

Abstract
Friedreich ataxia (FA) is currently an incurable inherited mitochondrial disease caused by reduced levels of frataxin (FXN). Cardiac dysfunction is the main cause of premature death in FA. Adeno-associated virus (AAV)-mediated gene therapy constitutes a promising approach for FA, as demonstrated in cardiac and neurological mouse models. While the minimal therapeutic level of FXN protein to be restored and biodistribution have recently been defined for the heart, it is unclear if FXN overexpression could be harmful. Indeed, depending on the vector delivery route and dose administered, the resulting FXN protein level could reach very high levels in the heart, cerebellum, or off-target organs such as the liver. The present study demonstrates safety of FXN cardiac overexpression up to 9-fold the normal endogenous level but significant toxicity to the mitochondria and heart above 20-fold. We show gradual severity with increasing FXN overexpression, ranging from subclinical cardiotoxicity to left ventricle dysfunction. This appears to be driven by impairment of the mitochondria respiratory chain and ultrastructure, which leads to cardiomyocyte subcellular disorganization, cell death, and fibrosis. Overall, this study underlines the need, during the development of gene therapy approaches, to consider appropriate vector expression level, long-term safety, and biomarkers to monitor such events.
Keywords: Friedreich ataxia, frataxin, mitochondria, adeno-associated virus, gene therapy, mouse model, cardiomyopathy, transgene overexpression, toxicity, cardiotoxicity
Graphical Abstract
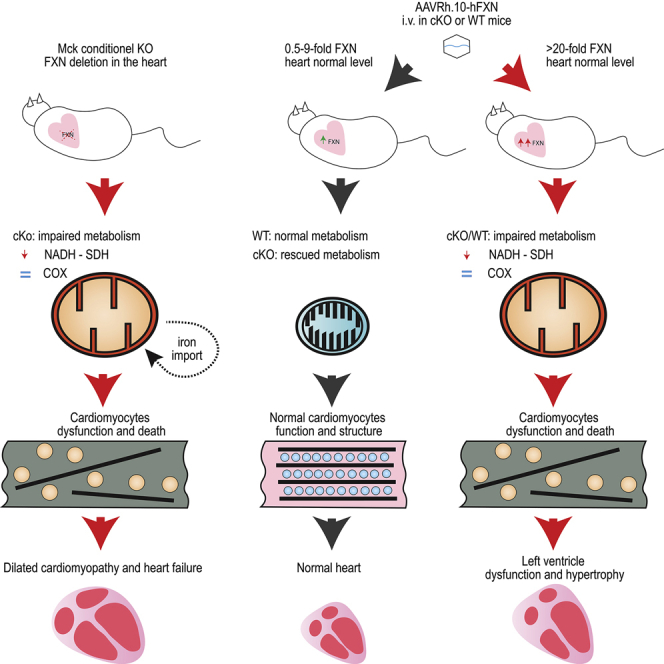
Deficiency in frataxin, an essential mitochondrial protein, leads to Friedreich ataxia, a neurological disease associated with cardiomyopathy. While gene therapy is a promising approach, for clinical translation it is essential to determine the toxicity threshold. In mice, frataxin overexpression below 9-fold was safe, but frataxin overexpression was toxic for the mitochondria and heart beyond 20-fold.
Introduction
Friedreich ataxia (FA) is a rare neurodegenerative disease characterized by spinocerebellar and sensory ataxia associated with hypertrophic cardiomyopathy (HCM).1 Cardiac dysfunction is the main cause of premature death in FA patients.2 95% of FA patients present a homozygous (GAA) expansion within the first intron of the frataxin gene (FXN).3 This pathological expansion causes the heterochromatinization of the locus leading to reduced transcription of the FXN gene.4 FXN is a highly conserved mitochondrial protein regulating the biosynthesis of iron sulfur clusters (Fe-S) through an interaction with the de novo Fe-S complex assembly machinery.5 Fe-S are prosthetic groups crucial for many biological functions, including the mitochondrial respiratory chain; heme, lipoid acid, and molybdenum biosynthesis; tRNA thiolation; and iron metabolism.6, 7, 8, 9 Frataxin deficiency leads to impaired Fe-S biogenesis, impairment of Fe-S enzymes, mitochondrial dysfunction, iron metabolism dysregulation, and eventually cellular dysfunction and death.10,11
The therapeutic potential of AAV-mediated in vivo gene therapy in preventing and rescuing mitochondrial dysfunction associated with FXN deficiency, both in cardiac and neurological tissues, has clearly been demonstrated in several mouse studies.12, 13, 14 To successfully translate these initial proof-of-concept studies to the clinic, we undertook to define the therapeutic thresholds conditioning the rescue of the cardiac phenotype as well as the potential toxic effects associated with FXN overexpression. This first point was addressed in a recent publication describing the dose-response studies we have conducted in the cardiac Mck conditional knockout mouse model (or Mck mouse), which recapitulates most features of the FA cardiomyopathy.10,15 We demonstrated that the therapeutic outcome of AAV-FXN gene therapy is directly and proportionally correlated with the vector biodistribution in the heart. The correction of only half the cardiomyocyte was sufficient to restore fully the cardiac morphology and function.15 Regarding the maximum level of FXN protein expression tolerated, earlier mouse studies have reported that the constitutive overexpression of FXN in muscle and heart up to 6-fold the normal level was innocuous.16,17 In contrast, several recent studies have suggested otherwise when constitutive or induced FXN overexpression reach higher levels in yeast (>14-fold),18 HEK293 inducible cell line (>13-fold),19 and transgenic Drosophila (>9-fold).20,21 However, the possible toxicity of similar levels of FXN overexpression were never investigated in preclinical relevant animal models (i.e., rodent or large animal models) or in the context of AAV-mediated gene therapy.
In the present study, we investigated whether in vivo AAV-mediated gene transfer could be detrimental in wild-type (WT) mice or frataxin-deficient Mck mice. We evaluated FXN overexpression in vivo and its functional and morphological consequences on mitochondria and the heart. FXN protein expression over 20-fold the endogenous level is toxic for the mitochondria and results in severe impairment of complexes I and II enzymatic activity, alteration of the mitochondria ultrastructure, cardiomyocyte cell death, and fibrosis leading to heart dysfunction. This was also associated with an increased mitochondria biomass in cardiomyocytes and the induction of the integrated stress response (ISR). Overexpression of frataxin up to 9-fold the endogenous level in the cardiac tissue was uneventful, suggesting that the maximum safe level of FXN overexpression is between 9- and 20-fold the normal level. Strikingly, liver overexpression of FXN > 90-fold the normal level did not result in any detectable toxicity, suggesting organs specific susceptibility to FXN overexpression in mice.
Results
FXN Overexpression Leads to Impairment of Mitochondria Succinate Dehydrogenase (SDH) Activity
Previously, we conducted a dose-response study in 5-week-old Mck mice injected intravenously with decreasing doses of AAVRh.10-CAG-hFXN-HA vector, from 5 × 1013 down to 1 × 1012 vector genomes (vg)/kg.15 Upon sacrifice at 12 weeks of age, the human FXN (hFXN) protein level measured in the heart by ELISA assay ranged from 2 to 10,927 ng of FXN per mg of total heart protein (ng/mg). In WT 9-week-old C57/B6J mice (n = 6), the mouse FXN protein level measured by ELISA in the heart was 147 ± 42 ng/mg. Despite more than 70-fold overexpression, the cardiac function and morphology of treated Mck mice was rescued. In the present study, we performed additional histological analysis on the heart tissue sections from the same Mck mice to further investigate the relationship between FXN overexpression in the heart and the rescue of mitochondrial metabolism (Table S1). Unexpectedly, the heart tissue sections from Mck mice treated at the highest dose of vector (i.e., 5 × 1013 vg/kg) displayed regions with very high level of hFXN-HA expression, where SDH enzymatic activity was systematically impaired (Figures 1A and S1). This was particularly obvious in the mouse with the highest cardiac vector biodistribution (7.68 per diploid genome on average) and protein concentration (10,927 ng/mg, or 74-fold the endogenous level) (Figure 1A, first row). No SDH impairment was observed in the cardiomyocytes expressing much lower hFXN-HA levels or neighboring these hotspots of expression. Quantification of the heart surface positive for SDH enzymatic activity in the Mck mice treated with the highest dose (n = 3; 73% ± 2%) in comparison to NaCl-injected WT mice (n = 7; 83% ± 4%) revealed that less than 12% of the total heart surface was affected. These hotspots were not associated with cardiomyocyte cell death, fibrosis, or cell infiltrations, as assessed by hematoxylin and eosin (H&E) and wheat germ agglutinin (WGA) staining of adjacent heart tissue sections, in comparison to untreated Mck mice (Figure 1B). Interestingly, the cardiomyocytes with high hFXN-HA expression and SDH activity impairment did not show any iron accumulation following Perls-DAB (3,3′-Diaminobenzidine) staining of Fe3+ (Figure 1C), as this would be expected following FXN deficiency and/or Fe-S biosynthesis impairment.10,22
Figure 1.

