

Abstract

Perfluorocarbon-loaded nanoparticles are powerful theranostic agents, which are used in the therapy of cancer and stroke and as imaging agents for ultrasound and 19F magnetic resonance imaging (MRI). Scaling up the production of perfluorocarbon-loaded nanoparticles is essential for clinical translation. However, it represents a major challenge as perfluorocarbons are hydrophobic and lipophobic. We developed a method for continuous-flow production of perfluorocarbon-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles using a modular microfluidic system, with sufficient yields for clinical use. We combined two slit interdigital micromixers with a sonication flow cell to achieve efficient mixing of three phases: liquid perfluorocarbon, PLGA in organic solvent, and aqueous surfactant solution. The production rate was at least 30 times higher than with the conventional formulation. The characteristics of nanoparticles can be adjusted by changing the flow rates and type of solvent, resulting in a high PFC loading of 20–60 wt % and radii below 200 nm. The nanoparticles are nontoxic, suitable for 19F MRI and ultrasound imaging, and can dissolve oxygen. In vivo19F MRI with perfluoro-15-crown-5 ether-loaded nanoparticles showed similar biodistribution as nanoparticles made with the conventional method and a fast clearance from the organs. Overall, we developed a continuous, modular method for scaled-up production of perfluorocarbon-loaded nanoparticles that can be potentially adapted for the production of other multiphase systems. Thus, it will facilitate the clinical translation of theranostic agents in the future.
Keywords: nanoparticles, perfluorocarbon, microfluidics, multimodal imaging, 19F MRI, ultrasound, oxygen carriers, cell tracking
Introduction
Perfluorocarbon (PFC)-loaded nanoparticles (NPs) emerged recently as powerful theranostic agents that could lead to the treatment of major public health problems. Owing to the ability to carry oxygen, PFC-loaded NPs find application in photodynamic1−4 and other hypoxia-affected treatments of cancer5,6 and treatment of stroke.7,8 Additionally, they act as multimodal imaging agents for 19F magnetic resonance imaging (19F MRI) and ultrasound.9−12 PFCs, however, have already seen a long and much more varied history of clinical use. They were first used in the 1980s as blood substitutes because of their ability to dissolve oxygen. However, most of them did not succeed in clinic because of issues with stability and handling or were even retracted by the FDA.8,13 Development of stable NPs and scaling up their production are essential requirements for the new generation of PFC-based nanotheranostic agents to succeed in clinic.14,15 However, challenges in both arise from the poor solubility of PFCs that are simultaneously hydrophobic and lipophobic.16
Frequently used formulations are PFC emulsions that are stabilized by surfactants, such as phospholipids or nonionic poloxamers.17−20 PFC emulsions, however, often suffer from shortcomings, such as coalescence or Ostwald ripening.19,21 The encapsulation of PFC in biocompatible polymers, such as poly(lactic-co-glycolic acid) (PLGA), leads to colloidally stable NPs with controllable size, PFC content, and long shelf life.3,22−25 Moreover, the use of a polymer enhances the flexibility in loading and release of drugs and modification with targeting ligands. For example, perfluorooctyl bromide (PFOB)–PLGA nanocapsules were recently applied as nanotheranostics for the delivery of paclitaxel along with 19F MRI imaging.23,26,27 Our group developed perfluoro-15-crown-5 ether (PFCE)–PLGA NPs for multimodal imaging with 19F MRI,9,24 ultrasound,9 and photoacoustics.23 These NPs display a fractal multicore structure, which is atypical for PFC. This structure leads to a faster in vivo clearance of PFCE, overcoming the organ accumulation issues of current PFCE formulations.9,28 PFCE–PLGA NPs were approved for a clinical trial for labeling dendritic cells (DCs) during the cell therapy of melanoma patients (ClinicalTrials.gov Identifier: NCT02574377).
Scaling up the production is an essential step for the clinical translation of PFC-loaded polymeric NPs. Hence, it was our goal in this study. PFC-loaded polymeric NPs are usually produced using an emulsion–solvent evaporation method, typically a miniemulsion.9,25 Because of the poor solubility of PFC, emulsification requires mixing of three phases that are all nonmiscible with each other: the hydrophobic polymer in an organic solvent, the liquid PFC phase, and the aqueous surfactant phase. To overcome the miscibility issues of PFC, we decided to use microfluidics. The microfluidic toolbox provides high flexibility in mixing of different components and was established for the continuous-flow synthesis of various types of theranostic particles in the recent years.15,29−33 Moreover, the production can be done continuously, which is currently recommended by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).34,35
In microfluidics, droplets or particles are generated in micrometer-sized channels, enabling the precise control of particle properties by device geometry, mixing ratios, and flow regimes.29,30 Polymeric particles can be produced in microfluidic devices using various approaches including miniemulsion, nanoprecipitation, self-assembly, or novel approaches such as polymerization-induced self-assembly (PISA).15,36−38 Homogenization can be done by active mixing that requires an external energy input, for example, sonication, or more commonly by passive mixing, where mixing is achieved by channel geometry.31 The last group can be roughly subdivided in droplet-based microfluidics that generate single droplets at high precision in a single channel and multichannel systems that split and recombine the flow in multiple channels. Within the last group, multilamination interdigital micromixers can achieve high flow rates and correspondingly high mixing efficiencies and high throughput. These systems have been successfully used to scale up the production of polymeric particles, including PLGA.15,39−41 Therefore, we selected the interdigital mixing to develop a continuous scaling-up method for PFC-loaded polymeric NPs.
To mix the three phases and to achieve additional stabilization of PFC, we combined the interdigital mixing and a flow sonication. We first studied the impact of several process parameters that allowed us to tailor the features of NPs obtaining high PFC encapsulation. Afterward, we demonstrated in vitro that microfluidic NPs are suitable as multimodal imaging agents for 19F MRI, ultrasound, and fluorescence. Moreover, they can dissolve oxygen and thus can potentially act in therapy as oxygen carriers. Finally, we injected our NPs in vivo and showed that they can be imaged with 19F MRI and display the same biodistribution fast-clearance behavior as multicore NPs made with the conventional method. These results further indicate that NPs produced with microfluidics display the multicore structure, preserving the properties of original formulation. Overall, we developed a modular continuous method for scaling up the production, which will take us a step closer to bring PFC NPs from the bench to clinic.
Results and Discussion
Combination of Slit Interdigital Micromixers with a Sonication Flow Cell for Continuous-Flow Production
The synthesis of PFC-loaded PLGA NPs involves mixing of three different phases: the liquid PFC phase, the organic phase with a dissolved polymer, and the aqueous phase with the surfactant. To mix these three phases stepwise in a continuous flow, we combined two slit interdigital micromixers (type SIMM V2) and an ultrasonic flow cell (Figure 1a). The core part of the micromixer is the interdigital multichannel inlay (Figure 1a, inset). Each micromixer has two inlets allowing to simultaneously mix two different liquids. The mixing takes place by alternating the arrangements of both phases in the microchannels.42 This micromixer is advantageous for scaling up the production as it can be operated at high flow rates, up to 50 mL min–1. After both micromixers, we included an additional active mixing step with a sonication flow cell to reduce the size of particles and to achieve better miscibility of PFCs. The whole process can be split into three steps (Figure 1a):
-
1.
Premixing of PLGA in the organic solvent with a liquid PFC in the first micromixer that leads to an unstable emulsion of PFC in the PLGA/organic solvent. The obtained mixture after this first step is denoted as mixture 1 (M1, flow rate QM1).
-
2.
Mixing mixture 1 with the aqueous solution of the surfactant in the second micromixer. During this step, the formation of emulsion droplets takes place. This first emulsion is named M2 for mixture 2 further in the manuscript. The flow rate of M2 is the sum of the individual flow rates, which corresponds to the total flow rate through the microfluidic system Qtotal.
-
3.
Active mixing in a sonication flow cell and the formation of a miniemulsion. This step provides an additional energy input for sufficient stabilization of the PFC phase.
Figure 1.
