

Abstract
Rare genetic variants are abundant across the human genome, and identifying their function and phenotypic impact is a major challenge. Measuring aberrant gene expression has aided in identifying functional, large-effect rare variants (RVs). Here, we expanded detection of genetically driven transcriptome abnormalities by analyzing gene expression, allele-specific expression, and alternative splicing from multitissue RNA-sequencing data, and demonstrate that each signal informs unique classes of RVs. We developed Watershed, a probabilistic model that integrates multiple genomic and transcriptomic signals to predict variant function, validated these predictions in additional cohorts and through experimental assays, and used them to assess RVs in the UK Biobank, the Million Veterans Program, and the Jackson Heart Study. Our results link thousands of RVs to diverse molecular effects and provide evidence to associate RVs affecting the transcriptome with human traits.
Graphical Abstract
INTRODUCTION:
The human genome contains tens of thousands of rare (minor allele frequency <1%) variants, some of which contribute to disease risk. Using 838 samples with whole-genome and multitissue transcriptome sequencing data in the Genotype-Tissue Expression (GTEx) project version 8, we assessed how rare genetic variants contribute to extreme patterns in gene expression (eOutliers), allelic expression (aseOutliers), and alternative splicing (sOutliers). We integrated these three signals across 49 tissues with genomic annotations to prioritize high-impact rare variants (RVs) that associate with human traits.
RATIONALE:
Outlier gene expression aids in identifying functional RVs. Transcriptome sequencing provides diverse measurements beyond gene expression, including allele-specific expression and alternative splicing, which can provide additional insight into RV functional effects.
RESULTS:
After identifying multitissue eOutliers, aseOutliers, and sOutliers, we found that outlier individuals of each type were significantly more likely to carry an RV near the corresponding gene. Among eOutliers, we observed strong enrichment of rare structural variants. sOutliers were particularly enriched for RVs that disrupted or created a splicing consensus sequence. aseOutliers provided the strongest enrichment signal when evaluated from just a single tissue.
We developed Watershed, a probabilistic model for personal genome interpretation that improves over standard genomic annotation–based methods for scoring RVs by integrating these three transcriptomic signals from the same individual and replicates in an independent cohort.
To assess whether outlier RVs identified in GTEx associate with traits, we evaluated these variants for association with diverse traits in the UK Biobank, the Million Veterans Program, and the Jackson Heart Study. We found that transcriptome-assisted prioritization identified RVs with larger trait effect sizes and were better predictors of effect size than genomic annotation alone.
CONCLUSION:
With >800 genomes matched with transcriptomes across 49 tissues, we were able to study RVs that underlie extreme changes in the transcriptome. To capture the diversity of these extreme changes, we developed and integrated approaches to identify expression, allele-specific expression, and alternative splicing outliers, and characterized the RV landscape underlying each outlier signal. We demonstrate that personal genome interpretation and RV discovery is enhanced by using these signals. This approach provides a new means to integrate a richer set of functional RVs into models of genetic burden, improve disease gene identification, and enable the delivery of precision genomics. ■
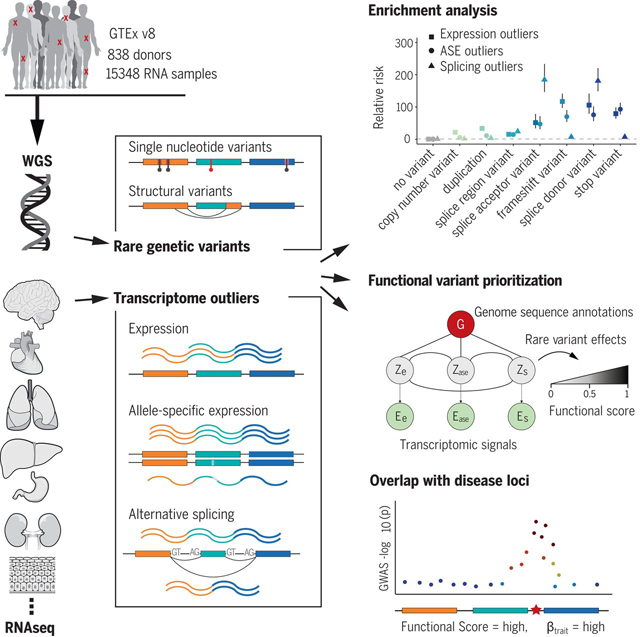
Transcriptomic signatures identify functional rare genetic variation. We identified genes in individuals that show outlier expression, allele-specific expression, or alternative splicing and assessed enrichment of nearby rare variation. We integrated these three outlier signals with genomic annotation data to prioritize functional RVs and to intersect those variants with disease loci to identify potential RV trait associations.
Background
The human genome contains tens of thousands of rare [minor allele frequency (MAF) <1%] variants (1), some of which contribute to rare and common disease risks (2, 3). However, identifying functional rare variants (RVs), especially in the noncoding genome, remains difficult because of their low frequency and the lack of a regulatory genetic code. Outlier gene expression aids in identifying functional, large-effect RVs (4–8). Furthermore, transcriptome sequencing provides diverse measurements beyond gene expression level, including allele-specific expression (ASE) and alternative splicing, that have yet to be systematically evaluated and integrated into variant effect prediction (9–11).
Using 838 samples with both whole-genome and transcriptome samples in the Genotype-Tissue Expression (GTEx) project version 8 (v8), we assessed how rare genetic variants contribute to outlier patterns in total expression (hereafter referred to simply as “expression”), allelic expression, and alternative splicing deep into the allele frequency (AF) spectrum. We integrated these three transcriptomic signals across 49 tissues, along with diverse genomic annotations to prioritize high-impact RVs, and assessed their relationship to complex traits in the UK Biobank (UKBB) (12), the Million Veterans Program (MVP) (13), and the Jackson Heart Study (JHS) (14). We further identified dozens of candidate RVs influencing well-studied disease genes, including APOE, FAAH, and MAPK3.
Results
Detection of aberrant gene expression across multiple transcriptomic phenotypes
We quantified three transcriptional phenotypes for each gene to capture a wide range of functional effects caused by regulatory genetic variants. Briefly, to identify expression outliers (eOutliers), we generated Z scores from corrected expression data per tissue to determine whether a gene in an individual has extremely high or low expression (fig. S1) (15, 16). To identify genes with excessive allelic imbalance [allele-specific expression (ASE) outliers (aseOutliers)] we used ANEVA-DOT (analysis of expression variation–dosage outlier test; figs. S2 and S3) (16, 17). This method uses estimates of genetic variation in dosage of each gene in a population to identify genes for which an individual has a heterozygous variant with an unusually strong effect on gene regulation (17). Splicing outliers (sOutliers) were detected using SPOT (splicing outlier detection), an approach introduced here that fits a Dirichlet-Multinomial distribution directly to counts of reads split across alternatively spliced exon-exon junctions for each gene. SPOT then identifies individuals that deviate significantly from the expectation on the basis of this fitted distribution (figs. S4 to S6) (16). Each of the three methods was applied across all GTEx samples. An individual was called a multitissue outlier for a given gene if its median outlier statistic across all measured tissues exceeded a chosen threshold (Fig. 1A) (16). Using this multitissue approach for each phenotype, we found that each individual had a median of four eOutlier, four aseOutlier, and five sOutlier genes.
Fig. 1. Enrichment of RVs underlying aberrant expression, splicing, and ASE.
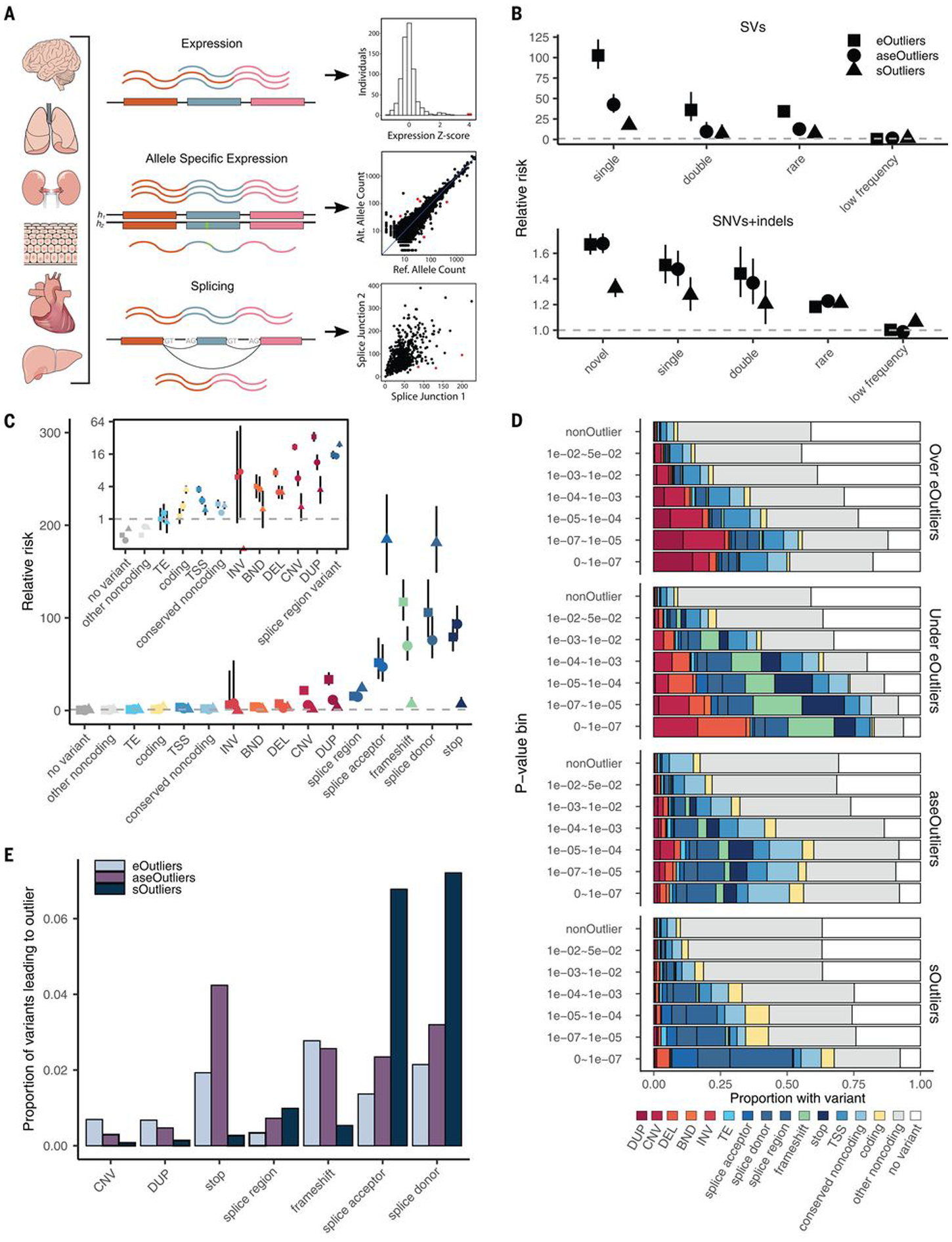
(A) RNA-seq data in 838 individuals were combined across 49 tissues and used to identify shared tissue expression, ASE, and alternative splicing outliers. (B) Relative risk of new (not in gnomAD), singleton, doubleton, rare (MAF <1%), and low-frequency (MAF 1 to 5%) variants in a 10-kb window around the outlier genes across all data types compared with nonoutlier individuals for the same genes. Outliers were defined as those with values >3 SDs from the mean (|median Z| > 3) or, equivalently, a median P < 0.0027. Bars represent the 95% confidence interval. (C) Assigning each outlier its most consequential nearby RV, the relative risk for different categories of RVs falling within 10 kb of each outlier type. The inset panel shows enrichments for a subset of variant categories on a log(2)-transformed y-axis scale for better visibility. (D) Proportion of outliers at a given threshold that have a nearby RV in the given category. eOutlier |median Z scores| were converted to P values using the cumulative probability density function for the normal distribution. TE, transposable element; INV, inversion; BND, break end; DEL, deletion; DUP, duplication. (E) Proportion of RVs in a given category that lead to an outlier at a P-value threshold of 0.0027 across types.
