

Abstract
Mitochondrial quality is controlled by the selective removal of damaged mitochondria through mitophagy. Mitophagy impairment is associated with aging and many pathological conditions. An iron loss induced by iron chelator triggers mitophagy by a yet unknown mechanism. This type of mitophagy may have therapeutic potential, since iron chelators are clinically used. Here, we aimed to clarify the mechanisms by which iron loss induces mitophagy. Deferiprone, an iron chelator, treatment resulted in the increased expression of mitochondrial ferritin (FTMT) and the localization of FTMT precursor on the mitochondrial outer membrane. Specific protein 1 and its regulator hypoxia‐inducible factor 1α were necessary for deferiprone‐induced increase in FTMT. FTMT specifically interacted with nuclear receptor coactivator 4, an autophagic cargo receptor. Deferiprone‐induced mitophagy occurred selectively for depolarized mitochondria. Additionally, deferiprone suppressed the development of hepatocellular carcinoma (HCC) in mice by inducing mitophagy. Silencing FTMT abrogated deferiprone‐induced mitophagy and suppression of HCC. These results demonstrate the mechanisms by which iron loss induces mitophagy and provide a rationale for targeting mitophagic activation as a therapeutic strategy.
Keywords: hepatocellular carcinoma, iron chelator, mitochondria, mitochondrial ferritin, mitophagy
Subject Categories: Autophagy & Cell Death, Molecular Biology of Disease,
Iron loss induces mitochondrial ferritin via hypoxia inducible factor 1α‐specific protein 1 axis, and triggers mitophagy for the damaged mitochondria.
Introduction
Mitochondria are essential to cellular metabolism and physiology. Mitochondrial dysfunction has been associated with a broad variety of human diseases. Mitochondrial quality is controlled by the selective removal of depolarized or damaged mitochondria through mitophagy (Kim et al, 2007; Youle, 2019). Mitophagy is the only known pathway through which whole mitochondria can be selectively removed. Mitophagy occurring in the physiological context is classified as basal, stress‐induced, or programmed (McWilliams et al, 2016; Esteban‐Martinez et al, 2017; Palikaras et al, 2018; Sekine & Youle, 2018). On the other hand, mitophagy impairment is associated with aging and many pathological conditions, such as neurodegenerative diseases, myopathies, metabolic disorders, inflammation, and cancer (Palikaras et al, 2018; Lou et al, 2020). Thus, the induction of mitophagy, i.e., targeting clearance of damaged mitochondria, has potential as a therapeutic strategy for various diseases.
Mitophagy regulatory pathways are classified as ubiquitin‐dependent or ubiquitin‐independent (Khaminets et al, 2016). The best described mitophagic pathway is the phosphate and tensin homologue (PTEN)‐induced putative kinase 1 (PINK1)/Parkin E3 ubiquitin ligase pathway, which is activated in response to the mitochondrial membrane depolarization (Narendra et al, 2008). The PINK1/Parkin pathway remains inactivate in pathophysiological conditions and can be triggered by carbonyl cyanide 3‐chlorophenylhydrazone (CCCP), an oxidative phosphorylation uncoupler that causes the depolarization of the entire mitochondrial network. In addition to Parkin, several other ubiquitin E3 ligases such as Gp78, SMURF1, SIAH1, MUL1, and ARIH1 function in mitophagy regulation (Orvedahl et al, 2011; Lokireddy et al, 2012; Fu et al, 2013; Szargel et al, 2016; Villa et al, 2017). In ubiquitin‐independent mitophagy, some mitochondrial outer membrane proteins serve as mitophagy receptors and interact directly with LC3 without ubiquitination and/or autophagic adaptor proteins. As mitophagy receptors, BCL2‐interacting protein 3 (BNIP3), NIP3‐like protein X (NIX), FUN14 domain‐containing protein 1 (FUNDC1), BCL‐2 like protein 13 (BCL2L13), and FK506‐binding protein 8 (FKBP8) have been identified so far (Schweers et al, 2007; Quinsay et al, 2010; Liu et al, 2012; Murakawa et al, 2015; Bhujabal et al, 2017).
Interestingly, iron loss induced by an iron chelator can also trigger mitophagy, although independently of the PINK1/Parkin pathway (Allen et al, 2013) with the underlying mechanisms remaining unknown. This type of mitophagy seems to be fascinating as therapeutic potential for intervention targeting mitophagy since iron chelators can be easily used as therapeutic drugs unlike endogenous proteins such as Parkin or mitophagy receptors. Additionally, there have been several lines of evidence demonstrating that iron chelators work against cancer as the therapeutic agents (Heath et al, 2013), including the anticancer effect of an iron chelator, deferoxamine in patients with hepatocellular carcinoma (HCC) (Yamasaki et al, 2011). Therefore, iron loss‐induced mitophagy may have the potential of anticancer treatment. Here, we aimed to clarify the mechanisms by which iron loss induces mitophagy and further evaluate whether iron loss‐induced mitophagy can suppress the development of HCC.
Results
Deferiprone (DFP) induces mitophagy more vigorously than deferoxamine (DFO) and deferasirox (DFX) in human liver cells
An iron chelator, DFP, has been shown to induce mitophagy in vitro (Allen et al, 2013). First, we assessed which iron chelator triggers mitophagy most vigorously among the three iron chelators (DFP, DFO, and DFX). Treatment with DFP resulted in the highest increase in LC3‐II expression (Fig 1A). The number of mitophagosome‐like structures in Huh7 cells induced by DFP was significantly higher than that induced by DFO (Fig EV1A). We defined a mitophagosome‐like structure as a spherical structure with an isolation membrane containing high electron density cellular components with a membranous structure. Therefore, we used DFP as an iron chelator in subsequent experiments.
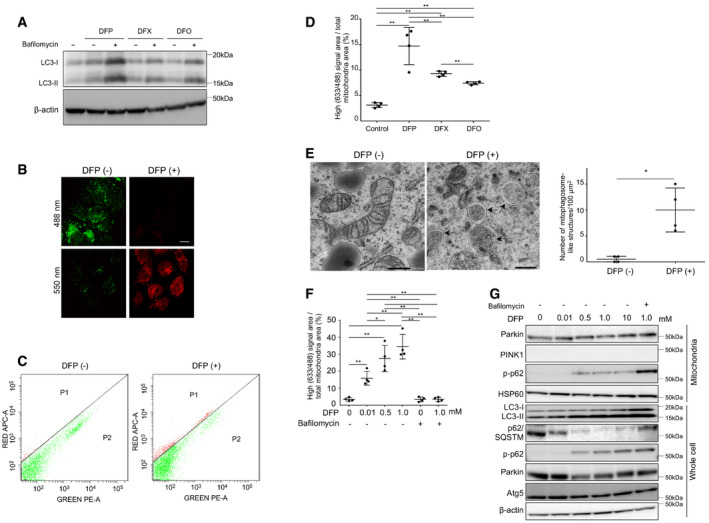
Figure 1. Induction of mitophagy by iron chelators (DFP, DFX, or DFO).
-
AImmunoblots for LC3 using whole cell lysates of Huh7 cells before and after treatment with iron chelators (DFP, DFX, or DFO). β‐actin was used as loading control. Bafilomycin (−): without bafilomycin, bafilomycin (+): with 100 mM of bafilomycin.
-
BDual excitation (550/488 nm) of mt‐mKeima‐transfected Huh7 cells shows red fluorescence indicating mitophagy with DFP treatment at 24 h and green fluorescence without DFP treatment. Scale bar: 10 μm.
-
CThe high ratio signal cells were quantified by flow cytometry with dual excitation of mt‐mKeima at 633/488 nm for untreated (left panel) and DFP‐treated (24 h, right panel) Huh7 cells.
-
DUntreated Huh7 cells or those treated with DFP (10 μM), DFX (10 μM), or DFO (10 μM) were quantified for mitophagy using mt‐mKeima transfection and FACS analysis (n = 4, biological replicates). A high signal (633/488) area/cell area indicates the proportion of cells undergoing mitophagy. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. **P < 0.01.
-
EElectron microscopic images of untreated (left panel) and DFP‐treated (right panel) Huh7 cells. Arrows indicate mitophagosome‐like structures. Scale bar: 500 nm. The number of mitophagosome‐like structures/100 μm2 was compared for four randomly selected areas (technical replicates) between cells treated with DFP (1.0 mM) and those without DFP. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05.
-
FQuantification of mitophagy using mt‐mKeima transfection and FACS analysis for Huh7 cells treated with DFP (0, 0.01, 0.5, or 1.0 mM as indicated) in the absence or presence of bafilomycin (n = 4, biological replicates). The central horizontal bar and the error bars indicate mean ± SD. *P < 0.05, **P < 0.01.
-
GImmunoblots for Parkin, PINK1, phosphorylated p‐62 (p‐p62), HSP60, LC3, p62/SQSTM, and Atg5 using the mitochondrial fraction and whole cell lysates of Huh7 cells. HSP60 and β‐actin were used as loading controls for mitochondria and whole cell lysates, respectively.
Source data are available online for this figure.
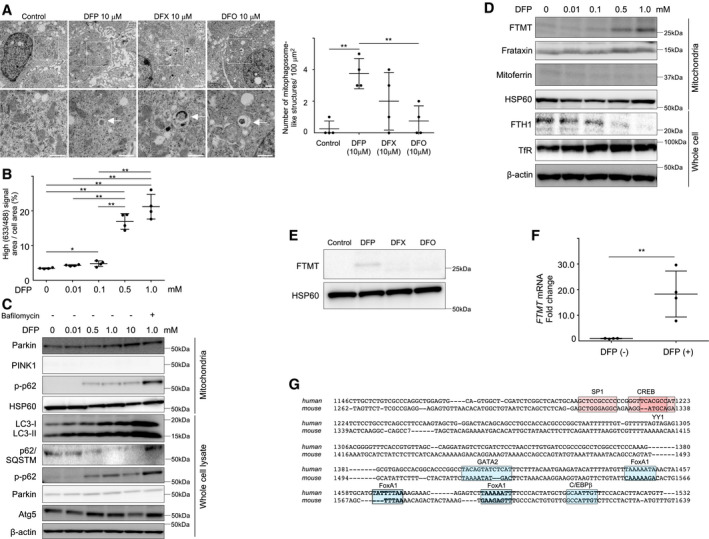
Figure EV1. Induction of mitophagy and FTMT by deferiprone, and a putative promoter region in FTMT .
-
AElectron microscopic images of Huh7 cells that were untreated or treated with DFP, DFX, or DFO. Boxed areas (×2,000) are enlarged below (×6,000). The white arrows indicate mitophagosome‐like structures. Scale bar: 2.0 nm. The number of mitophagosome‐like structures/100 μm2 was compared for four randomly selected areas (technical replicates) between groups. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. **P < 0.01.
-
BQuantification of mitophagy using mt‐mKeima transfection and FACS analysis for HepG2 cells treated with DFP (0, 0.01, 0.5, or 1.0 mM as indicated) (n = 4, biological replicates). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
CImmunoblots for Parkin, PINK1, phosphorylated p‐62 (p‐p62), HSP60, LC3, p62/SQSTM, and Atg5 using the mitochondrial fraction and whole cell lysates of HepG2 cells. HSP60 and β‐actin were used as loading controls for mitochondrial and whole cell lysates, respectively.
-
DImmunoblots for mitochondria ferritin (FTMT), frataxin, mitoferrin, ferritin heavy chain (FTH1), and transferrin receptor (TfR) using the mitochondrial fraction and whole cell lysates of HepG2 cells following treatment with DFP (0, 0.01, 0.1, 0.5, or 1.0 mM as indicated). HSP60 and β‐actin were used as loading controls for mitochondrial and whole cell lysates, respectively.
-
EImmunoblots for FTMT using the mitochondrial fraction lysate of Huh7 cells treated without or with iron chelators (DFP, DFX, or DFO). HSP60 was used as loading control.
-
FThe mRNA expression of FTMT was quantified by real‐time RT‐PCR before and after DFP (1.0 mM) treatment (n = 4, biological replicates) in HepG2 cells. The mRNA expression of FTMT was normalized to GAPDH. The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. **P < 0.01.
-
GA representation of the reported transcription factors for the mitochondrial ferritin promoter (Guaraldo et al, 2016). The locations of putative binding sites for transcription are boxed: Positive transcription factors (SP1, CREB, and YY1) are in red, while negative transcription factors (GATA2, FoxA1, and C/EBPβ) are in blue.
Source data are available online for this figure.
To visualize the process by which impaired mitochondria are delivered to lysosomes and quantify mitophagy, we transfected Huh7 and HepG2 cells with a mitochondria‐targeted coral‐derived acid‐stable fluorescent protein, mitochondrial monometric Keima (mt‐mKeima), that has a pH‐dependent bimodal excitation spectrum (Katayama et al, 2011), with short‐wavelength excitation under neutral conditions for detection of undamaged mitochondria and long‐wavelength excitation for detection of acidified mitophagosomes. The quantifiable proportion of cells with a high fluorescence intensity ratio (633/488 nm) (red) to the total mitochondria area is an indicator of mitophagy.
Treatment of mt‐mKeima‐transfected Huh7 cells with DFP resulted in an increase in the proportion of cells with a high signal ratio (633/488 nm) (Fig 1B and C), indicating mitophagic induction and a higher ratio signal (633/488 nm) area than with DFX or DFO (Fig 1D). This result was consistent with a significant increase in mitophagosome‐like structures upon treatment with DFP in Huh7 cells (Fig 1E). DFP induced mitophagy in a dose‐dependent manner, as indicated in high ratio signal (663/438 nm) area in Huh7 cells (Fig 1F) and HepG2 cells (Fig EV1B). DFP treatment also resulted in increased LC3‐II formation and phosphorylated p62 (p‐p62) and decreased p62 in a dose‐dependent manner, but did not affect PINK1, Parkin, and Atg5 in Huh7 cells (Fig 1G) or HepG2 cells (Fig EV1C). These results indicated that DFP‐induced mitophagy was independent of the PINK1/Parkin signal, which is consistent with a previous report by Allen et al (2013).