High Expression of FXN-HA in Cardiomyocytes Is Associated with Impaired Succinate Dehydrogenase (SDH) Enzymatic Activity despite Functional Rescue of Mck Mice Treated with AAVRh.10-CAG-hFXN-HA
(A–C) Histological analysis of heart tissue sections collected from the Mck mice treated at 5 weeks of age with the AAVRh.10-CAG-hFXN-HA vector and sacrificed at 12 weeks. Representative images are from the Mck mouse treated with 5 × 1013 vg/kg and expressing the highest level of hFXN-HA (10,927 ng/mg) and from another Mck mouse treated with 2.5 × 1013 vg/kg and with lower hFXN-HA level (695 ng/mg). For controls, heart tissue sections from 12-week-old NaCl-injected wild-type (WT) mice and 9-week-old untreated Mck mice were also analyzed. The corresponding vector copies per diploid genome (VCN) and tissue concentration in human FXN ([hFXN] in ng per mg of total protein) are reported. The same time exposure was used for all animals. (A) Co-staining and co-localization analysis of hFXN-HA overexpression and SDH enzymatic activity. Acquisitions of a single microscopic field at low and high magnification are shown, to compare the distribution of hFXN-HA expression hotspots and SDH activity impairment. (B) Hematoxylin and eosin (H&E) and wheat germ agglutinin (WGA) staining. (C) DAB-enhanced Perls labeling of iron deposits with methyl green counterstaining. (D–F) Longitudinal echocardiography analysis of the same Mck mice, represented here as individual kinetics. Data are represented as mean ± SD for WT control mice (n = 7) and untreated Mck mice (n = 10). For untreated Mck mice, historical data were plotted. Statistical analyses are reported in Table S2. (D) Left ventricle (LV) shortening fraction (SF). (E) Cardiac blood output (CO) measured at the aorta and normalized to body weight (BW). (F) LV mass normalized to BW. See also Figures S1A–S1C for the extended echocardiography analysis of this cohort of mice. See also Figures S1D–S1H for the echocardiography follow-up, until 25 weeks of age, of a second cohort of Mck mice treated at 5 weeks of age at 5 × 1013 vg/kg. Figure partially adapted from Belbellaa et al.15
Importantly, the left ventricle (LV) function of these mice did not appear substantially impaired at rest at 12 weeks of age (Figures 1D–1F and S1A–S1C). Mck mice treated at 5 × 1013 vg/kg displayed LV shortening fraction (SF) and cardiac blood output normalized to body weight (CO/BW) corrected to WT levels (Figures 1D and 1E), similar to Mck mice treated at 2.5 × 1013 vg/kg. Importantly, Mck mice treated at 5 × 1013 vg/kg displayed a sustained functional rescue at long term (Figures S1D–S1H) when evaluated by echocardiography until 25 weeks of age in a separate cohort of mice (n = 5; Table S1). Most likely, this is explained by the relative small proportion of cardiomyocytes and heart surface affected overall (<12%). Indeed, we showed previously that the functional rescue of the cardiac phenotype is achieved with as low as 50%–60% of heart surface corrected.15 It is worth noting that these mice did not display significant LV hypertrophy (Figure 1F), but the LV diameters at the end systole (LV-ESD) and diastole (LV-EDD) appeared slightly higher in the mouse treated at 5 × 1013 vg/kg and expressing 74-fold the normal level of FXN (Figures S1A and S1B). Overall, these results suggested that a very high level of hFXN-HA protein in cardiomyocytes could lead to impaired mitochondrial SDH enzymatic activity but without notable iron accumulation.
Overexpression of human FXN in WT Mice Leads to Mitochondrial and Cardiac Toxicity
In order to rule out any potential confounding effects from the Mck mouse cardiac phenotype, we undertook to replicate these experimental conditions in WT C57/B6J mice. WT mice received 5 × 1014 vg/kg (n = 4) or 5 × 1013 vg/kg (n = 3) of the same AAVRh.10-CAG-hFXN-HA vector at 7 weeks of age (Table S1). At the 5 × 1014 vg/kg dose, we were able to replicate similar vector copy number (VCN) (≥7) and hFXN-HA expression (≥10,000 ng/mg) (Figures 2A–2C), as reported above in Mck mice treated at 5 × 1013 vg/kg. However, one-log higher of vector dose was required in WT mice to achieve the same vector biodistribution and overexpression as in Mck mice. This might suggest a difference of transgene expression between WT and Mck mice. However, no significant difference in hFXN-HA expression when normalized to the vector copy number was observed between treated WT mice (n = 7) and Mck mice (n = 35) (Figure S2). At the study endpoint, 21 weeks of age, all treated WT mice were alive, with normal BW growth curve (Figure 2D) and no behavioral sign of stress or suffering. At necropsy, no gross anatomical anomaly was observed at the levels of the heart, lung, liver, kidney, digestive tract, or skeletal muscles.
Figure 2.

High Level of FXN-HA Overexpression in the Heart of WT Mice Treated with AAVRh.10-CAG-hFXN-HA Vector Is Associated with Cardiac Fibrosis and Subclinical Impairment of Heart Function and Morphology
WT C57/B6J mice were treated at 7 weeks of age at 5 × 1014 (n = 4, red) or 5 × 1013 (n = 3, blue) vg/kg and sacrificed at 21 weeks. Unless stated otherwise, individual data points are plotted, with mean and SD. (A) qPCR quantification of the VCN in the heart. Light orange area represents the VCN range in Mck mice treated at 5 × 1013 vg/kg (n = 3) with the same vector from a previous study.15 (B) qRT-PCR analysis of the transgene mRNA level normalized to mouse Fxn mRNA level. (C) ELISA assay quantification of hFXN-HA protein concentration ([hFXN]) in the heart, normalized to mg of total heart protein. Light orange area represents the [hFXN] range in Mck mice treated at 5 × 1013 vg/kg (n = 3) from a previous study and reported in Figure 1.15 Black dotted line represents the endogenous level of FXN in untreated WT mice. (D) BW reported as individual, with males in blue and females in red. For untreated WT mice, historical data were plotted as mean ± SD. (E) Representative images from the histological analysis of adjacent heart tissue section collected from WT mice (5 × 1014, n = 4; 5 × 1013, n = 3) and stained with HA or WGA. Red arrows indicate fibrosis and cell infiltrates and yellow arrows indicate cardiomyocytes displaying subcellular disorganization. VCN and [hFXN] values are reported above for each animal. See also Figure S3 for the extended histological analysis. (F) Quantification of heart surface labeled with WGA. NaCl-injected WT mice sacrificed at 15 or 22 weeks of age (n = 11) were used as control. Brown-Forsythe and Welch one-way ANOVA statistical test, p values are reported with n.s. p > 0.05. (G–H) qRT-PCR quantification of cardiac gene expression normalized to 18S and reported as percentage of NaCl-treated WT mice (n = 9). (G) Col1a1, Col3a1, and Tgfβ1 mRNA level. Two-way ANOVA and FDR 5% statistical test, p values are reported with n.s. p > 0.05. (H) Nppa mRNA level. Brown-Forsythe and Welch one-way ANOVA test, p values are reported with n.s. p > 0.05. (I and J) Echocardiography at 21 weeks of age. For control, historical data from 21-week-old WT mice treated with NaCl are reported. Brown-Forsythe and Welch one-way ANOVA test, p values are reported with n.s. p > 0.05. (I) LV SF. (J) LV mass normalized to BW. (K) Heart ventricle weight measured upon necropsy at 21 weeks of age, normalized to BW. (L) Correlation analysis between [hFXN] and LV SF. Spearman non-parametric correlation coefficient and p value are reported.
The consequences of hFXN overexpression on the heart histology were evaluated following H&E and WGA staining, performed on adjacent heart frozen tissue sections (Figures 2E and S3). Both stains revealed and confirmed sparse fibrotic patches, but only in the animals treated at 5 × 1014 vg/kg and more visibly in the three mice with the highest transgene expression, ranging from 5,206 up to 12,773 ng/mg (i.e., 34- and 85-fold the normal FXN level, respectively) (Figure 2C). Heart tissue sections from WT mice treated at 5 × 1013 vg/kg and NaCl-injected WT mice appeared similar (Figures 2C and S3). While the overall total extent of heart fibrosis was not statistically different between NaCl-injected WT mice and WT mice treated at 5 × 1014 or 5 × 1013 vg/kg (Figure 2F), the qRT-PCR analysis of Col1a1, Col3a1, and Tgfβ1 revealed significant increased expression of Col1a1 and Col3a1 in the hearts of WT mice treated at 5 × 1014 vg/kg, indicating ongoing fibrosis (Figure 2G).
While WT mice treated at 5 × 1014 vg/kg displayed significant increased Nppa gene expression (Figure 1H), indicative of volume/pressure overload, none of them developed severe cardiac dysfunction at 21 weeks of age (Figures 1I–1K). Nonetheless, the three mice with the highest level of transgene expression in the 5 × 1014 vg/kg group displayed meaningful reduction of LV SF at rest (Figure 2I; Video S1) and increased heart hypertrophy (Figures 2J and 2K). Importantly, no functional or morphologic heart alteration was observed in the mice treated with vehicle or at 5 × 1013 vg/kg. The inverse correlation between hFXN-HA expression and the LV SF was statistically significant (Figure 2L).
To assess the mitochondria function in relation to the level of hFXN-HA expression, semiquantitative histoenzymatic assays were performed on adjacent heart tissue sections. We probed the activity of the respiratory chain complexes I, II, and IV (Figures 3 and S3). As reported in Mck mice (Figure 1A), the co-labeling and co-localization analysis of SDH enzymatic activity and hFXN-HA revealed hotspots of transgene expression where the SDH enzymatic activity was strongly reduced (Figure 3). However, the overall heart surface affected was relatively limited (Figure S3), and this was observed only in mice treated at 5 × 1014 vg/kg. SDH enzymatic activity in WT mice treated at 5 × 1013 vg/kg appeared similar to NaCl-injected WT mice (Figures 3 and S3). When quantified, the total surface positive for SDH activity was, respectively, 95.0% ± 2.1% (n = 4) and 98.5% ± 0.3% (n = 3) in mice treated at 5 × 1014 and 5 × 1013 vg/kg. Of note, the SDH staining and image acquisition and quantification were performed in batch, with all individuals from a given study processed in a single run, including untreated WT and Mck controls. However, there were some variations from one run to another, at different steps and especially during the image quantification, which thresholding result led to bias in the background signal. This explains the difference in WT mice values between the analysis of this cohort of mice and the one reported earlier. WT mice treated at 5 × 1014 vg/kg also displayed sparse impairment of the enzymatic activity of complex I (NADH-ubiquinone oxidoreductase [NADH]), contrary to WT mice treated at 5 × 1013 vg/kg. In contrast, the enzymatic activity of complex IV (cytochrome C oxidase [COX]) seemed unaffected in both dose groups (Figures 3 and S3). Strikingly, these results were reminiscent of what is observed in the heart of untreated Mck mice, in which FXN deficiency leads to the impairment of Fe-S enzymes, including SDH and NADH, but not COX, dependent on heme cofactors and not Fe-S cofactors.23
Figure 3.

High Level of FXN-HA Overexpression in WT Mice Cardiomyocytes Leads to Mitochondrial Function and Structure Impairment
Representative histological observations from the correlative analysis of adjacent heart tissue sections/sample from WT mice treated at 7 weeks of age with AAVRh.10-CAG-hFXN-HA vector (5 × 1014 vg/kg, n = 4; 5 × 1013 vg/kg, n = 3) and sacrificed at 21 weeks. For controls, 21-week-old NaCl-injected WT mouse (n = 1) and 9-week-old untreated Mck mice (n = 2) were also analyzed. Each image series corresponds to the same individual for which dose of vector, VCN, and [hFXN] are reported. From left to right, co-staining and co-localization analysis of hFXN-HA protein expression and SDH enzymatic activity, cytochrome C oxidase (COX), and NADH-ubiquinone oxidoreductase (NADH) enzymatic activities assessed by in situ histoenzymatic assay are shown. Transmission electron microscopy (TEM) observations at low and high magnification of the LV myocardium is shown, following negative stain performed to assess cardiomyocyte and mitochondria ultrastructure. Finally, TEM observations of adjacent ultrathin sections at high magnification following bismuth sodium tartrate labeling of iron-ferritin complexes are shown. White arrows indicate non-iron mitochondrial electron-dense bodies, blue arrowheads indicate collapsed mitochondrial cristae, orange arrowheads indicate mitochondrial iron deposits, yellow arrowheads indicate mitochondrial iron-ferritin complex accumulation in mitochondria, and red arrowheads indicate ferritin sequestrated in the lysosome. myo, myofibrils; mito, mitochondria; nuc, nucleus. See also Figure S3 for extended histological analysis.
