da Costa L, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol. 2016. August 18;14:69. [DOI] [PMC free article] [PubMed] [Google Scholar]; * TBK1 localization is key to antiviral response.
- 58.O’Loughlin T, Kruppa AJ, Ribeiro ALR, et al. OPTN recruitment to a Golgi-proximal compartment regulates immune signalling and cytokine secretion. J Cell Sci. 2020. June 15;133(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao W Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. 2013. March 18;587(6):542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matthys V, Gorbunova EE, Gavrilovskaya IN, et al. The C-terminal 42 residues of the Tula virus Gn protein regulate interferon induction. J Virol. 2011. May;85(10):4752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Otsuka M, Kato N, Moriyama M, et al. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology. 2005. May;41(5):1004–12. [DOI] [PubMed] [Google Scholar]
- 62.Siu KL, Kok KH, Ng MH, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009. June 12;284(24):16202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Unterholzner L, Sumner RP, Baran M, et al. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 2011. September;7(9):e1002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verpooten D, Ma Y, Hou S, et al. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem. 2009. January 9;284(2):1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Unterstab G, Ludwig S, Anton A, et al. Viral targeting of the interferon-{beta}-inducing Traf family member-associated NF-{kappa}B activator (TANK)-binding kinase-1. Proc Natl Acad Sci U S A. 2005. September 20;102(38):13640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gruber C Impaired interferon signature in severe COVID-19. Nat Rev Immunol. 2020. April 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.al. HJe. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. MedRxiv. 2020. (Pre-print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020. May 28;181(5):1036–1045 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020. April 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen X, Yang X, Zheng Y, et al. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell. 2014. May;5(5):369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ivanov KA, Thiel V, Dobbe JC, et al. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J Virol. 2004. June;78(11):5619–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect. 2020. June;80(6):607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hung IF, Lung KC, Tso EY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020. May 30;395(10238):1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sallard E, Lescure FX, Yazdanpanah Y, et al. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res. 2020. April 7;178:104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herman M, Ciancanelli M, Ou YH, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. 2012. August 27;209(9):1567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differ. 2020. March;27(3):858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017. September;17(9):528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cruz VH, Brekken RA. Assessment of TANK-binding kinase 1 as a therapeutic target in cancer. J Cell Commun Signal. 2018. March;12(1):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011. July 8;333(6039):228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Newman AC, Scholefield CL, Kemp AJ, et al. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-kappaB signalling. PLoS One. 2012;7(11):e50672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matsumoto G, Shimogori T, Hattori N, et al. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015. August 1;24(15):4429–42. [DOI] [PubMed] [Google Scholar]
- 82.Kumar S, Gu Y, Abudu YP, et al. Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev Cell. 2019. April 8;49(1):130–144 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pickles S, Vigie P, Youle RJ. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol. 2018. February 19;28(4):R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015. October 1;60(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A. 2016. June 14;113(24):E3349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heo JM, Ordureau A, Swarup S, et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. 2018. November;4(11):eaav0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sarraf SA, Sideris DP, Giagtzoglou N, et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019. October 1;29(1):225–235 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; * connecting TBK1’s role in innate immunity, mitophagy, and cell cycle regulation.
- 88.Onorati M, Li Z, Liu F, et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016. September 6;16(10):2576–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]; * connecting TBK1’s role in innate immunity, mitophagy, and cell cycle regulation.
- 89.Yang S, Imamura Y, Jenkins RW, et al. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol Res. 2016. June;4(6):520–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018. April 13;37(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016. September 1;30(17):1913–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Germic N, Frangez Z, Yousefi S, et al. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019. March;26(4):715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nixon RA, Yang DS. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol. 2012. October 1;4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pillai S, Nguyen J, Johnson J, et al. Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat Commun. 2015. December 10;6:10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weil R, Laplantine E, Genin P. Regulation of TBK1 activity by Optineurin contributes to cell cycle-dependent expression of the interferon pathway. Cytokine Growth Factor Rev. 2016. June;29:23–33. [DOI] [PubMed] [Google Scholar]
- 96.Heger K, Dixit VM. TBK1 and IKKepsilon restrain cell death. Nat Cell Biol. 2018. December;20(12):1330–1331. [DOI] [PubMed] [Google Scholar]
- 97.Lafont E, Draber P, Rieser E, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol. 2018. December;20(12):1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** TBK1 and IKKε restrain cell death signaling by phosphorylating RIPK1.
