

Abstract
Poly(ADP‐ribose) polymerase 1 (PARP1) facilitates DNA damage response (DDR). While the Ewing's sarcoma breakpoint region 1 (EWS) protein fused to FLI1 triggers sarcoma formation, the physiological function of EWS is largely unknown. Here, we investigate the physiological role of EWS in regulating PARP1. We show that EWS is required for PARP1 dissociation from damaged DNA. Abnormal PARP1 accumulation caused by EWS inactivation leads to excessive Poly(ADP‐Ribosy)lation (PARylation) and triggers cell death in both in vitro and in vivo models. Consistent with previous work, the arginine‐glycine‐glycine (RGG) domain of EWS is essential for PAR chain interaction and PARP1 dissociation from damaged DNA. Ews and Parp1 double mutant mice do not show improved survival, but supplementation with nicotinamide mononucleotides extends Ews‐mutant pups’ survival, which might be due to compensatory activation of other PARP proteins. Consistently, PARP1 accumulates on chromatin in Ewing's sarcoma cells expressing an EWS fusion protein that cannot interact with PARP1, and tissues derived from Ewing's sarcoma patients show increased PARylation. Taken together, our data reveal that EWS is important for removing PARP1 from damaged chromatin.
Keywords: DNA damage response, EWS, PAR, PARP‐1, PARylation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
EWS removes PARP1 from damaged DNA, and abnormal retention of PARP1 due to the loss of EWS triggers cell death.
Introduction
Maintenance of genomic integrity is important to preserve genetic information and ensure cellular and organismal survival. Failure to maintain genomic integrity leads to inherited disease and cancer. The integrity of the genome is constantly challenged by exogenous and endogenous insults. DNA damage, such as single‐strand breaks (SSB) induced by endogenous oxidative stress (Tubbs & Nussenzweig, 2017), is sensed by DNA damage response (DDR) pathways which activate various DNA repair pathways. Besides inactivation, the inappropriate activation of DNA repair pathways can equally cause genome instability (Hingorani et al, 2005).
The activation of the DDR is controlled by a variety of post‐translational modifications such as phosphorylation, ubiquitination, methylation, acetylation, and poly(ADP‐ribosy)lation (PARylation) (Hottiger et al, 2010; Teloni & Altmeyer, 2016). The initial activation of the DDR requires the recruitment of poly(ADP‐ribose) polymerase 1 (PARP1) to damaged DNA (Krishnakumar & Kraus, 2010). PARP1 is the first PARP family protein to be identified and shares a conserved catalytic domain. Each PARP protein has one or more additional domains required for its unique function (Li & Chen, 2014). PARP1 is evolutionarily conserved in all mammals and is comprised of a N‐terminal zinc finger DNA‐binding domain (DBD), a nuclear localization signal (NLS), a central auto‐modification domain, and a C‐terminal catalytic domain that binds to nicotinamide adenine dinucleotide (NAD+). PARP1 acts by sensing DNA damage and PARylating proteins localized at damaged DNA using NAD+ as a cofactor for PARylation to promote base excision‐, SSB‐, and double‐strand break (DSB) repair (Krishnakumar & Kraus, 2010; Khoury‐Haddad et al, 2014; Luijsterburg et al, 2016; Kruger et al, 2020). Poly ADP‐ribose (PAR) appended to PARP1 and other proteins at damaged DNA is composed of linear or branched repeats of ADP ribose (Kim et al, 2005; Teloni & Altmeyer, 2016). PAR chains are thought to activate the DDR and recruit DNA repair proteins to damaged DNA (Durkacz et al, 1980; Haince et al, 2008; Luo & Kraus, 2012). PARylated PARP1 quickly dissociates from damaged DNA after the recruitment of other DNA repair proteins allowing DNA repair proteins to directly access damaged DNA (Krishnakumar & Kraus, 2010). The stringent regulation of the recruitment and dissociation of PARP1 from damaged DNA is considered as a key step for proper DDR activation and DNA repair. Consistently, depletion of PARP1 sensitizes cells to alkylating agents (Trucco et al, 1998). Alternatively, hyper‐PARylation caused by abnormal activation of PARP1 exhausts cellular NAD+ level leading to defects in diverse biological processes (Luo & Kraus, 2012). The current understanding of PARP1 in DDR and DNA repair is mainly focused on PARP1′s ability to sense DNA damage and to PARylate target proteins. However, the mechanisms by which PARP1 dissociates from damaged DNA to promote downstream DDR and DNA repair are underexplored.
The Ewing's sarcoma breakpoint region 1 (EWSR1, herein termed EWS) protein is a member of the FET family of RNA‐binding proteins, which includes the fused in sarcoma RNA‐binding protein (FUS; also known as translocated in liposarcoma) and the TATA‐box binding protein associated factor 15. The EWS gene was originally identified at the breakpoint of the Ewing sarcoma chromosomal translocation t(11;22)(q24;q12), which results in a fusion protein of the EWS and the Friend leukemia virus integration site 1 (FLI1) protein (Delattre et al, 1992). The EWS‐FLI1 fusion protein initiates sarcoma during childhood and puberty. Research on Ewing's sarcoma has been focused on the “gain‐of‐function” properties of the fusion in genomic integrity. However, the physiological functions of EWS in normal untransformed cells are not fully understood. We reported previously that the loss of endogenous EWS is associated with defects in the lineage determination of adipocytes, with dysfunctional mitochondrial homeostasis and with postnatal lethality in Ews‐deficient (Ews −/−) mice (Park et al, 2013, 2015). Interestingly, mouse embryonic fibroblasts derived from Ews −/− mice were hypersensitive to ionizing radiation (IR) and showed DNA damage induced cellular senescence (Li et al, 2007). Consistently, several studies have suggested that EWS confers cellular resistance to DNA damage and genomic instability in both in vitro and in vivo models (Klevernic et al, 2009; Paronetto et al, 2011; Park et al, 2016). Recently, a potential link between EWS and PARP1 in the DNA damage response has been suggested (Jungmichel et al, 2013; Mastrocola et al, 2013; Altmeyer et al, 2015). However, the detailed molecular mechanism of how EWS regulates PARP1 is not fully understood.
Here, we report that the loss of EWS leads to the accumulation of PARP1 at DNA damage sites. Trapped PARP1 induces hyper‐PARylation, and we provide evidence that is correlated with the exhaustion of cellular NAD+. EWS recruitment to PARP1 regulates the dissociation of PARylated PARP1 from damaged DNA and is mediated by the interaction between the EWS arginine‐glycine‐glycine (Arg‐Gly‐Gly, RGG) domains and PAR chains formed by PARP1. In line with this model, we observed an excessive accumulation of PARP1 and hyper‐PARylation in Ews −/− mouse embryos and in cancer cells derived from human Ewing's sarcoma patients. We provide genetic evidence that the accumulation of chromatin‐associated PARP1 caused by the loss of EWS results in hypersensitivity to DNA damaging agents; the hypersensitivity of EWS mutants being suppressed by PARP1 deficiency.
Results
The steady state level of DDR proteins is increased in the nucleus of Ews knockout cells
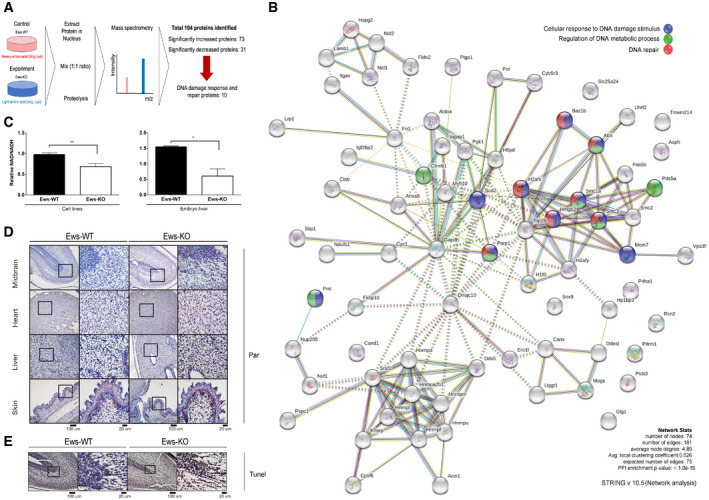
Previous studies had demonstrated that Ews knockout mice are associated with multiple defects including compromised B‐cell development and meiosis, excessive cellular senescence, mitochondrial dysfunction, increased serum lactate levels, and hypersensitivity to ionizing radiation (IR) (Li et al, 2007; Park et al, 2015). Since the combination of these defects were frequently observed in mouse models of genomic instability (Fang et al, 2014, 2016; Scheibye‐Knudsen et al, 2014), we hypothesized that EWS may function in genomic integrity. We started by investigating whether the abundance of proteins required for genome integrity is altered in mouse brown adipocyte (mBA) Ews −/− cells when compared to wild‐type (WT). We chose a proteomics‐based approach employing stable isotope labeling with amino acids in cells (SILAC) combined with mass spectrometry. Analyzing nuclear extracts, we found that out of the 927 proteins detected 73 proteins were significantly upregulated and 31 were significantly downregulated in Ews −/− mBA cells compared to WT (Fig 1A). In addition to proteins involved in regulating gene expression, the biosynthesis of cellular macromolecules and the assembly of cellular components, the level of DDR and DNA repair proteins including Parp1, H2ax, Hmgb2, Smc1a, Smc3, Baz1b, and Atrx (Fig 1B, Table EV1, and Dataset EV1) was increased. Consistent with the abnormal expression of DDR and DNA repair proteins, employing cell viability assay we found that Ews −/− mBA cells were hypersensitive to a variety of DNA damaging agents including hydroxyurea, cisplatin, ultraviolet irradiation, hydrogen peroxide (H2O2), and methyl methanesulfonate (MMS). The sensitivity to MMS, H2O2, DNA alkylating, and oxidative agents were particularly strong (Fig EV1A). To corroborate our findings in cells other than adipocytes and to demonstrate effects in human cell lines, we used HEK‐293 cells and generated an EWS mutant in this cell line. EWS knockout cells were hypersensitive to the long‐term exposure with MMS and H2O2 as determined by clonogenic survival assays (Fig EV1B and C). Consistent with the hypersensitivity being caused by DNA repair defects, using alkaline comet assays we found an increased level of SSBs upon MMS or H2O2 treating knockout cells (Fig EV1D and E). In addition, the DNA damage response as measured by checkpoint kinase 1 (CHK1) and histone H2A.X (γH2AX) phosphorylation was hyper‐induced upon silencing of EWS expression in MMS‐treated HEK‐293 cells (siRNA, Fig EV1F). The same experiments also suggested that the loss of EWS did not lead to increased DNA damage in cells not treated with DNA‐damaging agents (Fig EV1D–F). Taken together, the level of DNA repair proteins including PARP1 is increased in Ews −/− cell lines and the knockout cell lines are hypersensitive to DNA damaging agents and show excessive SSB formation upon MMS or H2O2 treatments.
