

Abstract
The human UBE3A gene, which is essential for normal neurodevelopment, encodes three Ubiquitin E3 ligase A (UBE3A) protein isoforms. However, the subcellular localization and relative abundance of these human UBE3A isoforms are unknown. We found, as previously reported in mice, that UBE3A is predominantly nuclear in human neurons. However, this conserved subcellular distribution is achieved by strikingly distinct cis-acting mechanisms. A single amino-acid deletion in the N-terminus of human hUBE3A-Iso3, which is homologous to cytosolic mouse mUBE3A-Iso2, results in its translocation to the nucleus. This singe amino-acid deletion is shared with apes and Old World monkeys and was preceded by the appearance of the cytosolic hUBE3A-Iso2 isoform. This hUBE3A-Iso2 isoform arose after the lineage of New World monkeys and Old World monkeys separated from the Tarsiers (Tarsiidae). Due to the loss of a single nucleotide in a non-coding exon, this exon became in frame with the remainder of the UBE3A protein. RNA-seq analysis of human brain samples showed that the human UBE3A isoforms arise by alternative splicing. Consistent with the predominant nuclear enrichment of UBE3A in human neurons, the two nuclear-localized isoforms, hUBE3A-Iso1 and -Iso3, are the most abundantly expressed isoforms of UBE3A, while hUBE3A-Iso2 maintains a small pool of cytosolic UBE3A. Our findings provide new insight into UBE3A localization and evolution and may have important implications for gene therapy approaches in Angelman syndrome.
Introduction
Ubiquitin E3 ligase (UBE3A) is a HECT (Homologous to E6AP C-Terminus) E3 ligase, required for normal neurodevelopment (1). Although expressed biallelically in most tissues, UBE3A is expressed exclusively from the maternal allele in neurons (2) and changes in neuronal UBE3A activity has major impact on neurodevelopment (3). Loss of functional UBE3A in the brain leads to Angelman syndrome (AS) (1,4,5). Conversely, increased levels of UBE3A appear to function critically in the pathophysiology of Dup15q syndrome and have been associated with autism spectrum disorder (ASD) (6,7). Yet, despite the extensive evidence implicating UBE3A in severe neurodevelopmental disorders, relatively little is known about the distinct isoforms of UBE3A.
Several studies have shown that UBE3A is highly enriched in the nucleus of mouse and human neurons (8,9), but protein labeling has also been observed in the cytosol, in particular within dendritic spines and axon terminals (10,11). Recent work has revealed a mechanism guiding the subcellular localization of UBE3A (9,12). Mice express two distinct UBE3A protein isoforms, which differ in their N-terminus. The shorter, more abundantly expressed, mouse isoform 3 (mUBE3A-Iso3) is predominantly localized to the nucleus, whereas the longer, less abundant, mUBE3A-Iso2 isoform, which possesses a N-terminal extension of 21 amino acids, is mainly localized in the cytosol (9). Importantly, only the selective loss of the nuclear-localized mUBE3A-Iso3 in mice results in behavioral and electrophysiological deficits resembling AS (9,13). In humans, differential splicing generates three protein isoforms (14), two of which, hUBE3A-iso1 and hUBE3A-iso3, are highly homologous to the mouse isoforms 3 and 2, respectively (Fig. 1A). In addition, a third human UBE3A isoform has been reported (hUBE3A-Iso2), which has no homologue in mice (14). Similar to mice, loss of the most abundantly expressed short isoform (hUBE3A-Iso1) in human results in a AS-like phenotype (15), but the localization of this human isoform has not been established.
Figure 1.
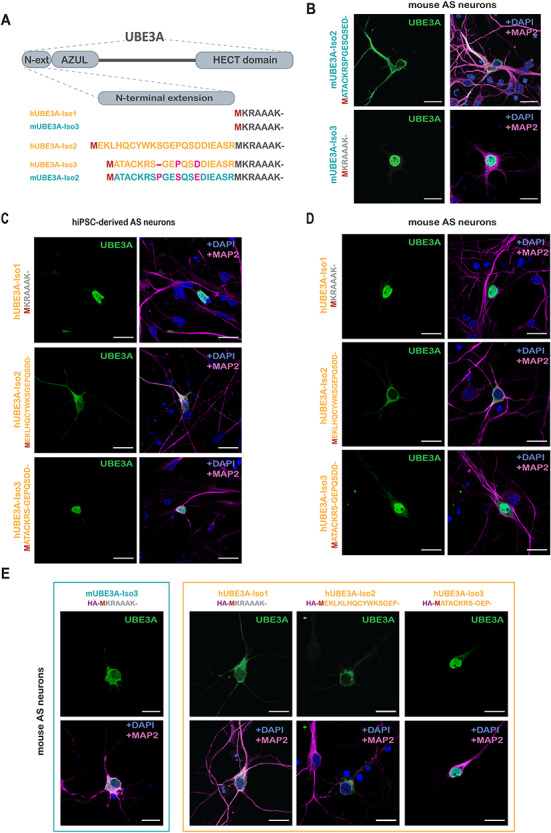
Localization of mouse and human UBE3A isoforms. (A) Schematic overview (top) of the domains of UBE3A and the N-terminal sequences (bottom) of the different mouse (blue) and human (orange) UBE3A isoforms. (B) Localization of mUBE3A isoforms in hippocampal neurons derived from E16.5 Ube3am−/p+ (AS) embryos. Neurons transfected at DIV7 with untagged mUBE3A-Iso2 or mUBE3A-Iso3 were fixed and stained at DIV10. Neurons were stained for UBE3A (green, left panels) together with MAP2 (pink) and DAPI (blue) (right panels). (C) Localization of hUBE3A isoforms in hIPSC-derived AS neurons. Neurons transfected after 6 weeks of differentiation with untagged hUBE3A-Iso1, hUBE3A-Iso2 or hUBE3A-Iso3 were fixed and stained 2 days after transfection. Staining was as described for (B). (D) Localization of hUBE3A isoforms in mouse hippocampal AS neurons. Neurons transfected at DIV7 with hUBE3A-Iso1, hUBE3A-Iso2 and hUBE3A-Iso3 were fixed and stained as described for (B). (E) Localization of HA-tagged mUBE3A-Iso3 and human UBE3A isoforms in mouse hippocampal AS neurons. Neurons were fixed and stained as described for (B). Scale bars, 20 μm.
Although the human UBE3A isoforms were described over two decades ago (14), still very little is known about how these isoforms are generated, their relative abundance and their subcellular localization. Here we report that, although the overall subcellular distribution of UBE3A in human-derived neurons resembles that of mouse primary neurons, the localization of human UBE3A is accomplished through a distinct mechanism. The human homologues of the two mouse UBE3A isoforms are both localized in the nucleus, whereas the hUBE3A-Iso2, which is shared with Old- and New World monkeys, is localized in the cytosol.
Results
Human UBE3A-iso1 and -iso3 are enriched in the nucleus while hUBE3A-Iso2 is cytosolic
We have previously shown that human UBE3A is highly enriched in the nucleus, as observed in cultured human iPSC-derived neurons and in postmortem human brains (9) (Supplementary Material, Figure S1A). To investigate how this overall subcellular distribution of UBE3A is achieved, we determined the subcellular localization of each of the three human UBE3A isoforms. We generated constructs that express each individual (untagged) isoform under the control of the CMV early enhancer/chicken β-actin/β-globin (CAG) promoter, and a tdTomato fluorescence reporter under the control of the PGK promoter to identify transfected neurons (16) (Supplementary Material, Figure S1B). In the constructs of the long isoforms (hUBE3A-Iso2 and -Iso3), the second ATG codon was mutated to ensure the expression of only the long isoforms from each of these constructs (Supplementary Material, Figure S1B).