Genes with aberrant expression, ASE, and splicing are enriched for functionally distinct RVs
We observed that multitissue outliers for any of the three transcriptomic phenotypes were significantly more likely to carry a RV (MAF <1%) in the gene body or ±10 kb than individuals without outliers, assessed among 714 individuals with European ancestry. These enrichments were progressively more pronounced for rarer variants and were stronger for structural variants (SVs) than for single-nucleotide variants (SNVs) and indels (Fig. 1B). These trends were not reliant on the specific choice of the threshold used to define outliers (figs. S7 and S8).
We found only 35 cases in which an individual gene was a multitissue outlier for all three transcriptional phenotypes. All but one of these had a nearby RV, and most were annotated as splice variants. Among genes that were outliers for two transcriptional phenotypes in an individual (n = 465), the greatest overlap occurred between aseOutliers and eOutliers (n = 319; fig. S9A). We found that aseOutliers with modest expression changes (1 < |median Z| < 3) showed stronger enrichment for nearby RVs than those without any expression change (fig. S9), highlighting an important benefit of combining these phenotypes to discover diverse RV effects. We found that genes for which no outlier individuals were identified were enriched for Gene Ontology biological process terms relating to sensory perception and detection of chemical stimuli for all outlier types (fig. S10) (16), which is consistent with enrichments seen for genes that do not have any cis-expression quantitative trait loci (eQTLs) discovered in GTEx (18).
We found that different types of genetic variants contribute to outliers for the three molecular phenotypes, although rare splice donor variants were enriched near all outlier types (Fig. 1C). The largest differences in variant type enrichment among the three outlier types were copy number variations (CNVs) and duplications, which were almost exclusively associated with eOutliers, and splice acceptor variants, which were enriched considerably more within sOutliers (fig. S11).
For all phenotypes, the proportion of outliers with a nearby RV of any category increased with threshold stringency (Fig. 1D). For eOutliers, aseOutliers, and sOutliers, at the strictest threshold of median outlier P < 1.1 × 10−7, most individuals were carrying at least one RV nearby the outlier gene (82 to 94%). When looking further at RVs with functional annotations (from the annotations listed in Fig. 1C), we found that underexpressed eOutliers were the most interpretable, with 88% of outlier-associated RVs having an additional functional annotation, whereas aseOutliers had the lowest proportion at 56% (Fig. 1D). This analysis provides further insight into expectations for causal RV types when an outlier effect of a specific magnitude is observed in an individual.
Conversely, a large proportion of genes with nearby rare genetic variants did not appear as outliers, even for the most predictive classes such as loss-of-function variants. The largest proportion of variants leading to any outlier status were rare splice donor and splice acceptor variants, of which only 7.2 and 6.8%, respectively, led to an sOutlier (Fig. 1E and fig. S11). Overall, whereas some transcriptomic effects may have been missed, the low frequency with which RVs of these classes led to large transcriptome changes reinforces the utility of incorporating functional data in variant interpretation even for specific variant classes already used in clinical interpretation.
Genomic position of RVs predicts the impact on expression
Although we primarily assessed RVs that occur either within an outlier gene or in a 10-kb surrounding window, gene regulation can occur at greater distances (19, 20). Because we observed the strongest enrichments for the lowest-frequency variants, we intersected singleton variants [(SVs); i.e., those appearing only once in GTEx and SNVs and/or indels that do not appear in the Genome Aggregation Database (gnomAD) (21)] with 200-kb-length windows exclusive of other windows and upstream from outlier genes and compared their frequency in outlier versus nonoutlier individuals. SNV enrichments dropped off quickly at greater distances from the gene but remained weakly enriched for eOutliers out to 200 kb. The same was true for rare indels, with enrichment at 200 kb only for sOutliers. SVs remained enriched at much longer distances, being enriched 2.33-fold as far as 800 kb to 1 Mb upstream and up to 600 kb downstream of the gene body (Fig. 2A and fig. S12A).
Fig. 2. RV enrichments in specific genomic positions.
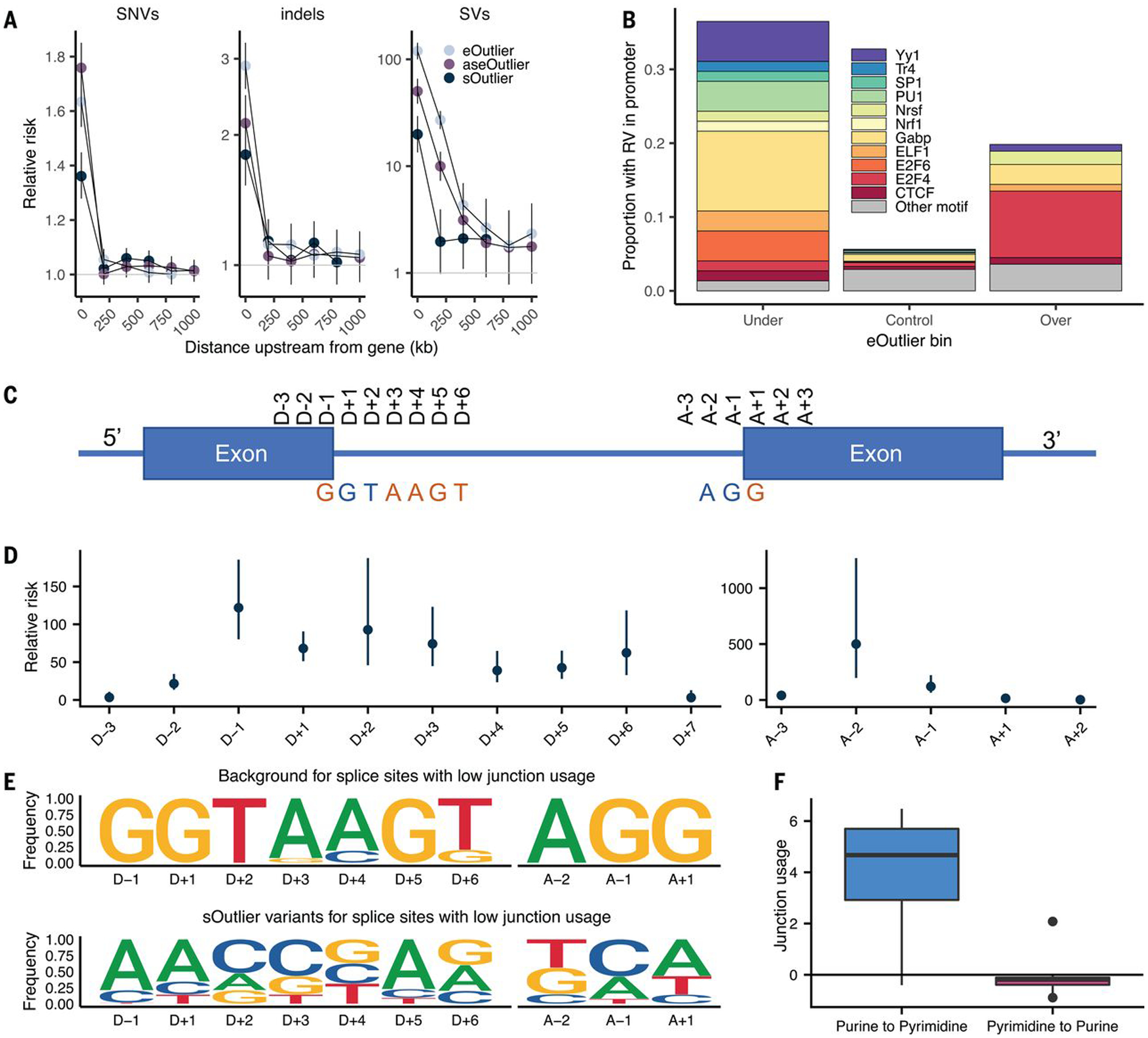
(A) Relative risk of SNVs and indels (not found in gnomAD), and SVs (singleton in GTEx) at varying distances upstream of outlier genes (bins exclusive) across data types. (B) Proportion of eOutliers with TSS RVs in promoter motifs within 1000 bp. Under and over bins are defined with a median Z score threshold of 3, and controls are all individuals with a median Z score <3 for the same set of outlier genes. (C) Graphic summarizing positional nomenclature relative to observed donor and acceptor splice sites. (D) Relative risk (y-axis) of an sOutlier (median LeafCutter cluster P < 1 × 10−5) RV being located at a specific position relative to the splice site (x-axis) compared with nonoutlier RVs. Relative risk calculation was done separately for donor and acceptor splice sites. (E) Independent position weight matrices showing mutation spectra of sOutlier (median LeafCutter cluster P < 1 × 10−5) RVs at positions relative to splice sites with negative junction usage (i.e., splice sites used less in outlier individuals than in nonoutliers). (F) Junction usage of a splice site is the natural log of the fraction of reads in a LeafCutter cluster mapping to the splice site of interest in sOutlier (median LeafCutter cluster P < 1 × 10−5) samples relative to the fraction in nonoutlier samples aggregated across tissues by taking the median (16). Junction usage (y-axis) of the closest splice sites to RVs that lie within a polypyrimidine tract (A − 5, A − 35) binned by the type of variant (x-axis).
RVs in promoter regions have been previously linked to outlier expression (5, 15). To extend these observations and to assess the types of transcription factor (TF)–binding sites that could lead to outliers, we tested enrichment of rare transcription start site (TSS) proximal variants in specific TF motifs near under- and over-eOutliers. For under-eOutliers, we saw an enrichment of variants in GABP, a TF that activates genes that control the cell cycle, differentiation, and other critical functions (22). For over-eOutliers, we saw an enrichment of RVs intersecting the E2F4 motif, a TF that has been reported as a transcriptional repressor (23). In both under- and over-eOutliers, we saw RVs in YY1, which can act as either an activator or repressor, depending on context (24), and has been associated with GABP in coregulatory networks (Fig. 2B and fig. S12B) (25). Thus, these naturally occurring RV perturbations can provide information about how specific TFs can strongly up- or down-regulate their target genes.
RVs can affect multiple genes and lead to new gene fusion
We observed that RVs can also affect multiple genes in an individual. We found a strong enrichment for multigene effects among eOutliers and, to a lesser degree, aseOutliers (fig. S13). As expected, we did not see enrichment for nearby sOutlier pairs, which are less subject to coregulation (26). Within a 100-kb window, neighboring eOutlier genes were 70 times more frequent than would be expected by chance if drawing outlier pairs at random. They were also significantly enriched for rare CNVs, duplications, and TSS variants nearby one or both genes compared with individuals who had outlier expression but for only one of the genes (fig. S13). We also found that rare SV enrichments were present near eOutliers regardless of whether the SV overlapped the gene itself (fig. S14). We observed 27 examples of rare SVs, including deletions, duplications, and break ends, associated with eOutliers in at least two genes in the same individual (fig. S15 and table S1). For one of these, we observed evidence of a fusion transcript resulting from a deletion spanning the end of the gene SPTBN1 and the TSS of EML6. This deletion led to underexpression of SPTBN1 (median Z score = −4.67) and overexpression of EML6 (median Z score = 8.12) compared with all other individuals. Supporting the presence of a new germline fusion transcript, we found evidence of a specific transcript spanning both SPTBN1 and EML6 in multiple tissues for the individual with the deletion (fig. S16). For both of these genes, this individual also showed sOutlier signal (median SPOT P = 0.0005 for EML6 and 0.0035 for SPTBN1). The identification of fusion transcripts has been of particular interest in cancer diagnosis and prognosis (27–30), and both EML genes and SPTBN1 have been previously implicated in cancer-associated fusions (31, 32).
