

Abstract
Streptococcus mutans, a significant contributor to dental caries, is occasionally isolated from the blood of patients with infective endocarditis. We previously showed that S. mutans strains expressing collagen-binding protein (Cnm) are present in the oral cavity of approximately 10–20% of humans and that they can effectively invade human umbilical vein endothelial cells (HUVECs). Here, we investigated the potential molecular mechanisms of HUVEC invasion by Cnm-positive S. mutans. The ability of Cnm-positive S. mutans to invade HUVECs was significantly increased by the presence of serum, purified type IV collagen, and fibrinogen (p < 0.001). Microarray analyses of HUVECs infected by Cnm-positive or -negative S. mutans strains identified several transcripts that were differentially upregulated during invasion, including those encoding the small G protein regulatory proteins ARHGEF38 and ARHGAP9. Upregulation of these proteins occurred during invasion only in the presence of serum. Knockdown of ARHGEF38 strongly reduced HUVEC invasion by Cnm-positive S. mutans. In a rat model of infective endocarditis, cardiac endothelial cell damage was more prominent following infection with a Cnm-positive strain compared with a Cnm-negative strain. These results suggest that the type IV collagen–Cnm–ARHGEF38 pathway may play a crucial role in the pathogenesis of infective endocarditis.
Subject terms: Bacterial pathogenesis, Bacteriology
Introduction
Oral bacteria affect the pathogenesis of cardiovascular diseases1, including the life-threatening disease infective endocarditis (IE)2. Among oral streptococci, mitis group streptococci such as Streptococcus sanguinis and Streptococcus mitis are recognized as the major causative microorganisms of IE3,4. These bacteria are thought to invade the bloodstream following invasive dental treatments, such as tooth extraction, endodontic treatment, and periodontal surgery5. The bacteria subsequently adhere to abnormal heart valves to initiate IE6. If the bacteria are capable of invading vascular endothelial cells, the pathology of IE worsens considerably because of the cellular damage7,8.
Although Streptococcus mutans present in the oral cavity is a major causative pathogen of dental caries, it is rarely associated with the development of IE9. Collagen-binding protein (Cnm) has been characterized as a novel LPXTG-anchored protein of S. mutans10. Cnm-positive S. mutans strains are detected in the oral cavity of 10–20% of healthy subjects11. In a rat IE model, large bacterial masses were detected on heart valve tissue following infection with Cnm-positive S. mutans12. Interestingly, Cnm-positive S. mutans strains are also associated with the deterioration of intracerebral hemorrhages, which are a major complication of IE13.
Activation and inactivation of small G proteins involves the cytoskeletal rearrangement of human cells14. Various bacterial species, such as members of the genera Staphylococcus, Salmonella, Shigella, and Yersinia, invade human cells via cytoskeletal rearrangement by expressing Rho guanine nucleotide exchange factors (ARHGEFs, also known as RhoGEF), which are involved in small G protein activation, and Rho GTPase-activating proteins (ARHGAPs, also known as RhoGAP), which are involved in small G protein inactivation15–19. Although Cnm-positive S. mutans strains are capable of effectively invading human venous endothelial cells20, whether this occurs in an analogous manner via collagen–Cnm interactions is unknown.
In the present study, we investigated this question by analyzing the involvement of Cnm and major serum extracellular matrix (ECM) proteins in the invasion of human umbilical vein endothelial cells (HUVECs) by S. mutans strains, exploring genes that are differentially regulated during Cnm-dependent invasion of HUVECs, and employing a rat IE model to verify the pathophysiological relevance of our findings in vivo. The results support a potentially important role for Cnm–collagen IV interactions and the regulation of small G proteins in the development of IE following S. mutans infection.
Results
Invasion of HUVECs by Cnm-positive S. mutans is dependent on a serum component(s)
We first examined the ability of Cnm-positive and Cnm-negative S. mutans strains to invade HUVECs in the presence or absence of serum by using a HUVEC invasion assay. MT8148, a Cnm-negative clinical strain21, was unable to invade the cells regardless of the presence of serum (Fig. 1A). In contrast, TW295, a Cnm-positive S. mutans strain isolated from a subject with bacteremia after tooth extraction22, displayed strong invasion of HUVECs in the presence of serum and significantly weaker invasion in the absence of serum (p < 0.001). Notably, invasion was not observed following the incubation of HUVECs with TW295CND, a Cnm-defective isogenic mutant strain23, in either the presence or absence of serum (Fig. 1A), while TW295comp, a Cnm-complemented mutant strain12, displayed recovered cell invasion ability in the presence of serum (Fig. 1A). Thus, S. mutans invades HUVECs in a serum- and Cnm-dependent manner. Confocal laser scanning microscopy observations revealed that a Cnm-specific antibody specifically stained the Cnm-positive S. mutans strains (Fig. 1B) and confirmed that TW295 exhibited high levels of invasion only in the presence of serum (Fig. 1C). Additional experiments using an anti-S. mutans antibody that reacted with MT8148 confirmed that the Cnm-negative S. mutans strain MT8148 did not invade vascular endothelial cells (Supplementary Fig. 1A,B).
Figure 1.

Invasion of HUVECs by S. mutans strains in the presence or absence of serum. (A) Invasion ratios of HUVECs after their incubation with the indicated S. mutans strains for 2 h at a multiplicity of infection of 100. Data are presented as the means ± SD of four technical replicates. ***p < 0.001 by ANOVA followed by Bonferroni’s post hoc test. (B) Representative confocal laser scanning microscopy images of S. mutans. Bacteria cells were stained red by using Alexa Fluor 555-conjugated anti-Cnm antibody. Scale bar, 10 µm. (C) Representative confocal laser scanning microscopy images of S. mutans TW295 invading HUVECs. Nuclei are stained blue (DAPI), bacteria cells are stained red (Alexa Fluor 555-conjugated anti-Cnm antibody), and actin filaments are stained green (Alexa Fluor 448-labelled phalloidin). Scale bar, 10 µm. All confocal laser scanning microscope images were taken using LSM510 (Carl Zeiss, Oberkochem, Germany).
Cnm-positive S. mutans binds to collagen and fibrinogen
We next sought to determine which major serum ECM component(s) was crucial for S. mutans invasion of HUVECs by examining bacterial adhesion to purified collagen, fibrinogen, fibronectin, and vitronectin in vitro. While MT8148 did not adhere to any of the tested ECM proteins, TW295 bound to both type IV collagen and fibrinogen, with type IV collagen being the preferred substrate (p < 0.001, Fig. 2A). This binding activity was dependent on Cnm expression, as illustrated by the lack of binding to either protein by TW295CND and the recovered binding demonstrated by TW295comp. We also evaluated S. mutans binding to type IV collagen and fibrinogen by confocal laser scanning microscopy of hexidium iodide-stained bacteria. This analysis confirmed the adhesion assay results, revealing that TW295 and TW295comp both formed thicker biofilms on type IV collagen than on fibrinogen, whereas no binding was observed by MT8148 or TW295CND (Fig. 2B,C).
Figure 2.