The consequences of FXN overexpression on the cardiomyocyte ultrastructure were also assessed by transmission electron microscopy (TEM). In contrast to WT mice treated at 5 × 1013 vg/kg or NaCl, WT mice treated at 5 × 1014 vg/kg displayed a substantial proportion of cardiomyocytes with severe alterations of their subcellular organization, with scattered and disordered myofibrils (Figure 3). Their mitochondria were swollen with sparse cristae, whose stacking is crucial for mitochondria bioenergetic efficiency.24 The total volume occupied by mitochondria in cardiomyocytes also appeared significantly increased, suggesting altered mitochondrial proliferation or turnover. We also frequently observed the presence of electron-dense bodies inside the mitochondria matrix. These were not reminiscent of collapsed cristae or iron deposits, as commonly observed in FA patients and untreated Mck mouse cardiomyocytes.10,25,26 To rule out the presence of mitochondrial iron deposits, adjacent ultrathin sections were stained with bismuth subnitrate to label iron complexed with ferritin, in the mitochondria as well as in lysosomes.25,27, 28, 29 All WT mice displayed lysosomal labeling, as expected, since this organelle is central to ferritin turnover, but none showed mitochondrial labeling in contrast to untreated Mck mice (Figure 3). Collectively, these observations rule out iron accumulation as part of the cardio- and mitochondrial toxicity mechanism, which is in line with the lack of Perls labeling observed in treated Mck mice (Figure 1C).
To further investigate the mitochondria biomass, heart tissue sections from the same WT mice were co-labeled for SDH enzymatic activity and for prohibitin (PHB) (Figure 4A). PHB is a ubiquitous protein predominately located in the mitochondrial inner membrane,30 which we used previously to assess mitochondria biomass.15 In line with the above TEM observations, WT mice treated at 5 × 1014 vg/kg displayed a substantial increase of PHB labeling, but less than untreated Mck mice. In WT mice treated at 5 × 1014 vg/kg, we also observed a substantial co-localization between increased PHB labeling and reduction of SDH enzymatic activity. In contrast, WT mice treated at 5 × 1013 vg/kg appeared similar to NaCl-injected WT mice. Interestingly, WT mice treated at 5 × 1014 vg/kg also displayed increased cardiac gene expression for Asns, Mthfd2, and Ddit3, hallmark genes of the ISR (Figure 4C). The expression of Fgf21 and Gdf15, which are downstream target genes of the ISR, were also strongly increased, although only statistically significant for Fgf21 (Figures 4D and 4E). Interestingly, Fgf21 and Gdf15 have been proposed as biomarkers of mitochondria and heart dysfunction.31,32
Figure 4.

Cardiotoxic Overexpression of FXN-HA Is Associated with Increased Mitochondria Biomass and the Induction of the Integrated Stress Response
(A) Co-labeling and co-localization observation for prohibitin (Phb) and the enzymatic activity of SDH, on heart tissue sections from WT mice treated with AAVRh.10-hFXN-HA (5 × 1014, n = 4; 5 × 1013, n = 3). For controls, NaCl-injected WT and untreated Mck mice are represented. For each image series, the dose of vector, VCN, and [hFXN] are reported. (B-D) qRT-PCR analysis of heart mRNA levels reported to 18S or Hprt (depending on the abundance of the target gene) and reported as percentage of NaCl-injected WT mice level. Individual data points are reported, with mean and SD. Brown-Forsythe and Welch ANOVA test, p values are reported with n.s. p > 0.05. (B) Integrated stress response (ISR) hallmark genes. (C and D) Downstream ISR-target genes encoding for secreted proteins (C) Fgf21 and (D) Gdf15.
Altogether, these results suggest that cardiac overexpression of hFXN-HA ≥ 20–30-fold the normal endogenous level is toxic for the heart and mitochondria. The mitochondria toxicity is characterized by severe alterations of the mitochondria ultrastructure and bioenergetics and the induction of ISR.
High Level of FXN Overexpression Results in Acute Cardiotoxicity and Compromised the Gene Therapy Outcome
While the study in WT mice recapitulated and confirmed the mitochondrial toxicity observed initially in Mck mice, it was unclear if this toxicity could also be explained by the high dose of vector administered, the resulting high number of vector copies per cell, and/or the HA tag fused to transgene (Figure 5A). To address these questions, we designed an optimized expression cassette and vector, AAVRh.10-hFXN (Figure 5B). The 5′ UTR and Kozak sequences were optimized. The hFXN cDNA was not codon optimized, but the HA tag was removed. The same promoter and polyA sequences were used in both vectors (Figures 5A and 5B). In this third mouse study, Mck mice at 7 weeks old (upon heart failure) received 2.5 × 1013 vg/kg of the optimized AAVRh.10-hFXN vector (n = 8) or 2.5 × 1013 vg/kg of the non-optimized AAVRh.10-CAG-hFXN-HA vector (n = 6) and were then sacrificed at 15 weeks of age (Table S1). For controls, 7-week-old WT mice received NaCl (n = 10) or the optimized AAVRh.10-hFXN vector at doses of 2.5 × 1013 (n = 2) or 5 × 1012 vg/kg (n = 1).
Figure 5.

Cardiotoxicity FXN Overexpression Is Independent of the Dose Administered and Vector Biodistribution and Compromised the Success of Cardiac Gene Therapy in Mck Mouse Heart
Mck mice were treated at 7 weeks of age with the non-optimized vector AAVRh.10-CAG-hFXN-HA (n = 6) or with the optimized vector AAVRh.10-hFXN (n = 8) at 2.5 × 1013 vg/kg, followed up by echocardiography, and then sacrificed at 15 weeks of age to perform molecular analysis. NaCl-injected WT (n = 8–10) mice were used as control. (A and B) Schematic description of the vector construct for (A) the non-optimized AAVRh10-CAG-hFXN-HA vector, and (B) the optimized AAVRh.10-hFXN vector. (C) qPCR quantification of the number of VCN in the heart. Individual data points are reported, with mean and SD. Welch’s t test, n.s. for p > 0.05. (D) ELISA assay quantification of hFXN protein concentration in the heart, normalized to mg of total heart protein. Black dotted line represents the endogenous mouse FXN level in untreated C57/B6J WT mice (i.e., 147 ± 42 ng/mg) (n = 6). Individual data points are reported, with mean and SD. Welch’s t test, p values are reported. (E) Western blot (WB) analysis of total heart protein extract from Mck mice treated with the non-optimized vector (n = 3) or the optimized vector (n = 5). Immunoblotting against FXN, SDHb, GAPDH, and beta-tubulin (β-Tub). (F and G) WB quantification of the relative protein levels of FXN (F) and SDHb (G), normalized to GAPDH. Welch’s t test, p values are reported. (H) WB analysis of FXN protein maturation, in the heart of Mck mice treated with the optimized vector (n = 5) and expressing up to 179-fold the normal level of FXN. The dose of vector, VCN, and [hFXN] corresponding to each sample are reported. (I–L) Longitudinal echocardiography analysis. For control, WT mice were injected with NaCl (n = 8), and historical data are plotted for NaCl-injected Mck mice (n = 10). Data are reported as mean ± SD. Statistical analyses are presented in Table S4. (I) LV SF. (J) CO normalized to BW. (K) LV mass normalized to BW. (L) LV end-systole diameter (LV-ESD) reported as individual kinetic for treated Mck mice. See also Figure S4 for LV end-diastole measurement and BW. (M) qRT-PCR quantification of the cardiac gene expression of Nppa and Nppb, normalized to 18S and reported as percentage of NaCl-injected WT mice. Brown-Forsythe and Welch ANOVA test, p values are reported with n.s. p > 0.05.
When compared to the non-optimized vector, the optimized vector led to much higher levels of hFXN expression in the heart and liver despite similar VCN (Figures 5C–5F, 8A, and 8C–8E). The resulting levels of hFXN protein in the heart were on average 23,376 ± 9,336 ng/mg and 441 ± 276 ng/mg, respectively, corresponding to 156-fold and 2.9-fold the endogenous FXN level (Figure 5D). This large difference in expression was confirmed by western blot analysis (Figures 5E and 5F). Despite the very high overexpression in Mck mice treated with the optimized vector, no significant accumulation of hFXN precursor (23 kDa) was observed (Figure 5H). While we observed some accumulation of the hFXN intermediary form (19 kDa), the vast majority was processed into the mature form (14 kDa). These results suggest that the majority of the transgenic hFXN protein was targeted to the mitochondria, without severe saturation or impairment of the mitochondria import and processing capacity.33 Of note, the biodistribution and expression of the optimized AAVRh.10-hFXN vector was also quantified in WT mice treated at 2.5 × 1013 (n = 2) or 5 × 1012 vg/kg (n = 1). The vector biodistribution was similar to Mck mice treated at identical doses,15 with, respectively, 0.983 and 0.475 VCN at 2.5 × 1013 vg/kg and 0.078 VCN at 5 × 1012 vg/kg. In contrast, the vector expression was much lower compared to Mck mice treated with the same vector, with, respectively, 5,963 and 3,526 ng/mg at 2.5 × 1013 vg/kg and 355 ng/mg at 5 × 1012 vg/kg.
Figure 8.