RVs in splicing consensus sequence drive splicing outliers
Previous studies have shown RVs disrupting splice sites result in outlier alternative splicing patterns (33, 34). We used sOutlier calls made for each LeafCutter cluster (16, 35) to assess enrichment of splicing-related variants more precisely. We observed extreme enrichment of RVs near splice sites in sOutliers. An sOutlier was 333 times more likely than a nonoutlier to harbor a RV within a 2-bp window around a splice site (fig. S17A) (16), with signal decaying at greater distances but still enriched up to 100 bp away (relative risk = 7.43). To obtain base pair resolution enrichments, we computed the relative risk of sOutlier RVs located at specific positions relative to observed donor and acceptor splice sites (16). Ten positions near the splice site showed significant enrichment for RVs in sOutliers compared with controls (Fig. 2, C and D). These positions corresponded precisely to positions that have also been shown to be intolerant to mutations because of their conserved role in splicing (we will refer to these positions as the splicing consensus sequence) (34). Among the most enriched positions within the splicing consensus sequence were the four essential splice site positions (D + 1, D + 2, A − 2, A − 1) (36), which showed an average relative risk of 195.
sOutliers further captured the transcriptional consequences both for variants that disrupted a reference splicing consensus sequence and those that created a new splicing consensus sequence. Individuals with sOutlier variants in which the rare allele deviated away from the splicing consensus sequence showed decreased junction usage of the splice site near the variant, whereas individuals with variants in which the rare allele created a splicing consensus sequence showed increased junction usage of the splice site near the variant relative to nonoutliers (Fig. 2E and figs. S17B and S18) (16). We saw a related enrichment pattern after separating annotated and new (unannotated) splice sites (fig. S19). sOutliers were also enriched for RVs positioned within the polypyrimidine tract (PPT), a highly conserved, pyrimidine-rich region, ~5 to 35 bp upstream from acceptor splice sites (37). A RV was 6.25 times more likely to be located in the PPT near an sOutlier relative to a nonoutlier. sOutliers with a RV that changed a position in the PPT from a pyrimidine to a purine (i.e., disrupting an existing PPT) showed decreased junction usage of the splice site near the variant, whereas the inverse was true for variants that changed a position in the PPT from a purine to pyrimidine (Fig. 2F and fig. S20).
RVs in tissue-specific regulatory regions can lead to tissue-specific outlier expression
Although multitissue outliers offer improved power to detect RV effects, we also evaluated RVs from outliers detected in individual tissues. Single-tissue measurements are subject to greater variation than repeat measurements across tissues but are representative of most experimental designs. First, we performed replication analysis across all individuals with data available for the three methods to evaluate the degree to which outlier status detected in one tissue of an individual was replicated in other tissues (16). On average, we found that eOutlier, aseOutlier, and sOutlier status in a discovery tissue was detected in a test tissue 5.1, 10.7, and 8.7% of the time, respectively (Fig. 3A and fig. S21). This is consistent with other findings that measurements of ASE are more consistent across tissues (18). Considering clinically accessible tissues, namely whole blood, fibroblasts, and lymphoblastoid cells, if we consider outliers observed for a gene in at least two of these tissues in the same individual, we saw average replication rates across all other tissues of 14.1, 20.9, and 15.0% for eOutliers, aseOutliers, and sOutliers, respectively (fig. S22). Both the higher replication rate for aseOutliers and the increase in outlier replication in non-accessible tissues when considering more than one accessible measurement are informative for the analysis of functional data from easily accessible tissues to understand disease states most relevant to other tissues.
Fig. 3. Single-tissue outlier enrichments and replication.
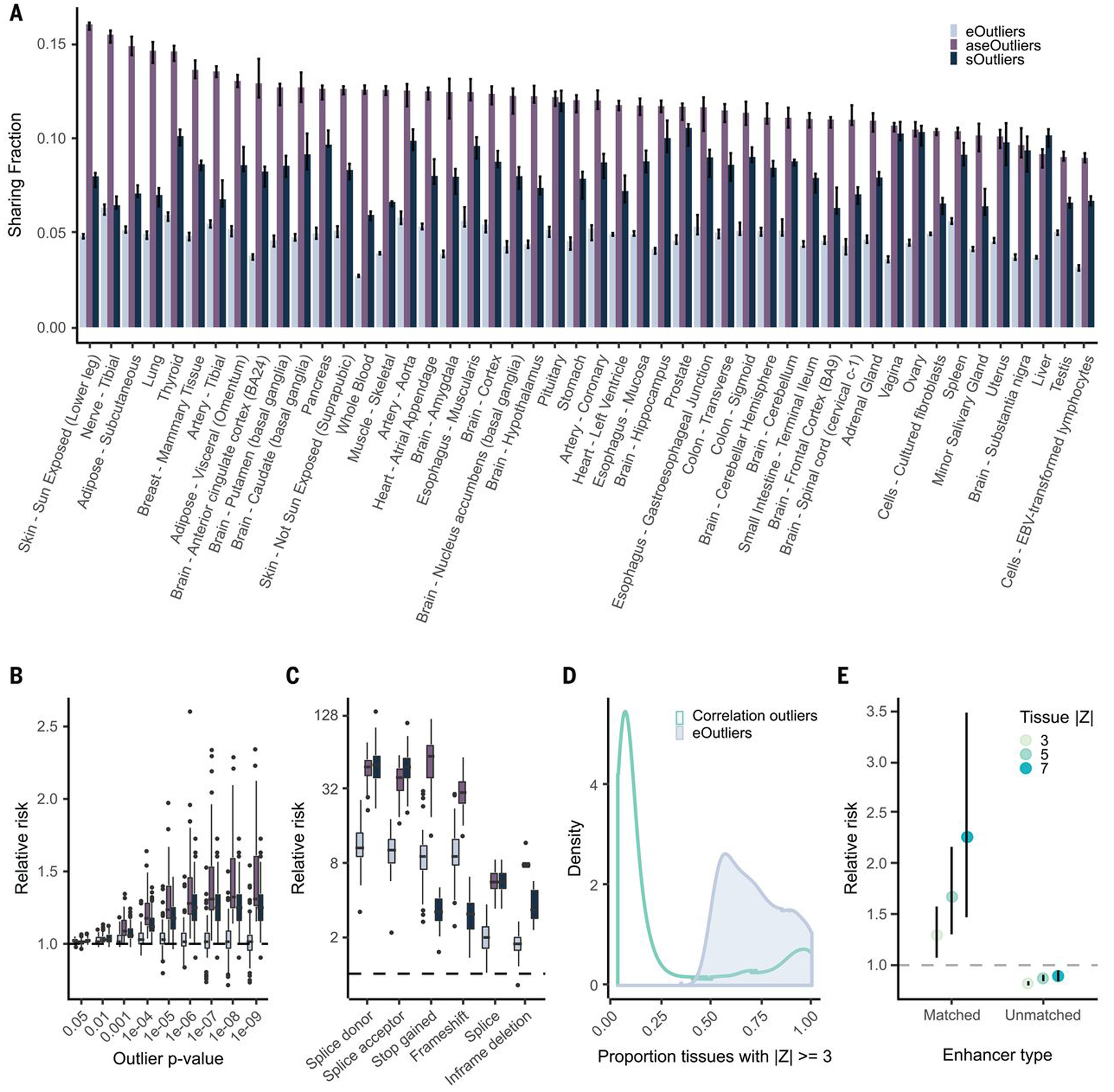
(A) Median replication of outliers identified per tissue across every other tissue for each outlier type. (B) Relative risk point estimate for nearby rare SNVs for outliers across all tissues individually. (C) Relative risk enrichments for likely gene disrupting RVs nearby single-tissue outliers at a threshold of |Z| > 4 (equivalently SPOT or ANEVA-DOT P < 0.000063), with one point per tissue. (D) Distribution of number of tissues with aberrant expression underlying expression outliers defined by median Z score (eOutliers) or Mahalanobis distance P value (correlation). (E) Relative risk of correlation outliers driven by a single tissue, defined as significant correlation outliers for which an expression change of the degree indicated by the point color is observed in only a single tissue (16) carrying a RV in enhancers annotated to that tissue within a 500-kb window of the outlier gene. Unmatched are defined as all tissue-specific enhancer regions regardless of outlier tissue.
We next evaluated the ability of single-tissue outliers from each method to prioritize RVs near outlier genes. Single-tissue aseOutliers were most enriched for nearby RVs, followed by sOutliers and then eOutliers, across all outlier cutoff thresholds (Fig. 3B and fig. S21 and S23A). We also observed enrichment of variants likely triggering nonsense-mediated decay among single-tissue eOutliers, aseOutliers, and sOutliers (Fig. 3C and fig. S23B). Additionally, we found that single-tissue sOutliers still showed strong enrichment for RVs in the splicing consensus sequence and the PPT (fig. S24).
Except for rare SVs that notably were enriched at comparable thresholds to multitissue eOutliers, single-tissue eOutliers show far weaker enrichments relative to multitissue outliers for nearby rare SNVs and indels across all thresholds (fig. S25). To improve discovery of tissue-specific outliers, we leveraged the breadth of tissue data available and used observed patterns of correlation across tissues to detect outliers that deviate from the expected covariance of expression in a subset of tissues (16). A similar approach has been implemented to identify functional RVs on the basis of the correlation of expression among genes in a single tissue (5). We found that outliers identified using this approach were often driven by expression changes in one or a few tissues compared with multitissue eOutliers based on median Z scores (Fig. 3D). The correlation tissue-specific outliers were also enriched for nearby RVs in a 10-kb window around the gene (fig. S26C). However, these outliers were also enriched for RVs in enhancers that were active in the tissue(s) driving the outlier effect (table S2), as determined by single-tissue Z score and within a 500-kb window around the gene (Fig. 3E). Notably, these tissue-specific outliers were depleted for rare variation in enhancers annotated in other, unmatched tissues.
Prioritizing RVs by integrating genomic annotations with diverse personal transcriptomic signals
To incorporate diverse transcriptome signals into a method to prioritize RVs, we developed Watershed, an unsupervised probabilistic graphical model that integrates information from genomic annotations of a personal genome (table S3) with multiple signals from a matched personal transcriptome. Watershed provides scores that can be used for personal genome interpretation or for cataloging potentially impactful rare alleles, quantifying the posterior probability that a variant has a functional effect on each transcriptomic phenotype based on both whole-genome–sequencing (WGS) and RNA-sequencing (RNA-seq) signals (Fig. 4A). The Watershed model can be adapted to any available collection of molecular phenotypes, including different assays, different tissues, or different derived signals. Further, Watershed automatically learns Markov random field (MRF) edge weights reflecting the strength of the relationship between the different tissues or phenotypes included that together allow the model to predict functional effects accurately.
Fig. 4. Prioritizing functional RVs with Watershed.