ECM binding of S. mutans strains. (A) Binding of the indicated S. mutans strains to type IV collagen, fibrinogen, fibronectin, or vitronectin. Data are presented as the means ± SD of three technical replicates. ***p < 0.001 by ANOVA followed by Bonferroni’s post hoc test. (B, C) Confocal laser scanning microscopy images (upper panels) and schematics (lower panels) of S. mutans binding to type IV collagen (B) or fibrinogen (C). Color coding of the biofilm thickness: 0–50 µm, blue; 50–100 µm, light blue; 100–150 µm, green; 150–200 µm, yellow; and 200–250 µm, red. All confocal laser scanning microscope images and schematics were made using LSM510.
Type IV collagen promotes invasion of HUVECs by S. mutans
To determine whether the ECM components are necessary for HUVEC invasion by S. mutans, we examined cell invasion by TW295 in serum-free medium individually supplemented with one of four ECM components at concentrations corresponding to those detected in healthy human blood [type IV collagen (140 ng/ml), fibrinogen (2 mg/ml), fibronectin (0.2 mg/ml), and vitronectin (500 µg/ml)24–27]. As expected, the level of HUVEC invasion by TW295 was higher in the presence of type IV collagen as compared with in the presence of the other tested proteins (Fig. 3A). When type IV collagen was added 2 h before bacterial infection, cell invasion by TW295 was inhibited, and the invasion ratio was significantly lower than that when type IV collagen and TW295 were added simultaneously (p < 0.01) (Fig. 3B). Additionally, the degree of invasion was positively associated with the type IV collagen concentration (Fig. 3C). Cnm expression was required for HUVEC invasion, as illustrated by the inability of S. mutans strains MT8148 and TW295CND to invade HUVECs, even in the presence of high type IV collagen concentrations. Confocal laser scanning microscopy of fluorescently labeled HUVECs and TW295 confirmed the presence of internalized bacteria in endothelial cells (Fig. 3D).
Figure 3.

Invasion of HUVECs by S. mutans in the presence of purified ECM proteins. (A) Invasion ratios of HUVECs after their incubation with S. mutans strain TW295 for 2 h at a multiplicity of infection of 100 in the presence of type IV collagen (140 ng/ml), fibrinogen (2 mg/ml), fibronectin (0.2 mg/ml), vitronectin (500 µg/ml), or bovine serum albumin (1 mg/ml). Data are presented as the means ± SD of three technical replicates. *p < 0.05, **p < 0.01 versus TW295 and #p < 0.05, ##p < 0.01 versus the protein-negative control, using ANOVA followed by Bonferroni’s post hoc test. (B) Ability of TW295 to invade HUVECs when type IV collagen and bacteria were added simultaneously or when type IV collagen was added 2 h before the bacterial infection. Data are presented as the means ± SD of three technical replicates. **p < 0.01 using a Student’s t-test. (C) HUVEC invasion ratio (as described for A) of the indicated strains in the presence of various concentrations of type IV collagen. Data are presented as the means ± SD of four technical replicates. ***p < 0.001 versus no collagen using ANOVA followed by Bonferroni’s post hoc test. (D) Representative confocal laser scanning microscopy images of S. mutans TW295 invading HUVECs. Bacteria are stained red (Alexa Fluor 555-conjugated anti-Cnm antibody), type IV collagen is stained green (Alexa Fluor 488), and nuclei are stained blue (DAPI). Arrowheads indicate areas were type IV collagen and bacteria colocalize. Scale bar, 10 µm. All confocal laser scanning microscope images were taken using LSM510.
Identification of differentially expressed genes upon Cnm-dependent invasion of HUVECs by S. mutans
To identify key regulators of Cnm-dependent S. mutans internalization, we performed a comparative DNA microarray analysis on three pairs of HUVECs: TW295CND-infected vs uninfected cells (to identify genes related to S. mutans invasion independently of Cnm); TW295-infected vs uninfected cells (to identify genes related to Cnm-positive S. mutans invasion); and TW295-infected vs TW295CND-infected cells (to identify genes specifically related to the involvement of Cnm in HUVEC invasion). Lists of genes with altered expression between each of these groups are shown in Supplementary Tables 1, 2 and 3. Among these variable genes, a total of 82 genes were significantly upregulated in HUVECs infected by TW295 in a Cnm-dependent manner (Fig. 4A, Supplementary Table 4). As expected, none of the 82 genes was overexpressed in TW295CND-infected HUVEC as compared with uninfected cells.
Figure 4.
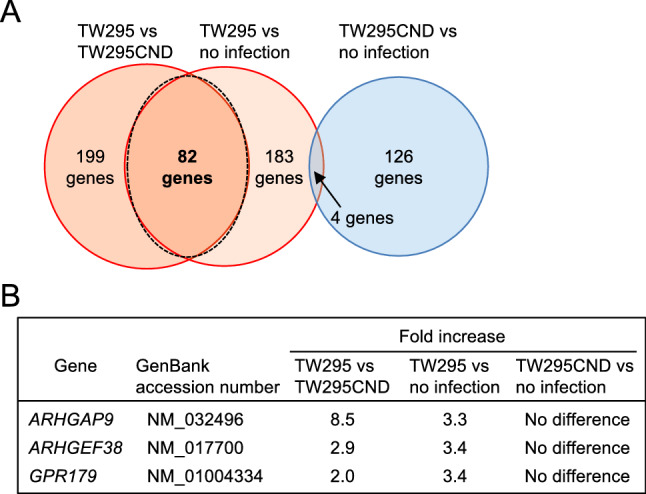
DNA microarray analysis of HUVEC genes involved in invasion by S. mutans. (A) Venn diagram showing the selection of genes by their differential expression (> 2.0-fold difference) in the indicated infected cells. A total of 82 genes were significantly upregulated both in TW295-infected HUVECs compared with TW295CND-infected HUVECs and in TW295-infected HUVECs compared with uninfected HUVECs but were not differentially expressed in TW295CND-infected HUVECs compared with uninfected HUVECs. (B) Three of the 82 differentially expressed genes were identified as regulators of small G proteins. Microarray data was analyzed using Agilent Feature Extraction (Agilent Technologies, Santa Clara, CA, USA).
Various bacterial species invade human cells via cytoskeletal rearrangement by expressing ARHGEFs, which are involved in small G protein activation, and ARHGAPs, which are involved in small G protein inactivation15–19. To determine if genes related to the activation or inactivation of small G proteins might play a role here, we examined the list of differentially expressed genes for ARHGEFs and ARHGAPs, and we identified three: Rho GTPase-activating protein 9 (ARHGAP9), Rho guanine nucleotide exchange factor 38 (ARHGEF38), and G protein-coupled receptor 179 (GPR179) (Fig. 4B).
Transcripts of small G protein regulators are involved in HUVEC invasion by S. mutans invasion
To confirm the involvement of the three small G protein regulatory genes in the internalization of Cnm-positive S. mutans into HUVECs, we knocked down the expression of each gene in HUVECs via transfecting the cells with specific small interfering RNA (siRNA). Effective silencing of ARHGAP9, ARHGEF38, and GPR179 expression was confirmed by RT-PCR analyses of the transfected cells (Fig. 5A; full-length is shown in Supplementary Fig. 2). As shown in Fig. 5B (ARHGEF38) and Supplementary Fig. 3 (ARHGAP9 and GPR179), all three genes were strongly upregulated during TW295 infection of HUVECs in the presence of serum, confirming their potential involvement in invasion. However, only the ARHGEF38 knockdown resulted in a significant inhibition of HUVEC invasion by TW295 S. mutans (p < 0.01, Fig. 5C). Interestingly, the ARHGAP9 knockdown in HUVECs significantly increased the invasion ratio as compared with cells transfected with the negative control siRNA (p < 0.05), whereas the GPR179 knockdown had no significant effect on invasion (Fig. 5C).