These constructs were transfected to human iPSC-derived AS neurons and localization in mature neurons was assessed by immunocytochemistry (Fig. 1C). Analogous to the nuclear localization of the short mUBE3A-Iso3 in mouse (9) (Fig. 1B), the hUBE3A-Iso1 orthologue was predominantly located in the nucleus (Fig. 1C). Remarkably, hUBE3A-Iso3, which is highly homologous to the cytosolic mUBE3A-Iso2, also localized predominantly to the nucleus (Fig. 1C). In contrast, the unique hUBE3A-Iso2 showed a clear cytosolic localization (Fig. 1C). Overexpression of the human UBE3A isoforms in primary murine AS hippocampal neurons (Fig. 1D) yielded the same localization as in AS iPSC-derived neurons and in non-neuronal human Bone Osteosarcoma Epithelial (U2OS) cells (Fig. 1C; Supplementary Material, Figure S1C). This indicates that the isoform-specific neuronal subcellular localization of UBE3A is an intrinsic property of the protein and not affected by possible differences in trans-acting factors between two species (i.e. human versus mouse), and independent of cell type (i.e. neuronal versus non-neuronal) (Fig. 1 and Supplemental Material, Figure S1C). Thus, in both humans and mice, the overall distribution of UBE3A appears to be determined exclusively by the distinct isoforms, each of which has a defined subcellular localization.
The N-terminal extension of UBE3A dictates subcellular localization
The above experiments indicate that the N-terminal amino acid sequence of UBE3A is critical for determining its subcellular localization. To test whether the addition of a short N-terminal tag affects the subcellular distribution of the nuclear-localized isoforms (mUBE3A-Iso3 and hUBE3A-Iso1), we N-terminally tagged both isoforms with the HA (hemagglutinin) tag and overexpressed them in AS mouse primary neurons. Strikingly, addition of an N-terminal HA-tag to the (short) nuclear mUBE3A-Iso3 or hUBE3A-Iso1 homologues resulted in their relocation to the cytosol. However, adding the tag to the nuclear hUBE3A-Iso3 or to the cytosolic hUBE3A-Iso2 did not affect the localization of either of these proteins (Fig. 1E).
It is remarkable that although mUBE3A-Iso2 and hUBE3A-Iso3 share an almost identical N-terminal amino acid extension that differs at only three positions, their localization is strikingly different (mUBE3A-Iso2, cytoplasmic; hUBE3A-Iso3, nuclear; Fig. 1). To address whether the distinct localization of these two homologs is solely dictated by their N-terminal extensions (and not by other differences in the UBE3A protein), we swapped the N-terminal extensions of mUBE3A-Iso2 and hUBE3A-Iso3, thereby creating chimeric mext2hUBE3A-Iso3 and hext3mUBE3A-Iso2 proteins (Fig. 2A). The localization of these chimeric proteins was determined in both primary murine AS hippocampal neurons (Fig. 2B) and hiPSC-derived AS neurons (Fig. 2C). In both human and mouse neurons, the localization of the two isoforms was inverse, with hext3mUBE3A-Iso2 showing a nuclear localization and mext2hUBE3A-Iso3 a cytosolic localization. Collectively these results indicate that the differential localization of these UBE3A isoforms is strongly determined by the sequence of their N-terminal extensions.
Figure 2.
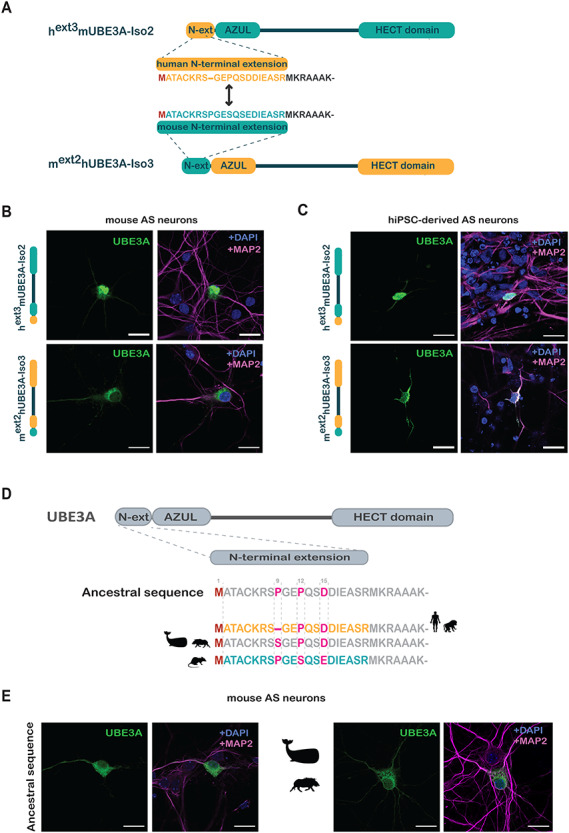
The N-terminal extension of UBE3A dictates its localization. (A) Schematic overview of the chimeric UBE3A constructs in which the N-terminal extensions of mUBE3A-Iso2 and hUBE3a-Iso3 were swapped. (B and C) Localization of chimeric hext3mUBE3A-Iso2 and mext2hUBE3A-Iso3 constructs after transfection in mouse hippocampal AS neurons (B) or into hIPSC-derived AS neurons (C). (D) Schematic overview of the ancestral ‘MATACK’ N-terminal extension (see Fig. 3) and the sequences found in Homo sapiens and Cercopithecidae (Old World Monkeys), Cetartiodactyla (wild boar and whale) and Mus musculus (mouse). (E) Localization of chimeric UBE3A constructs in mouse hippocampal AS neurons. Neurons were transfected at DIV7 with hUBE3A-Iso3 harboring the ancestral ‘MATACK’ extension or the extension of the Cetartiodactyla. All neurons were stained for UBE3A (green) together with MAP2 (pink) and DAPI (blue). Scale bars, 20 μm.
A unique deletion of a Proline in the N-terminal extension of hUBE3A-Iso3 results in its translocation to the nucleus
The N-terminal ‘MATACK’ leader sequences of mUBE3A-Iso2 and hUBE3A-Iso3, differ only at three positions: Proline (P) 9 in the mUBE3A-Iso2 is missing in hUBE3A-Iso3, Serine (S) 12 in mUBE3A-Iso2 is a Proline in hUBE3A-Iso3 and Glutamate (E) 15 in mUBE3A-Iso2 is an Aspartate (D) in hUBE3A-Iso3 (Figs 1A and2D). To decipher which of these changes might be responsible for determining the differential subcellular location, we first investigated their molecular evolution. A basic local alignment search tool (BLAST) analysis on protein sequences was performed, using residues 1–16 of hUBE3A-Iso3 as a query sequence. A multiple sequence alignment of the identified sequences indicated an ancestral sequence of the placental mammals (Placentalia) that possesses a Proline at positions 9 and 12 and an Aspartate at position 15, which is shared amongst most branches of the mammalian evolutionary tree (Fig. 3, Supplementary Material, Figure S2A). Notably, we also found that the majority of evolutionary changes that subsequently occurred in the ‘MATACK’ leader sequence are confined to these three residues (amino acids 9, 12 and 15).
Figure 3.