Figure 1. Loss of EWS regulates expression of DDR proteins and causes PARP1 activation.
-
ASchematic of SILAC‐mass spectrometry. Wild‐type (WT) and Ews −/− mBA cells were labeled by heavy or light amino acid, respectively. Differently labeled proteins were analyzed by mass spectrometry.
-
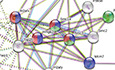
BNetwork analysis of mass spectrometry results.
-
CNAD+/NADH ratio was measured in mBA cell lines and 17.5 days embryo liver. Data represented as mean ± SEMs, and obtained from three different cell line and embryo liver. Significance determined by Student's t‐test, two‐tailed, *P < 0.05, **P < 0.01.
-
DImmunostaining of 17.5 days embryo tissues (mid‐brain, heart, liver, and skin) with anti‐PAR antibody. Insets show higher magnification. Scale bar indicates 100 μm and 20 μm.
-
EWild‐type (WT) and Ews −/− embryos mid‐brain at E17.5 days were subjected to TUNEL assay. Scale bar indicates 100 μm and 20 μm.
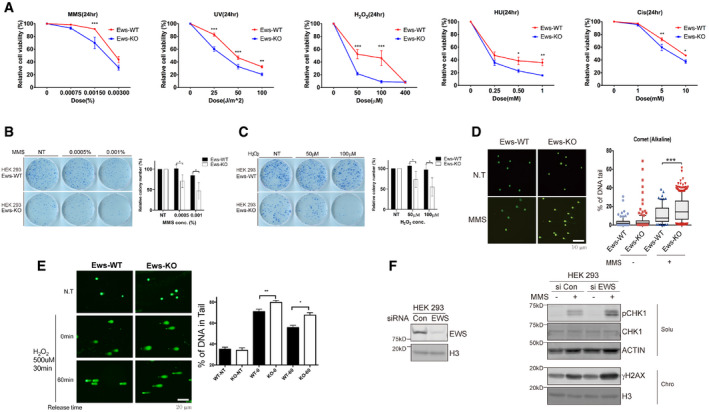
Figure EV1. Loss of Ews induces hypersensitivity to DNA‐damaging agents.
-
ARelative cell viability was measured in wild‐type (WT) and Ews‐KO mBA cells after treatment with various DNA‐damaging agents. MMS: Methyl methane sulfonate, H2O2: Hydrogen peroxide, Cis: Cisplatin, UV: Ultraviolet, and HU: Hydroxyurea. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
-
B, CAfter 10 days of low dose MMS (B) and H2O2 (C) treatment, survived colony were measured using clonogenic assay. Data represented as mean ± SEMs, n > 3. Significance determined by Student's t‐test, two‐tailed, *P < 0.05.
-
DDNA breaks in wild‐type (Ews‐WT) and Ews‐KO mBA cells were measured using Alkaline Comet assay after MMS treatment (0.0015%, 24 h). Error bars represent as mean ± SEMs, n > 50. Significance determined by ***P < 0.001.
-
EAlkaline Comet assay were conducted in Ews‐WT and Ews‐KO mBA cells after treatment and release of H2O2. Error bars represent ± SEMs, n > 50. Significance determined by two‐way ANOVA, *P < 0.05, **P < 0.01.
-
FUpon inactivation of EWS in HEK‐293 cells, expression of DNA damage markers (pCHK1 and γH2AX) were measured using Western blotting after MMS (0.02%, 1 h) treatment.
Loss of Ews leads to PARP1 activation
Ews −/− mice die within 12 h after birth showing mitochondrial abnormalities and increased serum lactate levels (Park et al, 2015). Lactate accumulation is a common feature of mitochondrial disease. For example, Cockayne syndrome cells defective for transcription‐coupled nucleotide excision repair show similar mitochondrial abnormalities, associated with a shift of the NAD+/NADH ratio toward decreased NAD+ and increased lactate production (Scheibye‐Knudsen et al, 2014). NAD+ is a key co‐substrate for multiple cellular processes, and NAD+ levels are depleted by the activities of sirtuins (SIRT1, 3, and 6), PARP1, and cyclic ADP‐ribose hydrolase (CD38) (Kim et al, 2005; Chini, 2009; Krishnakumar & Kraus, 2010). Importantly, the excessive activation of PARP in Cockayne syndrome cells was associated with decreased NAD+ levels (Scheibye‐Knudsen et al, 2014). We therefore measured if the NAD+/NADH ratio is equally altered in Ews −/− mBA cells and in embryonic livers derived from E17.5 day mice and found that the NAD+/NADH ratio was significantly decreased in both (Fig 1C). Since the enzymatic activity of PARP1 is the major cause of NAD+ depletion and consumes up to 80% of cellular NAD+ to produce PAR chains in response to DNA damage (Verdin, 2015), we decided to determine whether the loss of EWS increased the level of PAR chains in mouse embryonic tissues. In line with increased PARP1 mRNA (Fig EV2A and B) and protein expression (Fig EV2C), PAR polymers were significantly enriched in Ews −/− mid‐brains, hearts, and livers (Fig 1D). Since excessive synthesis of PAR chains and depletion of NAD+ induce neurotoxicity and cell death (Eliasson et al, 1997; Krishnakumar & Kraus, 2010), we examined whether hyper‐PARylation is associated with excessive apoptosis. We therefore analyzed the embryonic brain of Ews −/− mice and found excessive apoptosis using terminal deoxynucleotidyl transferase dUTP nick end labeling (Fig 1E). Taken together, these findings indicate that the loss of EWS leads to hyperactivation of PARP1 leading to excessive PARylation, which may be associated with cell death in vivo.
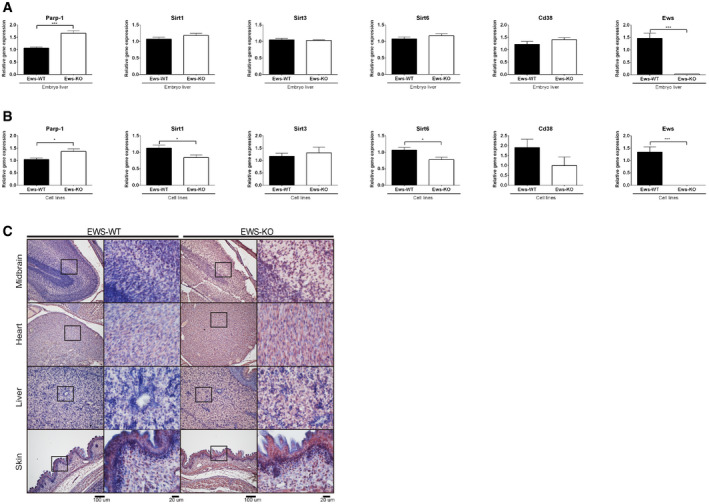
Figure EV2. EWS depletion induces PARP1 expression in mouse embryo and cell lines.
-
A, BQuantitative real‐time PCR was carried out to investigate the expression level of NAD+ salvage pathway genes using E17.5 day embryos liver (A) and cell lines (B). Data represented as mean ± SEMs, and obtained from three different cell line and embryo liver. Significance determined by Student's t‐test, two‐tailed, *P < 0.05, ***P < 0.001.
-
CImmunostaining of 17.5 days embryo tissues (mid‐brain, heart, liver, and skin) with anti‐PARP1‐antibody. Insert shows higher magnification. Scale bar indicates 200 μm (left).
Loss of EWS leads to accumulation of PARP1 on damaged DNA
PARP1 accumulates on damaged DNA at the earliest stages of the DDR and synthesizes PAR chains. Since the depletion of EWS lead to the accumulation of PARP1 and caused sensitivity to DNA alkylating and oxidative DNA damaging agents, we hypothesized that EWS might regulate the physiological function of PARP1 at damaged DNA. To start testing this hypothesis, we fractionated cell extracts into chromatin‐bound and soluble fractions and used total cell extracts as a control. We found that PARP1 was enriched in the chromatin fraction upon DNA damage (Fig 2A, top panel) but neither in the soluble fraction (lower panel) nor in the corresponding total cell extracts (Fig EV3A, left panel); PARP1 levels being highest in MMS‐treated Ews −/− cells chromatin‐bound fraction. The accumulation of PARP1 in the chromatin fraction correlated with the accumulation of pRPA32 and γH2Ax protein. The induction of Chk1 phosphorylation in soluble extracts further confirms that DDR signaling is activated in Ews −/− cells (Fig 2A, lower panel). The accumulation of PARP1 on chromatin in Ews −/− cells was corroborated by immunostaining (Fig EV3B).
Figure 2. Loss of EWS induces accumulation of PARP1 at damaged DNA sites.
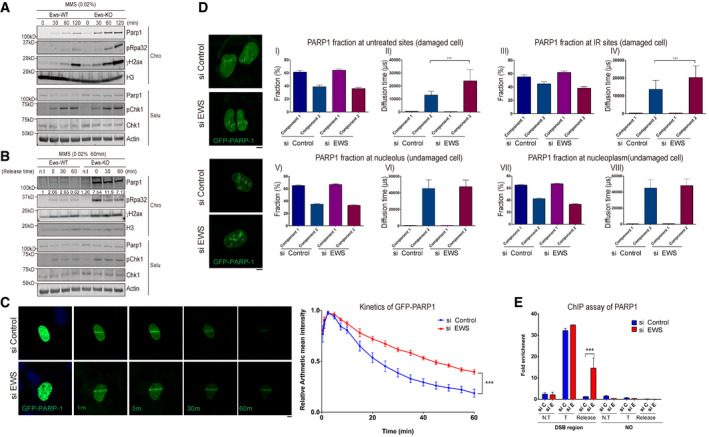
-
AWestern blot analysis of Parp1 and DNA damage markers in wild‐type (WT) and Ews −/− mBA cells. Cells were treated with MMS (0.02%) in a time dependent manner. Proteins were fractionated into two groups, chromatin‐bound (Chro) proteins and soluble (Solu) proteins.
-
BAfter treatment with MMS (0.02%, 1 h), the media was replaced to release the DNA damage. The proteins were fractionated and the specific protein kinetics at chromatin were analyzed by Western blot.
-
CGFP‐PARP1 U2OS cells were transfected with siControl and siEWS. Local DNA damage was induced by micro‐irradiation using a 405 nm laser. Data represented as mean ± SEMs and more than six cells were analyzed from six independent experiments. The statistical significance determined by one‐way ANOVA, ***P < 0.001. Scale bar indicates 5 μm.
