

Abstract
With any new disease a framework for the development of preventative or treatment therapeutics is key; the absence of such in COVID‐19 has enabled ineffective and potentially unsafe treatments to be taken up by governments and clinicians desperate to have options for patients. As we still have few therapies and nil vaccines yet available, the void of a clear framework for research and practice is increasingly clear. We describe a framework that has been used to prioritise therapeutic research in previous pandemics which could be used to progress clinical pharmacology and therapeutics research in COVID‐19.
This is particularly relevant as discussion has already moved on from antiviral therapeutics to delineating the treatment of the host from treatment and elimination of the infective agent. Focussing on the host brings together three concepts: host treatment, the damage response framework and therapeutic repurposing. The integration of these three areas plays to the traditional strength of pharmaceuticals in providing a period of stabilization to permit time for the development of novel antiviral drugs and vaccines. In integrating approaches to repurposing, host treatment and damage response we identified three key properties that a potentially effective repurposed drug must possess by way of a framework. There must be homology, i.e., the same or similar relation with the pathogenesis of the disease, ideally targeted to the conserved pathophysiological outcomes of the viral attack; there must be a defined locus within the spectrum to prevention to severe disease and the framework must draw upon the historical dose and safety experience of the repurposed drug. To illustrate, we have mapped therapeutics that impact upon a key dysregulated pathway in COVID‐19 – the renin angiotensin system – using this approach. Collectively this type of analysis reveals the importance of existing data (repurposed information and administrative observational data) and the importance of the details of the known pathophysiological response to viruses in approaches to treating the host.
Keywords: COVID‐19, damage response framework, dose and safety, innate immune system, renin‐angiotensin system, repurposing, target homology, treating the host
1. INTRODUCTION
Recently Sanders et al. 1 highlighted both that no therapies for treating COVID‐19 were available at their time of writing and the need to have high‐quality evidence readily available in a pandemic. In a solution to this problem, we note there exists the potential confluence of two approaches that may prove beneficial in this pandemic setting. The first seeks agents that treat the host, not necessarily the virus, and builds on the pioneering studies of David Fedson in this field 2 , 3 , 4 , 5 ; the second is the concept of repurposing existing therapeutics for new indications. Both concepts are enshrined in pharmacological principles.
In this Review we explore these concepts and provide a potential path for efficient evaluation of candidate drugs using inhibitors of the renin angiotensin system (RAS) as an example.
2. TREATING THE HOST
One of the key values of therapeutics is their potential in stabilizing patients prior to subsequent interventions or their use as adjuncts to those interventions. The settings where this may occur are varied and include response to traumatic injury to control damage, treat infection and alleviate pain. Other notable examples include the development of anaesthesiology and sedation to enable the performance of surgery and complex procedures, as well the development of nondepolarizing neuromuscular‐blocking drugs as adjuncts to those surgical procedures and of adjunctive chemotherapy to improve quality of life and survival time. Of particular interest in this pandemic context has been the centuries‐old development of agents to promote survival with infections. Historically the focus was on identifying agents that would kill the invading species and not damage the host. In more recent times the focus has moved to delineating the treatment of the host from treatment and elimination of the infective agent.
In 2016, Fedson highlighted the treating of the host concept based on an understanding that a beneficial clinical phenotype has been observed with a treatment and this can be employed for the purpose of treating the host. 3 This was done in the context of emerging virus diseases, and more recently in the context of severe COVID‐19 infection. 5 Consistent with this view Yan et al. 6 drew attention to the need for agents to alleviate H5N1 virus‐induced lung injury due to the limited strategies for treating influenza virus infection.
The concept of treating the host is entirely complementary to opportunities arising in the area of drug repurposing that we have highlighted recently. 7 In the case of COVID‐19 disease this would mean identifying drugs that are focused not on the elimination of the virus, but on the survival of the patient with severe COVID‐19 disease. This includes drugs approved for other indications that have the beneficial phenotype for treating severe COVID‐19 disease. The fundamental advantages of this approach are, first, to buy needed time for the concurrent development of antiviral drugs or vaccines, and, second, to provide health authorities with a treatment insurance as they begin to release the public from social isolation. We have highlighted this key aspect recently in the context of COVID‐19 disease 8 supported by a recent UK study showing a mortality benefit of dexamethasone in patients undergoing invasive mechanical ventilation and among those receiving oxygen without invasive mechanical ventilation. Note that the data suggest that dexamethasone might be harmful if used earlier, confirming the need for clinical trials to understand not just the dose, but the pathophysiology to guide the timing of therapies 9 (Figure 1).
FIGURE 1.
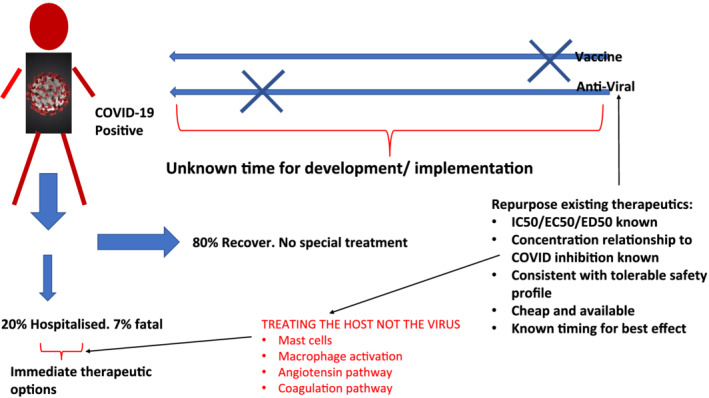
Treating the host not the virus
However, this very obvious but distinct suite of proposed solutions to reduce morbidity and mortality from COVID‐19 has the potential risk of cutting across fixed interests and previously effective methods of succeeding in grant funding rounds, including silos of ‘ology’ disciplines, and pressures from news cycles and campaigning for ineffective policies.
What is needed is the appropriate framework for treating the host, a strategy for positioning this approach, and then implementation.
3. A FRAMEWORK FOR TREATING THE HOST WITH REPURPOSED DRUGS
Treating the host in COVID‐19 brings together two key pharmacological principals. The first involves the understanding of how a proposed therapeutic agent integrates within the pathogenic response. This approach thus involves understanding initially the integration of the host into the consequences of viral pathogenesis. This relationship has been well described for microbial pathogenesis in the Damage–Response Framework (DRF) in infectious diseases. 10 The damaging host factors described in this microbial model, namely cytokine release (storm), chemical mediators signalling, oxygen radical production and immune complexes leading to aberrant inflammation, are not specific and in fact apply equally to viruses such as influenza and the current COVID‐19 disease.