Liver Overexpression up to 87-Fold the Normal Level Does Not Result in Similar Cellular and Mitochondria Acute Toxicity
(A–C) The vector biodistribution and expression were assessed in the liver of Mck mice treated at 7 weeks of age with AAVRh.10-CAG-hFXN-HA (n = 6) or AAVRh.10-hFXN (n = 8) at 2.5 × 1013 vg/kg and sacrificed at 15 weeks of age. Individual data points are reported, with mean and SD. Welch’s t test, p values are reported with n.s. p > 0.05. (A) qPCR analysis of VCN. (B) qRT-PCR quantification of the transgene mRNA level normalized to mouse FXN mRNA level. (C) ELISA quantification of FXN reported as ng per mg of total liver protein. Black dotted line represents the normal level of mouse FXN in WT mouse liver (i.e., 49.6 ± 7.8 ng/mg) (n = 5). (D–F) WB analysis of total liver protein extract from Mck mice treated with the non-optimized (n = 3) and optimized (n = 5) AAVRh.10 vector. (D) Immunoblotting against FXN, SDHb, GAPDH, and beta-Tubulin (β-Tub). (E) Relative FXN protein level normalized to GAPDH. (F) Relative SDHb protein level normalized to GAPDH. Individual data points are reported, with mean and SD. Welch’s t test, p values are reported. (G) Representative histological observations of liver tissue section and ultrathin sections from Mck mice treated at 7 weeks of age with AAVRh.10-hFXN (n = 2–3) at 2.5 × 1013 vg/kg, WT treated with NaCl (n = 2) or with optimized AAVRh.10-hFXN at 5 × 1012 vg/kg (n = 1) and sacrificed at 15 weeks of age. The corresponding dose, VCN, and [hFXN] are reported next to each image series. Upper row, H&E staining. Lower row, TEM observations following negative stain.
The expression normalized to the VCN of the optimized vector was 35-fold higher than the expression of the non-optimized vector in the heart of Mck mice (Figure S2). As mentioned above, the expression of the non-optimized AAVRh.10-CAG-hFXN-HA vector was similar in the heart of WT and Mck mice (Figure S2), despite the significant transcriptional and metabolic dysregulations occurring upon mouse FXN depletion.10,12,15,34 In contrast, the expression normalized to the VCN of the optimized AAVRh.10-hFXN vector was on average 5-fold higher in the heart of Mck mice than in WT mice (Figure S2). This increased expression is possibly driven by the strong induction of the ISR and EIF2α phosphorylation in Mck mouse heart,34 which might favorably impact the transcription and translation of the optimized expression cassette, in part through the optimized 5′ UTR and Kozak sequence. In line with this hypothesis, both vectors displayed similar VCN and transcription levels in the liver of Mck mice (Figures 8A and 8B), which is not depleted in FXN and does not present any functional or molecular phenotype. Nonetheless, the optimization of the 5′ UTR and Kozak sequences still resulted in a 48-fold increase of the average hFXN protein levels in the liver (Figures 8C–8E). Furthermore, the optimized AAVRh.10-hFXN vector led to much higher hFXN protein expression in the heart than liver of the same Mck mice (Figures 5D and 8C).
To assess the consequences of hFXN overexpression on the outcome of FA cardiac gene therapy, the cardiac function and morphology of the same Mck mice was assessed by echocardiography (Table S1). Longitudinal echocardiography analyses were performed from 7 to 15 weeks of age (Figures 5I–5L and S4). By 12 weeks of age, Mck mice treated with either vector displayed full correction of the heart function and morphology. However, mice treated with the optimized AAVRh.10-hFXN vector started deteriorating afterward, while sustained full correction was achieved in mice treated with the non-optimized AAVRh.10-CAG-hFXN-HA vector. This deterioration was characterized by a decrease of the LV SF (Figure 5I) and CO/BW (Figure 5J) and an increase of the LV mass (Figure 5K) and LV-ESD (Figure 5L). The LV-EDD was also increased but in a delayed manner compared to the other parameters measured (Figure S4A). Overall, this suggest a primary impairment of the contractile and systolic function of the LV, in line with our previous observation in WT mice treated with the non-optimized AAVRh.10-CAG-hFXN-HA (Figures 2I and 2J; Video S1). The impairment of the cardiac function in Mck mice treated with the optimized vector is supported by the increased gene expression of Nppa and Nppb, indicative of pressure/volume overload (Figure 5M).
Histological analysis of the heart fibrosis and cell infiltration was also performed on heart tissue sections (Figures 6A and 6B). Following H&E stain, Mck mice treated with the optimized vector displayed substantial heart fibrosis, distributed throughout the LV and associated with numerous cell infiltrates (Figure 6A). Mck mice treated with the non-optimized vector displayed a few fibrotic patches. Importantly, heart tissue sections from WT mice treated with the optimized vector at 2.5 × 1013 (n = 2) or 5 × 1012 vg/kg (n = 1) looked similar to heart tissue section from NaCl-injected WT mice (Figure 6A). This suggests the lack of intrinsic toxicity of the optimized vector. To quantify the accumulation of extracellular matrix associated with fibrosis, adjacent heart tissue sections were stained with WGA (Figure 6B). Mck mice treated with the non-optimized vector displayed more labeling than WT mice treated with NaCl, but at a much lower level than Mck mice treated with the optimized vector (Figure 6C). Furthermore, these observations were in agreement with the increased gene expression of Col1a1, Col3a1, and Tgfβ, which are indicative of ongoing fibrosis (Figure 6D), but also of Il1b and Il6, which are indicative of ongoing inflammatory response (Figure 6E). To further investigate the nature of these cell infiltrates, adjacent heart tissue sections were immunolabeled for cluster of differentiation 14 (CD14), a maker of monocytes and macrophages (Figures 6F and S5A); CD45, a general marker of leukocytes including lymphocytes T, B, and monocytes (Figure S5B); and CD3, a general marker of lymphocytes including regulatory T cells and cytotoxic/natural killer (NK) T cells (Figure S5C). Spleen tissue sections were used as positive labeling controls for all these markers (Figures S5A–S5C). In line with the inflammatory and fibrotic response measured, Mck mice treated with the optimized vector showed extensive infiltrations of CD14+ and CD45+ cells, while mice treated with non-optimized vector showed much lower CD14+ and CD45+ labeling. Importantly, WT mice treated with the optimized vector did not show any cell infiltrates, supporting further the lack of intrinsic immunogenicity of the vector or its formulation (Figures S5A–S5C). In contrast, we were unable to identify CD3+ cell infiltrates in the heart tissue sections from animals treated with either vector, which would be expected in case of cytotoxic immune response (NK cells), as shown previously.35,36
Figure 6.

FXN Cardiotoxic Overexpression Is Associated with Acute Heart Fibrosis and Inflammation
(A) Histological analysis of fibrosis and cell infiltrates following H&E staining of heart tissue sections. Analysis of Mck mice treated with the AAVRh.10-hFXN (n = 8) or the AAVRh.10-hFXN-HA vector (n = 6) and WT mice treated with the AAVRh.10-hFXN vector at 2.5 × 1013 (n = 2) or 5 × 1012 (n = 1) vg/kg. The VCN and [FXN] in ng/mg are reported for each animal. (B) WGA staining of the extracellular matrix on heart tissue sections. Analysis of Mck mice injected with AAVRh.10-hFXN (n = 3) or AAVRh.10-CAG-hFXN-HA (n = 6) at 2.5 × 1013 vg/kg, WT mice injected with NaCl (n = 11) and untreated 9-week-old Mck mice (n = 6). (C) Quantification of heart surface labeled with WGA. Brown-Forsythe and Welch ANOVA test, p values are reported with n.s. p > 0.05. (D and E) qRT-PCR analysis of the heart mRNA level normalized to 18S and reported as percentage of NaCl-WT level. Brown-Forsythe and Welch ANOVA test, p values are reported with n.s. p > 0.05. (D) Col1a1, Col3a1, and Tgfb1 mRNA levels. (E) Il1b, Il6, and Tnfα. (F) Representative observations at low and high magnification of heart tissue sections immunolabeled for the monocyte cell marker CD14 (same exposure time). Analysis of 15-week-old NaCl-injected mice (n = 3) and Mck mice treated with AAVRh.10-hFXN (n = 3) or AAVRh.10-CAG-hFXN-HA (n = 3) at 2.5 × 1013 vg/kg. See also Figure S5 for supplementary histological analysis of CD14, CD45, and CD3.
The investigation of the mitochondria function in Mck mice treated with the optimized vector at 2.5 × 1013 vg/kg (Figures 7A, 7B, and S6A) revealed similar alterations as reported above in the Mck and WT mice treated with the non-optimized vector at higher doses (Figures 1A and 3). However, the severity and extent were much higher in mice treated with the optimized vector, in line with the much higher hFXN overexpression across the heart. In Mck mice treated with the optimized vector at 2.5 × 1013 vg/kg, co-labeling and co-localization analysis revealed numerous cardiomyocytes throughout the heart, with very high levels of FXN expression and impaired SDH enzymatic activity (Figure 7A). These hotspots of expression covered most of the heart surface (Figure S6A). In contrast, Mck mice treated with the non-optimized vector were rescued for SDH enzymatic activity throughout the heart, except for a few fibrotic patches (Figure S6B). Interestingly, the protein level of SDH subunit b (SDHb) was significantly decreased in the heart of the Mck mice treated with the optimized vector, when compared with Mck mice treated with the non-optimized vector (Figures 5E and 5G). Moreover, Mck mice treated with the optimized vector also showed impaired NADH enzymatic activity throughout the heart (Figures 7B and S6A), in line with previous observations in WT mice treated with the non-optimized vector at 5 × 1014 vg/kg (Figures 3 and S3). In contrast, the WT mouse treated with the optimized vector at 2.5 × 1013 vg/kg and expressing 9.2-fold the endogenous FXN level displayed normal SDH and NADH enzymatic activity (Figures 7A, 7B, and S6A). The second WT mouse treated similarly but overexpressing 38.7-fold the endogenous level of FXN displayed few and sparse cardiomyocytes, with reduced SDH and NADH enzymatic activity.
Figure 7.

FXN Cardiotoxic Overexpression Is Associated with Impaired Mitochondrial Function and Ultastructure, but Not with Iron Overload
Representative histological observations from the analysis of adjacent heart tissue sections/sample collected from mice treated at 7 weeks of age with the AAVRh.10-hFXN vector and sacrificed at 15 weeks of age. Analysis of Mck mice treated at 2.5 × 1013 vg/kg (n = 3), WT mice treated at 2.5 × 1013 (n = 2) or 5 × 1012 vg/kg (n = 1), NaCl-injected WT mice (n = 2) and untreated 9-week-old Mck mice (n = 2). See also Figure S6 for low-magnification imaging. (A) Co-staining and co-localization analysis of hFXN-HA protein expression and SDH enzymatic activity, respectively, labeled by immunofluorescence and in situ histoenzymatic assay. (B) NADH enzymatic activities assessed by in situ histoenzymatic assay. (C) TEM observations at low and high magnification of the LV myocardium, following negative stain. White arrows indicate non-iron mitochondrial electron-dense bodies; myo, myofibrils; mito, mitochondria; nuc, nucleus. (D) DAB-enhanced Perls labeling of iron deposits.