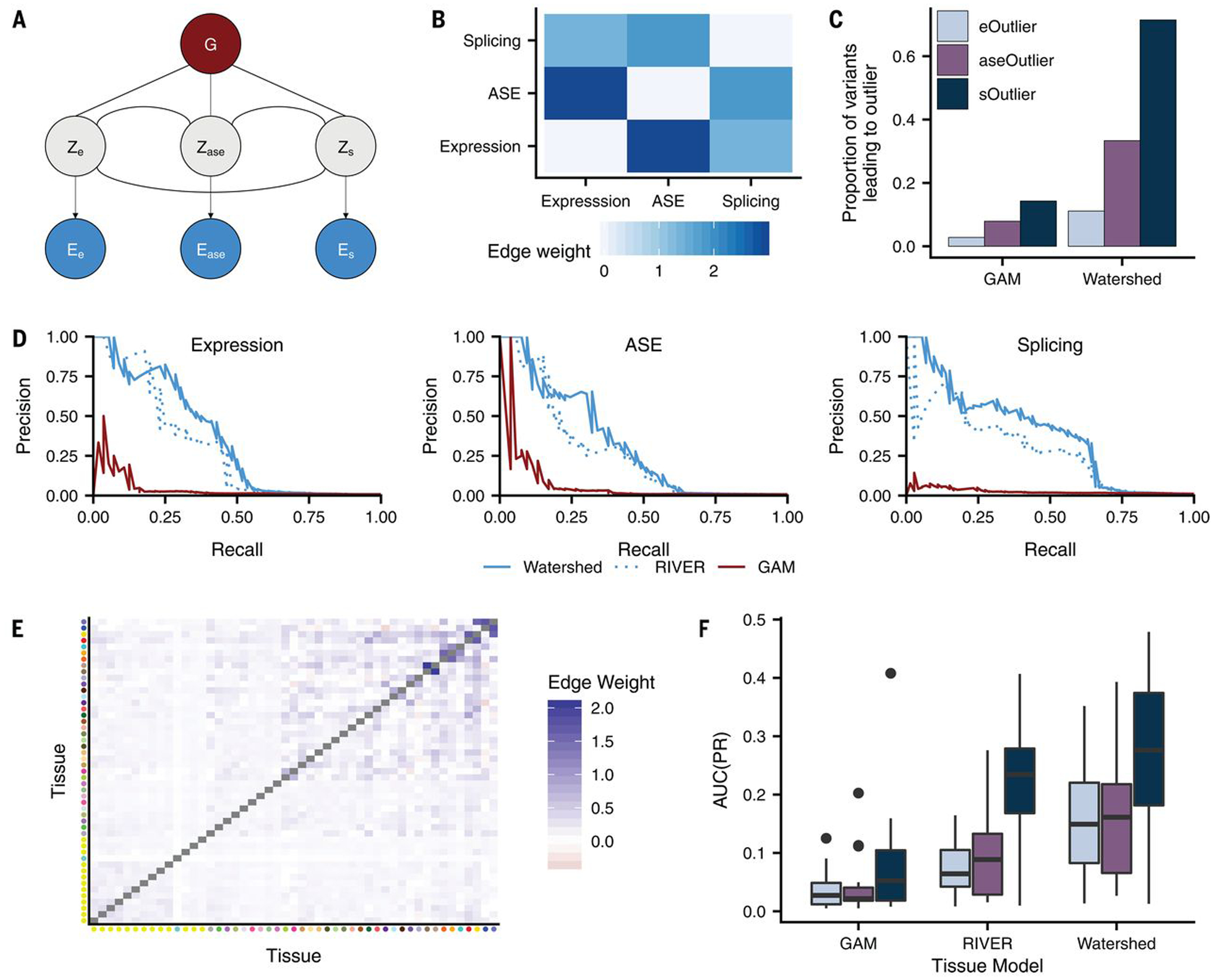
(A) Graphic summarizing plate notation for the Watershed model when it is applied to three median outlier signals (expression, ASE, and splicing). (B) Symmetric heatmap showing learned Watershed edge parameters (weights) between pairs of outlier signals after training Watershed on three median outlier signals. (C) The proportion of RVs with Watershed posterior probability >0.9 (right) and with GAM probability greater than a threshold set to match the number of Watershed variants for each outlier signal (left) that lead to an outlier at a median P-value threshold of 0.0027 across three outlier signals (colors). Watershed and GAM models were evaluated on held-out pairs of individuals. (D) Precision-recall curves comparing performance of Watershed, RIVER, and GAM (colors) using held-out pairs of individuals for three median outlier signals. (E) Symmetric heatmap showing learned tissue-Watershed edge parameters (weights) between pairs of tissue outlier signals after training tissue-Watershed on eOutliers across single tissues. Tissue color to tissue name mapping can be found in fig. S21D. (F) Area under precision recall curves [AUC(PR); y-axis] in a single tissue between tissue-GAM, tissue-RIVER, and tissue-Watershed (x-axis) when applied to outliers across single tissues in all three outlier signals (colors). Precision recall curves in each tissue were generated using held-out pairs of individuals.
We first applied Watershed to the GTEx v8 data using the three outlier signals examined here, expression, ASE, and splicing (Fig. 4A) (16), for which each was first aggregated by taking the median across tissues for the corresponding individual. In agreement with existing evidence of similarity between outlier signals (fig. S9), the learned Watershed edge parameters were strongest between ASE and expression, followed by ASE and splicing, but strictly positive for all pairs of outlier signals (i.e., each outlier signal was informative of all other signals; Fig. 4B). To evaluate our model, we used held-out pairs of individuals that shared the same RV, making Watershed predictions in the first individual and evaluating those predictions using the second individual’s outlier status as a label (15, 16). Watershed outperforms methods based on genome sequence alone [our genomic annotation model (GAM) and combined annotation-dependent depletion (CADD); Fig. 4C and fig. S27] (38, 39). We also compared performance of Watershed with RIVER [RNA-informed variant effect on regulation (15)], a simplification of the Watershed model in which each outlier signal is treated independently. We found that explicitly modeling the relationship between different molecular phenotypes provided a performance gain for Watershed (Fig. 4D, figs. S28 and S29, and table S4) (16). We observed that even the most predictive genomic annotations only resulted in eOutliers, aseOutliers, and sOutliers 2.8, 7.9, and 14.3% of the time, respectively (Figs. 1E and 4C). However, integrating transcriptomic signals with genomic annotations from Watershed (at a posterior threshold of 0.9) detected SNVs that resulted in eOutliers, aseOutliers, and sOutliers with greater frequency 11.1, 33.3, and 71.4% of the time, respectively (Fig. 4C and fig. S30).
We further extended the Watershed framework to prioritize variants on the basis of their predicted tissue-specific impact. We trained three “tissue-Watershed” models (one for each of expression, ASE, and splicing separately), in which each model considers the effects in all tissues jointly, sharing information in the MRF, and ultimately outputs 49 tissue-specific scores for each RV (figs. S29 and S31) (16). We observed that the parameters learned for each of the three tissue-Watershed models resembled known patterns of tissue similarity (Fig. 4E and fig. S32) (18). Further, using held-out individuals, the tissue-Watershed model outperformed a RIVER model in which each tissue is treated completely independently (P = 2.00 × 10−5, P = 2.00 × 10−5, and P = 5.90 × 10−3 for expression, ASE, and splicing, respectively; one-sided binomial test; Fig. 4F and figs. S33 and S34) and a collapsed RIVER model trained with single median outlier statistics (P = 0.0577, P = 0.251, and P = 0.00128 for expression, ASE, and spicing, respectively; one-sided binomial test; figs. S35 and 36). Critically, integrative models that incorporated transcriptomic signal and genomic annotations from a single tissue still outperformed methods based only on genome sequence annotations (Fig. 4F), supporting the benefit of collecting even a single RNA-seq sample to improve personal genome interpretation.
Replication and experimental validation of predicted RV transcriptome effects
We first assessed the replication of “candidate causal RVs” previously identified by the SardiNIA Project (6), using GTEx Watershed prioritization. Of five SardiNIA candidate causal RVs that were also present in a GTEx individual, four had high (>0.7) GTEx Watershed expression posterior probabilities (table S5). Next, we tested replication of GTEx RVs, prioritized by Watershed, in an independent cohort evaluating 97 whole-genome and matched transcriptome samples from the Amish Study of Major Affective Disorders (ASMAD) (40). We evaluated GTEx RVs also present in this cohort at any frequency, quantifying eOutlier, aseOutlier, and sOutlier signal in each ASMAD individual harboring one of the GTEx variants (16). For all three phenotypes, ASMAD individuals with variants having high (>0.8) Watershed posterior probability based on GTEx data had significantly more extreme outlier signals at nearby genes compared with individuals with variants having low (<0.01) GTEx Watershed posterior probability (expression: P = 2.729 × 10−6, ASE: P = 2.86 × 10−3, and splicing: P = 5.86 × 10−13; Wilcoxon rank-sum test; fig. S37). Every variant with a high GTEx Watershed splicing posterior probability (>0.8) resulted in an sOutlier (P ≤ 0.01) in the ASMAD cohort. Furthermore, ASMAD individuals with variants having high (>0.8) GTEx Watershed posterior probability had significantly larger outlier signals relative to equal size sets of variants prioritized by GAM (expression: P = 0.00129, ASE: P = 0.0287, and splicing: P = 0.00058; Wilcoxon rank-sum test; fig. S37). Overall, RVs prioritized by Watershed using GTEx data displayed evidence of functional effects in ASMAD individuals.
We further applied both a massively parallel reporter assay (MPRA) and a CRISPR-Cas9 assay to assess the impact of Watershed-prioritized RVs. We experimentally tested the regulatory effects of 52 variants with moderate Watershed expression posterior (≥0.5) and 98 variants with low Watershed expression posterior (<0.5) using MPRA (16). We observed increased effect sizes for RVs with high Watershed expression posterior relative to variants with low expression posterior (P = 0.025; one-sided Wilcoxon rank-sum test; fig. S38 and table S6). Next, we assessed the functional effects of 20 variants by editing them into inducible-Cas9 293T cell lines. These included 14 rare stop-gained variants and six non-eQTL common variants as negative controls. Of the 14 rare stop-gained variants, 13 had expression or ASE Watershed posterior >0.8, with the remaining variant [previously tested in (41)] having a posterior of 0.22. All control variants had Watershed posteriors <0.03. Of the 13 variants with a Watershed posterior >0.8, 12 showed a significant decrease in expression of the rare allele (P < 0.05, Bonferroni corrected; fig. S39 and table S7) (16).
Aberrant expression informs RV trait associations
We found that each individual had a median of three eOutliers, aseOutliers, and sOutliers (median outlier P < 0.0027) with a nearby RV. When filtering by moderate Watershed posterior probability (>0.5) of affecting expression, ASE, or splicing, individuals had a median of 17 genes with RVs predicted to affect expression, 27 predicted to affect ASE, and nine predicted to affect splicing (Fig. 5A). From the set of outlier calls, we found multiple instances of RVs influencing well-known and well-studied genes, including APOE and FAAH (table S8). In particular, for APOE, which has been associated with numerous neurological diseases and psychiatric disorders (42), we found two aseOutlier individuals both carrying a rare, missense variant, rs563571689, with ASE Watershed posteriors >0.95, not previously reported. For FAAH, which has been linked to pain sensitivity in numerous contexts (43, 44), we found two eOutlier individuals with a rare 5′ untranslated region variant, rs200388505, with ASE and expression Watershed posteriors >0.9.
Fig. 5. Trait associations for RVs underlying outlier genes.