-
DFluorescence correlation spectroscopy (FCS) was measured after 15 min later following micro‐irradiation (Upper) or under normal condition in GFP‐PARP1 U2OS cells with either siControl or siEWS. Data represented as mean ± SEMs, more than 50 cells were analyzed, and significance determined by one‐way ANOVA, ***P < 0.001. Scale bar indicate 5 μm.
-
EAsiSI endonuclease‐integrated U2OS cells were transfected with siControl and siEWS. Cells were incubated with doxycycline for 4 h to induce DSBs. With or without changing the Dox‐added media to fresh media for 2 h (for release samples), the amount of chromatin‐associated PARP1 was measured using ChIP assay. N.T: Non‐treat, T: AsiSI treat, Release: Damage released samples. Data represented as mean ± SEMs, and technical repeats (n = 3), significance determined by two‐way ANOVA, ***P < 0.001.
Figure EV3. EWS protein is recruited to DNA damage site in a PARP1 dependent manner and regulates PARP1 dissociation from DNA damage sites.
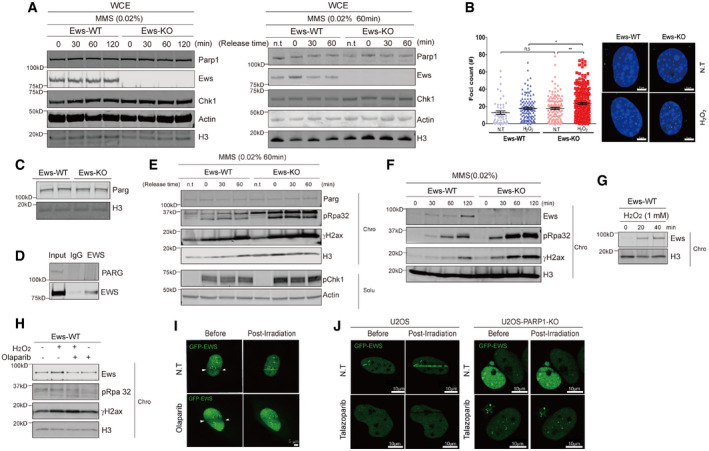
-
AWhole cell extract of wild‐type and Ews‐KO cells following MMS treatment and releasement were subjected to Western blotting.
-
B, C(B) Immunofluorescence with anti‐PARP1 antibody in wild‐type and Ews‐KO cells following H2O2 treatment. Scale bar indicate 5 μm. n > 150 (C) PARG expression was measured by Western blot in wild‐type and Ews‐KO mBA cells.
-
DEndogenous interaction between EWS and PARG were analyzed by co‐immunoprecipitation followed by Western blot analysis with anti‐EWS antibody.
-
EAfter release from MMS treatment, proteins in cells were fractionated into chromatin‐bound and soluble forms to measure PARG kinetics in wild‐type and Ews‐KO mBA cells.
-
F, GChromatin‐bound levels of EWS were measured after treatment of MMS (F) or H2O2 (G).
-
HWild‐type cells were treated with 1 mM H2O2 for 20 min with or without PARPi (5 μM Olaparib for 7 h). The kinetics of chromatin‐bound EWS was analyzed by Western blot.
-
ILocalized specific DNA damage was induced in GFP‐EWS cell lines with or without Olaparib using micro‐irradiation. Scale bar represents 5 μm.
-
JDNA damage was induced by micro‐irradiation in U2OS and U2OS‐PARP1‐KO cells with or without Talazoparib (5 μM, 24 h). Scale bar represents 10 μm. N > 10.
Source data are available online for this figure.
It had previously been shown that trapping of PARP1 on chromatin causes genomic instability and reduces cellular viability to a greater extend as PARP1 loss of function mutations; trapped PARP1 being an obstacle for DNA repair and for eliciting a proper DNA damage response (Murai et al, 2012). We therefore wished to analyze PARP1 turnover on chromatin and started by assessing if the rate of PARP1 chromatin dissociation is differentially affected after washing out MMS from WT and Ews −/− cells. We indeed found that PARP1 rapidly disappeared from chromatin when MMS was washed out from WT cells, while it persisted in mutant cells even 60 min after washing out MMS (Fig 2B). In contrast, the level of PARP1 was the same in the corresponding whole cell extracts derived from both WT and mutant lines (Fig EV3A, right panels).
To confirm that PARP1 is trapped on DNA upon Ews siRNA depletion, we stably expressed GFP‐PARP1 in U2OS cells and precisely monitored the kinetics of GFP‐PARP1 after micro‐irradiation (Smith et al, 2019). We found that while GFP‐PARP1 equally accumulated on micro‐irradiated sites both in WT and Ews −/− cells, it persisted longer when EWS was depleted (Fig 2C). To further support our findings, we used fluorescence correlation spectroscopy (FCS), a method that measures the dynamics of fluorescently tagged proteins based on frequency fluctuations in a defined small focal area in live‐cell image analysis at DNA damage sites (Jeyasekharan et al, 2010; Pack et al, 2014). We micro‐irradiated WT and EWS‐depleted cells to cause the same level of DNA damage and analyzed GFP‐PARP1 dynamics at micro‐irradiated sites which we refer to as “irradiated (IR) sites” and at untreated areas of sample nuclei (“untreated sites”). In our analysis, components 1 and 2 indicate the movement of fast (free) and slow (trapped) forms of GFP‐PARP1, respectively (Fig 2D). The fraction of free and trapped forms was similar at IR sites and untreated sites, irrespective of the status of EWS, suggesting that the loss of EWS did not increase endogenous DNA damage and that the localization of GFP‐PARP1 to damaged DNA was not affected by EWS depletion (Fig 2D, panels I and III). In contrast, the diffusion time which is the inverse of the velocity of GFP‐PARP1 trapped at the untreated sites and IR site was greatly increased in the absence of EWS (component 2, panels II and IV), indicating that the velocity of trapped GFP‐PARP1 is decreased in EWS depleted cells. Differential GFP‐PARP1 diffusion/velocity was not observed in the absence of DNA damage; both in the nucleolus and the nucleoplasm (Fig 2D, lower panels). In summary, our FCS measurements indicate that the mobility of GFP‐PARP1 localized at damaged chromatin is decreased in EWS‐depleted cells.
PARP1 binds to SSB and double‐strand breaks (DSB) (Kim et al, 2005). To further corroborate that PARP1 dissociation is delayed in EWS depleted cells, we induced a DSB at a defined site using the estrogen inducible expression of the ER‐AsiSI meganuclease enzyme fused to the estrogen response element (ER). Using the chromatin immunoprecipitation (ChIP) procedures, we found that PARP1 equally loaded to the DSB site when the DSB is induced. However, once estrogen is washed out, PARP1 disappears in wild‐type cells but remains bound to the site in EWS‐depleted cells (Fig 2E “release”). Collectively, these findings suggest that EWS facilitates the dissociation of PARP1 from damaged DNA.
In response to DNA damage, chromatin‐associated PARP1 catalyzes the production of PAR chains using NAD+ as a substrate (Kim et al, 2005). Since depletion of EWS caused accumulation of PARP1 on chromatin, and loss of Ews‐induced PAR hyperaccumulation in mouse embryos, we investigated the dynamics of PAR accumulation in cells treated with DNA damaging agents. Treatment with H2O2 increased PAR accumulation in Ews −/− mBA cells, PAR accumulation being blocked by Olaparib treatment serving as a control (Fig 3A). Western blot analysis and ADP‐ribose staining also revealed that the dissociation kinetics of PAR from chromatin was significantly delayed in Ews −/− mBA cells after H2O2 (Fig 3B and C), MMS treat, and removal (Fig 3D and E). Supporting these results, the NAD+/NADH ratio upon H2O2 treatment was further decreased in the Ews −/− cell line (Fig 3F). The delayed PAR dissociation is unlikely due to a reduced level of the PAR glycohydrolase (PARG), which is the major enzyme needed for PAR degradation (Luo & Kraus, 2012) as we did not detect any change in PARG levels in Ews −/− mBA cells (Fig EV3C). Furthermore, EWS did not interact with PARG and did not affect the level of PARG on chromatin (Fig EV3D and E). All together these results indicate that PAR accumulation induced by the loss of EWS is caused by trapping PARP1 on chromatin and not by PARG activity.
Figure 3. Loss of EWS induces PARP1‐dependent PARylation.
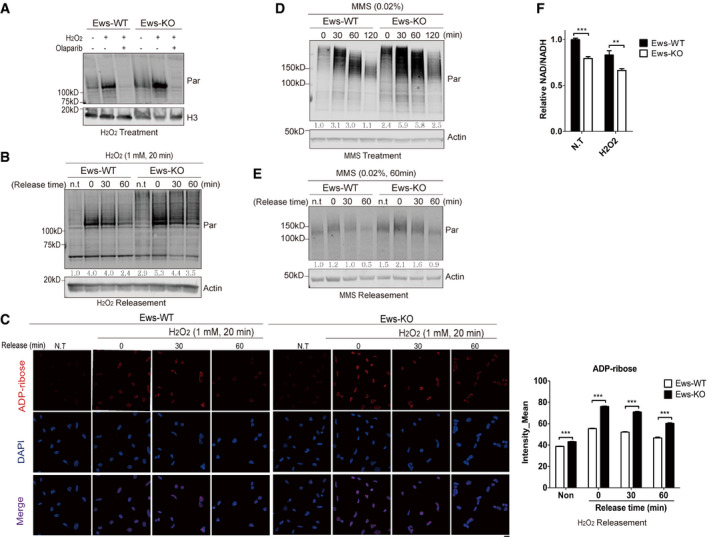
-
ATotal levels of PAR in whole cell lysate were measured using Western blot analysis. Wild‐type (WT) and Ews −/− mBA cells were treated with H2O2, (1 mM, 20 min) with or without Olaparib (5 μM, 7 h).
-
B, C(B) The kinetics of PAR accumulation was analyzed by Western blotting. WT and Ews −/− cells were treated with H2O2 (1 mM, 20 min), followed by incubation in fresh media for the indicated times to allow release of DNA damage. (C) Immunohistochemistry detection of PAR by ADP‐ribose antibody in WT and Ews −/− cells after H2O2 treatment (1 mM, 20 min) and recovery from DNA damage. Right graph displays mean of intensities measured from 500 cells. Data represented as mean ± SEMs, significance determined by one‐way ANOVA, ***P < 0.001. Scale bar indicate 20 μm.