The second principal involves the appropriate use of therapeutics approved for one indication but used effectively in a different setting within the DRF. Approaches used for drug repurposing, the challenges and the ways in which these challenges may be tackled have been described recently in detail. 11 The application to COVID‐19 involves identification of drugs that in a new indication and in the correct dosage for the new disease have a substantial impact on the consequences of infection in the host.
A logical framework based on the above considerations for identifying repurposed drugs for treatment of the host would potentially include:
Pathogenesis. Homology with the novel action of the repurposed drug and the molecular events that drive the consequences of viral pathogenesis in COVID‐19 disease.
Target homology of COVID‐19 and therapeutic agents. Ideally the repurposed drug should target the pathophysiology of COVID‐19 that is tightly linked to the conserved mutational properties of the virus and its class.
Prevention to treatment. An understanding of how the repurposed drug provides benefit in treating the host across the cascade from prevention to the life‐threatening consequences of other viral pathogenesis in COVID‐19 disease.
Dose and safety. Ideally an application of the repurposed drug either within the approved dose ranges for its initial indications for which the pharmacokinetics and toxicology are documented as part of the registration for the drug's initial indication or a new application of an already registered drug in another indication, using doses based on the pharmacological and physiological attributes of the new disease and the specific chosen effect of the drug at different dosages and/or plasma concentrations.
Using similar deduction, in 2020 Fedson et al. proposed clinical trials should be undertaken with combinations of the RAS agents (ATC C09) and statins in patients with severe COVID‐19 infection. 5 Fedson's proposal provides an excellent opportunity to use our framework in evaluating repurposed RAS drugs for use in COVID‐19. In the following example we focus on drugs targeted to the RAS, but the procedure would equally apply to statins either alone or in combination with angiotensin receptor blockers (ARBs). 12
4. EXPLORATION OF TREATING THE HOST CONCEPT USING REPURPOSED AGENTS ACTING ON THE RAS ATC C09
From the preceding discussion, a framework for treating the host and repurposing can be employed to explore the eligibility of drugs that influence the RAS as candidates for clinical trials. In particular, the framework will identify what is known about these agents in regard to (a) pathogenesis, (b) target homology of COVID‐19, (c) prevention/treatment and (d) the dose–response curve in relation to safety.
4.1. Pathogenesis
To treat the host requires an understanding of the physiology of this response to that condition (in this case COVID‐19). In addition, knowledge is needed of chemical, structural and immunological homology with the novel action of the repurposed drug and the molecular events that drive the consequences of viral pathogenesis in COVID‐19 disease. As the main causes of death in COVID‐19 are pulmonary vasoconstriction, pulmonary oedema, thrombosis and fibrosis, a focus on RAS inhibition, which is a key driver of pulmonary complications (prothrombotic and inflammation in the lung), innate immune response and vascular response (vasoconstriction, inflammation and coagulation), is key.
4.1.1. Pulmonary complications: Prothrombotic and inflammatory lung disease
During early 2020, an enormous amount was written on the pathogenesis of COVID‐19, including comprehensive summary review articles with strong emphasis on pulmonary pathophysiology in this condition (see Zhang et al., Li et al. and Kreutz et al., 12 , 13 , 14 inter alia). The relationship between the dysregulation of the pulmonary RAS and the progression to severe acute respiratory syndrome (SARS), including the intermediate stages of infections and pneumonia, has been discussed at length. By way of summary the fundamental initiation is the role of angiotensin‐converting enzyme 2 (ACE2) as the receptor for SARS coronavirus infection. 13 It is the impact of this initiation that drives a pulmonary dysregulation of the finely balanced RAS. This balance involves the generation of angiotensin II by ACE and the inactivation of that peptide by ACE2. The functional role of ACE2, its catalytic product angiotensin1–7 and the Mitochondrial Assembly Receptor (MAS) receptor are the counterbalance moieties for angiotensin II and its Angiotensin Type 1 Receptor (AT1R) receptor. 14 The interaction of angiotensin 1–7 with MAS receptors promotes an anti‐inflammatory response, vasodilatation and an antifibrotic response and is seen as protective. Downregulation of ACE2 in acute lung injury models with SARSCoV infection supports a protective role for RAS blockade in this form of pulmonary infection. 14 The core hypothesis is that a dysregulated RAS is core to the pathophysiology of COVID‐19‐driven lung damage and acute respiratory distress syndrome. 15 This builds on much earlier experimentation showing the angiotensin II receptor blocker (ARB) losartan could effectively reduce acute lung injury induced by SARS‐CoV. 16 The summary statement is most apt: “Therefore, in acute lung injury, ACE, Ang II, and AT1R function as lung‐injury‐promoting factors, while ACE2 protects from lung injury”. 17 This pathophysiological pathway is described in significant detail elsewhere. 16 , 18 , 19 , 20 , 21
It follows that from a repurposing standpoint that there exists a strong target homology with agents that promote RAS inhibition and the pathogenesis of COVID‐19 disease in the lung in terms of pathophysiology and pharmacology.