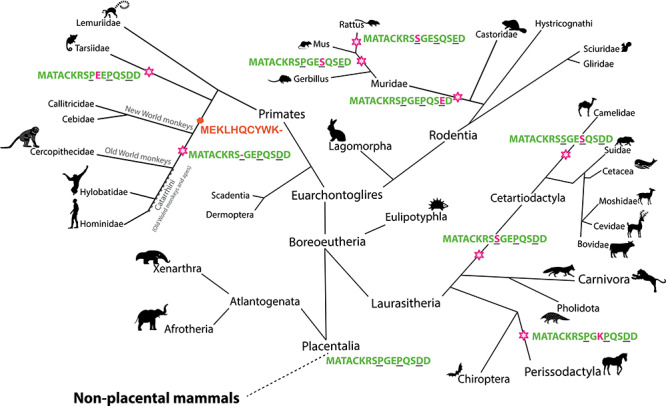
Molecular evolution of UBE3A isoforms. Molecular evolution of the (N-terminal extension of) UBE3A isoforms in placental mammals. Stars indicate changes to the ancestral N-terminal sequence, MATACKRSPGEPQSDD, and are indicated in magenta. The three amino acid positions different between the mUBE3A-Iso2 and hUBE3A-Iso3 are underlined in black. The orange hexagon marks the time in evolution when hUBE3A-Iso2 first is predicted to start being expressed.
The conservative change of D15E in mouse mUBE3A-Iso2 was also observed in other rodents (rats, gerbils) of the Muridae family, suggesting that this substitution occurred late in rodent evolution. The P12 to mouse S12 substitution occurred even later in rodent evolution, but it was additionally observed in other minor branches of mammals (e.g. Camelidae) (Fig. 3). The Proline at position 9 has been substituted for a Serine at several instances during evolution but is most notably frequent in the branch of all Cetartiodactyla, which includes all even-toed ungulates (hoofed animals), whales and dolphins. However, the deletion of this amino-acid in human hUBE3A-Iso3 is highly unique and shared only with Great Apes and Old World Monkeys (collectively referred to as Catarrhini), but not New World Monkeys. These findings suggest that the Proline 9 deletion arose during recent primate evolution, around the time of divergence of Catarrhini and New World monkeys, less than 25–40 million years ago (Mya) (Fig. 3).
To identify the critical substitutions responsible for the differential subcellular localization of mUBE3A-Iso2 and hUBE3A-Iso3, we constructed chimeric proteins in which the ‘MATACK’ leader sequence of hUBE3A-Iso3 was replaced by the ancestral ‘MATACK’ sequence along with the substitutions found in Cetartiodactyla (Fig. 2D). Since the localization of overexpressed UBE3A isoforms is not dependent on the host cell species (Fig. 1), the localization of these chimeric isoforms was investigated in primary mouse AS neurons. Chimeric isoforms harboring the ancestral ‘MATACK’ leader sequence or the ‘MATACK’ sequence as found in Cetartiodactyla showed a predominantly cytosolic localization (Fig. 2E), mirroring the subcellular distribution of mouse mUBE3A-Iso2. Hence, the deletion of Proline 9 (mUBE3A-Iso2 numbering) in the ‘MATACK’ leader sequence is responsible for the nuclear localization of hUBE3A-Iso3, an isoform that is selectively present among Catarrhini.
Appearance of the cytosolic hUBE3A-Iso2 during primate evolution preceded relocation of hUBE3A-iso3 to the nucleus
The above experiments establish that humans possess two UBE3A isoforms (hIso1 and hIso3) that are both predominantly nuclear and one cytosolic isoform (hUBE3A-Iso2). Given the conserved overall subcellular distribution of UBE3A in rodents and human, we hypothesized that the highly unusual deletion of the Proline in the Catarrhini ‘MATACK’ leader sequence which relocates UBE3A to the nucleus, was only tolerated in evolution after the cytosolic hUBE3A-Iso2 had arisen and took over the role of UBE3A in the cytosol. To address this intriguing evolutionary question, we investigated the genomes of selected species for the presence of the exon that encodes the ‘MEKL’ leader sequence (Supplementary Material, Figure S2B). We found that indeed different variations of the ‘MEKL’ exon can be traced back to all mammalian clades (both in placental and non-placental mammals). However, it is a non-coding exon in most species since it possesses a nucleotide insertion 3′ of the start codon (ATG) that results in an out-of-frame exon. We also identified several species in which the exon had acquired mutations that disrupt the open reading frame by altering the start codon (ATG), or in which it had lost critical splicing sites (or a combination of these changes). Thus, there has been little (to none) selective pressure on this non-coding exon, and consequently, many additional mutations in this sequence can be observed in various species (Supplementary Material, Figure S2B and Supplementary Material, Table S1). Only after the lineage of New World monkeys and Old World monkeys separated from the Tarsiers (Tarsiidae) (40–58 Mya), a single nucleotide in this non-coding exon was lost, which then allowed the translation of the in-frame ‘MEKL’ encoding exon. Hence, the cytosolic hUBE3A-Iso2 ‘MEKL’ isoform arose before the deletion of the Proline in the human ‘MATACK’ leader sequence occurred, which relocates the hUBE3A-Iso3 isoform to the nucleus.
Human UBE3A isoforms are generated by alternative splicing, while mice isoforms arise via alternative translation
In a previous study, we demonstrated that the nuclear short mUBE3A-Iso3, as well as its nuclear homologue hUBE3A-Iso1, are the most abundantly expressed protein isoforms in mouse and human brain, respectively, constituting approximately 80% of all UBE3A protein (9). Since hUBE3A-Iso2 and hUBE3A-Iso3 isoforms cannot be separated on a western blot, their combined contribution was estimated to be 10–20% of total UBE3A protein. To gain more insight into the relative expression level of each human UBE3A isoform, we analyzed a previously published human cortex RNA-seq dataset (17) (human cortex autopsy tissue from individuals with no history of neurological disorders; cause of death was an acute cardiorespiratory failure) and determined the abundance of each distinct UBE3A transcript based on an analysis of alternatively spliced exons. We used the Integrated Genome Viewer (IGV) software to generate Sashimi plots for every sample of the dataset (Supplementary Material, Figure S3A) and quantified the exon junctions occurring in the UBE3A transcripts (Fig. 4A and B). Similar to what has been observed previously by sequencing of human UBE3A transcripts (14), we found two UTRs preceding the coding UBE3A sequence. In addition, we observed a non-coding exon between coding exons 4 and 6 that has been reported previously (14) and is now also annotated in the latest version of the human reference genome. Its presence was observed in the majority of the transcripts and its inclusion prevents translation of UBE3A from the upstream exons (exons 3 and 4) that encode the ‘MATACK’ and ‘MEKL’ N-terminal extensions. Hence, even though these ‘MATACK’ and ‘MEKL’ coding exons are present in a large number of transcripts, the presence of the non-coding exon results in the synthesis of the short (nuclear) hUBE3A-Iso1, which initiates translation at the methionine of the MKRAAAK sequence encoded by exon 6/7 (Fig. 4C). Based on the frequency of exon junctions as indicated in the Sashimi plots (Fig. 4B), we identified all possible splice variants that potentially encode any of the three isoforms and calculated their relative abundance (as % of total UBE3A transcripts in the data set) (Fig. 4C). According to these calculations the majority of transcripts encodes for the short nuclear isoform, hUBE3A-Iso1 (82%), which is entirely consistent with our protein data (9). Transcripts potentially encoding nuclear hUBE3A-Iso3 are characterized by the presence of the ‘MATACK’ exon, but with the obligatory absence of the ‘MEKL’ and non-coding exon (incorporation of either exon would result in premature termination of translation), which accounts for 17% of the total transcripts. Lastly, transcripts that could potentially encode the cytosolic hUBE3A-Iso2 are characterized by the presence of the ‘MEKL’ exon and absence of the non-coding exon. We only observed 1% of such transcripts, which also consistently included the ‘MATACK’ exon. Given that the translation initiation codon of the ‘MEKL’ exon has a more favorable Kozak sequence (18) compared to the ‘MATACK’ exon, and that its presence renders the ‘MATACK’ exon out-of-frame, it is likely that translation of this transcript starts at the ‘MEKL’ exon to generate the hUBE3A-Iso2 protein (Fig. 4C and Supplementary Material, Figure S3D). Using the same methodology, we quantified the expression levels of each UBE3A isoform in an in vitro model, an already published RNA-seq dataset of forebrain-patterned neural progenitor cells (NPCs) derived from human embryonic stem cells (hESC), at week 6 of differentiation (19). The ratio of UBE3A isoforms in these cells is similar to the brain samples, with transcripts that would encode hUBE3A-Iso1 making up the majority of total UBE3A (80.6%), hUBE3A-Iso3 being the second most abundant UBE3A isoform (17%), and the remaining 2.4% of transcripts encoding for the cytosolic hUBE3A-Iso2 (Supplementary Material, Figure S3B and C).