-
D, EWhole cell level of PAR was measured in WT and Ews −/− mBA cells upon treatment with (D) MMS (0.02%, 60 min) (E) with or without recovery from DNA damage.
-
FNAD+/NADH ratio was measured in mBA cell lines following treatment of H2O2 (1 mM, 10 min). Data represented as mean ± SEMs, and technical repeats (n = 3). Significance determined by Student's t‐test, two‐tailed, **P < 0.01, ***P < 0.001.
Previous studies had reported that EWS localizes to DNA damage sites in a PARP1‐ and PAR‐dependent manner (Mastrocola et al, 2013; Rulten et al, 2014; Altmeyer et al, 2015). To gain additional insight on how EWS functions in the DDR, we first confirmed the localization of endogenous EWS in response to DNA damage. As expected, EWS protein accumulated on chromatin following treatment with MMS or H2O2 (Fig EV3F and G). This accumulation was blocked by Olaparib treatment (Fig EV3H) indicating that EWS accumulation depends on PARylation. In line with this notion, we found that the recruitment of an EWS‐green fluorescent protein (GFP) fusion protein to micro‐irradiated DNA damage sites was equally compromised upon Olaparib and Talazoparib treatment, or upon PARP1 depletion (Fig EV3I and J). Given that Talazoparib treatment more effectively traps PARP1 compared to Olaparib treatment (Murai et al, 2012), EWS location at DNA damaged sites is unlikely to be linked to PARP1 trapping.
EWS and PAR directly interact via EWS RGG domains
The fact that EWS is recruited to the DNA damage sites by PARP1‐ and PAR‐dependent manner (Mastrocola et al, 2013; Altmeyer et al, 2015), and EWS regulate PARP1 chromatin dissociation (Fig 2) made us to investigate whether PARP1 and EWS directly interact with each other. We employed the cell‐based unidentified protein interaction discovery (CUPID) assay to probe for interaction. In this assay, a bait is fused to protein kinase C‐δ (PKC‐δ) and mRCD tag that migrate to the nuclear membrane during treatment with phorbol‐12‐myristate 13‐acetate (PMA) (Lee et al, 2011). Using this technique, we found that a PKC‐δ‐mRCD‐EWS fusion tethered EGFP‐PARP1 to the nuclear membrane upon PMA treatment only after H2O2 treatment (Fig 4A), suggesting that PAR chains on PARylated PARP1 interacted with EWS.
Figure 4. EWS interacts with PARylated PARP1 using RGG domains at DNA damage sites.
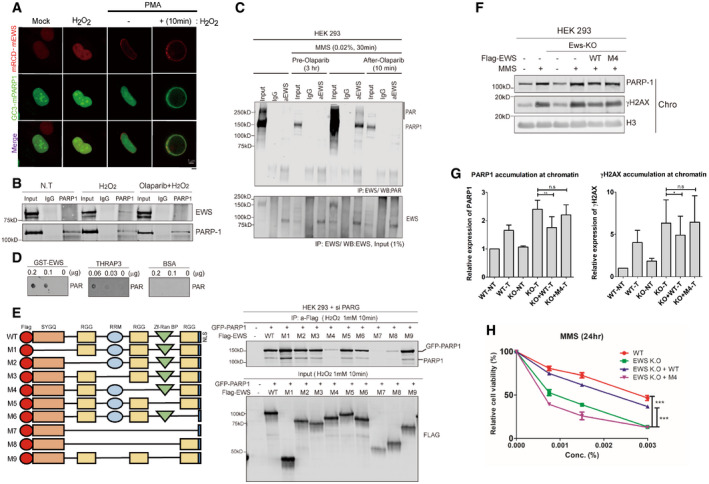
-
APARP1 and EWS interaction was visualized in cells using the CUPID system. After transfection of mRCD‐EWS and GC3‐PARP1 (EGFP‐PARP1) plasmid, the cells were treated with either H2O2 (1 mM, 10 min) or PMA. Scale bar indicates 5 μm.
-
BEndogenous interaction between EWS and PARP1 was measured using immunoprecipitation Western blot analysis. Cells were treated with H2O2 (1 mM, 10 min) with or without Olaparib (5 μM, 7 h).
-
CMMS‐treated cells, with or without Olaparib, were immunoprecipitated using an anti‐EWS antibody and immunoblotted by anti‐PAR antibody (upper). Pre‐Olaparib group was treated with Olaparib for 3 h before MMS treatment (0.02%, 30 min) and after‐Olaparib group treated Olaparib after 10 min following treatment of MMS. Lower blot indicates input of anti‐EWS antibody.
-
DIn vitro PAR‐binding assay. THRAP3 binding to PAR was used as a positive control and BSA binding to PAR was used as a negative control.
-
ESchematic map of nine EWS mutants (left) and IP‐Western blot analysis (right). GFP‐PARP1 and siPARG were transfected into HEK‐293 cells with each EWS‐mutant plasmid. After H2O2 treatment, the cells were conducted to IP with anti‐Flag antibody.
-
FWestern blot analysis of PARP1 accumulation on chromatin. EWS‐WT and M4 mutants were transfected into EWS‐depleted HEK‐293 cells. After treatment of MMS (0.02%, 1 h), the proteins were fractionated to either chromatin‐bound or soluble fraction.
-
G, H(G) Quantification of relative amount of PARP1 and γH2AX on chromatin, divided by chromatin H3, from three independent Western experiments is presented. Data represented as mean ± SEMs, were measured from 3 independent experiments. Significance determined by t‐test. *P < 0.05, **P < 0.01. n.s indicate non‐significance, (H) WT and Ews −/− cells were transfected with EWS‐WT and –M4 mutant and cell viability was measured 24 h after MMS treatment (indicated concentration). Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, **P < 0.01. n.s indicate non‐significance.
We next investigated whether endogenous EWS interacts with PARylated PARP1 resulting from H2O2 and MMS treatment. We found that EWS coimmunoprecipitated with PARP1 derived from H2O2‐treated cells and that this interaction was compromised when cells were pretreated with Olaparib, indicating that PARP1‐mediated PARylation is important for the interaction (Figs 4B and EV4A). Likewise, we found that the EWS‐PARP1 interactions only occurred after MMS treatment and that this interaction was lost when cells were treated with Olaparib both before and after MMS treatment, further confirming that the interaction requires active PARylation (Fig 4C). This interaction was not DNA‐dependent as it remained even after benzonase treatment (Fig EV4B). Consistent with this, recombinant EWS interacted with PAR in vitro, the well‐known PAR‐binding protein Thyroid Hormone Receptor Associated Protein 3 serving as a positive control (THRAP3, Fig 4D; Jungmichel et al, 2013). EWS contains a N‐terminal SYGQ‐rich domain, three RGG motifs, a RNA‐recognition motif (RRM), and a Zinc finger domain (Izhar et al, 2015). To identify which EWS domain mediates binding to PARylated PARP1, we expressed nine EWS mutants missing one or more functional domains in HEK 293 cell (Fig 4E, left panel). Of all truncations tested, the loss of SYGQ‐rich domain, first and third RGG, RRM or Zinc finger domain was not affected EWS‐PARylated PARP1 binding. Interestingly, mutant 4 (M4) that lacks the second RGG domain did not interact with GFP‐PARP1. Consistently, mutants 7 and 8 that lacked the second RGG domain and other domains also showed a remarkably reduced interaction (Fig 4E, right panels). Consistent with previous works (Krietsch et al, 2013; Altmeyer et al, 2015), RGG domain is important for EWS‐PARP1 interaction, however, in our results strongly suggested that not all RGG domains have same function and the second RGG domain is essential for EWS‐PARP1 interaction. Analyzing the same blot, we found that endogenous PARP1 showed similar interaction patterns with the EWS truncations (Fig 4E).
Figure EV4. RGG domain of Ews interact with PARP1.
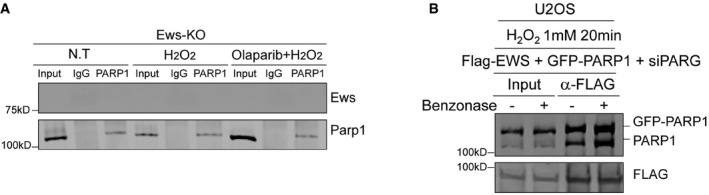
-
AInteraction between EWS and PARP1 was analyzed by Immunoprecipitation assay in Ews‐KO cells. Cells were treated with H2O2 (1 mM, 10 min) with or without Olaparib (5 μM, 7 h).
-
BWith or without Benzonase‐treated cells were immune‐precipitated by anti‐FLAG antibody and immunoblotted by anti‐PARP1 and FLAG antibody.
Source data are available online for this figure.
To determine whether the interaction between EWS and PARP1 is important for PARP1 dissociation from damaged DNA, PARP1 accumulation in response to DNA damaging agents was investigated in HEK‐293‐EWS −/− cells expressing either WT or the M4 EWS truncation that lacks the second RGG domain. In contrast to WT EWS, expressing the M4 EWS mutant could not reduce PARP1 from chromatin and DNA damage (Fig 4F and G). Consistently, M4 EWS expression did not restore cellular resistance to DNA damaging agents in contrast to WT EWS (Fig 4H). Taken together, these findings indicate that the second RGG domain of EWS has an essential role for the interaction with PARylated PARP1 and the regulation of the PARP1 dissociation from damaged DNA.
EWS suppresses DNA damage by preventing excessive PARP1 accumulation
Accumulation of PARP1 and excessive PARylation enhance the sensitivity to genotoxic agents (Koh et al, 2004) and lead to increased genomic instability (Fujimoto et al, 2017; Michelena et al, 2018). We performed genetic epistasis experiments to test whether the excessive accumulation of PARP1 observed in EWS‐deficient lines leads to increased hypersensitivity to DNA damaging agent and lethality. We therefore compared the viability of WT cells, to cells which were RNAi depleted for either EWS or PARP1 alone, or for both. We found that cells depleted for PARP1 were hypersensitive for MMS indicating that PARP1 is required for efficient DNA repair. The EWS single depletion was even more sensitive to MMS. However, the hypersensitivity of EWS‐depleted cells was suppressed by concomitant PAPR1 depletion, consistent with the idea that hyperaccumulation of PARP1 at DNA damage sites in EWS‐depleted cells causes increased MMS sensitivity (Figs 5A and EV5A). We verified our findings by generating and analyzing the corresponding single and double mutant combinations in mBA cells (Figs 5B and EV5B). Furthermore, we used both experimental setups to measure the level of phosphorylation of γH2ax and Chk1 which served as quantitative markers for DNA damage and damage‐dependent checkpoint activation, respectively. We found that excessive DNA damage induction observed in EWS‐deficient cells is suppressed by PARP1 deficiency (Fig 5C and D). To directly determine whether the double mutant shows a reduced level of DNA damage in response to MMS, we performed the alkaline comet assays (Fig EV5C) confirming that the level of DNA damage was reduced in the double mutant compared to the Ews single mutant. Collectively these data show that the DNA damage hypersensitivity associated with EWS deficiency depends on PARP1.