Iron loss enhances expression of mitochondrial ferritin (FTMT) via hypoxia‐inducible factor‐1α (HIF1α)‐specific protein 1 (SP1) axis
We next examined the underlying mechanism behind iron chelator‐induced mitophagy. Both cytoplasmic and mitochondrial chelatable iron contents that were measured by a ferrozine‐based assay decreased following DFP treatment, while only cytoplasmic iron decreased following DFX or DFO treatment (Fig 2A). Additionally, the extent of the decrease in iron content following DFP treatment was greater in the cytoplasm than in the mitochondria (Fig 2B). Among the mitochondria‐associated iron regulatory proteins examined, DFP treatment resulted in increased expression of FTMT, decreased expression of ferritin heavy chain (FTH1), and increased expression of transferrin receptor (TfR) in both Huh7 cells (Fig 2C) and HepG2 cells (Fig EV1D) in a dose‐dependent manner. Neither DFX nor DFO treatment increased expression of FTMT in the same dose as DFP (Fig EV1E). Decreased expression of FTH1 and increased expression of TfR upon iron loss are assumed to occur through the iron regulatory protein/iron‐responsive element (IRE) regulatory system (Hentze et al, 2004). However, it remained uncertain why the expression of FTMT increased upon iron loss, since FTMT mRNA does not contain any functional IRE sequence (Drysdale et al, 2002). DFP‐induced increase in protein expression of FTMT was associated with a significant increase in the mRNA expression of FTMT in Huh7 cells (Fig 2D) and HepG2 cells (Fig EV1F), suggesting DFP‐induced regulation of FTMT at the transcription level. A time course of mRNA expression of FTMT revealed that DFP‐induced transcriptional regulation of FTMT is a late response that requires incubation with DFP at least for 24 h (Fig 2E).
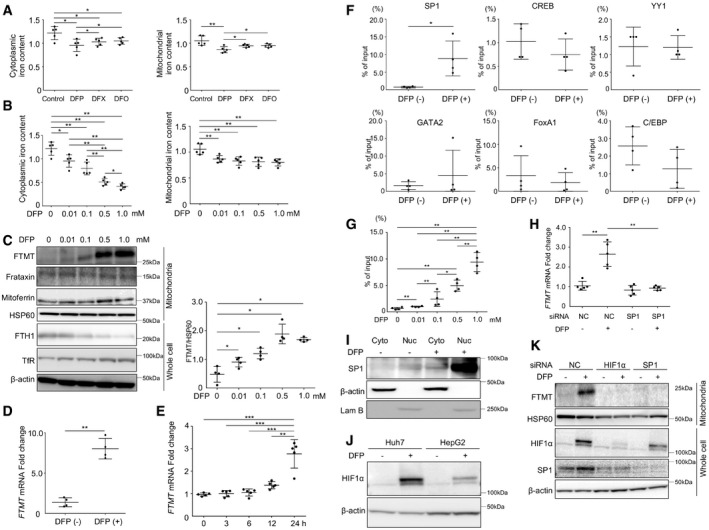
Figure 2. Induction of mitochondrial ferritin via hypoxia‐inducible factor 1α‐specific protein 1 axis.
-
ACytoplasmic and mitochondrial iron content was measured by a ferrozine‐based assay for Huh7 cells with DFP, DFX, or DFO treatment (0.01 mM) for 24 h (n = 5, biological replicates). The cytoplasmic iron content was measured by subtracting the iron content of the mitochondrial fraction from the iron content of the whole lysate. The central horizontal bar and the error bars indicate mean ± SD. *P < 0.05, **P < 0.01.
-
BCytoplasmic and mitochondrial iron content was measured for Huh7 cells with DFP treatment (0, 0.01, 0.1, 0.5, or 1.0 mM as indicated) for 24 h. (n = 5, biological replicates) The central horizontal bar and the error bars indicate mean ± SD. *P < 0.05, **P < 0.01.
-
CImmunoblots (left panel) for mitochondrial ferritin (FTMT), frataxin, mitoferrin, heat shock protein 60 (HSP60), ferritin heavy chain (FTH1), and transferrin receptor (TfR) and β‐actin using the mitochondrial fraction or whole cell lysate of Huh7 cells following treatment with DFP (0, 0.01, 0.1, 0.5, or 1.0 mM as indicated). HSP60 and β‐actin were used as loading controls for mitochondrial and whole cell lysates, respectively. The expression level of FTMT was normalized to HSP60 (n = 4, biological replicates, right panel). The central horizontal bar and the error bars indicate mean ± SD. *P < 0.05.
-
DThe mRNA expression of FTMT in Huh7 cells was quantified by real‐time RT–PCR before and after DFP (1.0 mM) treatment (n = 4, biological replicates). The expression of FTMT mRNA level was normalized to GAPDH mRNA. The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. **P < 0.01.
-
EThe mRNA expression of FTMT in Huh7 cells was quantified by real‐time RT–PCR before DFP (1.0 mM) treatment and 3, 6, 12, and 24 h after DFP treatment (n = 5, biological replicates). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. **P < 0.01, ***P < 0.001.
-
FDNA‐binding activity of six transcription factors [specific protein 1 (SP1), cAMP response element‐binding protein (CREB), Ying yang 1 (YY1), GATA2, forkhead box protein A1 (FoxA1), and CCAAT enhancer‐binding protein β (C/EBPβ)] to the FTMT promoter region was compared by chromatin immunoprecipitation before and after DFP (1.0 mM) treatment in Huh7 cells (n = 4, biological replicates). The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. *P < 0.05.
-
GThe DNA‐binding activity of SP1 to the FTMT promoter region was assayed by chromatin immunoprecipitation following treatment with DFP (0, 0.01, 0.1, 0.5, or 1.0 mM) as indicated in Huh7 cells (n = 4, biological replicates). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
HFTMT mRNA expression was quantified for Huh7 cells by real‐time RT‐PCR before and after DFP (1.0 mM) treatment with or without SP1 knockdown by siRNA (n = 5, biological replicates). mRNA expression of FTMT level was normalized to GAPDH. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. NC: negative control siRNA, **P < 0.01.
-
IImmunoblots for SP1 using the cytoplasmic fraction and nuclear fraction before and after DFP (1.0 mM) treatment. β‐actin and Lam B were used as loading control for the cytoplasmic and nuclear fractions, respectively.
-
JImmunoblots for HIF1α using Huh7 cells and HepG2 cells before and after DFP (0.1 mM) treatment. β‐actin was used as the loading control.
-
KImmunoblots for FTMT, HIF1α, and SP1 using the mitochondrial fraction lysate and whole cell lysate of Huh7 cells before and after DFP (1.0 mM) treatment with or without HIF1α or SP1 knockdown by siRNA. HSP60 and β‐actin were used as loading controls for the mitochondrial fraction and whole cell lysates, respectively.
Source data are available online for this figure.
Recently, a putative promoter region in FTMT, and positive regulators [SP1, cAMP response element‐binding protein (CREB), and Ying Yang 1 (YY1)] and negative regulators [GATA2, forkhead box protein A1 (FoxA1), and CCAAT enhancer‐binding protein β (C/EBPβ)] of FTMT transcription have been identified (Guaraldo et al, 2016; Fig EV1G). We examined the effect of DFP on the DNA‐binding activity of these regulators to the FTMT promoter using chromatin immunoprecipitation (ChIP) assay. Among the regulators, only SP1 displayed significantly increased DNA‐binding activity following DFP treatment (Fig 2F) in a dose‐dependent manner (Fig 2G). SP1 knockdown by siRNA abolished the DFP‐induced increase in the mRNA levels of FTMT (Fig 2H), indicating SP1‐mediated regulation of FTMT expression in the presence of DFP. Treatment with DFP increased the expression of cytoplasmic and nuclear SP1 with predominant localization in the nucleus (Fig 2I).
To examine the mechanism behind SP1‐mediated regulation of FTMT expression, we next examined whether DFP enhances the expression of HIF1α since knockdown of HIF1α has been reported to downregulate the SP1 expression (Jeong & Park, 2012). The protein expression of HIF1α was enhanced by DFP treatment in Huh7 cells and HepaG2 cells (Fig 2J). Notably, knockdown of HIF1α by siRNA suppressed the DFP‐induced increase in the expression of both FTMT and SP1. On the other hand, knockdown of SP1 by siRNA abolished the DFP‐induced increase in the expression of FTMT but not in the expression of HIF1α (Fig 2K). These results suggested that HIF1α acts upstream of SP1 in the DFP‐induced regulation of FTMT expression. Thus, the iron loss was associated with HIF1α‐SP1 axis‐mediated regulation of FTMT expression.
FTMT is required for iron loss‐induced mitophagy
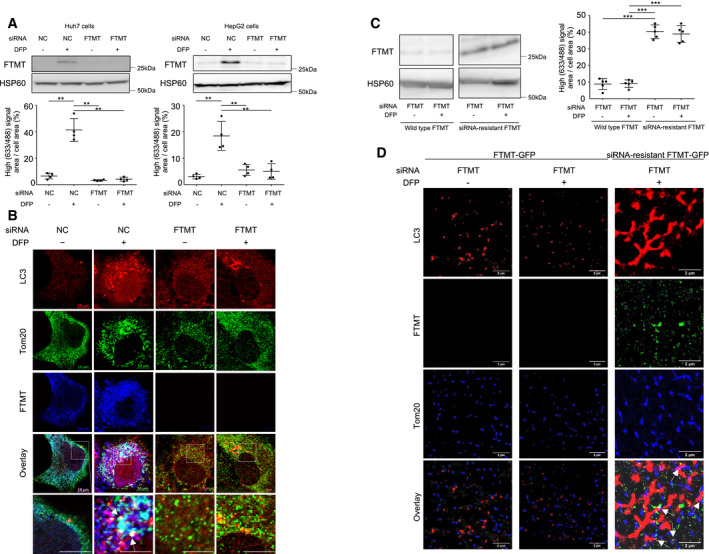
We next investigated whether FTMT is required for DFP‐induced mitophagy. Quantification of mitophagy using mt‐mKeima revealed that FTMT knockdown by small interfering RNA (siRNA) (upper panel, Fig 3A) abolished DFP‐induced mitophagy in Huh7 cells and HepG2 cells (lower panel, Fig 3A). Although the ratio of cells undergoing mitophagy in Fig 3A (approximately 40%) is much higher than that of cells undergoing mitophagy in Fig 1D (approximately 15%) regardless of the same cell line, this may be explained by the difference in DFP dose between two experiments (1 mM in Fig 3A and 10 μM in Fig 1D, respectively). Immunofluorescence analysis for LC3, Tom20, and FTMT also showed that FTMT knockdown by siRNA abrogated DFP‐induced mitophagy in Huh7 cells (Fig 3B). As this is the first evidence for FTMT playing a critical role in iron loss‐induced mitophagy, we further monitored the rescue of mitophagy using siRNA‐resistant FTMT. We stably expressed siRNA‐resistant FTMT, or C‐terminally tagged wild‐type FTMT (FTMT‐GFP) or siRNA‐resistant FTMT (siRNA‐resistant FTMT‐GFP) in Huh7 cells. Quantification of mitophagy using mt‐mKeima revealed that FTMT knockdown by siRNA did not result in suppression of mitophagy regardless of DFP in Huh7 cells transfected with siRNA‐resistant FTMT (Fig 3C). A single‐molecule fluorescence microscopy enabled us to detect fluorescent proteins in spatial resolution of 23–40 nm. Despite FTMT knockdown by siRNA, cells transfected with siRNA‐resistant FTMT‐GFP demonstrated the colocalization of LC3, FTMT, and Tom20, suggesting the induction of mitophagy. In contrast, cells transfected with FTMT‐GFP exhibited puncta of LC3 not colocalized with Tom 20 (Fig 3D). Additionally, FTMT knockdown rescued reactive oxygen species (ROS) production to almost the same level as the control, even though treatment with DFP significantly suppressed ROS production in Huh7 cells and HepG2 cells (Fig EV2A). These results indicate that FTMT is required for DFP‐induced mitophagy, which contributes to the suppression of cellular oxidative stress.
Figure 3. Mitochondrial ferritin is required for iron loss‐induced mitophagy.
-
AImmunoblots for mitochondrial ferritin (FTMT) with HSP60 as the loading control using the mitochondrial fraction of Huh7 cells and HepG2 cells (upper panels) and the quantification of mitophagy using mt‐mKeima transfection and FACS analysis (n = 4, biological replicates) (lower panels) before and after DFP (1.0 mM) treatment with or without FTMT knockdown by siRNA. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. NC: negative control siRNA, **P < 0.01.
-
BImmunofluorescence staining for LC3 (red), the mitochondrial marker Tom20 (green), and FTMT (blue) in Huh7 cells before and after DFP (1.0 mM) treatment with or without FTMT knockdown by siRNA. FTMT colocalizes with LC3 at the mitochondria (white puncta indicated by arrows). Boxed areas are enlarged below. Scale bar: 5 μm.
-
CImmunoblots for mitochondrial ferritin (FTMT) following FTMT knockdown in the absence or presence of DFP using the mitochondrial fraction of Huh7 cells and those transfected with FTMT siRNA‐resistant FTMT (left panel). HSP60 as the loading control. Those cells were subjected to the quantification of mitophagy using mt‐mKeima transfection and FACS analysis before and after DFP (1.0 mM) treatment (n = 5, biological replicates) (right panel). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. ***P < 0.001.
-
DSuper‐resolution images of immunofluorescence staining for LC3 (red), FTMT (green), and Tom20 (blue) in FTMT knockdown‐Huh7 cells transfected with FTMT‐GFP or FTMT siRNA‐resistant FTMT‐GFP before and after DFP (1.0 mM) treatment. White arrows indicate colocalization of LC3, FTMT, and Tom20.
Source data are available online for this figure.