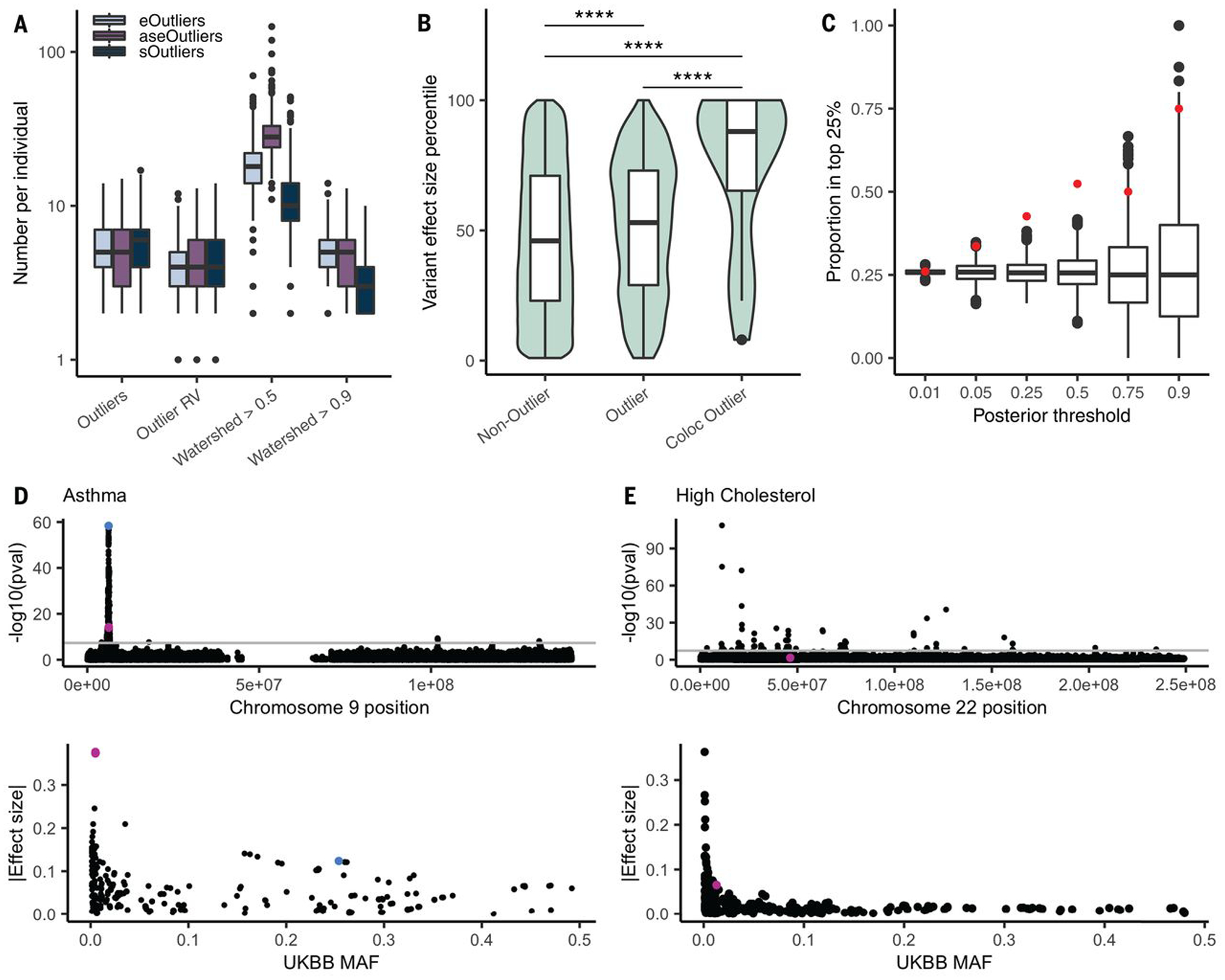
(A) Distribution of the number of outlier genes, outlier genes with a nearby RV, and genes with a high Watershed posterior variant per data type. We added one to all values so that individuals with 0 are included. (B) Distribution of effect sizes, transformed to a percentile, for the set of GTEx RVs that appear in UKBB and are not outlier variants, those that are outlier variants, and those outlier variants that fall in colocalizing genes for the matched trait across 34 traits. Percentiles were calculated on the set of rare GTEx variants that overlap UKBB. The set of genes was restricted to those with at least one outlier individual in any data type and a nearby variant included in the test set (4787 variants and 1323 genes). P values were calculated from a one-sided Wilcoxon rank-sum test. (C) Proportion of variants filtered by Watershed posterior that fell in the top 25% of effect sizes for a colocalized trait (red) and the proportion of randomly selected variants of an equal number that also fall in these regions over 1000 iterations (black). (D) Manhattan plot (top) across chromosome 9 for asthma in the UKBB, filtered for non–low-confidence variants, with two high-Watershed variants, rs149045797 and rs146597587, shown in pink and the lead colocalized variant, rs3939286, shown in blue. The variants’ effect size ranks were similarly high for both self-reported and diagnosed asthma, but the summary statistics are shown for asthma diagnosis here. The UKBB MAF versus absolute value of the effect size for all variants within 10 kb of the Watershed variant is also shown (bottom). (E) Manhattan plot across chromosome 22 for self-reported high cholesterol in the UKBB, filtered to remove low confidence variants, with the high-Watershed variant rs564796245 shown in pink. The UKBB MAF versus absolute value of the effect size for all variants within 10 kb of the Watershed variant is also shown (bottom).
To assess whether identified rare functional variants from GTEx associate with traits, we intersected this set with variants present in the UKBB (12). We focused on a subset of 34 traits for which GWAS association for a UKBB trait had evidence of colocalizations with eQTLs and/or alternative splicing QTLs (sQTLs) in any tissue (table S9) (16, 45). GTEx has demonstrated that genes with RV associations for a trait are strongly enriched for their eQTLs colocalizing with GWAS signals for the same trait (18), indicating that QTL evidence can be used to guide RV analysis. Furthermore, RVs near GTEx outliers had larger trait association effect sizes than background RVs near the same set of genes in the UKBB data (P = 3.51 × 10−9; one-sided Wilcoxon rank-sum test), with a shift in median effect size percentile from 46 to 53%. Notably, outlier variants that fell in or nearby genes with an eQTL or sQTL colocalization had even larger effect sizes (median effect size percentile 88%) than nonoutlier variants (P = 1.93 × 10−5; one-sided Wilcoxon rank-sum test) or outlier variants falling near any gene not matched to a colocalizing trait (P = 4.88 × 10−5; one-sided Wilcoxon rank-sum test; Fig. 5B).
Although most variants tested in UKBB had low Watershed posterior probabilities of affecting the transcriptome (fig. S40A), we hypothesized that filtering for those variants that do have high posteriors would yield variants in the upper end of the effect size distribution for a given trait. For each variant tested in UKBB, we took the maximum Watershed posterior per variant and compared this with a genomic annotation-defined metric, CADD (38, 39). We found that Watershed posteriors were a better predictor of variant effect size than CADD scores for the same set of RVs in a linear model (Table 1). Across different Watershed posterior thresholds, we found that the proportion of variants falling in the top 25% of RV effect sizes in colocalized regions exceeded the proportion expected by chance (Fig. 5C). Whereas filtering by CADD score did return some high effect size variants, this proportion declined at the highest thresholds (fig. S40D). Furthermore, there was very little overlap between variants with high Watershed posteriors and high CADD variants (fig. S40D), with CADD variants more likely to occur in coding regions and Watershed variants more frequent in noncoding regions (fig. S40D). Thus, the approaches largely identified distinct and complementary sets of variants for these traits.
Table 1. Watershed and CADD as predictors of variant effect size percentile.
Shown are the coefficient estimates and 95% confidence intervals from separate linear models with variant effect size percentile as the response and CADD score or Watershed posterior (scaled to have a mean of 0 and an SD of 1 so that values are of comparable range) as the predictor for all tested variants in colocalized regions (n = 5277).
| Predictor | Beta | P value | 95% confidence interval |
|---|---|---|---|
| Watershed posterior | 1.61 | 2.12 × 10−6 | 0.95–2.27 |
| CADD score | 0.77 | 2.41 × 10−2 | 0.10–1.43 |
We identified 33 rare GTEx variant trait combinations in which the variant had a Watershed posterior >0.5 and fell in the top 25% of variants by effect size for the given trait (table S10). We highlight two such examples, for asthma and high cholesterol (Fig. 5, D and E), showing that although RVs usually do not have the frequency to obtain genome-wide significant P values, when they are prioritized by the probability of affecting expression, we could identify those with greater estimated effect sizes on the trait (table S11). In the case of asthma, the RV effect sizes in UKBB were three times greater than the lead colocalized variant. These variants included rs146597587, which is a high-confidence loss-of-function splice acceptor with an overall gnomAD AF of 0.0019, and rs149045797, an intronic variant with a frequency of 0.0019, both of which were associated with the gene IL33, the expression of which has been implicated in asthma (46, 47). Previous work has identified the protective association between rs146597587 and asthma (48, 49), and we found that this is potentially mediated by outlier allelic expression of IL33 leading to moderate decreases in total expression, with median Z scores ranging from −1.08 to −1.77 in individuals with the variant, and median single-tissue Z scores across the six individuals exceeding −2 in 10 tissues. An asthma association had also been reported recently for the other high Watershed asthma-associated variant rs149045797 and was in perfect linkage disequilibrium with rs146597587 (50). An additional high Watershed variant, rs564796245, an intronic variant in TTC38 with a gnomAD AF of 0.0003, had a high effect size for self-reported high cholesterol in the UKBB but was not previously reported. We were able to test this variant against four related blood lipids traits in the MVP (51). We found that for these traits, which included high-density lipoprotein (HDL), low-density lipoprotein, total cholesterol, and triglycerides, among rare (gnomAD AF <0.1%) variants within a 250-kb window of rs564796245, this variant was in the top 5% of variants by effect size; for HDL specifically, it was in the top 1% (fig. S41). We also assessed this variant’s association with the same four traits in the JHS (14), an African American cohort in which four individuals carried the RV. Here, we found that the direction of effect was consistent with MVP and UKBB for all four traits (tables S11 and S12), and the variant fell in the top 28th to 38th percentile of all rare (gnomAD AF <0.1%) variants in this region (fig. S42). Only four of the variants tested in UKBB had Watershed posterior probabilities >0.9 for colocalized genes, but of those, three showed high effect sizes for a relevant trait (table S10).
Discussion
RVs are abundant in human genomes, yet they have remained difficult to study systematically. Using multitissue transcriptome and whole-genome data from GTEx v8, we have been able to identify and assess the properties of RVs, including SVs, that underlie extreme changes in expression, alternative splicing, and ASE.
We observed that each signal informs distinct classes of RVs, demonstrating the benefit of integrating multiple sources of personal molecular data to improve variant interpretation. We expanded characterization of the properties of RVs in multiple contexts, including structural variants affecting multiple genes, rare splice variants that disrupt or create splicing consensus sequences, and RVs occurring in tissue-specific enhancers leading to tissue-specific eOutliers. Together, these provide a map of the properties of large-effect RVs, aiding their identification and evaluation in future studies. We note that although our approach can be used to identify some large-effect RVs underlying disease, it is unlikely to capture the full spectrum of functional RVs contributing to heritability because some effects will not manifest as clear transcriptome aberrations (8).
We further developed a probabilistic model for personal genome interpretation, Watershed, which improves standard methods by integrating multiple transcriptomic signals from the same individual. Relevant to ongoing efforts to identify RVs affecting human traits, we found that in RVs within trait-colocalized regions, filtering by Watershed posteriors can identify variants with larger trait effect sizes better than relying on genomic annotations alone. As further demonstrated by our discovery of outlier RVs in well-studied disease genes, application of Watershed and other integrative methods will prove increasingly helpful for cataloging and prioritizing RVs affecting traits, especially those at the lowest ends of the AF spectrum. Our results provide a means to improve the quality and extent of RV prioritization, with potential future impacts enhancing RV association testing and disease gene identification.
Materials and methods summary
Detailed materials and methods are available in the supplementary materials. Briefly, we used RNA-seq and WGS data from the v8 release of the GTEx project, which contains 49 biological tissues with at least 70 samples per tissue.
For the set of RVs analyzed, we retained all SNVs and indels that passed quality control in the GTEx v8 variant call format file using the hg38 genome build. Structural variants were called on the subset of individuals available in the GTEx v7 release. We defined RVs as those with <1% MAF within GTEx and, for SNVs and indels, also occurring at <1% frequency in non-Finnish Europeans within gnomAD (21). Annotation of protein-coding regions and TF-binding site motifs was generated by running Ensembl VEP (v88).
We next used the RNA-seq data to make outlier calls in each tissue. Briefly, we log2-transformed the expression values [log2(TPM + 2)], where TPM is the number of transcripts per million mapped reads, restricted to lincRNA and protein-coding genes with at least six reads and TPM >0.1 in at least 20% of individuals. We scaled the expression of each gene to mean of 0 and a standard deviation of 1 to avoid the deflation of outlier values caused by quantile normalization. We corrected for hidden factors using PEER [probabilistic estimation of expression residuals (52)] to account for unmeasured technical confounders, as well as the top three genotype principal components, sex, and the genotype of the strongest cise-QTL per gene in each tissue. We rescaled the residual values per gene and used the resulting corrected Z scores to determine eOutliers.
ASE outlier calls in a single tissue were made using ANEVA-DOT to identify genes in each individual that showed excessive allelic imbalance of ASE relative to the population. Briefly, ANEVA-DOT relies on tissue-specific estimates of genetic variation in gene dosage, VG, derived by ANEVA on a reference population’s ASE data to identify genes in individual test samples that are likely affected by RVs with unusually large regulatory effects.