Figure 5. EWS regulates genomic integrity in a PARP1‐dependent manner.
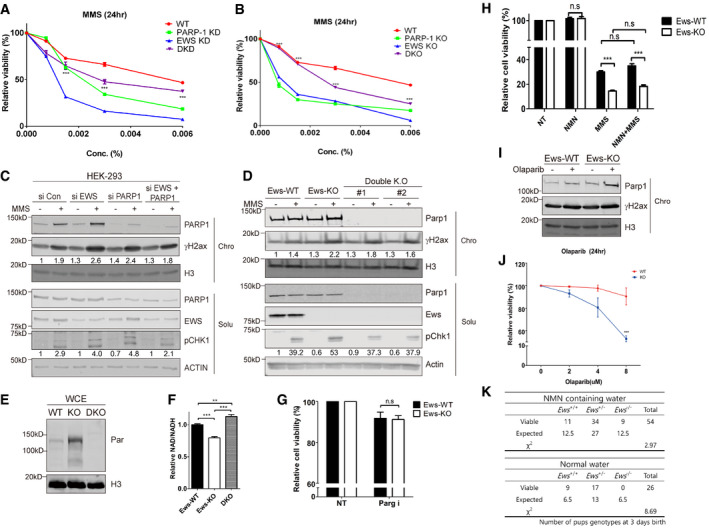
-
ARelative cell viability of cells transfected with siCon (WT), siEWS (EWS KD), PARP1 (PARP‐1 KD), and double siRNA (DKD) after MMS treatment. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, ***P < 0.001. Analysis (***) indicates differences between DKD with EWS KD.
-
BAdditional Parp1 knockout in Ews knockout cells (DKO) were subjected to viability test after MMS treatment. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, ***P < 0.001. *** indicated differences between DKO with EWS‐KO.
-
CAfter transfection of siCon, siEWS, PARP1, and double siRNA, the effect of co‐depletion of PARP1 and EWS on the γH2AX and phosphorylated CHK1 upon MMS treatment was analyzed by Western blot.
-
DAfter treatment of MMS, the effect of additional Parp1 knockout in Ews knockout cells (Double KO) to the γH2AX and phosphorylated CHK1 were analyzed using Western blot.
-
EWhole cell expression of PAR in mBA (WT: Ews‐WT, KO: Ews‐KO, DKO: double KO).
-
FRelative NAD+/NADH ratios were measured in WT, Ews −/− and DKO cells. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, ***P < 0.001.
-
GPARGi (5 μM, 24 h) treated WT and Ews −/− cells were subjected to cellular viability assay. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA.
-
HRelative cellular viability was measured after NMN treatment, with or without MMS pretreatment, in WT and Ews −/− cells. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, ***P < 0.001.
-
IChromatin‐bound PARP1 was quantified by Western blot in WT and Ews −/− cells treated with Olaparib (5 μM, 24 h) treatment.
-
JRelative viability was measured in WT and Ews −/− cells upon treatment of Olaparib for 24 h. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, ***P < 0.001.
-
KTable of Ews −/− pups survival number in control (normal water) or NMN supplements (NMN containing water) groups 3 days after birth. Statistics (χ2) were determined by chi‐square test.
Source data are available online for this figure.
Figure EV5. EWS regulates genomic integrity in PARP1‐dependent manner.
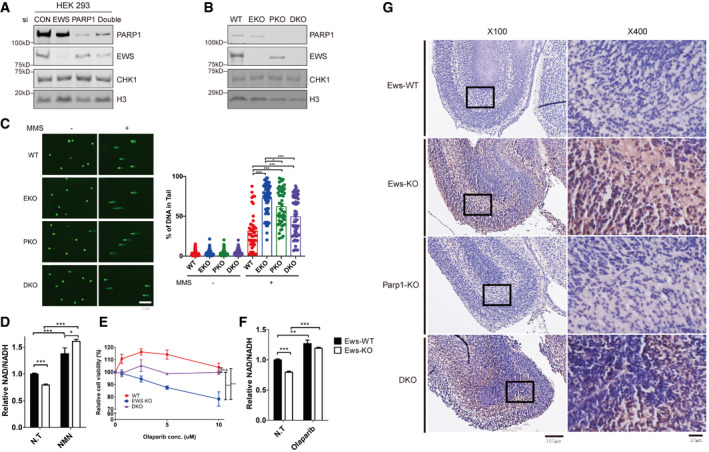
- A, B
-
CDNA breaks were measured using the Alkaline Comet assay in WT, EKO, PKO, and DKO cells treated with MMS (0.0015%, 24 h). WT: Wild‐type, EKO: Ews‐KO, PKO: Parp1‐KO and DKO: double KO. Error bars represent ± SEMs, n > 35. Significance determined by two‐way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
-
DRelative NAD+/NADH ratios were measured in wild‐type (Ews‐WT) and Ews‐KO cells following treatment of NMN (20 μM, 24 h). Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
-
ERelative cell viability was measured in HEK293 WT, EWS‐KO and EWS‐PARP1 KO (DKO) cell lines upon Olaparib treatment. Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
-
FNAD+/NADH ratios were measured in wild‐type (Ews‐WT) and Ews‐KO cells after treatment of Olaparib (5 μM 24 h). Error bars represent as mean ± SEMs, and technical repeats (n = 3). Significance determined by two‐way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001.
-
GCell death was observed in wild‐type (Ews‐WT), Ews‐KO, Parp1‐KO, and DKO embryos mid‐brain at E17.5 days using the TUNEL assay. ×100 and ×400 image scale bar represents 100 μm and 20 μm, respectively.
Since hyper‐PARylation is associated with reduced cellular viability, we measured the total level of PAR and the ratio of NAD+/NADH in the single and double knockout lines. We found that the Ews −/− Parp −/− double mutant showed a reduced level of PAR chains and a higher NAD+/NADH ratio compared to the Ews −/− single mutant (Fig 5E and F).
To further investigate the mechanism by which EWS regulates abnormal DDR and cell death, we treated WT and Ews −/− mBA cells with the PARG inhibitor (PARGi) to induce PARylation, the NAD+ precursor nicotinamide mononucleotide (NMN) to restore energy balance, or Olaparib to block PARylation and induce the trapping of PARP on damaged chromatin. Cellular viability of wild‐type and Ews −/− cells was not affected by PARGi treatment (Fig 5G). Supplementing wild‐type and Ews −/− cells with NMN restored a high NAD+/NADH ratio (Fig EV5D), but did not alter the viability of MMS‐treated cells (Fig 5H). These results suggest that the depletion of NAD+ may be a product of hyper‐PARylation. Olaparib is known to trap PARP1 on chromatin and at the same time blocks PARylation (Murai et al, 2012). Strikingly, Ews −/− cells treated with Olaparib showed an increased level of PARP1 in the chromatin‐bound fraction (Fig 5I) and cellular viability decreased proportionally with the dose of Olaparib (Fig 5J). PARPi sensitivity of EWS‐deficient cells were rescued by EWS‐PARP1 double depletion suggesting that trapped PARP1 in EWS‐deficient cells mainly cause cell death (Fig EV5E). In contrast, cellular NAD+ levels were restored by Olaparib treatment (Fig EV5F). Thus, our results suggest that PARP1 trapped on damaged DNA sites in Ews −/− cells cause excessive damage and ensuing cell death.
Given that Ews −/− Parp1 −/− double mutant cells restored cell viability following DNA damage, we wished to analyze whether Parp1 −/− mutations rescue the postnatal lethality of Ews −/− mice. Ews −/− mutant mice typically die within 12 h after birth. We bred Ews +/− Parp1 −/− mice and analyzed the genotypes of newborn pups. Of the 50 pups tested only two (well below the expected Mendelian ratio) were Ews −/− Parp1 −/− double mutants, and these died 3 days after birth. Directly testing for apoptosis induction in the brain using the TUNEL assay, we found evidence for reduced apoptosis analyzing tissue sections derived from embryonic brains (Fig EV5G). Given possible differences in exact developmental staging, or the positions of the cross sections examined, we could not be absolutely certain that apoptosis was reduced in the double mutant. We thus decided to attempt bypassing the lethality of Ews −/− mutant mice by supplementing pups with NAD+ precursors, a procedure that has recently been shown to alleviate defects caused by PARP hyperactivation (Scheibye‐Knudsen et al, 2014). Supplying 1 mg/ml NMN to the drinking water of pregnant mice, we found that the proportion of Ews −/− mutant litter (alive 3 days after birth) approximates the Mendelian ratio (Fig 5K). Most mice succumbed to death at an age of 4–5 days, while two survived up to 8 days. Collectively, we provide evidence that PARP1 hyperactivation linked to NAD+ depletion contributes to the postnatal lethality in Ews −/− mice.
Accumulation of PARP1 and hyper‐PARylation were observed in human Ewing sarcoma
The Ewing sarcoma fusion protein includes the SYGQ but not the RGG domain (Yang et al, 2000), the latter being important for PAR interaction (Fig 4). We thus hypothesized that PARP1 may hyperaccumulate at the chromatin and that PARP1 might be hyper‐PARylated in Ewing sarcoma cells. We found that in the CHP100 and A4573 Ewing's Sarcoma lines PARP1 hyperaccumulated on chromatin after MMS treatment and that PARP1 was no released from chromatin after washing out MMS (Fig 6A). We furthermore, performed immunostaining using a chromatin pre‐extraction method (Lee et al, 2013). We found that the two Ewing's sarcoma cell lines showed significantly increased level of chromatin‐bound PARP1 following MMS treatment compared to U2OS cells which served as a negative control (Fig 6B). In line with these results, histological samples derived from human Ewing sarcoma patients showed high levels of PAR compared to undifferentiated chondrosarcoma samples (Fig 6C). Taken together, these findings suggest that the EWS fusion in Ewing sarcoma compromises the physiological function of EWS which is to dissociate PARP1 from damaged DNA.
Figure 6. Accumulation of PARP1 or hyper‐PARylation in human Ewing Sarcoma patient‐derived samples.