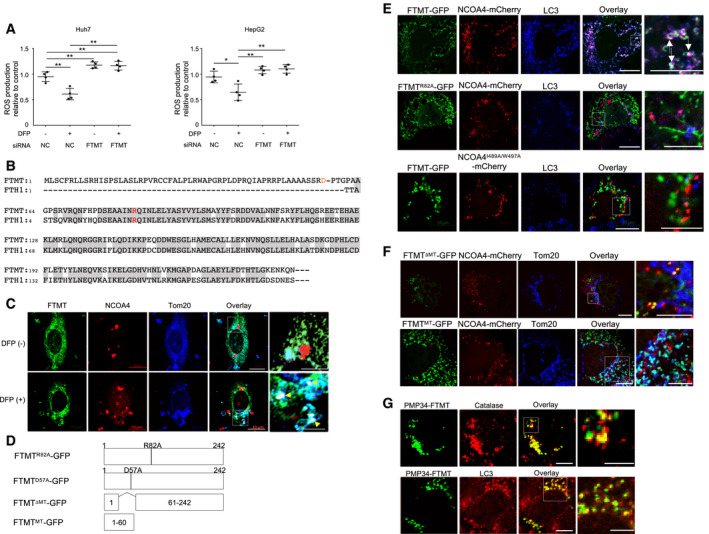
Figure EV2. Effects of FTMT knockdown on reactive oxygen species (ROS) production in vitro, the interaction of FTMT with NCOA4, and peroxisome‐specific autophagy by FTMT with a peroxisome‐targeting sequence.
-
AThe cellular reactive oxygen species (ROS) levels were measured by oxidation of the cell‐permeable and oxidation‐sensitive fluorogenic precursor, 2,7‐dihydrodichlorofluorescein diacetate for Huh7 cells (left), and HepG2 cells (right) with or without silencing of FTMT by siRNA (n = 4, biological replicates). DFP (−): without DFP, DFP (+): with 1 mM of DFP. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
BReported alignment of human FTMT and FTH1 amino acids (Levi et al, 2001). FTH1 R23 corresponds to R82 of FTMT. The orange “D” indicates the predicted cleavage site of the leader peptide of FTMT. The gray boxes represent the overlapping coding region where amino acid sequence of FTMT is identical to FTH1.
-
CImmunofluorescence staining for Tom20 (blue), NCOA4 (red), and endogenous FTMT (green) in normal human hepatocytes before and after DFP (1 mM) treatment. Boxed area is enlarged on the right. Scale bar: 10 and 5 μm in enlarged images. The white puncta by yellow arrowheads indicate the colocalization of FTMT, NCOA4, and Tom20.
-
DSchematic representation of GFP‐tagged FTMTR82A, FTMTD57A, FTMTΔMT, and FTMTMT.
-
EImmunofluorescence staining for NCOA4‐mCherry (red), NCOA4I489A/W497A‐mCherry (red), FTMT‐GFP (green), FTMTR82A‐GFP (green), and LC3 (blue) in Huh7 cells. The white puncta shown by white arrows in the overlaid picture indicate the colocalization of FTMT‐GFP, NCOA4‐mCherry, and LC3. Boxed areas are enlarged on the right. Scale bar: 10 and 5 μm in enlarged images.
-
FImmunofluorescence staining for the mitochondrial target sequence‐deleted FTMT‐GFP (FTMTΔMT‐GFP) (green), FTMT mitochondrial target sequence‐GFP (FTMTMT‐GFP) (green), NCOA4‐mCherry (red), and Tom20 (blue) in Huh7 cells. Boxed areas are enlarged on the right. The yellow puncta and cyan puncta in the overlaid pictures indicate the colocalization of FTMTΔMT‐GFP and NCOA4‐mCherry and the colocalization of FTMTMT‐GFP and Tom20, respectively. Scale bar: 10 μm and 5 μm in enlarged images.
-
GImmunofluorescence staining for catalase (red), PMP34‐FTMT-GFP that has the peroxisome‐targeting sequence instead of the mitochondria‐targeting sequence (green), and LC3 (red) in Huh7 cells. The yellow puncta in the overlaid pictures indicate the colocalization of catalase and PMP34‐FTMT-GFP or the colocalization of LC3 and PMP34‐FTMT-GFP. Boxed areas are enlarged below. Scale bar: 10 and 5 μm in enlarged images.
FTMT interacts with the autophagic cargo receptor, nuclear receptor coactivator 4 (NCOA4) at the specific residues
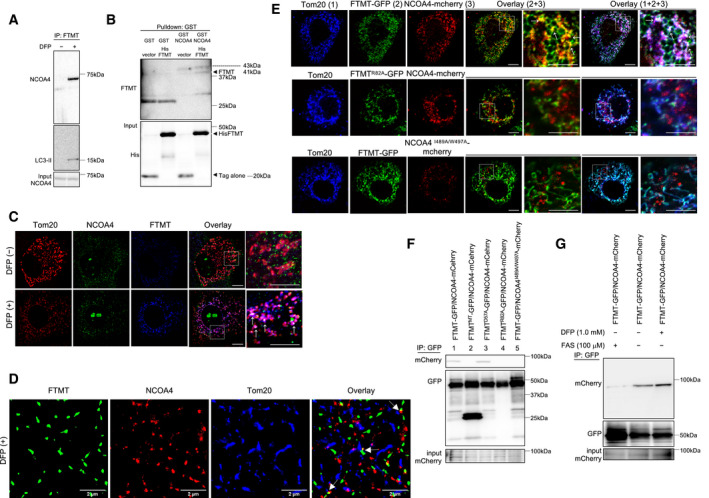
In selective autophagy, autophagic cargo receptors specifically bind the cargo material and the autophagosomal membrane (Zaffagnini & Martens, 2016). NCOA4 has been identified as a selective cargo receptor for autophagic turnover of the cytosolic ferritin (ferritinophagy) (Mancias et al, 2014). The protein sequence of FTMT is 79% identical to FTH1 in overlapping coding regions (Levi et al, 2001) (Fig EV2B). Therefore, we examined whether FTMT interacts with NCOA4 under iron loss‐induced mitophagy. Co‐immunoprecipitation of endogenous proteins in Huh7 cells revealed that FTMT was associated with NCOA4 in the presence of DFP (Fig 4A). We subsequently tested for direct interactions between the two proteins using GST pull‐down assays. Upon the construction of recombinant proteins, a residue (382–522) of NCOA4 was chosen referring to the pull‐down assays of NCOA4 and FTH1 (Mancias et al, 2015). Recombinant GST‐NCOA4 (382–522) specifically recognized recombinant His‐FTMT (61–242) (Fig 4B). Immunofluorescence study showed that endogenous NCOA4 colocalized with FTMT at the mitochondria in the presence of DFP in Huh7 cells (Fig 4C) and normal human hepatocytes (Fig EV2C). Further, observation with the single‐molecule fluorescence microscopy ensured these results by demonstrating clear colocalization of endogenous NCOA4 and FTMT at the mitochondria in the presence of DFP (Fig 4D).
Figure 4. Interaction of mitochondrial ferritin with nuclear receptor coactivator 4.
-
AExtracted proteins from Huh7 cells that were untreated or treated with DFP were mixed with anti‐FTMT-coupled magnetic beads and immunoblotted with the indicated antibodies (anti‐NCOA4 or anti‐LC3).
-
BGST pull‐down assay using recombinant GST‐NCOA4 mixed with recombinant His‐FTMT. Bound FTMT was analyzed by immunoblotting using either anti‐FTMT or anti‐His antibodies.
-
CImmunofluorescence staining for Tom20 (red), NCOA4 (green), and FTMT (blue) in Huh7 cells before and after DFP (1 mM) treatment. Boxed areas are enlarged on the right. The white puncta indicated by white arrows show the colocalization of FTMT, NCOA4, and Tom20. Scale bar: 10 and 5 μm in enlarged images.
-
DSuper‐resolution images of immunofluorescence staining for Tom20 (blue), NCOA4 (red), and FTMT (green) in Huh7 cells after DFP (1 mM) treatment, using the single‐molecule fluorescence microscope. The white arrows indicate colocalization of these proteins.
-
EImmunofluorescence staining for Tom20 (blue), FTMT‐GFP (green), recombinant FTMTR82A‐GFP (green), NCOA4‐mCherry (red), and NCOA4I489A/W497A‐mCherry (red) in Huh7 cells. Boxed areas are enlarged on the right. The white puncta pointed by white arrows in the overlaid picture indicate the colocalization of Tom20, FTMT‐GFP, and NCOA4‐mCherry. The yellow puncta shown by white arrows in the overlaid picture indicate the colocalization of FTMT‐GFP and NCOA4‐mCherry. Scale bar: 10 and 5 μm in enlarged images.
-
FExtracted proteins from Huh7 cells expressing the recombinant proteins as indicated were mixed with anti‐GFP-coupled magnetic beads and immunoblotted with anti‐mCherry and anti‐GFP antibodies. Co‐immunoprecipitation revealed a specific interaction of NCOA4‐mCherry with FTMT‐GFP or FTMTD57A‐GFP, which has a substitution of alanine (A) for aspartic acid (D) at position 57 in the FTMT mitochondrial leader sequence (lanes 1 and 3).
-
GFollowing DFP (1.0 mM) or ammonium iron sulfate (FAS) treatment as indicated, extracted proteins from Huh7 cells expressing FTMT‐GFP and NCOA4‐mCherry were mixed with anti‐GFP-coupled magnetic beads and immunoblotted with anti‐mCherry and anti‐GFP antibodies.
Source data are available online for this figure.
The two amino acids, isoleucine (I) at position 489 and tryptophan (W) at position 497 of NCOA4, have been identified to be critical for FTH1 binding (Mancias et al, 2015). Arginine (R) at position 23 of FTH1 is also essential for the ferritin association with NCOA4 (Mancias et al, 2015). R23 of FTH1 corresponds to R82 of FTMT (Fig EV2B). Therefore, we studied whether I489 and W497 in NCOA4 and R82 in FTMT were involved in the interaction between these two proteins. To this end, we stably expressed C‐terminally tagged wild‐type NCOA4 (NCOA4‐mCherry) or NCOA4I489A/W497A (NCOA4I489A/W497A‐mCherry), and wild‐type FTMT (FTMT‐GFP) or FTMTR82A (FTMTR82A‐GFP) in Huh7 cells. NCOA4‐mCherry colocalized with FTMT‐GFP at the mitochondria that were stained with Tom20 (Fig 4E) in the absence of DFP. In contrast, colocalization was not observed between FTMTR82A‐GFP and NCOA4‐mCherry or between FTMT‐GFP and NCOA4I489A/W497A‐mCherry (Fig 4E). Both NCOA4‐mCherry and FTMT‐GFP colocalized with LC3 (Fig EV2E). However, FTMTR82A‐GFP did not colocalize with NCAO4‐mCherry or LC3, and NCOA4I489A/W497A‐mCherry did not colocalize with FTMT‐GFP or LC3 (Fig EV2E). These results identify the residues that are essential for the colocalization of FTMT and NCOA4 at the mitochondria and with LC3.
To determine the role of mitochondria targeting in colocalization and interaction between FTMT and NCOA4, we next generated an FTMT construct lacking the mitochondrial target sequence and bearing a GFP tag at the C‐terminal (FTMTΔMT‐GFP). We also generated a construct harboring only the mitochondrial target sequence of FTMT and bearing a GFP tag at the C‐terminal (FTMTMT‐GFP) (Fig EV2D). FTMTΔMT‐GFP associated with NCOA4‐mCherry, which is likely due to the presence of the R82 residue in the FTMTΔMT sequence, but did not colocalize with NCOA4‐mCherry at the mitochondria. FTMTMT‐GFP, on the other hand, localized at the mitochondria, but did not associate with NCOA4‐mCherry (Fig EV2F).
We finally examined the interaction between the wild type and mutated constructs of FTMT and NCOA4 using co‐immunoprecipitation assays. Consistent with the results from the immunofluorescence analysis (Figs 4E, and EV2E and F), while FTMT‐GFP bound to NCOA4‐mCherry, FTMTMT‐GFP and FTMTR82A‐GFP did not bind to NCOA4‐mCherry (Fig 4F, lanes 1, 2, and 4). Additionally, NCOA4I489A/W497A‐mCherry did not bind to FTMT‐GFP (Fig 4F, lane 5). FTMTD57A‐GFP, which has a substitution of aspartic acid (D) for alanine (A) at position 57 in the mitochondrial target sequence (Fig EV2D), bound to NCOA4‐mCherry (Fig 4F, lane 3). Notably, treatment with iron (ferrous ammonium sulfate) inhibited the binding between FTMT‐GFP and NCOA4‐mCherry, while treatment with DFP increased binding between the two proteins (Fig 4G), though underlying mechanisms remain unknown.
Based on these results, we hypothesized that replacing the mitochondrial target sequence of FTMT with a peroxisome‐targeting sequence while retaining R82 (NCOA4‐binding site) would induce peroxisome‐specific autophagy. We expressed PMP34‐FTMT‐GFP, which has a peroxisome‐targeting sequence (Kim et al, 2008) instead of a mitochondrial target sequence, in Huh7 cells. PMP34‐FTMT‐GFP colocalized with LC3 at peroxisomes stained with catalase, suggesting the peroxisome‐specific autophagy (Fig EV2G). Thus, FTMT specifically interacts with NCOA4 and iron/DFP affects this interaction. It appears that the mitochondrial‐targeting sequence does not mediate the interaction between FTMT and NCOA4, and only plays a role in targeting FTMT to the mitochondria.
Iron loss‐induced mitophagy increases as mitochondrial damage progresses
We next examined how DFP‐mediated iron loss selectively targeted defective mitochondria for mitophagy. It is reported that iron chelation caused depolarization of some mitochondria but not entire mitochondria (Allen et al, 2013). Thus, we examined the proportion of the depolarized mitochondria to total mitochondria isolated from Huh7 cells and HepG2 cells using flow cytometry. Flow cytometric analysis revealed that DFP treatment increased the proportion of the depolarized mitochondria determined by JC1 in Huh7 cells [3.01% vs. 0.34% (control)] (Fig 5A) and HepG2 cells [2.41% vs. 0. 42% (control)] (Fig EV3A), but the degree of increase was not as high as that seen with CCCP treatment (49.2%) (Fig 5A).