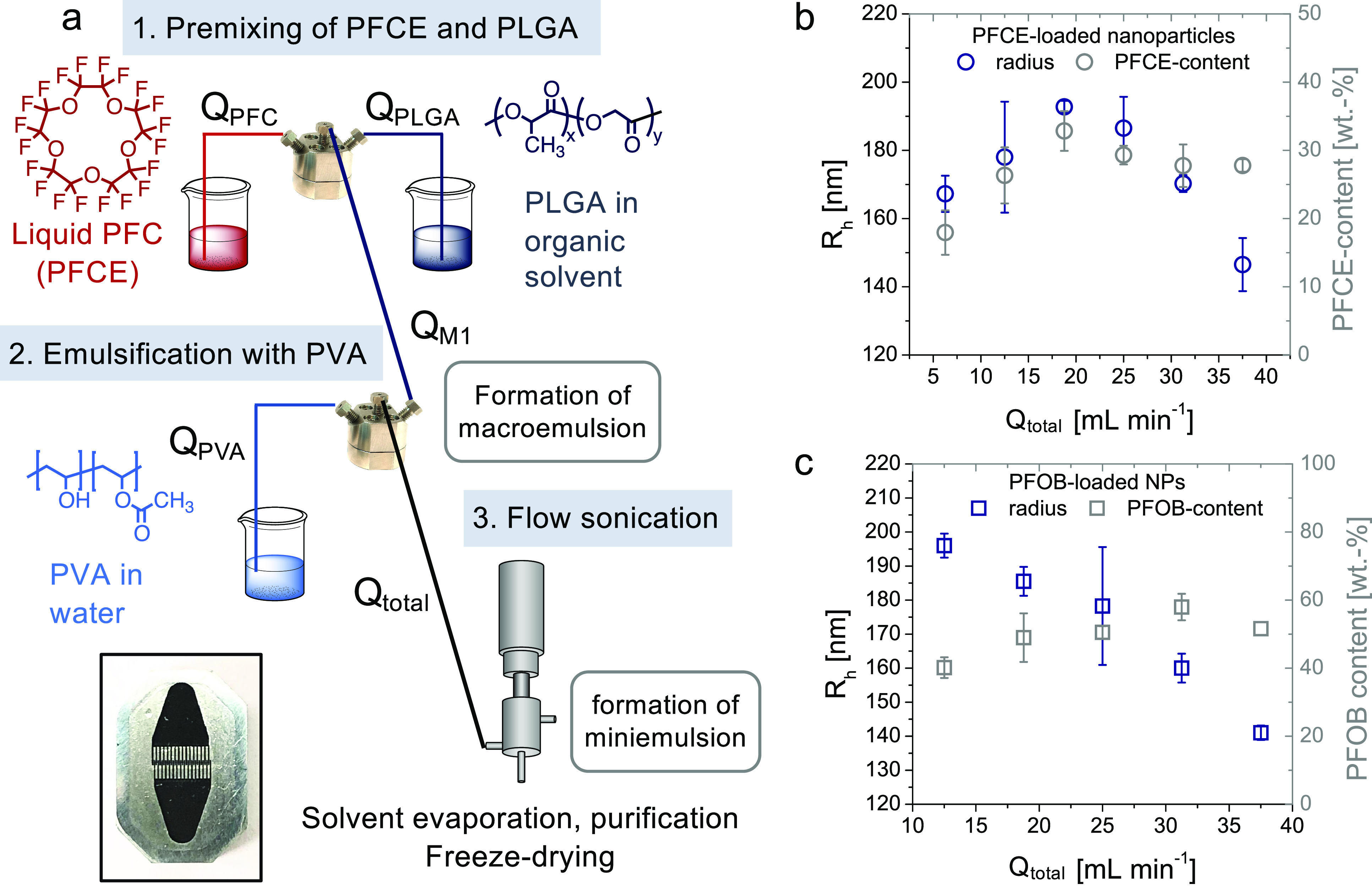
(a) Schematic overview of the microfluidic setup. The first step is mixing of the PFC (flow rate QPFC) with the organic solution of the polymer (flow rate QPLGA) in the first micromixer. The resulting mixture M1 then proceeds to the second micromixer, where it is emulsified with an aqueous surfactant solution (here PVA, flow rate QPVA). This emulsion (flow rate Qtotal) flows to the sonication flow cell for active mixing. Ultracompact high-pressure pumps are not shown for simplicity. Inset: Mixing inlay of SIMM V2. (b,c) Encapsulation of PFCE (b) and PFOB (c) with the microfluidic setup. Hydrodynamic radius (DLS, blue, c(NP) = 0.1 mg mL–1) and PFC content [NMR, grey, in D2O with trifluoroacetic acid (TFA) as an internal reference, 5–10 mg in 600 μL of D2O, 378 MHz] at different total flow rates Qtotal are shown. The flow rate ratios between single phases were kept constant (compare Table S3). For encapsulation of PFCE, (b) size and PFCE content first increase and then decrease with increasing flow rate. For PFOB (c), an increasing flow rate results in the decrease in hydrodynamic radius and in the increase of PFOB encapsulation (compare Table S4). Error bars represent the standard deviation between the results obtained from two independent batches of particles that were produced on different days (see also Table S9 for additional results of NPs produced with the addition of a fluorophore).
The Total Flow Rate Is More Important than the Individual Flow Rates for Size and PFC Encapsulation in the Three-Phasic System
To develop a continuous-flow production, the first step is to find the optimal flow parameters. The main focus here was on the encapsulation of PFCE and PFOB as both compounds are used for imaging, and PFOB is established as an oxygen carrier.21,25,43 As a starting point for our study, we used the composition of our conventional batch formulation of PFCE–PLGA-NPs that were approved for a clinical trial.9,24 At the beginning, we kept all main parameters, such as the concentration of the surfactant [poly(vinyl alcohol) (PVA)] and PLGA, the same as in the batch method, with dichloromethane (DCM) as a solvent for the organic phase, focusing on the effect of flow parameters.22−24 The ratios between the three phases, which correspond to the volumes used in the conventional method, can be adjusted directly in the microfluidic system by changing the flow rates. At the beginning, we studied the effect of flow parameters on the particle size and PFCE encapsulation to find optimal settings for the microfluidic synthesis.
The following flow parameters can affect the characteristics of the particles:
-
1.
flow rate ratio between PFCE and organic PLGA solution (QPFC/QPLGA).
-
2.
flow rate ratio between PFCE/PLGA mixture M1 and aqueous surfactant solution (QM1/QPVA).
-
3.
The total flow rate (Qtotal) that is determined by individual flow rates of each component.
After production, NPs were isolated by centrifugation, washed, and freeze-dried prior to further use.
Changing the flow rate ratio between the organic PLGA solution and PFCE or between PFCE–PLGA mixture (M1 in Figure 1) and the aqueous surfactant solution did not lead to any noteworthy effects on the size or PFCE content (compare Tables S1 and S2). In contrast, for the encapsulation of PFCE, the total flow rate turned out to be the most important parameter that determines the characteristics of NPs (Figure 1b).
When the total flow rate was varied between 6 and 36 mL min–1 while keeping flow rate ratios between individual phases fixed, the encapsulation of PFCE first increased from 18 to 33 wt % PFCE content, reaching the maximum at a flow rate of 19 mL min–1 (Figure 1b, compare also Table S3 for exact flow parameters). After further increase of the flow rate, the PFCE content changed only slightly and remained almost constant with an increasing flow rate. Moreover, the size of NPs first slightly increased from a hydrodynamic radius of 167 to 190 nm, with the maximum between flow rates of 19 and 25 mL min–1. Further increase in the flow rate above 25 mL min–1 resulted in a decrease in the radius to 147 nm (Figure 1b). All NPs showed moderate polydispersity with a polydispersity index (PDI) around 0.2, which is well-suitable for the in vivo or clinical application of PLGA NPs (Table S3).
After establishing the parameters with the PFCE system, we applied the method for the encapsulation of PFOB. In the PFOB system, an increase of flow rates also resulted in the decrease of particle size and polydispersity (Figures 1c and S1). Opposite to PFCE, no increase in size between 6 and 25 mL min–1 was observed. The PFOB encapsulation increased up to a flow rate up of 30 mL min–1 and remained almost constant at higher flow rates. The resulting PFOB–PLGA NPs had a high PFOB encapsulation, up to 60 wt %, hydrodynamic radii between 150 and 200 nm depending on the flow rate, and a medium to low polydispersity (Figure 1c and Table S4).