Splicing outlier calls were made in a single tissue using SPOT to identify genes in each individual that show abnormal splicing patterns. Briefly, For a given LeafCutter cluster in a given tissue, we defined a matrix, X (dim NxJ), where each row corresponds to one of N samples, each column corresponding to one of J exon-exon junctions mapped to the LeafCutter cluster, and each element was the number of raw split read counts corresponding to that row’s sample and that column’s exon-exon junction. We were able to compute a P value representing how abnormal a given sample’s splicing patterns were for the given LeafCutter cluster as follows:
Fitted parameters of Dirichlet-Multinomial distribution based on observed data X to capture the distribution of split read counts mapping to this LeafCutter cluster;
Used the fitted Dichlet-Multinomial distribution to compute the Mahalanobis distance for each of the N samples; and
Computed the Mahalanobis distance for 1,000,000 samples simulated from the fitted Dirichlet-Multinomial distribution and used these 1,000,000 Mahalanobis distances as an empirical distribution to assess the significance of the N real Mahalanobis distances.
To generate multitissue outlier calls for each gene and outlier type, we calculated an individual’s median outlier score across all tissues for which data were available, restricting the analysis to individuals with measurements in at least five tissues. To account for situations in which widespread extreme expression might occur in an individual because of nongenetic influences, we excluded individuals in whom the proportion of tested genes that were multitissue outliers, at a P-value threshold of 0.0027, exceeded 1.5 times the interquartile range of the distribution of proportion of outlier genes across all individuals.
For the correlation-aware outlier calls, we determined a subset of individuals and tissues with <75% missingness, leading to 762 individuals and 29 tissues. We imputed missing expression values to improve our estimate of the tissue-by-tissue covariance matrix per gene that would be used in outlier calling. We used K-nearest neighbors in the impute R package (53) with k = 200 to impute values for missing tissues per individual on a gene-by-gene basis. From the imputed matrix, we estimated the tissue covariance matrix for each gene. We calculated the Mahalanobis distance for each gene-individual pair and assigned a P value to each gene individual from the chi-squared distribution, with degrees of freedom equal to the number of tissues available for that individual.
Watershed is a hierarchical Bayesian model that predicts the regulatory effects of RVs on a specific outlier signal based on the integration of multiple transcriptomic signals along with genomic annotations describing the RVs. Watershed models instances of gene-individual pairs to predict the regulatory effects of RVs nearby the gene. The Watershed model for a particular gene-individual pair, assuming K outlier signals, consists of three layers (Fig. 4A):
A set of variables G, representing the P observed genomic annotations aggregated over all RVs in the individual that are nearby the gene.
A set of binary latent variables Z = Z1, …, ZK representing the unobserved functional regulatory status of the RVs on each of the K outlier signals.
A set of categorical nodes E = E1, …, EK representing the observed outlier status of the gene for each of the K outlier signals.
A fully connected conditional random field (CRF) is defined over variables Z given G. Variables Ei are each connected only to the corresponding latent variable Zi. Specifically, the following conditional probability distributions together define the full Watershed model:
Z|G ~ CRF(α, β1, …, βk, θ)
Ek|Zk ~ Categorical(ϕk) ∀ k ∈ K
ϕk ~ Dirichlet(C, …,C)
where βk ∈ RP ∀ k ∈ K are the parameters defining the contribution of the genomic annotations to the CRF for each outlier signal (k), α ∈ RK are the parameters defining the intercept of the CRF for each outlier signal (k), θ ∈ R(Kchoose2) are the parameters defining the edge weights between pairs of outlier signals, ϕk ∀ k ∈ K are the parameters defining the categorical distributions of each outlier signal, and C and λ are hyperparameters of the model.
For the CRISPR assay, we selected 14 rare stop-gained variants that were good candidates, eight of which passed quality control through (1) filtering to rare stop-gained variants with expression and ASE watershed posterior >0.9, (2) filtering to multitissue outlier status in both, and (3) keeping four remaining candidates that lie in complex trait genes and the next 10 with the highest individual outlier signal and Watershed posterior. Variants were tested using the polyclonal editing assay described in (41). Briefly, inducible-Cas9 293T cells were transfected with a guide RNA and a single-stranded homologous template specific to each variant. After sequencing, the effect size was calculated as log2[(Alt/Ref in cDNA)/(Alt/Ref in gDNA)] (54). These results were combined with six previously tested stop-gained and six non-eQTL control variants for which Watershed posteriors were available.
For the MPRA, we designed a set of synthetic DNA fragments by retrieving the genomic sequence corresponding to a 150-bp window centered at each variant of interest for the set of eOutlier-associated RVs and controls. For each variant, a reference and alternative sequence was designed that corresponded to each allele. GM12878 cells were cultured, electroporated, and collected. MPRA plasmid library construction proceeded as described in (55). To assemble oligo-barcode pairings, we merged all paired-end reads using FLASH2 (56), requiring a minimum 10-bp overlap to retain each pair. Sequences corresponding to genomic fragments were mapped using STAR (57) against a reference assembled using the designed oligo library sequences. To count reads per individual barcode sequence, we took raw single-end reads, extracted the 20-bp region corresponding to the random barcode, and counted the number of reads per individual sequence. Finally, to generate oligo-level read counts, we computed the sum of all barcodes for each oligo within each sample. We used negative binomial regression with an interaction term, implemented using DESeq2 (58), to identify significant allele-independent and allele-dependent regulatory effects.
To connect outlier-associated RVs to traits, we assessed genome-wide association study (GWAS0 summary statistics from the UKBB phase 2, made available by the Neale laboratory (www.nealelab.is/uk-biobank/). We subsetted the variants, either genotyped or imputed, in UKBB phase 2 to those that also appeared in any GTEx individuals with a frequency of <1% in GTEx, resulting in 45,415 SNVs. We filtered the set of GTEx RVs in UKBB to those in trait-colocalized regions, defined as being in a colocalized gene or within a 10-kb window. Colocalization calls are detailed in (45).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Lappalainen, Mohammadi, Montgomery, and Battle laboratories for helpful discussions and feedback; J. Bonnie for providing comments on the manuscript; K. Tayeb and R. Ungar for reviewing code; the investigators and participants who provided biological samples and data for GTEx, ASMAD, MVP, and JHS Trans-Omics in Precision Medicine (TOPMed); and the staff and participants of the JHS.
Funding: This work was supported by the Common Fund of the Office of the Director, U.S. National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, NIA, NIAID, and NINDS through NIH contracts HHSN261200800001E (Leidos Prime contract with NCI: A.M.S., D.E.T., N.V.R., J.A.M., L.S., M.E.B., L.Q., T.K., D.B., K.R., A.U.), 10XS170 (NDRI: W.F.L., J.A.T., G.K., A.M., S.S., R.H., G.Wa., M.J., M.Wa., L.E.B., C.J., J.W., B.R., M.Hu., K.M., L.A.S., H.M.G., M.Mo., L.K.B.), 10XS171 (Roswell Park Cancer Institute: B.A.F., M.T.M., E.K., B.M.G., K.D.R., J.B.), 10X172 (Science Care, Inc.), 12ST1039 (IDOX), 10ST1035 (Van Andel Institute: S.D.J., D.C.R., D.R.V.), HHSN268201000029C (Broad Institute: F.A., G.G., K.G.A., A.V.S., X.Li., E.T., S.G., A.G., S.A., K.H.H., D.T.N., K.H., S.R.M., J.L.N.), 5U41HG009494 (F.A., G.G., K.G.A.) and through NIH grants R01 DA006227-17 (Univ. of Miami Brain Bank: D.C.M., D.A.D.), Supplement to University of Miami grant DA006227 (D.C.M., D.A.D.), R01 MH090941 (Univ. of Geneva), R01 MH090951 and R01 MH090937 (Univ. of Chicago), R01 MH090936 (Univ. of North Carolina-Chapel Hill), R01MH101814 (M.M-A., V.W., S.B.M., R.G., E.T.D., D.G-M., A.V., A.B.), U01HG007593 (S.B.M.), R01MH101822 (C.D.B.), U01HG007598 (M.O., B.E.S.), U01MH104393 (A.P.F.), extension H002371 to 5U41HG002371 (W.J.K.) as well as other funding sources: R01MH106842 (T.L., P.M., E.F., P.J.H.), R01HL142028 (T.L., Si.Ka., P.J.H.), R01GM122924 (T.L., S.E.C.), R01MH107666 (H.K.I.), P30DK020595 (H.K.I.), UM1HG008901 (T.L.), R01GM124486 (T.L.), R01HG010067 (Y.Pa.), R01HG002585 (G.Wa., M.St.), Gordon and Betty Moore Foundation GBMF 4559 (G.Wa., M.St.), 1K99HG009916-01 (S.E.C.), R01HG006855 (Se.Ka., R.E.H.), BIO2015-70777-P, Ministerio de Economia y Competitividad and FEDER funds (M.M-A., V.W., R.G., D.G-M.), la Caixa Foundation ID 100010434 under agreement LCF/BQ/SO15/52260001 (D.G-M.), NIH CTSA grant UL1TR002550-01 (P.M.), Marie-Skłodowska Curie fellowship H2020 Grant 706636 (S.K-H.), R35HG010718 (E.R.G.), FPU15/03635, Ministerio de Educación, Cultura y Deporte (M.M-A.), R01MH109905, 1R01HG010480 (A.B.), Searle Scholar Program (A.B.), R01HG008150 (S.B.M., A.B.), 5T32HG000044-22, NHGRI Institutional Training Grant in Genome Science (N.R.G.), EU IMI program (UE7-DIRECT-115317-1) (E.T.D., A.V.), FNS funded project RNA1 (31003A_149984) (E.T.D., A.V.), DK110919 (F.H.), F32HG009987 (F.H.), Massachusetts Lions Eye Research Fund Grant (A.R.H.), Mr. and Mrs. Spencer T. Olin Fellowship for Women in Graduate Study (A.J.S.), P30DK20595 (H.K.I.), UL1 TR001114 (P.M.), R01AG066490 (S.B.M.), R01HL142015 (S.B.M.), U01HG009431 (S.B.M.), U01HG009080 (S.B.M.), NIMH 1R01MH109905 (A.B.), National Science Foundation Graduate Research Fellowship, grant no. DGE - 1656518 (N.M.F.), graduate fellowship from the Stanford Center for Computational, Evolutionary and Human Genomics (N.M.F.), New York Center for Collaborative Research in Common Disease Genomics grant UM1HG008901 (J.E.), National Science Foundation of China grant 31970554 (X.L.), Shanghai Science and Technology Major Project IHPC 2017SHZDZX01 (X.L.), NIH T32 LM012409 (C.S.), Hewlett-Packard Stanford Graduate Fellowship and a doctoral scholarship from the Natural Science and Engineering Council of Canada (E.K.T.), Lucille P. Markey Stanford Graduate Fellowship (J.R.D.). We used data from the MVP, Office of Research and Development, Veterans Health Administration, supported by award no. MVP000. This publication does not represent the views of the Department of Veterans Affairs, the U.S. Food and Drug Administration, or the U.S. Government. Molecular Data for the TOPMed program was supported by the National Heart, Lung and Blood Institute (NHLBI). Genome sequencing for “NHLBI TOPMed: The Jackson Heart Study” (phs000964.v1.p1) was performed at the Northwest Genomics Center (HHSN268201100037C). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). This research was also supported by funding from: the Department of Veterans Affairs awards nos. I01-BX03340 and I01-BX003362 (T.L.A.). P.N. and G.M.P. are supported by R01HL142711 from the National Heart, Lung, and Blood Institute (NHLBI). The JHS is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD).