- 98.Gerondakis S, Grumont R, Gugasyan R, et al. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene. 2006. October 30;25(51):6781–99. [DOI] [PubMed] [Google Scholar]
- 99.Kuai J, Wooters J, Hall JP, et al. NAK is recruited to the TNFR1 complex in a TNFalpha-dependent manner and mediates the production of RANTES: identification of endogenous TNFR-interacting proteins by a proteomic approach. J Biol Chem. 2004. December 17;279(51):53266–71. [DOI] [PubMed] [Google Scholar]
- 100.Burstein E, Duckett CS. Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr Opin Cell Biol. 2003. December;15(6):732–7. [DOI] [PubMed] [Google Scholar]
- 101.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002. March;3(3):221–7. [DOI] [PubMed] [Google Scholar]
- 102.Xu D, Jin T, Zhu H, et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell. 2018. September 6;174(6):1477–1491 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** RIPK1 signaling is required for cell death TBK1-null cells.
- 103.Matsui K, Kumagai Y, Kato H, et al. Cutting edge: Role of TANK-binding kinase 1 and inducible IkappaB kinase in IFN responses against viruses in innate immune cells. J Immunol. 2006. November 1;177(9):5785–9. [DOI] [PubMed] [Google Scholar]
- 104.Newton K RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015. June;25(6):347–53. [DOI] [PubMed] [Google Scholar]
- 105.Peltzer N, Darding M, Walczak H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016. June;26(6):445–461. [DOI] [PubMed] [Google Scholar]
- 106.Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015. May;18(5):631–6. [DOI] [PubMed] [Google Scholar]
- 107.Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017. February 2;10(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yu H, Cleveland DW. Tuning Apoptosis and Neuroinflammation: TBK1 Restrains RIPK1. Cell. 2018. September 6;174(6):1339–1341. [DOI] [PubMed] [Google Scholar]
- 109.Cruz VH, Arner EN, Wynne KW, et al. Loss of Tbk1 kinase activity protects mice from diet-induced metabolic dysfunction. Mol Metab. 2018. October;16:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu L, Chen X, Sun X, et al. The Glycolytic Switch in Tumors: How Many Players Are Involved? J Cancer. 2017;8(17):3430–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Uhm M, Bazuine M, Zhao P, et al. Phosphorylation of the exocyst protein Exo84 by TBK1 promotes insulin-stimulated GLUT4 trafficking. Sci Signal. 2017. March 21;10(471). [DOI] [PubMed] [Google Scholar]
- 112.Munoz MC, Giani JF, Mayer MA, et al. TANK-binding kinase 1 mediates phosphorylation of insulin receptor at serine residue 994: a potential link between inflammation and insulin resistance. J Endocrinol. 2009. May;201(2):185–97. [DOI] [PubMed] [Google Scholar]
- 113.Tan Y, Kagan JC. Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell. 2019. April 4;177(2):384–398 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Regulation of TBK1 as a part of supramolecular organizing complexes.
- 114.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017. April 6;169(2):361–371. [DOI] [PubMed] [Google Scholar]
- 115.Bodur C, Kazyken D, Huang K, et al. The IKK-related kinase TBK1 activates mTORC1 directly in response to growth factors and innate immune agonists. EMBO J. 2018. January 4;37(1):19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cooper JM, Ou YH, McMillan EA, et al. TBK1 Provides Context-Selective Support of the Activated AKT/mTOR Pathway in Lung Cancer. Cancer Res. 2017. September 15;77(18):5077–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hasan M, Gonugunta VK, Dobbs N, et al. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci U S A. 2017. January 24;114(4):746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Antonia RJ, Castillo J, Herring LE, et al. TBK1 Limits mTORC1 by Promoting Phosphorylation of Raptor Ser877. Sci Rep. 2019. September 17;9(1):13470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ou YH, Torres M, Ram R, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011. February 18;41(4):458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011. March 4;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 121.Chien Y, Kim S, Bumeister R, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006. October 6;127(1):157–70. [DOI] [PubMed] [Google Scholar]
- 122.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009. November 5;462(7269):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]; * TBK1 is synthetic lethal target in KRAS mutant non-small cell lung cancer.