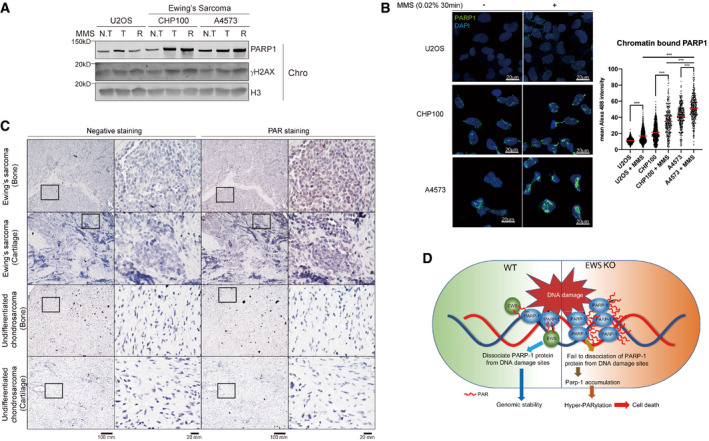
-
AWestern blot analysis of PARP1 in chromatin. Two Ewing Sarcoma cells (CHP100 and A4573) were treated with MMS (0.02%, 1 h) with or without release (1 h) from MMS (N.T: non‐treat, T: treat, R: release sample).
-
BU2OS and two Ewing Sarcoma cells were treated MMS (0.02%, 30 min) and subjected to immunofluorescence by PARP1 antibody after chromatin‐bound fraction. Scale bar indicates 20 μm (left). Red line indicates mean and more than 300 cells were analyzed. Significance determined by two‐way ANOVA, ***P < 0.001.
-
CHuman Ewing Sarcoma histology samples were analyzed by immunohistochemistry using PAR antibody. Scale bar represents 100 mm.
-
DModel of EWS role in genomic integrity.
Source data are available online for this figure.
Discussion
The function of PARP1 in DDR has been extensively studied because the inhibition of PARP1 specifically kills ovarian and breast cancer cells carrying BRCA1 or BRCA2 mutations (Kim et al, 2005; Krishnakumar & Kraus, 2010; Luo & Kraus, 2012; Li & Chen, 2014; Teloni & Altmeyer, 2016; Ray Chaudhuri & Nussenzweig, 2017). However, the regulatory mechanism of PARP1 under physiological conditions is still poorly understood. In this study, we identified a new role of Ewing sarcoma protein (EWS) facilitating the dissociation of PARP1 from damaged DNA. Our data are consistent with a model where loss of EWS inhibits the dissociation of PARP1 leading to the excessive synthesis of PAR chains and the concomitant depletion of cellular NAD+ in Ews −/− mBA cells and mouse embryo. Excessive PARylated PARP1 accumulated on DNA damage sites leads to excessive DNA damage (Fig 6D).
We confirmed that the accumulation of PARP1 on chromatin is more detrimental to genomic stability and cellular homeostasis than genetic inactivation of PARP1 (Helleday, 2011; Murai et al, 2012). We propose that the increased sensitivity to DNA damaging agents and the reduced cellular viability observed in Ews −/− mBA cells and embryonic brains is linked to PARP1 accumulation on chromatin. Our observation of PARP1 accumulation in chromatin in Ews −/− cells might be due to (i) EWS's direct role to remove PARP1 from damaged DNA (DIRECT) or (ii) the absence of EWS caused endogenous DNA damage resulting PARP1 accumulation in DNA (INDIRECT). The current data cannot completely rule out the both possibilities. However, our data favor a direct role of EWS for removing PARP1 from damaged DNA. Consistently, FCS and ChIP analysis supports that EWS directly regulates the physiological function of PARP1 at DNA damage sites by removing PARP1 from damaged DNA (Fig 2). If EWS deficiency phenotypes are secondary effects, double knockout (DKO) should show a similar or worse phenotype compared to a single knockout. However, the cell viability and levels of DNA break in single knockout were partially rescued by double knockout (Figs 5 and EV5). Furthermore, under normal condition, we showed that loss of EWS did not increase the level of DNA damage markers such as γH2AX, phospho‐CHK1, and the level of single‐ and double‐strand breaks measured by COMET assay (Figs 2A and B, and EV1F).
PARylation of PARP1 inhibits its DNA binding and catalytic activity, leading to changes in chromatin structure and transcription (D'Amours et al, 1999; Tulin & Spradling, 2003; Kim et al, 2004). It appears that PARylated PARP1 rapidly dissociates from DNA damage sites and that this dissociation is essential for proper DNA damage response. Our data suggest that EWS facilitates the timely release of PARylated PARP1.
The aberrant trapping PARP1 at DNA damage sites explains the hypersensitivity to genotoxic agents observed in Ews −/− mBA cells. In addition, hyperactivation of PARP1 causes excessive synthesis of PAR and this exhausts cellular pools of NAD+, resulting in the disruption of cellular homeostasis (Krishnakumar & Kraus, 2010; Luo & Kraus, 2012). Consistent with this notion, the postnatal lethality in Ews −/− newborn pups was partially rescued by supplementing with an NAD+ precursor. However, Parp1 mutation in Ews −/− background did not significantly prolong the survival of Ews −/− newborn pups. We postulate that this might be due to the compensatory activation of other PARP enzymes such as PARP2. However, supplementation of NMN could not rescue cellular viability of ews null cells to MMS. Such inconsistence between in vivo and in vitro may be due to the level of NAD complemented by NMN may not be sufficient for rescuing survival following a high level of exogenous DNA damage when compared to endogenous levels of damage in the mice. Alternatively, trapping compared to NAD depletion may be more dominate in cultured cell lines, where replication fork collapses by trapped lesions dominate to produce cellular toxicity compared to in vivo embryos.
The mitochondrial dysfunction and the increased levels of lactate in Ews −/− mutants might not be rescued by the PARP1 inactivation. Maintaining sufficient levels of cellular NAD+ is critical for mitochondrial function, aging, neurodegeneration, adipocyte differentiation, and cell death (Kim et al, 2005; Fauzee et al, 2010; Krishnakumar & Kraus, 2010; Luo et al, 2017). Our previous studies have shown that EWS regulates mitochondrial homeostasis and brown adipocyte differentiation (Park et al, 2013, 2015). Thus, EWS appears to play an important role in all these cellular processes by regulating PARP1 activity and maintaining cellular NAD+ levels. Additionally, the loss of EWS altered the expression of DDR proteins and proteins involved in gene regulation and macromolecule biosynthetic processes. It is possible that increased PARP1 accumulation and hyperactivity on chromatin in the absence of EWS could be responsible for the altered expression of DDR proteins, gene regulation, and macromolecule biosynthesis.
We found that the second RGG domain of EWS interacts with PARP1 via their appended PAR chains, following exposure to DNA damaging agents. The positively charged Arg residues in the RGG domain are crucial for PAR‐dependent EWS‐PARP1 interaction. This electrostatic interaction between EWS and PAR could lead to spatial proximity of PARP1 and EWS proteins. Subsequently, PARylated EWS as demonstrated by Jungmichel et al (2013) may act as an amplifier of charge repulsion between DNA and PARylated PARP1, leading to dissociation of PARP1 from chromatin. It has been demonstrated that the RGG domains of FET family proteins are involved in “PAR‐seeded liquid demixing” (Altmeyer et al, 2015). Liquid demixing, also known as phase separation, is an important phenomenon driven by intrinsically disordered proteins that participate in the maintenance of many cellular mechanisms such as cellular signal transduction and DNA damage repair (Altmeyer et al, 2015; Chong & Forman‐Kay, 2016; Strom et al, 2017). Accumulation of PARP1 and hyper‐PARylation due to loss of EWS could regulate the liquid demixing process, resulting PARP1 retention at DNA damage sites leading to excessive DNA damage and cell death.
Recent studies showed that inhibition of PARP1 activity increased sensitivity of Ewing sarcoma tumor cells to various therapies (Boro et al, 2012; Brenner et al, 2012), but the precise mechanism of how PARPi increases sensitivity has not been clearly demonstrated. It is known that the EWS‐fusion protein inhibits the physiological function of EWS, likely in a dominant‐negative manner (Spahn et al, 2003). Our study showing that endogenous EWS is required for the effective dissociation of PARP1 from damaged DNA could explain why Ewing sarcoma tumors become sensitive to PARP1 inhibitor. Thus, our data suggest that using PARP1 inhibitors in conjunction with current clinically used therapies may be an effective way to treat Ewing sarcoma.
Materials and Methods
Cell culture and transfection
Mouse brown pre‐adipocytes, U2OS, and HEK‐293 cells were cultured in DMEM containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin antibiotics (Invitrogen). Transfection of cells was performed using Lipofectamine 3000 or RNAiMAX (Invitrogen) following the manufacturer's protocol. Olaparib was purchased from Cayman Chemical.
Western blot
Nuclear soluble fraction was prepared by incubating harvested cells in buffer A (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES (pH 6.8), 1 mM EGTA, 0.2% Triton X‐100, 100 μM NaVO4, 50 mM NaF, and protease inhibitors (Roche)) on ice for 10 min, followed by centrifugation. The chromatin‐bound fraction was isolated by resuspending the insoluble pellet in RIPA buffer (150 mM NaCl, 1% NP‐40 or Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris (pH 8.0) 10 mM NaF, 1 mM Na3VO4, and protease inhibitors (Roche)) on ice for 30 min, followed by sonication and centrifugation. The whole cell extract was prepared by lysis of harvested cells with RIPA buffer. Proteins in prepared samples were separated by SDS–PAGE and transferred to nitrocellulose membrane for Western blot analysis. Each protein was detected with an antibody listed below. The images were detected by Odyssey Imaging System (Li‐COR Biosciences). Quantification of western bands was performed using Odyssey imaging system (Li‐COR Biosciences).
Antibodies
The following antibodies were used; Rabbit polyclonal PARP1 (Abcam, ab32071), EWS (Bethyl, A300‐418A), pRPA32 s4/8 (Bethyl, A300‐245A), pRPA32 s33 (Bethyl, A300‐246A), pCHK1 (Cell signaling, 2348L), H3 (Cell signaling, 4499L), PAR (Trevigen, 4336‐BPC‐100), PARG (Cell signaling, 66564S), Rabbit IgG (Santa Cruz, sc2027), mouse monoclonal phosphor‐Histone H2AX (Ser139) (Millipore, #05‐636), β‐ACTIN (Thermo Fisher, MA5‐15739), FLAG (Sigma, F3165), and PAR antibody (Trevigen, 4335‐MC‐100).
Animal care
All animal procedures were approved and performed according to the guidelines provided by the Ulsan National Institute of Science and Technology's Institutional Animal Care and Use Committee.