GTEx Consortium
Laboratory and Data Analysis Coordinating Center (LDACC): François Aguet1, Shankara Anand1, Kristin G. Ardlie1, Stacey Gabriel1, Gad A. Getz1,2,3, Aaron Graubert1, Kane Hadley1, Robert E. Handsaker4,5,6, Katherine H. Huang1, Seva Kashin4,5,6, Xiao Li1, Daniel G. MacArthur5,7, Samuel R. Meier1, Jared L. Nedzel1, Duyen T. Nguyen1, Ayellet V. Segrè1,8, Ellen Todres1
Analysis Working Group Funded by GTEx Project Grants: François Aguet1, Shankara Anand1, Kristin G. Ardlie1, Brunilda Balliu9, Alvaro N. Barbeira10, Alexis Battle11,12, Rodrigo Bonazzola10, Andrew Brown13,14, Christopher D. Brown15, Stephane E. Castel16,17, Donald F. Conrad18,19, Daniel J. Cotter20, Nancy Cox21, Sayantan Das22, Olivia M. de Goede20, Emmanouil T. Dermitzakis13,23,24, Jonah Einson16,25, Barbara E. Engelhardt26,27, Eleazar Eskin28, Tiffany Y. Eulalio29, Nicole M. Ferraro29, Elise D. Flynn16,17, Laure Fresard30, Eric R. Gamazon21,31,32,33, Diego Garrido-Martín34, Nicole R. Gay20, Gad A. Getz1,2,3, Michael J. Gloudemans29, Aaron Graubert1, Roderic Guigó34,35, Kane Hadley1, Andrew R. Hamel8,1, Robert E. Handsaker4,5,6, Yuan He11, Paul J. Hoffman16, Farhad Hormozdiari1,36, Lei Hou1,37, Katherine H. Huang1, Hae Kyung Im10, Brian Jo26,27, Silva Kasela16,17, Seva Kashin4,5,6, Manolis Kellis1,37, Sarah Kim-Hellmuth16,17,38, Alan Kwong22, Tuuli Lappalainen16,17, Xiao Li1, Xin Li30, Yanyu Liang10, Daniel G. MacArthur5,7, Serghei Mangul28,39, Samuel R. Meier1, Pejman Mohammadi16,17,40,41, Stephen B. Montgomery20,30, Manuel Muñoz-Aguirre34,42, Daniel C. Nachun30, Jared L. Nedzel1, Duyen T. Nguyen1, Andrew B. Nobel43, Meritxell Oliva10,44, YoSon Park15,45, Yongjin Park1,37, Princy Parsana12, Abhiram S. Rao46, Ferran Reverter47, John M. Rouhana1,8, Chiara Sabatti48, Ashis Saha12, Ayellet V. Segrè1,8, Andrew D. Skol10,49, Matthew Stephens50, Barbara E. Stranger10,51, Benjamin J. Strober11, Nicole A. Teran30, Ellen Todres1, Ana Viñuela13,23,24,52, Gao Wang50, Xiaoquan Wen22, Fred Wright53, Valentin Wucher34, Yuxin Zou54
Analysis Working Group Not Funded by GTEx Project Grants: Pedro G. Ferreira55,56,57,58, Gen Li59, Marta Melé60, Esti Yeger-Lotem61,62
Leidos Biomedical Project Management: Mary E. Barcus63, Debra Bradbury63, Tanya Krubit63, Jeffrey A. McLean63, Liqun Qi63, Karna Robinson63, Nancy V. Roche63, Anna M. Smith63, Leslie Sobin63, David E. Tabor63, Anita Undale63
Biospecimen Collection Source Sites: Jason Bridge64, Lori E. Brigham65, Barbara A. Foster66, Bryan M. Gillard66, Richard Hasz67, Marcus Hunter68, Christopher Johns69, Mark Johnson70, Ellen Karasik66, Gene Kopen71, William F. Leinweber71, Alisa McDonald71, Michael T. Moser66, Kevin Myer68, Kimberley D. Ramsey66, Brian Roe68, Saboor Shad71, Jeffrey A. Thomas71,70, Gary Walters70, Michael Washington70, Joseph Wheeler69
Biospecimen Core Resource: Scott D. Jewell72, Daniel C. Rohrer72, Dana R. Valley72
Brain Bank Repository: David A. Davis73, Deborah C. Mash73
Pathology: Mary E. Barcus63, Philip A. Branton74, Leslie Sobin63
ELSI Study: Laura K. Barker75, Heather M. Gardiner75, Maghboeba Mosavel76, Laura A. Siminoff75
Genome Browser Data Integration and Visualization: Paul Flicek77, Maximilian Haeussler78, Thomas Juettemann77, W. James Kent78, Christopher M. Lee78, Conner C. Powell78, Kate R. Rosenbloom78, Magali Ruffier77, Dan Sheppard77, Kieron Taylor77, Stephen J. Trevanion77, Daniel R. Zerbino77
eGTEx Group: Nathan S. Abell20, Joshua Akey79, Lin Chen44, Kathryn Demanelis44, Jennifer A. Doherty80, Andrew P. Feinberg81, Kasper D. Hansen82, Peter F. Hickey83, Lei Hou1,37, Farzana Jasmine44, Lihua Jiang20, Rajinder Kaul84,85, Manolis Kellis1,37, Muhammad G. Kibriya44, Jin Billy Li20, Qin Li20, Shin Lin86, Sandra E. Linder20, Stephen B. Montgomery20,30, Meritxell Oliva10,44, Yongjin Park1,37, Brandon L. Pierce44, Lindsay F. Rizzardi87, Andrew D. Skol10,49, Kevin S. Smith30, Michael Snyder20, John Stamatoyannopoulos84,88, Barbara E. Stranger10,51, Hua Tang20, Meng Wang20
NIH Program Management: Philip A. Branton74, Latarsha J. Carithers74,89, Ping Guan74, Susan E. Koester90, A. Roger Little91, Helen M. Moore74, Concepcion R. Nierras92, Abhi K. Rao74, Jimmie B. Vaught74, Simona Volpi93
1Broad Institute of MIT and Harvard, Cambridge, MA, USA. 2Cancer Center and Department of Pathology, Massachusetts General Hospital, Boston, MA, USA. 3Harvard Medical School, Boston, MA, USA. 4Department of Genetics, Harvard Medical School, Boston, MA, USA. 5Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA. 6Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, MA, USA. 7Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA. 8Ocular Genomics Institute, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, USA. 9Department of Biomathematics, University of California, Los Angeles, CA, USA. 10Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, IL, USA. 11Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA. 12Department of Computer Science, Johns Hopkins University, Baltimore, MD, USA. 13Department of Genetic Medicine and Development, University of Geneva Medical School, Geneva, Switzerland. 14Population Health and Genomics, University of Dundee, Dundee, Scotland, UK. 15Department of Genetics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA. 16New York Genome Center, New York, NY, USA. 17Department of Systems Biology, Columbia University, New York, NY, USA. 18Department of Genetics, Washington University School of Medicine, St. Louis, MO, USA. 19Division of Genetics, Oregon National Primate Research Center, Oregon Health & Science University, Portland, OR, USA. 20Department of Genetics, Stanford University, Stanford, CA, USA. 21Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA. 22Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA. 23Institute for Genetics and Genomics in Geneva (iGE3), University of Geneva, Geneva, Switzerland. 24Swiss Institute of Bioinformatics, Geneva, Switzerland. 25Department of Biomedical Informatics, Columbia University, New York, NY, USA. 26Department of Computer Science, Princeton University, Princeton, NJ, USA. 27Center for Statistics and Machine Learning, Princeton University, Princeton, NJ, USA. 28Department of Computer Science, University of California, Los Angeles, CA, USA. 29Program in Biomedical Informatics, Stanford University School of Medicine, Stanford, CA, USA. 30Department of Pathology, Stanford University, Stanford, CA, USA. 31Data Science Institute, Vanderbilt University, Nashville, TN, USA. 32Clare Hall, University of Cambridge, Cambridge, UK. 33MRC Epidemiology Unit, University of Cambridge, Cambridge, UK. 34Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Barcelona, Catalonia, Spain. 35Universitat Pompeu Fabra (UPF), Barcelona, Catalonia, Spain. 36Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA, USA. 37Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA. 38Statistical Genetics, Max Planck Institute of Psychiatry, Munich, Germany 39Department of Clinical Pharmacy, School of Pharmacy, University of Southern California, Los Angeles, CA, USA. 40Scripps Research Translational Institute, La Jolla, CA, USA. 41Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA. 42Department of Statistics and Operations Research, Universitat Politècnica de Catalunya (UPC), Barcelona, Catalonia, Spain. 43Department of Statistics and Operations Research and Department of Biostatistics, University of North Carolina, Chapel Hill, NC, USA. 44Department of Public Health Sciences, The University of Chicago, Chicago, IL, USA. 45Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA. 46Department of Bioengineering, Stanford University, Stanford, CA, USA. 47Department of Genetics, Microbiology and Statistics, University of Barcelona, Barcelona, Spain. 48Departments of Biomedical Data Science and Statistics, Stanford University, Stanford, CA, USA. 49Department of Pathology and Laboratory Medicine, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, USA. 50Department of Human Genetics, University of Chicago, Chicago, IL, USA. 51Center for Genetic Medicine, Department of Pharmacology, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA. 52Department of Twin Research and Genetic Epidemiology, King’s College London, London, UK. 53Bioinformatics Research Center and Departments of Statistics and Biological Sciences, North Carolina State University, Raleigh, NC, USA. 54Department of Statistics, University of Chicago, Chicago, IL, USA. 55Department of Computer Sciences, Faculty of Sciences, University of Porto, Porto, Portugal. 56Instituto de Investigação e Inovação em Saúde, University of Porto, Porto, Portugal. 57Institute of Molecular Pathology and Immunology, University of Porto, Porto, Portugal. 58Laboratory of Artificial Intelligence and Decision Support, Institute for Systems and Computer Engineering, Technology and Science, Porto, Portugal. 59Mailman School of Public Health, Columbia University, New York, NY, USA. 60Life Sciences Department, Barcelona Supercomputing Center, Barcelona, Spain. 61Department of Clinical Biochemistry and Pharmacology, Ben-Gurion University of the Negev, Beer-Sheva, Israel. 62National Institute for Biotechnology in the Negev, Beer-Sheva, Israel. 63Leidos Biomedical, Rockville, MD, USA. 64Upstate New York Transplant Services, Buffalo, NY, USA. 65Washington Regional Transplant Community, Annandale, VA, USA. 66Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA. 67Gift of Life Donor Program, Philadelphia, PA, USA. 68LifeGift, Houston, TX, USA. 69Center for Organ Recovery and Education, Pittsburgh, PA, USA. 70LifeNet Health, Virginia Beach, VA. USA. 71National Disease Research Interchange, Philadelphia, PA, USA. 72Van Andel Research Institute, Grand Rapids, MI, USA. 73Department of Neurology, University of Miami Miller School of Medicine, Miami, FL, USA. 74Biorepositories and Biospecimen Research Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. 75Temple University, Philadelphia, PA, USA. 76Virginia Commonwealth University, Richmond, VA, USA. 77European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, UK. 78Genomics Institute, University of California, Santa Cruz, CA, USA. 79Carl Icahn Laboratory, Princeton University, Princeton, NJ, USA. 80Department of Population Health Sciences, The University of Utah, Salt Lake City, UT, USA. 81Departments of Medicine, Biomedical Engineering, and Mental Health, Johns Hopkins University, Baltimore, MD, USA. 82Department of Biostatistics, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, USA. 83Department of Medical Biology, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia. 84Altius Institute for Biomedical Sciences, Seattle, WA, USA. 85Division of Genetics, University of Washington, Seattle, WA, USA. 86Department of Cardiology, University of Washington, Seattle, WA, USA. 87HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA. 88Genome Sciences, University of Washington, Seattle, WA, USA. 89National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD, USA. 90Division of Neuroscience and Basic Behavioral Science, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA. 91National Institute on Drug Abuse, Bethesda, MD, USA. 92Office of Strategic Coordination, Division of Program Coordination, Planning and Strategic Initiatives, Office of the Director, National Institutes of Health, Rockville, MD, USA. 93Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD, USA.
Footnotes
Competing interests: F.A. is an inventor on a patent application related to TensorQTL. S.E.C. is a cofounder, chief technology officer, and stock owner at Variant Bio. E.R.G. is on the editorial board of Circulation Research and does consulting for the City of Hope/Beckman Research Institute. E.T.D. is chairman and member of the board of Hybridstat Ltd. B.E.E. is on the scientific advisory boards of Celsius Therapeutics and Freenome. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, POLYSOLVER and TensorQTL. S.B.M. is on the scientific advisory board of MyOme. D.G.M. is a cofounder with equity in Goldfinch Bio and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer, and Sanofi-Genzyme. H.K.I. has received speaker honoraria from GSK and AbbVie. T.L. is a scientific advisory board member of Variant Bio with equity and of Goldfinch Bio. P.F. is member of the scientific advisory boards of Fabric Genomics, Inc., and Eagle Genomes, Ltd. P.G.F. is a partner of Bioinf2Bio. G.G. is a founder, consultant, and holds privately held equity in Scorpion Therapeutics. P.N. reports investigator-initiated research grants from Amgen, Apple, and Boston Scientific and is a scientific adviser to Apple and Blackstone Life Sciences. The remaining authors have no competing interests.