- 123.Zhu Z, Aref AR, Cohoon TJ, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014. April;4(4):452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim JK, Jung Y, Wang J, et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013. September;15(9):1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019. February;20(2):69–84. [DOI] [PubMed] [Google Scholar]
- 127.Cruz VH, Arner EN, Du W, et al. Axl-mediated activation of TBK1 drives epithelial plasticity in pancreatic cancer. JCI Insight. 2019. April 2;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y, Unnithan RVM, Hamidi A, et al. TANK-binding kinase 1 is a mediator of platelet-induced EMT in mammary carcinoma cells. FASEB J. 2019. July;33(7):7822–7832. [DOI] [PubMed] [Google Scholar]
- 129.Cheng C, Ji Z, Sheng Y, et al. Aphthous ulcer drug inhibits prostate tumor metastasis by targeting IKKvarepsilon/TBK1/NF-kappaB signaling. Theranostics. 2018;8(17):4633–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu W, Huang YJ, Liu C, et al. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3beta and repression of ZEB1. Lab Invest. 2014. April;94(4):362–70. [DOI] [PubMed] [Google Scholar]
- 131.Eskiocak B, McMillan EA, Mendiratta S, et al. Biomarker Accessible and Chemically Addressable Mechanistic Subtypes of BRAF Melanoma. Cancer Discov. 2017. August;7(8):832–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jenkins RW, Barbie DA. Refining Targeted Therapy Opportunities for BRAF-Mutant Melanoma. Cancer Discov. 2017. August;7(8):799–801. [DOI] [PubMed] [Google Scholar]
- 133.Yang KM, Jung Y, Lee JM, et al. Loss of TBK1 induces epithelial-mesenchymal transition in the breast cancer cells by ERalpha downregulation. Cancer Res. 2013. November 15;73(22):6679–89. [DOI] [PubMed] [Google Scholar]
- 134.Hu L, Xie H, Liu X, et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2020. Mar;10(3):460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]; * TBK1 is synthetic lethal target in renal cell carcinoma with VHL loss
- 135.Bakouny Z, Barbie DA. TBK1 Activation by VHL Loss in Renal Cell Carcinoma: A Novel HIF-Independent Vulnerability. Cancer Discov. 2020. March;10(3):348–350. [DOI] [PubMed] [Google Scholar]
- 136.Zhou T, Erber L, Liu B, et al. Proteomic analysis reveals diverse proline hydroxylation-mediated oxygen-sensing cellular pathways in cancer cells. Oncotarget. 2016. November 29;7(48):79154–79169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018. May;24(5):541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Darvin P, Toor SM, Sasidharan Nair V, et al. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018. December 13;50(12):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015. April 9;161(2):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018. September;8(9):1069–1086. [DOI] [PubMed] [Google Scholar]
- 141.Kitajima S, Asahina H, Chen T, et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell. 2018. September 10;34(3):439–452 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Czabanka M, Korherr C, Brinkmann U, et al. Influence of TBK-1 on tumor angiogenesis and microvascular inflammation. Front Biosci. 2008. May 1;13:7243–9. [DOI] [PubMed] [Google Scholar]
- 143.Korherr C, Gille H, Schafer R, et al. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc Natl Acad Sci U S A. 2006. March 14;103(11):4240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xiao Y, Zou Q, Xie X, et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J Exp Med. 2017. May 1;214(5):1493–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Loss of TBK1 in dendritic cells enhances antitumor immune responses.