Figure 4.
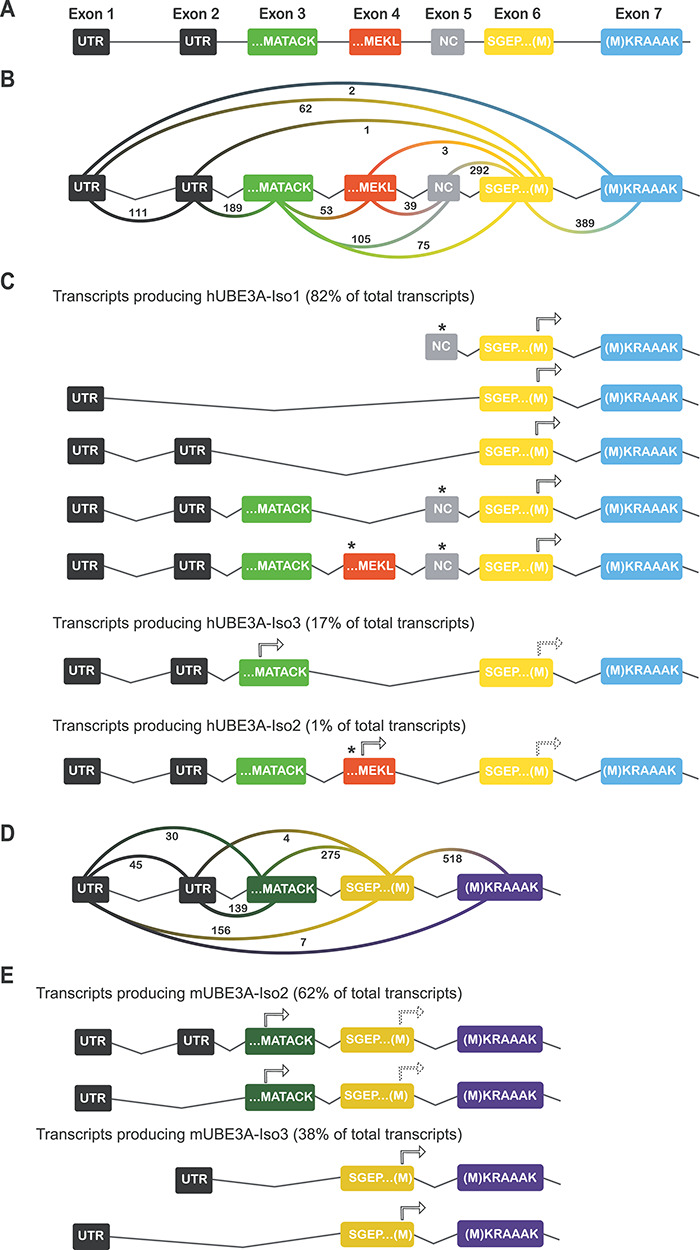
5′ coding region of the UBE3A gene and predicted splicing pattern of transcripts. (A) Schematic representation of the first seven exons of UBE3A. UTR stands for untranslated region, NC for non-coding. Exon numbering according to LaSalle et al. (2). (B) Schematic representation of the number of UBE3A exon junctions found in the human cortex RNA-seq dataset. (C) Splicing pattern of predicted UBE3A transcripts. Splicing pattern and relative abundance of each isoform producing transcript are based on the quantification of exon junctions in the human cortex RNA-seq dataset shown in (B). Putative initiation ATGs (arrows) and stop codons (asterisks) upstream of the initiation ATG are indicated for each predicted transcript. The predicted transcripts producing hUBE3A-Iso3 and hUBE3A-Iso2 contain a downstream initiation codon (dashed arrows) that could potentially also result in the synthesis of hUBE3A-Iso1. (D) Schematic representation of the number of Ube3a exon junctions found in the mouse RNA-seq dataset. (E) Splicing pattern of predicted Ube3a transcripts. Splicing pattern and relative abundance of each isoform producing transcript are based on the quantification of exon junctions in the mouse RNA-seq dataset shown in (D).
Previous studies have suggested that the expression of distinct mouse UBE3A isoforms is controlled by alternative splicing as well (12,20,21). However, those findings relied exclusively on RT-PCR analyses, which may fail to capture changes in relative abundance due to the use of different primer sets. Hence, we quantified the RNA-seq data to investigate the relative abundance of the mouse UBE3A isoforms. Surprisingly, we observed that the majority of transcripts (62%) contained the ‘MATACK’ exon of the longer cytosolic mUBE3A-Iso2 (Fig. 4D and E). In contrast, only 38% of the transcripts was specific for the short (nuclear) mUBE3A-Iso3. Given that at the protein level 75–80% of the total UBE3A protein present in mouse brain is the short isoform (9), it must be concluded that regulation of the mouse protein isoforms is heavily regulated by alternative translation, in which transcripts that contain both the ‘MATACK’ and ‘MKRAAAK’ exons favor the translation of the short (nuclear) mUBE3A-Iso3.
Discussion
In the present study, we have determined the subcellular localization of the three human UBE3A isoforms using both human iPSC-derived neurons and mouse primary neurons. We showed that, similar to mouse (9), the shortest and most abundantly expressed isoform (hUBE3A-Iso1) is predominantly nuclear. This is in line with previous studies that showed that human UBE3A immunoreactivity is highly enriched in the nucleus of human brain and human iPSC-derived neurons (9,11).