To assess the cardiomyocytes and mitochondria ultrastructure following the administration of the optimized vector, samples from the LV myocardium of these same Mck (n = 3) and WT (n = 1) mice were collected and observed under TEM after negative staining (Figure 7C). Mck mice treated at 2.5 × 1013 vg/kg displayed severe alterations of cardiomyocyte subcellular organization, with scattered and disordered myofibrils and accumulation of swollen mitochondria, with very few visible cristae (Figure 7C). We also observed electron-dense bodies, inside the matrix of several mitochondria. These did not resemble collapsed cristae or iron deposits but appeared similar to what we observed in WT mice treated with the non-optimized vector at 5 × 1014 vg/kg (Figure 3). The absence of iron accumulation in the heart of these Mck mice was confirmed with DAB-Perls staining (Figure 7D).
Finally, we also analyzed the liver of the same Mck mice treated with the optimized vector at 2.5 × 1013 vg/kg to assess possible toxic effects beyond the heart. The level of hFXN protein in the liver ranged from 1,419 to 4,890 ng/mg when quantified by ELISA assay (Figure 8C). When measured in a distinct cohort of 9-week-old WT C57/B6J mice, the normal level of mouse FXN protein in the liver is 49.6 ± 7.8 ng/mg (n = 5). Thus, Mck mice treated with the optimized vector at 2.5 × 1013 vg/kg express up to 97-fold the endogenous level in the liver. The high expression was confirmed by western blot analysis (Figures 8D and 8E). Despite this high level of overexpression ([hFXN] = 2,565 ± 1,123 ng/mg; n = 8) and the high VCN (31 ± 10; n = 8) measured (Figures 8A–8C), the liver histology of the three mice evaluated appeared all normal following H&E staining, with no obvious sign of fibrosis or cell infiltrations (Figure 8G). The latter would have been expected if an adaptive immune response was raised against the transgene and driving the cardiotoxicity. Moreover, TEM observation of adjacent liver samples collected from the same mice (n = 3) did not reveal any obvious alterations of hepatocyte subcellular organization or alterations of mitochondria ultrastructure and/or biomass (Figure 8G). These results support further the lack of intrinsic toxicity of the AAVRh.10-hFXN vector or its formulation, when administered in Mck or WT mice (Figure 8G). The lack of similar mitochondrial toxicity in the liver was also supported by quantification of SDHb protein level (Figure 8F), which was similar to Mck mice treated with the non-optimized vector and expressing much lower levels of FXN ([hFXN] = 53 ± 14 ng/mg; n = 3). Altogether, these results support the lack of acute liver toxicity, despite very high levels of FXN overexpression, which contrast with the observed cardiotoxicity in the same mice.
Discussion
Here, we showed unequivocally the cardiotoxicity of FXN overexpression when expressed >20-fold the endogenous level and its safety when expressed <9-fold. This was demonstrated in three independent studies, using two different animal models and two different AAVRh.10 vector constructs. The severity of this cardiotoxicity appeared to be proportional to the level of hFXN overexpression but also to the proportion and extent of cardiomyocytes affected throughout the heart. This led to more- or less-severe impairment of LV function and morphology, which then evolved either toward compensated heart hypertrophy or severe impairment of heart function. As demonstrated here, this cardiotoxicity might also compromise the therapeutic outcome of cardiac gene therapy for FA. The identification of this toxic threshold will help define more precisely the safe and efficient range of FXN expression for sustained correction of the FA cardiac phenotype. Our results also suggest that this toxicity might not be generalizable to all organs and/or cell types, as the mouse liver appeared to be tolerant to hFXN overexpression up to 90-fold the normal level.
The [hFXN] values and fold-overexpression numbers reported here correspond to average values reflecting unequal expression of FXN among the cardiomyocytes, some with relatively low or moderate levels and hotspots with very high levels. As shown previously,37 the inhomogeneous distribution of the FXN expression in the heart reflects most likely the inhomogeneous diffusion of the AAV vectors throughout the cardiac vasculature and then into the parenchyma. This is most likely an inherent characteristic of AAV and intravenous administration. Nonetheless, the safe level of hFXN overexpression in the heart (≤9-fold the endogenous level) is in line with previous studies conducted in yeast, mammalian cell lines, transgenic Drosophila, and mouse models constitutively overexpressing FXN. Collectively, these studies reported toxicity when FXN overexpression was higher than 6- to 10-fold the endogenous level18, 19, 20, 21,38,39 but good tolerance for lower levels.16,17,40, 41, 42, 43 Importantly, the current study did not investigate if the expression of the hFXN in the mouse organism would overestimate or underestimate the toxic threshold to be identified if a similar study were to be conducted with the overexpression of the mouse FXN protein. Indeed, the precursor and mature form of the mouse and hFXN present, respectively, 70% and 91% identity. Previous studies have shown that the human ortholog can complement the knockout of the mouse Fxn gene, through the genomic insertion and the constitutive expression of the hFxn gene locus in transgenic mouse models.16,44 These transgenic mice have a normal phenotype and lifespan, therefore suggesting that the hFXN protein has the same functionalities as its mouse ortholog. However, it remained to be shown whether the human and mouse FXN protein have the same affinity and kinetics of activation on the mouse Fe-S cluster assembly complex. It is also unclear if a large excess of FXN could stifle and impair the Fe-S cluster assembly complex and how potential difference in affinity would be impactful. As non-human primates (NHP) have closer physiology to humans and express FXN ortholog with higher identity, we expect toxicology studies conducted in NHP to be more predictive of future clinical trials in FA patients. Indeed, the FXN precursor proteins in cynomolgus and rhesus monkeys are 91% and 92% identical to the hFXN, respectively.
Our results do not support the hypothesis of a potential intrinsic toxicity of the vector construct or production lot used in these mouse studies, as WT mice injected with the same vectors did not show signs of toxicity, unless the hFXN protein level was beyond the toxic threshold. Several studies have shown cytotoxicity or genotoxicity following the administration of high-dose vector, particularly in large animal models.45,46 Here, we have partially ruled out this possibility, as a much lower dose of the optimized AAVRh.10-hFXN vector also induced high levels of FXN expression and cardiotoxicity. In addition, previous mouse studies have shown that similar doses of vector, but with different DNA cargo, were well tolerated up to 5 × 1014 vg/kg, without noticeable cardiotoxicity at long term.47, 48, 49, 50, 51 In the current study, we did not investigate the potential involvement of the innate or adaptive immune response against the vector, transgene, and/or transduced cells.35,36,52,53 It is likely that the treated mice developed a humoral response against the capsid, including neutralizing antibodies, as shown previously in many mouse gene therapy studies.50,51 However, the emergence of antibody against the capsid has not been shown to preclude the long-term expression of transgene or to drive cardiac inflammation but only the re-administration of the same vector.54 Here, we did not explore the potential presence of antibodies against the transgene and, more importantly, the development of a potential cytotoxic cell response targeting the transduced cardiomyocytes. The cytotoxic immune response was shown previously to result in loss of transgene expression in animal models and patients.35,36,52,53 In the present study, the AAVRh.10 vectors were delivered intravenously, leading to broad biodistribution and strong expression in many organs and muscles.12,55 Therefore, we would expect a potential cytotoxic immune response to lead to cell infiltration, not only in the heart but also in all transduced organs, especially the liver, for which the AAVRh.10 capsid has the highest tropism. We will investigate these questions in future studies, as they are critical for future clinical development.
Cardiotoxic hFXN overexpression appeared to be driven by cell autonomous impairment of the mitochondria function and structure, most likely followed by cardiomyocyte cell death, heart fibrosis, and LV contractile dysfunction. Strikingly, the mitochondria toxicity affected NADH and SDH enzymatic activities but not COX. The severe ultrastructure anomalies of the mitochondria most likely contributed to their functional impairment,24 which altogether leads to higher mitochondria biomass. These features are reminiscent of the cardiac, mitochondrial, and biochemical phenotype observed in untreated Mck mice and are hallmarks of FXN deficiency.10,12,15,56 However, it is unclear if hFXN toxic overexpression was associated with or caused by (partial) impairment of Fe-S biogenesis or handling. Indeed, these two cardiac phenotypes differ from one another by the accumulation of mitochondrial iron upon FXN deficiency,22 which was consistently not observed upon hFXN overexpressing in the three mouse studies conducted here. Indeed, cellular iron metabolism and dysregulation are mediated by IRP1, which is a cytosolic Fe-S protein.22 These results are in line with previous overexpression studies conducted in yeast,18 human cells,19 and Drosophila,20 where only a modest increase in labile iron was observed. Future studies will be needed to address the underlying mechanism, through time course analysis of pathological events, including exhaustive biochemical analysis of the Fe-S biosynthesis, handling, and Fe-S enzymes.
These findings would likely apply to all in vivo gene replacement strategies for hFXN, independently of the AAV serotype or other type of viral vector used. Besides gene replacement strategies, this would also be of concern for alternative approaches if presenting the same risk of acute hFXN tissue concentration, either due to their mechanism of action or their delivery modality. This would include synthetic mRNA,57 FXN protein replacement,58,59 and in vivo gene transfer of strong artificial transcription factors.60,61 While the current study was focused on the heart, this mitochondrial toxicity is likely to affect other organs. The dorsal root ganglia, the spinal cord, the cerebellum, and the dentate nucleus are important targets to address the neurological symptoms and would be of particular interest for future studies. The spleen and kidney are major off-target sites for AAV vectors following intravenous delivery and should also be considered carefully. Importantly, the FXN level varies largely among these organs,3,10 most likely along cell-type specific metabolism and abundance of mitochondria. In the present and previous studies,14,15 we have shown that the normal mouse FXN level is 147 ng/mg in the heart, 49 ng/mg in the liver, 12–25 ng/mg in dorsal root ganglia, and 28 ng/mg in the cerebellum. Therefore, we can hypothesize a different therapeutic index for each one of these organs. To ensure the safety of future clinical trials, AAV vectors will need to be designed to manage appropriately their expression profile across these different tissues. This could be achieved with (1) synthetic weak promoter or the endogenous human frataxin promoter,62, 63, 64 (2) vector de-targeting strategies,65,66 and/or (3) the design of expression cassette responsive to negative cellular feedback loop. Leveraging our knowledge of the cellular response to toxic FXN overexpression might help achieve the self-regulation of the vector expression and avoid toxic overexpression. For example, the 3′ UTR sequence could be engineered to be targeted by inhibitory endogenous microRNA, specifically induced by the ISR,67,68 or other cellular stress response such as the oxidative stress response.69,70 In addition, specific routes of drug delivery might also present risk of high local concentration, such as intracardiac injection71 or local delivery in the central nervous system.72,73 Due diligence in the design of appropriate pharmacokinetic and toxicity studies, in relevant large animal models, will be crucial for the development of safe clinical therapeutic protocols. Finally, the identification of biomarkers to monitor subclinical toxicity and manage such events would be very advantageous. In this regard, the current study has identified potential candidates, including FGF21, to be explored in future studies.