Figure 5.
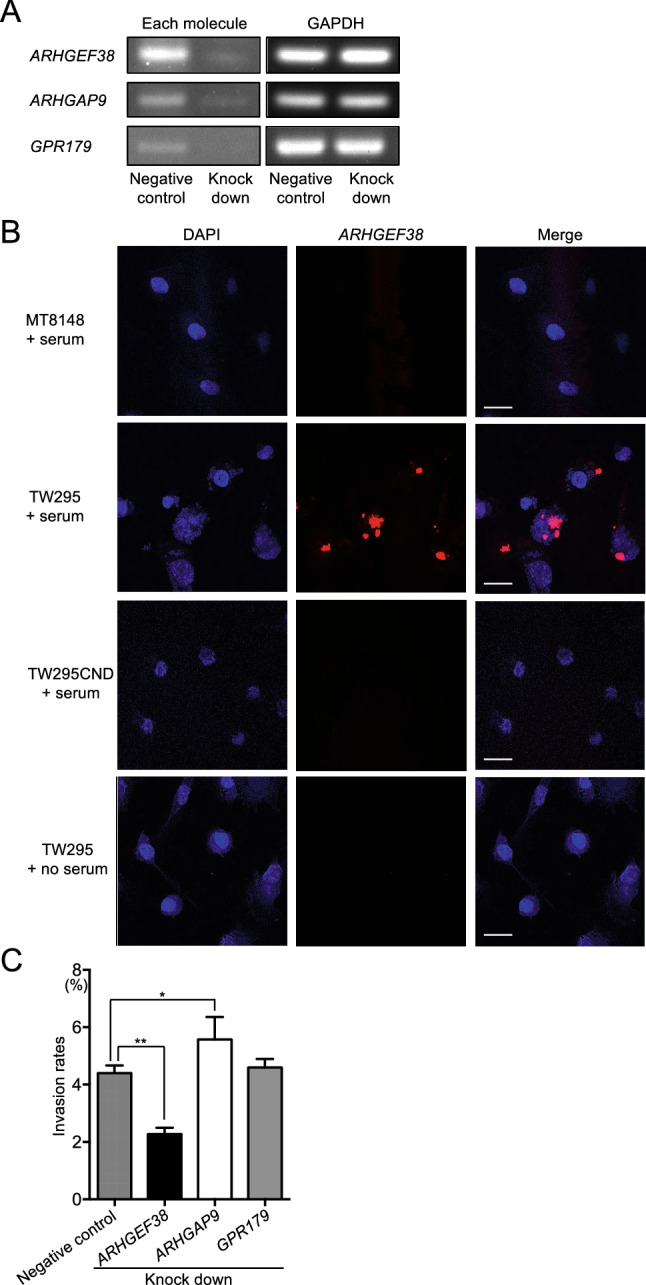
Effect of silencing genes involved in HUVEC invasion by S. mutans. (A) RT-PCR analysis of HUVECs transfected with siRNAs targeting ARHGAP9, ARHGEF38, or GPR179. GAPDH mRNA expression was analyzed as an internal control. (B) Representative confocal laser scanning microscopy images of HUVECs invaded by S. mutans in the presence or absence of serum. Nuclei are stained blue (DAPI) and each transcript (ARHGAP9, ARHGEF38, and GPR179) is stained red (Alexa Fluor 555). Arrowheads indicate the expression of each transcript. (C) HUVECs were transfected with ARHGEF38, ARHGAP9, GPR179, or control siRNAs. Invasion ratios of these HUVECs after their incubation with S. mutans strain TW295 for 2 h at a multiplicity of infection of 100. Data are presented as the means ± SD of four technical replicates. *p < 0.05, **p < 0.01 using ANOVA followed by Bonferroni’s post hoc test. Scale bar, 10 µm. All confocal laser scanning microscope images were taken using LSM510.
Evaluations of vascular endothelial cell damage and histopathological findings in a rat IE model
Finally, we verified our findings in vivo by evaluating the effects of S. mutans infection on vascular endothelial cells using a rat IE model. An aortic valve injury was induced in rats by using a sterile polyethylene catheter, which was inserted through the right carotid artery and then immediately removed after causing the heart valve injury. The rats were then infected with one strain of S. mutans via the jugular vein. All rats survived for 7 days after the bacterial infection, after which they were euthanized, and their hearts were extirpated. To visualize healthy (CD31-positive) and damaged (CD31-negative) endothelial cells, sections of the rat hearts were stained for CD31 expression by using immunohistochemistry (IHC). The lengths of the damaged and healthy endocardium were then measured and compared using imaging software. Representative IHC images displaying the total lengths of the endocardium are shown in Fig. 6A. No significant differences were detected in the total endocardium length between hearts from rats infected with MT8148, TW295, or TW295CND (Fig. 6B). Representative images of vascular endothelial cell damage in the heart sections are shown in Fig. 6C–F. An evaluation of the proportions of undamaged and damaged endothelial cells between rat groups revealed a significantly greater percentage of damaged endocardium in the hearts of rats infected with TW295 compared with those infected with TW295CND (p < 0.01, Fig. 6F). Collectively, these data underscore the potential pathological relevance of Cnm-dependent invasion of endothelial cells by S. mutans in the development of IE.
Figure 6.

Evaluation of vascular endothelial cell damage in a rat IE model. (A) Representative IHC images of anti-CD31-stained sections of extirpated heart valves from rats infected with S. mutans strain TW295. Black lines indicate the total length of the endocardium. (B) Total length of the endocardium of extirpated hearts from rats infected with the indicated S. mutans strain. Data are presented as the mean ± SD of six biological replicates per strain. (C) Magnified images of the box outlined in (A). Right panel shows endothelial cell damage. Blue and red lines indicate non-damaged (CD31-positive) and damaged (CD31-negative) endocardium, respectively. (D) Representative magnified images of normal (CD31-positive; arrowheads in left panel) and deficient (CD31-negative; arrowheads in right panel) endocardium. (E) Representative images of vascular endothelial cell damage in hearts from rats infected with the indicated S. mutans strains. Blue and red lines indicate non-damaged (CD31-positive) and damaged (CD31-negative) endocardium, respectively. (F) Rates of damaged endocardium following infection by each S. mutans strain. Data are presented as the mean ± SD of six biological replicates per strain. *p < 0.05, **p < 0.01 using ANOVA followed by Bonferroni’s post hoc test. The lengths of total, CD31-positive, and CD31-negative endocardium in the heart valves were measured using WinROOF Vol 5.0 software (Mitsuya Co., Ltd., Tokyo, Japan).
In addition to evaluating the vascular endothelial cell damage, we assessed the histopathology by performing hematoxylin–eosin staining of tissue sections (Supplementary Fig. 4), then scoring them with the following scale: 0 (none), 1 (mild), 2 (moderate), and 3 (severe). Among the six evaluation points by which the histopathological findings were assessed, five in the TW295-infected group and two in the TW295comp-infected group showed significantly higher values compared with those for the non-infected group (p < 0.05) (Table 1). In contrast, only one evaluation point had a significantly higher score for the MT8148 strain-infected group as compared with the non-infected group (p < 0.05), and none of the evaluation point scores were significantly different between the TW295CND-infected group and non-infected group.