A rather surprising finding from our study is that hUBE3A-Iso3, which is highly homologous to the cytosolic localized mUBE3A-Iso2, is also predominantly targeted to the nucleus. By swapping the N-terminal extensions of the mouse isoform 2 and human isoform 3, we show that the N-terminal extension dictates UBE3A isoform localization, irrespective of the cell-type or species origin in which the UBE3A isoform is expressed. This observation allowed us to use the mUBE3A-Iso2/hUBE3A-Iso3-like extensions of a variety of species to identify the crucial change in the hUBE3A-Iso3 extension that caused the protein to switch from the cytosol to the nucleus: loss of the Proline residue at position 9. Evolutionary analysis of the mUBE3A-Iso2/hUBE3A-Iso3-like extensions revealed that the variant lacking the proline at position 9 is only present in human, Apes and Old World monkeys (Catarrhini), and must therefore have arisen 25–40 Mya. Although the exact mechanism by which the N-terminal extension mediates differential localization remains to be established, our previous work in mice suggested that the cytosolic mUBE3A-Iso2, following initial targeting to the nucleus through interaction of the UBE3A AZUL domain with PSMD4 (9,22), is actively transported out of the nucleus. Since there is no nuclear export signal in either of the UBE3A extensions, our results suggest that loss of Proline 9 interferes with nuclear export by affecting protein folding and/or interaction with other proteins, ultimately resulting in nuclear retention of UBE3A. The importance of the N-terminal extension for UBE3A localization is supported by the observation that the addition of an N-terminal HA-tag to the mouse or human short nuclear isoform results in a dramatic shift of UBE3A into the cytosol. This observation provides a warning regarding the interpretation of functional studies that use N-terminal tagged UBE3A, since a change in UBE3A localization is also likely to change its function. Further investigations are required to unravel the precise mechanism by which the N-terminal extension regulates the subcellular localization and function of UBE3A.
While a transcript encoding hUBE3A-Iso2 was reported more than two decades ago (14), its abundance has only now been quantified, based on the analysis of human cortical RNA-seq data. While the majority of UBE3A transcripts encode hUBE3A-Iso1 (82%) and hUBE3A-Iso3 (17%), we find that approximately 1% of the UBE3A transcripts encode hUBE3A-Iso2. These transcript levels correspond well with the reported protein levels of hUBE3A-Iso1 (~80%), and the combined protein levels of hUBE3A-Iso2 and UBE3A-Iso3 (~20%) in human brain (9). These findings imply that the novel hUBE3A-Iso2 isoform has a low abundance in human brain. This is of particular relevance because this isoform has been frequently used in studies of UBE3A function, most likely since it encodes the longest UBE3A protein isoform [e.g. (23–25)].
Our evolutionary analysis of the UBE3A isoforms indicates that the nuclear localization of hUBE3A-Iso3 followed the emergence of the evolutionarily novel cytosolic isoform, hUBE3A-Iso2. At the DNA level, this exon can be found in all investigated mammals, but it only became an in-frame, protein-coding, exon approximately 40–60 My ago, when a single nucleotide was lost after New World monkeys and Catarrhini separated from Tarsiers. The finding that the emergence of the novel cytosolic isoform preceded the mutation that caused hUBE3A-Iso3 to move to the nucleus is suggestive of an evolutionary pressure to maintain at least a small cytosolic pool of UBE3A, and points toward an important role for UBE3A in the cytosol. Whether this cytosolic UBE3A activity is required for neuronal function, as well as for the pathophysiology of AS, remains to be established. A study in which specifically the cytosolic mUBE3A-Iso2 was deleted in mice, showed no behavioral phenotypes and changes in synaptic function, and the behavioral deficits of AS mice could be fully recapitulated by selective deletion of the nuclear mUBE3A-Iso3 isoform (9). Recently three individuals with AS have been described who specifically lack the nuclear hUBE3A-Iso1 (15). Based on the isoform localization, we determined in this study, we would predict that this mutation causes a loss of most of the nuclear UBE3A and that the cytosolic UBE3A levels remain the same. These AS patients showed the typical facial features of AS and severe neurodevelopmental delay, as well as different combinations of the behavioral features, such as happy disposition, easily provoked laughter, hand-flapping, hyperkinesia, short attention span, easy excitability, sleep issues and seizures. In terms of speech and motor skills, the individuals performed at a developmental level of 2–6 years, which is appreciably better than a typical AS patient with a 15q11–13 deletion and somewhat better than the higher functioning patients with a UBE3A mutation. It is likely that this milder profile is caused by the presence of residual (nuclear) hUBE3A-Iso3 and (cytosolic) hUBE3A-Iso2 proteins.
In an elegant study published alongside our own, Chamberlain and co-workers successively deleted each of the three UBE3A isoforms from human iPS-derived neurons, in turn enabling the focused study of the remaining UBE3A proteins (26). Despite our different experimental approaches (i.e. deletion of endogenous isoforms versus exogenous overexpression of isoforms), our studies produced several convergent findings: first, Chamberlain and co-workers showed that hUBE3A-Iso1 is the predominant protein (84–88%), which matches very well with our calculation of 82%, based on mRNA quantification. Second, in agreement with our work and previous studies (8–11), the authors further found that UBE3A immunoreactivity is highly enriched in the nucleus of human neurons, and to a much lower extent, in neuronal processes. Finally, they show that nuclear UBE3A enrichment in undifferentiated hESC cells is lower compared to differentiated neurons, similar to what we previously found for immature mouse neurons (9). But notably, despite predominant UBE3A immunoreactivity in the nucleus of differentiated neurons, they found only 30% of UBE3A protein in the nuclear/mitochondrial pellet upon subcellular fractionation. Given our finding that the ‘cytosolic’ hUBE3A-Iso2 isoform constitutes only approximately 1% of the UBE3A transcripts, the biochemical and immunohistochemistry data appear to be discrepant, but maybe reconciled by a number of plausible explanations. First, possibly, the low mRNA transcript level of the cytosolic hUBE3A-Iso2 may not faithfully reflect the abundance of the protein; however, this does not appear to be the case, as the amount of hUBE3A-Iso2 protein in Chamberlain study correlates very well with the amount of hUBE3A-Iso2 mRNA as determined in our study. Second, as suggested by Chamberlain and co-workers, it could be that the volume of the neuropil is so much larger than the nucleus, that a cytosolic signal can be hardly observed by immunohistochemistry. Considering that the nucleus represents 10% of the total neuronal volume, and 30% of total UBE3A resides in the nucleus, it follows that the nuclear enrichment as assessed by immunohistochemistry would approximately be 3-fold. However, immunohistochemistry data from mature neurons—in our study and in all previously studies to date—indicates a much greater concentration of nuclear UBE3A (8–11). Moreover, following transfection with UBE3A isoform constructs, strong nuclear UBE3A enrichment manifested even in U2OS cells in which the cytoplasm to nucleus ratio is closer to 1:1 (Supplementary Material, Figure S1C). Third, overexpression of the hUBE3A-Iso1 and Iso3 (nuclear) isoforms could conceivably exaggerate their nuclear localization, but this seems unlikely as UBE3A is dependent on binding PSMD4 to translocate to the nucleus (9). Thus, we carefully conclude that biochemical fractionation may underestimate levels of UBE3A in the nucleus relative to other compartments. This notion is also supported by the finding of Chamberlain and co-workers of similarly low amounts of UBE3A protein in the nuclear fractions of (undifferentiated) ESCs and neurons, despite comparable cytosolic and nuclear volumes of the former. Hence, one would expect (at least) equivalent UBE3A protein levels in the nuclear fraction compared to the cytosolic fraction of ESCs. Thus, further studies are needed to address how much UBE3A is truly in the nucleus, and to address its role in this subcellular compartment.