In summary, the overexpression of hFXN is safe when ≤9-fold the normal endogenous level but cardiotoxic when greater than 20-fold. The pathological mechanism was partially elucidated and is most likely primed by the alteration of the mitochondria structure and function. Depending on the percentage of cardiomyocytes affected, this resulted in either subclinical or acute cardiotoxicity. The toxic threshold and potential readout for toxicity identified here will support the design of robust and meaningful toxicity studies, for safer FA gene therapy clinical trials.
Materials and Methods
Adeno-Associated Viral Vector Constructs and Production
The AAVRh.10-CAG-hFXN-HA and AAVRh.10-hFXN vectors are described in Figures 5A and 5B and encode, respectively, the human frataxin cDNA, including the mitochondrial targeting sequence, and fused or not in C-terminal to the HA tag. These vectors were produced by triple transfection method in HEK293 cells,74 respectively, at the Vector Core at the University Hospital of Nantes (France) and the Belfer Gene Therapy Core facility at Weill Cornell Medical College. Their respective concentration was measured as 8.7 × 1013 and 1.96 × 1014 vg/mL, by qPCR. Their purity was checked with a combination of the following assay: potential protein contamination by SDS-PAGE and Coomassie Blue Staining, endotoxin contamination with Endosafe-PTS LAL (<0.5 EU/m), sterility by growth in liquid and agar-based test media (no contamination detected at 48 h), and qPCR quantification of residual DNA material from AAV Rep or Adenovirus plasmids.
Animal Procedures
WT C57/B6J mice were supplied by Charles River Laboratory, France. Mck mice were generated in 100% C57BL/6J background, with a conditional deletion of Fxn gene in cardiac and skeletal muscle (Mck-Cre-FxnL3/L) and genotyped, as described previously.10 This results in the complete depletion of frataxin protein in these organs. Note that Mck mice develop a cardiac dysfunction at 5 weeks of age, progressively worsening into heart failure at 7 weeks of age, eventually causing their death around 10 weeks of age.10 Housing animal facility was controlled for temperature and humidity, with a 12-h light/dark cycle and free access to water and a standard rodent chow (D03, SAFE, Villemoisson-sur-Orge, France). All animal procedures and experiments were approved by the local ethical committee for animal care and use (Com’Eth HP/2012/09/27 and 5344). They were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). Five- and seven-week-old animals were anesthetized with isoflurane prior to the retro-orbital injection of vector, in 100 μL bolus. Control littermate or WT mice were injected with equivalent volume of NaCl 0.9% solution. Survival and comorbidities were evaluated daily, and BW was evaluated weekly. Echocardiography was performed with the Sonos 5500 system (Hewlett Packard) with a 15 MHz linear transducer (15L6) or with the Vevo 2100 system (Fujifilm Visualsonics) with a 25–55 MHz transducer, as described previously.15 Briefly, echocardiography was performed under isoflurane anesthesia (0.5%–1.5%) and 0.8 mL/min O2, to maintain the heart rhythm between 450 and 550 bpm. Historical data were used as reference for untreated Mck mouse survival, BW, and cardiac function
Following intraperitoneal injection of ketamine-xylazine and blood sampling, mice were perfused with cooled NaCl 0.9%, and the heart and liver were retrieved quickly.
Briefly, the apex of the heart was sampled in cryotubes and flash frozen in liquid nitrogen for subsequent DNA and RNA extraction and analysis, as described previously.15 Two small pieces from the LV anterior wall (less than 1 mm3) were sampled in Karnovsky’s fixative and processed for TEM morphological analysis. A thin transverse section (around 2 mm thick) was cut at the middle level of the heart for protein analysis. The middle to the base of the heart was embedded in OCT and frozen in isopentane chilled with liquid nitrogen for histological analysis.
The liver medial left lobe was sampled and fixed in PBS 1× formaldehyde 10%, dehydrated, and then embedded in paraffin for histological analysis. Two small pieces from the left lateral lobe (less than 2 mm3) were sampled and fixed in Karnovsky’s fixative for transmitted electron microscopy (TEM) analysis. The remaining lobes—lateral left, medial right, lateral right, and caudate lobes—were sampled in a cryotube and flash frozen in liquid nitrogen for subsequent DNA, RNA, and protein analysis.
The spleen was fixed in PBS 1× formaldehyde 4%, embedded in OCT, and frozen in isopentane chilled with liquid nitrogen for histological analysis.
Histochemistry
Frozen sections from the heart and the spleen, and liver paraffin sections, were collected on Superfrost glass slides. For each staining, the group and number of animals analyzed is reported in the respective figure legend (1–3 tissue sections stained per animal for each histological analysis). Heart and liver tissue sections were stained with H&E. In addition, heart tissue sections were stained with WGA conjugated with Alexa 488 nm, or stained with DAB-enhanced Perls labeling, or used to perform in situ histoenzymatic activity assay for SDH or cytochrome C oxidase, as previously described.15 NADH dehydrogenase in situ histoenzymatic activity was performed on heart tissue sections accordingly to Luna et al.75
Immunofluorescent labeling, with or without SDH co-labeling, was performed as described previously,15 with the following antibodies and dilutions: frataxin (anti-FXN, 1/50, IGBMC; FXN935 and anti-HA, 1/100, Abcam, Ab9110), prohibitin (1/300, Ab28172), Sqstm1 (1/300, 2C11, H00008878-M01, Millipore). The FXN antibodies recognize both the human and mouse FXN, including the precursor, intermediate, and mature forms.
For labeling of CD3-, CD14-, and CD45-positive cells, tissue sections were fixed in 4% paraformaldehyde (PFA) for 5 min, permeabilized in PBS 1× 0.3% Triton X-100 at room temperature (RT) for 10 min, and then blocked with PBS, 1% NGS, 5% BSA, 0.2% Tween (PBS-NBT) for 30 min at RT. Subsequently, tissue sections were incubated overnight (O/N) at 4°C with the rabbit antibody against CD3 (1/100, ab16669, Abcam) or CD14 (1/100, ab183322, Abcam,) or CD45 (1/100, ab10558, Abcam) and afterward with Alexa fluor 488 nm conjugated goat anti-rabbit antibody (1/300, Molecular Probes).
Imaging of the heart, liver, and spleen tissue sections were performed with the Hamamatsu NanoZoomer 2.0 slide scanner.
Electron Microscopy Analysis
The samples were fixed with 2.5% glutaraldehyde and 2.5% PFA in cacodylate buffer (0.1 M [pH 7.4]), washed 30 min in cacodylate buffer, post-fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 h at 4°C, and dehydrated through graded alcohol (50%, 70%, 90%, and 100%) and propylene oxide for 30 min each. Samples were oriented and embedded in Epon 812. Semithin sections were cut at 2 μm with the Leica Ultracut UCT ultramicrotome and stained with 1% toluidine blue and 1% sodium borate. Ultrathin sections were cut at 70 nm and contrasted with uranyl acetate and lead citrate. Electron microscopy observation and image acquisition were performed at 70 kv with the Morgagni 268D electron microscope (FEI Electron Optics, Eindhoven, the Netherlands) and equipped with the Mega View III camera (Soft Imaging System). The bismuth sodium tartrate staining of ferritin was performed as described previously.27,76
DNA, RNA, and Protein Analysis
DNA, RNA, and protein extraction and quantification, as well as cDNA synthesis, were performed as described previously.15
Briefly, absolute quantification of vector biodistribution was performed in triplicate and the qRT-PCR in duplicate, using the Quantitect Sybr Green CR kit (QIAGEN, 1037795) and the Light Cycler 480 II (Roche Biosciences). Primers were used at 0.4 μM final concentration, and their sequences are reported in Table S5. qPCR amplification program: 1 cycle, 15 min 95°C; 50 cycles, 15 s 94°C–30 s 60°C–30 s 72°C; 1 cycle for melting curve. Depending on the expression level of the target genes quantified, cDNA dilutions ranging from 1/50 to 1/2,000 were performed. As working Ct (threshold cycle) values ranged between 12 and 34 cycles, 18S and Hprt were used for normalization, respectively, when the target genes required high and low cDNA dilution for the qPCR assay.
SimpleStep Human Frataxin ELISA Kit (Abcam, ab176112) and SimpleStep Mouse Frataxin ELISA kit (Abcam, ab199078) were used to quantify the heart and liver tissue concentration in human and mouse frataxin, respectively, following manufacturer instructions.
For western blot analysis, between 10 and 30 μg of total protein extract were loaded and migrated in 14% or 4%–12% SDS-PAGE gels (Novex, Nupage 4%–12% Bis-Tris Midi Gels, 26 wells). Following transfer onto nitrocellulose membranes (Amersham Protran 0.45 μm NC), Red Ponceau staining (Sigma Aldrich, P7170-1L), and blocking, the membranes were immunoblotted O/N at 4°C with the following antibodies and dilution: mouse anti-frataxin (1/10,000, 4F9, IGBMC), mouse anti-complex II 30 kDa subunit SDHb (1/10,000, 21A11AE7, Novex), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (1/30,000, MAB374, Millipore), and/or mouse anti-beta-tubulin (1/10,000, IGBMC). After incubation for 2 h at 4°C with goat anti-mouse IgG H+L (1/10,000, 115-035-146, Jackson Immunoresearch), the signal was revealed using the SuperSignal West Dura Extended Duration Substrate (ThermoSienctific, 34075) and imaged with the Amersham Imager 600 (GE Healthcare).
Statistical, Correlation, and Regression Analysis
Unless otherwise specified, data are reported as mean ± standard deviation (SD). Statistical analyses were carried out using GraphPad Prism 8.3 (GraphPad Software, La Jolla CA, USA), and methods are described in the figure legends.
Author Contributions
B.B. and H.P. conceived the project. B.B. conducted and analyzed the experiments. L.R. managed mouse production. N.M. performed electron microscopy. L.M. provided logical support for echocardiography. B.B. and H.P. wrote the manuscript.