Table 1.
Histopathological evaluation of extirpated heart tissues from IE model rats.
| Histopathological finding | MT8148 (n = 6) | TW295 (n = 6) | TW295CND (n = 6) | TW295comp (n = 6) | No infection (n = 6) |
|---|---|---|---|---|---|
| Infiltration of inflammatory cells | 1.17 ± 0.28** | 1.83 ± 0.40** | 0.17 ± 0.17# | 2.17 ± 0.40** | 0.17 ± 0.17## |
| Hypertrophy of the endocardium | 0.75 ± 0.11## | 1.83 ± 0.31* | 0.33 ± 0.21# | 0.50 ± 0.34## | 0.33 ± 0.33# |
| Hypertrophy of the annulus | 1.08 ± 0.42 | 1.33 ± 0.49 | 0.00 ± 0.00# | 0.17 ± 0.17 | 0.33 ± 0.33 |
| Acceleration of fibrosis | 1.25 ± 0.34 | 1.50 ± 0.41* | 0.00 ± 0.00# | 1.67 ± 0.33** | 0.17 ± 0.17# |
| Fibrin-like deposition | 1.00 ± 0.34 | 1.58 ± 0.27* | 0.08 ± 0.08## | 1.00 ± 0.52 | 0.33 ± 0.33# |
| Bacterial mass | 0.08 ± 0.08# | 1.33 ± 0.42* | 0.00 ± 0.00# | 0.17 ± 0.17# | 0.00 ± 0.00# |
Values in each column indicate mean ± standard error. There were significant differences between no infection (*P < 0.05, **P < 0.01) and TW295 groups (#P < 0.05, ##P < 0.01).
Discussion
The invasion of S. mutans into vascular endothelial cells is considered an exacerbating factor in the development of IE28. Although we previously showed that Cnm-positive, but not Cnm-negative, S. mutans strains exhibited high levels of invasion into vascular endothelial cells20, the invasion mechanism was unknown. Because Cnm-positive S. mutans form aggregates when mixed with serum12, we suspected that a serum component may be crucial to the internalization of Cnm-positive S. mutans strains into vascular endothelial cells.
Bacterial adhesion to ECM proteins is important for host cell invasion29. Fibronectin-binding proteins are the major adhesins involved in the invasion of vascular endothelial cells by S. aureus, a major pathogen of acute IE30. In the present study, however, we found that type IV collagen was the major serum ECM protein involved in HUVEC invasion by Cnm-positive S. mutans. Thus, Cnm-positive S. mutans and fibronectin-binding proteins-positive S. aureus use different ECM proteins to invade vascular endothelial cells.
Most streptococcal species are isolated from the oral cavity or nasopharynx31, and adherence is a major virulence factor for streptococcal species. Collagen-binding proteins have been reported to be present in some streptococcal species, including Streptococcus pyogenes, Streptococcus pneumoniae, Streptococcus equi, and Streptococcus gallolyticus31–34. S. mutans Cnm has no homology with the collagen-binding proteins of S. pyogenes and S. pneumoniae, which are commensal bacteria in humans that are sometimes associated with severe systemic diseases. In contrast, S. mutans Cnm has 40–50% homology with the collagen-binding proteins of S. equi and S. gallolyticus10,35, which are infrequently detected in humans. S. gallolyticus, which is rarely involved in the onset of IE, can adhere to and invade vascular endothelial cells36; however, the mechanism by which these bacteria invade into vascular endothelial cells has not been elucidated. The bacterial species that encode a collagen-binding protein with homology to S. mutans Cnm, including S. gallolyticus, may have an invasion mechanism similar to that of Cnm-positive S. mutans.
In our previous study, Cnm was found to be an important factor for the invasion of S. mutans into vascular endothelial cells37. Cnm is co-transcribed with PgfS, which is located immediately downstream and encodes a putative glycosyltransferase; notably, the deletion of PgfS reduces the cell’s ability to invade vascular endothelial cells38. Therefore, a lack of PgfS expression in the Cnm-complemented mutant strain may be one reason why the pathogenicity of the complemented mutant strain was not completely restored to wildtype levels. Furthermore, in a two-component system of S. mutans, CovR is known as a positive regulator of Cnm, while VicRKX is a negative regulator of Cnm39, and both a VicRKX overexpression strain and a CovR inactivation mutant strain have a reduced ability to invade vascular endothelial cells. These proteins that regulate Cnm expression may affect the mechanism by which Cnm-positive S. mutans invade into vascular endothelial cells.
Bacterial invasion of host cells is accompanied by an increased expression of host genes involved in the regulation of small G proteins, leading to actin cytoskeleton remodeling15,40. Here, we performed a microarray analysis to identify genes related to Cnm-dependent HUVEC invasion by S. mutans, and we used RNA interference and invasion assays to verify the microarray results. These analyses revealed a crucial role for ARHGEF38 and ARHGAP9 in endothelial cell invasion by Cnm-positive S. mutans. Although our understanding of the detailed functions of the ARHGEF family is incomplete41, these proteins are guanine nucleotide exchange factors that promote the activation of Rho family G proteins by inducing GDP dissociation, thereby enabling GTP binding14. In turn, the GDP–GTP exchange promotes a cytoskeletal rearrangement that facilitates bacterial internalization, as demonstrated with Yersinia species, which are Gram-negative bacteria associated with digestive diseases18,19. In the present study, we found that an ARHGEF38 knockdown significantly inhibited HUVEC invasion by TW295, but this inhibition was incomplete, suggesting that other pathways also support the invasion of vascular endothelial cells by Cnm-positive S. mutans.
Intriguingly, we found that an ARHGAP9 knockdown in Cnm-positive S. mutans significantly increased their ability to invade HUVECs. ARHGAP9 is a member of the ARHGAP family of GTPase-activating proteins, which promote the inactivation of Rho family G proteins through the hydrolysis of bound GTP42. A previous study showed that ARHGAP9 overexpression in human leukemia cells significantly reduced their level of adhesion to type IV collagen43. Thus, our results suggest that ARHGAP9 normally prevents the invasion of Cnm-positive S. mutans by inhibiting type IV collagen binding.
Several animal models of IE have been developed, including dog, pig, rabbit, and rat models; of these, the rat model is the most commonly used44,45. We recently examined the virulence of S. mutans strains using the rat IE model12, and histopathological analyses revealed increased bacterial masses in abnormal heart valves. In the present study, we evaluated endothelial cell damage in S. mutans-infected rats by staining heart sections with anti-CD31 antibody, which allowed us to quantify the damage to endothelial cells.
Recent studies have reported correlations between Cnm-positive S. mutans infection and intracerebral hemorrhage13,46, which is a major complication of IE that occurs in approximately 10% of cases47. We previously demonstrated in a mouse model that Cnm-positive S. mutans strains caused deterioration of intracerebral hemorrhages that involved photochemical damage to the endothelium of the middle cerebral artery13. Cnm-positive S. mutans were observed around the damaged blood vessels, suggesting that they may contribute to the hemorrhage. This possibility should be analyzed in more detail in future studies.