Immunohistochemistry
Wild‐type and mutant embryos (E17.5) were euthanized and fixed in 10% neutral buffered formalin (NBF, Sigma). Fixed embryos were embedded in paraffin and sectioned for immunostaining. Human Ewing sarcoma patient histology samples were purchased from U. S. Biomax. Immunohistochemistry was performed using PAR antibody (Trevigen), ABC elite kit, and ImmPACT NovaRED (Vector laboratories) following manufacturer's protocols. Samples were counterstained by hematoxylin GS (Vector labs), dehydrated, and mounted for light microscopic analysis (Olympus).
Fluorescence correlation spectroscopy
GFP‐PARP1 U2OS cells were used in FCS measurement. FCS measurement was all performed at room temperature with a Confocal microscope (Carl Zeiss) following published methods (Pack et al, 2014).
Laser micro‐irradiation
Cells were sensitized with BrdU at 37°C for 24 h. After washing with PBS, cells were incubated with media containing Hoechst 33342 and BrdU for 30 min at 37°C. Cells in fresh media were micro‐irradiated using a LSM 880 confocal microscope (Carl Zeiss). A region of interest (ROI) was selected and irradiated by 405 nm laser with 100% power for 1 iteration. Time‐lapse images were acquired in every 30 s after micro‐irradiation. The intensities at the ROI were analyzed by ZEN blue software (Carl Zeiss).
SILAC LC‐MS/MS analysis
Wild‐type (Ews‐WT, Heavy) and Ews‐KO (light) cells were grown continuously in SILAC media containing arginine and lysine with light isotopes of carbon, hydrogen and nitrogen (i.e., 12C14N) (light), or with media containing l‐lysine‐13C6 and l‐arginine‐13C6‐15N4 (heavy). Proteins extracted from labeled cell nucleus were analyzed by mass spectrometry to determine the incorporation ratios, which indicated an enrichment of > 95%, as above. 1D SDS–PAGE was performed with 1:1 amount mixed protein samples prepared from both labeled cells. For LC‐MS/MS analyses, the gel was de‐stained and bands were cut and processed as follows. Briefly, excised proteins bands were divided into 10 mm sections and subjected to in‐gel digestion with trypsin. The tryptic digests were separated by online reversed‐phase chromatography using a Thermo Scientific Eazy nano LC 1200 UHPLC equipped with an autosampler using a reversed‐phase peptide trap Acclaim PepMap™ 100 (75 μm inner diameter, 2 cm length) and a reversed‐phase analytical column PepMap™ RSLC C18 (75 μm inner diameter, 15 cm length, 3 μm particle size), both from Thermo Scientific, followed by electrospray ionization at a flow rate of 300 nl/min. The chromatography system was coupled in line with an Orbitrap Fusion Lumos mass spectrometer. Spectra were searched against the Uniprot‐human DB using the Proteome Discoverer Sorcerer 2.1 with SEQUEST‐based search algorithm and a comparative analysis of the identified proteins identified was performed using the Scaffold 4 Q+S software. The initial mass tolerance was set to 10 ppm, and MS/MS mass tolerance was 0.8 Da. Enzyme was set to Trypsin/P with two missed cleavages. Carbamidomethylation of cysteine was searched as a fixed modification, whereas 13C6‐Lys, 13C6‐15N4, 13C N‐acetyl protein, and oxidation of methionine were searched as variable modifications. Identification was set to a false discovery rate of 1%.
Viability assay
After seeding cells in 96‐well plates, cell viability was determined by the Cell Titer‐Glo assay kit following the manufacturer's protocol (Promega).
NAD+/NADH assay
The NAD+/NADH ratios were determined using the NAD+/NADH assay kit (Abcam) following the manufacturer's protocol. Briefly, NAD+/NADH were extracted with extraction buffer by two freezing and thawing cycles in dry ice. After centrifugation, supernatants (containing extracted total NAD+/NADH) were divided in half. NAD in half of supernatant was decomposed to NADH by heating samples to 60°C for 30 min in water bath (NADH decomposed samples). The remaining half of total NAD/NADH, and the NADH decomposed sample was incubated with NAD cycling buffer mixture at room temperature for 5 min. After adding NADH developer, the relative NAD+/NADH ratios were measured using plate reader (Biotek).
In vitro PAR binding assay
2 μl of recombinant proteins (as indicated concentration) were blotted onto nitrocellulose membrane and air‐dried for 30 min. The membrane was incubated with PAR polymer solution (ChromoTek) for 1 h and washed with TBST. PAR was detected by incubating the membrane with the PAR antibody for 1 h at room temperature, and then secondary antibody for 30 min at room temperature. The images were developed by the Odyssey imaging system (Li‐COR Biosciences).
Immunoprecipitation
Harvested cells were resuspended with IP buffer (150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 5 mM EDTA, 0.5% NP‐40, 1% Triton X‐100) for 1 h at 4°C with gentle agitation. The lysed proteins were immunoprecipitated by adding 1 μg/ml of the indicated antibody and magnetic beads (Invitrogen) followed by gentle rotation overnight at 4°C. The bead‐antibody‐protein complexes were washed for three times, and then, the proteins were released by boiling for 10 min at 100°C in 2XLaemmli sample buffer. Recovered proteins were resolved by SDS–PAGE.
Clonogenic assay
Mouse pre‐brown adipocytes were counted (LUNA™ automated Cell Counter) and seeded in an Eppendorf 6‐well culture plate. Culture media was changed every 3 days with or without MMS (0.0005%). Cells were cultured for about 2 weeks. For staining, cells were washed with PBS, stained with 2% methylene blue in 70% EtOH solution for 15 min, and then dried after thorough washing. For quantification, stained cell colonies were directly counted.
TUNEL assay
To analyze cell death in mouse tissues, we used DeadEnd colorimetric TUNEL system (Promega) following the manufacturer's protocol. Briefly, after fixation and permeabilization using 4% paraformaldehyde and 0.25% Triton X‐100, respectively, cells were incubated with proteinase K solution for 10 min. rTdT reaction cocktail were added to coverslips at 37°C for 1 h. After DAB substrate reaction and counterstaining by hematoxylin GS (Vector laboratories), the slides were mounted permanent mounting medium (Vector laboratories). The images were obtained using a light microscope (Olympus).
Immunofluorescence
After permeabilization using CSK buffer [10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA (pH 7.5)] with 0.5% Triton X‐100, cells were fixed with 10% NBF for 20 min at room temperature and then incubated with 100% methanol at −20°C for 10 min, followed by 1 h blocking with goat serum at 4°C. The fixed cells were subjected to stain with indicated antibodies overnight at 4°C. Cells were then incubated with fluorescence‐conjugated second antibodies (Invitrogen) and mounted in permanent mounting medium with DAPI (Vector Laboratories).
Plasmids and cloning
All EWS‐mutant constructs were generated using p3xFLAG‐CMV‐10 vector (Sigma), pCDH‐Hyg vector (SBI) as backbone with In‐Fusion cloning kit (Takara). CRISP‐Cas9 plasmid was purchased from Santa Cruz biotechnology.
ChIP assay
ChIP assay was carried out as described previously (Park et al, 2013) using PARP trapping beads (ChromoTek). After transfection of control (Dutertre et al, 2010) or Ews (siE) siRNAs to AsiSI endonuclease‐integrated U2OS cell lines, doxycycline was added to induce DNA double‐strand breaks for 4 h. Cells were cultured in either Dox‐containing media or fresh media (for release samples) for 2 h and then fixed with formaldehyde at room temperature for 15 min. To quench formaldehyde crosslinking, 1.25 M glycine was added and the solution was incubated an additional 5 min at room temperature. Cells were lysed using IP buffer and sonicated in a Qsonica water bath (60% power for 40/20 s on/off intervals for 30 min). For immunoprecipitation, PARP trapping beads were added to the soluble chromatin for 1 h at 4°C with agitation. The bead‐antibody–protein–chromatin complexes were washed for three times, and DNA was isolated using Chelex‐100 (Bio‐rad). qPCR was performed using SYBR green (Invitrogen) mixture with primers at or adjacent to the AsiSI sites.
Cell‐based un/identified protein interaction discovery (CUPID) assay
U2OS cells were transfected with either mRCD‐EWS or EGFP‐tagged PARP1 and exposed to H2O2 with or without translocation signal activator (Phorbol 12‐myristate 13‐acetate, PMA).
Alkaline Comet assay
Comet assays were performed using the Comet Assay Kit (Trevigen) following manufacturer's protocol. Cells were treated with mock, MMS, or H2O2 with indicated concentration and times. Harvested cells were combined with 1% molten LMAagarose at 37°C. The agarose plugs were immediately loaded onto comet slides and incubated at 4°C for 10 min. The slides were incubated with lysis solution, followed by alkaline unwinding. Alkaline electrophoresis of the slides was conducted at 21 V at 4°C for 15 min. To stain DNA, SYBR gold solution was added and the images were taken by microscope (Olympus) and analyzed by OpenComet software using ImageJ.
Quantitative real‐time PCR (qRT–PCR)
Total RNA was isolated using RNeasy mini kit (Qiagen). cDNAs were prepared using SuperScript IV cDNA synthesis kit (Invitrogen) and analyzed by real‐time PCR using SYBR Green Master Mix (Applied Biosystems). Primer sequences are listed below Table 1.
Table 1.
Real‐time PCR primers
| Gene | Sequence |
|---|---|
| Ews | Forward: CTC CTA CCA GCT ACT CCT CCT CAC |
| Reverse: TGC TTT GTT GAC CAT AGC TAC TCT | |
| Parp1 | Forward: GGC AGC CTG ATG TTG AGG |
| Reverse: GCG ATC TCC GCT AAA AAG | |
| Sirt1 | Forward: CAG TGT CAT GGT TCC TTT GC |
| Reverse: CAC CGA GGA ACT ACC TGA T | |
| Sirt3 | Forward : AAC ATC GAC GGG CTT GAG |
| Reverse: ACA GAC CGT GCA TGT AGC TG | |
| Sirt6 | Forward: CAG AGC TGC ACG GAA ACA T |
| Reverse: ACC GTG TCT CTG ACG TAC TGC | |
| Cd38 | Forward: TCT CTA GGA AAG CCC AGA TCG |
| Reverse: GTC CAC ACC AGG AGT GAG C |
Statistical analysis
Statistical analysis was performed by two‐way ANOVA or t‐test using GraphPad Prism 5 software (GraphPad Software). Data are represented as means ± SEM and significance was set at *P < 0.05, **P < 0.01, ***P < 0.001.
Author contributions
S‐gL, NK, S‐mK, IBP, HK, SK, B‐gK, JMH, I‐JB, and JHP performed experiments; KM guided the project; AG contributed to reviewing and editing the manuscript; S‐gL and JHP designed experiments, analyzed data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 5
{kind=link}
Source Data for Figure 6
{kind=link}
Acknowledgements
We thank the members of IBS Center for Genomic Integrity. Dr. Groehler IV Arnold Scott, professor Hongtae Kim (UNIST), and Chan‐Gi Pack (Asan Institute for Life Sciences) for helpful comments and discussion. This research was supported by the Institute for Basic Science (IBS‐R022‐D1) to KJ Myung.