4.1.2. Innate immune response
Angiotensin II, via activation of the AT1R receptor, is pivotal in modulating immune responses. A key immune cell involved with the RAS pathway – the mast cell – is found in the submucosal tissue in human bronchi in the upper and lower respiratory tree. 22 Mast cells rapidly recognize pathogens and mount an immune response that includes plasma extravasation, immunological and cytokine modulation, leukocyte recruitment and ultimately cellular inflammation. 23 What is critical is the role of mast cells in the bronchi in the release of renin and the initiation of the production of the angiotensin II that acts through AT1R receptors. 22 Moreover, in an experimental model of hypersensitivity, anaphylactic mast cell degranulation has been shown to result in angiotensin bronchoconstriction. 22 Of significance is the observation that mast cell chymase can generate angiotensin II independently of ACE activity, 24 the salient point being that ACE is membrane bound and chymase on release moves within interstitial spaces. ACE also modulates macrophage and neutrophil function in its role in the innate and adaptive response. 25
Angiotensin II mediates its pro‐inflammatory responses by signalling through AT1R receptor. These responses include leukocyte recruitment and increased production of reactive oxygen species by receptor (AT1R) activation of NADPH oxidase, contributing to inflammation and the stimulation of the innate immune responses (summarized in Veerappan et al. 22 ). As mentioned above, angiotensin 1–7 serves as a counter to the pro‐inflammatory influence of angiotensin II. Angiotensin 1–7 achieves this by acting through the MAS receptors expressed at the surface of the bronchial smooth muscle and alveolar epithelium. 18 With the viral infection in COVID‐19 the cytokine storm is a key characteristic. The release of large amounts of pro‐inflammatory cytokines and chemokines (IFNα, IFNγ, IL‐1β, IL‐6, IL‐12, IL‐18, IL‐33, TNFα, TGFβ, CXCL10, CXCL8, CXCL9, CCL2, CCL3, CCL5) by immune effector cells drives the aberrant inflammatory response. 26
Many cells of the innate immune system have been shown to have an important role in the development of COVID‐19 lung. Pulmonary RAS has a key role in bronchoconstriction consistent with the release of renin and subsequent angiotensin II formation with mast cell degranulation. 22 Moreover, in an animal model of hypersensitivity mast cell degranulation was associated with angiotensin II‐mediated constriction, inhibited with an AT1R blocker or an inhibitor of renin. 22 From a repurposing standpoint that there exists a strong target similarity with agents that promote RAS inhibition and the modulation of innate immune responses in the lung.
4.1.3. Vascular damage: Vasoconstriction, inflammation and coagulation
There is growing evidence that there exists a confluence of vascular and diffuse alveolar damage with SARS and possibly also COVID‐19 disease. In some of the data with COVID‐19 pneumonia, McGonagle et al. 27 have drawn attention to the similarities of blood vessel wall oedema, modest vessel wall immune cell infiltration, hyaline thrombosis and haemorrhagic change described earlier for patients with SARS, consistent with the observation that disseminated intravascular coagulation may develop late in COVID‐19 disease. Consistent with this view Teuwen et al. 28 suggested that pulmonary endothelial cells (ECs) have been largely overlooked as a target in COVID‐19, as the attending ECs alter the blood vessel barrier function, promoting a pro‐coagulative vascular inflammatory state and inflammatory cell infiltration. They also highlight the fact that SARS‐CoV‐2 binds to the ACE2 receptor, impairing the enzyme that is the counterbalance for the vasoactive angiotensin II. The lung pathology of COVID‐19 disease displays microvascular thrombosis and haemorrhage with extensive alveolar and interstitial inflammation termed lung‐restricted vascular immunopathology. 27 In the early stage this is distinct from diffuse pulmonary intravascular coagulopathy. Cao and Li 29 also similarly highlighted the two hallmarks of critical patients with COVID‐19 disease, namely progressive inflammation and the trend to hypercoagulation. Furthermore, they draw attention not only to the inflammatory state as a trigger for coagulation, but endothelial cell disseminated intravascular coagulation, characterizing the COVID‐19 hypercoagulable state as displaying elevated D‐dimer (a fibrin degradation product) and fibrinogen concentrations, and prolonged prothrombin time. 29
Previous studies have shown that in pulmonary hypertension there is a reduced ACE2 activity and that augmentation of ACE2 reduced oxidant and inflammatory markers as well as improving pulmonary hemodynamics. 30 Thirty years ago, our team established that in hypertensive blood vessels the endothelial cell‐mediated relaxation was impaired and this could be reversed with impairment of the prostanoid system or treatment with the Angiotensin Converting Enzyme Inhibitor (ACEI) captopril. 31 This collectively demonstrates a potential key role of the vascular endothelium in coagulation, inflammation and vasoconstriction and the capacity for abnormal function driven by the RAS. The RAS influence on the vasculature also extends to trophic factors. The neurotrophic peptide nerve growth factor (NGF) is believed to be a pro‐inflammatory mediator in airways and promote bronchial hyperresponsiveness. 32 Of significance was our demonstration that the AT1R blocking agent losartan reversed the abnormal concentrations of NGF in blood vessels in hypertension. 33
As indicated earlier there is growing evidence that there exists a confluence of vascular and diffuse alveolar damage with SARS and possibly COVID‐19. Gonzalez‐Jaramillo et al., in a commentary on the double burden of COVID‐19, also implicate the RAS in chronic pulmonary diseases such as pulmonary fibrosis and pulmonary arterial hypertension. 34 Similarly, pulmonary arterial hypertension (PAH) is characterized by reduced ACE2 activity and the production of angiotensin‐1–7 by ACE2 activating MAS receptors improves experimental models of PAH. 30
As pointed out by Cao and Li, 29 SARS‐CoV‐2 can initially replicate in the upper respiratory track and later in the disease course replicate in the lower respiratory tract, generating a secondary viremia with a subsequent attack on organ targets expressing ACE2. This is a dynamic consistent with the possibility outlined by Kreutz et al. 14 and other groups that ACEIs and ARBs may be associated with lower incidence and/or improved outcome in patients with lower respiratory tract infections. From a repurposing standpoint there exists a strong target similarity with agents that promote RAS inhibition that involves blood vessel and vascular endothelial cell damage.
4.2. Target homology of COVID‐19 and therapeutic agents
SARS‐CoV‐2 is an RNA virus. These viruses have high mutation rates, up to a million times higher than their hosts, and it is this high rate that enables them to avoid host immunity and achieve drug resistance. 35 From the standpoint of drug repurposing the selected drug should target not the virus, but that aspect of the pathophysiology of COVID‐19 that is tightly linked to the conserved mutational characteristics of the virus and its class.
4.2.1. Conserved target for SARS‐CoV‐2
It is now generally accepted that ACE2 is the receptor for SARS‐CoV and SARS‐CoV‐2 to enable entry to the host. 19 Moreover, South et al. 20 suggested that angiotensin may promote both lung injury and ARDS in SARS‐CoV, SARS‐CoV‐2/COVID‐19 and possibly Middle East respiratory syndrome coronavirus (MERS‐CoV).
Using crystal structures for NL63 coronavirus (NL63‐CoV) and SARS coronavirus (SARS‐CoV) receptor binding domains Wu et al. demonstrated virus binding hot spots on their common receptor, the human ACE2. 36 From that experimentation Wu suggested that the hot spot features were one of the driving forces for the convergent evolution of these two viruses. It follows that this would infer multiple binding opportunities for differing or mutated viruses, restricted to a common protein target. The targeted physiological process for driving pulmonary inflammation will be hard for a virus to abandon without significant penalty and can be viewed as conserved. From a repurposing standpoint there exists a strong target similarity with agents that promote RAS inhibition and the common conserved site of the pathogenesis for COVID‐19 and related viruses.