Conflicts of Interest
H.P. is the owner of patents pertaining to cardiac and neurological gene therapy methods for the treatment of FA (WO2016150964A1, EP2950824A1, US20150313969A1, US9066966B2, and CA2900135A1) and is one of the scientific co-founders of AAVlife SAS. H.P. is an advisor and beneficiary of sponsored research agreement from Adverum Biotechnologies and Voyager Therapeutics. B.B. is currently employed by Adverum Biotechnologies, although the work was done previous to employment. H.P. and B.B. own equities in Adverum Biotechnologies. All other authors declare no competing financial interests.
Acknowledgments
We thank Dr. Hugues Jacob, Karim Hnia, Nicolas Charlet-Berguerand, and Pr. Jean-Louis Mandel for scientific discussions and Dr. Wyatt Delphino and Mehdi Gasmi for critical review of the manuscript. We thank Dr. Ronald G. Crystal (Department of Genetic Medicine, Weill Cornell Medicine, NY, USA) for the production of the optimized construct and vector and Dr. Mustapha Oulad-Abdelghani for providing the 4F9 mouse monoclonal antibodies against FXN. We thank Dr. Tania Sorg, Ghina Bou-About, Josiane Hergueux, the mouse clinic institute phenotyping platform, and Dr. Luc Dupuis for logistical and technical assistance. This work was supported by the Lefoulond Delalande Foundation–Institut de France (post-doctoral research fellowships, 2016); the Friedreich Ataxia Research Alliance (Keith Michael Andrus Cardiac Research Award, 2016); the French Muscular Dystrophy Association AFM-Téléthon (research grant year 2014); and AAVLIFE SAS (sponsored research agreement years 2014–2016). This study was also supported by the grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissement d’Avenir ANR-10-IDEX-0002-02.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.08.018.
Contributor Information
Brahim Belbellaa, Email: bbelbellaa@adverum.com.
Hélène Puccio, Email: helene.puccio@inserm.fr.
Supplemental Information
References
- 1.Bürk K. Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias. 2017;4:4. doi: 10.1186/s40673-017-0062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsou A.Y., Paulsen E.K., Lagedrost S.J., Perlman S.L., Mathews K.D., Wilmot G.R., Ravina B., Koeppen A.H., Lynch D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011;307:46–49. doi: 10.1016/j.jns.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 3.Campuzano V., Montermini L., Moltò M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 4.Saveliev A., Everett C., Sharpe T., Webster Z., Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature. 2003;422:909–913. doi: 10.1038/nature01596. [DOI] [PubMed] [Google Scholar]
- 5.Maio N., Jain A., Rouault T.A. Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 2020;55:34–44. doi: 10.1016/j.cbpa.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheftel A., Stehling O., Lill R. Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab. 2010;21:302–314. doi: 10.1016/j.tem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Mayr J.A., Feichtinger R.G., Tort F., Ribes A., Sperl W. Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 2014;37:553–563. doi: 10.1007/s10545-014-9705-8. [DOI] [PubMed] [Google Scholar]
- 8.Leimkühler S. Shared function and moonlighting proteins in molybdenum cofactor biosynthesis. Biol. Chem. 2017;398:1009–1026. doi: 10.1515/hsz-2017-0110. [DOI] [PubMed] [Google Scholar]
- 9.Braymer J.J., Lill R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017;292:12754–12763. doi: 10.1074/jbc.R117.787101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puccio H., Simon D., Cossée M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 11.Chandran V., Gao K., Swarup V., Versano R., Dong H., Jordan M.C., Geschwind D.H. Inducible and reversible phenotypes in a novel mouse model of Friedreich’s Ataxia. eLife. 2017;6:e30054. doi: 10.7554/eLife.30054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perdomini M., Belbellaa B., Monassier L., Reutenauer L., Messaddeq N., Cartier N., Crystal R.G., Aubourg P., Puccio H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014;20:542–547. doi: 10.1038/nm.3510. [DOI] [PubMed] [Google Scholar]
- 13.Gérard C., Xiao X., Filali M., Coulombe Z., Arsenault M., Couet J., Li J., Drolet M.C., Chapdelaine P., Chikh A., Tremblay J.P. An AAV9 coding for frataxin clearly improved the symptoms and prolonged the life of Friedreich ataxia mouse models. Mol. Ther. Methods Clin. Dev. 2014;1:14044. doi: 10.1038/mtm.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piguet F., de Montigny C., Vaucamps N., Reutenauer L., Eisenmann A., Puccio H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018;26:1940–1952. doi: 10.1016/j.ymthe.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belbellaa B., Reutenauer L., Monassier L., Puccio H. Correction of half the cardiomyocytes fully rescue Friedreich ataxia mitochondrial cardiomyopathy through cell-autonomous mechanisms. Hum. Mol. Genet. 2019;28:1274–1285. doi: 10.1093/hmg/ddy427. [DOI] [PubMed] [Google Scholar]
- 16.Pook M.A., Al-Mahdawi S., Carroll C.J., Cossée M., Puccio H., Lawrence L., Clark P., Lowrie M.B., Bradley J.L., Cooper J.M. Rescue of the Friedreich’s ataxia knockout mouse by human YAC transgenesis. Neurogenetics. 2001;3:185–193. doi: 10.1007/s100480100118. [DOI] [PubMed] [Google Scholar]
- 17.Miranda C.J., Santos M.M., Ohshima K., Tessaro M., Sequeiros J., Pandolfo M. Frataxin overexpressing mice. FEBS Lett. 2004;572:281–288. doi: 10.1016/j.febslet.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Wang Y., Marcus S., Busenlehner L.S. The role of frataxin in fission yeast iron metabolism: implications for Friedreich’s ataxia. Biochim. Biophys. Acta. 2014;1840:3022–3033. doi: 10.1016/j.bbagen.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 19.Vannocci T., Notario Manzano R., Beccalli O., Bettegazzi B., Grohovaz F., Cinque G., de Riso A., Quaroni L., Codazzi F., Pastore A. Adding a temporal dimension to the study of Friedreich’s ataxia: the effect of frataxin overexpression in a human cell model. Dis. Model. Mech. 2018;11:dmm032706. doi: 10.1242/dmm.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edenharter O., Clement J., Schneuwly S., Navarro J.A. Overexpression of Drosophila frataxin triggers cell death in an iron-dependent manner. J. Neurogenet. 2017;31:189–202. doi: 10.1080/01677063.2017.1363200. [DOI] [PubMed] [Google Scholar]
- 21.Navarro J.A., Llorens J.V., Soriano S., Botella J.A., Schneuwly S., Martínez-Sebastián M.J., Moltó M.D. Overexpression of human and fly frataxins in Drosophila provokes deleterious effects at biochemical, physiological and developmental levels. PLoS ONE. 2011;6:e21017. doi: 10.1371/journal.pone.0021017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richardson D.R., Huang M.L., Whitnall M., Becker E.M., Ponka P., Suryo Rahmanto Y. The ins and outs of mitochondrial iron-loading: the metabolic defect in Friedreich’s ataxia. J. Mol. Med. (Berl.) 2010;88:323–329. doi: 10.1007/s00109-009-0565-x. [DOI] [PubMed] [Google Scholar]
- 23.Rich P.R., Maréchal A. The mitochondrial respiratory chain. Essays Biochem. 2010;47:1–23. doi: 10.1042/bse0470001. [DOI] [PubMed] [Google Scholar]
- 24.Dudkina N.V., Kouril R., Peters K., Braun H.P., Boekema E.J. Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta. 2010;1797:664–670. doi: 10.1016/j.bbabio.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Koeppen A.H., Ramirez R.L., Becker A.B., Bjork S.T., Levi S., Santambrogio P., Parsons P.J., Kruger P.C., Yang K.X., Feustel P.J., Mazurkiewicz J.E. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS ONE. 2015;10:e0116396. doi: 10.1371/journal.pone.0116396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward P.G.D., Harding I.H., Close T.G., Corben L.A., Delatycki M.B., Storey E., Georgiou-Karistianis N., Egan G.F. Longitudinal evaluation of iron concentration and atrophy in the dentate nuclei in friedreich ataxia. Mov. Disord. 2019;34:335–343. doi: 10.1002/mds.27606. [DOI] [PubMed] [Google Scholar]
- 27.Michael S., Petrocine S.V., Qian J., Lamarche J.B., Knutson M.D., Garrick M.D., Koeppen A.H. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum. 2006;5:257–267. doi: 10.1080/14734220600913246. [DOI] [PubMed] [Google Scholar]
- 28.Koeppen A.H. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011;303:1–12. doi: 10.1016/j.jns.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arosio P., Elia L., Poli M. Ferritin, cellular iron storage and regulation. IUBMB Life. 2017;69:414–422. doi: 10.1002/iub.1621. [DOI] [PubMed] [Google Scholar]
- 30.Tatsuta T., Model K., Langer T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Planavila A., Redondo-Angulo I., Villarroya F. FGF21 and Cardiac Physiopathology. Front. Endocrinol. (Lausanne) 2015;6:133. doi: 10.3389/fendo.2015.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montero R., Yubero D., Villarroya J., Henares D., Jou C., Rodríguez M.A., Ramos F., Nascimento A., Ortez C.I., Campistol J. GDF-15 Is Elevated in Children with Mitochondrial Diseases and Is Induced by Mitochondrial Dysfunction. PLoS ONE. 2016;11:e0148709. doi: 10.1371/journal.pone.0148709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmucker S., Argentini M., Carelle-Calmels N., Martelli A., Puccio H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008;17:3521–3531. doi: 10.1093/hmg/ddn244. [DOI] [PubMed] [Google Scholar]
- 34.Huang M.L., Sivagurunathan S., Ting S., Jansson P.J., Austin C.J., Kelly M., Semsarian C., Zhang D., Richardson D.R. Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: activation of the integrated stress response, eIF2α phosphorylation, and the induction of downstream targets. Am. J. Pathol. 2013;183:745–757. doi: 10.1016/j.ajpath.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 35.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 36.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E., Ragni M.V., Manno C.S., Sommer J., Jiang H. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 37.Prasad K.M., Xu Y., Yang Z., Acton S.T., French B.A. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18:43–52. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vannocci T., Dinarelli S., Girasole M., Pastore A., Longo G. A new tool to determine the cellular metabolic landscape: nanotechnology to the study of Friedreich’s ataxia. Sci. Rep. 2019;9:19282. doi: 10.1038/s41598-019-55799-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Llorens J.V., Navarro J.A., Martínez-Sebastián M.J., Baylies M.K., Schneuwly S., Botella J.A., Moltó M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007;21:333–344. doi: 10.1096/fj.05-5709com. [DOI] [PubMed] [Google Scholar]