The observed trends in size and PFC encapsulation can be explained by mixing energy in the micromixer. The total flow rate Qtotal determines the energy during emulsification in the micromixer.42 A higher flow rate typically results in higher mixing energy, which correspondingly should lead to smaller particles.42 In our setup, the sonication flow cell provides an additional energy input to obtain monodisperse NPs with high PFC encapsulation. While the increase of flow rates leads to a higher mixing energy in micromixers, it also reduces the time that the final mixture M2 needs to pass thought the sonication flow cell. Thus, it leads to lower energy input from the sonication.
For the PFCE–PLGA system, the increasing size at flow rates between 5 and 25 mL min–1 may indicate that the sonication time became too short as the flow rate increased. When the flow rates increase above 25 mL min–1, the mixing energy in the micromixer also increases and can countervail the shortening of the sonication time. However, even at the highest flow rate, an active mixing step seems important for NP production. Without flow sonication, particles had sizes in the micrometer range and were difficult to isolate because of the phase separation of the PFC phase during the subsequent washing steps, indicating lower stability (data not shown). Based on the observed trend in size and PFCE content (Figure 1b), the balance between the mixing energy in the micromixer and flow cell appears to determine the size and PFCE content of NPs. The differences between the encapsulation of PFCE and PFOB could be the result of different solubilities of both PFCs. Because of the presence of a bromine atom, PFOB has more lipophilic character and better solubility in organic solvents.16,44
Both PFCE- and PFOB-loaded NPs produced with our method displayed sizes and PFC content in a range that should be suitable for biomedical use, for example cell labeling.45
Impact of Different Solvents on NP Size and PFC Encapsulation
Next to flow parameters, the solvent is another important factor that can affect the features of NPs. To gain systematic information about the effect of organic solvents on the microfluidic synthesis, we tested different solvents for the PLGA phase for the encapsulation of PFCE (Figure 2 and Table S5). Based on polarity, we have chosen chloroform, ethyl acetate, and DCM/acetonitrile (MeCN) mixture (1:1, v/v). The last solvent mixture was selected because we have shown previously that in the conventional batch method, it resulted in smaller NPs compared to DCM.22
Figure 2.

Effect of the organic solvent on size and PFCE encapsulation. (a) Radius [DLS, blue, c(NP) = 0.1 mg mL–1] and (b) PFCE encapsulation versus total flow rate through the system Qtotal [NMR, grey, TFA as an internal reference, D2O, 378 MHz] of NPs synthesized with chloroform, ethyl acetate, and a mixture of DCM/MeCN in comparison with DCM are shown. For all solvents, three different flow rates were tested. The sizes of NPs are decreasing with increasing polarity of the solvent. The encapsulation of PFCE was higher in chloroform compared to DCM. In contrast, the use of DCM/MeCN mixture or ethyl acetate resulted in a lower PFCE encapsulation.
The size of NPs prepared with the DCM/MeCN mixture was indeed lower compared to the size of the particles prepared with DCM, similar to the batch method. However, the PFCE encapsulation was around 10 wt % and thus lower than the 20–30 wt % that we obtained using DCM. This difference in the PFCE content was not observed in our conventional batch method.22 Ethyl acetate (AcOEt), which also has a higher polarity compared to DCM, resulted in a strong decrease in the size of the NPs at different flow rates (Figure 2a). However, the PFCE encapsulation was even lower than with the DCM/MeCN mixture as a solvent (Figure 2b). In contrast, in the conventional batch method, similar encapsulation of PFCE was obtained using DCM or AcOEt as a solvent.22 The reduced size of the NPs prepared with the DCM/MeCN mixture and AcOEt can be explained by the higher polarity of these solvents and correspondingly lower surface tensions.22
Finally, using chloroform, which also has a higher polarity than DCM, resulted in particles with a slightly smaller size compared to DCM. Furthermore, the encapsulation of PFCE using chloroform was higher compared to all other solvents, up to 42 wt % (Figure 2a,b). A possible reason for higher encapsulation could be the better solubility of PFCE in chloroform compared to the other solvents. However, literature data on the solubility of PFCE in both solvents are not available. Overall, chloroform provided the highest PFCE encapsulation and thus the best results at all flow rates.
Microfluidic System is Suitable for Continuous Large-Scale Production of PFC-Loaded NPs
After establishing the optimal synthesis parameters, we investigated the performance of the system for the actual scaling up of the synthesis. Batch-to-batch variation is a major problem with conventional batch sonication. Therefore, we collected several fractions with a volume of a typical batch during the constant operation of the setup using DCM or chloroform as an organic solvent for the production of PFCE–PLGA NPs.
The total volume which was collected was approximately 150 mL. The variation between fractions was negligible for both DCM and chloroform, as shown by DLS and NMR spectroscopy (Figure 3a,b, compare also Tables S6 and S7). In particular, the standard deviation of the radius between the fractions was 1.3% for chloroform and 2.2% for DCM. For the PFCE content, we obtained a standard deviation of 1.1% in chloroform and 3.6% for DCM (compare Tables S6 and S7 for individual values). Thus, the deviation between both the size and PFCE encapsulation is in the range that one would expect for errors because of, for example, sample preparation. In contrast, a typical deviation of the PFCE content between different batches prepared with conventional sonication is larger, typically around 10%.9 Scanning electron microscopy (SEM) further confirmed that the NPs were spherical in shape and displayed some polydispersity, which is generally typical for PLGA particles. The majority of NPs had a radius between 100 and 200 nm, similar to DLS results. Afterward, we accessed the production of PFOB-loaded NPs, showing that the system can be operated over a run time of 1 h (see Supporting Information Section 1.6 for discussion and Figure S2).
Figure 3.
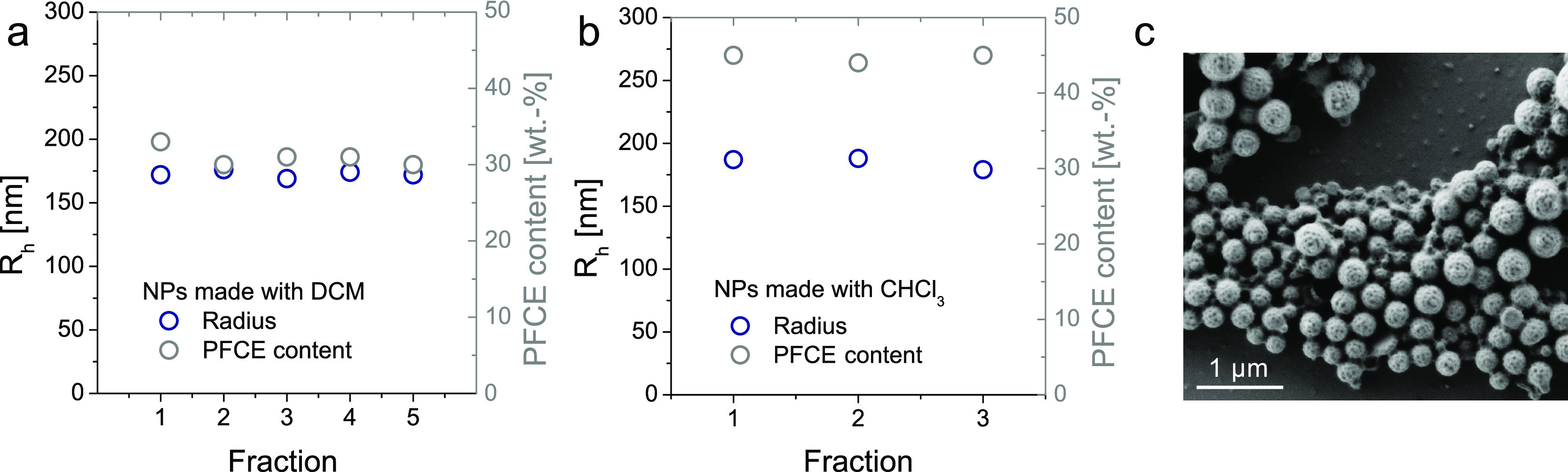
Reproducibility of the production of PFCE–PLGA NPs with the microfluidic system. (a) Radius [DLS, blue, c(NP) = 0.1 mg mL–1] and (b) PFCE encapsulation (NMR, grey TFA as an internal reference, 5–10 mg in 600 μL of D2O, 378 MHz) of single fractions. (c) SEM micrograph of NPs prepared using chloroform revealed that the majority of NPs have a radius between 100 and 200 nm. Scale bar 1 μm.