A schematic of the mechanism by which Cnm-positive S. mutans may invade vascular endothelial cells during IE is shown in Fig. 7. We speculate that Cnm-positive S. mutans in the blood adhere to type IV collagen in serum and attach to endothelial cell surfaces, inducing an upregulation of ARHGEF38 expression in the endothelial cells. In turn, ARHGEF38 induces the activation of Rho family G proteins, leading to cytoskeletal rearrangement, which facilitates bacterial internalization. The invasion of Cnm-positive S. mutans results in vascular endothelial cell damage that subsequently contributes to many cardiovascular and cerebrovascular diseases.
Figure 7.

Proposed model of the interaction between Cnm-positive S. mutans and vascular endothelial cells. Cnm-positive S. mutans strains adhere to type IV collagen in the serum, and (2) then adhere to vascular endothelial cell surfaces. (3) The binding of bacteria activates ARHGEF38 in the endothelial cells, leading to GDP–GTP exchange and the activation of Rho family G proteins, cytoskeletal rearrangement, and bacterial internalization. (4) Invasion by Cnm-positive S. mutans induces vascular endothelial cell damage.
In summary, we showed here that S. mutans Cnm, its interaction with type IV collagen in serum, and the upregulation of ARHGEF38 in endothelial cells are all important for the invasion of Cnm-positive S. mutans into vascular endothelial cells. These events may therefore contribute to the deterioration of IE associated with Cnm-positive S. mutans infection.
Methods
Ethics statement
All rats were treated humanely, in accordance with the guidelines of the National Institutes of Health and the AERI-BBRI Animal Care and Use Committee. Animal experiments were approved by the Institutional Animal Care and Use Committee of Osaka University Graduate School of Dentistry (approval number 24-019-0).
S. mutans strains
MT8148 (serotype c) is a Cnm-negative strain isolated from the oral cavity of a healthy child21, TW295 (serotype k) is a Cnm-positive strain isolated from an individual with bacteremia after tooth extraction, TW295CND is the Cnm-defective isogenic mutant strain of TW29522,23, and TW295comp is the Cnm-complemented mutant strain of TW29512. All strains were confirmed to be S. mutans on the basis of their biochemical properties and the observation of rough colony morphology on Mitis-Salivarius agar plates (Difco Laboratories, Detroit, MI, USA) containing bacitracin (0.2 U/ml; Sigma-Aldrich, St. Louis, MO, USA) and 15% (wt/vol) sucrose (MSB agar), as well as the results of a sequence analysis targeting 16S rRNA, with the primers 8UA (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1540R (5′-AAGGAGGTGATCCAGCC-3′), as described previously48. For routine growth, all strains were cultured overnight in brain heart infusion (BHI) broth (Difco Laboratories). BHI broth containing erythromycin (10 µg/ml) was used to culture TW295CND, and BHI broth containing erythromycin (10 µg/ml) and spectinomycin (1 mg/ml) was used to culture TW295comp.
The presence or absence of cnm was determined by applying the PCR method, using primers cnm-1F (5′-GACAAAGAAATGAAAGATGT-3′) and cnm-1R (5′-GCAAAGACTCTTGTCCCTGC-3′), which can amplify the entire length of the cnm gene11. The putative amino acid sequence of Cnm from S. mutans strain Z1 (GenBank accession number AB102689), found in a previous study10, had a region with homology with the collagen-binding domain of the Staphylococcus aureus collagen-binding protein Cna with 54.8% identity, and this region of Cnm had collagen-binding ability10. The Cnm-positive S. mutans strain TW295 used in the present study also had the Cnm collagen-binding domain, which was previously registered in GenBank (accession number AB469913).
The Cnm-defective isogenic mutant strain TW295CND was generated in our previous study23. Briefly, the erythromycin resistance gene was inserted into the gene encoding the collagen-binding domain, located in the central part of cnm. A plasmid containing a complex of the erythromycin resistance gene and the collagen-binding gene was then introduced into a Cnm-positive S. mutans strain using a homologous recombination method. The appropriate insertional inactivation of the isogenic mutant was confirmed by Southern hybridization23 and western blotting49. The absence of collagen-binding ability in the isogenic mutant strain was confirmed by a collagen-binding assay23, in which the strain was attached to the collagen-coated plate using a previously described method11.
The Cnm-complemented strain TW295comp was generated in a previous study12. Briefly, the entire length of cnm was amplified and cloned into the shuttle vector pDL278. The plasmid containing cnm was then added to bacterial broth of TW295CND, which was cultured in Todd–Hewitt medium supplemented with 10% heat-inactivated horse serum (Invitrogen, Carlsbad, CA, USA). After being incubated for 2 h at 37 °C, the cells were plated on Mitis-salivarius agar containing spectinomycin (1 mg/ml) and erythromycin (10 µg/ml), followed by anaerobic incubation at 37 °C for 48 h, to select for the complemented mutant TW295comp strain. The successful generation of TW295comp was confirmed in our previous study by performing a PCR analysis for the presence of pDL278 in the cnm gene and collagen-binding assays12.
HUVEC invasion assay
HUVECs were purchased from Lonza (Walkersville, MD, USA), and bacterial invasion was evaluated as previously described37,50, with some modifications. In brief, 1 × 105 HUVECs were seeded in 24-well plates (Corning Inc., Corning, NY, USA) and washed three times with phosphate-buffered saline (PBS). Antibiotic-free EBM-2 medium (Lonza) and S. mutans (1 × 107 colony-forming units [CFU]) were added to the HUVECs. When purified ECM proteins were present, serum was omitted, and type IV collagen (Cellmatrix Type IV; Nitta Gelatin, Osaka, Japan), fibrinogen, fibronectin, and vitronectin (all from human plasma; Sigma-Aldrich) were added to the medium at final concentrations equivalent to those found in healthy human blood24–27. The cells were incubated for 2 h, after which the supernatant was removed, and the infected cells were washed three times with PBS. Medium containing penicillin (50 µg/ml) and gentamycin (300 µg/ml) was added to the wells, and the plates were then incubated for an additional 3 h. The preliminary experiments for this study revealed that this mixture of antibiotics could kill 1 × 109 CFU of each S. mutans strain used in the present study within 3 h. Additionally, all invasion assays included controls (no HUVECs), in which antibiotics were used to verify the killing of each S. mutans strain. The cells were lysed by the addition of sterile distilled water, and dilutions of the resulting lysates were plated onto MSB plates and cultured at 37 °C for 2 days. The invasion ratio was calculated as the ratio of recovered bacteria to the initial inoculum. Data are expressed as the mean ± standard deviation (SD) of triplicate experiments.
Fluorescence microscopy of S. mutans and HUVECs
The observation of S. mutans strains using confocal laser scanning microscopy was performed as follows. Each S. mutans strain, adjusted to a concentration of 1 × 109 CFU using PBS, was streaked on cover glasses, and fixed with 3% paraformaldehyde (Wako Pure Chemical Industries, Osaka, Japan) for 10 min. The S. mutans strains were then stained with rabbit anti-Cnm serum35 or anti-S. mutans antibody (whole cell as immunogen; Abcam, Cambridge, MA, USA) followed by detection with Alexa Fluor 555-conjugated goat anti-rabbit IgG (Molecular Probes, Life Technologies, Eugene, OR, USA).