- 145.Yu T, Yi YS, Yang Y, et al. The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm. 2012;2012:979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yu J, Zhou X, Chang M, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun. 2015. January 21;6:6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jenkins RW, Aref AR, Lizotte PH, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018. February;8(2):196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhu Y, An X, Zhang X, et al. STING: a master regulator in the cancer-immunity cycle. Mol Cancer. 2019. November 4;18(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang H, Hu S, Chen X, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017. February 14;114(7):1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014. November 20;41(5):830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018. January 25;553(7689):467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cai H, Yan L, Liu N, et al. IFI16 promotes cervical cancer progression by upregulating PD-L1 in immunomicroenvironment through STING-TBK1- NF-kB pathway. Biomed Pharmacother. 2020. March;123:109790. [DOI] [PubMed] [Google Scholar]
- 153.Hasan M, Yan N. Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharmacol Res. 2016. September;111:336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bai LY, Chiu CF, Kapuriya NP, et al. BX795, a TBK1 inhibitor, exhibits antitumor activity in human oral squamous cell carcinoma through apoptosis induction and mitotic phase arrest. Eur J Pharmacol. 2015. December 15;769:287–96. [DOI] [PubMed] [Google Scholar]
- 155.Chen W, Luo K, Ke Z, et al. TBK1 Promote Bladder Cancer Cell Proliferation and Migration via Akt Signaling. J Cancer. 2017;8(10):1892–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu Y, Lu J, Zhang Z, et al. Amlexanox, a selective inhibitor of IKBKE, generates anti-tumoral effects by disrupting the Hippo pathway in human glioblastoma cell lines. Cell Death Dis. 2017. August 31;8(8):e3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bordonaro M, Lazarova D. Amlexanox and UPF1 Modulate Wnt Signaling and Apoptosis in HCT-116 Colorectal Cancer Cells. J Cancer. 2019;10(2):287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bishop RT, Marino S, de Ridder D, et al. Pharmacological inhibition of the IKKepsilon/TBK-1 axis potentiates the anti-tumour and anti-metastatic effects of Docetaxel in mouse models of breast cancer. Cancer Lett. 2019. May 28;450:76–87. [DOI] [PubMed] [Google Scholar]
- 159.Wei C, Cao Y, Yang X, et al. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proc Natl Acad Sci U S A. 2014. February 4;111(5):E601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Deng T, Liu JC, Chung PE, et al. shRNA kinome screen identifies TBK1 as a therapeutic target for HER2+ breast cancer. Cancer Res. 2014. April 1;74(7):2119–30. [DOI] [PubMed] [Google Scholar]
- 161.Yin M, Wang X, Lu J. Advances in IKBKE as a potential target for cancer therapy. Cancer Med. 2020. January;9(1):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu S, Marneth AE, Alexe G, et al. The kinases IKBKE and TBK1 regulate MYC-dependent survival pathways through YB-1 in AML and are targets for therapy. Blood Adv. 2018. December 11;2(23):3428–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Balka KR, Louis C, Saunders TL, et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep. 2020. April 7;31(1):107492. [DOI] [PubMed] [Google Scholar]
- 164.Barbie TU, Alexe G, Aref AR, et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J Clin Invest. 2014. December;124(12):5411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rajurkar M, Dang K, Fernandez-Barrena MG, et al. IKBKE Is Required during KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2017. January 15;77(2):320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Crew AP, Raina K, Dong H, et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem. 2018. January 25;61(2):583–598. [DOI] [PubMed] [Google Scholar]
- 167.Vu HL, Aplin AE. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol Cancer Res. 2014. October;12(10):1509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kitajima S, Ivanova E, Guo S, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019. January;9(1):34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018. July;8(7):822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Barbie DA, Spira A, Kelly K, et al. Phase 1B Study of Momelotinib Combined With Trametinib in Metastatic, Kirsten Rat Sarcoma Viral Oncogene Homolog-Mutated Non-Small-Cell Lung Cancer After Platinum-Based Chemotherapy Treatment Failure. Clin Lung Cancer. 2018. November;19(6):e853–e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ng K, Hendifar A, Starodub A, et al. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2019. February;37(1):159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal. 2011. August 9;4(187):pe39. [DOI] [PubMed] [Google Scholar]
- 173.Oral EA, Reilly SM, Gomez AV, et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017. July 5;26(1):157–170 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dienstmann R, Jang IS, Bot B, et al. Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov. 2015. February;5(2):118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tsoi J, Robert L, Paraiso K, et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell. 2018. May 14;33(5):890–904 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jerby-Arnon L, Shah P, Cuoco MS, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018. November 1;175(4):984–997 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fallahi-Sichani M, Becker V, Izar B, et al. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol Syst Biol. 2017. January 9;13(1):905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Russo M, Crisafulli G, Sogari A, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019. December 20;366(6472):1473–1480. [DOI] [PubMed] [Google Scholar]