Finally, we obtained around 1.5 g of PFCE–PLGA NPs after running the setup for 6 min, followed by purification and freeze-drying. This amount is sufficient for the use of NPs in 19F MRI applications, such as labeling of immune cells. For instance, labeling the DCs requires typically 30–60 mg of NPs per patient. Based on the yield from a 6 min run, our system is capable of producing 15 g of PFCE–PLGA NPs per hour. Extrapolated for an 8 h work day, this would mean that one operator can produce roughly 100 g of NPs per day with a single setup. Clearly, a longer operation of the setup requires optimization of the purification procedure that is currently done using a laboratory-scale centrifuge. Ideally, purification should also be done with a flow filtration system, such as dynamic dialysis or tangential-flow filtration. In contrast to the continuous method, the realistic amount that we can produce with the conventional batch method is around 2–3 g of particles per day. The last value is estimated based on our experience on the batch formulation over the last 8 years, including the cleanroom production for a clinical trial, for production with a single ultrasonic homogenizer. Thus, with this microfluidic setup, the yield increased at least 30-fold compared to the conventional batch method.
NPs Are Suitable for Multimodal Imaging and Oxygen Loading
After successful development of the microfluidic production method, the next step was to confirm that the NPs produced using a microfluidic setup can be used as multimodal imaging agents and as oxygen carriers. Labeling the immune cells for 19F MRI to monitor the cell therapy is one of the biggest applications of conventional PFCE emulsions and PFC-based NPs.21,45,46 In this application, PFCE–PLGA NPs made with the conventional batch method can act not just as imaging agents for 19F MRI but also as multimodal imaging agents for ultrasound and optical methods.9,22,24 On the therapeutic side, PFC-based colloids show promising results in the transportation of oxygen for the treatment of cancer1−3,5,6 or stroke.7 Therefore, in the current study, after basic toxicity testing, we focused on imaging applications and the oxygen loading in vitro.
Viability testing was done using a standard viability assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, MTT). We used primary human monocyte-derived DCs (moDC) as labeling moDC ex vivo for cell therapy is one of the main applications of PFCE–PLGA NPs in our group. The dose used here is typically applied for cell labeling with NPs made with the conventional method (see Table S8 for characteristics of the NPs). The viability values of cells incubated with NPs were slightly higher compared to the living cells (Figure 4a DCM and Figure S3 chloroform). This increase can be attributed to an increased phagocytic activity of moDCs in the presence of NPs. Neither NPs made with chloroform nor those made with DCM showed any toxic effects on moDC, additionally confirming the removal of organic solvents (Figure 4a DCM and Figure S3 chloroform).
Figure 4.

NPs can act as multimodal imaging agents and oxygen carriers. (a) Cell viability assay after incubation with PFCE–PLGA NPs for 24 h (prepared using DCM) showed that PFCE–PLGA NPs did not affect cell viability. DCM1/2/3 corresponds to different batches of NPs produced with DCM as a solvent (see Table S8 for characteristics of NPs and Figure S3 for NPs made with chloroform). (b) confocal microscopy images of NP uptake by moDCs. Fluorescent signal coming from the NPs (red) partially overlaps with the signal of the early endosomal marker EEA1. Scale bar 25 μm (c) ultrasound of NP dispersion (sample PFCE16) in gel phantom shows acoustic contrast to the surrounding gel (see Figure S6 for different samples & Table S10 for details on NPs). The settings were similar to the settings that are used for the imaging of PFCE–PLGA NPs prepared with the conventional method indicating that NPs should be suitable for in vivo imaging. c(NP) = 10 mg mL–1, 21 MHz, 50 dB. (d) 19F longitudinal relaxivity R1 changes with oxygen pressure indicating loading with oxygen. Relaxivity of three different 19F-groups at different oxygen pressures is shown. NPs at pO2 = 0 mm Hg were saturated with Ar; another sample was measured at ambient pressure and the third one was saturated with oxygen. Lines correspond to linear fits of the data points, demonstrating the linear trend of the data (R2 ≥ 0.999). NPs in D2O, 378 MHz. Compare SI for further images and NPs characteristics (Tables S8–S11, Figures S3–S7).
To study the cell uptake of NPs, we used confocal microscopy. For this purpose, we produced PFCE–PLGA NPs that additionally encapsulated the Atto647 dye. The addition of the dye did not affect the size or PFCE content, indicating that coloading of NPs with PFCE and other cargos should be possible (Table S9). After 24 h of incubation with NPs, cells were stained for the early (EEA1) and late (LAMP1) endosomal markers. The red fluorescent signal from the NPs overlaps mostly with the signals of both markers indicating the intracellular presence of the NPs and colocalization with either early or late endosomes (Figures 4b and S3 and S4). However, part of the NP signal did not colocalize with either of the two markers, which can be caused by the potential escape of NPs out of the endosomal pathway or release of the dye from the NPs, similar to NPs made with the conventional method.22,47 Thus, these results indicate that microfluidic NPs should be suitable for labeling cells as used in cell-tracking studies.
To confirm that the NPs made with microfluidics are suitable for multimodal imaging, we performed imaging experiments with 19F MRI and ultrasound (Figure 4c,d, see Tables S10 and S11 for details on used NPs). As already expected based on high PFC content, both PFCE– and PFOB–PLGA NPs can be imaged with 19F MRI using a conventional 3D RARE imaging sequence at concentrations relevant for in vivo imaging (Figure S7). Ultrasound imaging of aqueous dispersions of NPs in a gel phantom on a preclinical high-resolution scanner confirmed that NPs can be imaged with ultrasound using the B-mode (in Figures 4c and S5). Usually, NPs are too small to be detected with ultrasound.9 Oppositely, we have recently shown that PFCE–PLGA NPs made with the conventional method can be imaged with ultrasound and are long-term stable in the acoustic field.9 Both properties appeared to be related to the multicore structure of PFCE–PLGA NPs.9 Thus, the fact that PFCE–PLGA NPs made with microfluidics can be detected with ultrasound may indicate that they also display the multicore structure.
To show that microfluidic NPs are potentially applicable as therapeutic oxygen carriers, we investigated their oxygen-binding properties. We used PFOB NPs as PFOB is often used in oxygen-carrying systems (sample PFOB7, Table S4).13,43 As dissolved oxygen changes the relaxation properties of the 19F nucleus, the oxygen binding was accessed by NMR spectroscopy (Figure 4d). The 19F longitudinal relaxivity R1 increased with increasing the oxygen pressure, as shown for three different groups of the PFOB molecule in Figure 4d. Based on a few data points, the increase seemed linear as one would expect for a liquid PFC, and in particular, PFOB loaded with oxygen that follows Henry’s law.21,44 Further modification and characterization of drug-loaded products is needed prior to the actual use as a therapeutic oxygen carrier. Nevertheless, these data show the potential of using the microfluidic synthesis to produce PFC-loaded NPs for the therapeutic use.
In summary, PFCE–PLGA NPs produced with the microfluidic method are nontoxic and suitable for multimodal imaging and potentially for oxygen delivery. Thus, the method could be used to produce larger amounts of NPs that are needed for clinical translation.
Microfluidic NPs Display Fast in Vivo Clearance and Biodistribution Similar to NPs Produced with a Conventional Batch Method
Biodistribution and clearance behavior are crucial parameters for the application of PFC–PLGA NPs as imaging or theranostic agents in vivo and in the clinic. Therefore, we determined their biodistribution in vivo and followed the injected animals longitudinally to observe clearance by quantitative 19F MRI.
We injected PFCE–PLGA NPs intravenously (i.v.) and imaged organs at several time points with 19F and 1H MRI (Figures 5 and S8, compare Table S6 for the characteristics of NPs). NPs were detected mainly in the liver and the spleen (Figure 5b). One week post-injection, NPs were cleared out from the spleen, and the NP signal in the liver reduced 75% after 2 weeks (Figures 5a and S7). Both biodistribution and clearance are similar to those of NPs made with the conventional method, as we have shown in our recent study.28
Figure 5.