Figure 5. Induction of mitochondrial damage by deferiprone and localization of mitochondrial ferritin precursor in the outer mitochondrial membrane.

-
AFlow cytometric analysis of the mitochondrial membrane potential of the isolated mitochondria. The mitochondria isolated from Huh7 cells with CCCP treatment and with or without DFP treatment were incubated with JC‐1 for 1 h at 30°C and subjected to flow cytometric analysis (n = 4, biological replicates) by excitation with a blue laser (488 nm) with simultaneous emission assessment using a 585/42 detector for JC‐1 red and a 530/30 for JC‐1 green. The trapezoid areas show the population of depolarized mitochondria. Quantification of the proportion of depolarized mitochondria in Huh7 cells with or without DFP treatment is shown in the lower panel. The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. **P < 0.01.
-
BThe oxygen consumption rate was measured using a Seahorse XF24 Extracellular Flux Analyzer (n = 4, technical replicates). Huh7 cells were incubated with 1 mM of DFP for different lengths of time (0, 3, 6, 18, and 24 h). Oxygen consumption was measured, and the respiration rate was analyzed by injecting of 1 μM oligomycin, 1 μM carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP), and 10 μM rotenone/antimycin A. OCR: oxygen consumption rate. The central point and the error bars indicate mean ± SD.
-
CImmunofluorescence staining for FTMT‐GFP (green) and LC3 (red) in Huh7 cells transfected with FTMT‐GFP. The proportion of colocalization was quantified for four randomly selected areas (technical replicates) using ImageJ software version 1.46 (NIH, Bethesda, MD). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. **P < 0.01.
-
DImmunofluorescence staining for ferritin‐GFP heavy chain (FTH1) (green) and LC3 (red) in Huh7 cells transfected with FTH1‐GFP. The proportion of colocalization was quantified as described in (C). *P < 0.05, **P < 0.01.
-
EImmunoblots for the precursor form and processed form of FTMT‐GFP, endogenous FTMT, NCOA4, LC3, and Tom20 using the mitochondrial fractions of Huh7 cells expressing FTMT‐GFP with or without digestion with proteinase K in the presence or absence of DFP. mtHSP70 was used as the loading control.
-
FImmunoblots for FTMT, VDAC, and mtHSP70 using the mitochondrial membrane from Huh7 cells with or without DFP treatment. The mitochondrial membrane was fractionated from Huh7 cells by means of sodium carbonate treatment. mtHSP70 was used as the non‐membrane‐integral protein control and VDAC as the membrane‐integral protein control. mtHSP70 at the right‐hand side in the third lane indicates the positive control obtained from the mitochondrial fraction of Huh7 cells without DFP treatment.
Source data are available online for this figure.
Figure EV3. Induction of mitochondrial damage by deferiprone, cytoplasmic localization of NCOA4, and FTMTD 57A precursor form in the mitochondrial membrane isolated from FTMTD 57A‐expressing cells.

-
AFlow cytometric analysis of the mitochondrial membrane potential of the isolated mitochondria. The isolated mitochondria from HepG2 cells with or without DFP treatment were incubated with JC‐1 for 1 h at 30°C and subjected to the flow cytometric analysis (n = 4, biological replicates) by excitation with a blue laser (488 nm) with emission assessed simultaneously using a 585/42 for JC‐1 red and a 530/30 for JC‐1 green. The trapezoid areas show the population of depolarized mitochondria. Quantification of the proportion of depolarized mitochondria is presented in the lower panel. The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. **P < 0.01.
-
BThe oxygen consumption rate was measured using a Seahorse XF24 Extracellular Flux Analyzer. Huh7 cells were incubated with different concentrations of DFP (0, 0.01, 0.1, 0.5, and 1 mM) for 24 h (n = 4, technical replicates). Oxygen consumption was measured, and the respiration rate was analyzed by injecting of 1 μM oligomycin, 1 μM carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP), and 10 μM rotenone/antimycin A. The central point and the error bars indicate mean ± SD. OCR: oxygen consumption rate.
-
CImmunofluorescence staining for NCOA4‐mCherry, GM130, protein disulfide isomerase (PDI), early endosome antigen 1 (EEA1), and Lamp2 in Huh7 cells. White arrowheads in the overlaid picture indicate the colocalization of NCOA4‐mCherry and GM130. Boxed areas are enlarged on the right. Scale bar: 10 and 5 μm in enlarged images.
-
DImmunoblots for FTMTD57A and VDAC using the mitochondrial membrane from Huh7 cells expressing FTMTD57A with or without DFP treatment. The mitochondrial membrane was fractionated from cells by sodium carbonate treatment. mtHSP70 was used as the non‐membrane‐integral protein control and VDAC as the membrane‐integral protein control. mtHSP70 at the right‐hand side in the third lane indicates the positive control obtained from the mitochondrial fraction of Huh7 cells without DFP treatment.
Source data are available online for this figure.
Mitochondrial basal and maximal respiration was reduced by treatment with DFP concentration of ≥ 0.1 mM for 24 h (Fig EV3B), suggesting that the DFP dose (1 mM) used for mitophagy induction in vitro in this study is enough to damage the mitochondria. When examined over 24 h, the basal and maximal respiration was repressed over time with DFP treatment (Fig 5B). The proportion of colocalization of FTMT with LC3 increased over time and reached a maximal level at 24 h after DFP treatment (Fig 5C). In contrast, colocalization of ferritin with LC3 occurred at the maximum level at 6 h after DFP treatment regardless of the progression of mitochondrial damage (Fig 5D). These results suggested that iron loss‐induced mitophagy increased as mitochondrial damage progressed.
Localization of the FTMT precursor form on the mitochondrial outer membrane
FTMT is first synthesized as a precursor on cytosolic ribosomes, then targeted to mitochondria, and finally processed into a functional protein (Levi et al, 2001). The translocases that catalyze the import of precursor proteins into mitochondria are driven by the membrane potential, ATP, or redox reactions (Chacinska et al, 2009). We found that NCOA4 colocalized with GM130 (Nakamura et al, 1995), a cytoplasmic protein that is tightly bound to the Golgi apparatus (Fig EV3C), which suggests that FTMT needs to localize in cytoplasm at least partially to interact with NCOA4. We hypothesized that FTMT precursor localizes in the outer mitochondria membrane without being processed in the damaged mitochondria, thus enabling NCOA4 to bind to FTMT and subsequently induce mitophagy. Proteinase K digestion of mitochondria from Huh7 cells expressing FTMT‐GFP removed the outer membrane protein Tom20 without digesting the matrix protein mtHSP70, irrespective of DFP treatment (Fig 5E, lanes 2 and 4). We detected two forms of endogenous FTMT only in DFP‐treated cells based on those different molecular weights (Fig 5E, lane 3), a precursor form (~ 30‐kDa), and an appropriately processed form (~ 27‐kDa) (Levi et al, 2001; Corsi et al, 2002), and a precursor form was digested with proteinase K (Fig 5E, lane 4). Similarly, two forms of FTMT‐GFP were detected regardless of DFP treatment (Fig 5E, lanes 1 and 3) and a precursor form was digested with proteinase K (Fig 5E, lanes 2 and 4). Under these conditions, NCOA4, LC3, and Tom20 were also digested with proteinase K, suggesting that NCOA4 and LC3 were associated with the outer mitochondrial membrane, along with FTMT.
Next, we isolated a mixture of mitochondrial inner and outer membranes from mitochondrial fractions of Huh7 cells employing sodium carbonate treatment that can isolate intracellular membranes of subcellular organelles (Fujiki et al, 1982). Following DFP treatment, a FTMT precursor form (~ 30‐kDa) was detected in the isolated mitochondrial membrane along with voltage‐dependent anion channel (VADC), an outer membrane protein. mtHSP70 was not detected, regardless of DFP treatment (Fig 5F). These results indicate that iron loss by DFP treatment results in the localization of the FTMT precursor form selectively on the mitochondrial membrane. As the D57 residue in FTMT is predicted to be a cleavage site of mitochondrial target sequence (Levi et al, 2001), FTMTD57A potentially localizes on the mitochondrial outer membrane without being processed. FTMTD57A precursor form (~ 30‐kDa) was weakly detected in the mitochondrial membrane isolated from FTMTD57A‐expressing cells even without DFP treatment (Fig EV3D). Taken together, FTMT seems to localize to the mitochondrial outer membrane with DFP treatment, which promotes access to NCOA4 in the cytosolic side. Thus, the role of FTMT in iron loss‐induced mitophagy may be like that of PINK1 in PINK1/Parkin‐associated mitophagy (Hasson et al, 2013) in terms of mitochondrial protein trafficking.
Damaged mitochondria selectively undergo iron loss‐induced mitophagy
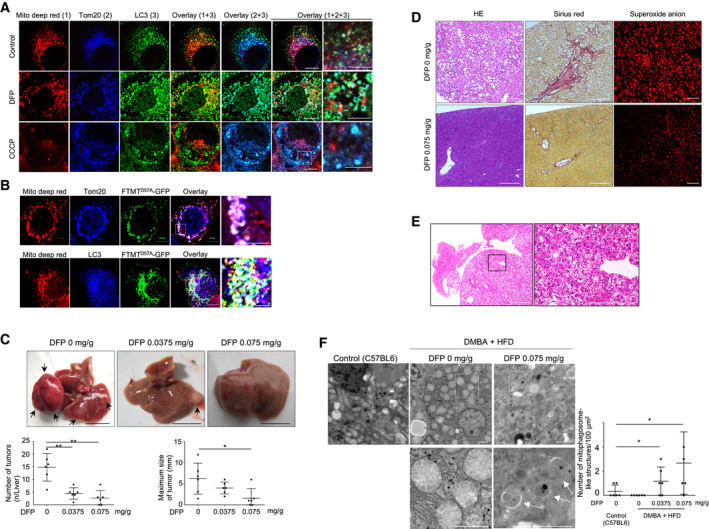
To examine whether iron loss‐induced depolarized mitochondria undergo mitophagy, we assessed polarized and depolarized mitochondria in DFP‐treated Huh7 cells using MitoTracker® Deep Red and Tom20‐based immunofluorescence staining. The polarized mitochondria shown by MitoTracker® Deep Red accumulation were not colocalized with LC3 following DFP treatment or CCCP treatment. The whole mitochondria (polarized and depolarized) shown by Tom20 staining were not colocalized with LC3 in the untreated cells but were partially associated with LC3 following DFP treatment or CCCP treatment (Fig 6A). These results suggest that depolarized mitochondria selectively undergo mitophagy upon DFP‐induced iron loss. We also observed that FTMTD57A‐GFP was associated with LC3 in the polarized mitochondria without DFP treatment (Fig 6B). This implies the importance of FTMT localization in the outer membrane of mitochondria in the induction of mitophagy.
Figure 6. Selective induction of iron loss‐induced mitophagy for damaged mitochondria and suppression of liver tumor, fibrosis, and reactive oxygen species production by deferiprone.
-
AImmunofluorescence staining for the polarized mitochondria (red), Tom20 (blue), and LC3 (green) in Huh7 cells untreated or treated with DFP (1 mM) for 24 h, or with CCCP (30 μM) for 6 h. Boxed areas are enlarged on the right. Scale bar: 10 and 5 μm in enlarged images.
-
BImmunofluorescence staining for the polarized mitochondria (red), Tom20 (blue), LC3 (blue), and FTMTD57A (green) using Huh7 cells expressing FTMTD57A. Boxed areas are enlarged on the right. The white puncta in overlaid pictures indicate the colocalization of the polarized mitochondria, Tom20, and FTMTD57A or the colocalization of the polarized mitochondria, FTMTD57A, and LC3. Scale bar: 10 μm and 5 μm in enlarged images.
-
CSTAM mice were fed a high‐fat diet and orally administered 0.0375 mg/g body weight of DFP, 0.075 mg/g body weight of DFP, or distilled water from 4 weeks of age to 16 weeks of age (n = 6). Arrows indicate liver tumors. Scale bar: 1.0 cm. The number and maximum size of liver tumors were compared among the three groups. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
DHistology of the nontumorous liver (H&E and Sirius red stain, original magnification ×40) and superoxide anion production in the livers of STAM mice fed a high‐fat diet with or without DFP (0.075 mg/g) administration. Frozen liver sections of mice were stained with dihydroethidium for assessment of superoxide anion. Scale bar: 100 μm.
-
EHistology of the hepatocellular carcinoma using H&E staining in STAM mice fed a high‐fat diet. The boxed area is enlarged on the right (×40 in left panel and ×400 in right panel). The tumor has increased cell density, an increased ratio of nucleus to cytoplasm, and thickened trabeculae Scale bar: 100 μm.
-
FElectron microscopic images of livers in control (C57BL/6) and DMBA + HFD mice that were orally administered 0.075 mg/g of DFP or distilled water. Scale bars: 2.0 μm. The boxed areas (×1,500) are enlarged below (×6,000 on left and ×10,000 on right). The white arrow indicates mitophagosome‐like structures. The mean number of mitophagosome‐like structures/100 μm2 was compared for four randomly selected areas (technical replicates) among the four groups (control, DMBA + HFD mice treated with 0, 0.0375, or 0.075 mg/g of DFP) (n = 6). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05.
Iron loss‐induced mitophagy correlates with suppression of liver tumor
Clearance of damaged mitochondria via mitophagy is critical for cellular fitness since dysfunctional mitochondria can impair electron transport chain function and increase oxidative stress. In the process of tumorigenesis, decreased mitophagy may allow for a permissive threshold of dysfunctional mitochondria to persist, generating increased tumor‐promoting ROS or other tumorigenic mitochondrial signals. Thus, interventions targeting mitophagy may have therapeutic potential for cancer prevention.