- 179.Manguso RT, Pope HW, Zimmer MD, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017. July 27;547(7664):413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pan D, Kobayashi A, Jiang P, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018. February 16;359(6377):770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vredevoogd DW, Kuilman T, Ligtenberg MA, et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell. 2019. July 25;178(3):585–599 e15. [DOI] [PubMed] [Google Scholar]
- 182.Mpekris F, Voutouri C, Baish JW, et al. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc Natl Acad Sci U S A. 2020. February 18;117(7):3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Neal JT, Li X, Zhu J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell. 2018. December 13;175(7):1972–1988 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Friedman AA, Letai A, Fisher DE, et al. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015. December;15(12):747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Grandori C, Kemp CJ. Personalized Cancer Models for Target Discovery and Precision Medicine. Trends Cancer. 2018. September;4(9):634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Letai A Functional precision cancer medicine-moving beyond pure genomics. Nat Med. 2017. September 8;23(9):1028–1035. [DOI] [PubMed] [Google Scholar]
- 187.Yuki K, Cheng N, Nakano M, et al. Organoid Models of Tumor Immunology. Trends Immunol. 2020. August;41(8):652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Reilly SM, Chiang SH, Decker SJ, et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013. March;19(3):313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Barrons RW. Treatment strategies for recurrent oral aphthous ulcers. Am J Health Syst Pharm. 2001. January 1;58(1):41–50; quiz 51–3. [PubMed] [Google Scholar]
- 190.Tamai H, Yamaguchi H, Miyake K, et al. Amlexanox Downregulates S100A6 to Sensitize KMT2A/AFF1-Positive Acute Lymphoblastic Leukemia to TNFalpha Treatment. Cancer Res. 2017. August 15;77(16):4426–4433. [DOI] [PubMed] [Google Scholar]
- 191.Cho CC, Chou RH, Yu C. Amlexanox Blocks the Interaction between S100A4 and Epidermal Growth Factor and Inhibits Cell Proliferation. PLoS One. 2016;11(8):e0161663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Landriscina M, Prudovsky I, Mouta Carreira C, et al. Amlexanox reversibly inhibits cell migration and proliferation and induces the Src-dependent disassembly of actin stress fibers in vitro. J Biol Chem. 2000. October 20;275(42):32753–62. [DOI] [PubMed] [Google Scholar]
- 193.Beyett TS, Gan X, Reilly SM, et al. Design, synthesis, and biological activity of substituted 2-amino-5-oxo-5H-chromeno[2,3-b]pyridine-3-carboxylic acid derivatives as inhibitors of the inflammatory kinases TBK1 and IKKepsilon for the treatment of obesity. Bioorg Med Chem. 2018. November 1;26(20):5443–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jaishankar D, Yakoub AM, Yadavalli T, et al. An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci Transl Med. 2018. February 14;10(428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Xu X, Kalac M, Markson M, et al. Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL). Cell Death Dis. 2020. February 5;11(2):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015. May 1;290(18):11376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Clark K, MacKenzie KF, Petkevicius K, et al. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc Natl Acad Sci U S A. 2012. October 16;109(42):16986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pardanani A, Lasho T, Smith G, et al. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009. August;23(8):1441–5. [DOI] [PubMed] [Google Scholar]
- 199.Pardanani A, Laborde RR, Lasho TL, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013. June;27(6):1322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lu Z, Hong CC, Jark PC, et al. JAK1/2 Inhibitors AZD1480 and CYT387 Inhibit Canine B-Cell Lymphoma Growth by Increasing Apoptosis and Disrupting Cell Proliferation. J Vet Intern Med. 2017. November;31(6):1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Thomson DW, Poeckel D, Zinn N, et al. Discovery of GSK8612, a Highly Selective and Potent TBK1 Inhibitor. ACS Med Chem Lett. 2019. May 9;10(5):780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Louis C, Ngo D, D’Silva DB, et al. Therapeutic Effects of a TANK-Binding Kinase 1 Inhibitor in Germinal Center-Driven Collagen-Induced Arthritis. Arthritis Rheumatol. 2019. January;71(1):50–62. [DOI] [PubMed] [Google Scholar]
- 203.Hasan M, Dobbs N, Khan S, et al. Cutting Edge: Inhibiting TBK1 by Compound II Ameliorates Autoimmune Disease in Mice. J Immunol. 2015. November 15;195(10):4573–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ren Z, Ding T, Zuo Z, et al. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front Immunol. 2020;11:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]