In vivo biodistribution and clearance of PFCE–PLGA NPs by 19F MRI (sample produced during the longer run of the system, compare Table S6). (a) Transversal 19F MRI images (red hot) overlaid on 1H MRI (grey scale) images of the liver and spleen 2 h (upper row) and 2 weeks (lower row) after the i.v. injection of 20 mg of PFCE–PLGA NPs. After 2 h NPs were mainly located in the liver and spleen. After 2 weeks, NPs show significant clearance from the organs. Note the varying reference tube signal because of the partial volume effect, and images are scaled identical. (b) Graph showing a biodistribution of NPs in the liver, spleen, and bone marrow of the thoracoabdominal part of the spine (BM) at day 1. The signal of the imaging agent is reported in corrected arbitrary units based on the signal from the reference tube. 11.7 T.
PFCE is advantageous for 19F MRI, as it displays a single MR resonance frequency resulting in the absence of chemical shift artifacts and high sensitivity.48 However, the biological half-life of PFCE formulated in emulsions is often very long, that is, 250 days.49 Such long organ-retention times can hamper the clinical translation. Moreover, slow clearance is a big disadvantage for imaging applications that require repeated injections of the imaging agent. PFCE–PLGA NPs made with the conventional method display an atypical multicore structure9 that results in a fast clearance with a half-life t1/2 of 16 days is typical for PFCE–PLGA.28 Thus, the fast-clearance PFCE–PLGA NPs produced with the microfluidic method indicate that they also display the multicore structure. The encapsulation of PFCE in multicore particles could overcome the limitations of core–shell systems in clinical use. Therefore, the finding that the half-life of microfluidic PFCE–PLGA NPs matches the short half-life of the batch-made PFCE–PLGA NPs underlines the potential of our method to be used for the production of PFCE–PLGA NPs for clinical use.28
Conclusions and Outlook
The translation of liquid PFC-loaded polymeric NPs to clinical use requires high amounts of the product, with low polydispersity and high PFC content. However, conventional formulation methods often yield only milligrams of the product per batch. The immiscible, highly hydrophobic, and lipophobic PFC phase makes scaling up the process challenging.
In this work, we developed the microfluidic miniemulsion method to scale up the synthesis of PFC–PLGA NPs and explored how different parameters affect the formulation of these three-phasic systems. By changing the flow rates and the type of organic solvents, we were able to adjust the radius of the NPs, between 90 and 200 nm, and achieved high PFC content, up to 60 wt %. Smaller sizes and higher PFC encapsulation were obtained at the higher flow rates. Furthermore, a fluorescent dye could be encapsulated, demonstrating that loading with additional cargo is generally feasible.
After the development of the scaled-up production method, we assessed the performance of NPs as imaging and oxygen delivery agents. We have shown that NPs are suitable for multimodal imaging with 19F MRI, ultrasound, and fluorescence. Moreover, 19F NMR relaxation time measurements indicated that these NPs should be able to act as oxygen carriers in therapeutic applications. Furthermore, in vivo19F MRI revealed that PFCE–PLGA NPs produced with microfluidics have similar biodistribution as the batch-made PFCE–PLGA NPs, demonstrating that our method allows for the scaled-up production of NPs maintaining the imaging performance. Finally, they were cleared within 2 weeks, which is typical for NPs that display a multicore structure and is beneficial for their clinical translation.
Overall, we developed a modular microfluidic method that can be used for the synthesis of PFC-loaded NPs and different types of polymeric nanotheranostics in imaging and therapy in the future fostering their translation from the bench to the clinic.
Materials and Methods
Materials
Chemicals for Synthesis and Characterization
Water was purified with a Synergy water purification system from Merck. The following chemicals were used as received: PLGA Resomer RG 502H, acid-terminated, Mw 7000–17,000 g mol–1, with a lactide/glycolide molar ratio of 50:50 was obtained from Evonik Industries AG (Essen, Germany), DCM, acetonitrile (MeCN), chloroform, and AcOEt from VWR, (Netherlands) or Merck (Germany) in at least p.a. quality, PFCE from Exfluor (USA), PFOB from Fluorochem, (UK), PVA, Mw 9000–1000 g mol–1, 80% hydrolyzed and poly(propylene oxide) (PPO) Mn 2700 g mol–1, deuterium oxide (99.9% D), and TFA (reagent plus ) were purchased from Sigma-Aldrich (St. Louis, MO, USA), and ATTO647n dye from Atto-tec (Siegen, Germany).
Chemicals for Cell Study
X-VIVO 15 medium (Lonza, Belgium) and phosphate-buffered saline (PBS) (Braun, Germany) were used. (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT tetrazolium reagent) purchased from Sigma-Aldrich, St Louis, MO, USA, and dimethyl sulfoxide (DMSO) purchased from WAK-Chemie, Steindbach, Germany, were used to kill the cells. EEA1 late endosomal marker was purchased from BD biosciences, LAMP1 early endosomal marker was purchased from Sigma-Aldrich, St Louis, MO, USA, and paraformaldehyde (PFA) was purchased from Merck/Sigma.
Note that the organic solvents used in this study have to be handled and disposed according to the safety regulations provided by the manufacturer and further specified by regulations of a country where the experiments are performed.
Synthesis of NPs
PFC–PLGA NPs were prepared by a continuous-flow microfluidic setup that was assembled as shown schematically on Figure 1a. The setup was placed in the fume hood. The setup consisted of three ultracompact high-pressure pumps, Azura P 4.1S, with a pressure sensor and 50 mL min–1 stainless-steel pump heads (Knauer, Germany), two slit stainless-steel interdigital micromixers (type SIMM V2) (Micro4Industries GmbH, Mainz, Germany), a probe sonifier, tip 13 mm, with a continuous-flow attachment (Branson Sonic Power, St. Louis, USA), operated at 10% amplitude. The different parts were connected via PTFE tubing, 1/16″ outer diameter (OD), (Separations, Hendrik-Ido-Ambacht, Netherlands), 0.2 mm inner diameter (ID) for the PFCE and PLGA phase, and 0.25 mm ID for PVA. The tube between the first and second SIMM was only 6.5 cm long (0.2 mm ID), as it is important that PFCE and PLGA are mixed with each other when entering the second SIMM, and PFCE is very poorly miscible with the PLGA phase without the use of a fluorinated surfactant. However, we wanted to avoid the use of the fluorinated surfactant, as they are often nonbiocompatible and would generate additional signals in 19F MRI, leading to artifacts. The tube between the second SIMM and flow sonifier was 5.5 cm (0.5 mm ID), followed by a Swagelock adaptor.
Three phases of liquids, PFC phase, organic phase, and aqueous phase, were prepared and kept in glass bottles. The PFC phase contained pure PFCE or PFOB. The organic phase consisted of a 1.75 wt % solution of the polymer PLGA in an organic solvent (DCM, AcOEt, MeCN, or chloroform) with the addition of PPO (3.33 mg in 1 mL DCM). For cellular uptake experiments, the Atto-647n dye was also included in the organic phase. The aqueous phase contained 1.96 wt % PVA dissolved in ultrapure water.
In the first mixing step, a liquid PFC was premixed with the organic polymer solution, followed by emulsification with the aqueous PVA solution in the second micromixer. The primary emulsion that was obtained in this step further flowed to the sonifier flow cell, where it was sonicated at 10% amplitude and 1 mm tip height. The emulsion was collected and stirred overnight at room temperature or 4 °C in an open vial to evaporate the solvent. Next, NPs were collected and washed three times with ultrapure water using centrifugation (16,000g for 35 min), followed by freeze-drying.
Characterization Methods
Dynamic Light Scattering
Dynamic light scattering (DLS) for size determination was done using a Malvern Zetasizer Nano ZS at a scattering angle of 173°. Freeze-dried, purified NPs were dispersed in ultrapure water and diluted to a concentration of 0.1 mg mL–1. Malvern software (Zetasizer software, Ink) was used to evaluate the data. The Z-average and the polydispersity of three independent measurements are reported.
Nuclear Magnetic Resonance
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker AVANCE III 400 MHz instrument, equipped with a BBFO probe. A known amount of NPs, typically 5–10 mg, was resuspended in 500 μL of deuterium oxide and mixed with 100 μL of 1 vol % solution of TFA in D2O, which served as an internal reference. The mixture was transferred to NMR tubes and measured with eight scans with an interscan relaxation delay of 25 s. Data were analyzed with Mestrenova 10.0.2.