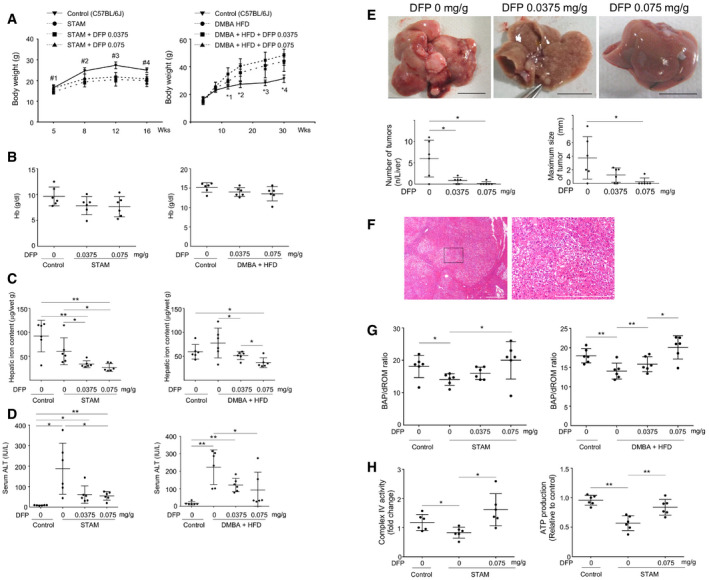
Accordingly, we assessed whether iron loss‐induced mitophagy can suppress the development of HCC using two hepatocarcinogenic mouse models: (i) the STAM model in which 2‐day‐old mice were treated with streptozotocin and then fed a high‐fat diet (HFD) from the age of 4 weeks (Fujii et al, 2013), and (ii) 7,12‐dimethylbenz(a)anthracene (DMBA) treatment to mice fed a high‐fat diet (DMBA + HFD) (Yoshimoto et al, 2013). DFP administration did not significantly change body weight (Fig EV4A) and hemoglobin levels in serum (Fig EV4B), but decreased hepatic iron content and serum ALT levels (Fig EV4C and D). Notably, DFP treatment at the equivalent dose (0.075 mg/g mouse body weight) used for patients with iron overload (Hider & Hoffbrand, 2018) resulted in a significant decrease in the number and maximum size of liver tumors in STAM mice (Fig 6C) and in DMBA + HFD mice (Fig EV4E) and also resulted in suppression of hepatic steatosis, fibrosis, and mitochondrial ROS (superoxide) production in STAM mice (Fig 6D). Liver tumors showed increased cell density, increased ratio of nucleus to cytoplasm, and thickened trabeculae in both STAM mice (Fig 6E) and DMBA + HFD mice (Fig EV4F), suggesting histology of HCC.
Figure EV4. Effects of DFP on body weight, hemoglobin level, hepatic iron content, ATL level, oxidative stress, and tumor growth in mice.
-
AChanges in body weights of untreated control (C57BL/6J, n = 6) mice, STAM mice (n = 6 for each group), and DMBA + HFD mice (n = 6 for each group) that were untreated or orally administered DFP as indicated (0, 0.0375, or 0.075 mg/g body weight) for 12 weeks (STAM mice) and for 26 weeks (DMBA + HFD mice). The central point and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. #1: P < 0.05, control vs. STAM + DFP (0.0375 or 0.075), #2: P < 0.01, control vs. other 3 groups, #3: P < 0.01, control vs. other 3 groups, #4: P < 0.01, control vs. other 3 groups, *1: P < 0.05, control vs. DMBA HFD + DFP (0.0375), *2: P < 0.05, control vs. other 3 groups, *3: P < 0.01, control vs. other 3 groups, *4: P < 0.01, control vs. other 3 groups.
-
BHemoglobin levels in untreated control (C57BL/6J) (n = 6) mice, STAM mice (n = 6 for each group), and DMBA + HFD mice (n = 6 for each group) that were orally administered DFP as described in (A). The central horizontal bar and the error bars indicate mean ± SD.
-
CHepatic iron content in untreated control (C57BL/6J) mice (n = 6), STAM mice (n = 6 for each group), and DMBA + HFD mice (n = 6 for each group) that were untreated or orally administered DFP as described in (A). Hepatic iron content was determined by atomic absorption spectrometry and expressed as micrograms Fe per gram of tissue (wet weight). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
DSerum ALT levels in untreated control (C57BL/6J) mice (n = 6), STAM mice (n = 6 for each group), and DMBA + HFD mice (n = 6 for each group) that were untreated or administered DFP as described in (A). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
EThe number and maximum size of liver tumors in DMBA + HFD mice (n = 6 for each group) that were untreated or orally administered DFP as described in (A). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. Representative images are shown in the upper panel. Scale bar: 1.0 cm. *P < 0.05.
-
FH&E staining of hepatocellular carcinoma in DMBA + HDF mice without DFP treatment. The tumor shows an increased cell density, an increased ratio of the nucleus to the cytoplasm, and thickened trabeculae (H&E stain). Scale bar: 100 μm.
-
GThe ratio of biological antioxidant potential (BAP) to diacron reactive oxygen metabolites (dROM), which indicates systemic antioxidant potential, was measured in untreated control (C57BL/6J) mice (n = 6), STAM mice (n = 6 for each group), and DMBA + HFD mice (n = 6 for each group) that were untreated or orally administered DFP as described in (A). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
HMitochondrial complex IV activity and ATP production in untreated control (C57BL/6J) mice (n = 6) and STAM mice (n = 6 for each group) that were untreated or administered with 0.75 mg/g body weight of DFP for 12 weeks. Mitochondrial complex IV activity (left panel) was measured using the hepatic mitochondrial fraction. ATP production (right panel) was measured in liver homogenates. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
The ratio of BAP to dROM, which indicates systemic antioxidant potential, was increased upon DFP administration in both mouse models (Fig EV4G). The complex IV activity in the mitochondrial electron transport chain and ATP production was significantly lower in STAM mice than control mice (C57BL/6J) (Fig EV4H). Restoration of complex IV activity and ATP production were observed in DFP‐treated STAM mice (Fig EV4H), suggesting the improvement of mitochondrial function by DFP in vivo. Although these results are seemingly contradictory to the reduced mitochondrial function by DFP in vitro (Figs 5A and B, and EV3A and B), it appears to be reasonable that clearance of damaged mitochondria by DFP‐induced mitophagy for a certain period of time (12 or 26 weeks in vivo) leads to the improvement of mitochondria function. This interpretation is also supported by the following observations by electron microscopy; an increase in the number of mitophagosome‐like structures that were identified by electron microscopy was observed in DMBA+HFD mice (Fig 6F) in a dose‐dependent manner.
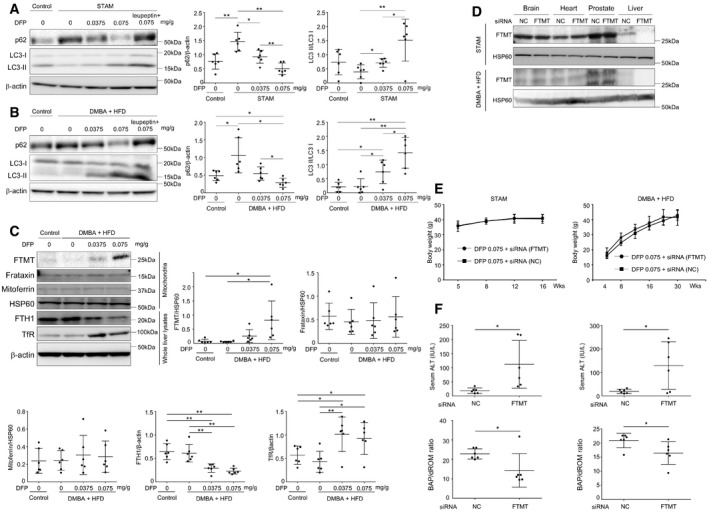
Next, we quantitatively assessed autophagic flux in mouse liver using a leupeptin (a membrane‐permeable thiol‐protease inhibitor)‐based assay (Haspel et al, 2011). DFP treatment resulted in a dose‐dependent decrease in p62 and an increase in LC3‐II, with a significant increase in both autophagy substrates upon treatment with leupeptin, suggesting DFP‐induced acceleration of autophagic flux in both STAM mice (Fig EV5A) and DMBA + HFD mice (Fig EV5B). These results suggested mitophagy induction in vivo by DFP.
Figure EV5. Effects of DFP on autophagic flux and FTMT induction, and effects of FTMT knockdown on body weight, ATL level, and oxidative stress in mice.
-
AImmunoblots for p62 and LC3 using whole liver lysates from untreated control (C57BL/6J) mice and STAM mice treated with DFP (0, 0.0375, or 0.075 mg/g body weight) or DFP (0.075) + leupeptin (n = 6 for each group, biological replicates). The expression levels of p62 were normalized to β‐actin, and LC3‐II was normalized to LC3‐I. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
BImmunoblots for p62 and LC3 using whole liver lysates from untreated control (C57BL/6J) mice and DMBA + HFD mice treated with DFP (0, 0.0375, or 0.075 mg/g body weight) or DFP (0.075) + leupeptin (n = 6 for each group, biological replicates). The expression levels of p62 were normalized to β‐actin, and LC3‐II was normalized to LC3‐I. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
CImmunoblots for FTMT, frataxin, mitoferrin, HSP60, FTH1, and TfR using the hepatic mitochondrial fractions and whole liver lysates in control (C57BL/6J) mice and DMBA + HFD mice treated with DFP (0, 0.0375, or 0.075 mg/g body weight) (n = 6 for each group, biological replicates). The expression levels of FTMT, frataxin, and mitoferrin were normalized to HSP60, and those of FTH1 and TfR were normalized to β‐actin. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
DImmunoblots for FTMT using the mitochondrial fractions of brain, heart, prostate, and liver obtained from STAM mice and DMBA + HFD mice. Mice were orally administered 0.075 mg/g body weight of DFP for 12 weeks (STAM mice) or 26 weeks (DMBA + HFD mice) with administration of control siRNA or FTMT siRNA.
-
EChanges in body weights of STAM mice (n = 6) and DMBA + HFD mice (n = 6); both mouse models were treated as indicated in (D). The central point and the error bars indicate mean ± SD.
-
FALT levels and the ratio of BAP to dROM in serum were measured in STAM mice (n = 6 for each group) (left panels) and DMBA + HFD mice (n = 6 for each group) (right panels). Both mouse models were treated as indicated in (D). The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. *P < 0.05.
Source data are available online for this figure.
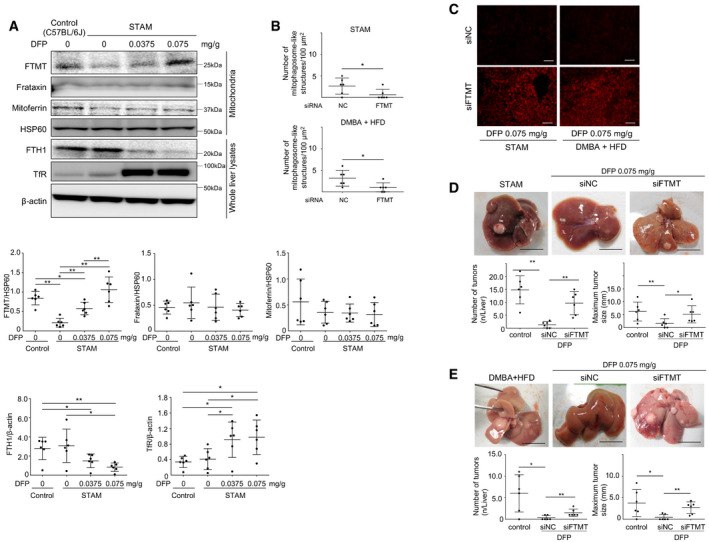
The DFP‐induced increase in the expression of FTMT was also observed in vivo in the liver of STAM mice, along with a decrease in the expression of FTH1 and an increase in the expression of TfR (Fig 7A) in a dose‐dependent manner. Similar DFP‐induced changes in the expression of FTMT, FTH1, and TfR were also observed in the livers of DMBA + HFD mice (Fig EV5C).
Figure 7. Induction of mitochondrial ferritin (FTMT) by deferiprone in vivo and FTMT is required for suppression of hepatocellular carcinoma.
-
AImmunoblots for FTMT, frataxin, mitoferrin, HSP60, FTH1, and TfR using the hepatic mitochondrial fraction lysate and whole liver lysate in control (C57BL/6J) mice and STAM mice treated with DFP (0, 0.0375, or 0.075 mg/g body weight) (n = 6). The expression of FTMT, frataxin, mitoferrin, FTH1, and TfR was normalized to HSP60 or β‐actin, respectively. The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01.
-
BThe mean number of mitophagosome‐like structures/100 μm2 from four randomly selected areas in the livers of DFP (0.075 mg/g body weight)‐administered mice was compared between control and FTMT siRNA‐treated groups for STAM mice (n = 6) and DMBA + HFD mice (n = 6). The central horizontal bar and the error bars indicate mean ± SD. Two‐sample t‐test was used for statistical analysis. NC: negative control siRNA, *P < 0.05.
-
CFrozen liver sections of DFP (0.075 mg/g body weight)‐administered mice (STAM mice and DMBA + HFD) with or without FTMT knockdown were stained with dihydroethidium. Red fluorescence indicates the ROS production. Scale bar: 100 μm. siNC: negative control siRNA.
-
D, ESTAM mice (D) were fed a high‐fat diet without DFP treatment or orally administered with 0.075 mg/g body weight of DFP from the age of 4 weeks, injected with FTMT siRNA (10 μl/g body weight) or negative control siRNA through the tail vein every 2 weeks for 8 weeks prior to sacrifice, and followed until 16 weeks of age (n = 6). DMBA + HFD mice (E) were fed a high‐fat diet, treated with DFP and siRNA in the same manner with STAM mice, and followed until 30 weeks of age (n = 6). The number and maximum size of liver tumors were compared among the three groups (control without DFP, negative control siRNA, and FTMT siRNA with DFP). The central horizontal bar and the error bars indicate mean ± SD. Tukey's honestly significant test was used for statistical analysis. *P < 0.05, **P < 0.01. Representative liver images are shown in the upper panel. Scale bar: 1 cm.
Source data are available online for this figure.