Bacterial invasion of HUVECs was confirmed using confocal laser scanning microscopy as described previously20. In brief, invaded cells were washed three times with PBS, fixed with 3% paraformaldehyde for 10 min, washed with PBS, and incubated with primary antibodies for 1 h. After being washed, the cells were incubated with secondary antibodies for 30 min. If necessary, the cells were permeabilized by the addition of 0.4% Triton X-100 for 5 min. The following antibodies were used: rabbit anti-Cnm serum, detected with Alexa Fluor 555-conjugated goat anti-rabbit IgG; anti-ARHGEF38, anti-ARHGAP9, or anti-GPR179 (all Funakoshi Corporation, Tokyo, Japan), detected with Alexa Fluor 555-conjugated goat anti-rabbit IgG (Molecular Probes); and anti-type IV collagen (Abcam), detected with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes). The cytoskeleton was visualized by staining actin filaments with Alexa Fluor 488-conjugated phalloidin (Molecular Probes). Nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Wako Pure Chemical Industries). All antibodies were diluted 1:500 in 0.5% bovine serum albumin (BSA) in PBS. Cells were examined using a confocal laser scanning microscope (LSM510; Carl Zeiss, Oberkochem, Germany) with a 63 × oil immersion objective.
Bacterial ECM-binding assay
The ECM-binding properties of S. mutans were evaluated as described by Nomura et al.11, using a protocol originally developed by Waterhouse and Russell51 with some modifications. Type IV collagen, fibrinogen, fibronectin, and vitronectin (the major ECM components of serum) were added to the wells of 96-well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ, USA) and incubated overnight at 4 °C. The plates were then washed three times with PBS, blocked for 1.5 h with 5% BSA in PBS at 37 °C, and washed again with PBS containing 0.01% Tween 20. S. mutans were collected by centrifugation from overnight cultures in BHI broth, diluted with PBS, and added to the coated wells (concentration: 2 × 109 CFU/well). After a 3-h incubation at 37 °C, non-adherent cells were removed by washing the plates with PBS three times, and the adherent cells were fixed with 25% formaldehyde (200 µl) at room temperature for 30 min. The cells were then washed three times with PBS and stained by the addition of 0.05% crystal violet (Wako Pure Chemical Industries) in water (200 µl) for 1 min. After the plates were washed again, the dye was dissolved by the addition of 7% acetic acid (200 µl), and the absorbance at 595 nm was recorded. Data are expressed as the mean ± SD of triplicate experiments.
Microscopy of ECM-bound cells
Streptococcus mutans binding to type IV collagen and fibrinogen was also assessed using confocal laser scanning microscopy as described previously52 with some modifications. Type IV collagen or fibrinogen were added to chambered coverglass wells (CultureWell; Grace Bio Labs, Bend, OR, USA) and incubated overnight at 4 °C. The coated wells were washed three times with PBS, blocked for 1.5 h with 5% BSA in PBS at 37 °C, and washed again with PBS containing 0.01% Tween 20. S. mutans cells were collected, stained with hexidium iodide (Molecular Probes), and added to the coated wells (2 × 109 CFU/well) in PBS. The cells were cultured anaerobically at 37 °C for 18 h in the dark. Non-attached S. mutans cells were removed by washing with PBS, and the adherent cells were observed using a confocal laser scanning microscope (LSM510; Carl Zeiss, Oberkochem, Germany) with a 63 × oil immersion objective.
DNA microarray assays
Genes involved in HUVEC invasion by Cnm-positive S. mutans were identified by DNA microarray assays as described previously53. Amino-allyl-amplified RNA was obtained from total RNA using an Amino-allyl MessageAmp aRNA kit (Ambion, Austin, TX, USA), and samples were labeled with Cy3 or Cy5 using FluoroLink Cy3 or Cy5 monofunctional Dye 5-Packs (Amersham Bioscience, London, UK). Samples were applied to a gene chip system (Sigma Genosis Japan, Sapporo, Japan), gene expression was detected by Cy3 and Cy5 fluorescence intensity, and signal transduction-related genes whose transcripts were considered to be important for bacterial internalization were identified. Genes significantly differentially expressed between (1) uninfected and TW295-infected HUVECs and (2) TW295- and TW295CND-infected HUVECs were identified, and overlapping genes were noted (Fig. 4A). Genes significantly differentially expressed between TW295CND-infected and uninfected HUVECs were excluded to remove genes involved in infection by Cnm-negative S. mutans strains. Genes were considered significantly differentially expressed if the change in expression was > 2.0-fold.
Silencing of gene expression
siRNA transfection of HUVECs was performed as described previously54 with some modifications. siRNAs targeting ARHGEF38, ARHGAP9, GPR179, or a negative control sequence (siRNA AllStars Negative Control siRNA) were purchased from Qiagen (Düsseldorf, Germany). HUVECs (1 × 105 cells/well) were seeded in 24-well plates and incubated for 24 h. The cell culture medium was then discarded and replaced with serum- and antibiotic-free medium containing siRNAs (final concentration: 40 nM) and Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA).
RT-PCR assay
Silencing of gene expression was assessed by RT-PCR at 24 h post-transfection. Total RNA was prepared from the HUVECs after the cells had been transfected with siRNA using a Pure Link RNA Mini Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions. A High Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific) was then used to amplify cDNA synthesized from the mRNA. The primers used to detect the transcription of each gene are shown in Supplementary Table 5. PCR assays were performed in 20-µl reaction mixtures containing TaKaRa Ex Taq polymerase (TAKARA BIO, Inc., Otsu, Shiga, Japan) in accordance with the manufacturer’s protocols. PCR amplification was performed under the following conditions: initial denaturation at 95 °C for 4 min, followed by 30 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, with a final extension step at 72 °C for 7 min. The PCR products were fractionated using a 1.5% (w/v) agarose gel containing Tris-acetate-EDTA buffer, stained with ethidium bromide (0.5 µg/ml), and visualized under UV illumination.
Rat model of IE
A previously described rat IE model was used55 with some modifications. Briefly, 30 Sprague-Dawley male rats (weighing 250–300 g) were anesthetized via injection of xylazine and midazolam, and a sterile polyethylene catheter with a guide wire was inserted through the right carotid artery to injure the aortic valve. The catheter was immediately removed after the heart valve injury. A bacterial suspension (108 cells/rat in PBS) was then intravenously administered through the jugular vein. All rats survived for 7 days after the bacterial infection. At this time, the rats were euthanized by anesthesia overdose, and their hearts were extirpated, sectioned transversely, and stained with anti-CD31 antibody (Abcam) to visualize vascular endothelial cells. The lengths of total, CD31-positive, and CD31-negative endocardium in the heart valves were measured using WinROOF Vol 5.0 software (Mitsuya Co., Ltd., Tokyo, Japan). The rate of endocardium damage was calculated as: ([length of CD31-negative endocardium in each heart valve of infected rats]/[length of total endocardium in each heart valve of infected rats]) − ([average length of CD31-negative endocardium in heart valves of uninfected rats]/[average length of total endocardium in heart valves of uninfected rats]) × 100. Histopathological features were evaluated by performing hematoxylin–eosin staining of tissue sections and scoring them as 0 (none), 1 (mild), 2 (moderate), or 3 (severe), as previously described55. All scoring evaluations were performed in a double-blinded fashion by a pathologist (Sept. Sapie Co., Ltd., Tokyo, Japan).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Intergroup differences were compared using an analysis of variance (ANOVA) with Bonferroni’s post hoc test. Comparisons between two groups were performed using a Student’s t test. A p value of < 0.05 was considered to indicate a significant difference.