For relaxation time measurements, 10 mg mL–1 of NP samples in deuterium oxide was transferred to gastight NMR tubes (Wilmad, quick pressure valve NMR tube) and were saturated with oxygen or argon or were left untreated. Either oxygen or argon was bubbled through the solution for 3–4 min. Relaxation times were measured using the inversion recovery sequence after prior determination of the 19F 90° pulse (12.9 μs). The total sweep width ranged from −60 to −135 ppm. Spectra were acquired with eight scans per increment, an acquisition time of 1.15 s, and a relaxation delay dependent on the sample: 8.8 s (Ar), 6 s (atmospheric O2), and 2 s (purged with O2). Mestrenova 10.0.2. was used to analyze the data.
Scanning Electron Microscopy
SEM was done using a LEO Gemini 1530, with a landing voltage (ETH) of 100 V. Aqueous dispersion of purified, freeze-dried NPs (10 mg L–1) was drop-casted on an Si wafer (as deliv., PLANO-EM#G3390).
Cell Culture and Labeling
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy individuals after informed consent using Ficoll density centrifugation (Lymphoprep, STEMCELL Technologies, Vancouver, Canada). Adherent monocytes were cultured in X-VIVO 15 medium supplemented with 2% human serum or in RPMI-1640 medium supplemented with 10% fetal bovine serum and in the presence of interleukin-4 (300 U mL–1) and granulocyte-monocyte colony stimulating factor (450 U/mL) to obtain immature DCs. On day 3, cells were harvested, counted, and labeled with PLGA NPs (resuspended in PBS shortly after the labeling) at a concentration of 2 mg of NPs/1 × 106 cells and incubated at 37 °C for 24 h.
Cell Staining and Confocal Microscopy
The cell uptake of NPs containing the Atto-647 dye was tested with confocal microscopy. Day 3 moDCs were incubated together with NPs at 37 °C for 24 h on glass coverslips (approximately 20,000 cells per coverslip). After 24 h, the excess label was removed, the coverslips with cells were washed gently with PBS, and then, the cells were fixed with 2% PFA. The labeled cells on the coverslips were permeabilized in CLSM buffer + 0.1% Saponin. First, cells were stained with LAMP-1 or EEA1 specific primary antibody, followed by staining with isotype-specific AlexaFluor-conjugated secondary antibodies for intracellular compartments. DAPI was used to stain the nucleus. The stained NP-loaded cells were imaged with an Olympus FV1000 confocal laser scanning microscope. The obtained images were processed in ImageJ.
Cell Viability
To investigate the influence of the particles on cells, an MTT viability assay was performed. A volume of 0.5 × 106 cells were incubated in the presence of different types of NPs (10 mg mL–1) for 24 h. Each condition was performed in triplicate. After incubation, the excess of NPs was removed by gentle washing with PBS. After washing, the cells were collected and plated in a flat-bottom 96-well plate and washed again with PBS by centrifugation for 2 min between each wash. Next, 60 μL of supplemented X-VIVO medium with 10 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (concentration of 4 mg mL–1) was added to each well, followed by 60 min of incubation in the dark at 37 °C. After incubation, the plate was centrifuged for 2 min and 100 μL of lysis buffer (isopropanol, 10% SDS, 2 N HCL, deionized water) was added to each well, followed by 15 min of incubation in the dark at room temperature. Before the measurements, the samples were resuspended to remove any precipitate of crystals. The plate was measured with an iMark microplate reader (Bio-Rad, The Netherlands) at 595 nm. The cells not loaded with NPs were used as the control for living viable cells. The cells treated with DMSO were used as the control for dead cells.
19F Magnetic Resonance Imaging
19F MRI was performed on a preclinical 11.7 T MRI scanner (Biospec 117/16, 500 MHz, Bruker, Ettlingen, Germany). 250 μL microcentrifuge tubes were filled with NPs in ultrapure water (10 mg mL–1) and were placed in a Styrofoam holder. A tube with ultrapure water was added as a control. The tubes were imaged with a 3D RARE sequence with the following parameters: TR = 1500 ms, TE 4.9 ms, turbo factor 32, matrix size = 64 × 90 × 32, FOV = 32 × 45 × 32 mm, number of averages = 2, and scan time = 96 s. Images were made with a narrow excitation bandwidth to selectively excite PFCE at 470.7440706 MHz or the PFOB CF3 group at 470.7480723 MHz.
Ultrasound
For US imaging, NPs were resuspended in ultrapure water at 10 mg/mL concentration followed by sonication in an ultrasonic bath to ensure homogenous resuspension. A volume of 200 μL of resuspended NPs was placed in a well in a gelatin phantom [8% gelatin, Fluka Sigma-Aldrich (St. Louis, MO, USA)]. Images were acquired using a Vevo 2100 ultrasound scanner form FUJIFILM Visualsonics (Toronto, Canada) with a linear array 21 MHz transducer (MS-250, MicroScan). The gain was set to 50 dB and power to 100%.
In Vivo19F MRI
Wild-type 10–14 week-old C57Bl6 female mice weighing 18-22 g were obtained from Charles River Laboratory and maintained under specific pathogen-free conditions at the Central Animal Laboratory in Nijmegen, Netherlands. Experiments were performed according to the guidelines for animal care of the Nijmegen Animal Experiments Committee (DEC AVD1030020173444). Mice (n = 10) were injected intravenously with 20 mg of NPs resuspended in PBS and imaged at various time points after injection (2 h, 1 day, 2 days, 1 week, and 2 weeks).
Mice were anesthetized using isoflurance, 4% induction and 1.5–2% maintenance, to achieve a stable breathing rate of 60–90/min. The core temperature was kept between 36.0 and 37.5 °C using an in-house built water-heated matrass. 19F MRI was performed on a 11.7 T MRI scanner (Biospec, Bruker, Ettlingen, Germany). The fluorine images were acquired using a 3D RARE sequence with a 12:48 min acquisition time and 0.5 × 0.5 × 2 mm voxel size. Imaging parameters were TR 1500 ms, TE 6.62 ms, turbo factor 44, and 32 averages. Narrow excitation bandwidth was used to entirely avoid isofluorane artifacts. For anatomical colocalization, a 1H respiratory gated 2D flash with overlapping FOV was used. The obtained data were processed, and images were made in OsiriX. 19F images are displayed in a red-metal color scheme and proton images are displayed in grey scale.
Acknowledgments
The authors kindly acknowledge Daniel Metzke (Micro4Industries), Wim van der Waal (Branson), Sybille von Bomhard (Fraunhofer IMM), Christian Hofmann (Fraunhofer IMM), Alexandre Colard (ICS), Dennis Löwik (RU), Helene Amatdjais-Groenen (RU), Peter van Dijk (RU), Daniela Wilson (RU), Eric van Dinther (Radboudumc), Oya Tagit (Radboudumc), Massis Krekorian (Radboudumc), Carl G. Figdor (Radboudumc), and I. Jolanda M. de Vries (Radboudumc).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c12020.
Flow parameters, additional characterization, and imaging data (PDF)
Author Contributions
# E.S. and A.H.J.S. contributed equally. O.K. and M.S. contributed equally.
The authors are thankful for the funding from ERC-2014-StG-336454-CoNQUeST, TTW-NWO open technology grant STW-14716, Erasmus staff travel grant for support of OK’s visit at the Institut Charles Sardon, CNRS, Max Plank University of Twente Center for Complex Fluid Dynamics, ERC-2015-PoC-713524-CONQUEST, ERC-2019-PoC-862089-CENYA.
The authors declare no competing financial interest.
Due to production error, this paper was published on the Web on October 21, 2020 with an incorrect funding number and additional sentence added to Figure 3 caption. The corrected version was reposted on October 23, 2020.