4.3. Prevention to treatment
An understanding of how the repurposed drug provides benefit in treating the host across the cascade from prevention to the life‐threatening consequences of other viral pathogenesis in COVID‐19 disease is needed. Cao and Li summarize the clinical course of SARS‐CoV‐2 infection by dividing it into three phases: the viremia phase, the acute phase (eg, with pneumonia) and the severe or recovery phase. 29 From the standpoint of drug repurposing in COVID‐19 we have adopted a similar approach, with particular focus on the severe phase, where the host system has switched on its innate immune system, affecting inflammation, oedema and vasconstriction. Our treatment focus in this phase is on RAS inhibition and (a) predisposition and age, (b) predisposition with hypertension and co‐morbidities, (c) treatment and secondary infection, and (d) intubation.
4.3.1. Predisposition and age
Increasing age has been shown to be a strong correlative factor with morbidity and mortality with COVID‐19. As summarized by Shahid et al. studies have highlighted the relatively higher mortality rates in older adults. 37 While a variety of reasons may underpin this predisposition, it would appear that the RAS has a strong input to aging by way of mitochondrial function, specifically a strong relationship between free radicals, the mitochondria and aging, with excessive free radical production linked to dysregulation of RAS. 38 Key in this dysregulation is the angiotensin receptor AT1R and its linkage to NADPH and increased production of reactive oxygen species (ROS), and the role of RAS in decreasing ROS scavenging enzymes. As highlighted by Wilson et al. 39 in mitochondria, the receptors for angiotensin (AT1R and AT2R) are linked to respiration and nitric oxide production. Furthermore, the presence of angiotensin 1–7 in purified mitochondria and the conversion of angiotensin I to angiotensin 1–7 by endopeptidases suggests an intramitochondrial pathway for formation of angiotensin 1–7. 39 Wang et al. 21 have shown that overexpression of ACE2 through mitochondrial function enhances the protective effects of endothelial progenitor cells (EPC) on endothelial cell injury. It remains to be determined whether the dysregulation of RAS described in COVID‐19 also similarly has a deleterious effect on mitochondrial function, as there appears to be an emerging possibility that a complex intersection between aging, mitochondrial function, apoptosis, the RAS and coronaviruses exists. Lai et al. 40 conducted a proteome analysis on human promonocyte HL‐CZ cells expressing SARS CoV3CL and found that 36% of the upregulated proteins were located in the mitochondria, including the apoptosis‐inducing factor ATP synthase beta chain and cytochrome c oxidase. They suggested that SARS CoV3CLpro could induce mitochondrial‐mediated apoptosis. 40 Additionally, it is possible that mitochondrial dysregulated RAS and free radical production in aging acts in concert with viral RAS dysregulation in COVID‐19 disease. From the standpoint of repurposing, what is important is the observation that inhibition of RAS with enalapril and losartan preserves mitochondrial function from the effects of aging. 41
4.3.2. Predisposition and comorbidities, eg, hypertension, other vascular disease and diabetes
Much has been written recently on the relationship between hypertension and COVID‐19 disease. Patients with severe COVID‐19 are often older and have a history of hypertension, for example, 75% of patients who died in the COVID‐19 pandemic in Italy had hypertension. 14
As indicated earlier, ACE2 is not only the target for SARS‐CoV‐2 but functionally balances the pulmonary and possibly vascular RAS/inflammation axis. Therefore, a potential increase in ACE2 mediated with antihypertensives, which act to inhibit RAS, raised potential concerns with patients with COVID‐19. However there was insufficient data to determine whether these findings translated to humans; clinical trials are underway to test these inhibitors in COVID‐19. 42 Furthermore, Kreutz et al. 14 indicated that after a review of the evidence there was currently no reason to discontinue RAS blockers in stable patients in the COVID‐19 pandemic.
Consistent with this view there are now a growing number of studies based on medical records of hospitalized COVID‐19 patients that do not indicate a deterioration of patients with RAS inhibition and to varying extents show an improvement in outcomes when compared to patients not treated with RAS inhibitors. 43 , 44 , 45 , 46 From these studies, however, it is not apparent whether the RAS inhibitors were maintained during hospitalization or possibly ceased on admission. This may be an important consideration based on the results of previous studies with patients with viral pneumonia. However, an increasing number of observational studies examining RAS agents as therapies to reduce hospital admission or ICU deterioration are being published. 47 , 48 , 49 Mortensen et al. 50 showed that both prior and continued ACEI was associated with decreased mortality and length of stay in hospitalized patients with pneumonia. Henry et al. 47 determined the effects of ACEIs and ARBs in patients with viral pneumonia, finding that patients on ACEIs prior to admission and subsequently discontinued had a higher mortality than those not on ACEIs prior to admission and there was a decrease in intubation or mortality in patients with continued use of ACE inhibitors during hospital admission. More recent publications show similar responses in the non COVID ICU setting. 51 , 52 Khera et al. showed there is no mortality decrement of ACE inhibitors and angiotensin receptor blockers with risk of hospitalization and death in hypertensive patients with COVID‐19, 53 and no increased risk of COVID‐19 causing infection requiring admission to hospital was seen with RAS agents. 54 , 55 These results illustrate the importance of administrative/observational data consistent with the view that observational studies based on both big and small data sets are useful in understanding how to treat viral disease. 4 They also call into question the need to keep doing observational studies of these agents when it is now reasonably compelling that RAS agents do not increase the risk of COVID‐19, do not increase mortality and in fact are very likely to reduce mortality significantly. In the time of a pandemic it is unclear why we are using off‐label agents with less data than we have for RAS agents where we know everything about the dose and the side effect profile, high potassium, allergic phenomena and low blood pressure, all of which can actually be managed in the hospital setting.
What is of significance is the emerging understanding that cardiovascular disease, similar to COVID‐19, is a partially inflammatory state. In addition to the extensive discussion on hypertension and cardiovascular disease in COVID‐19 there is an increasing body of evidence for a role of inflammation and immunity in hypertension and cardiovascular disease. 56 For example, there is a growing appreciation of the role of oxidative stress in the arterial wall, with inflammatory infiltration, regulatory and interleukin 17 producing T‐cells leading the development of hypertension. 57
A role for inflammation in hypertension is documented, as is a powerful role for inflammation in COVID‐19 disease. 58 It is not surprising with this confluence that hypertension and related vascular disorders enhance susceptibility to this disease. It follows that from a repurposing standpoint a growing view would be not to abandon RAS inhibitors but rather explore their potential as repurposing agents in COVID‐19.