We next examined the in vivo effects of FTMT knockdown by examining serum ALT, oxidative stress, mitophagy, and HCC development in both STAM mice and DMBA + HFD mice that were administered DFP (0.075 mg/g body weight). We first confirmed that FTMT was knocked down specifically in the liver (Fig EV5D). The knockdown of FTMT did not affect the change in body weight in either mouse model (Fig EV5E). The number of mitophagosome‐like structures in the liver was decreased upon FTMT knockdown in both mouse models (Fig 7B). Conversely, mitochondrial ROS production (superoxide assessed with dihydroethidium) in the liver was increased upon FTMT knockdown (Fig 7C), which was consistent with increased serum ALT and decreased serum BAP/dROM ratio (systemic antioxidant potential) following FTMT knockdown (Fig EV5F). Notably, the suppressive effect of DFP on HCC development that we observed previously (Figs 6C and EV4E) was almost completely abrogated by FTMT knockdown in STAM mice (Fig 7D) and DMBA + HFD mice (Fig 7E). Thus, the hepatic iron decrease itself did not suppress HCC development since FTMT knockdown abrogated DFP‐induced suppression of HCC.
Discussion
In the present study, we revealed the molecular mechanisms behind iron loss‐induced mitophagy and its association with the suppression of liver tumors. As shown in Fig 8, iron loss is associated with HIF1α‐SP1 axis‐mediated regulation of FTMT expression, which is required for iron loss‐induced mitophagy. FTMT localization on the outer membrane of mitochondria and the interaction of FTMT with NCOA4 lead to the induction of mitophagy.
Figure 8. A schematic diagram depicting the mechanisms underlying the iron loss‐induced mitophagy.

Iron loss induced by DFP is associated with HIF1α‐SP1 axis‐mediated regulation of FTMT expression. The FTMT accumulation across the outer membrane of defective mitochondria leads to induction of mitophagy through the specific interaction between FTMT and nuclear receptor coactivator 4 (NCOA4), autophagic cargo receptor. PHD; propyl hydrogenase domain protein. VHL: von Hippel–Lindau protein.
Iron chelation‐mediated stabilization of HIF1α is deduced from the evidences that the propyl hydrogenase domain proteins (PHDs) require Fe2+ as a cofactor to be activated and activated PHDs degrade HIF1α through the binding of hydroxylated HIF1α by the von Hippel–Lindau protein (VHL) (Baek et al, 2011). The finding that iron loss activated FTMT transcription is seemingly contradictory to published studies that have shown the processed form of FTMT to have ferroxidase activity, similar to the H‐subunit of cytosolic ferritin. However, unlikely cytoplasmic ferritin, the levels of FTMT are not increased by exogenous iron (Corsi et al, 2002). Although exogenous iron readily available to mitochondria, excess iron is not usually retained in FTMT but is sequestered in cytosolic ferritin (Corsi et al, 2002). Thus, FTMT may not normally be required to detoxify excess iron. On the other hand, FTMT retained more iron than cytosolic ferritin even when iron stores were depleted by iron chelation (Corsi et al, 2002), which was consistent with present results that the extent of decrease in iron content was greater in the cytoplasm than in the mitochondria upon FTMT induction by DFP (Fig 2B). From these points of view, iron loss‐induced activation of FTMT protein synthesis may not necessarily be contradictory to its ferroxidase activity, even though a rationale for this cannot be completely explained.
DFP chelated iron from the mitochondria, but DFX and DFO did not (Fig 2A). These results were consistent with a previous report that DFP readily enters cells and efficiently reaches the major intracellular sites of iron accumulation, while DFO slowly enters cells via endocytosis (Glickstein et al, 2005). On the other hand, DFP increased the expression of FTMT, but DFX and DFO did not (Fig EV1E). These results suggested that mitochondrial iron deficiency may be required for FTMT induction apart from the stabilization of HIF1α‐SP1 axis, even though the underlying mechanisms remained uncertain. Although DFP‐induced transcriptional regulation of FTMT was a late response in this study, the expression of HIF1α has been shown to increase following 2‐h treatment with DFP (Zhao et al, 2020). This difference raised a possibility that there exists another factor other than HIF1α‐SP1 signaling for DFP‐regulated FTMT induction. Furthermore, the DFP concentration of ≥ 0.1 mM damaged the mitochondria (Fig EV3B); however, the lower DFP dose (0.01 mM) did not damage the mitochondria (Fig EV3B) but increased the expression of FTMT (Fig 2C). These results suggested that mitochondria damage may not be necessarily required for FTMT induction.
In addition to the activation of FTMT expression, the interaction of FTMT with NCOA4 increases the colocalization of FTMT with LC3 as mitochondrial damage progressed. Prior to this, the localization of FTMT occurs on the mitochondrial outer membrane. It seems that FTMT might act like PINK1 accumulation on depolarized mitochondria, even though FTMT is not a kinase, like PINK1. Further studies will be required to elucidate whether FTMT localizes on the mitochondrial outer membrane due to mitochondrial depolarization by DFP treatment.
The hepatic iron decrease itself did not suggest the suppression of HCC development, since FTMT knockdown abrogated DFP‐induced suppression of HCC in this study. To the best of our knowledge, this is the first report showing that the induction of mitophagy can reduce HCC development in a hepatocarcinogenic mouse model. This is supported by a previous report shown that autophagy‐deficient mice developed multiple liver tumors (Takamura et al, 2011). There have been also several lines of evidence demonstrating that iron chelators work against cancer as the potential therapeutic agents. This concept is based on inhibitory effects of iron chelation on cell cycle and DNA synthesis via ribonucleotide reductase (Heath et al, 2013). Interestingly, FTMT is reported to downregulate cell proliferation and affect cell cycle, causing G1/S arrest (Shi et al, 2015). These results are consistent with the present results that FTMT knockdown abrogated DFP‐induced suppression of HCC, and suggest a role of FTMT in anticancer effects of iron chelators. Further, FTMT overexpression has been reported to inhibit xenograft tumors in nude mice via cytosolic iron deprivation (Nie et al, 2006). We also found in vitro that the extent of decrease in iron content was greater in the cytoplasm than in the mitochondria upon FTMT induction by DFP. These results suggest that FTMT induction may suppress tumor development by affecting tumor iron homeostasis via shunting iron into mitochondria.
Mitophagy plays an essential role in reducing mitochondrial ROS production (Kurihara et al, 2012). We observed decreased mitochondrial ROS production upon induction of mitophagy. The increase in p‐p62 expression may also account for cellular antioxidative conditions because p‐p62 activates nuclear factor erythroid 2, which induces the expression of genes encoding antioxidant and detoxifying enzymes (Ichimura et al, 2013). While cellular antioxidative conditions may, in part, contribute to the suppression of HCC development, we believe that this alone cannot explain our observation of suppression of HCC development. Further analyses are required to elucidate more detailed mechanisms underlying suppressive effect of mitophagy on HCC development.
Materials and Methods
Cell culture and iron chelator treatment
Huh7 and HepG2 hepatoma cells were purchased from the Cell Resource Center for Biomedical Research for the Institute of Development, Aging and Cancer at Tohoku University and Cellular Engineering Technologies Inc. (Coralville, IA), respectively. Normal human hepatocytes were purchased from Zen‐Bio Inc. (Research Triangle Park, NC). All three types of cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin–streptomycin solution (Wako Pure Chemical Industries, Ltd., Osaka, Japan) at 37°C in a humidified atmosphere with 5% CO2. DFP (Deferiprone; Sigma‐Aldrich, St Louis, MO) was dissolved in H2O at 95°C and subjected to sterile filtration. DFO (Deferoxamine, Sigma‐Aldrich) was dissolved in DMSO at room temperature, and DFX (Deferasirox, kindly provided from Novartis Pharmaceutical Corp.) was dissolved in H2O at room temperature and subjected to sterile filtration. Cells were treated with DFP, DFO, or DFX for 24 h at the following final concentrations with/without 100 mM of bafilomycin: DFP at 0.01, 0.1, 0.5, and 1.0 mM; DFO at 0.01 mM; and DFX at 0.01 mM. Cells were washed twice with PBS on ice and subsequently scraped into ice‐cold lysis buffer [50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP‐40, 1 mM 1,4 dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitors], incubated on ice for 10 min, and centrifuged at 20,000 × g for 10 min at 4°C. Supernatants were retained, and protein concentration was measured using Direct Detect™ (Merk Millipore, Burlington, MA).
Detection of mitophagy using mt‐mKeima
Huh7 and HepG2 cells were plated on 6‐well culture plates, transfected with the plasmid, pMT‐mKeima‐Red (MBL, Aichi, Japan), using Lipofectamine™ 3000 (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer's instructions, and then treated with DFP for 24 h for the detection of mitophagy. Cells were excited with both red (550 nm) and blue laser (488 nm) with emission assessed simultaneously using a 660 ± 20 nm and a 585 ± 42 nm detector. Cells were also trypsinized, washed once with PBS buffer, resuspended in 500 μl of PBS, and subjected to FACS analysis for the quantification of mitophagy using a BD FACS Canto™ flow cytometer (BD Biosciences, Franklin Jersey, NJ). Cells were excited with both red (633 nm) and blue laser (488 nm) with emission assessed simultaneously using a 660 ± 20 nm and a 585 ± 42 nm detector.
Electron microscopy
A small portion of mouse liver specimens and cells which were treated with 1 mM DFP for 24 h were fixed in 3% glutaraldehyde, post‐fixed in 1% OSO4 in 0.1 M phosphate buffer for 15 min at 4°C, stained with 1% uranyl acetate, dehydrated in a graded series of ethanol, and embedded in Epon‐Araldite. Serial ultrathin sections, each 70–80 nm in thickness, were stained with uranyl acetate and lead acetate, and observed using an electron microscope (JEOL JEM1400).
Isolation of mitochondria
The cells were lysed by mechanical homogenization using a small pestle, and mitochondrial extraction was performed using a Qproteome Mitochondria Isolation Kit (Qiagen, Hilden, Germany). Liver mitochondria were isolated as described previously (Korenaga et al, 2005). In brief, the livers were minced on ice and homogenized by five strokes with a Dounce homogenizer and a tight‐fitting pestle in isolation buffer (70 mM sucrose, 1 mM KH2PO4, 5 mM HEPES, 220 mM mannitol, 5 mM sodium succinate, and 0.1% bovine serum albumin, pH 7.4). The homogenate was centrifuged at 500 × g for 5 min at 4°C. The supernatant fraction was retained, whereas the pellet was washed with isolation buffer and centrifuged again. The combined supernatant fractions were centrifuged at 10,000 × g for 10 min at 4°C to obtain crude mitochondrial fractions.
Immunoblotting
Cell lysates or mitochondrial fractions were subjected to immunoblot analysis using an iBlot® Gel Transfer Device (Thermo Fisher Scientific). Membranes were incubated with the following primary antibodies: a rabbit anti‐LC3 polyclonal antibody (Novus Biologicals, Littleton, CO), a rabbit anti‐p62/SQSTM1 polyclonal antibody (MBL, Nagoya, Japan), a mouse anti‐phospho‐p62 monoclonal antibody (MBL), a rabbit anti‐Parkin polyclonal antibody (Abcam, Cambridge, UK), a rabbit anti‐PINK1 monoclonal antibody (Cell Signaling Technology, Danvers, MA), a rabbit anti‐Atg5 polyclonal antibody (MBL), a mouse anti‐mitochondrial heat shock protein (HSP)60 monoclonal antibody (Abcam), a rabbit anti‐mitochondrial ferritin polyclonal antibody (Abcam), a mouse anti‐NCOA4 monoclonal antibody (Abcam), a rabbit anti‐FTH1 polyclonal antibody (Cell Signaling Technology), a rabbit anti‐TfR polyclonal antibody (Abcam), a rabbit anti‐Frataxin polyclonal antibody (Bioss Inc., Woburn, MA), a rabbit anti‐Mitoferrin polyclonal antibody (Abcam), a rabbit anti‐SP1 polyclonal antibody (Abcam), a rabbit anti‐HIF1α monoclonal antibody (Abcam), a rabbit anti‐VDAC monoclonal antibody (Cell Signaling Technology), a rabbit anti‐Tom20 polyclonal antibody (Santa Cruz Inc., Santa Cruz, CA), a mouse anti‐mitochondrial HSP70 monoclonal antibody (Thermo Fisher Scientific), a rabbit anti‐β‐actin monoclonal antibody (Cell Signaling Technology), a rabbit anti‐Lamin B1 polyclonal antibody (Abcam), and a rabbit anti‐His‐tag polyclonal antibody (MBL). All antibodies were used in 1:1,000 dilution except for anti‐Tom20 polyclonal antibody (1:100).
Measurement of iron content
Before measuring iron content, any traces of iron chelator were removed from the samples (whole cell lysates or mitochondrial fractions) by mixing with nitrate (1:5) and heating for 20–30 min at 100–110°C, followed by the addition of 30% hydrogen peroxide and heating at 200°C. Then, the samples were dissolved in 0.01 M HNO3, and the iron content was measured using a Metallo Assay Kit based on the Ferrozine method (Riemer et al, 2004) according to the manufacturer's instructions (MG Metallogenics, Chiba, Japan). The cytoplasmic iron content was measured by subtracting the iron content of the mitochondrial fractions from the iron content of the whole lysate.
Real‐time reverse transcription polymerase chain reaction
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). Total RNA (2 μg) was reverse‐transcribed to cDNA using the High‐Capacity RNA to cDNA Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. TaqMan® Gene Expression Assays for FTMT and GAPDH were purchased from Thermo Fisher Scientific, and mRNA levels were quantified in triplicate using a 7500 Real‐Time PCR system (Thermo Fisher Scientific) according to the supplier's recommendation. The expression level for FTMT was normalized to that of GAPDH.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed with Huh7 cells equivalent to 1 × 106, using a LowCell# ChIP kit (Diagenode Inc., Denville, NJ) according to the manufacturer's instructions. Chromatin samples were immunoprecipitated by rotating overnight at 4°C and incubated with the following antibodies: a rabbit anti‐CREB monoclonal antibody (Abcam), a rabbit anti‐SP1 polyclonal antibody (Abcam), a rabbit anti‐YY1 polyclonal antibody (Abcam), a rabbit anti‐FOXA1 polyclonal antibody (Abcam), a rabbit anti‐GATA2 polyclonal antibody (Abcam), and a rabbit anti‐CEBP Beta (Abcam). DNA samples were purified by GE Healthcare PCR Purification Kit (GE Healthcare, CA). DNA was eluted in 50 μl of H2O, and 2 μl of elution was subjected to the quantitative PCR. Quantitative PCR was performed by TaqMan® with primers designed to specifically amplify the human FTMT promoter region using a 7500 Real‐Time PCR system (Thermo Fisher Scientific). Quantification of chromatin immunoprecipitation was performed using % of input. Input DNA (control) is the DNA that went through the process without any specific selection for fragments related to binding of transcription factors. % of input was measured by dividing DNA signal obtained from chromatin immunoprecipitation by input DNA signal.