Supplementary Material
References
- Cheng Y.; Cheng H.; Jiang C.; Qiu X.; Wang K.; Huan W.; Yuan A.; Wu J.; Hu Y. Perfluorocarbon Nanoparticles Enhance Reactive Oxygen Levels and Tumour Growth Inhibition in Photodynamic Therapy. Nat. Commun. 2015, 6, 8785. 10.1038/ncomms9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Chen Q.; Feng L.; Liu Z. Nanomedicine for Tumor Microenvironment Modulation and Cancer Treatment Enhancement. Nano Today 2018, 21, 55–73. 10.1016/j.nantod.2018.06.008. [DOI] [Google Scholar]
- Hu D.; Zhong L.; Wang M.; Li H.; Qu Y.; Liu Q.; Han R.; Yuan L.; Shi K.; Peng J.; Qian Z. Perfluorocarbon-Loaded and Redox-Activatable Photosensitizing Agent with Oxygen Supply for Enhancement of Fluorescence/Photoacoustic Imaging Guided Tumor Photodynamic Therapy. Adv. Funct. Mater. 2019, 29, 1806199. 10.1002/adfm.201806199. [DOI] [Google Scholar]
- Ren H.; Liu J.; Su F.; Ge S.; Yuan A.; Dai W.; Wu J.; Hu Y. Relighting Photosensitizers by Synergistic Integration of Albumin and Perfluorocarbon for Enhanced Photodynamic Therapy. ACS Appl. Mater. Interfaces 2017, 9, 3463–3473. 10.1021/acsami.6b14885. [DOI] [PubMed] [Google Scholar]
- Wang W.; Cheng Y.; Yu P.; Wang H.; Zhang Y.; Xu H.; Ye Q.; Yuan A.; Hu Y.; Wu J. Perfluorocarbon Regulates the Intratumoural Environment to Enhance Hypoxia-based Agent Efficacy. Nat. Commun. 2019, 10, 1580. 10.1038/s41467-019-09389-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Xue C.; Hou Y.; Li M.; Hu Y.; Chen Q.; Li Y.; Li K.; Song G.; Cai K.; Luo Z. Oxygenated Theranostic Nanoplatforms with Intracellular Agglomeration Behavior for Improving the Treatment Efficacy of Hypoxic Tumors. Biomaterials 2019, 197, 129–145. 10.1016/j.biomaterials.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Deuchar G. A.; Brennan D.; Holmes W. M.; Shaw M.; Macrae I. M.; Santosh C. Perfluorocarbon Enhanced Glasgow Oxygen Level Dependent (GOLD) Magnetic Resonance Metabolic Imaging Identifies the Penumbra Following Acute Ischemic Stroke. Theranostics 2018, 8, 1706–1722. 10.7150/thno.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Kheir J. N.; Polizzotti B. D. Injectable Oxygen: Interfacing Materials Chemistry with Resuscitative Science. Chem.—Eur. J. 2018, 24, 18820–18829. 10.1002/chem.201802054. [DOI] [PubMed] [Google Scholar]
- Koshkina O.; Lajoinie G.; Baldelli Bombelli F.; Swider E.; Cruz L. J.; White P. B.; Schweins R.; Dolen Y.; van Dinther E. A. W.; van Riessen N. K.; Rogers S. E.; Fokkink R.; Voets I. K.; van Eck E. R. H.; Heerschap A.; Versluis M.; de Korte C. L.; Figdor C. G.; de Vries I. J. M.; Srinivas M. Multicore Liquid perfluorocarbon-loaded Multimodal Nanoparticles for Stable Ultrasound and 19F MRI Applied to in vivo Cell Tracking. Adv. Funct. Mater. 2019, 29, 1806485. 10.1002/adfm.201806485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-López R.; Tsapis N.; Fattal E. Liquid Perfluorocarbons as Contrast Agents for Ultrasonography and 19F-MRI. Pharm. Res. 2010, 27, 1–16. 10.1007/s11095-009-0001-5. [DOI] [PubMed] [Google Scholar]
- Kee A. L. Y.; Teo B. M. Biomedical Applications of Acoustically Responsive Phase shift Nanodroplets: Current Status and Duture Directions. Ultrason. Sonochem. 2019, 56, 37–45. 10.1016/j.ultsonch.2019.03.024. [DOI] [PubMed] [Google Scholar]
- Jahromi A. H.; Wang C.; Adams S. R.; Zhu W.; Narsinh K.; Xu H.; Gray D. L.; Tsien R. Y.; Ahrens E. T. Fluorous-Soluble Metal Chelate for Sensitive Fluorine-19 Magnetic Resonance Imaging Nanoemulsion Probes. ACS Nano 2019, 13, 143–151. 10.1021/acsnano.8b04881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C. I.; Briceno J. C. Perfluorocarbon-Based Oxygen Carriers: Review of Products and Trials. Artif. Organs 2010, 34, 622–634. 10.1111/j.1525-1594.2009.00944.x. [DOI] [PubMed] [Google Scholar]
- Ferrari R.; Sponchioni M.; Morbidelli M.; Moscatelli D. Polymer Nanoparticles for the Intravenous Delivery of Anticancer Drugs: the Checkpoints on the Road from the Synthesis to Clinical Translation. Nanoscale 2018, 10, 22701–22719. 10.1039/c8nr05933k. [DOI] [PubMed] [Google Scholar]
- Li X.; Jiang X. Microfluidics for Producing Poly(lactic-co-glycolic acid)-based Pharmaceutical Nanoparticles. Adv. Drug Delivery Rev. 2018, 128, 101–114. 10.1016/j.addr.2017.12.015. [DOI] [PubMed] [Google Scholar]
- Riess J. G. Fluorous Micro- and Nanophases with a Biomedical Perspective. Tetrahedron 2002, 58, 4113–4131. 10.1016/s0040-4020(02)00262-4. [DOI] [Google Scholar]
- Ruiz-Cabello J.; Barnett B. P.; Bottomley P. A.; Bulte J. W. Fluorine (19F) MRS and MRI in Biomedicine. NMR Biomed. 2011, 24, 114–129. 10.1002/nbm.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirotta I.; Dichiarante V.; Pigliacelli C.; Cavallo G.; Terraneo G.; Bombelli F. B.; Metrangolo P.; Resnati G. 19F Magnetic Resonance Imaging (MRI): from Design of Materials to Clinical Applications. Chem. Rev. (Washington, DC, U. S.) 2014, 115, 1106–1129. 10.1021/cr500286d. [DOI] [PubMed] [Google Scholar]
- Janjic J. M.; Ahrens E. T. Fluorine-containing Nanoemulsions for MRI Cell Tracking. WIREs Nanomed. Nanobiotechnol. 2009, 1, 492–501. 10.1002/wnan.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas M.; Boehm-Sturm P.; Figdor C. G.; de Vries I. J.; Hoehn M. Labeling cells for in vivo tracking using 19F MRI. Biomaterials 2012, 33, 8830–8840. 10.1016/j.biomaterials.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Tirotta I.; Dichiarante V.; Pigliacelli C.; Cavallo G.; Terraneo G.; Bombelli F. B.; Metrangolo P.; Resnati G. F-19 Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem. Rev. (Washington, DC, U. S.) 2015, 115, 1106–1129. 10.1021/cr500286d. [DOI] [PubMed] [Google Scholar]
- Swider E.; Staal A. H. J.; Koen van Riessen N.; Jacobs L.; White P. B.; Fokkink R.; Janssen G.-J.; Van Dinther E.; Figdor C. G.; de Vries I. J. M.; Koshkina O.; Srinivas M. Design of Triphasic Poly(lactic-co-glycolic acid) Nanoparticles Containing a Perfluorocarbon Phase for Biomedical Applications. RSC Adv. 2018, 8, 6460–6470. 10.1039/c7ra13062g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swider E.; Daoudi K.; Staal A. H. J.; Koshkina O.; van Riessen N. K.; van Dinther E.; de Vries I. J. M.; de Korte C. L.; Srinivas M. Clinically-Applicable Perfluorocarbon-Loaded Nanoparticles For In vivo Photoacoustic, 19F Magnetic Resonance And Fluorescent Imaging. Nanotheranostics 2018, 2, 258–268. 10.7150/ntno.26208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas M.; Cruz L. J.; Bonetto F.; Heerschap A.; Figdor C. G.; De Vries I. J. M. Customizable, Multi-functional Fluorocarbon Nanoparticles for Quantitative in vivo Imaging Using 19F MRI and Optical Imaging. Biomaterials 2010, 31, 7070–7077. 10.1016/j.biomaterials.2010.05.069. [DOI] [PubMed] [Google Scholar]