4.3.3. Secondary pulmonary infection
Acute respiratory distress syndrome (ARDS) is a severe form of acute lung injury with a high mortality rate; infections such as COVID‐19 and sepsis are predisposing factors together with aspiration (summarized in Verdecchiaa et al. 18 ). Twenty years before COVID‐19, it was suggested that RAS agents had a role in the pathogenesis of ARDS and it was demonstrated that ACE insertion/deletion polymorphism was associated with susceptibility and outcome in ARDS. 59 This has been shown in human data with COVID‐19, with mortality disproportionally affecting blacks, who commonly have DD mutation. Even in the past this team suggested that ACE inhibitors may lower the risk of developing ARDS or reduce the severity of the disease. Subsequently, it was shown that in animal experiments in acute lung injury angiotensin II is upregulated and mediates acute lung failure by way of the AT1 receptor, and ACE2 and the AT2 receptor are protective against this injury. 16 Consistent with these findings Yan et al., using influenza A H5N1 virus infection in mice, showed that the ARB losartan improved animal survival and ameliorated acute lung injury. 6
Based on a retrospective cohort non‐COVID‐19 pneumonia study Mortensen et al. 50 examined the influence of statins and inhibitors of the RAS on outcomes. They concluded that statins and to a lesser extent ACE inhibitors and ARBs improved these outcomes. The potential benefits of the pulmonary effects of ARBs have been noted, with ACE2 expression from pre‐existing ARB treatment speculated to be of benefit in the course of SARS‐CoV‐2 infection. 58 In a similar fashion, a recent review on COVID‐19 14 concluded that in patients with lower respiratory tract infections ACEIs and ARBs may be associated with improved outcomes. From a repurposing standpoint the prevailing view would be that inhibitors of RAS may be of benefit in lower respiratory tract infections.
4.3.4. Intubation
Severe gas exchange problems requiring mechanical ventilation can occur with COVID‐19 disease. As indicated by Kim et al., ARDS is refractory hypoxemic respiratory failure with a high mortality rate, reported to range from 30% to 70%. 60 Animals studies by Jerng et al. suggested a link between mechanically induced proinflammatory responses and the RAS, particularly the observation that mechanical ventilation can produce a proinflammatory response in the lungs. 61 Moreover, this study demonstrated that RAS is involved in the pathogenesis of ventilator‐induced lung injury. Of particular importance is the observation that pretreatment with captopril or concomitant infusion of losartan attenuated the lung injury and inflammation. 61 A role for angiotensin 1–7 in improving key responses in an experimental model of ARDS was described by Zambelli et al. 62 They demonstrated that angiotensin 1–7 reduced the severity of lung injury and inflammation following pulmonary acid aspiration and high stretch mechanical ventilation.
Kim et al. 60 examined the medical records of patients admitted to a medical intensive care unit for mechanical ventilation support. They concluded that an ACE inhibitor or ARB may have a beneficial effect on ARDS patients. From the standpoint of repurposing, what is important is the observation that inhibition of RAS is implicated in ventilation proinflammatory response in the lungs, that COVID‐19 is a disease of RAS dysregulation and that observational data with RAS inhibitors suggests they may be beneficial.
4.4. Dose, timing and safety (Figure 2)
FIGURE 2.
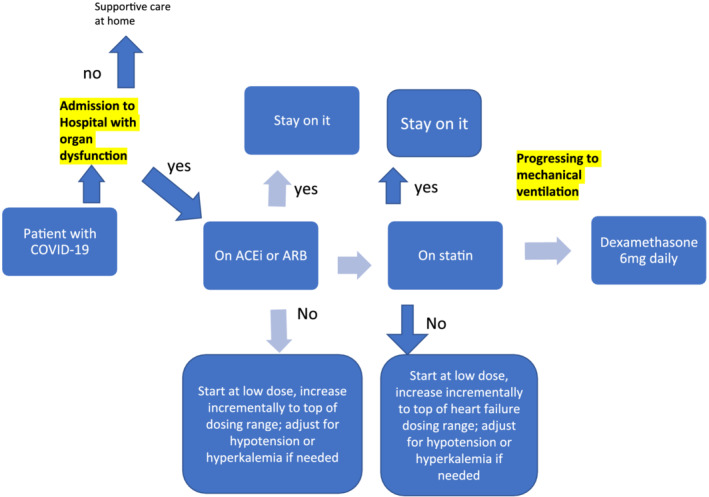
Possible clinical algorithm for treating the host with moderate to severe COVID
An application of the repurposed drug within the approved dose ranges for its initial indications for which the pharmacokinetics and toxicology are documented as part of the registration for the drug's initial indication is needed. Using the RAS as an example, a process could identify what is known about RAS inhibitors in regard to (a) observational data based upon administrative records, (b) the relationship between the existing Cmax and EC50 for new indication, (c) the historical safety of the proposed repurposed drug including knowledge of clinical maximal dose.
4.4.1. Observational data based on administrative records
The key question here is not just around the evidence that the agents that inhibit the RAS improve patient outcomes in COVID‐19 disease but at what doses, for both efficacy and for toxicity.
During 2020 there have been at least four clinical observational‐based studies 43 , 44 , 45 , 46 derived from medical records of patients hospitalized for COVID‐19. As indicated above these studies did not indicate a deterioration of patients with RAS inhibition. Conversely to varying extents an improvement in outcomes was seen when compared to patients with COVID‐19 disease not treated with RAS inhibitors. By inference these patients were likely being treated prior to hospitalization for hypertension or diabetes on standard drug regimens. Based on observational data there would be an argument to explore further the possibility of a new indication for RAS inhibitors as repurposed drugs in COVID‐19.
4.4.2. The relationship between the existing C max and EC 50 for new indications
The specific question of interest here is whether there is evidence that RAS agents are effective at doses below the approved Cmax in attenuating pulmonary responses and preventing or reversing the aberrant inflammatory pathophysiology. This is also important dosing knowledge to reduce toxicity.