RNA interference
The siRNA oligonucleotides for the knockdown of FTMT, SP1, and HIF1α were obtained from Thermo Fisher Scientific. Huh7 cells and HepG2 cells were grown to 50–60% confluency and transfected with 100 pM siRNA oligonucleotides using Lipofectamine 3000 (Thermo Fisher Scientific). Cells were analyzed 72 h after transfection. Knockdown of FTMT in vivo was performed using Invivofectamine® 3.0 Reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. In brief, the FTMT siRNA (10 μl/g body weight) or negative control siRNA was injected through the tail vein every 2 weeks for a total of 8 weeks, followed by sacrificing the mice for analysis.
Rescue experiments were performed as described previously (Tabuchi et al, 2010). To generate encoding FTMT siRNA‐resistant wild‐type FTMT, inverse PCR was performed with primers (the forward primer: 5′‐TTG GTA AAA ATG GGG GCC CCG GAT GCT GG‐3′; the reverse primer: 5′‐ATT ATG TAC ATG ATC ACC TAG TTC TTT GAT AG‐3′; substituted nucleotides without an amino acid change are underlined) corresponding to the sequence of FTMT siRNA using pUC19‐FTMT plasmid as a template, and the PCR product was self‐ligated with T4 DNA ligase and T4 polynucleotide kinase. The resulting clone was transferred into a pIRES neo‐GFP vector or pIRES‐puro vector by EcoRI‐BgIII digestion.
Immunofluorescence microscopy
Cells were fixed, permeabilized, and immunostained using the following primary antibodies: rabbit anti‐mitochondrial ferritin polyclonal antibody (Abcam), goat anti‐Tom20 polyclonal antibody (Santa Cruz, Inc.), rabbit anti‐LC3 monoclonal antibody (MBL), mouse anti‐NCOA4 monoclonal antibody (Abcam), rabbit anti‐catalase polyclonal antibody (Abcam), mouse anti‐GM130 monoclonal antibody (BD Transduction Laboratories, San Jose, CA), mouse anti‐protein disulfide isomerase (PDI) monoclonal antibody (Daiichi Fine Chemical, Toyama, Japan), mouse anti‐early endosome antigen 1 (EEA1) monoclonal antibody (BD Transduction Laboratories), and mouse anti‐Lamp2 monoclonal antibody (Developmental Studies Hybridoma Bank, Baltimore, MD). The following secondary antibodies were used: DyLight 405‐conjugated goat anti‐rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., Baltimore, PA), Alexa Fluor 488‐conjugated donkey anti‐mouse IgG (Jackson ImmunoResearch Laboratories, Inc.), Alexa Fluor 488‐conjugated goat anti‐rabbit IgG (Thermo Fisher Scientific), Alexa Fluor 488‐conjugated donkey anti‐rabbit IgG (Thermo Fisher Scientific), Alexa Fluor 594‐conjugated goat anti‐rabbit IgG (Thermo Fisher Scientific), or Alexa Fluor 647‐conjugated donkey anti‐mouse IgG (Jackson ImmunoResearch Laboratories, Inc.). The cell images were captured using a confocal microscope (model LSM700, Zeiss, Jena, Germany) equipped with 488‐, 555‐, and 639‐nm diodes. The images were acquired in a sequential mode using a 63 × Plan Apochromat NA/1.4 oil objective and the appropriate filter combinations. All images were saved as tagged image file format files. The contrast was adjusted using Photoshop version CS5 (Adobe, San Jose, CA), and the images were imported into Illustrator version CS5 (Adobe). Pixel‐by‐pixel colocalization analysis was performed using ImageJ software (NIH, Bethesda, MD).
To analyze images of colocalized proteins with high accuracy, some immunofluorescent images were analyzed using the single‐molecule fluorescence microscopy (HM‐1000, Sysmex, Hyogo, Japan). Briefly, cells were prepared according to the manufacturer's instructions, and the sample was continuously irradiated with a laser so that a fluorescence dye that blinks repeatedly began to blink, and the bright spots were extracted from thousands of acquired images. The individual position of the fluorescence dye was processed through image analysis to construct a super‐resolution image.
Co‐immunoprecipitation
Co‐immunoprecipitation was performed using a Dynabeads® Co‐Immunoprecipitation Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Magnetic beads (Dynabeads M‐270 Epoxy) were conjugated to anti‐FTMT (Abcam), anti‐GFP (Novus Biologicals), or anti‐mCherry (Abcam) antibodies by rotating overnight at 37°C. The antibody–Dynabeads complex was then treated with coupling buffer. Beads coupled to anti‐FTMT, anti‐GFP, or anti‐mCherry antibodies were incubated with cell lysates for 30 min at 4°C and then washed with coupling buffer. Collected protein complexes were subjected to immunoblot analysis using anti‐NCOA4 (Abcam), anti‐LC3 (Novus Biologicals), or anti‐mCherry antibodies for detecting co‐immunoprecipitated mCherry‐tagged proteins.
Recombinant protein expression
The amino acid coding regions of human full‐length FTMT, FTMT (61–242), the mitochondrial target sequence of FTMT (1–61), NCOA4, NCOA4 (382–522), and FTH1 were amplified using total RNA from human HEp2 cells. FTMTR82A, FTMTD57A, mitochondrial target sequence‐deleted FTMT (FTMT∆MT), and NCOA4I489A/W497A were generated by site‐directed mutagenesis using KOD Plus DNA polymerase (Toyobo, Osaka, Japan). Full‐length of FTMT (wild type or point mutant), FTMT∆MT, FTMTMT, and FTH1 were cloned into the pIRES neo‐GFP plasmid, as described previously (Tabuchi et al, 2010). NCOA4 (wild type or point mutant) was cloned into the pREST‐B‐mCherry vector (kindly provided by Roger Y. Tsien, University of California, San Diego). PMP34 (kindly gifted by Peter Kim), which is encoding the leader sequence of peroxisome membrane, GFP, and FTMT∆MT, was cloned into pcDNA3.4‐TOPO (Thermo Fisher Scientific). Huh7 cells were grown to 50–60% confluence and transfected with GFP‐tagged FTMT (wild type or point mutant), mCherry‐tagged NCOA4 (wild type or point mutant), or GFP‐tagged FTMT∆MT with PMP34 using Lipofectamine™ 3000 (Thermo Fisher Scientific). Cells were analyzed 72 h after transfection. Additionally, FTMT (61–242) was cloned into the pET‐32a vector (Merck Millipore), and NCOA4 (382–522) was cloned into the pGEX2T vector (GE Healthcare).
Purification of recombinant proteins
For the purification of the GST‐fusion proteins and the His‐fusion proteins, Escherichia coli Lemo cells and E. coli Lemo21 (DE3) cells transformed with protein expression plasmids were grown in LB broth with 100 μg/ml ampicillin at 37°C to a OD600 of 0.8, respectively. Then, recombinant GST and recombinant His gene expression were induced by the addition of isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) (0.3 mM for GST and 0.5 mM for His at final concentration) for 3 h at 37°C for GST and 30°C for His. Cells were pelleted, resuspended in buffer A (1.0% Triton X‐100 in PBS) containing 1 mM PMSF, disrupted by sonication, and incubated with glutathione–Sepharose 4B beads (GE Healthcare) for 30 min at 37°C. The beads were centrifuged at 3,000 × g for 1 min at 4°C.
GST pull‐down assay
GST‐tagged proteins bound to beads were washed five times with buffer A. The GST‐fusion protein was incubated with His‐FTMT protein in pull‐down buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.4, 2 mM DTT, and 0.5% NP40) for 30 min at 4°C. The Sepharose beads were eluted with elution buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.4, 2 mM DTT, 0.5% NP40, and 50 mM glutathione) after washing. The recombinant fusion proteins were analyzed by immunoblotting.
Measurement of the membrane potential of isolated mitochondria
Mitochondria were isolated from untreated or DFP‐treated Huh7 cells as described above and adjusted to 30 μg/ml of the mitochondrial protein as a final concentration. The isolated mitochondria were incubated with JC‐1 (Setareh Biotech, LLC., Eugene, OR) in a mitochondria isolation buffer (3 mM HEPES‐KOH pH 7.4, 210 mM mannitol, 70 mM sucrose, and 0.2 mM EGTA) for 1 h at 30°C, and subjected to flow cytometric analysis, as described previously (Hendgen‐Cotta et al, 2017). The isolated mitochondria were excited with a blue laser (488 nm) with emission assessed simultaneously using a 585/42 nm for JC‐1 red and a 530/30 nm detector for JC‐1 green.
Oxygen consumption rate
Oxygen consumption was measured using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA). Cells were cultured in 24‐well seahorse plates at a density of 4 × 104 cells/well and incubated with several concentrations of DFP (0, 0.01, 0.1, 0.5, and 1 mM) for 24 h or with 1 mM of DFP for various lengths of time (0, 3, 6, 18, and 24 h). The cell culture medium was replaced with unbuffered DMEM containing 200 mM GlutaMax‐1, 100 mM Na pyruvate, 25 mM glucose, 32 mM NaCl, and 40 μM Phenol Red. Oxygen consumption was measured, and the respiration rate was analyzed with an injection of 1 μM oligomycin (Sigma‐Aldrich), 1 μM carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP), and 10 μM rotenone/antimycin A (Sigma‐Aldrich).
Isolation of the mitochondrial membrane
The mitochondrial membrane was isolated by means of sodium carbonate treatment, as described previously (Fujiki et al, 1982). In brief, the crude mitochondrial fractions were diluted 50‐ to 1,000‐fold with 100 mM sodium carbonate, pH 11.5, to bring the protein concentration to 0.02–1 mg/ml, and incubated for 30 min at 0°C. The suspensions were centrifuged for 30 min at 4°C, and the membrane pellets were gently rinsed with ice‐cold distilled water. The pellets were dissolved in RIPA buffer (50 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, and 0.1% SDS).
Staining of polarized mitochondria
Cells cultured on glass‐bottomed dishes were incubated with 200 nM MitoTracker® Deep Red FM (Thermo Fisher Scientific) for 30 min at 37°C.
Animals and experimental design
A NASH‐related HCC mouse model (STAM mouse, SMC Laboratories, Inc, Tokyo, Japan) was developed as described previously (Fujii et al, 2013). Briefly, 2‐day‐old male C57BL/6J (Clea, Tokyo, Japan) mice were injected with streptozotocin (200 μg/mouse) and fed a high‐fat diet (HFD‐32, Clea) from the age of 4 weeks, which was followed by the development of NASH and HCC at 8 and 18 weeks of age, respectively. We also used DMBA‐treated mice fed an HFD as an obesity‐related HCC mouse model (Yoshimoto et al, 2013). Briefly, 4‐ to 5‐day‐old male C57BL/6J mice were administered 50 μl of 0.5% DMBA (chemical carcinogen) solution to their dorsal surface and fed an HFD from the age of 4 weeks, which was followed by the development of HCC until 30 weeks of age. Four‐week‐old mice were orally administered with 0.0375 mg/g body weight of DFP, 0.075 mg/g body weight of DFP, or distilled water for 12 weeks for STAM mice and for 26 weeks for DMBA + HFD mice. The mice were bred and maintained according to the guidelines approved by the Institutional Animal Care Use Committee (Kawasaki Medical School).
Hepatic iron concentrations
Hepatic iron concentrations were determined by atomic absorption spectrometry (Z‐6100, Hitachi, Tokyo, Japan) as described previously (Furutani et al, 2006) and expressed as micrograms Fe per gram of tissue (wet weight).
Analysis of alanine aminotransferase (ALT) levels in serum
Serum ALT levels were measured using an automated analyzer for clinical chemistry SPOTCHEM EZ SP‐4430 (Arkray, Kyoto, Japan).
Measurement of ROS production
The cellular ROS level was measured by oxidation of the cell‐permeable and oxidation‐sensitive fluorogenic precursor, 2,7‐dihydrodichlorofluorescein diacetate (Thermo Fisher Scientific). Fluorescence was measured using a Varioskan Flash Multimode Reader (Thermo Fisher Scientific) at 494/535 nm (excitation/emission). In situ ROS production in the liver was assessed by staining with dihydroethidium, as described previously (Nishina et al, 2008). Fluorescence intensity was quantified using ImageJ (NIH) for three randomly selected areas of digital images in each mouse.
Measurement of the ratio of biological antioxidant potential (BAP) to diacron oxygen metabolites (dROM)
The dROM and BAP levels were measured using a FREE Carpe Diem device (WISMERLL, Tokyo, Japan) according to the manufacturer's instructions, as described previously (Korenaga et al, 2015).
Measurement of mitochondrial complex IV activity
Liver mitochondria of STAM mice were obtained as described in the section “Isolation of mitochondria in Materials and Methods”. Mitochondrial complex IV activity was measured using a MitoCheck® Complex IV Activity Assay Kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
Measurement of ATP production
One hundred milligrams of the liver from STAM mice was homogenized and centrifuged at 1,000 × g for 10 min at 4°C. The supernatant was assayed for ATP production using a Tissueno ATP Assay Kit (TOYO B‐Net Co., Ltd., Tokyo, Japan) according to the manufacturer's instructions.