Data and materials availability: The data analyzed for this study are available to authorized users through dbGaP under accession no. phs000424.v8 and on the GTEx portal (https://gtexportal.org/). The complete set of multitissue outlier statistics can be found on the GTEx portal (https://gtexportal.org/). Reference variance estimates and blacklisted genes for all GTEx v8 tissues (59), ANEVA-DOT code (60), SPOT code (61), code for correlation eOutlier (62), Watershed (63), and the code used to generate all figures in this manuscript (64) are available at Zenodo. Data underlying each figure are available to download from https://drive.google.com/open?id=1dCxoYDPjKE7qTUQhHN5Z-e6hiS5BQCG. J.H.S.’s data were accessed through dbGaP application no. 6213 for the TOPMed Exchange Area and were supported by secondary-use institutional review board approval from the Massachusetts General Hospital. VA MVP data were accessed through dbGaP application no. 2638.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/369/6509/eaaz5900/suppl/DC1
Materials and Methods
Figs. S1 to S42
Tables S1 to S12
References (65–91)
MDAR Reproducibility Checklist
REFERENCES AND NOTES
- 1.Keinan A, Clark AG, Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 336, 740–743 (2012). doi: 10.1126/science.1217283; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright CF, FitzPatrick DR, Firth HV, Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet 19, 325 (2018). doi: 10.1038/nrg.2018.12; [DOI] [PubMed] [Google Scholar]
- 3.Bomba L, Walter K, Soranzo N, The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 18, 77 (2017). doi: 10.1186/s13059-017-1212-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montgomery SB, Lappalainen T, Gutierrez-Arcelus M, Dermitzakis ET, Rare and common regulatory variation in population-scale sequenced human genomes. PLOS Genet. 7, e1002144 (2011). doi: 10.1371/journal.pgen.1002144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng Y et al. , Aberrant gene expression in humans. PLOS Genet. 11, e1004942 (2015). doi: 10.1371/journal.pgen.1004942; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pala M et al. , Population- and individual-specific regulatory variation in Sardinia. Nat. Genet 49, 700–707 (2017). doi: 10.1038/ng.3840; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X et al. , Transcriptome sequencing of a large human family identifies the impact of rare noncoding variants. Am. J. Hum. Genet 95, 245–256 (2014). doi: 10.1016/j.ajhg.2014.08.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez RD et al. , Ultrarare variants drive substantial cis heritability of human gene expression. Nat. Genet 51, 1349–1355 (2019). doi: 10.1038/s41588-019-0487-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Battle A et al. , Genomic variation. Impact of regulatory variation from RNA to protein. Science 347, 664–667 (2015). doi: 10.1126/science.1260793; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li YI et al. , RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604 (2016). doi: 10.1126/science.aad9417; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frésard L et al. , Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med 25, 911–919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bycroft C et al. , The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). doi: 10.1038/s41586-018-0579-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaziano JM et al. , Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol 70, 214–223 (2016). doi: 10.1016/j.jclinepi.2015.09.016; [DOI] [PubMed] [Google Scholar]
- 14.Taylor HA Jr. et al. , Toward resolution of cardiovascular health disparities in African Americans: Design and methods of the Jackson Heart Study. Ethn. Dis 15 (Suppl 6), S6–S4, 17 (2005). [PubMed] [Google Scholar]
- 15.Li X et al. , The impact of rare variation on gene expression across tissues. Nature 550, 239–243 (2017). doi: 10.1038/nature24267; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Materials and methods are available as supplementary materials.
- 17.Mohammadi P et al. , Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science 366, 351–356 (2019). doi: 10.1126/science.aay0256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguet F et al. , The GTEx Consortium atlas of genetic regulatory effects across human tissues. bioRxiv 787903 [Preprint]. 3 October 2019. https://doi.org/10.1101/787903. doi: 10.1101/787903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spitz F, Gene regulation at a distance: From remote enhancers to 3D regulatory ensembles. Semin. Cell Dev. Biol 57, 57–67 (2016). doi: 10.1016/j.semcdb.2016.06.017; [DOI] [PubMed] [Google Scholar]
- 20.Krijger PHL, de Laat W, Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol 17, 771–782 (2016). doi: 10.1038/nrm.2016.138; [DOI] [PubMed] [Google Scholar]
- 21.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). doi: 10.1038/s41586-020-2308-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Z-F, Mott S, Rosmarin AG, The Ets transcription factor GABP is required for cell-cycle progression. Nat. Cell Biol 9, 339–346 (2007). doi: 10.1038/ncb1548; [DOI] [PubMed] [Google Scholar]
- 23.Takahashi Y, Rayman JB, Dynlacht BD, Analysis of promoter binding by the E2F and pRB families in vivo: Distinct E2F proteins mediate activation and repression. Genes Dev. 14, 804–816 (2000). [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon S, Akopyan G, Garban H, Bonavida B, Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 25, 1125–1142 (2006). doi: 10.1038/sj.onc.1209080; [DOI] [PubMed] [Google Scholar]
- 25.Han T, Oh S, Kang K, ETS family protein GABP is a novel co-factor strongly associated with genomic YY1 binding sites in various cell lines. Genes Genomics 38, 119–125 (2016). doi: 10.1007/s13258-015-0358-2 [DOI] [Google Scholar]
- 26.Saha A et al. , Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 27, 1843–1858 (2017). doi: 10.1101/gr.216721.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mittal VK, McDonald JF, De novo assembly and characterization of breast cancer transcriptomes identifies large numbers of novel fusion-gene transcripts of potential functional significance. BMC Med. Genomics 10, 53 (2017). doi: 10.1186/s12920-017-0289-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.López-Nieva P et al. , Detection of novel fusion-transcripts by RNA-Seq in T-cell lymphoblastic lymphoma. Sci. Rep 9, 5179 (2019). doi: 10.1038/s41598-019-41675-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neckles C, Sundara Rajan S, Caplen NJ, Fusion transcripts: Unexploited vulnerabilities in cancer? Wiley Interdiscip. Rev. RNA 11, e1562 (2020). doi: 10.1002/wrna.1562; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baty F, Brutsche M, Fusion transcripts in lung cancer. Lung Cancer (2017). [Google Scholar]
- 31.Chen S, Li J, Zhou P, Zhi X, SPTBN1 and cancer, which links? J. Cell. Physiol 235, 17–25 (2020). doi: 10.1002/jcp.28975; [DOI] [PubMed] [Google Scholar]
- 32.Fry AM, O’Regan L, Montgomery J, Adib R, Bayliss R, EML proteins in microtubule regulation and human disease. Biochem. Soc. Trans 44, 1281–1288 (2016). doi: 10.1042/BST20160125; [DOI] [PubMed] [Google Scholar]
- 33.Burset M, Seledtsov IA, Solovyev VV, Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 28, 4364–4375 (2000). doi: 10.1093/nar/28.21.4364; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S et al. , Base-specific mutational intolerance near splice sites clarifies the role of nonessential splice nucleotides. Genome Res. 28, 968–974 (2018). doi: 10.1101/gr.231902.117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li YI et al. , Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet 50, 151–158 (2018). doi: 10.1038/s41588-017-0004-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapiro MB, Senapathy P, RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 15, 7155–7174 (1987). doi: 10.1093/nar/15.17.7155; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coolidge CJ, Seely RJ, Patton JG, Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res. 25, 888–896 (1997). doi: 10.1093/nar/25.4.888; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kircher M et al. , A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). doi: 10.1038/ng.2892; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M, CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47 (D1), D886–D894 (2019). doi: 10.1093/nar/gky1016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Georgi B et al. , Genomic view of bipolar disorder revealed by whole genome sequencing in a genetic isolate. PLOS Genet. 10, e1004229 (2014). doi: 10.1371/journal.pgen.1004229; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brandt M, Gokden A, Ziosi M, Lappalainen T, A polyclonal allelic expression assay for detecting regulatory effects of transcript variants. bioRxiv 794081 [Preprint]. 7 October 2019. 10.1101/794081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forero DA et al. , APOE gene and neuropsychiatric disorders and endophenotypes: A comprehensive review. Am. J. Med. Genet. B. Neuropsychiatr. Genet 177, 126–142 (2018). doi: 10.1002/ajmg.b.32516; [DOI] [PubMed] [Google Scholar]
- 43.Habib AM et al. , Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br. J. Anaesth 123, e249–e253 (2019). doi: 10.1016/j.bja.2019.02.019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H, Mittal DP, Iadarola MJ, Dionne RA, Genetic predictors for acute experimental cold and heat pain sensitivity in humans. J. Med. Genet 43, e40 (2006). doi: 10.1136/jmg.2005.036079; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barbeira AN et al. , Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. bioRxiv 814350 [Preprint]. 23 May 2020. 10.1101/814350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Préfontaine D et al. , Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol 125, 752–754 (2010). doi: 10.1016/j.jaci.2009.12.935; [DOI] [PubMed] [Google Scholar]
- 47.Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC, Decoding asthma: Translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J. Allergy Clin. Immunol 131, 856–865 (2013). doi: 10.1016/j.jaci.2012.11.028; [DOI] [PubMed] [Google Scholar]
- 48.Smith D et al. , A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma. PLOS Genet. 13, e1006659 (2017). doi: 10.1371/journal.pgen.1006659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mousas A et al. , Rare coding variants pinpoint genes that control human hematological traits. PLOS Genet. 13, e1006925 (2017). doi: 10.1371/journal.pgen.1006925; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olafsdottir TA et al. , Eighty-eight variants highlight the role of T cell regulation and airway remodeling in asthma pathogenesis. Nat. Commun 11, 393 (2020). doi: 10.1038/s41467-019-14144-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klarin D et al. , Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet 50, 1514–1523 (2018). doi: 10.1038/s41588-018-0222-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stegle O, Parts L, Piipari M, Winn J, Durbin R, Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc 7, 500–507 (2012). doi: 10.1038/nprot.2011.457; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hastie T, Tibshirani R, Narasimhan B, Chu G, impute: Imputation for microarray data (2020); http://www.bioconductor.org/packages/release/bioc/manuals/impute/man/impute.pdf.
- 54.Mohammadi P, Castel SE, Brown AA, Lappalainen T, Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884 (2017). doi: 10.1101/gr.216747.116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tewhey R et al. , Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 165, 1519–1529 (2016). doi: 10.1016/j.cell.2016.04.027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Magoč T, Salzberg SL, FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011). doi: 10.1093/bioinformatics/btr507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobin A et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). doi: 10.1093/bioinformatics/bts635; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). doi: 10.1186/s13059-014-0550-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferraro NM et al. , Reference variance estimates and blacklisted genes for all GTEx v8 tissues for: Transcriptomic signatures across human tissues identify functional rare genetic variation, Zenodo; (2020); 10.5281/zenodo.3899574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferraro NM et al. , ANEVA-DOT code for: Transcriptomic signatures across human tissues identify functional rare genetic variation, Zenodo; (2020); 10.5281/zenodo.3406690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferraro NM et al. , SPOT code for: Transcriptomic signatures across human tissues identify functional rare genetic variation, Zenodo; (2020); https://zenodo.org/badge/latestdoi/209325700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferraro NM et al. , eOutlier code for: Transcriptomic signatures across human tissues identify functional rare genetic variation, Zenodo; (2020); https://zenodo.org/badge/latestdoi/210649448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferraro NM et al. , A. Battle, Watershed model for: Transcriptomic signatures across human tissues identify functional rare genetic variation, Zenodo; (2020); https://zenodo.org/badge/latestdoi/210165360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferraro NM et al. , Code used in all figures for: Transcriptomic signatures across human tissues identify functional rare genetic variation, Zenodo; (2020); https://zenodo.org/badge/latestdoi/265935957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.