In a comprehensive review on repurposing 11 potential differences in dosing amounts and schedules in the area of repurposing have apparently been overlooked. Our experience has indicated that if the ED50 for the new indication for the repurposed drug is equal to or less than the Cmax achieved by the drug in registered dosing schedules for its original indication, then dosing issues become less complex and the arguments for registration of the repurposed drug for a new indication are much easier. Thus, the bioavailability, toxicology and pharmaco‐surveillance of the drug at that dose is well established as part of the original drug approval process, albeit the disease being treated is different. This latter point may introduce issues around dose–response and dose‐toxicity, but they can be surveyed from an a priori basis rather than from no knowledge.
By inference in the studies based on observational data described immediately above, COVID‐19 patients were likely being treated prior to hospitalization for hypertension or diabetes on standard drug regimens. If that were so, then it could be assumed that the RAS inhibitors would have been employed at or below their approved Cmax obtained from the top of the dosing range for the RAS inhibitors.
As there is no human information to date on these preclinical aspects, we need to analyse known pharmacological data from cellular systems and animals, and use supporting data from human studies of other viruses that cause ARDS in a pathophysiological process resembling COVID‐19. Based on the observational data there would be an argument to explore further the possibility of a new indication for RAS inhibitors as repurposed drugs in COVID‐19 initially within the existing approved Cmax of the drugs.
4.4.3. The historical safety of the proposed repurposed drug
We need to know the historical safety of proposed RAS inhibitors in exploring their potential role as repurposed drugs for treating COVID‐19. Losartan, telmisartan and olmesartan (and additional AT1R antagonists) have been widely applied in the clinic since the 1990s for control of hypertension, diabetes and some kidney disorders, and are known as safe drugs that are rarely implicated in adverse drugs events. 63 , 64
Based on historical safety experience data there would be an argument to explore further the possibility of a new indication for RAS inhibitors as repurposed drugs in COVID‐19.
5. CONCLUSION
The importance of the host response in infectious disease has been highlighted very recently, including the suggestion that ARBs and statins, both very cheap, off‐patent and in our pharmacies currently, could be evaluated to improve outcomes including survival in COVID‐19 patients. 5 This approach builds on the pioneering earlier, 4 treating of the host concept, based on an understanding that a beneficial clinical phenotype has been observed with a treatment and this can be employed for the purpose of treating the host.
The approach brings together three concepts: the value of therapeutics in stabilizing patients prior to subsequent interventions, repurposing therapeutics for new indications and the potential of treating the host. During this review the following became evident:
There is a need to comprehensively determine if there exists an alignment of the proposed repurposed drug with the known pathophysiology of the viral infection. In the case of RAS what is remarkable is the interplay between an existing predisposition to RAS induced inflammation with an overlay of severe RAS dysregulation with COVID‐19 disease.
The fundamental importance of observational (administrative) data as a pathfinder source of information from which to select candidate drugs for evaluation in a repurposed context.
The importance of adhering to the principals of repurposing as they relate to dose and safety.
We have described a framework for treating the host with repurposed drugs and illustrated its use using RAS inhibitors. We would caution that even though there exists observational data highlighting a potential protective effect of RAS inhibition consistent with a pathophysiological alignment, this outcome may still be due to chance. Therefore, based on our analysis we believe a clinical trial for RAS inhibitors would be justified. However, it is noted that at least eight trials of this type have already been registered, with quality observational data on infection rate, severity of disease, admission to ICU and death reported. This data at least shows noninferiority with RAS, and in some studies suggests significant mortality benefit. As these drugs have plausible biology, together with the now available mechanistic data from currently available doses, and a side effect profile that is well known and able to be managed, use in clinical practice could be justified. This line of thinking is in line with critical trial design thinkers emerging in this pandemic. 65 , 66 , 67 This reinforces the concurrent move in the pharmacology world to delineate the treatment of the host, from treatment and elimination of the infective agent, as outlined and pioneered by David Fedson many years ago.
COMPETING INTERESTS
There are no competing interests to declare.
Martin JH, Head R. A pharmacological framework for integrating treating the host, drug repurposing and the damage response framework in COVID‐19. Br J Clin Pharmacol. 2021;87:875–885. 10.1111/bcp.14551
REFERENCES
- 1. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID‐19): a review. JAMA. 2020;323(18):1824‐1836. 10.1001/jama.2020.6019 [DOI] [PubMed] [Google Scholar]
- 2. Fedson DS, Jacobson JR, Rordam OM, Opal SM. Treating the host response to Ebola virus disease with generic statins and angiotensin receptor blockers. MBio. 2015;6:e00716‐e00715. 10.1128/MBio00716-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fedson DS. Treating the host response to emerging virus diseases: lessons learned from sepsis, pneumonia, influenza and Ebola. Ann Transl Med. 2016;4(21):421. 10.21037/atm.2016.11.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fedson DS. Observational studies help us understand how to treat pandemic influenza and other emerging virus diseases. J Emerg Crit Care Med. 2017;1(6):1–10. 10.21037/jeccm.2017.05.03 [DOI] [Google Scholar]