Primers
All primer sequences are shown in the following table.
| Primer | Sequence |
|---|---|
| FTMT forward (for) | GAATTCATGCTGTCCTGCTTCAGGCTCCTC |
| FTMT reverse (rev) | AGATCTGCGTAGTTCTGCTTGTTTTCATTT |
| FTMTR82A for | GCCCAGATCAACCTCGAGCTCTATGC |
| FTMTR82A rev | GTTGATGGCAGCCTCGGAGT |
| FTMTR57A for | GCCCCTACCGGGCCCGCGGCC |
| FTMTR57A rev | CCGGGAGGAGGCGGCTGCGGC |
| FTMTMT for | GAATTCATGCTGTCCTGCTTCAGGCTCCTC |
| FTMTMT rev | AGATCTATCCGAGAGGGGCCGGCGGCGGG |
| FTMTΔMT for | CCCGCCGCCGGCCCCTCTCGGGT |
| FTMTΔMT rev | CATGAATTCGGGGATCCTCTAGAGTC |
| siRNA‐resistant FTMT for | TTGGTAAAAATGGGGGCCCCGGATGCTGG |
| siRNA‐resistant FTMT for | ATTATGTACATGATCACCTAGTTCTTTGATAG |
| FTMT (w/o tag) ecor1 for | GAATTCATGCTGTCCTGCTTCAGGCTCCTCTCC |
| FTMT (w/o tag) stop bgl2 | AGATCTGCTTAGTTCTGCTTGTTTTCATTTCCAAG |
| Ferritin for | GAATTCATGACGACCGCGTCCACCTCGCA |
| Ferritin rev | AGATCTGCGCTTTCATTATCACTGTCTCCC |
| NCOA4 for | GGATCCATGAATACCTTCCAAGACCAGAGT |
| NCOA4 rev | GGATCCGCGCACATCTGTAGAGGAGTTCGA |
| NCOA4I489A/W497A for | CTTGTCGGAGGCGCTTATCAGG |
| NCOA4I489A/W497A rev | GGGCTGTTCTTTGCGACTTGGA |
| FTMT‐His (for pull‐down assay) for | GAATTCATGCTGTCCTGCTTCAGGCTCCTC |
| FTMT‐His (for pull‐down assay) rev | GTCGACTTAGTTCTGCTTGTTTTCATTT |
| NCOA4382–522 (for pull‐down assay) for | GGATCCAGCATGGTTACAGAGGATTGGC |
| NCOA4382–522 (for pull‐down assay) rev | GAATTCTACTTCTGTTTGCCAGCTCTGT |
| siRNA in vitro | |
| Silence® Select | |
| siFTMT | |
| Thermo Fisher Scientific s41237 sense | GGUGAAGUCUAUCAAAGAAtt |
| Antisense | UUCUUUGAUAGACUUCACCtg |
| siHIF1α | |
| Thermo Fisher Scientific s6591 sense | CCUCAGUGUGGGUAUAAGAtt |
| Antisense | UCUUAUACCCACACUGAGGtt |
| Negative control No. 1 siRNA | Sequence is not revealed |
| Stealth™ RNAi | |
| siFTMT | |
| Thermo Fisher Scientific HSS132591 | ACCACGUGCACAACUUAGUGAAGAU |
| siSP1 | |
| Thermo Fisher Scientific HVS40865 | GCAGACACAGCAGCAACAAAUUCUU |
| Negative control 452002 | Low GC duplex (sequence is not revealed) |
| siRNA in vivo | |
| siFTMT | |
| Ambion S85393 sense | GGCUUAGCGGAAUAUCUUUtt |
| Antisense | AAAGAUAUUCCGCUAAGCCcg |
| Ambion™ In vivo negative control #1 siRNA 4457289 | Sequence is not revealed |
| RT–PCR | |
| FTMT HS00893202_s1 | Sequence is not revealed |
| GAPDH HS02758991_g1 | Sequence is not revealed |
Statistics
Quantitative values are expressed as the mean ± standard deviation. Two groups among the multiple groups were compared by Tukey's honestly significant test. Data of 2 groups were compared by the two‐sample t‐test for continuous variables.
Author contributions
YH designed the study, acquired data, analyzed and interpreted the data, drafted the manuscript, involved in statistical analysis, and acquired funding; IY designed the study, acquired data, analyzed and interpreted the data, and drafted the manuscript; AT designed the study, acquired data, analyzed and interpreted the data, drafted the manuscript, involved in statistical analysis, and acquired funding; FK analyzed and interpreted the data; JJL analyzed and interpreted the data, and drafted the manuscript; SN analyzed and interpreted the data; KS analyzed and interpreted the data; KH supervised the study, designed the study, analyzed and interpreted the data, wrote the manuscript, and acquired funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We thank Shinichi Tatsumi of Sysmex Corporation for technical assistance for the single‐molecule fluorescence microscope, HM‐1000. This work was supported by a Grant‐in‐Aid for Scientific Research (B) (26293179) from the Japan Society for the Promotion of Science and the Research Program on Hepatitis (18fk0210016 h0003) from the Japan Agency for Medical Research and Development (K.H.), a Grant‐in‐aid for Scientific Research (C) (16K09384) from the Japan Society for the Promotion of Science (Y. H.), a Grant‐in‐Aid for Young Scientists (B) (26870067/16K19047), and a Grant‐in‐Aid for Research Activity Start‐up (23870008) from the Japan Society for the Promotion of Science, GSK Japan Research Grant, the Cell Science Research Foundation, the Uehara memorial Foundation, and the Sumitomo Foundation (A. T.). We also thank Nuria Andreu, senior editor of scientific editing nature research editing service, for English language editing.
EMBO Reports (2020) 21: e50202
See also: Y Aman et al (November 2020)
Contributor Information
Atsushi Tanaka, Email: atsushi.tanaka@med.id.yamagata-u.ac.jp.
Keisuke Hino, Email: khino@med.kawasaki-m.ac.jp.
Data availability
No data were deposited in a public database.
References
- Allen GFG, Toth R, James J, Ganley IG (2013) Loss of iron triggers PINK1/Parkin‐independent mitophagy. EMBO Rep 14: 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek JH, Reiter CE, Manalo DJ, Buehler PW, Hider RC, Alayash AI (2011) Induction of hypoxia inducible factor (HIF‐1α) in rat kidneys by iron chelation with the hydroxypyridione, CP94. Biochem Biophys Acta 1809: 262–268 [DOI] [PubMed] [Google Scholar]
- Bhujabal Z, Birgisdottir A, Sjottem E, Brenne HB, Overvatn A, Habisov S, Kirkin V, Lamark T, Johansen T (2017) FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Rep 18: 947–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138: 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsi B, Cozzi A, Arosio P, Drysdale J, Santambrogio P, Campenella A, Biasiotto G, Albertini A, Levi S (2002) Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron homeostasis. J Biol Chem 277: 22430–22437 [DOI] [PubMed] [Google Scholar]
- Drysdale J, Arosio P, Invermizzi R, Cazzola M, Volz A, Corsi B, Biasiotto G, Levi S (2002) Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis 29: 376–383 [DOI] [PubMed] [Google Scholar]
- Esteban‐Martinez L, Sierra‐Filardi E, McGreal RS, Salazar‐Roa M, Marino G, Seco E, Durand S, Enot D, Grana O, Malumbres M et al (2017) Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 36: 1688–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, St‐Pierre P, Shankar J, Wang PTC, Joshi B, Nabi IR (2013) Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 24: 1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, Arumugam S, Watanabe K, Ichida T, Asakura H et al (2013) A murine model for non‐alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol 46: 141–152 [DOI] [PubMed] [Google Scholar]
- Fujiki Y, Fowler S, Shio H, Hubbard AL, Lazarow PB (1982) Polypeptide and phospholipid composition of the membrane of rat liver peroxisomes: comparison with endoplasmic reticulum and mitochondrial membranes. J Cell Biol 93: 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furutani T, Hino K, Okuda M, Gondo T, Nishina S, Kitase A, Korenaga M, Xiao SY, Weinman SA, Lemon SM et al (2006) Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 130: 2087–2098 [DOI] [PubMed] [Google Scholar]
- Glickstein H, Ben EIR, Shvartsman M, loav Cabantchik Z (2005) Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood 106: 3242–3250 [DOI] [PubMed] [Google Scholar]
- Guaraldo M, Santambrogio P, Rovelli E, Di Savino A, Saglio G, Cittaro D, Roetto A, Levi S (2016) Characterization of human mitochondrial ferritin promoter: identification of transcription factors and evidences of epigenetic control. Sci Rep 6: 33432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinary T, Englert JA, Choi AMK (2011) Characterization of macroautophagic flux in vivo using a leupeptin‐based assay. Autophagy 7: 629–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman‐Ackah S, Hessa T, Guha R et al (2013) High‐content genome‐wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504: 291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath JL, Weiss JM, Lavau CP, Wechsler DS (2013) Iron deprivation in cancer‐potential therapeutic implications. Nutrients 5: 2836–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendgen‐Cotta UB, Esfeld S, Rudi K, Miinalainen I, Klare JP, Rassaf T (2017) Cytosolic BNIP3 dimer interacts with mitochondrial BAX forming heterodimers in the mitochondrial outer membrane under basal conditions. Int J Mol Sci 18: E687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297 [DOI] [PubMed] [Google Scholar]
- Hider RC, Hoffbrand AV (2018) The role of deferiprone in iron chelation. N Engl J Med 379: 2140–2150 [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T et al (2013) Phosphorylation of p62 activates the Keap1‐Nrf2 pathway during selective autophagy. Mol Cell 51: 618–631 [DOI] [PubMed] [Google Scholar]
- Jeong JK, Park SY (2012) Transcriptional regulation of specific protein 1 (SP1) by hypoxia‐inducible factor 1 alpha (HIF1α) leads to PRNP expression and neuroprotection from toxic prion peptide. Biochem Biophys Res Commun 429: 93–98 [DOI] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Kim I, Rodriguez‐Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PK, Hailey DW, Mullen RT, Lippincott‐Schwartz J (2008) Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA 105: 20567–20574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J, Weinman SA (2005) Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem 280: 37481–37488 [DOI] [PubMed] [Google Scholar]
- Korenaga M, Nishina S, Korenaga K, Tomiyama Y, Yoshioka N, Hara Y, Sasaki Y, Shimonaka Y, Hino K (2015) Branched‐chain amino acids reduce hepatic iron accumulation and oxidative stress in hepatitis C virus polyprotein‐expressing mice. Liver Int 35: 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D (2012) Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quality in yeast. J Biol Chem 287: 3265–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, Arosio P, Drysdale J (2001) A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem 276: 24437–24440 [DOI] [PubMed] [Google Scholar]
- Liu L, Feng DF, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R (2012) The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle‐wasting stimuli. Cell Meatb 16: 613–624 [DOI] [PubMed] [Google Scholar]
- Lou G, Palikaras K, Lautrup S, Scheibye‐Knudsen M, Tavernarakis N, Fang EF (2020) Mitophagy and neuroprotection. Trends Mol Med 26: 8–20 [DOI] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, Harper JW (2015) Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2‐mediated proteolysis. Elife 4: e10308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Allen GFG, Tamjar J, Munson MJ, Thomson C, Muqit MMK, Ganley IG (2016) Mito‐QC illuminates mitophagy and mitochondrial architectures in vivo . J Cell Biol 214: 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H et al (2015) Bcl2‐2‐like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6: 7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G (1995) Characterization of a cis‐Golgi matrix protein, GM130. J Cell Biol 131: 1715–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie G, Chen G, Sheftel AD, Pantopoulos K, Ponka P (2006) In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood 108: 2428–2434 [DOI] [PubMed] [Google Scholar]
- Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, Furutani T, Sasai A, Okuda M, Hidaka I et al (2008) Hepatitis C virus‐induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 134: 226–238 [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M et al (2011) Image‐based genome‐wide siRNA screen identifies selective autophagy factors. Nature 480: 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20: 1013–1022 [DOI] [PubMed] [Google Scholar]
- Quinsay MN, Thomas RL, Lee Y, Gustafsson AB (2010) Bnip3‐mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6: 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer J, Hoepken HH, Czewinska H, Robinson SR, Dringen R (2004) Calorimetric ferrozine‐based assay for the quantification of iron in cultured cells. Anal Biochem 331: 370–375 [DOI] [PubMed] [Google Scholar]
- Schweers R, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL et al (2007) Nix is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104: 19500–19505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Youle RJ (2018) PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 16: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi ZH, Shi FF, Wang YQ, Sheftel AD, Nie G, Zhao YS, You LH, Gou YJ, Duan XL, Zhao BL et al (2015) Mitochondrial ferritin, a new target for inhibiting neuronal tumor cell proliferation. Cell Mol Life Sci 72: 983–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szargel R, Shani V, Abd Elghani F, Mekies LN, Liani E, Rott R, Engelender S (2016) The PINK1, synphilin‐1 and SIAH‐1 complex constitutes a novel mitophagy pathway. Hum Mol Genet 25: 3476–3490 [DOI] [PubMed] [Google Scholar]
- Tabuchi M, Yanatori I, Kawai Y, Kishi F (2010) Retromer‐mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. J Cell Sci 123: 756–766 [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy‐deficient mice develop multiple liver tumors. Genes Dev 25: 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa E, Proics E, Rubio‐Patino C, Obba S, Zunino B, Bossowski JP, Rozier RM, Chiche J, Mondragon L, Riley JS et al (2017) Parkin‐independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep 20: 2846–2859 [DOI] [PubMed] [Google Scholar]
- Yamasaki T, Terai S, Sakaida I (2011) Deferoxamine for advanced hepatocellular carcinoma. N Engl J Med 365: 576–578 [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M et al (2013) Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101 [DOI] [PubMed] [Google Scholar]
- Youle RJ (2019) Mitochondria‐striking a balance between host and endosymbiont. Science 365: eaaw9855 [DOI] [PubMed] [Google Scholar]
- Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428: 1714–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JF, Rodger CE, Allen GFG, Weidlich S, Ganley IG (2020) Hif1α‐dependent mitophagy facilitates cardiomyoblast differentiation. Cell Stress 5: 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
No data were deposited in a public database.