Considering the very good match of the transcriptome data (this study) and proteome data (9,26), the expression of distinct human UBE3A isoforms appears to be entirely governed by alternative splicing. But unlike human UBE3A isoforms, our data suggest that mouse UBE3A protein isoforms arise to a large extent by alternative translation. Although the short (nuclear) mUBE3A-Iso3 constitutes almost 80% of the protein (9), the transcripts that uniquely encode mUBE3A-Iso3 represent less than 40% of total Ube3a transcripts. Hence, it is likely that the most frequently observed transcript, which possesses both the ‘MATACK’ and ‘MKRAAAK’ exons, generates both the short and long isoforms by alternative translation. Indeed, we observed that if a Ube3a cDNA containing both exons is placed behind an exogenous promoter, it efficiently generates both isoforms, and only upon mutating the ATG of the mUBE3A-Iso3 ‘MKRAAAK’ exon does it uniquely generate the longer mUBE3A-Iso2 ‘MATACK’ containing protein (9). Surprisingly, the Kozak sequence appears to be a bit less optimal for the ‘MKRAAAK’ exon compared to the one for the ‘MATACK’ exon, hence it is unclear why the ATG of ‘MKRAAAK’ is used so efficiently. Possibly, tertiary structure of the mRNA affects the accessibility of the different initiation codons, as well as the commitment of the ribosome to an initiation codon, after the ribosome has recognized it (27,28).
In conclusion, our current data, together with previous observations in mice (9,12), show that the conserved subcellular distribution of human and murine UBE3A is mediated by non-homologous isoforms. The finding that the two most abundant human UBE3A isoforms are both nuclear emphasize an important nuclear function of UBE3A in human, as was also shown in mice (9). However, the observation that the mutation that caused the hUBE3A-Iso3 to become nuclear occurred after the appearance of the cytosolic hUBE3A-Iso2, could suggest an evolutionary pressure to maintain a cytosolic pool of UBE3A.
Materials and Methods
Cell lines
Mouse hippocampal primary neurons used for localization studies were isolated from E16.5 male and female Ube3amE115X/p+ embryo brains [Ube3atm2Yelg (MGI: 5911277)] (29) in the FvB/NHanHsd background (designated as ‘AS’ mice). Cells were cultured on poly-d-lysine coated glass coverslips at 37°C in a 5% CO2 humidified incubator in Neurobasal™ medium (NBM; Thermoscientific, 2 110 304) supplemented with 1% penicillin and streptomycin, 1% Glutamax (Invitrogen) and 2% B27 (NBM+++).
For transfections, iPSCs derived from a Dutch AS patient (UBE3A maternal nonsense mutation W577*) were differentiated to neurons as previously described (30) with slight modifications. Namely, on day 15 cells were passaged 1:4 using collagenase or ACCUTASE™ (Stemcell technologies, #07920), passage 1, and further expanded in NPC medium. After passage 3, the NPC culture was purified using fluorescence-activated cell sorting (FACS), as described previously (31). NPCs were detached from the culture plate and resuspended into a single cell solution. CD184+/CD44−/CD271−/CD24+ cells were collected using a FACSaria III (BD bioscience) and expanded.
In order to visualize endogenous UBE3A expression in iPSC derived neurons, a commercial cell line (GM25256, Coriell Institute) was differentiated with the same protocol (30) and stained on week 8 of differentiation.
U2OS cells (ATCC HTB-96) were cultured in DMEM GlutaMAX (Thermo Scientific, 10569010) supplemented with 10% fetal calf serum and 1% penicillin and streptomycin.
Plasmid construction
The original sequence for hUBE3A-Iso1 was obtained from Addgene (Plasmid # 8657). This sequence was used for the construction of hUBE3A-Iso2 and hUBE3A-Iso3, by addition of the N-terminal extensions using PCR. Primers and plasmids used in this study are listed in Supplemental Material, Tables S2 and S3, respectively. The exchange of the N-terminal extensions between mUBE3A-Iso2 and hUBE3A-Iso3 was generated using site-directed mutagenesis via PCR amplification and using as template hUBE3A-Iso3 and mUBE3A-Iso2, respectively. UBE3A N-terminal variation constructs were generated taking advantage of the same approach and using as template hUBE3A-Iso3. Amplified fragments were originally cloned into the TOPO vector using a TOPO™ TA Cloning™ Kit (Life Technologies) and sequence verified. For all neuronal transfections UBE3A constructs were cloned into a dual promoter expression vector, expressing the gene of interest under a CAG promoter and tdTomato under a PGK promoter, ensuring independent expression of the reporter gene (16). UBE3A isoforms were cloned in the multiple cloning site of the vector downstream of the CAG promoter and followed by a transcription terminator sequence (Supplementary Material, Figure S1B). mUBE3A-Iso2 and mUBE3A-Iso3 constructs have been previously described (9).
Transfections
On DIV7, mouse hippocampal primary neurons were transferred in NBM supplemented with glutamine (500 μM), and their conditioned medium was temporarily stored at 37°C, 5% CO2. A total of 1.8 μg of the desired construct was complexed with Lipofectamine 2000 (Invitrogen, 11668-019) and added to the neurons. After 1 h of incubation at 37°C in 5% CO2, the medium was replaced by the previously stored conditioned medium. Neurons were fixed 3 days post-transfection.
iPSC-derived neural network cultures were transfected after 6 weeks of differentiation using lipofectamine 3000 (ThermoFisher, L3000001) according to manufacturer’s instructions. Cells were fixed 2 days post-transfection.
U2OS cells were seeded 24 h prior to transfection in non-coated coverslips in a 12-well plate (~150.000 cells per well). On the day of transfection, a total of 1.8 μg of the desired construct was mixed with Polyethylemine (PEI) in a ratio of 3:1 (v/v) and added to the cells. After a 6 h incubation the PEI containing medium was removed and replaced by fresh medium. Cells were fixed 2 days post-transfection.
Immunofluoresence
Primary hippocampal neurons were fixed by incubation with 0.5 mL of 4% paraformaldehyde (PFA)/4% sucrose at room temperature for 10 min. After three washes with PBS at room temperature, a primary antibody mix, containing anti-UBE3A (1:750, Sigma Aldrich, SAB1404508) and anti-MAP2 (1:500, Synaptic Systems, 188 004), dissolved in GDB solution (0.1% (w/v) gelatin, 0.3% (v/v) Triton X-100, 450 mM NaCl, 16 mM phosphate buffer (pH 7.4)) was added to the coverslips, which were then incubated overnight at 4°C, in a dark and moist chamber. Notably, the UBE3A antibody is a monoclonal antibody directed against amino acid residues 318-417 of UBE3A (hUBE3A-Iso1). This sequence is 100% conserved between all three isoforms. The use of this antibody has previously been optimized and validated in the lab for immunocytochemistry applications (Fig. 1). The next day, following three washes with PBS at room temperature, secondary antibody mix dissolved in GDB solution was added to the coverslips, consisting of anti-mouse Alexa 488 (1:200, Jackson Immunoresearch, 715-545-150) and anti-guinea pig Alexa 647 (1:200, Jackson Immunoresearch, 706-605-148), for 1 h at room temperature, in a dark and moist chamber. Subsequently, coverslips were washed three times with PBS, and DAPI (ThermoFisher Scientific, D1306) was used to visualize the nuclei. Ultimately the coverslips were washed three times with PBS and mounted on glass slides, using Mowiol 4–88 (Sigma-Aldrich, 81 381). The same procedure was followed when staining U2OS cells, but without including MAP2 antibody staining.
Human neurons were fixed using 4% formaldehyde in PBS and stained using immunocytochemistry. Primary antibody incubation was done overnight at 4°C, secondary antibody incubation was done for 2 h at room temperature, both in a staining buffer [0.05 M Tris, 0.9% NaCl, 0.25% gelatin, and 0.5% Triton X-100 (Sigma, T8787) in PBS (pH 7.4)]. Primary antibodies used were UBE3A (1:750) and MAP2 (1:500), secondary antibodies conjugated to Alexa-488 and Alexa-647 were used at a dilution of 1: 200. Nuclei were visualized using DAPI (ThermoFisher Scientific, D1306). Samples were embedded in Mowiol 4-88 (Sigma-Aldrich, 81 381). All antibodies used in this study are listed in the Reagents and Tools Table. All images were acquired with a LSM700 confocal microscope (Zeiss).