- Pisani E.; Tsapis N.; Galaz B.; Santin M.; Berti R.; Taulier N.; Kurtisovski E.; Lucidarme O.; Ourevitch M.; Doan B. T.; Beloeil J. C.; Gillet B.; Urbach W.; Bridal S. L.; Fattal E. Perfluorooctyl Bromide Polymeric Capsules as Dual Contrast Agents for Ultrasonography and Magnetic Resonance Imaging. Adv. Funct. Mater. 2008, 18, 2963–2971. 10.1002/adfm.200800454. [DOI] [Google Scholar]
- Boissenot T.; Fattal E.; Bordat A.; Houvenagel S.; Valette J.; Chacun H.; Gueutin C.; Tsapis N. Paclitaxel-loaded PEGylated Nanocapsules of Perfluorooctyl Bromide as Theranostic Agents. Eur. J. Pharm. Biopharm. 2016, 108, 136–144. 10.1016/j.ejpb.2016.08.017. [DOI] [PubMed] [Google Scholar]
- Boissenot T.; Bordat A.; Larrat B.; Varna M.; Chacun H.; Paci A.; Poinsignon V.; Fattal E.; Tsapis N. Ultrasound-induced Mild Hyperthermia Improves the Anticancer Efficacy of both Taxol® and Paclitaxel-loaded Nanocapsules. J. Controlled Release 2017, 264, 219–227. 10.1016/j.jconrel.2017.08.041. [DOI] [PubMed] [Google Scholar]
- Staal A. H. J.; Becker K.; Tagit O.; Koen van Riessen N.; Koshkina O.; Veltien A.; Bouvain P.; Cortenbach K. R. G.; Scheenen T.; Flögel U.; Temme S.; Srinivas M. , In Vivo Clearance of 19F MRI Imaging Nanocarriers is Strongly Influenced by Nanoparticle Ultrastructure. Biomaterials 2020, 261, 120307. 10.1016/j.biomaterials.2020.120307. [DOI] [PubMed] [Google Scholar]
- Li W.; Zhang L.; Ge X.; Xu B.; Zhang W.; Qu L.; Choi C.-H.; Xu J.; Zhang A.; Lee H.; Weitz D. A. Microfluidic Fabrication of Microparticles for Biomedical Applications. Chem. Soc. Rev. 2018, 47, 5646–5683. 10.1039/c7cs00263g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins J. P.; Torrieri G.; Santos H. A. The Importance of Microfluidics for the Preparation of Nanoparticles as Advanced Drug Delivery Systems. Expert Opin. Drug Delivery 2018, 15, 469–479. 10.1080/17425247.2018.1446936. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Zhu Y.; Shen Y. Microfluidics for Cancer Nanomedicine: From Fabrication to Evaluation. Small 2018, 14, 1800360. 10.1002/smll.201800360. [DOI] [PubMed] [Google Scholar]
- Sheeran P. S.; Matsuura N.; Borden M. A.; Williams R.; Matsunaga T. O.; Burns P. N.; Dayton P. A. Methods of Generating Submicrometer Phase-shift Perfluorocarbon Droplets for Applications in Medical Ultrasonography. IEEE Trans. Ultrason. Ferroelectrics Freq. Contr. 2017, 64, 252–263. 10.1109/tuffc.2016.2619685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte A. R. C.; Ünal B.; Mano J. F.; Reis R. L.; Jensen K. F. Microfluidic Production of Perfluorocarbon-Alginate Core–Shell Microparticles for Ultrasound Therapeutic Applications. Langmuir 2014, 30, 12391–12399. 10.1021/la502822v. [DOI] [PubMed] [Google Scholar]
- EMA . Regulatory Science to 2025 Strategic Reflection, 2020.
- FDA . Quality Considerations for Continuous Manufacturing, 2019.
- Buckinx A. L.; Verstraete K.; Baeten E.; Tabor R. F.; Sokolova A.; Zaquen N.; Junkers T. Kinetic Control of Aggregation Shape in Micellar Self-Assembly. Angew. Chem., Int. Ed. 2019, 58, 13799–13802. 10.1002/anie.201907371. [DOI] [PubMed] [Google Scholar]
- Zaquen N.; Yeow J.; Junkers T.; Boyer C.; Zetterlund P. B. Visible Light-Mediated Polymerization-Induced Self-Assembly Using Continuous Flow Reactors. Macromolecules 2018, 51, 5165–5172. 10.1021/acs.macromol.8b00887. [DOI] [Google Scholar]
- Bleul R.; Thiermann R.; Maskos M. Techniques To Control Polymersome Size. Macromolecules 2015, 48, 7396–7409. 10.1021/acs.macromol.5b01500. [DOI] [Google Scholar]
- de Solorzano I. O.; Uson L.; Larrea A.; Miana M.; Sebastian V.; Arruebo M. Continuous Synthesis of Drug-loaded Nanoparticles Using Microchannel Emulsification and Numerical Modeling: Effect of Passive Mixing. Int. J. Nanomed. 2016, 11, 3397. 10.2147/IJN.S108812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischke C.; Borchert H.-H. Influence of the Primary Emulsification Procedure on the Characteristics of Small Protein-loaded PLGA Microparticles for Antigen Delivery. J. Microencapsulation 2006, 23, 435–448. 10.1080/02652040600612512. [DOI] [PubMed] [Google Scholar]
- Kuchler-Bopp S.; Larrea A.; Petry L.; Idoux-Gillet Y.; Sebastian V.; Ferrandon A.; Schwinté P.; Arruebo M.; Benkirane-Jessel N. Promoting Bioengineered Tooth Innervation Using Nanostructured and Hybrid Scaffolds. Acta Biomater. 2017, 50, 493–501. 10.1016/j.actbio.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Löb P.; Pennemann H.; Hessel V.; Men Y. Impact of Fluid Path Geometry and Operating Parameters on l/l-dispersion in Interdigital Micromixers. Chem. Eng. Sci. 2006, 61, 2959–2967. 10.1016/j.ces.2005.10.076. [DOI] [Google Scholar]
- Zhang F.; Zhuang J.; Esteban Fernández de Ávila B.; Tang S.; Zhang Q.; Fang R. H.; Zhang L.; Wang J. A Nanomotor-Based Active Delivery System for Intracellular Oxygen Transport. ACS Nano 2019, 13, 11996–12005. 10.1021/acsnano.9b06127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riess J. G. Understanding the Fundamentals of Perfluorocarbons and Perfluorocarbon Emulsions Relevant to In Vivo Oxygen Delivery. Artif. Cells, Blood Substitutes, Biotechnol. 2005, 33, 47–63. 10.1081/bio-200046659. [DOI] [PubMed] [Google Scholar]
- Chapelin F.; Capitini C. M.; Ahrens E. T. Fluorine-19 MRI for Detection and Quantification of Immune Cell Therapy for Cancer. J. Immunother. Cancer 2018, 6, 105. 10.1186/s40425-018-0416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas M.; Boehm-Sturm P.; Figdor C. G.; de Vries I. J.; Hoehn M. Labeling Cells for in vivo Tracking Using 19F MRI. Biomaterials 2012, 33, 8830–8840. 10.1016/j.biomaterials.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Cruz L. J.; Tacken P. J.; Bonetto F.; Buschow S. I.; Croes H. J.; Wijers M.; de Vries I. J.; Figdor C. G. Multimodal Imaging of Nanovaccine Carriers Targeted to Human Dendritic Cells. Mol. Pharm. 2011, 8, 520–531. 10.1021/mp100356k. [DOI] [PubMed] [Google Scholar]
- Srinivas M.; Heerschap A.; Ahrens E. T.; Figdor C. G.; Vries I. J. M. d. 19F MRI for Quantitative in vivo Cell Tracking. Trends Biotechnol. 2010, 28, 363–370. 10.1016/j.tibtech.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby C.; Temme S.; Mayenfels F.; Benoit N.; Krafft M. P.; Schubert R.; Schrader J.; Flögel U. Probing Different Perfluorocarbons for in vivo Inflammation Imaging by F-19 MRI: Image Reconstruction, Biological Half-lives and Sensitivity. NMR Biomed. 2014, 27, 261–271. 10.1002/nbm.3059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.