- 40.Seguin A., Bayot A., Dancis A., Rogowska-Wrzesinska A., Auchère F., Camadro J.M., Bulteau A.L., Lesuisse E. Overexpression of the yeast frataxin homolog (Yfh1): contrasting effects on iron-sulfur cluster assembly, heme synthesis and resistance to oxidative stress. Mitochondrion. 2009;9:130–138. doi: 10.1016/j.mito.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Ristow M., Pfister M.F., Yee A.J., Schubert M., Michael L., Zhang C.Y., Ueki K., Michael M.D., 2nd, Lowell B.B., Kahn C.R. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc. Natl. Acad. Sci. USA. 2000;97:12239–12243. doi: 10.1073/pnas.220403797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shoichet S.A., Bäumer A.T., Stamenkovic D., Sauer H., Pfeiffer A.F., Kahn C.R., Müller-Wieland D., Richter C., Ristow M. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum. Mol. Genet. 2002;11:815–821. doi: 10.1093/hmg/11.7.815. [DOI] [PubMed] [Google Scholar]
- 43.Runko A.P., Griswold A.J., Min K.T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008;582:715–719. doi: 10.1016/j.febslet.2008.01.046. [DOI] [PubMed] [Google Scholar]
- 44.Sarsero J.P., Li L., Holloway T.P., Voullaire L., Gazeas S., Fowler K.J., Kirby D.M., Thorburn D.R., Galle A., Cheema S. Human BAC-mediated rescue of the Friedreich ataxia knockout mutation in transgenic mice. Mamm. Genome. 2004;15:370–382. doi: 10.1007/s00335-004-3019-3. [DOI] [PubMed] [Google Scholar]
- 45.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nault J.-C., Datta S., Imbeaud S., Franconi A., Mallet M., Couchy G., Letouzé E., Pilati C., Verret B., Blanc J.F. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015;47:1187–1193. doi: 10.1038/ng.3389. [DOI] [PubMed] [Google Scholar]
- 47.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., Kaspar B.K. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Bočkor L., Bortolussi G., Iaconcig A., Chiaruttini G., Tiribelli C., Giacca M., Benvenuti F., Zentilin L., Muro A.F. Repeated AAV-mediated gene transfer by serotype switching enables long-lasting therapeutic levels of hUgt1a1 enzyme in a mouse model of Crigler-Najjar Syndrome Type I. Gene Ther. 2017;24:649–660. doi: 10.1038/gt.2017.75. [DOI] [PubMed] [Google Scholar]
- 49.Xu R., Jia Y., Zygmunt D.A., Martin P.T. rAAVrh74.MCK.GALGT2 Protects against Loss of Hemodynamic Function in the Aging mdx Mouse Heart. Mol. Ther. 2019;27:636–649. doi: 10.1016/j.ymthe.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zygmunt D.A., Xu R., Jia Y., Ashbrook A., Menke C., Shao G., Yoon J.H., Hamilton S., Pisharath H., Bolon B., Martin P.T. rAAVrh74.MCK.GALGT2 Demonstrates Safety and Widespread Muscle Glycosylation after Intravenous Delivery in C57BL/6J Mice. Mol. Ther. Methods Clin. Dev. 2019;15:305–319. doi: 10.1016/j.omtm.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manso A.M., Hashem S.I., Nelson B.C., Gault E., Soto-Hermida A., Villarruel E., Brambatti M., Bogomolovas J., Bushway P.J., Chen C. Systemic AAV9.LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease. Sci. Transl. Med. 2020;12:eaax1744. doi: 10.1126/scitranslmed.aax1744. [DOI] [PubMed] [Google Scholar]
- 52.Zhu J., Huang X., Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest. 2009;119:2388–2398. doi: 10.1172/JCI37607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ashley S.N., Somanathan S., Giles A.R., Wilson J.M. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell. Immunol. 2019;346:103997. doi: 10.1016/j.cellimm.2019.103997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leborgne C., Barbon E., Alexander J.M., Hanby H., Delignat S., Cohen D.M., Collaud F., Muraleetharan S., Lupo D., Silverberg J. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020;26:1096–1101. doi: 10.1038/s41591-020-0911-7. [DOI] [PubMed] [Google Scholar]
- 55.Hu C., Busuttil R.W., Lipshutz G.S. RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J. Gene Med. 2010;12:766–778. doi: 10.1002/jgm.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagner G.R., Pride P.M., Babbey C.M., Payne R.M. Friedreich’s ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum. Mol. Genet. 2012;21:2688–2697. doi: 10.1093/hmg/dds095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nabhan J.F., Wood K.M., Rao V.P., Morin J., Bhamidipaty S., LaBranche T.P., Gooch R.L., Bozal F., Bulawa C.E., Guild B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016;6:20019. doi: 10.1038/srep20019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vyas P.M., Tomamichel W.J., Pride P.M., Babbey C.M., Wang Q., Mercier J., Martin E.M., Payne R.M. A TAT-frataxin fusion protein increases lifespan and cardiac function in a conditional Friedreich’s ataxia mouse model. Hum. Mol. Genet. 2012;21:1230–1247. doi: 10.1093/hmg/ddr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Britti E., Delaspre F., Feldman A., Osborne M., Greif H., Tamarit J., Ros J. Frataxin-deficient neurons and mice models of Friedreich ataxia are improved by TAT-MTScs-FXN treatment. J. Cell. Mol. Med. 2018;22:834–848. doi: 10.1111/jcmm.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chapdelaine P., Gérard C., Sanchez N., Cherif K., Rousseau J., Ouellet D.L., Jauvin D., Tremblay J.P. Development of an AAV9 coding for a 3XFLAG-TALEfrat#8-VP64 able to increase in vivo the human frataxin in YG8R mice. Gene Ther. 2016;23:606–614. doi: 10.1038/gt.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cherif K., Gérard C., Rousseau J., Ouellet D.L., Chapdelaine P., Tremblay J.P. Increased Frataxin Expression Induced in Friedreich Ataxia Cells by Platinum TALE-VP64s or Platinum TALE-SunTag. Mol. Ther. Nucleic Acids. 2018;12:19–32. doi: 10.1016/j.omtn.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puspasari N., Rowley S.M., Gordon L., Lockhart P.J., Ioannou P.A., Delatycki M.B., Sarsero J.P. Long range regulation of human FXN gene expression. PLoS ONE. 2011;6:e22001. doi: 10.1371/journal.pone.0022001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarsero J.P., Holloway T.P., Li L., Finkelstein D.I., Ioannou P.A. Rescue of the Friedreich ataxia knockout mutation in transgenic mice containing an FXN-EGFP genomic reporter. PLoS ONE. 2014;9:e93307. doi: 10.1371/journal.pone.0093307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J., Li Y., Wang J., Gonzalez T.J., Asokan A., Napierala J.S., Napierala M. Defining Transcription Regulatory Elements in the Human Frataxin Gene: Implications for Gene Therapy. Hum. Gene Ther. 2020;31:839–851. doi: 10.1089/hum.2020.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang D., Li S., Gessler D.J., Xie J., Zhong L., Li J., Tran K., Van Vliet K., Ren L., Su Q. A Rationally Engineered Capsid Variant of AAV9 for Systemic CNS-Directed and Peripheral Tissue-Detargeted Gene Delivery in Neonates. Mol. Ther. Methods Clin. Dev. 2018;9:234–246. doi: 10.1016/j.omtm.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie J., Xie Q., Zhang H., Ameres S.L., Hung J.-H., Su Q., He R., Mu X., Seher Ahmed S., Park S. MicroRNA-regulated, systemically delivered rAAV9: a step closer to CNS-restricted transgene expression. Mol. Ther. 2011;19:526–535. doi: 10.1038/mt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chitnis N.S., Pytel D., Bobrovnikova-Marjon E., Pant D., Zheng H., Maas N.L., Frederick B., Kushner J.A., Chodosh L.A., Koumenis C. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol. Cell. 2012;48:353–364. doi: 10.1016/j.molcel.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye H., Xie M., Xue S., Hamri G.C.-E., Yin J., Zulewski H., Fusseneggar M. Self-adjusting synthetic gene circuit for correcting insulin resistance. Nat. Biomed. Eng. 2016;1:0005. doi: 10.1038/s41551-016-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shah N.M., Rushworth S.A., Murray M.Y., Bowles K.M., MacEwan D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget. 2013;4:1130–1142. doi: 10.18632/oncotarget.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun W., Yi Y., Xia G., Zhao Y., Yu Y., Li L., Hua C., He B., Yang B., Yu C. Nrf2-miR-129-3p-mTOR Axis Controls an miRNA Regulatory Network Involved in HDACi-Induced Autophagy. Mol. Ther. 2019;27:1039–1050. doi: 10.1016/j.ymthe.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Merentie M., Lottonen-Raikaslehto L., Parviainen V., Huusko J., Pikkarainen S., Mendel M., Laham-Karam N., Kärjä V., Rissanen R., Hedman M., Ylä-Herttuala S. Efficacy and safety of myocardial gene transfer of adenovirus, adeno-associated virus and lentivirus vectors in the mouse heart. Gene Ther. 2016;23:296–305. doi: 10.1038/gt.2015.114. [DOI] [PubMed] [Google Scholar]
- 72.Hordeaux J., Hinderer C., Goode T., Buza E.L., Bell P., Calcedo R., Richman L.K., Wilson J.M. Toxicology Study of Intra-Cisterna Magna Adeno-Associated Virus 9 Expressing Iduronate-2-Sulfatase in Rhesus Macaques. Mol. Ther. Methods Clin. Dev. 2018;10:68–78. doi: 10.1016/j.omtm.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hordeaux J., Hinderer C., Goode T., Katz N., Buza E.L., Bell P., Calcedo R., Richman L.K., Wilson J.M. Toxicology Study of Intra-Cisterna Magna Adeno-Associated Virus 9 Expressing Human Alpha-L-Iduronidase in Rhesus Macaques. Mol. Ther. Methods Clin. Dev. 2018;10:79–88. doi: 10.1016/j.omtm.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rabinowitz J.E., Rolling F., Li C., Conrath H., Xiao W., Xiao X., Samulski R.J. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luna L.G. American Histolabs; 1992. Histopathologic Methods and Color Atlas of Special Stains and Tissue Artifacts. [Google Scholar]
- 76.Ainsworth S.K., Karnovsky M.J. An ultrastructural staining method for enhancing the size and electron opacity of ferritin in thin sections. J. Histochem. Cytochem. 1972;20:225–229. doi: 10.1177/20.3.225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.