Basic local alignment search tool (BLAST) analysis
In order to identify all reported protein isoforms of UBE3A across evolution, a series of BLAST analyses were performed. Using the NCBI platform of National Institute of Health (32), and cross-referencing in the Ensembl platform (33), we performed a BLAST analysis against protein sequences (BLASTP) using as query the residues 1–16 of hUBE3A-Iso3 (Fig. 1A). The results of the BLASTP were arranged in an order corresponding to the evolutionary tree of mammals and ClustalOmega (34) and Jalview (35) were used to visualize the multiple sequence alignment, the degree of sequence conservation and the consensus sequence.
Using the DNA sequence of exon 4 of hUBE3A as query, we performed BLAST analyses against DNA sequences at the Ensembl platform with the search sensitivity set at ‘short sequences.’ The output sequences were manually inspected and all exon 4-like sequences were mapped on the genome of the respective organisms to confirm that the sequences were present in the UBE3A locus.
RNA-seq dataset and transcripts analysis
For the analysis of human UBE3A transcripts in brain cortex, we utilized an RNA-seq dataset of cortical tissue specimens (17). The raw sequencing data were imported to the Galaxy web platform for mapping the reads to the genome (36). CutAdapt (Galaxy Version 11.16.5) was used to trim reads of low quality (threshold set at 20) and consecutively filter out reads with a read length lower than 75 nucleotides. Read quality was assessed by FastQC (Galaxy Version 0.72 + galaxy1). Using the STAR algorithm (37) (Galaxy Version 2.7.2b), within the GALAXY platform, paired-end reads were aligned to the human reference genome (GRCh38). Transcript assembly was guided by the reference annotation file Gencode v34 (38). The mapped dataset was viewed in the Integrative Genomic Viewer IGV 2.3 (39) and an exon junction analysis was performed, extracting the respective Sashimi curves. We counted, for each exon, the total number of junctions in the whole dataset. Considering that exon 6 (Fig. 4A) should be present in all protein-coding UBE3A transcripts, we calculated the junctions of each upstream exon with exon 6. Of these, we summed all exon junction counts that would encode for each individual isoform and normalized this against the total number of exon junctions between exon 6 and upstream exons, in order to calculate the percentage in which each isoform is represented. Considering all junctions between exons upstream of exon 6, we list all possible combinations of alternative exon splicing that are possible to occur according to the Sashimi analysis. Using the same method, we quantified the relative amounts of UBE3A isoforms in an RNA-seq dataset of human hES-derived NPCs, after 6 weeks of differentiation, as previously described (19).
For the quantification of mUBE3A-Iso2 and mUBE3A-Iso3, an RNA-seq dataset of four mouse hippocampi, of mice previously described (40), was utilized. The samples were processed as described above. CutAdapt (Galaxy Version 11.16.5) was used to trim reads of low quality (threshold set at 20) and consecutively filter out reads with a read length lower than 75 nucleotides. Using the STAR algorithm (37) (Galaxy Version 2.7.2b), within the GALAXY platform, paired-end reads were aligned to the mouse reference genome (M24). Subsequently, the exon junctions and transcript percentages were quantified in the same way as with the human datasets.
Data Availability
All human RNA-seq datasets analyzed during this study are included in the following published articles (and their supplementary information files):
Human cortex RNA-seq data: DOI: 10.1038/s41598-017-06145-8
Human embryonic cell derived neurons RNA-seq data: DOI: 10.1186/s13073-016-0347-3
The mouse RNA-seq data analyzed during this study have not been deposited in a public repository, due to their inclusion in an unpublished dataset, but are available from the lead contact upon reasonable request.
Supplementary Material
Acknowledgements
The authors are grateful to the following individuals and groups: M. Elgersma, C. de Konink and M. Aghadavoud Jolfaei for colony management and genotyping; I. Wallaard for setting up mouse primary neuron cultures; S.N.V. Bossuyt for providing the HA-tagged constructs; B. Planterose Jimenez for providing insight on the evolutionary analysis; the Erasmus MC iPS Core Facility for reprogramming and characterization of human iPSC lines; and M. Elgersma for reviewing of the manuscript.
Conflict of Interest statement. The authors declare that they have not a conflict of interest.
Contributor Information
F Isabella Zampeta, Department of Neuroscience, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; ENCORE Expertise Center for Neurodevelopmental Disorders, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Monica Sonzogni, Department of Neuroscience, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; ENCORE Expertise Center for Neurodevelopmental Disorders, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Eva Niggl, Department of Neuroscience, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; ENCORE Expertise Center for Neurodevelopmental Disorders, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Bas Lendemeijer, Department of Psychiatry, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Hilde Smeenk, Department of Psychiatry, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Femke M S de Vrij, Department of Psychiatry, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Steven A Kushner, ENCORE Expertise Center for Neurodevelopmental Disorders, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; Department of Psychiatry, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Ben Distel, Department of Neuroscience, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; ENCORE Expertise Center for Neurodevelopmental Disorders, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; Department of Medical Biochemistry, Amsterdam UMC, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands.
Ype Elgersma, Department of Neuroscience, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands; ENCORE Expertise Center for Neurodevelopmental Disorders, Erasmus University Medical Center, 3015 GD Rotterdam, The Netherlands.
Funding
This work was supported by a ZonMw TOP grant (40-00812-98-16045 to S.A.K., B.D., Y.E.) and the Netherlands Organ-on-Chip Initiative (NOCI), an NWO Gravitation project funded by the Ministry of Education, Culture and Science of the government of the Netherlands (024.003.001; to S.A.K., Y.E.). S.A.K., F.M.S.de V, B.L. and H.S. are funded by Hersenstichting Nederland TNS2016-42, Horizon 2020/NWO NEURON-JTC2018-024, Horizon 2020/NWO ERA-PerMed2018-127 grants. M.S. was supported by grants from Associazione Angelman and FROM.
Author Contributions
M.S. generated UBE3A constructs. I.F.Z. cultured the mouse neurons and U2OS cells, performed confocal imaging experiments. I.F.Z. and M.S. performed BLAST analysis. B.L. and H.S. performed iPSC-derived neurons culturing and immunofluorescence experiments. I.F.Z. and E.N. performed analysis of RNA-seq data. Y.E., B.D., S.A.K. and F.M.S.de V. supervised the research. Y.E., B.D. and I.F.Z. wrote the first draft of the manuscript. All authors reviewed the manuscript.