- 5. Fedson DS, Opal SM, Rordam OM. Hiding in plain sight: an approach to treating patients with severe COVID‐19 infection. MBio. 2020;11(2):e00398‐e00320. 10.1128/mBio.00398-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan Y, Liu Q, Li N, et al. Angiotensin II receptor blocker as a novel therapy in acute lung injury induced by avian influenza a H5N1 virus infection in mouse. Sci China Life Sci. 2015;58(2):208‐211. 10.1007/s11427-015-4814-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martin JH, Bowden NA. Drug repurposing in the era of COVID‐19: a call for leadership and government investment. Med J Aust. 2020;212(10):450‐452.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin JH, Clark J, Head R. Buying time: drug repurposing to treat the host in COVID‐19H. Pharmacol Res Perspect. 2020;00(4):e00620. 10.1002/prp2.620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. The RECOVERY Collaborative Group . Dexamethasone in hospitalized patients with Covid‐19 — preliminary report. N Engl J Med. 2020;1‐11. 10.1056/NEJMoa2021436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pirofski L, Casadevalli A. The damage–response framework as a tool for the physician‐scientist to understand the pathogenesis of infectious diseases. J Infect Dis. 2018;218(S1):S7‐S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41‐58. [DOI] [PubMed] [Google Scholar]
- 12. Zhang XJ, Qin JJ, Cheng X, et al. In‐hospital use of statins is associated with a reduced risk of mortality among individuals with COVID‐19. Cell Metab. 2020;32(2):176‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li W, Moore MJ, Vasilieva N, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450‐454. 10.1038/nature02145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kreutz R, Algharably E, Azizi M, et al. Hypertension, the renin–angiotensin system, and the risk of lower respiratory tract infections and lung injury: implications for COVID‐19: European Society of Hypertension COVID‐19 Task Force Review of Evidence. Cardiovascular Research. 2020;116(10):1688‐1699. 10.1093/cvr/cvaa097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brandon MH, Jens V, Giuseppe L. Response to the emerging novel coronavirus outbreak. BMJ. 2020;368:m406. [DOI] [PubMed] [Google Scholar]
- 16. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS‐coronavirus‐induced lung injury. Nat Med. 2005;11(8):875‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imai Y, Kuba K, Penninger J. The discovery of angiotensin‐converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol. 2008;93.5:543‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verdecchiaa P, Cavallinia C, Spanevellob A, et al. The pivotal link between ACE2 deficiency and SARS‐CoV‐2 infection. Eur J Intern Med. 76:14‐20. 10.1016/j.ejim.2020.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—lessons from available evidence and insights into COVID‐19. Hypertens Res. 2020;43(7):648‐654. 10.1038/s41440-020-0455-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. South A, Diz D, Chappell M. COVID‐19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ Physiol. 2020;318(5):H1084‐H1090. 10.1152/ajpheart.00217.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J, Chen S, Bihl J, et al. Exosome‐mediated transfer of ACE2 (angiotensin‐converting enzyme 2) from endothelial progenitor cells promotes survival and function of endothelial cell. Oxid Med Cell Longev. 2020;2020:Article ID 4213541. 10.1155/2020/4213541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veerappan A, Reid A, Estephan R, et al. Mast cell renin and a local renin–angiotensin system in the airway: role in bronchoconstriction. PNAS. 2008;105(4):1315‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jonas S, Erjefa J. Mast cells in human airways: the culprit? Eur Respir Rev. 2014;23:299‐307. 10.1183/09059180.00005014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker BF. All because of the mast cell: blocking the angiotensin receptor‐1 should be better than inhibiting ACE (theoretically). Cardiovasc Res. 2011;92:7‐9. 10.1093/cvr/cvr214 [DOI] [PubMed] [Google Scholar]
- 25. Bernstein K, Khan Z, Giani J, Cao D‐Y, Bernstein E, Shen X. Angiotensin‐converting enzyme in innate and adaptive immunity. Nat Rev Nephrol. 2018;14(5):325‐336. 10.1038/nrneph.2018.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coperchinia F, Chiovatoa L, Crocea L, et al. The cytokine storm in COVID‐19: an overview of the involvement of the chemokine/chemokine‐receptor system. Cytokine and Growth Factor Reviews. 53:25‐32. 10.1016/j.cytogfr.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McGonagle D, O'Donnell J.S., Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID‐19 pneumonia. The Lancet Rheumatology. 2020;2(7):E437‐E445. 10.1016/S2665-9913(20)30121-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teuwen L‐A, Geldhof V, Pasut A, Carmeliet P. COVID‐19: the vasculature unleashed nature reviews. Immunology. 2020;20(7):389‐391. 10.1038/s41577-020-0343-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wei Cao and Taisheng Li Research highlight COVID‐19 . Towards understanding of pathogenesis. Cell Res. 2020;30(5):367‐369. 10.1038/s41422-020-0327-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hemnes A, Rathinasabapathy A, Austin E, et al. A potential therapeutic role for angiotensin converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J. 2018;51(6):1702638–1702638. 10.1183/13993003.02638-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dyer S, Frewin D, Head R. The influence of chronic captopril treatment and its withdrawal on endothelium‐dependent relaxation. Blood Press. 1992;1(4):247‐253. 10.3109/08037059209077670 [DOI] [PubMed] [Google Scholar]
- 32. Freund‐Michel V, Cardoso Dos Santos M, Guignabert C, et al. Role of nerve growth factor in development and persistence of experimental pulmonary hypertension. Am J Respir Crit Care Med. Aug 1, 2015;192(3):342‐355. [DOI] [PubMed] [Google Scholar]
- 33. Jeffreson S, Rush R, Zettler C, Frewin DB, Head RJ. The influence of the renin angiotensin system on abnormal expression of nerve growth factor in the spontaneously hypertensive rat. July 1995;22(6‐7):478‐480. 10.1111/j.1440-1681.1995.tb02050.x [DOI] [PubMed] [Google Scholar]
- 34. Gonzalez‐Jaramillo N, Low N, Franco O. The double burden of disease of COVID‐19 in cardiovascular patients: overlapping conditions could lead to overlapping treatments. Eur J Epidemiol. 2020;35(4):335‐337. 10.1007/s10654-020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pachetti M, Marini B, Benedetti F, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. J Transl Med. 2020;18(1):179. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu K, Lang Chen LG, et al. A virus‐binding hot spot on human angiotensin‐converting enzyme 2 is critical for binding of two different coronaviruses. J Virol. 2011;85(11):5331‐5337. 10.1128/JVI.02274-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shahid Z, Kalayanamitra R, McClafferty B, et al. COVID‐19 and older adults: what we know. J Am Geriatr Soc. 2020;68(5):926‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vajapey R, Rini D, Walston J, Abadir P. The impact of age‐related dysregulation of the angiotensin system on mitochondrial redox balance. Frontiers in Physiology|Mitochondrial Research. 2014;5:439. 10.3389/fphys.2014.00439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson B, Nautiyal M, Tan Ya M. Evidence for a mitochondrial angiotensin‐(1–7) system in the kidney. Am J Physiol Renal Physiol. 2016;310:F637‐F645. 10.1152/ajprenal.00479.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lai C, Jou M, Huang S, et al. Proteomic analysis of up‐regulated proteins in human promonocyte cells expressing severe acute respiratory syndrome coronavirus 3C‐like protease. Proteomics. 2007;7(9):1446‐1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Cavanagh E, Piotrkowski B, Basso N, et al. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB Jexpress article. 2003;17(9):1096‐1098. 10.1096/fj.02-0063fje [DOI] [PubMed] [Google Scholar]