Supplementary information
Acknowledgements
The authors thank Prof. Howard K. Kuramitsu (State University of New York at Buffalo) for editing the manuscript. We also thank Ms. Rewa Yanagisawa, Department of Pediatric Dentistry, Osaka University Graduate School of Dentistry, for technical support with the molecular analyses. This work was supported by JSPS KAKENHI (Grant nos. 15K11363, 18H03010, 18K09831, and 18K17254).
Author contributions
R.N. designed the entire study under the supervision of K.N. R.N., M.O., M.H., S.M., N.T., S.N., and M.M. performed the experiments, and data interpretation was conducted by R.N. and K.N. N.I. performed the histopathological evaluation. R.N. and K.N. wrote the manuscript, which all authors read and approved.
Data availability
All data generated or analyzed during the described study are included in this published article (and its “Supplementary Information” files).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Noboru Teramoto is deceased.
Supplementary information
is available for this paper at 10.1038/s41598-020-75933-6.
References
- 1.Li X, Kolltveit KM, Tronstad L, Olsen I. Systemic diseases caused by oral infection. Clin. Microbiol. Rev. 2002;13:547–558. doi: 10.1128/CMR.13.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moreillon P, Que YA. Infective endocarditis. Lancet. 2004;363:139–149. doi: 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- 3.Holland TL, et al. Infective endocarditis. Nat. Rev. Dis. Primers. 2017;2:16059. doi: 10.1038/nrdp.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakatani S, et al. Japanese Circulation Society Joint Working Group. JCS 2017 Guideline on prevention and treatment of infective endocarditis. Circ. J. 2019;83:1767–1809. doi: 10.1253/circj.CJ-19-0549. [DOI] [PubMed] [Google Scholar]
- 5.Roberts GJ, Lucas VS, Omar J. Bacterial endocarditis and orthodontics. J. R. Coll. Surg. Edinb. 2000;45:141–145. [PubMed] [Google Scholar]
- 6.Cabell CH. Cardiology patient page. Bacterial endocarditis: The disease, treatment, and prevention. Circulation. 2003;107:e185–e187. doi: 10.1161/01.CIR.0000071082.36561.F1. [DOI] [PubMed] [Google Scholar]
- 7.Chorianopoulos E, Bea F, Katus H, Frey N. The role of endothelial cell biology in endocarditis. Cell Tissue Res. 2009;335:153–163. doi: 10.1007/s00441-008-0687-4. [DOI] [PubMed] [Google Scholar]
- 8.Jung CJ, et al. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation. 2015;131:571–581. doi: 10.1161/CIRCULATIONAHA.114.011432. [DOI] [PubMed] [Google Scholar]
- 9.Nakano K, Ooshima T. Serotype classification of Streptococcus mutans and its detection outside the oral cavity. Future Microbiol. 2009;4:891–902. doi: 10.2217/fmb.09.64. [DOI] [PubMed] [Google Scholar]
- 10.Sato Y, et al. Streptococcus mutans strains harboring collagen-binding adhesin. J. Dent. Res. 2004;83:534–539. doi: 10.1177/154405910408300705. [DOI] [PubMed] [Google Scholar]
- 11.Nomura R, et al. Molecular and clinical analyses of the gene encoding the collagen-binding adhesin of Streptococcus mutans. J. Med. Microbiol. 2009;58:469–475. doi: 10.1099/jmm.0.007559-0. [DOI] [PubMed] [Google Scholar]
- 12.Otsugu M, et al. Contribution of Streptococcus mutans strains with collagen-binding proteins in the presence of serum to the pathogenesis of infective endocarditis. Infect. Immun. 2017;85:e00401–e00417. doi: 10.1128/IAI.00401-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakano K, et al. The collagen-binding protein of Streptococcus mutans is involved in haemorrhagic stroke. Nat. Commun. 2011;2:485. doi: 10.1038/ncomms1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 15.Rauch L, et al. Staphylococcus aureus recruits Cdc42GAP through recycling endosomes and the exocyst to invade human endothelial cells. J. Cell Sci. 2016;129:2937–2949. doi: 10.1242/jcs.186213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel JC, Galán JE. Differential activation and function of Rho GTPases during Salmonella–host cell interactions. J. Cell Biol. 2006;175:453–463. doi: 10.1083/jcb.200605144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Handa Y, et al. Shigella IpgB1 promotes bacterial entry through the ELMO–Dock180 machinery. Nat. Cell Biol. 2007;9:121–128. doi: 10.1038/ncb1526. [DOI] [PubMed] [Google Scholar]
- 18.Isberg RR, Barnes P. Subversion of integrins by enteropathogenic Yersinia. J. Cell Sci. 2001;114:21–28. doi: 10.1242/jcs.114.1.21. [DOI] [PubMed] [Google Scholar]
- 19.Roppenser B, Röder A, Hentschke M, Ruckdeschel K, Aepfelbacher M. Yersinia enterocolitica differentially modulates RhoG activity in host cells. J. Cell Sci. 2009;122:696–705. doi: 10.1242/jcs.040345. [DOI] [PubMed] [Google Scholar]
- 20.Nomura R, et al. Potential involvement of collagen-binding proteins of Streptococcus mutans in infective endocarditis. Oral Dis. 2013;19:387–393. doi: 10.1111/odi.12016. [DOI] [PubMed] [Google Scholar]
- 21.Ooshima T, Izumitani A, Sobue S, Hamada S. Cariostatic effect of palatinose on experimental dental caries in rats. Jpn. J. Med. Sci. Biol. 1983;36:219–223. doi: 10.7883/yoken1952.36.219. [DOI] [PubMed] [Google Scholar]
- 22.Nakano K, Nomura R, Nakagawa I, Hamada S, Ooshima T. Demonstration of Streptococcus mutans with a cell wall polysaccharide specific to a new serotype, k, in the human oral cavity. J. Clin. Microbiol. 2004;42:198–202. doi: 10.1128/JCM.42.1.198-202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakano K, et al. Molecular characterization of Streptococcus mutans strains containing the cnm gene encoding a collagen-binding adhesin. Arch. Oral Biol. 2010;55:34–39. doi: 10.1016/j.archoralbio.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Mosesson MW, Umfleet RA. The cold-insoluble globulin of human plasma. I. Purification, primary characterization, and relationship to fibrinogen and other cold-insoluble fraction components. J. Biol. Chem. 1970;245:5728–5736. [PubMed] [Google Scholar]
- 25.Boyd NA, Bradwell AR, Thompson RA. Quantitation of vitronectin in serum: Evaluation of its usefulness in routine clinical practice. J. Clin. Pathol. 1993;46:1042–1045. doi: 10.1136/jcp.46.11.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asselta R, Duga S, Tenchini ML. The molecular basis of quantitative fibrinogen disorders. J. Thromb. Haemost. 2006;4:2115–2129. doi: 10.1111/j.1538-7836.2006.02094.x. [DOI] [PubMed] [Google Scholar]