References
- 1. Buiting K., Williams C. and Horsthemke B. (2016) Angelman syndrome-insights into a rare neurogenetic disorder. Nat. Rev. Neurol., 12, 584–593. [DOI] [PubMed] [Google Scholar]
- 2. LaSalle J.M., Reiter L.T. and Chamberlain S.J. (2015) Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics, 7, 1213–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elgersma Y. (2015) Neurodevelopmental disease: a molecular tightrope. Nature, 526, 50–51. [DOI] [PubMed] [Google Scholar]
- 4. Matsuura T., Sutcliffe J.S., Fang P., Galjaard R.J., Jiang Y.H., Benton C.S., Rommens J.M. and Beaudet A.L. (1997) De novo truncating mutations in E6-Ap ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet., 15, 74–77. [DOI] [PubMed] [Google Scholar]
- 5. Kishino T., Lalande M. and Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet., 15, 70–73. [DOI] [PubMed] [Google Scholar]
- 6. Nurmi E.L., Bradford Y., Chen Y.H., Hall J., Arnone B., Gardiner M.B., Hutcheson H.B., Gilbert J.R., Pericak-Vance M.A., Copeland-Yates S.A. et al. (2001) Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics, 77, 105–113. [DOI] [PubMed] [Google Scholar]
- 7. Smith S.E.P., Zhou Y.D., Zhang G., Jin Z., Stoppel D.C. and Anderson M.P. (2011) Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med., 3, 103ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burette A.C., Judson M.C., Burette S., Phend K.D., Philpot B.D. and Weinberg R.J. (2017) Subcellular organization of UBE3A in neurons. J. Comp. Neurol., 525, 233–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avagliano Trezza R., Sonzogni M., Bossuyt S.N.V., Zampeta F.I., Punt A.M., Berg M., Rotaru D.C., Koene L., Munshi S.T., Stedehouder J. et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci., 22, 1235–1247. [DOI] [PubMed] [Google Scholar]
- 10. Dindot S.V., Antalffy B.A., Bhattacharjee M.B. and Beaudet A.L. (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet., 17, 111–118. [DOI] [PubMed] [Google Scholar]
- 11. Burette A.C., Judson M.C., Li A.N., Chang E.F., Seeley W.W., Philpot B.D. and Weinberg R.J. (2018) Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism., 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miao S., Chen R., Ye J., Tan G.H., Li S., Zhang J., Jiang Y.H. and Xiong Z.Q. (2013) The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci., 33, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rotaru D.C., Woerden G.M., Wallaard I. and Elgersma Y. (2018) Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of Angelman syndrome. J. Neurosci., 38, 8011–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto Y., Huibregtse J.M. and Howley P.M. (1997) The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics, 41, 263–266. [DOI] [PubMed] [Google Scholar]
- 15. Sadhwani A., Sanjana N.E., Willen J.M., Calculator S.N., Black E.D., Bean L., Li H. and Tan W.H. (2018) Two Angelman families with unusually advanced neurodevelopment carry a start codon variant in the most highly expressed UBE3A isoform. Am. J. Med. Genet. Part A, 176, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reijnders M.R.F., Kousi M., Van Woerden G.M., Klein M., Bralten J., Mancini G., Essen T., Proietti-Onori M., Smeets E., Gastel M. et al. (2017) Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun., 8, 1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mills J.D., Iyer A.M., Van Scheppingen J., Bongaarts A., Anink J.J., Janssen B., Zimmer T.S., Spliet W.G., Rijen P.C., Jansen F.E. et al. (2017) Coding and small non-coding transcriptional landscape of tuberous sclerosis complex cortical tubers: implications for pathophysiology and treatment. Sci. Rep., 7, 8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kozak M. (2005) Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene, 361, 13–37. [DOI] [PubMed] [Google Scholar]
- 19. Grabole N., Zhang J.D., Aigner S., Ruderisch N., Costa V., Weber F.C., Theron M., Berntenis N., Spleiss O., Ebeling M. et al. (2016) Genomic analysis of the molecular neuropathology of tuberous sclerosis using a human stem cell model. Genome Med., 8, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greer P.L., Hanayama R., Bloodgood B.L., Mardinly A.R., Lipton D.M., Flavell S.W., Kim T.K., Griffith E.C., Waldon Z., Maehr R. et al. (2010) The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell, 140, 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valluy J., Bicker S., Aksoy-Aksel A., Lackinger M., Sumer S., Fiore R., Wüst T., Seffer D., Metge F., Dieterich C. et al. (2015) A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci., 18, 666–673. [DOI] [PubMed] [Google Scholar]
- 22. Buel G.R., Chen X., Chari R., O'Neill M.J., Ebelle D.L., Jenkins C., Sridharan V., Tarasov S.G., Tarasova N.I., Andresson T. and Walters K.J. (2020) Structure of E3 ligase E6AP with a proteasome-binding site provided by substrate receptor hRpn10. Nat. Commun., 11, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yi J.J., Berrios J., Newbern J.M., Snider W.D., Philpot B.D., Hahn K.M., Zylka M.J. et al. (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell, 162, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yi J.J., Paranjape S.R., Walker M.P., Choudhury R., Wolter J.M., Fragola G., Emanuele M.J., Major M.B. and Zylka M.J. (2017) The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J. Biol. Chem., 292, 12503–12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu X., Li C., Gao X., Xia K., Guo H., Li Y., Hao Z., Zhang L., Gao D., Xu C. et al. (2017) Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res., 28, 48–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sirois C.L., Bloom J.E., Fink J.J., Gorka D., Keller S., Germain N.D., Levine E.S. and Chamberlain S.J. (2020) Abundance and localization of human UBE3A protein isoforms. Hum. Mol. Genet.. 10.1093/hmg/ddaa191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jackson R.J., Hellen C.U.T. and Pestova T.V. (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol., 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodnina M.V. (2016) The ribosome in action: tuning of translational efficiency and protein folding. Protein Sci., 25, 1390–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang T., Woerden G.M., Elgersma Y. and Borst J. (2018) Enhanced transmission at the calyx of held synapse in a mouse model for Angelman syndrome. Front. Cell. Neurosci., 11, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gunhanlar N., Shpak G., Kroeg M., Gouty-Colomer L.A., Munshi S.T., Lendemeijer B., Ghazvini M., Dupont C., Hoogendijk W., Gribnau J. et al. (2017) A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol. Psychiatry, 23, 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan S.H., Martin J., Elia J., Flippin J., Paramban R.I., Hefferan M.P., Vidal J.G., Mu Y., Killian R.L., Israel M.A. et al. (2011) Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One, 6, e17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. States D.J. and Gish W. (1994) Combined use of sequence similarity and codon bias for coding region identification. J. Comput. Biol., 1, 39–50. [DOI] [PubMed] [Google Scholar]
- 33. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S. et al. (2018) Ensembl 2019. Nucleic Acids Res., 47, 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A., Potter S.C., Finn R.D. and Lopez R. (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res, 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Waterhouse A.M., Procter J.B., Martin D.M., Clamp M. and Barton G.J. (2009) Sequence analysis Jalview version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics, 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Afgan E., Baker D., Batut B., Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A. et al. (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res., 46, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T.R. (2013) Sequence analysis STAR: ultrafast universal RNA-seq aligner. 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. et al. (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res., 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson J.T., Thorvaldsdóttir H., Wenger A.M., Zehir A. and Mesirov J.P. (2017) Variant review with the integrative genomics viewer. Cancer Res., 77, e31–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koene L., Grondelle S.E., Proietti Onori M., Wallaard I., Kooijman N., Oort A., Schreiber J. and Elgersma Y. (2019) Effects of antiepileptic drugs in a new TSC/mTOR-dependent epilepsy mouse model. Ann. Clin. Transl. Neurol., 6, 1273–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All human RNA-seq datasets analyzed during this study are included in the following published articles (and their supplementary information files):
Human cortex RNA-seq data: DOI: 10.1038/s41598-017-06145-8
Human embryonic cell derived neurons RNA-seq data: DOI: 10.1186/s13073-016-0347-3
The mouse RNA-seq data analyzed during this study have not been deposited in a public repository, due to their inclusion in an unpublished dataset, but are available from the lead contact upon reasonable request.