EMBO Reports (2020) 21: e48676
Contributor Information
Jun Hong Park, Email: jhpark@kiom.re.kr.
Kyungjae Myung, Email: kmyung@ibs.re.kr.
Data availability
The complete list of SILAC LC‐MS/MS analysis, which is available via ProteomeXchange with identifier PXD016145 (http://www.ebi.ac.uk/pride/archive/projects/PXD016145).
Additional information can be found in the supplemental data.
References
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MB, Streicher W, Jungmichel S, Nielsen ML et al (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP‐ribose). Nat Commun 6: 8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boro A, Pretre K, Rechfeld F, Thalhammer V, Oesch S, Wachtel M, Schafer BW, Niggli FK (2012) Small‐molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing's sarcoma. Int J Cancer 131: 2153–2164 [DOI] [PubMed] [Google Scholar]
- Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou‐Maroun LM, Liu M, Lonigro R, Prensner JR, Tomlins SA et al (2012) PARP‐1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res 72: 1608–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini EN (2009) CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des 15: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong PA, Forman‐Kay JD (2016) Liquid‐liquid phase separation in cellular signaling systems. Curr Opin Struct Biol 41: 180–186 [DOI] [PubMed] [Google Scholar]
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG (1999) Poly(ADP‐ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342(Pt 2): 249–268 [PMC free article] [PubMed] [Google Scholar]
- Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G et al (1992) Gene fusion with an ETS DNA‐binding domain caused by chromosome translocation in human tumours. Nature 359: 162–165 [DOI] [PubMed] [Google Scholar]
- Durkacz BW, Omidiji O, Gray DA, Shall S (1980) (ADP‐ribose)n participates in DNA excision repair. Nature 283: 593–596 [DOI] [PubMed] [Google Scholar]
- Dutertre M, Sanchez G, De Cian MC, Barbier J, Dardenne E, Gratadou L, Dujardin G, Le Jossic‐Corcos C, Corcos L, Auboeuf D (2010) Cotranscriptional exon skipping in the genotoxic stress response. Nat Struct Mol Biol 17: 1358–1366 [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH et al (1997) Poly(ADP‐ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095 [DOI] [PubMed] [Google Scholar]
- Fang EF, Scheibye‐Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA (2014) Defective mitophagy in XPA via PARP‐1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157: 882–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Kassahun H, Croteau DL, Scheibye‐Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA et al (2016) NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 24: 566–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauzee NJ, Pan J, Wang YL (2010) PARP and PARG inhibitors–new therapeutic targets in cancer treatment. Pathol Oncol Res 16: 469–478 [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Takii R, Takaki E, Katiyar A, Nakato R, Shirahige K, Nakai A (2017) The HSF1‐PARP13‐PARP1 complex facilitates DNA repair and promotes mammary tumorigenesis. Nat Commun 8: 1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG (2008) PARP1‐dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283: 1197–1208 [DOI] [PubMed] [Google Scholar]
- Helleday T (2011) The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5: 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483 [DOI] [PubMed] [Google Scholar]
- Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch‐Nolte F (2010) Toward a unified nomenclature for mammalian ADP‐ribosyltransferases. Trends Biochem Sci 35: 208–219 [DOI] [PubMed] [Google Scholar]
- Izhar L, Adamson B, Ciccia A, Lewis J, Pontano‐Vaites L, Leng Y, Liang AC, Westbrook TF, Harper JW, Elledge SJ (2015) A systematic analysis of factors localized to damaged chromatin reveals PARP‐dependent recruitment of transcription factors. Cell Rep 11: 1486–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyasekharan AD, Ayoub N, Mahen R, Ries J, Esposito A, Rajendra E, Hattori H, Kulkarni RP, Venkitaraman AR (2010) DNA damage regulates the mobility of Brca2 within the nucleoplasm of living cells. Proc Natl Acad Sci USA 107: 21937–21942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML (2013) Proteome‐wide identification of poly(ADP‐Ribosyl)ation targets in different genotoxic stress responses. Mol Cell 52: 272–285 [DOI] [PubMed] [Google Scholar]
- Khoury‐Haddad H, Guttmann‐Raviv N, Ipenberg I, Huggins D, Jeyasekharan AD, Ayoub N (2014) PARP1‐dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double‐strand break repair. Proc Natl Acad Sci USA 111: E728–E737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL (2004) NAD+‐dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP‐1. Cell 119: 803–814 [DOI] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL (2005) Poly(ADP‐ribosyl)ation by PARP‐1: ‘PAR‐laying’ NAD+ into a nuclear signal. Genes Dev 19: 1951–1967 [DOI] [PubMed] [Google Scholar]
- Klevernic IV, Morton S, Davis RJ, Cohen P (2009) Phosphorylation of Ewing's sarcoma protein (EWS) and EWS‐Fli1 in response to DNA damage. Biochem J 418: 625–634 [DOI] [PubMed] [Google Scholar]
- Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM (2004) Failure to degrade poly(ADP‐ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci USA 101: 17699–17704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, Dawson VL, Masson JY, Poirier GG, Gagne JP (2013) Reprogramming cellular events by poly(ADP‐ribose)‐binding proteins. Mol Aspects Med 34: 1066–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39: 8–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger A, Burkle A, Hauser K, Mangerich A (2020) Real‐time monitoring of PARP1‐dependent PARylation by ATR‐FTIR spectroscopy. Nat Commun 11: 2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KB, Hwang JM, Choi IS, Rho J, Choi JS, Kim GH, Kim SI, Kim S, Lee ZW (2011) Direct monitoring of the inhibition of protein‐protein interactions in cells by translocation of PKCdelta fusion proteins. Angew Chem Int Ed Engl 50: 1314–1317 [DOI] [PubMed] [Google Scholar]
- Lee KY, Fu H, Aladjem MI, Myung K (2013) ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J Cell Biol 200: 31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Watford W, Li C, Parmelee A, Bryant MA, Deng C, O'Shea J, Lee SB (2007) Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J Clin Invest 117: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Chen J (2014) ADP‐ribosylation: activation, recognition, and removal. Mol Cells 37: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJL, Pines A, Vertegaal ACO, Jacobs JJL, Shah GM et al (2016) PARP1 links CHD2‐mediated chromatin expansion and H3.3 deposition to DNA repair by non‐homologous end‐joining. Mol Cell 61: 547–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Kraus WL (2012) On PAR with PARP: cellular stress signaling through poly(ADP‐ribose) and PARP‐1. Genes Dev 26: 417–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Ryu KW, Kim DS, Nandu T, Medina CJ, Gupte R, Gibson BA, Soccio RE, Yu Y, Gupta RK et al (2017) PARP‐1 controls the adipogenic transcriptional program by PARylating C/EBPbeta and modulating its transcriptional activity. Mol Cell 65: 260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS (2013) The RNA‐binding protein fused in sarcoma (FUS) functions downstream of poly(ADP‐ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 288: 24731–24741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelena J, Lezaja A, Teloni F, Schmid T, Imhof R, Altmeyer M (2018) Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat Commun 9: 2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y (2012) Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72: 5588–5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack CG, Yukii H, Toh‐e A, Kudo T, Tsuchiya H, Kaiho A, Sakata E, Murata S, Yokosawa H, Sako Y et al (2014) Quantitative live‐cell imaging reveals spatio‐temporal dynamics and cytoplasmic assembly of the 26S proteasome. Nat Commun 5: 3396 [DOI] [PubMed] [Google Scholar]
- Park JH, Kang HJ, Kang SI, Lee JE, Hur J, Ge K, Mueller E, Li H, Lee BC, Lee SB (2013) A multifunctional protein, EWS, is essential for early brown fat lineage determination. Dev Cell 26: 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kang HJ, Lee YK, Kang H, Kim J, Chung JH, Chang JS, McPherron AC, Lee SB (2015) Inactivation of EWS reduces PGC‐1alpha protein stability and mitochondrial homeostasis. Proc Natl Acad Sci USA 112: 6074–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Galbraith R, Turner T, Mehojah J, Azuma M (2016) Loss of Ewing sarcoma EWS allele promotes tumorigenesis by inducing chromosomal instability in zebrafish. Sci Rep 6: 32297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, Minana B, Valcarcel J (2011) The Ewing sarcoma protein regulates DNA damage‐induced alternative splicing. Mol Cell 43: 353–368 [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Nussenzweig A (2017) The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18: 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gomez‐Herreros F, Hafezparast M, Caldecott KW (2014) PARP‐1 dependent recruitment of the amyotrophic lateral sclerosis‐associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res 42: 307–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye‐Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan‐Olive MM et al (2014) A high‐fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20: 840–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Lebeaupin T, Juhasz S, Chapuis C, D'Augustin O, Dutertre S, Burkovics P, Biertumpfel C, Timinszky G, Huet S (2019) Poly(ADP‐ribose)‐dependent chromatin unfolding facilitates the association of DNA‐binding proteins with DNA at sites of damage. Nucleic Acids Res 47: 11250–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn L, Siligan C, Bachmaier R, Schmid JA, Aryee DN, Kovar H (2003) Homotypic and heterotypic interactions of EWS, FLI1 and their oncogenic fusion protein. Oncogene 22: 6819–6829 [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teloni F, Altmeyer M (2016) Readers of poly(ADP‐ribose): designed to be fit for purpose. Nucleic Acids Res 44: 993–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trucco C, Oliver FJ, de Murcia G, Menissier‐de Murcia J (1998) DNA repair defect in poly(ADP‐ribose) polymerase‐deficient cell lines. Nucleic Acids Res 26: 2644–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A (2017) Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulin A, Spradling A (2003) Chromatin loosening by poly(ADP)‐ribose polymerase (PARP) at Drosophila puff loci. Science 299: 560–562 [DOI] [PubMed] [Google Scholar]
- Verdin E (2015) NAD(+) in aging, metabolism, and neurodegeneration. Science 350: 1208–1213 [DOI] [PubMed] [Google Scholar]
- Yang L, Chansky HA, Hickstein DD (2000) EWS.Fli‐1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine‐arginine protein‐mediated RNA splicing. J Biol Chem 275: 37612–37618 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The complete list of SILAC LC‐MS/MS analysis, which is available via ProteomeXchange with identifier PXD016145 (http://www.ebi.ac.uk/pride/archive/projects/PXD016145).
Additional information can be found in the supplemental data.