- 27.Kinoshita J, et al. Type IV collagen levels are elevated in the serum of patients with peritoneal dissemination of gastric cancer. Oncol. Lett. 2010;1:989–994. doi: 10.3892/ol.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abranches J, et al. Invasion of human coronary artery endothelial cells by Streptococcus mutans OMZ175. Oral Microbiol. Immunol. 2009;24:141–145. doi: 10.1111/j.1399-302X.2008.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenkinson HF, Lamont RJ. Streptococcal adhesion and colonization. Crit. Rev. Oral Biol. Med. 1997;8:175–200. doi: 10.1177/10454411970080020601. [DOI] [PubMed] [Google Scholar]
- 30.Henderson B, Nair S, Pallas J, Williams MA. Fibronectin: A multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol. Rev. 2011;35:147–200. doi: 10.1111/j.1574-6976.2010.00243.x. [DOI] [PubMed] [Google Scholar]
- 31.Nobbs AH, Lamont RJ, Jenkinson HF. Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 2009;73:407–450. doi: 10.1128/MMBR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lannergard J, Frykberg L, Guss B. CNE, a collagen-binding protein of Streptococcus equi. FEMS Microbiol. Lett. 2003;222:69–74. doi: 10.1016/S0378-1097(03)00222-2. [DOI] [PubMed] [Google Scholar]
- 33.Sillanpää J, et al. A collagen-binding adhesin, Acb, and ten other putative MSCRAMM and pilus family proteins of Streptococcus gallolyticus subsp. gallolyticus (Streptococcus bovis Group, biotype I) J. Bacteriol. 2009;191:6643–6653. doi: 10.1128/JB.00909-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becke TD, et al. Pilus-1 backbone protein RrgB of Streptococcus pneumoniae binds collagen I in a force-dependent way. ACS Nano. 2019;13:7155–7165. doi: 10.1021/acsnano.9b02587. [DOI] [PubMed] [Google Scholar]
- 35.Nomura R, et al. Identification and characterization of a collagen-binding protein, Cbm, in Streptococcus mutans. Mol. Oral Microbiol. 2012;27:308–323. doi: 10.1111/j.2041-1014.2012.00649.x. [DOI] [PubMed] [Google Scholar]
- 36.Vollmer T, Hinse D, Kleesiek K, Dreier J. Interactions between endocarditis-derived Streptococcus gallolyticus subsp. gallolyticus isolates and human endothelial cells. BMC Microbiol. 2010;10:78. doi: 10.1186/1471-2180-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abranches J, et al. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect. Immun. 2011;79:2277–2284. doi: 10.1128/IAI.00767-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avilés-Reyes A, et al. Modification of Streptococcus mutans Cnm by PgfS contributes to adhesion, endothelial cell invasion, and virulence. J. Bacteriol. 2014;196:2789–2797. doi: 10.1128/JB.01783-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Araújo LA, et al. CovR and VicRKX regulate transcription of the collagen binding protein Cnm of Streptococcus mutans. J. Bacteriol. 2018;200:e00141–e1118. doi: 10.1128/JB.00141-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popoff MR. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases. 2014;5:e28209. doi: 10.4161/sgtp.28209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuroiwa M, Oneyama C, Nada S, Okada M. The guanine nucleotide exchange factor Arhgef5 plays crucial roles in Src-induced podosome formation. J. Cell Sci. 2011;124:1726–1738. doi: 10.1242/jcs.080291. [DOI] [PubMed] [Google Scholar]
- 42.Orlandi C, Cao Y, Martemyanov KA. Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Invest. Ophthalmol. Vis. Sci. 2013;54:7153–7161. doi: 10.1167/iovs.13-12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furukawa Y, et al. Isolation of a novel human gene, ARHGAP9, encoding a rho-GTPase activating protein. Biochem. Biophys. Res. Commun. 2001;284:643–649. doi: 10.1006/bbrc.2001.5022. [DOI] [PubMed] [Google Scholar]
- 44.Johnson CM, Bahn RC, Fass DN. Experimental porcine infective endocarditis: Description of a clinical model. Vet. Pathol. 1986;23:780–782. doi: 10.1177/030098588602300620. [DOI] [PubMed] [Google Scholar]
- 45.Wang ML, et al. A rabbit model of right-sided Staphylococcus aureus endocarditis created with echocardiographic guidance. Cardiovasc. Ultrasound. 2013;11:3. doi: 10.1186/1476-7120-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tonomura S, et al. Intracerebral hemorrhage and deep microbleeds associated with cnm-positive Streptococcus mutans; a hospital cohort study. Sci. Rep. 2016;6:20074. doi: 10.1038/srep20074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eishi K, et al. Surgical management of infective endocarditis associated with cerebral complications. Multi-center retrospective study in Japan. J. Thorac. Cardiovasc. Surg. 1995;110:1745–1755. doi: 10.1016/S0022-5223(95)70038-2. [DOI] [PubMed] [Google Scholar]
- 48.Nomura R, et al. Isolation and characterization of Streptococcus mutans in heart valve and dental plaque specimens from a patient with infective endocarditis. J. Med. Microbiol. 2006;55:1135–1140. doi: 10.1099/jmm.0.46609-0. [DOI] [PubMed] [Google Scholar]
- 49.Ito S, et al. Presence of specific strains of Streptococcus mutans, a pathogen of dental caries, in the tonsils is associated with IgA nephropathy. Sci. Rep. 2019;9:20130. doi: 10.1038/s41598-019-56679-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagata E, de Toledo A, Oho T. Invasion of human aortic endothelial cells by oral viridans group streptococci and induction of inflammatory cytokine production. Mol. Oral Microbiol. 2011;26:78–88. doi: 10.1111/j.2041-1014.2010.00597.x. [DOI] [PubMed] [Google Scholar]
- 51.Waterhouse JC, Russell RR. Dispensable genes and foreign DNA in Streptococcus mutans. Microbiology. 2006;152:1777–1788. doi: 10.1099/mic.0.28647-0. [DOI] [PubMed] [Google Scholar]
- 52.Kuboniwa M, et al. Streptococcus gordonii utilizes several distinct gene functions to recruit Porphyromonas gingivalis into a mixed community. Mol. Microbiol. 2006;60:121–139. doi: 10.1111/j.1365-2958.2006.05099.x. [DOI] [PubMed] [Google Scholar]
- 53.Hokamura K, et al. Molecular analysis of aortic intimal hyperplasia caused by Porphyromonas gingivalis infection in mice with endothelial damage. J. Periodontal Res. 2010;45:337–344. doi: 10.1111/j.1600-0765.2009.01242.x. [DOI] [PubMed] [Google Scholar]
- 54.Inaba H, et al. Upregulation of S100 calcium-binding protein A9 is required for induction of smooth muscle cell proliferation by a periodontal pathogen. FEBS. Lett. 2009;583:128–134. doi: 10.1016/j.febslet.2008.11.036. [DOI] [PubMed] [Google Scholar]
- 55.Nomura R, et al. Contribution of the interaction of Streptococcus mutans serotype k strains with fibrinogen to the pathogenicity of infective endocarditis. Infect. Immun. 2014;82:5223–5234. doi: 10.1128/IAI.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during the described study are included in this published article (and its “Supplementary Information” files).