- 42. Vaduganathan M, Vardeny O, Michel T, McMurray J, Pfeffer M, Solomon S. Renin–angiotensin–aldosterone system inhibitors in patients with COVID‐19. N Engl J Med. 2020;382(17):1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang P, Zhu L, Cai J, et al. Association of Inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID‐19. Circ Res. 126(12):1671‐1681. 10.1161/CIRCRESAHA.120.317134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mehra M, Desai S, Kuy S, Henry TD, Patel AN. Cardiovascular disease, drug therapy, and mortality in COVID‐19. N Engl J Med. 2020;382(25):e102. 10.1056/NEJMoa2007621 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Meng J, Xiao G, Zhang J, et al. Renin‐angiotensin system inhibitors improve the clinical outcomes of COVID‐19 patients with hypertension. Emerging Microbes & Infections. 2020;9(1):757‐760. 10.1080/22221751.2020.1746200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lam KW, Chow KW, Vo J, et al. Continued in‐hospital ACE inhibitor and ARB use in hypertensive COVID‐19 patients is associated with positive clinical outcomes. J Infect Dis. 2020;9(14):1256–1264, jiaa447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gustafson D, Raju S, Wu R, et al. Overcoming barriers: the endothelium as a linchpin of coronavirus disease 2019 pathogenesis? Arterioscler Thromb Vasc Biol. 2020;40(8):1818‐1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Roquetaillade C, Jamme M, Charpentier J, et al. Hemodynamic impact of cardiovascular antihypertensive medications in patients with sepsis‐related acute circulatory failure. Shock. 2020;54(3):315‐320. 10.1097/SHK.0000000000001524 [DOI] [PubMed] [Google Scholar]
- 49. Bean D, Kraljevic Z, Searle T. ACE‐inhibitors and Angiotensin‐2 receptor blockers are not associated with severe SARS‐ COVID‐19 infection in a multi‐site UK acute hospital trust. medRxiv. 2020;2020.04.07.20056788. 10.1101/2020.04.07.20056788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mortensen EM, Nakashima B, Cornell J, et al. Population‐based study of statins, angiotensin II receptor blockers, and angiotensin‐converting enzyme inhibitors on pneumonia‐related outcomes. Clin Infect Dis. 2012;55(11):1466‐1473. 10.1093/cid/cis733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim J, Kim YA, Hwangbo B, et al. Effect of antihypertensive medications on sepsis‐related outcomes: a population‐based cohort study. Crit Care Med. 2019;47(5):e386‐e393. 10.1097/CCM.0000000000003654 [DOI] [PubMed] [Google Scholar]
- 52. Hsieh MS, How CK, Hsieh VC, Chen PC. Preadmission antihypertensive drug use and sepsis outcome: impact of angiotensin‐converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs). Shock. 2019;53(4):407‐415. 10.1097/SHK.0000000000001382 [DOI] [PubMed] [Google Scholar]
- 53. Khera R, Clark C, Lu Y, et al. Association of angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers with the risk of hospitalization and death in hypertensive patients with coronavirus disease‐19. medRxiv. 2020;2020.05.17.20104943. 10.1101/2020.05.17.20104943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Abajo FJ, Rodríguez‐Martín S, Lerma V, et al. Use of renin–angiotensin–aldosterone system inhibitors and risk of COVID‐19 requiring admission to hospital: a case‐population study. Lancet. 2020;395(10238):1705‐1714. 10.1016/S0140-6736(20)31030-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dublin S, Walker RL, Floyd JS, et al. Renin‐angiotensin‐aldosterone system inhibitors and COVID‐19 infection or hospitalization: a cohort study. Preprint. medRxiv. 2020;2020.07.06.20120386. 10.1101/2020.07.06.20120386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. David G, Harrison TJ, Guzik HL, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132‐140. 10.1161/HYPERTENSIONAHA.110.163576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Solak Y, Afsar B, Nosratola D. Hypertension as an autoimmune and inflammatory disease. Hypertens Res. 2016;39(8):567‐573. [DOI] [PubMed] [Google Scholar]
- 58. Danser J, Epstein M, Batlle D. Renin‐angiotensin system blockers and the COVID‐19 pandemic. At present there is no evidence to abandon renin‐angiotensin system blockers. Hypertension. 2020;75. 10.1161/HYPERTENSIONAHA.120.15082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marshall R, Webb S, Bellingan GJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;166(5):646‐650. [DOI] [PubMed] [Google Scholar]
- 60. Kim J, Choi S‐M, Lee J, Sik Park Y, et al. Effect of renin‐angiotensin system blockage in patients with acute respiratory distress syndrome: a retrospective case control study. Korean J Crit Care Med. 2017;32(2):154‐163. 10.4266/kjccm.2016.00976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jerng J, Hsu Y‐C, Wu H‐D, et al. Role of the renin‐angiotensin system in ventilator‐induced lung injury: an in vivo study in a rat model. Thorax. 2007;62(6):527‐535. 10.1136/thx.2006.061945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zambelli V, Bellani G, Roberto Borsa R, et al. Angiotensin‐(1–7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental Acute Respiratory Distress Syndrome. Intensive Care Med Exp. 2015;3(1):44. 10.1186/s40635-015-0044-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deppe S, Böger RH, Weiss J, Benndorf RA. Telmisartan: a review of its pharmacodynamic and pharmacokinetic properties. Expert Opin Drug Metab Toxicol. 2010;6(7):863‐871. 10.1517/17425255.2010.494597 [DOI] [PubMed] [Google Scholar]
- 64. McIntyre M, Caffe SE, Michalak RA, Reid JL. Losartan, an orally active angiotensin (AT1) receptor antagonist: a review of its efficacy and safety in essential hypertension. Pharmacol Ther. 1997;74(2):181‐194. [DOI] [PubMed] [Google Scholar]
- 65. Angus DC. Optimizing the trade‐off between learning and doing in a pandemic. JAMA. 2020;19:1895–1896. 10.1001/jama.2020.4984 [DOI] [PubMed] [Google Scholar]
- 66. Vandenbroucke JP. Observational research, randomised trials, and two views of medical science. PLoS Med. 2008;5:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Henry C, Zaizafoun M, Stock E, Shamande S, Arroliga A, et al. Impact of angiotensin‐converting enzyme inhibitors and statins on viral pneumonia. PROC (BAYL UNIV Med Cent). 2018;31(4):419‐423. 10.1080/08998280.2018.1499293 [DOI] [PMC free article] [PubMed] [Google Scholar]